

Abstract
Key points
Gap junctional electrical coupling between neurons of the reticular thalamic nucleus (RTN) is critical for hypersynchrony in the thalamo‐cortical network.
This study investigates the role of electrical coupling in pathological rhythmogenesis in RTN neurons in a rat model of absence epilepsy.
Rhythmic activation resulted in a Ca2+‐dependent short‐term depression (STD) of electrical coupling between pairs of RTN neurons in epileptic rats, but not in RTN of a non‐epileptic control strain.
Pharmacological blockade of gap junctions in RTN in vivo induced a depression of seizure activity.
The STD of electrical coupling represents a mechanism of Ca2+ homeostasis in RTN aimed to counteract excessive synchronization.
Abstract
Neurons in the reticular thalamic nucleus (RTN) are coupled by electrical synapses, which play a major role in regulating synchronous activity. This study investigates electrical coupling in RTN neurons from a rat model of childhood absence epilepsy, genetic absence epilepsy rats from Strasbourg (GAERS), compared with a non‐epileptic control (NEC) strain, to assess the impact on pathophysiological rhythmogenesis. Whole‐cell recordings were obtained from pairs of RTN neurons of GAERS and NEC in vitro. Coupling was determined by injection of hyperpolarizing current steps in one cell and monitoring evoked voltage responses in both activated and coupled cell. The coupling coefficient (cc) was compared under resting condition, during pharmacological interventions and repeated activation using a series of current injections. The effect of gap junctional coupling on seizure expression was investigated by application of gap junctional blockers into RTN of GAERS in vivo. At resting conditions, cc did not differ between GAERS and NEC. During repeated activation, cc declined in GAERS but not in NEC. This depression in cc was restored within 25 s and was prevented by intracellular presence of BAPTA in the activated but not in the coupled cell. Local application of gap junctional blockers into RTN of GAERS in vivo resulted in a decrease of spike wave discharge (SWD) activity. Repeated activation results in a short‐term depression (STD) of gap junctional coupling in RTN neurons of GAERS, depending on intracellular Ca2+ mechanisms in the activated cell. As blockage of gap junctions in vivo results in a decrease of SWD activity, the STD observed in GAERS is considered a compensatory mechanism, aimed to dampen SWD activity.
Key points
Gap junctional electrical coupling between neurons of the reticular thalamic nucleus (RTN) is critical for hypersynchrony in the thalamo‐cortical network.
This study investigates the role of electrical coupling in pathological rhythmogenesis in RTN neurons in a rat model of absence epilepsy.
Rhythmic activation resulted in a Ca2+‐dependent short‐term depression (STD) of electrical coupling between pairs of RTN neurons in epileptic rats, but not in RTN of a non‐epileptic control strain.
Pharmacological blockade of gap junctions in RTN in vivo induced a depression of seizure activity.
The STD of electrical coupling represents a mechanism of Ca2+ homeostasis in RTN aimed to counteract excessive synchronization.
Abbreviations
- CIN
input capacitance
- Cx36
connexin 36
- cc
coupling coefficient
- GAERS
genetic absence epileptic rats from Strasbourg
- LFP
local field potential
- LTD
long‐term depression
- LTS
low threshold spike
- mGluR
metabotropic glutamate receptor
- NEC
non‐epileptic control rats
- RIN
input resistance
- RMP
resting membrane potential
- RTN
reticular thalamic nucleus
- STD
short‐term depression
- SWD
spike wave discharge
- VB
ventro‐basal complex of the thalamus
- WAG/Rij
Wistar Albino Glaxo rat from Rijswijk
Introduction
One of the most prototypical type of secondary generalized seizures are the bilateral synchronous spike wave discharges (SWDs), as diagnosed in childhood absence epilepsy, a non‐convulsive type of epilepsy seen in up to 10% of children with epilepsy. If untreated, SWDs spontaneously and unpredictably arise up to several hundred times per day, concomitant with a sudden drop in conscious experience and responsiveness of the child (Panayiotopoulos, 1999; Crunelli & Leresche, 2002). While there is a wide agreement that SWDs are generated within the cortico‐thalamo‐cortical synaptic network, seizure origin, either in cortex or in thalamus, is highly debated and the (network) mechanisms of SWD generation and maintenance are still poorly understood. In general, it is assumed that the generation of hypersynchronous seizures results from an interplay between several microcircuits and that, next to the ictal onset zone, there are so‐called ‘choke point’ or ‘hubs’, also outside the seizure onset zone, which are of crucial relevance for seizure generation. Specific targeting of such ‘choke points’ might open new ways for the treatment of epileptic seizures (Paz & Huguenard, 2015). For SWDs, one such ‘choke point’ might be the reticular thalamic nucleus (RTN). Situated as a shell‐like structure between thalamus and cortex, it receives excitatory input both from axon collaterals of thalamo‐cortical relay neurons and from axon collaterals of cortico‐thalamic neurons, projecting back from cortical layer 6 to the first‐order thalamic nuclei (Guillery & Harting, 2003; Pinault, 2004). In return, the RTN sends GABAergic projections onto thalamo‐cortical relay cells, enabling the network to act as a potential synchronizer of the seizure activity (Pinault & O'Brien, 2005; Huguenard & McCormick, 2007). A convergent line of evidence obtained in the well validated rat models of absence epilepsy, genetic absence epileptic rats from Strasbourg (GAERS) and Wistar Albino Glaxo rat from Rijswijk (WAG/Rij) rats (Depaulis & van Luijtelaar, 2006), indicates that a focal zone situated in the somatosensory cortex triggers input (i.e. activity from the seizure onset zone) to various parts of the thalamus, including the posterior nucleus and the sensory regions. Importantly, cortical input evokes both excitation and inhibition during SWDs, the latter resulting from feed‐forward synaptic inhibition of thalamo‐cortical cells via the RTN (Steriade, 1998; Meeren et al. 2002; Pinault & O'Brien, 2005; Huguenard & McCormick, 2007). Likewise, in children with childhood absence epilepsy, a focal cortical onset zone situated in orbito/medial frontal and medial/parietal cortical regions has been described (Westmijse et al. 2009; Bai et al. 2010) which interacts with the reciprocally connected thalamus (Moeller et al. 2010; Gupta et al. 2011; Tenney et al. 2013).
Communication within the RTN occurs to a great extent via electrical synapses (gap junctions), small (diameter of 1.2 nm) aqueous channels, which are homomerically composed from the transmembrane molecule connexin 36 (Cx36) (Landisman et al. 2002; Sohl et al. 2005). Bridging the membranes of neighbouring cells, these are thought to quickly transmit excitation from an active (depolarized) neuron to its post‐synaptic neighbour (Long et al. 2004) and, by this, to synchronize their activity. As a result of intra‐RTN synchrony, the output to thalamo‐cortical relay cells is synchronized. Activity of gap junctions has thus been suggested to be of crucial importance for regulating synchronicity of thalamo‐cortical neurons and networks (Slaght et al. 2002; Fuentealba & Steriade, 2005; Sohl et al. 2005; Huguenard & McCormick, 2007). Pathophysiological changes in the communication between reticular thalamic gap junctions, in turn, might render an individual more prone to develop abnormal synchronized activity such as SWDs (Paz & Huguenard, 2015).
In the current work, gap junctional coupling within the RTN was compared between GAERS, a well‐validated genetic rat model of absence epilepsy, and a non‐epileptic control strain (NEC) with the same genetic background (Marescaux et al. 1992; Depaulis & van Luijtelaar, 2006). Gap junctional coupling has been shown to be activity‐dependent (Haas et al. 2011; Haas & Landisman, 2012). Therefore, we analysed the properties of basal coupling under resting conditions using paired whole‐cell recordings of RTN neurons in vitro, as well as coupling during strong repetitive stimulation, mimicking a pattern of activation occurring during SWDs (Coulon et al. 2009). Membrane potential, proton gradient and intracellular Ca2+ influence gap junctional coupling in cells (Burr et al. 2005). Both intrinsic and synaptic activity in RTN cells result in changes in intracellular Ca2+ concentration (Kostyuk & Verkhratsky, 1994; Coulon et al. 2009; Neyer et al. 2016) prompting us to assess the calcium dependency of gap junctional coupling. In a final set of experiments, the relevance of changes in gap junctional coupling within the RTN for the expression of SWDs was investigated in vivo by local application of gap junction blockers.
Methods
Animals
GAERS and NEC rats of postnatal day (P)12–14 were used as experimental subjects for the in vitro studies (n = 51 NEC and 40 GAERS) and at an age of 3 month for the in vivo studies (n = 28).
Animals were born and raised at the Institute of Physiology I, Westfälische Wilhelms‐University, Münster, under standard laboratory conditions with a 12–12 h dark–light cycle and ad libitum access to water and food.
All experimental procedures were performed in accordance with the guidelines of the council of the European Union of 22 September 2010 (2010/63/EU) and approved by local authorities (review board institution: Landesamt für Natur, Umwelt und Verbraucherschutz Nordrhein‐Westfalen; approval ID numbers: 8.87‐51.05.20.10.117, 84‐02.04.2011.A177, 87‐51.04.2010.A322).
Tissue preparation
Animals were anaesthetized with a mixture of isoflurane (4%) and O2 and decapitated. The brain was quickly removed, a block containing RTN was placed in an ice‐cold oxygenated saline solution (composition in mm: sucrose, 200; glucose, 10; PIPES, 20; KCl, 2.5; MgSO4, 10; CaCl2, 0.5; adjusted to a pH of 7.35 with NaOH) and coronal brain slices of 350 μm thickness were obtained using a vibratome (Pelco Vibratome Series 1000 Sectioning System TPI Inc., St. Louis, MO, USA and Leica VT 1200, Leica, Wetzlar, Germany). Slices were incubated for 20 min at 30°C and for 40 min at room temperature in an incubation solution (composition in mm: NaCl, 125; KCl, 2.5; NaH2PO4, 1.25; NaHCO3, 24; MgSO4, 2; CaCl2, 2; glucose, 10; pH of 7.35 was adjusted by gassing with 5% CO2 and 95% O2). For electrophysiological experiments, individual slices were transferred to a bathing chamber and continuously superfused with artificial cerebrospinal solution at room temperature (composition in mm: NaCl, 120; KCl, 2.5; NaH2PO3, 1.25; NaHCO3, 22; glucose, 20; CaCl2, 2; MgSO4, 2; pH of 7.35, adjusted with CO2).
Electrophysiological recordings in vitro
Electrophysiological experiments were performed under whole‐cell current‐clamp conditions. Patch pipettes were pulled from borosilicate glass capillaries (GC150TF‐10, Clark Electromedical Instruments, Pangbourne, UK) on a vertical puller (L/M‐3P‐A, List‐Medical, Champaign, IL, USA) and connected to an EPC‐10 amplifier (HEKA Electronics, Lambrecht/Pfalz, Germany). Intracellular solutions were composed of (in mm): NaCl, 10; potassium gluconate, 105; K3‐citrate, 20; Hepes, 10; EGTA, 0.25; MgCl2, 0.5; Mg‐ATP, 3; Na‐GTP 0.5; or: NaCl, 10; potassium gluconate, 88; K3‐citrate, 20; Hepes, 10; K‐BAPTA, 3; Mg‐ATP, 3; Na‐GTP, 0.5; phosphocreatine, 15; MgCl2, 1; CaCl2, 0.5; pH adjusted to 7.25. Electrode resistances were 2–3 MΩ. Series resistances were 5–20 MΩ and monitored during experiments, and data were analysed only from recordings in which series resistance varied by less than 20%.
All electrophysiological recordings were controlled by the software PatchMaster (HEKA Electronics), installed on an IBM‐compatible computer.
Resting membrane potential (RMP) was quickly assessed after breaking the membrane patch, and cells showing an RMP more positive than −50 mV were discarded. The voltage shift occurring during injection of a negative current step at −60 mV (−250 pA; duration 1 s) was used to calculate the input resistance, RIN. The input capacitance CIN was calculated from τ=RIN x CIN (τ = the membrane time constant was obtained by a mono‐exponential fit of the hyperpolarizing voltage deflection).
The RTN was identified by its position and structure, and, after gaining whole‐cell access, RTN neurons were identified by their electrophysiological characteristics as described previously (Coulon et al. 2009). Simultaneous recordings were obtained from two neighbouring RTN neurons, infrared‐visualized through an upright microscope (BX51WI, Olympus, Hamburg, Germany).
Assessment of electrical coupling in vitro
Coupling of the two cells at resting conditions was assessed by holding the cells at −60 mV, injection of hyperpolarizing current steps (−250 pA; duration 1 s) in one cell (referred to as the activated cell) and monitoring evoked voltage responses in this cell and the second cell (referred to as the coupled cell). Voltage deflections in the two cells (ΔV H1 and ΔV H2, respectively) were analysed, and the coupling coefficient (cc) was calculated as ΔV H2/ΔV H1 as described previously (Landisman et al. 2002; Long et al. 2004; Haas et al. 2011; Wang et al. 2015).
To assess coupling under conditions of repeated activation, cells were activated with a series of current steps: a series of current steps consisted of a hyperpolarizing current step (−250 pA, 150 ms duration) followed by a depolarizing current step (100 pA, 250 ms), repeated 50 times with 100 ms inter‐stimulus interval, resulting in an overall series of stimuli at 2 Hz stimulus frequency and a duration of 25 s. While higher stimulation frequencies result in an inconsistent induction of LTS in the stimulated cell, the 2 Hz protocol allows reliable induction of low threshold spikes (LTS) with each stimulation pulse (Coulon et al. 2009). This series of stimuli was repeatedly applied to pairs of RTN neurons, starting with direct current injection in one cell (cell a) (repetition 1), followed by injection into the other cell (cell b) (repetition 2) in a given pair, and finally again in cell a (repetition 3) (a → b, b → a, a → b). The series of stimuli was repeated in this order in a given pair (a → b, b → a, a → b), and 10 min later in reversed order (b → a, a → b, b → a). Voltage responses were recorded in the directly injected (activated) and the coupled cell, and directionality of coupling (a → b; b → a) and activity‐dependent changes in cc during the time course of stimulation were monitored. Coupling coefficient was calculated from responses to hyperpolarizing stimuli in the two cells, with five responses averaged at three different time points of stimulation (1–5, designated 1; 21–25, designated 25; 46–50, designated 50), normalized to the cc of time point 1. Coupling coefficient was compared between different time points of the series of stimuli (1; 25; 50), at different directionality (a → b; b → a), between repetitions of the series of stimuli within the same directionality (a → b of repetition 1; a → b of repetition 3), between the two orders of stimulation (a → b, b → a, a → b vs. b → a, a → b, b → a) as well as between rat strains (GAERS, NEC).
Cells were excluded from analysis if current steps failed to evoke an LTS in more than 5% of stimuli within a given series in the activated cell.
Pharmacological intervention in vitro
To determine the contribution of gap junctions to coupling in RTN cells at resting condition, the gap junction blockers carbenoxolone and mefloquine were applied. Carbenoxolone (100 μm, Tocris Bioscience, Bristol, UK, dissolved in DMSO, final concentration of DMSO ≤ 0.2 %) was superfused with the bathing solution, and the cc was determined before application, 20 min after application and 30 min following washout. Mefloquine (25 μm, Sigma Aldrich, Hamburg, Germany, dissolved in DMSO, final concentration of DMSO ≤ 0.2%) was also superfused via the bathing solution, and the cc was determined before as well as 30, 60 and 90 min following application.
To investigate calcium dependencies for coupling during repeated activation, the calcium buffer BAPTA and the calmodulin antagonist ophiobolin A were applied during activation of the cells with the series of stimuli (see above). Ophiobolin A (25 μm; Enzo Life Science AG, Lausen, Switzerland) was superfused with the bathing solution and was present in both the activated and the coupled cell during all time points of the stimulus series (1, 25, 50), during each repetition of the stimulus series [a → b (repetition 1); b → a (repetition 2), a → b (repetition 3)] and at both orders (a → b, b → a, a → b; b → a, a → b, b → a). Calcium buffer BAPTA (3 mm, Sigma Aldrich) was applied intracellularly to one neuron of a given pair of RTN cells (cell a) within the internal recording solution, while the other neuron of that pair (cell b) was recorded with BAPTA‐free solution. This implies that BAPTA was present in the activated cell at repetitions 1 and 3 of the series of stimuli and in the coupled cell at repetition 2 of the series of stimuli for stimulus order [a → b (repetition 1), b → a (repetition 2), a → b (repetition 3)], while the reverse was the case for stimulus order [b → a (repetition 1), a → b (repetition 2), b → a (repetition 3)] (see Fig. 4).
Figure 4. Influence of intracellular application of Ca2+ buffer BAPTA on gap junctional coupling during repeated stimulation .
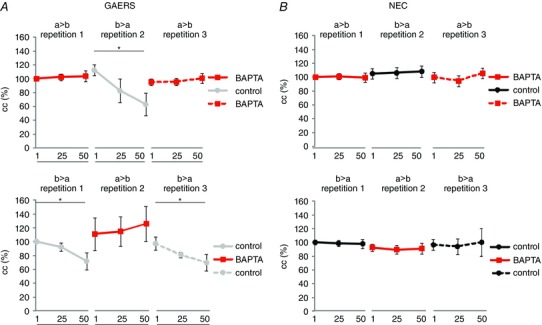
Neurons were repeatedly activated by injection of series of stimuli (50 alternating hyperpolarizing and depolarizing current steps). First, direct current was injected in one cell (cell a) (repetition 1) of a given pair, then by injection in the other cell (cell b) (repetition 2) of that pair, and finally in cell a again (repetition 3) (a → b, b → a, a → b). Ten minutes later, series of stimuli were injected in reverse order (b → a, a → b, b → a). Directionality and order of stimulation are indicated as a → b, b → a, a → b and b → a, a → b, b → a, where the cell in front of the arrow denotes the activated cell and the cell behind the arrow denotes the coupled cell in a given pair of RTN neurons. Note that in all cases the Ca2+ ‐buffer BAPTA was applied intracellularly to one neuron of a given pair of RTN cells (cell a) within the internal recording solution, while the other neuron of that pair (cell b) was recorded with BAPTA‐free solution. Average cc values taken at (1, 25, 50; as in Fig. 3) are illustrated for NEC (n = 6 cell pairs, six rats) (black, B) and GAERS (n = 6 cell pairs, six rats) (grey, A). Data points are displayed in red when the Ca2+ ‐buffer BAPTA was present in the activated cell, i.e. when the series of stimuli was injected into the cell containing the Ca2+ ‐buffer. Note the gradual decline in cc in GAERS that occurred when BAPTA was present in the coupled cell, which is not present when BAPTA was present in the activated cell. Asterisks indicate significant changes (*, p < 0.05).
Stereotactic surgery
Stereotactic surgery was performed in a stereotactic frame (David Kopf Instruments, Tujunga, CA, USA) under pentobarbital anaesthesia (Narcoren, 50 mg kg−1; Merial GmbH, Münster, Germany). The local anaesthetic lidocaine was used on all pressure and incision points. Holes were drilled into the skull for the insertion of epidural recording electrodes at right and left somatosensory cortex (A/P = 0, L = –/+4.6 mm, respectively), a depth recording electrode into the right ventro‐basal (VB) complex of the thalamus (A/P = −4.16 mm, L = −2.8 mm, depth = 6 mm) and a combined cannula/electrode system into the right RTN (A/P = −3.5 mm, L = −3.1 mm, depth = 5.6 mm). An epidural silver wire placed on top of the cerebellum served as ground and reference electrode. All coordinates were determined relative to bregma according to the stereotactic atlas of Paxinos & Watson (1998). The electrode assembly was fixed to the skull with the aid of dental acrylic cement (Pulpdent Glasslute, Watertown, MA, USA).
LFP recording and local pharmacological intervention in vivo
Local field potential (LFP) recordings in vivo were performed under Neurolept anaesthesia/analgesia, as described by Inoue et al. (1994) and Seidenbecher et al. (1998): following electrode implantation (see above) Neurolept anaesthesia/analgesia was induced and maintained via regular i.p. injection (every 15–20 min) of the opioid fentanyl (3.74 mg kg−1; Janssen, Neuss, Germany) and the D2‐Antagonist Droperidol (0.033 mg kg−1; Xomolix, ProStrakan, Düsseldorf, Germany). The depth of anaesthesia was constantly controlled by monitoring breathing rate, LFP recording (potential desynchronization) and responses to noxious stimulation of the hind paw. Upon signs of awakening, an additional low dose of fentanyl was administered.
Recording electrodes were connected to an amplifier (DPA‐2 FX, NPI Electronics, Tamm, Germany). Signals were filtered between 1 (HP) and 30 (LP) Hz and digitized using a data system (1401plus; Cambridge Electronic Design, Cambridge, UK) at a constant sampling rate of 1 kHz.
Following a stable SWD baseline recording of 1 h, either mefloquine (50 μm; n = 6 rats), ethosuximide (100 μm; n = 6 rats), carbenoxolone (100 μm; n = 6 rats) or saline (n = 6 rats) each diluted in 300 nl NaCl, 2% DMSO, was unilaterally pressure injected into the RTN via a guide cannula. In a fifth group (n = 4 rats), mefloquine (50 μm) was injected into the VB, as offset target control. Following injection, LFP recordings were continued for another 2.5 h.
The percentage of time in SWD during baseline and post‐injection recordings were scored offline with the aid of Spike2 analysis software (Version 7.08, Cambridge Electronic Design). To assess the time‐course of drug effects, baseline and post‐injection recordings were subdivided into 30 min bins, baseline values were set to 100% and the relative increase or decrease to that during the post‐injection recording bins was determined.
Histology
To verify the injection site and spread of the injected substance, all drugs were co‐applied with the fluorescent dye Alexa 594 (Molecular Devices, Sunnyvale, CA, USA). At the end of the experiment, animals were killed with an overdose of isoflurane and decapitated, the brain was quickly removed and placed in a 4% paraformaldehyde solution for at least 24 h. Brains were fixed in a 30% sucrose solution and cut into 60 μm slices with the aid of a microtome. Slices were mounted on microscope slides and inspected for fluorescent labelling with the aid of a fluorescence microscope (FV 300, Olympus). Only animals with a verified injection site within the RTN (or VB in the offset target control group) were considered for statistical analysis (see Fig. 6 C).
Figure 6. Local pharmacological intervention in vivo .
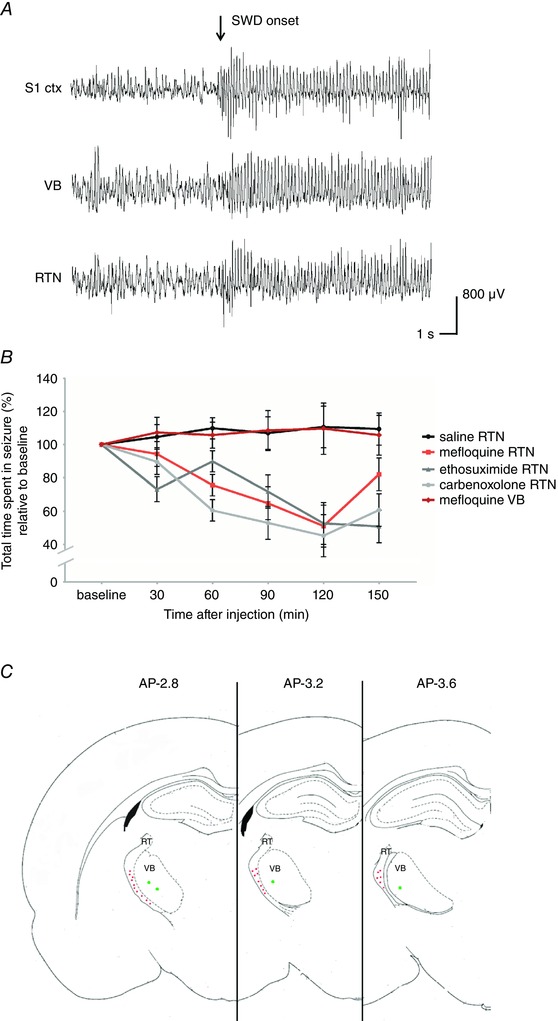
A, example local field potential recordings displaying the onset of a spike and wave discharge (SWD) (S1 ctx, somatosensory cortex; VB, ventrobasal complex of the thalamus; RTN, reticular thalamic nucleus). B, influence of local injection of saline (n = 6 rats; black line), carbenoxolone (n = 6 rats; light grey line), mefloquine (n = 6 rats; red line), ethosuximide (n = 6 rats; dark grey line) into the RTN and mefloquine into the VB (n = 4 rats; dark red line) on SWD expression in GAERS in vivo. Data are displayed as relative change to baseline (%, mean ± SEM). C, schematic overview of injection sites of either saline, mefloquine, carbenoxolone or ethosuximide, respectively, into the reticutar thalamic nucleus (RT) (red dots) or mefloquine into VB (offsite target group, green dots); this figure is adapted from Paxinos & Watson (1998).
Statistical analysis
All statistical analyses were performed using IBM‐SPSS 22‐software (SPSS Inc., Chicago, IL, USA). Passive membrane properties of recorded cells in vitro were analysed using a one‐way ANOVA with either RMP, input resistance (R IN) or capacitance (CIN) as dependent variable and strain (GAERS, NEC) as between‐subjects factor.
Gap junctional coupling in vitro at resting conditions was analysed by means of a repeated‐measures ANOVA, with cc as dependent variable, direction (a → b, b → a) as within‐subjects factor and strain (GAERS, NEC) as between‐subjects factor. The effects of carbenoxolone on coupling at resting conditions were analysed by means of a repeated‐measures ANOVA with cc as dependent variable, strain (GAERS, NEC) as between‐subjects factor and time (before drug application, 20 min after drug application, 30 min after washout) as within‐subjects factor. Effects of mefloquine on coupling at resting conditions were equally analysed by means of a repeated‐measures ANOVA with cc as dependent variable, strain (GAERS, NEC) as between‐subjects factor and time (before drug application, 30, 60, 90 min after drug application) as within‐subjects factor.
Gap junctional coupling during series of stimuli was analysed with repeated‐measures ANOVA, with cc as dependent variable, time point in a stimulus series (1, 25, 50) as within‐subjects factor 1, repetition and directionality of the series of stimuli (a → b, b → a, a → b) as within‐subjects factor 2, order of the series of stimuli (a → b, b → a, a → b; b → a, a → b, b → a) as within‐subjects factor 3 and strain (GAERS, NEC) as between‐subjects factor.
The same analysis was used to determine the effects of the calcium buffer BAPTA on coupling during series of stimuli. As BAPTA was intracellularly applied to only one cell (cell a) of each pair of RTN neurons, BAPTA was present in either the activated or the coupled cell depending on the repetition and the order of the series of stimuli (see above). Effects of the extracellularly applied ophiobolin A on coupling during series of stimuli was analysed with the aid of a repeated‐measures ANOVA, with cc as dependent variable, time point in a stimulus series (1, 25, 50) as within‐subjects factor 1, repetition and directionality of the series of stimuli (a → b, b → a, a → b) as within‐subjects factor 2, order of the series of stimuli (a → b, b → a, a → b; b → a, a → b, b → a) as within‐subjects factor 3, strain (GAERS, NEC) as between‐subjects factor 1 and drug (control, ophiobolin A) as between‐subjects factor 2. Effects of ophiobolin A and BAPTA on gap junctional coupling at resting condition were analysed with the aid of a repeated‐measures ANOVA, with cc as dependent variable, condition (control, drug) as within‐subjects factor and strain (GAERS, NEC) as between‐subjects factor. Absolute average cc values are given as mean ± SEM.
SWD expression in vivo was analysed using a repeated‐measures ANOVA with percentage of total time spent in SWD as dependent variable, group (saline, mefloquine, ethosuximide, carbenoxolone, offsite) as between‐subjects factor and time (baseline, 30, 60, 90, 120, 150 min after injection) as within‐subjects factor. Asterisks (*, **) in Figs. 2, 3, 4 represent significant changes, as specified in figure legends.
Figure 2. Influence of gap junctional blockers on gap junctional coupling at resting conditions .
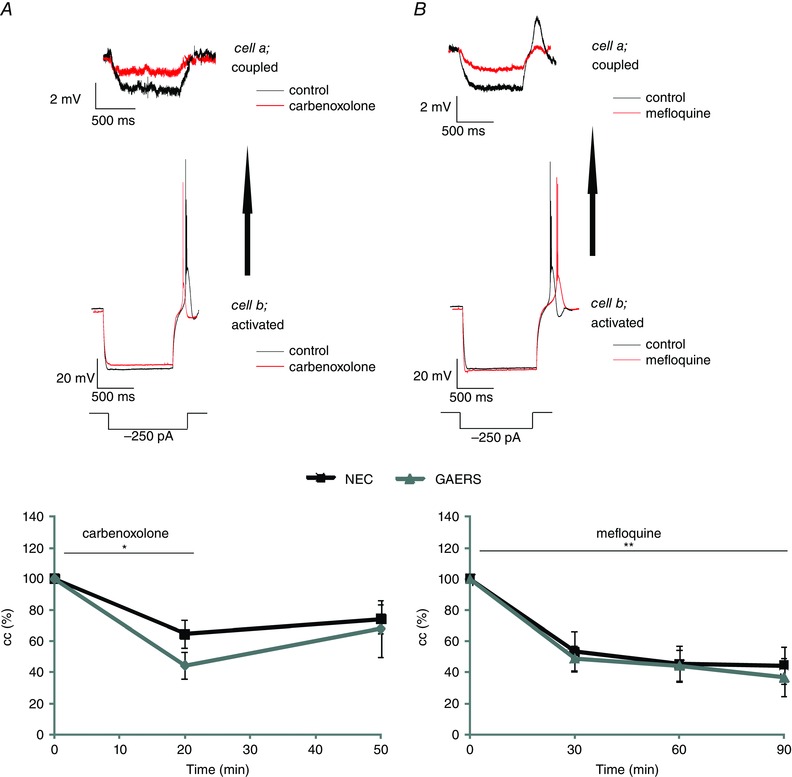
A, upper panel: example traces of voltage responses to hyperpolarizing current steps in the activated and coupled cell of a recorded pair of RTN neurons in NEC before (black traces) and during action of carbenoxolone (red traces). Note, three traces are averaged for clarity. Lower panel: average cc in NEC (n = 6 cell pairs, five rats) and GAERS (n = 6 cell pairs, five rats) before application, during application and after washout of carbenoxolone (100 μm; time of application indicated by horizontal bar). Note the significant decline of cc during action of carbenoxolone and partial recovery upon wash‐out. Black symbols and lines represent NEC, grey symbols and lines represent GAERS. B, upper panel: example traces of voltage responses to hyperpolarizing current steps in the activated and coupled cell of a recorded pair of RTN neurons in NEC before (black traces) and during action of mefloquine (red traces). Lower panel: average (mean ± SEM) cc in NEC (n = 6 cell pairs, five rats) and GAERS (n = 6 cell pairs, five rats) before and during (every 30 min) application of mefloquine (25 μm; time of application indicated by horizontal bar). Black symbols and lines represent NEC, grey symbols and lines represent GAERS. Asterisks indicate signifcant changes (*, p < 0.05; **, p < 0.01).
Figure 3. Gap junctional coupling of RTN neurons during repeated activation .
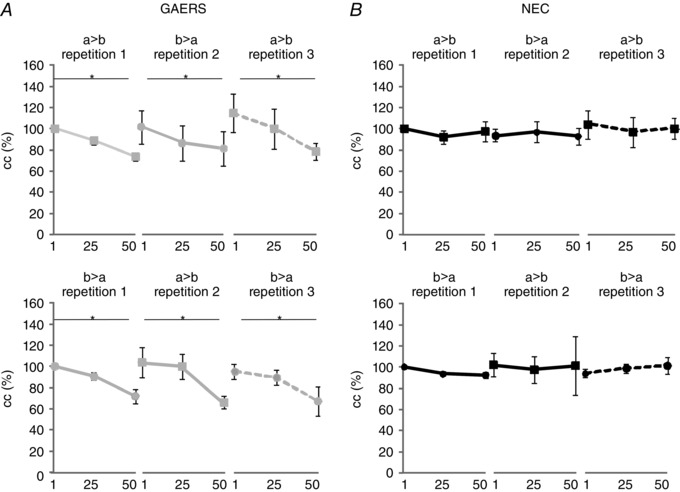
Neurons were repeatedly activated by injection of series of stimuli (50 alternating hyperpolarizing and depolarizing current steps; step protocol as in Fig. 1 B), starting with direct current injection in one cell (cell a) (repetition 1), followed by injection in the other cell (cell b) (repetition 2) in a given pair, and finally in cell a again (repetition 3) (a → b, b → a, a → b). The series of stimuli was repeated in this order in a given pair (a → b, b → a, a → b), and 10 min later in reversed order (b → a, a → b, b → a). Average cc values taken at three different time points of stimulation (1, 25, 50; see text) are illustrated for NEC (black, B) and GAERS (grey, A). Directionality and order of stimulation are indicated as a → b, b → a, a → b and b → a, a → b, b → a, where the cell in front of the arrow denotes the activated cell and the cell behind the arrow denotes the coupled cell in a given pair of RTN neurons. Note the gradual decline of cc in GAERS (n = 6 cell pairs, six rats) which is absent in NEC (n = 6 cell pairs, six rats). Asterisks indicate significant changes (*, P < 0.01).
Results
Basic properties of electrical coupling in RTN cells of GAERS and NEC
Whole cell recordings were obtained from RTN neurons, and RMP, CIN and R IN were assessed in 204 cells in NEC (n = 51 rats) and 155 cells in GAERS (n = 40 rats). In GAERS, the RMP was at −65.4 ± 0.55 mV, significantly more positive than in NEC, which had an RMP of −68.5 ± 0.49 mV (one‐way ANOVA: F 1,350 = 17.857, P < 0.001). No significant differences between GAERS and NEC were revealed for CIN (64.37 ± 2.33 pF in GAERS, 64.2 ± 2.04 pF in NEC; one‐way ANOVA: F 1,350 = 0.003, P > 0.05) and RIN (210.5 ± 8.2 MΩ in GAERS and 207.32 ± 7.1 MΩ in NEC; one‐way ANOVA: F 1,350 = 0.008, P > 0.05).
Simultaneous recordings were obtained from two neighbouring neurons in RTN, and coupling was assessed by holding the cells at −60 mV, injection of hyperpolarizing (−250 pA; 1 s duration) and depolarizing (+150 pA, 1 s) current steps in one cell (activated cell), and monitoring evoked voltage responses in both the activated and the second cell (coupled cell). A total of 30 and 28 pairs of neurons were recorded in this way in RTN of NEC (n = 10 rats) and GAERS (n = 9 rats), respectively. Upon termination of the current‐evoked membrane hyperpolarization, activated RTN neurons generated a rebound LTS, triggering a burst of fast action potentials. Injection of positive current steps from −60 mV evoked tonic series of single action potentials with little spike frequency adaptation. These properties are typical of RTN neurons (Avanzini et al. 1989; Contreras et al. 1993; Coulon et al. 2009). Coupled cells generated voltage responses in close temporal relation to voltage deflections in activated cells, apparent as membrane hyperpolarization followed by a rebound burstlet (Fig. 1). The cc was calculated from hyperpolarizing responses in each pair of cells, and was found not to differ between NEC and GAERS [cc of GAERS = 0.208 ± 0.015 (n = 28), cc of NEC = 0.200 ± 0.025 (n = 30); repeated‐measures ANOVA: main effect of strain: F 1,52 = 1.120, P > 0.05]. When the stimulation was repeated at reversed directionality, by direct current stimulation of the coupled neuron in a given pair, coupled responses were observed also in the previously stimulated cells. The cc was similar as before in a given pair, and no significant differences were found between NEC and GAERS [cc of GAERS at reversed direction = 0.216 ± 0.019 (n = 28), cc of NEC at reversed direction = 0.161 ± 0.032 (n = 30); repeated‐measures ANOVA, main effect of direction: F 1,52 = 1.118, P > 0.05; strain × direction interaction: F 1,52 = 2.741, P > 0.05].
Figure 1. Recording of coupled RTN neurons .
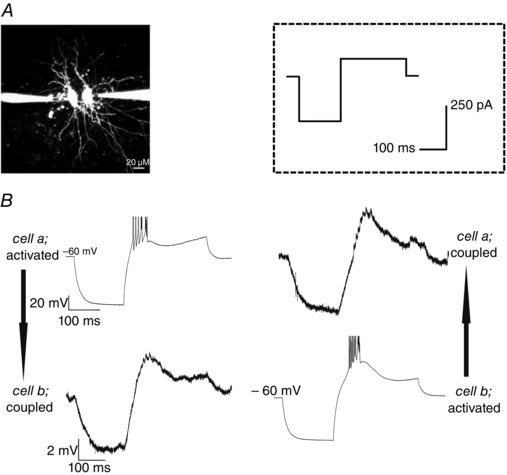
A, photomicrograph showing two simultaneously recorded RTN neurons, each filled via a patch pipette with fluorescent dye Alexa 594. Scale bar = 20 μm. B, example recording traces of coupled RTN neurons. Injection of a hyperpolarizing (−250 pA, 150 ms duration) followed by a depolarizing (100 pA, 250 ms) current step (from −60 mV; stimulation protocol shown in inset) produced a membrane hyperpolarization and rebound LTS in the activated neuron, and a concurrent hyperpolarization and burstlet in the coupled neuron. Electrical coupling was thus strong enough to produce a rebound LTS in the coupled cell. Spikes in traces are truncated for clarity.
These data indicate that RTN neurons are electrically coupled in both NEC and GAERS, with no apparent directionality and no differences in basal cc between the two strains.
To determine the contribution of gap junctions to coupling in RTN cells, the gap junction blockers carbenoxolone and mefloquine were applied. In both GAERS (n = 6 cell pairs, 5 rats) and NEC (n = 6 cell pairs, 5 rats), carbenoxolone resulted in a significant decrease in cc by 45% at 20 min after application (average absolute cc values: cc at baseline = 0.095 ± 0.016; cc 20 min after application = 0.052 ± 0.012). The cc partially recovered to 71% of the baseline value seen 30 min after washout (average absolute cc value = 0.061 ± 0.014; Fig. 2 A) (repeated‐measures ANOVA: main effect of time: F 2,13 = 16.615, P < 0.001; main effect of strain: F 1,8 = 0.811, P > 0.05; time × strain interaction F 2,13 = 1.053, P > 0.05).
Effects of mefloquine, a specific blocker of connexin 36 (Cruikshank et al. 2004), were assessed over 90 min, according to the reported time point of near‐maximal effect (Cruikshank et al. 2004). Mefloquine resulted in a significant decrease in cc by 48, 55 and 59% at 30, 60 and 90 min after application, respectively, with no differences occurring between GAERS (n = 6 cell pairs, 5 rats) and NEC (n = 6 cell pairs, 5 rats) (average absolute cc values at baseline and 30, 60 and 90 min after application = 0.138 ± 0.044, 0.084 ± 0.033, 0.0774 ± 0.032 and 0.0771 ± 0.034, respectively; Fig. 2 B) (repeated‐measures ANOVA: main effect of time: F 3,20 = 36.556, P < 0.001; main effect of strain: F 1,10 = 0.071, P > 0.05; time × strain interaction effect F 3,20 = 0.138, P > 0.05).
Activity‐dependent changes in electrical coupling
Activity‐dependent changes in cc were investigated by injection of a series of alternating current steps at −60 mV, consisting of a hyperpolarizing (−250 pA, 150 ms) followed by a depolarizing (100 pA, 250 ms) current step, which were repeated 50 times with 100 ms inter‐stimulus interval. Termination of membrane hyperpolarization reliably evoked an LTS crowned by a burst of fast action potentials (Fig. 1 B). This series of stimuli was repeatedly applied to pairs of RTN neurons, starting with direct current injection in one cell (cell a) (repetition 1), followed by injection in the other cell (cell b) (repetition 2) in a given pair and finally in cell a again (repetition 3) (a → b, b → a, a → b). The series of stimuli was repeated in this order in a given pair (a → b, b → a, a → b), followed by series of stimuli in that pair in reversed order (b → a, a → b, b → a). Voltage responses were recorded in the directly injected (activated) and the coupled cell, allowing us to assess the directionality of coupling (a → b vs. b → a), and activity‐dependent changes in cc during the time course of stimulation in a recorded pair of RTN neurons. The cc was calculated from responses to hyperpolarizing stimuli in the two cells, with five responses averaged at three different time points of stimulation (1–5, designated 1; 21–25, designated 25; 46–50, designated 50), and normalized to the cc of time point 1. Results are illustrated in Fig. 3.
In GAERS (n = 6 cell pairs, six rats), the series of stimuli resulted in a gradual decline in cc to approximately 80% of the baseline value (average absolute cc values: cc at 1st LTS = 0.078 ± 0.014; cc at 50th LTS = 0.043 ± 0.010) (Fig. 3 A; a → b). Reversing the directionality of stimulation between cells in a given pair resulted in a similar decline in cc with stimulation (Fig. 3 A; b → a) (average absolute cc values: cc at 1st LTS = 0.078 ± 0.019; cc at 50th LTS = 0.056 ± 0.021). When the stimuli were discontinued, cc returned to baseline values within 25 s, and repeating the series of stimuli at initial directionality resulted in a decline in cc as before (Fig. 3 A; a → b, compare repetition 1 and 3) (average absolute cc values at repetition 3: cc at 1st LTS = 0.077 ± 0.034; cc at 50th LTS = 0.064 ± 0.033). Furthermore, repeating the stimulation protocol at the same (a → b, b → a, a → b) or reversed order (b → a, a → b, b → a) resulted in a similar decline in cc with repeated stimuli, irrespective of directionality (Fig. 3 A, compare a → b, b → a, a → b with b → a, a → b, b → a) (repeated‐measures ANOVA: main effect of time point in stimulus series: F 2,20 = 10.485, P < 0.01; main effect of repetition and directionality of the series of stimuli: F 2,20 = 0.127, P > 0.05; main effect of order of the series of stimuli: F 1,10 = 0.145, P > 0.05). In NEC (n = 6 cell pairs, six rats), series of repetitive current stimuli were found not to affect cc, irrespective of the order or directionality of stimulation in recorded pairs of RTN neurons (Fig. 3 B; average absolute cc values: cc at 1st LTS = 0.089 ± 0.013; cc at 50th LTS = 0.088 ± 0.012) (repeated‐measures ANOVA: main effect of strain: F 1,10 = 1,447, P > 0.05; time point in stimulus series × strain interaction effect F 2,20 = 9.221, P < 0.01).
Pharmacological intervention with intracellular Ca2+ mechanisms
Next, we addressed the possibility that intracellular Ca2+ contributes to the activity‐dependent changes in cc. In a first series of experiments, BAPTA (3 mm) was included in the internal solution for recording one RTN neuron (cell a), while the other neuron in a given pair (cell b) was recorded with BAPTA‐free solution (n = 6 cell pairs, six GAERS; n = 6 cell pairs, six NEC). Changes in cc were probed using the stimulation protocol with series of alternating hyper‐ and depolarizing current steps. The results are illustrated in Fig. 4. During direct stimulation of the cell recorded with BAPTA‐containing solution, cc remained at the baseline value with repetitive stimulation (Fig. 4 A; a → b) (average absolute cc values: cc at 1st LTS = 0.119 ± 0.037; cc at 50th LTS = 0.117 ± 0.036). Upon reversal of stimulation directionality to the cell recorded with BAPTA‐free solution in a given RTN pair, a gradual decline in cc was observed with repetitive stimulation (Fig. 4 A, b → a) as before (Fig. 4 A, compare with Fig. 3 A) (average absolute cc values: cc at 1st LTS = 0.124 ± 0.043; cc at 50th LTS = 0.093 ± 0.034). In the activated cell BAPTA exerted a blocking effect on stimulus‐induced changes in cc, irrespective of the order of stimulation (Fig. 4 A; a → b, b → a, a → b; b → a, a → b, b → a). In NEC, cc remained at baseline values under all recording conditions (with or without inclusion of BAPTA) during repetitive stimulation (Fig. 4 B) (average absolute cc values: cc at 1st LTS = 0.095 ± 0.030; cc at 50th LTS = 0.098 ± 0.031) (repeated‐measures ANOVA: time point in stimulus series × strain interaction effect F 2,20 = 4.506, P < 0.05; order of the series of stimuli × repetition and directionality of the series of stimuli × time point in stimulus series × strain interaction effect F 4,40 = 3.921, P < 0.05).
Of note, for both GAERS and NEC, basal coupling (cc at resting condition) of cells recorded with BAPTA‐containing solutions and BAPTA‐free control solutions were not significantly different (cc of control cells in NEC = 0.17± 0.024, cc of BAPTA‐treated cells in NEC = 0.21 ± 0.036, cc of control cells in GAERS = 0.22 ± 0.056, cc of BAPTA‐treated cells in GAERS = 0.28 ± 0.044; repeated‐measures ANOVA: main effect of drug F 1,10 = 3.943, P > 0.05; drug × strain interaction effect F 1,10 = 0.165, P > 0.05).
Next, the calmodulin antagonist ophiobolin A (25 μm) was added to the bathing solution. Under these conditions, no significant changes were observed in cc of RTN neurons in GAERS (n = 6 cell pairs, six rats), as probed with repetitive stimulation at different directionality and order (Fig. 5 A) (average absolute cc values: cc at 1st LTS = 0.085 ± 0.029; cc at 50th LTS = 0.089 ± 0.028). Similarly in RTN neurons of NEC (n = 6 cell pairs, six rats), cc remained at baseline value with repetitive stimulation during presence of ophiobolin A (Fig. 5 B) (average absolute cc values: cc at 1st LTS = 0.076 ± 0.015; cc at 50th LTS = 0.079 ± 0.014). Of note, for the GAERS strain this reaction of the cc towards a series of stimuli was significantly different from that towards a series of stimuli under control conditions (displayed in Fig. 3) (repeated‐measures ANOVA: main effect of stimulus series F 2,40 = 5.072, P < 0.05; stimulus series × strain interaction effect: F 2,20 = 8.104, P < 0.01; stimulus series × drug interaction effect: F 2,40 = 11.862, P < 0.01; stimulus series × strain × drug interaction effect: F 2,40 = 6.130, P < 0.01).
Figure 5. Influence of calmodulin antagonist ophiobolin A on gap junctional coupling during repeated stimulation .

Ophiobolin A was superfused via the bathing solution and neurons were repeatedly activated by injection of series of stimuli (50 alternating hyperpolarizing and depolarizing current steps). First, direct current was injected in one cell (cell a) (repetition 1) of a given pair, then by injection in the other cell (cell b) (repetition 2) of that pair, and finally in cell a again (repetition 3) (a → b, b → a, a → b). Ten minutes later, series of stimuli were injected in reversed order (b → a, a → b, b → a). Directionality and order of stimulation are indicated as a → b, b → a, a → b and b → a, a → b, b → a, where the cell in front of the arrow denotes the activated cell and the cell behind the arrow denotes the coupled cell in a given pair of RTN neurons. Average cc values taken at three different time points of the stimulation (1, 25, 50; as in Fig. 3) are illustrated for GAERS (n = 6 cell pairs, six rats) (A) and NEC (n = 6 cell pairs, six rats) (B).
No difference was observed in the basal coupling values (cc at resting condition) between ophiobolin A‐treated cells and control cells in NEC and GAERS (cc ophiobolin A‐treated cell in NEC = 0.14 ± 0.02, cc control cell in NEC = 0.12 ± 0.02, cc ophiobolin A‐treated cell in GAERS = 0.15 ± 0.03, cc control cell in GAERS = 0.15 ± 0.04; repeated‐measures ANOVA: main effect of drug F 1,22 = 0.334, P > 0.05; drug × strain interaction effect F 1,22 = 0.586, P > 0.05).
Effects of local gap junctional blockage on SWD expression in vivo
To investigate the relevance of changes in gap junctional coupling for SWD expression in vivo, the gap junctional blockers carbenoxolone and mefloquine were locally pressure‐injected in the RTN of adult GAERS rats (six animals per substance), which show abundant spontaneously occurring SWDs. The percentage of time spent in SWD, as determined in the LFP recordings obtained in these rats, was determined for a baseline period before drug application as well as during five time blocks of 30 min each after drug application (0–30, 30–60, 60–90, 90–120 and 120–150 min after drug application) (Fig. 6). In a third group (n = 4) of GAERS, mefloquine was pressure injected into the VB, a brain structure virtually devoid of gap junctions, to assess unspecific effects of the gap junctional blocker on SWD expression. In a fourth group of GAERS (n = 6), the clinically used anti‐absence drug ethosuximide was locally injected into the RTN. This group served as a comparison of effect size. In a fifth group (n = 6), which served as control, saline was locally injected into the RTN. The total time spent in SWD was compared between time points (before drug application, 0–30, 30–60, 60–90, 90–120 and 120–150 min after drug application) as well as between groups (mefloquine‐RTN, carbenoxolone, mefloquine‐VB, ethosuximide, saline; Fig. 6).
Injection of saline into the RTN as well as injection of mefloquine into the VB did not result in significant changes of the percentage of time spent in SWD. During all time blocks after drug injection, the percentage of time spent in SWD equalled the percentage of time spent in SWD seen during baseline (i.e. before drug application) (average absolute time spent in SWD per time block = 26.9 ± 1.4 min). A different picture arose for the groups in which either mefloquine, carbenoxolone or ethosuximide was locally injected into the RTN. In all groups drug application resulted in a significant decrease in the percentage of time spent in SWD compared to baseline (before drug application), starting within the 30–60 min post‐injection interval (Fig. 6). On average, drug application resulted in a reduction of 24% compared to baseline within the 30–60 min post‐injection interval (average absolute time spent in SWD during baseline = 27.7 ± 0.9 min, during 30–60 min post‐injection interval = 20.8 ± 1.8 min), a reduction of 37% within the 60–90 min post‐injection interval (average absolute time = 17.6 ± 2.9 min), a reduction of 50% within the 90–120 min post‐injection interval (average absolute time = 14.1 ± 4.2 min) and a reduction by 36% within the 90–120 min post‐injection interval (average absolute time = 18.2 ± 3.1 min) (Fig. 6). For GAERS, in which mefloquine was injected into the RTN, the percentage of time was found to recover to baseline levels within the 90–120 min post‐injection interval (average absolute time = 23.9 ± 3.6 min). In total, no significant difference in effect size was revealed between the latter three groups (repeated‐measures ANOVA: significant main effect of drug F 4,23 = 10.178, P < 0.01; a significant main effect of time F 5,115 = 7.498, P < 0.01; significant drug × time interaction effect F 20,115 = 3.238, P < 0.05).
Discussion
One important function of gap junctional coupling between neurons is to correlate their activity, for instance to synchronize oscillatory activity in extended networks (Huguenard & McCormick, 2007). Gap junctions have been proposed to be crucially involved in the regulation of synchrony in thalamo‐cortical networks (Slaght et al. 2002; Fuentealba & Steriade, 2005; Sohl et al. 2005; Huguenard & McCormick, 2007). In fact in RTN, around 50% of neighbouring neurons interact via electrical coupling (Deleuze & Huguenard, 2006), which synchronizes electrogenic activity of the connected neurons (Long et al. 2004). Cx36‐containing gap junctions in RTN have been found to be long‐term modulated via stimulation of metabotropic glutamate receptors (mGluRs), resulting in reduced coupling strength, weakening of temporally correlated spike firing and reduction of coordinated activity (Long et al. 2004; Landisman & Connors, 2005). It was recently shown that long‐term depression (LTD) can be caused by group I mGluR subtypes, while long‐term potentiation of electrical synaptic strength occurs via stimulation of group II mGluRs (Wang et al. 2015), showing that a differential regulation of spike firing or burst firing correlation is possible via mGluR‐mediated gap junctional modulation. The RTN is considered a so‐called choke point within the brain network relevant for pathological forms of synchrony, in particular for the generation and maintenance of SWD, the electrophysiological hallmark of absence seizures (Steriade, 2005; Paz & Huguenard, 2015). The involvement of Cx36 connections in RTN has been suggested based largely upon neuropharmacological evidence in experimental models of absence epilepsy (Gareri et al. 2005; Gigout et al. 2006; Proulx et al. 2006), while a point mutation (single nucleotide polymorphism) in the gene coding for Cx36 was reported in 35% of juvenile myoclonic epilepsy patients in a genome‐wide association study (Hempelmann et al. 2006).
The present study significantly extends these observations by identifying a short‐term modulation in gap junctional coupling in RTN neurons, which occurred in rats with genetically determined absence epilepsy, but not in non‐epileptic controls. This modulation was evident as a decrease in coupling strength (short‐term depression, STD) upon slow‐rhythmic activation (2 Hz) of LTS‐evoked burst firing in one of a coupled pair of RTN neurons. There was no evidence of a difference in basic properties of electrical coupling in RTN neurons between the epileptic and non‐epileptic strain of rats. Activity‐dependent LTD of gap junctional coupling has also been reported by Haas et al. (2011) in RTN neurons recorded in slices of Sprague Dawley rats. This LTD developed over a time course of minutes, thereby differing from the rather rapid time course of the depression observed in the present study. Changes in gap junctional communication were asymmetrical between the two neurons of a pair in the previous work (Haas et al. 2011), indicating that regulation of connectivity was dependent on the direction of use. Asymmetrical stimuli allowed us to investigate whether the STD was expressed asymmetrically. Only coupling from the activated neuron to the coupled neuron (e.g. a → b) was depressed, while coupling in the opposite direction (b → a) was unchanged. Furthermore, the depression was found to be reversible in that coupling strength returned to baseline within 25 s. Pre‐activity asymmetry of coupling in any given pair was not observed, suggesting asymmetrical use of a gap junction as a potential source of plasticity. Intracellular presence of BAPTA in the activated cell prevented the modulation of electrical coupling, which persisted upon activation of the BAPTA‐free neuron in the same individual pair of RTN neurons. Along the same line, the extracellular presence of the calmodulin antagonist ophiobolin A prevented STD upon activation of either one in a pair of RTN neurons. These data indicate an intracellular Ca2+‐dependent modulatory mechanism residing in the presynaptic (activated) cell rather than the postsynaptic coupled cell. Previous studies have provided evidence for Ca2+‐dependent modulation of gap junctions. Rao et al. (1987) reported a decrease in electrical coupling upon increases of intracellular Ca2+ in rat hippocampal neurons in vitro. In Cx35‐expressing cell cultures from perch, Ca2+ has a higher binding probability to gap junctions if coupled to calmodulin (Burr et al. 2005). Furthermore, Cx36 proteins possess phosphorylation sites (Kothmann et al. 2007; Alev et al. 2008) and phosphorylation‐related changes in coupling mediated by either protein kinase A (Urschel et al. 2006; Kothmann et al. 2009) or CamKII (Alev et al. 2008) have been described. By contrast, De Vuyst et al. (2006) reported an opening of Cx32 hemichannels upon increases in cytoplasmic Ca2+ in cell cultures of bladder cancer epithelial cells, which was blocked by application of a calmodulin inhibitor.
What might be the functional relevance of this modulation of gap junctional communication within the RTN? Hypersynchrony of rhythmically re‐occurring low‐threshold Ca2+‐spikes and associated burst firing in RTN synaptic networks characterizes SWD (Crunelli & Leresche, 2002; Sohal & Huguenard, 2003; Coulon et al. 2009). Given the low‐pass properties of electrical synapses and their Ca2+ dependency (Long et al. 2004), the electrical coupling of neighbouring RTN neurons seems prone to these LTS‐induced bursts of activity. The I T channels mediating an LTS are mainly located in the proximal and intermediate parts of dendrites in RTN neurons (Crandall et al. 2010). Interestingly, a selective increase in I T current in RTN neurons of GAERS and an increase in mRNA levels of T‐type Ca2+ channel‐encoding genes were reported previously (Tsakiridou et al. 1995; Talley et al. 2000), which have been suggested to increase the propensity of burst activity in RTN neurons (Tsakiridou et al. 1995). Accumulation of intracellular Ca2+ upon repetitive LTS generation releases intracellular Ca2+ via ryanodine receptors (RyRs), resulting in a reduction of low threshold activity and thus potentially a reduction in hypersynchrony (Coulon et al. 2009). Gap junctions are assumed to be located at the proximal part of the dendrite (Haas et al. 2011), and increases in intracellular Ca2+ mediated via influx and release mechanisms during burst activity will result in a transient depression of electrical coupling. The STD may thus reflect a mechanism of Ca2+ homeostasis, dampening cell–cell coupling on a short time scale, in particular when an RTN neuron generates repeated burst discharges over an extended period.
In vivo, application to RTN neurons of both carbenoxolone or a specific blocker of Cx36, mefloquine, resulted in a significant decrease of time spent in SWD, demonstrating the overall relevance of gap junctional coupling for hypersynchronous activity during SWD expression. These results are in agreement with studies by Gigout et al. (2006) and Gareri et al. (2005), who demonstrated a reduction of ictal‐like activities in thalamo‐cortical slices of GAERS and a reduction of spontaneous SWD activity in freely moving GAERS and WAG/Rij rats, following administration of carbenoxolone. On the other hand, as the STD found in the present study is a characteristic of a rat strain developing SWD, it is apparent that the mechanisms of calcium homeostasis outlined above are not sufficient as an effective compensatory mechanism. A potential reason is the transient nature of the STD. The treatment with carbenoxolone and mefloquine results in a lasting depression of gap junctional coupling within the RTN. In fact, short lasting STD may even render the thalamo‐cortical system more prone to SWD generation. As a result of the intra‐RTN STD, also the synchronized output from RTN onto thalamic relays is temporarily reduced (Fuentealba & Steriade, 2005). Of note, such a transient decoupling of the caudal RTN from some thalamic nuclei (posterior thalamic nucleus and ventral‐postero‐medial thalamic nucleus) was described to occur immediately preceding the onset of an SWD in signal analytical studies (Lüttjohann & van Luijtelaar, 2012; Lüttjohann et al. 2013). These authors argued that such a decoupling is a prerequisite for SWD generation, as it creates an idle state of thalamic neurons, which can easily be recruited into the epileptic process by the cortical focus (Lüttjohann et al. 2013; Lüttjohann & van Luijtelaar, 2015). Therefore, the results of the present study can be seen as an intracellular extension of the recently formulated SWD generation scenario (Lüttjohann et al. 2013).
In summary, this study demonstrates a Ca2+‐dependent, asymmetric STD of gap junctional coupling between neurons of the RTN, which is induced in the absence epileptic GAERS upon SWD‐like activation of RTN neurons. This STD is likely to be a mechanism of Ca2+ homeostasis within the presynaptic cell, aimed to counteract excessive synchronization. Given the short lasting nature of the STD, however, it fails as a sufficient compensatory mechanism for SWD generation.
Additional information
Competing interests
None of the authors has any conflict of interest to disclose.
Author contributions
H.C.P. and P.C. conceived the study. D.K. performed the in vitro experiments, with contribution by P.C. A.L. performed the in vivo experiments, with contributions by T.S. and D.K. D.K., A.L. and H.C.P. analysed the data. A.L., D.K. and H.C.P. wrote the paper. All authors agreed to the final version of the paper.
Funding
This work reported of herein was supported by a grant from the programme Innovative Medical Research of the Medical Faculty of Münster to P.C. (grant no. I‐CO121008).
Acknowledgements
We thank Dr Kay Jüngling for initial experiments and scientific discussion as well as Elke Nass and Petra Berenbrock for expert technical assistance and animal care.
References
- Alev C, Urschel S, Sonntag S, Zoidl G, Fort AG, Hoher T, Matsubara M, Willecke K, Spray DC & Dermietzel R (2008). The neuronal connexin36 interacts with and is phosphorylated by CaMKII in a way similar to CaMKII interaction with glutamate receptors. Proc Natl Acad Sci USA 105, 20964–20969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avanzini G, de Curtis M, Panzica F & Spreafico R (1989). Intrinsic properties of nucleus reticularis thalami neurones of the rat studied in vitro . J Physiol 416, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Vestal M, Berman R, Negishi M, Spann M, Vega C, Desalvo M, Novotny EJ, Constable RT & Blumenfeld H (2010). Dynamic time course of typical childhood absence seizures: EEG, behavior, and functional magnetic resonance imaging. J Neurosci 30, 5884–5893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burr GS, Mitchell CK, Keflemariam YJ, Heidelberger R & O'Brien J (2005). Calcium‐dependent binding of calmodulin to neuronal gap junction proteins. Biochem Biophys Res Commun 335, 1191–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contreras D, Curro Dossi R & Steriade M (1993). Electrophysiological properties of cat reticular thalamic neurones in vivo . J Physiol 470, 273–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulon P, Herr D, Kanyshkova T, Meuth P, Budde T & Pape HC (2009). Burst discharges in neurons of the thalamic reticular nucleus are shaped by calcium‐induced calcium release. Cell Calcium 46, 333–346. [DOI] [PubMed] [Google Scholar]
- Crandall SR, Govindaiah G & Cox CL (2010). Low‐threshold Ca2+ current amplifies distal dendritic signaling in thalamic reticular neurons. J Neurosci 30, 15419–15429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruikshank SJ, Hopperstad M, Younger M, Connors BW, Spray DC & Srinivas M (2004). Potent block of Cx36 and Cx50 gap junction channels by mefloquine. Proc Natl Acad Sci USA 101, 12364–12369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V & Leresche N (2002). Childhood absence epilepsy: genes, channels, neurons and networks. Nat Rev Neurosci 3, 371–382. [DOI] [PubMed] [Google Scholar]
- De Vuyst E, Decrock E, Cabooter L, Dubyak GR, Naus CC, Evans WH & Leybaert L (2006). Intracellular calcium changes trigger connexin 32 hemichannel opening. EMBO J 25, 34–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deleuze C & Huguenard JR (2006). Distinct electrical and chemical connectivity maps in the thalamic reticular nucleus: potential roles in synchronization and sensation. J Neurosci 26, 8633–8645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaulis A & van Luijtelaar G (2006). Genetic models of absence epilepsy in the rat In Models of Seizures and Epilepsy, eds Pitkänen A, Schwartzkroin PA, Moshé SL. Elsevier, Amsterdam, pp. 233–248. [Google Scholar]
- Fuentealba P & Steriade M (2005). The reticular nucleus revisited: intrinsic and network properties of a thalamic pacemaker. Progr Neurobiol 75, 125–141. [DOI] [PubMed] [Google Scholar]
- Gareri P, Condorelli D, Belluardo N, Citraro R, Barresi V, Trovato‐Salinaro A, Mudo G, Ibbadu GF, Russo E & De Sarro G (2005). Antiabsence effects of carbenoxolone in two genetic animal models of absence epilepsy (WAG/Rij rats and lh/lh mice). Neuropharmacology 49, 551–563. [DOI] [PubMed] [Google Scholar]
- Gigout S, Louvel J & Pumain R (2006). Effects in vitro and in vivo of a gap junction blocker on epileptiform activities in a genetic model of absence epilepsy. Epilepsy Res 69, 15–29. [DOI] [PubMed] [Google Scholar]
- Guillery RW & Harting JK (2003). Structure and connections of the thalamic reticular nucleus: advancing views over half a century. J Comp Neurol 463, 360–371. [DOI] [PubMed] [Google Scholar]
- Gupta D, Ossenblok P & van Luijtelaar G (2011). Space–time network connectivity and cortical activations preceding spike wave discharges in human absence epilepsy: a MEG study. Med Biol Eng Comput 49, 555–565. [DOI] [PubMed] [Google Scholar]
- Haas JS & Landisman CE (2012). Bursts modify electrical synaptic strength. Brain Res 1487, 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas JS, Zavala B & Landisman CE (2011). Activity‐dependent long‐term depression of electrical synapses. Science 334, 389–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempelmann A, Heils A & Sander T (2006). Confirmatory evidence for an association of the connexin‐36 gene with juvenile myoclonic epilepsy. Epilepsy Res 71, 223–228. [DOI] [PubMed] [Google Scholar]
- Huguenard JR & McCormick DA (2007). Thalamic synchrony and dynamic regulation of global forebrain oscillations. Trends Neurosci 30, 350–356. [DOI] [PubMed] [Google Scholar]
- Inoue M, Ates N, Vossen JM & Coenen AM (1994). Effects of the neuroleptanalgesic fentanyl‐fluanisone (Hypnorm) on spike‐wave discharges in epileptic rats. Pharmacol Biochem Behav 48, 547–551. [DOI] [PubMed] [Google Scholar]
- Kostyuk P & Verkhratsky A (1994). Calcium stores in neurons and glia. Neuroscience 63, 381–404. [DOI] [PubMed] [Google Scholar]
- Kothmann WW, Li X, Burr GS, & O'Brien J (2007). Connexin 35/36 is phosphorylated at regulatory sites in the retina. Vis Neurosci 24, 363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kothmann WW, Massey SC & O'Brien J (2009). Dopamine‐stimulated dephosphorylation of connexin 36 mediates AII amacrine cell uncoupling. J Neurosci 29, 14903–14911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landisman CE & Connors BW (2005). Long‐term modulation of electrical synapses in the mammalian thalamus. Science 310, 1809–1813. [DOI] [PubMed] [Google Scholar]
- Landisman CE, Long MA, Beierlein M, Deans MR, Paul DL & Connors BW (2002). Electrical synapses in the thalamic reticular nucleus. J Neurosci 22, 1002–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long MA, Landisman CE & Connors BW (2004). Small clusters of electrically coupled neurons generate synchronous rhythms in the thalamic reticular nucleus. J Neurosci 24, 341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüttjohann A, Schoffelen JM & van Luijtelaar G (2013). Peri‐ictal network dynamics of spike‐wave discharges: phase and spectral characteristics. Exp Neurol 239, 235–247. [DOI] [PubMed] [Google Scholar]
- Lüttjohann A & van Luijtelaar G (2012). The dynamics of cortico‐thalamo‐cortical interactions at the transition from pre‐ictal to ictal LFPs in absence epilepsy. Neurobiol Dis 47, 49–60. [DOI] [PubMed] [Google Scholar]
- Lüttjohann A & van Luijtelaar G (2015). Dynamics of networks during absence seizure's on‐ and offset in rodents and man. Front Physiol 6, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marescaux C, Vergnes M & Depaulis A (1992). Genetic absence epilepsy in rats from Strasbourg–a review. J Neural Transm Suppl 35, 37–69. [DOI] [PubMed] [Google Scholar]
- Meeren HK, Pijn JP, Van Luijtelaar EL, Coenen AM & Lopes da Silva FH (2002). Cortical focus drives widespread corticothalamic networks during spontaneous absence seizures in rats. J Neurosci 22, 1480–1495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moeller F, LeVan P, Muhle H, Stephani U, Dubeau F, Siniatchkin M & Gotman J (2010). Absence seizures: individual patterns revealed by EEG‐fMRI. Epilepsia 51, 2000–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyer C, Herr D, Kohmann D, Budde T, Pape H‐C & Coulon P (2016). mGluR‐mediated Calcium Signalling in the Thalamic Reticular Nucleus. Cell Cal 59, 312–323. [DOI] [PubMed] [Google Scholar]
- Panayiotopoulos CP (1999). Typical absence seizures and their treatment. Arch Disease Childhood 81, 351–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G & Watson C (1998). The Rat Brain in Stereotaxic Coordinates. Academic Press, San Diego. [Google Scholar]
- Paz JT & Huguenard JR (2015). Microcircuits and their interactions in epilepsy: is the focus out of focus? Nat Neurosci 18, 351–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D (2004). The thalamic reticular nucleus: structure, function and concept. Brain Res Brain Res Rev 46, 1–31. [DOI] [PubMed] [Google Scholar]
- Pinault D, & O'Brien TJ (2005). Cellular and network mechanisms of genetically‐determined absence seizures. Thalamus Relat Syst 3, 181–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proulx E, Leshchenko Y, Kokarovtseva L, Khokhotva V, El‐Beheiry M, Snead OC, 3rd & Perez Velazquez JL (2006). Functional contribution of specific brain areas to absence seizures: role of thalamic gap junctional coupling. Eur J Neurosci 23, 489–496. [DOI] [PubMed] [Google Scholar]
- Rao G, Barnes CA & McNaughton BL (1987). Occlusion of hippocampal electrical junctions by intracellular calcium injection. Brain Res 408, 267–270. [DOI] [PubMed] [Google Scholar]
- Seidenbecher T, Staak R & Pape H‐CC (1998). Relations between cortical and thalamic cellular activities during absence seizures in rats. Eur J Neurosci 10, 1103–1112. [DOI] [PubMed] [Google Scholar]
- Slaght SJ, Leresche N, Deniau JM, Crunelli V & Charpier S (2002). Activity of thalamic reticular neurons during spontaneous genetically determined spike and wave discharges. J Neurosci 22, 2323–2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohal VS & Huguenard JR (2003). Inhibitory interconnections control burst pattern and emergent network synchrony in reticular thalamus. J Neurosci 23, 8978–8988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohl G, Maxeiner S & Willecke K (2005). Expression and functions of neuronal gap junctions. Nat Rev Neurosci 6, 191–200. [DOI] [PubMed] [Google Scholar]
- Steriade M (1998). Corticothalamic networks, oscillations, and plasticity. Adv Neurol 77, 105–134. [PubMed] [Google Scholar]
- Steriade M (2005). Sleep, epilepsy and thalamic reticular inhibitory neurons. Trends Neurosci 28, 317–324. [DOI] [PubMed] [Google Scholar]
- Talley EM, Solorzano G, Depaulis A, Perez‐Reyes E & Bayliss DA (2000). Low‐voltage‐activated calcium channel subunit expression in a genetic model of absence epilepsy in the rat. Brain Res Mol Brain Res 75, 159–165. [DOI] [PubMed] [Google Scholar]
- Tenney JR, Fujiwara H, Horn PS, Jacobson SE, Glauser TA & Rose DF (2013). Focal corticothalamic sources during generalized absence seizures: a MEG study. Epilepsy Res 106, 113–122. [DOI] [PubMed] [Google Scholar]
- Tsakiridou E, Bertollini L, de Curtis M, Avanzini G & Pape HC (1995). Selective increase in T‐type calcium conductance of reticular thalamic neurons in a rat model of absence epilepsy. J Neurosci 15, 3110–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urschel S, Hoher T, Schubert T, Alev C, Sohl G, Worsdorfer P, Asahara T, Dermietzel R, Weiler R & Willecke K (2006). Protein kinase A‐mediated phosphorylation of connexin36 in mouse retina results in decreased gap junctional communication between AII amacrine cells. J Biol Chem 281, 33163–33171. [DOI] [PubMed] [Google Scholar]
- Wang Z, Neely R & Landisman CE (2015). Activation of group I and group II metabotropic glutamate receptors causes LTD and LTP of electrical synapses in the rat thalamic reticular nucleus. J Neurosci 35, 7616–7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westmijse I, Ossenblok P, Gunning B & van Luijtelaar G (2009). Onset and propagation of spike and slow wave discharges in human absence epilepsy: a MEG study. Epilepsia 50, 2538–2548. [DOI] [PubMed] [Google Scholar]