

Abstract
Rapid eye movement (REM) sleep is a recurring part of the sleep–wake cycle characterized by fast, desynchronized rhythms in the electroencephalogram (EEG), hippocampal theta activity, rapid eye movements, autonomic activation and loss of postural muscle tone (atonia). The brain circuitry governing REM sleep is located in the pontine and medullary brainstem and includes ascending and descending projections that regulate the EEG and motor components of REM sleep. The descending signal for postural muscle atonia during REM sleep is thought to originate from glutamatergic neurons of the sublaterodorsal nucleus (SLD), which in turn activate glycinergic pre‐motor neurons in the spinal cord and/or ventromedial medulla to inhibit motor neurons. Despite work over the past two decades on many neurotransmitter systems that regulate the SLD, gaps remain in our knowledge of the synaptic basis by which SLD REM neurons are regulated and in turn produce REM sleep atonia. Elucidating the anatomical, cellular and synaptic basis of REM sleep atonia control is a critical step for treating many sleep‐related disorders including obstructive sleep apnoea (apnea), REM sleep behaviour disorder (RBD) and narcolepsy with cataplexy.
Abbreviations
- BLA
basolateral nucleus of the amygdala
- CeA
central nucleus of the amygdala
- DpMe
deep mesencephalic reticular nucleus
- DRN
dorsal raphe nucleus
- EEG
electroencephalogram
- EMG
electromyogram
- GAD
glutamic acid decarboxylase
- GG
genioglossus nucleus
- GiA
α gigantocellular reticular nucleus
- GiV
ventral gigantocellular reticular nucleus
- IPSP
inhibitory postsynaptic potential
- LC
locus coeruleus
- LDT
laterodorsal tegmental nucleus
- LH
lateral hypothalamus
- LPGi
lateral paragigantocellular nucleus
- LPT
lateral pontine tegmentum nucleus
- MCH
melanin‐concentrating hormone
- mPFC
medial prefrontal cortex
- OSA
obstructive sleep apnoea
- OxR1 and OxR2
orexin receptors 1 and 2
- PB
parabrachial nucleus
- PC
precoeruleus nucleus
- PD
Parkinson's disease
- peri‐LCα
peri‐locus coeruleus alpha nucleus
- PnO
oralis pontine nucleus
- PPT
pedunculopontine tegmental nucleus
- REM
rapid eye movement
- RBD
REM sleep behaviour disorder
- SLD
sublaterodorsal nucleus
- subLC
subcoeruleus nucleus
- SWS
slow‐wave sleep
- vGat
vesicular GABA transporter
- vGlut2
vesicular glutamate transporter 2
- vlPAG
ventrolateral periaqueductal grey matter
- VMM
ventromedial medulla
The behavioural state of rapid eye movement (REM) sleep itself is characterized by the appearance of fast, desynchronized rhythms in the cortical electroencephalogram (EEG), hippocampal theta activity, autonomic activation, muscle atonia and its eponymous hallmark feature, bursts of rapid eye movements. REM sleep is also associated with dream content, although the extent to which dream content, REM sleep and rapid eye movements are correlated remains unclear. Given the striking resemblance of the REM cortical EEG to that of the waking state, some investigators have instead preferred the term ‘paradoxical sleep’ or ‘active sleep’ to describe this behavioural state.
There is near consensus among sleep neuroscientists that REM sleep is important, if not necessary, for normal neurobehavioural and physiological function. In large part this consensus derives from the ubiquitous and recurring nature of REM sleep, its strong ‘rebound’ after deprivation (Beersma et al. 1990), and the finding that REM deprivation, over the course of several weeks, is lethal in rodents (Kushida et al. 1989). And while the latter finding is credited with inspiring greater interest in REM sleep function, it remains to date unclear whether the reported lethal outcomes associated with REM sleep deprivation were in fact due to REM deprivation per se, or were rather secondary to the stress of the deprivation intervention itself. In general support of a contributing stress covariate is the finding that chronic, e.g. pharmacological, suppression of REM sleep does not appear to have deleterious effects on the body or mind, including in humans. Hence a unified explanation for REM sleep remains elusive.
As indicated, REM sleep is characterized, in part, by changes in muscle activity, including a complete loss of muscle tone in axial postural muscles, phasic muscle twitches in distal limb and orofacial muscles and, of course, phasic bursting of oculomotor muscles. Respiratory‐related muscles are also tonically suppressed during REM sleep, but to a variable degree, ranging from nearly unaffected (diaphragm) to complete suppression (genioglossus muscle). The importance of postural atonia, in particular, during REM sleep is profoundly illustrated in human patients with REM sleep behaviour disorder (RBD). RBD is a parasomnia in which patients have excessive tonic and phasic electromyogram (EMG) activity during REM sleep, which can manifest behaviourally as involuntary movements including kicking, punching, shouting and screaming (Schenck et al. 1986, 2013). These unconscious movements can be violent, often resulting in injury to both the individual and bed partner alike, occasionally with life‐threatening outcomes. Emerging clinical data have also established an intriguing link between RBD and several degenerative neurological disorders including Parkinson's disease (PD) and other synucleinopathies, such as Lewy body dementia, multiple systems atrophy and pure autonomic failure (Boeve, 2013; Peever et al. 2014). Perhaps the most important aspect of this link is that RBD may appear decades prior to the motoric and cognitive symptoms of these neurodegenerative disorders. Hence the diagnosis of RBD may provide an early clinical predictor for some degenerative neurological disorders, including PD, (Boeve et al. 2007) and an early therapeutic window for delaying their full development.
Cataplexy, which affects approximately 70% of people with narcolepsy and is characterized by a bilateral loss of muscle tone during wake, is another example of pathological postural atonia control. Loss of muscle tone in cataplexy is triggered by strong, typically positive emotions (e.g. laughter, surprise) and can last from seconds to minutes (Overeem et al. 2011). While cataplexy can be partial, involving only individual muscle groups (often of the face or neck), it more typically includes postural muscle groups, resulting in patient collapse (Khoury & Doghramji, 2015). Respiratory muscles, on the other hand, are not affected. Interestingly, cataplexy occurs almost exclusively in narcolepsy, a neurological condition linked to low levels of orexin (also called hypocretin) peptides in the cerebrospinal fluid (Nishino et al. 2000; Peyron et al. 2000; Mignot et al. 2002), secondary to loss of orexin/hypocretin neurons in the lateral hypothalamus (Thannickal et al. 2000; Blouin et al. 2005; Crocker et al. 2005). Because of the shared, albeit directionally opposite, feature of dysregulated postural atonia control between cataplexy and REM sleep, it has been hypothesized that the failure of waking postural motor tone in cataplexy involves state‐inappropriate activation of the same circuitry controlling postural muscle atonia during REM sleep (Siegel, 2011; Burgess & Scammell, 2012).
Given the foregoing, defining the anatomical, cellular and synaptic control mechanisms of motor atonia is critical not only for elucidating the neural mechanisms of sleep movement disorders (RBD and other parasomnias and cataplexy) but also for aiding the identification of early stage loci for a host of neurodegenerative diseases. In this review, we highlight work leading to the identification of pontine and medullary circuitry controlling REM sleep and REM sleep muscle atonia. We next explore the synaptic inputs that modulate the ‘executive’ elements of the REM sleep atonia control circuit and conclude by detailing the synaptic output mechanisms that contribute to postural, orofacial and respiratory motor neuron suppression during REM sleep.
REM circuitry: the pontine REM atonia generator
Pioneering experimental work in the 1960s (Jouvet & Michel, 1960; Mouret et al. 1967) and mid (Henley & Morrison, 1974) and late (Sakai et al. 1979) 1970s identified a region of the dorsal rostral pons crucial for the generation of muscle atonia during REM sleep in cats. The specific pontine neurons that linked most strongly to the generation of REM atonia were found in the ventral and medial locus coeruleus (LC), termed the peri‐locus coeruleus α (peri‐LCα) in cats (Sakai et al. 1979, 1981, 2001). The pontine homologue of the cat peri‐LCα was subsequently identified in rats and mice and comprised a small region of the pontine tegmentum, just ventral to the caudal laterodorsal tegmental nucleus (LDT) and the LC. This region was termed the subcoeruleus (subLC; Pollock & Mistlberger, 2003; Brown et al. 2006) or sublaterodorsal nucleus (SLD; Boissard et al. 2002; Lu et al. 2006; Clement et al. 2011; Figs 1 and 2).
Figure 1. Anatomic representation of brain regions involved in the regulation of REM sleep and REM sleep atonia .
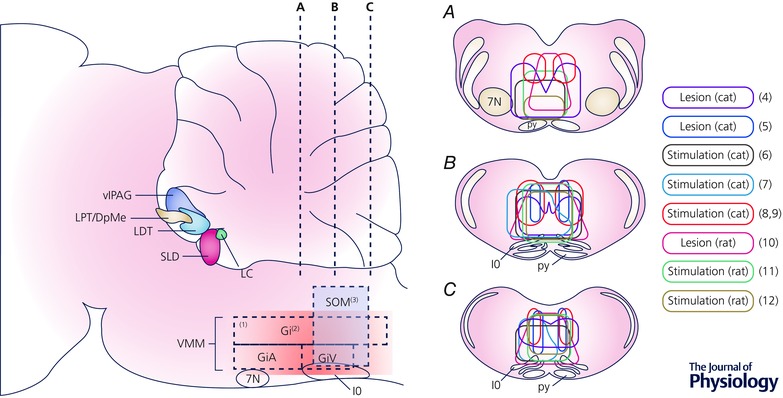
Sagittal representation of a rodent brain (left), with coronal views at three rostral–caudal levels (right, A–C). A zone within the medulla, specifically in the ventromedial medulla (VMM), is thought to play a key role in REM sleep atonia control. (1) The most rostral part of the VMM does not have a significant role in REM sleep atonia (Sastre et al. 1981; Lu et al. 2006). (2) The VMM near the inferior olive, near Magoun's inhibitory section, is the key zone for REM sleep atonia control (Magoun & Rhines, 1946; Kanamori et al. 1980; Chase et al. 1984; Schenkel & Siegel, 1989; Holmes & Jones, 1994; Lai & Siegel, 1997; Hajnik et al. 2000; Boissard et al. 2002; Morales et al. 2006; Sapin et al. 2009). (3) Caudal to the inhibitory zone, glutamatergic neurons regulate REM sleep atonia (Vetrivelan et al. 2009). Coronal views (A–C) show the zones of stimulation (continuous outlines) or lesion (dashed outlines) that either induce muscle atonia (stimulation) or decrease muscle atonia (lesion). (4) Lesions in the cat have targeted the VMM near the inferior olive (Holmes & Jones, 1994), while stimulation studies (6, 7) have also targeted the VMM as an atonia zone (Magoun & Rhines, 1946). Other studies, both lesion (5) studies (Schenkel & Siegel, 1989) and stimulation (8, 9) experiments (Takakusaki et al. 2001; Habaguchi et al. 2002) have targeted a more dorsal region just lateral to the midline, the caudal and dorsal extent of the nucleus gigantocellularis and nucleus magnocellularis or dorsal paragigantocellular region. In rats, both lesion (10) studies (Vetrivelan et al. 2009) and studies with electrical stimulation (11, 12) have targeted the ventromedial medulla as a key atonia zone (Lai and Siegel, 1988; Hajnik et al. 2000). Overall, despite the use of different methods and species, there is a significant overlap in the regions implicating the ventromedial medulla in atonia. Abbreviations: DpMe, deep mesencephalic reticular nucleus; Gi, gigantocellular nucleus; GiA, α gigantocellular nucleus; GiV, ventral gigantocellular nucleus; IO, inferior olive; LC, locus coeruleus; LDT, laterodorsal tegmental nucleus; LPT, lateral pontine tegmentum; py, pyramids; SLD, sublaterodorsal nucleus; SOM, supraolivary medulla; vlPAG, ventrolateral periaqueductal grey matter; VMM, ventromedial medulla; 7N, facial nucleus.
Figure 2. Schematic summary showing synaptic regulation of SLD REM‐atonia neurons .
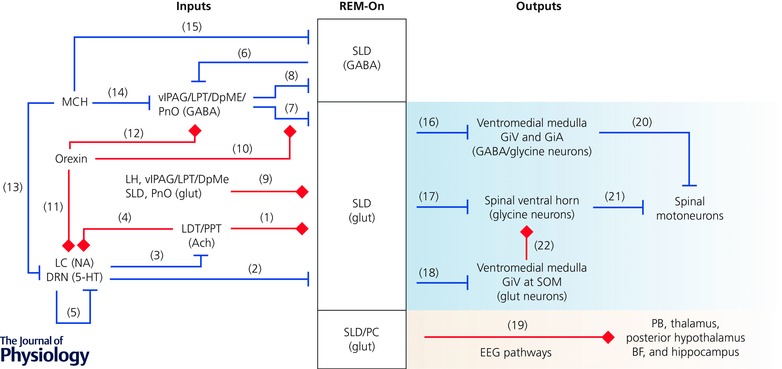
The activity of SLD neurons is likely to be controlled by coordinated cholinergic, monoaminergic, GABAergic, glutamatergic and peptidergic inputs. Key references for the represented pathways include (1) REM‐On pathway (Baghdoyan et al. 1987; Semba, 1993; Kubin, 2001; Weng et al. 2014); (2) REM‐Off pathway (Semba, 1993; Williams et al. 2012); (3) REM‐Off pathway (Hobson et al. 1975; McCarley & Hobson, 1975; Luebke et al. 1992; Semba & Fibiger, 1992; Williams & Reiner, 1993); (4) REM‐On pathway (Hobson et al. 1975; McCarley & Hobson, 1975; Svensson & Engberg, 1980; Egan & North, 1985; Satoh & Fibiger, 1986; Jones, 1990; Semba & Fibiger, 1992; Luppi et al. 1995); (5) REM‐Off pathway (Hobson et al. 1975; McCarley & Hobson, 1975); (6) REM‐On pathway (Lu et al. 2006; Fuller et al. 2007); (7) REM‐Off pathway (Boissard et al. 2003; Lu et al. 2006; Sapin et al. 2009); (8) REM‐Off pathway (Lu et al. 2006; Fuller et al. 2007); (9) REM‐On pathway (Shammah‐Lagnado et al. 1987; Lai et al. 1993; Semba, 1993; Boissard et al. 2003); (10) REM‐Off pathway (Peyron et al. 1998; Mileykovskiy et al. 2002; Zhang et al. 2004; Xi & Chase, 2010; Torterolo et al. 2013); (11) REM‐Off pathway (Peyron et al. 1998; Date et al. 1999; Ivanov & Aston‐Jones, 2000; Brown et al. 2002; Liu et al. 2002; Sakurai et al. 2005); (12) REM‐Off pathway (Boissard et al. 2003; Lu et al. 2006); (13) REM‐On pathway (Bittencourt et al. 1992; Del Cid‐Pellitero & Jones, 2012; Monti et al. 2013; Yoon & Lee, 2013; Devera et al. 2015); (14) REM‐On pathway (Luppi et al. 2013 a); (15) REM‐On pathway (Torterolo et al. 2009, 2013); (16) REM‐On pathway (Boissard et al. 2002; Morales et al. 2006; Sapin et al. 2009); (17) REM‐On pathway (Lu et al. 2006); (18) REM‐On pathway (Vetrivelan et al. 2009); (19) REM‐On pathway (Lu et al. 2006; Fuller et al. 2007); (20) REM‐On pathway (Chase et al. 1984, 1986; Soja et al. 1987 b; Lai & Siegel, 1988; Castillo et al. 1991 a,b; Holstege & Bongers, 1991; Kodama et al. 2003; Kato et al. 2006; Lai et al. 2010); (21) REM‐On pathway (Taal & Holstege, 1994; Alvarez et al. 2005); (22) REM‐On pathway (Takakusaki et al. 2001, 2003). Abbreviations: ACh, acetylcholine; BF, basal forebrain; DpMe, deep mesencephalic reticular nucleus; DRN, dorsal raphe nucleus; GiA, α gigantocellular nucleus; GiV, ventral gigantocellular nucleus; glut, glutamate; LC, locus coeruleus; LDT, laterodorsal tegmental nucleus; LH, lateral hypothalamus; LPT, lateral pontine tegmentum; MCH, melanin‐concentrating hormone; NA, noradrenaline; PB, parabrachial nucleus; PC, precoeruleus; PnO, oralis pontine; PPT, pedunculopontine tegmental nucleus; SLD, sublaterodorsal nucleus; SOM, supraolivary medulla; vlPAG, ventrolateral periaqueductal grey matter; 5‐HT, serotonin.
A large number of experimental studies in rats and mice have established that SLD neurons play a critical role in both the initiation and the maintenance of postural atonia during REM sleep. As examples, electrical stimulation of the SLD region produces bilateral loss of postural muscle tone (Hajnik et al. 2000), lesions of the SLD produce REM without atonia (Mouret et al. 1967; Hendricks et al. 1982; Morrison, 1988; Sanford et al. 2001; Lu et al. 2006), and rats killed during REM sleep exhibit dense c‐Fos immunolabelling in SLD neurons, with the number of c‐Fos‐labelled cells positively correlated with the percentage of time spent in REM sleep (Maloney et al. 1999, 2000; Verret et al. 2005; Lu et al. 2006; Sapin et al. 2009), the latter marking SLD neurons as ‘REM‐On’. Unit recording studies have provided additional evidence that REM‐On SLD neurons satisfy the criteria for REM‐generating neurons: SLD neurons increase firing rate in anticipation of the onset of REM sleep, fire maximally during REM sleep and maintain sustained tonic discharge throughout REM sleep, and become silent during the transitions from REM to slow‐wave sleep (SWS) or wakefulness (Sakai & Koyama, 1996; Sakai et al. 2001; Karlsson & Blumberg, 2005). Pharmacological activation of SLD neurons via microinjections of glutamate, glutamate agonists or bicuculline (the latter of which blocks GABAergic afferent inputs) produces, with short‐latency, a long‐lasting REM‐like state characterized by low voltage EEG and continuous muscle atonia (Lai & Siegel, 1991; Onoe & Sakai, 1995; Xi et al. 1999 a; Hajnik et al. 2000; Boissard et al. 2002; Pollock & Mistlberger, 2003; Sanford et al. 2003).
Human clinical reports have also revealed a strong correlation between structural damage to the dorsal pons and the development of RBD. For example, Mathis et al. (2007) reported a case of human RBD following an encephalitis‐induced lesion that was restricted to the dorsal pontine tegmentum (presumably involving the human SLD bilaterally). Development of RBD was also reported in a patient following a unilateral stroke affecting the right SLD region (Xi & Luning, 2009) and in another patient following a discrete dorsomedial pontine lesion due to vasculitis (St Louis et al. 2014). Taken together, the foregoing experimental and clinical data provide evidence that SLD neurons are both necessary and sufficient for generating postural atonia during REM sleep.
A theoretical model of REM sleep control
In 1975 Hobson and McCarley introduced a ‘reciprocal‐interaction’ model of behavioural state control. In this influential model, which was informed by the work of Jouvet (1972), these scientists proposed that reciprocal interactions between mesopontine cholinergic REM‐On neurons and aminergic REM‐Off neurons were responsible for the alternation of wakefulness, SWS and REM sleep (Hobson et al. 1975; McCarley & Hobson, 1975). The model predicted that, during wakefulness, activity of the aminergic system would inhibit the laterodorsal and pedunculopontine tegmental nuclei (LDT/PPT) cholinergic system. With the onset of SWS, aminergic inhibition would wane and cholinergic excitation would wax, reaching a reciprocal trough and peak during REM sleep. Consistent with this model, monoaminergic neurons project to and inhibit cholinergic LDT/PPT neurons (Kubota et al. 1992; Luebke et al. 1992; Semba & Fibiger, 1992; Williams & Reiner, 1993; Honda & Semba, 1994; Fig. 2, Pathway 3, REM‐Off), and LDT/PPT cholinergic neurons project back to and excite noradrenergic LC and serotoninergic dorsal raphe nucleus (DRN; Svensson & Engberg, 1980; Egan & North, 1985; Satoh & Fibiger, 1986; Jones, 1990; Semba & Fibiger, 1992; Luppi et al. 1995; Fig. 2, Pathway 4, REM‐On).
Over the years, however, it became apparent that the reciprocal inhibition model could not fully account for the complexity of behavioural state transitions. And to this end, more recent experimental work has informed several modifications and additions to the model (Pace‐Schott & Hobson, 2002; Luppi et al. 2006; McCarley, 2007). The first of these modifications to the model was the introduction of non‐cholinergic, possibly glutamatergic, neurons as REM‐generators (Sakai et al. 2001; Boissard et al. 2002; Lu et al. 2006; Luppi et al. 2006). In this conceptualization cholinergic inputs would directly activate putative glutamatergic REM‐generator neurons (Fig. 2, Pathway 1, REM‐On) and the strength of REM‐generator output would remain under the control of the aminergic‐cholinergic interplay (Fig. 2, Pathway 3, REM‐Off and Pathway 4, REM‐On). The second modification to the model was the addition of GABAergic synaptic inputs that, during wakefulness and SWS, and in addition to monoaminergic inputs (Fig. 2, Pathway 2, REM‐Off), would inhibit the glutamatergic REM‐generator (Fig. 2, Pathway 7, REM‐Off; Boissard et al. 2003; Lu et al. 2006).
More recent experimental work has resulted in further refinements to the original model, including the identification of two functionally segregated, but anatomically opposed sets of glutamatergic REM‐generating neurons. The first of these cell groups spans the SLD, caudal LDT and adjacent precoeruleus (PC) and promotes cortical activation through ascending inputs to the parabrachial nucleus and forebrain (Fig. 2, Pathway 19, REM‐On; Fuller et al. 2011; Krenzer et al. 2011), whereas the second cell group, which includes SLD REM‐atonia neurons, generates postural muscle atonia through descending projections to the medulla and spinal ventral horn (Figs 1 and 2, Pathway 16, 17, 20 and 21, all REM‐On; Boissard et al. 2002; Lu et al. 2006; Morales et al. 2006). The existence of separate pathways mediating the cortical and motoric components of REM sleep in fact provides a possible basis for the occasional dissociation of cortical activation and muscle atonia during pathological states such as cataplexy, sleep paralysis and RBD (Fuller et al. 2007; Vetrivelan et al. 2009; Luppi et al. 2011).
Taken together, work over the past two decades has identified glutamatergic SLD neurons as REM generators and further shown that these neurons are regulated by many neurotransmitter systems including not only acetylcholine and monoamines – as predicted by the original 1975 reciprocal‐interaction model – but also GABA, glutamate and peptides. In fact, contemporary models of REM circuit control now consider the cholinergic and aminergic components as modulatory, not central, elements of the REM sleep regulatory circuit network. We next discuss the synaptic regulation of SLD REM‐On atonia neurons by these systems.
Inputs: the cellular and synaptic SLD
Cholinergic regulation
The cholinergic hypothesis of REM sleep induction derives from the observations that systemic administration of the cholinergic antagonists atropine or cholinesterase inhibitor physostigmine block or enhance, respectively, REM sleep (Jouvet & Michel, 1960) and that microinjections of carbachol (a cholinergic agonist) into the pontine tegmentum produce a long‐lasting REM‐like state in cats and rodents (Baghdoyan et al. 1987; Hobson et al. 1993; Kubin, 2001). Although the amount and onset delay of REM sleep generated by carbachol in the pons varies by location and dose of injection, species and the type of preparation (Kubin, 2001; Grace & Horner, 2015), the hypothesis that pontine cholinergic neurons participate in REM genesis is still widely accepted. In support of this view, acetylcholine levels in the dorsal pons are high during REM sleep (Leonard & Lydic, 1997), depletion of acetylcholine inhibits REM sleep and blocking acetylcholine degradation promotes REM sleep (reviewed in Jones, 1991 a,b). In addition, LDT/PPT cholinergic neurons are active during REM sleep and wakefulness but silent during SWS (Boucetta et al. 2014), and when LDT/PPT cholinergic neurons are optogenetically activated during SWS they promote the transition from SWS to REM sleep (Van Dort et al. 2015). LDT/PPT cholinergic neurons also project to the SLD (Quattrochi et al. 1989; Jones, 1990; Semba, 1993), and carbachol excites spinally projecting SLD neurons (Fig. 2, Pathway 1, REM‐On; Weng et al. 2014). It is also the case that the medulla (see below) contains cholinergic neurons that may be REM‐On (Holmes & Jones, 1994); however, these neurons do not project to the SLD (Semba, 1993; Holmes et al. 1994). Taken as a whole these findings strongly support a role for LDT/PPT cholinergic neurons in not only activating monoaminergic REM‐Off neurons (Fig. 2, Pathway 4, REM‐On; Svensson & Engberg, 1980; Egan & North, 1985; Satoh & Fibiger, 1986; Jones, 1990; Semba & Fibiger, 1992), but also in activating the pontine REM generator(s) (Fig. 2, Pathway 1, REM‐On; Jones, 1993; Steriade, 2004).
Importantly, a recent paper has cast doubt over the necessity of cholinergic input to the SLD for generating REM sleep (Grace et al. 2014). In this study the authors specifically showed that microinjection of scopolamine – a competitive antagonist at muscarinic acetylcholine receptors – in the SLD region was without effect on the frequency or duration of REM bouts and REM muscle atonia. Scopolamine administration was, however, found to increase both REM duration and the failure rate of transitions from SWS to REM. Therefore, while the results from Grace and colleagues convincingly show that acetylcholine is dispensable for the induction of REM sleep and muscle atonia, cholinergic inputs may reinforce REM sleep once initiated. To this end, the authors proposed that cholinergic inputs to the REM‐generator(s) including the SLD REM‐atonia neurons could help ensure rapid transitions into REM sleep that are less likely to fail (Grace & Horner, 2015). For the interested reader, Grace and Horner recently published an elegant historical overview of the cholinergic system in REM control (Grace & Horner, 2015).
Monoaminergic regulation
Both noradrenergic and serotoninergic neurons of the LC and DRN, respectively, are silent during REM sleep, resume firing just before awakenings (McGinty & Harper, 1976; Trulson & Jacobs, 1979; Aston‐Jones & Bloom, 1981) and, importantly, cease discharging during periods of cataplexy (Wu et al. 1999, 2004). Silencing of LC and DRN neurons during REM sleep has been attributed to both recurrent inhibition (Fig. 2, Pathway 5, REM‐Off; Hobson et al. 1975; McCarley & Hobson, 1975) and GABAergic input from REM active neurons (Nitz & Siegel, 1997 a,b; Gervasoni et al. 2000; Verret et al. 2006; Goutagny et al. 2008; Clement et al. 2014). Both the original and the modified reciprocal inhibitory interaction models (see above) argue that this reduction of activity in brainstem monoaminergic neurons is permissive in the generation of REM sleep (Pace‐Schott & Hobson, 2002; McCarley, 2007). In other words, the models predict that noradrenergic and serotoninergic synaptic inputs inhibit both LDT/PPT cholinergic neurons (Fig. 2, Pathway 3, REM‐Off) and pontine REM‐generating neurons, including REM‐atonia neurons of the SLD (Fig. 2, Pathway 2, REM‐Off; Pace‐Schott & Hobson, 2002; McCarley, 2007).
To the foregoing, local application of noradrenaline or an α2‐adrenoceptor agonist, but not serotonin, in the peri‐LCα region in cats inhibits REM active neurons and induces REM sleep without atonia, robustly so when injections are placed in the caudal region of the peri‐LCα (Tononi et al. 1991; Sakai & Koyama, 1996; Crochet & Sakai, 1999 a,b). In agreement with the inhibitory response of noradrenaline on REM active peri‐LCα neurons, spinally projecting neurons of the SLD are also directly inhibited by noradrenaline (Williams et al. 2012). These findings suggest that the activity of SLD REM‐atonia neurons may be suppressed by noradrenaline released during wakefulness and, by extension, disinhibition of SLD neurons would permit the onset and maintenance of muscle atonia during REM (Fig. 2, Pathway 2, REM‐Off). Interestingly, antidepressants that are noradrenaline reuptake inhibitors, and hence increase noradrenergic tone, have been shown to be effective in reducing the occurrence of cataplexy attacks (Schachter & Parkes, 1980; Nishino & Mignot, 1997). Selective serotonin reuptake inhibitors and tricyclic antidepressants have likewise been used, albeit less frequently, to successfully suppress cataplexy (Gowda & Lundt, 2014).
GABAergic regulation
During wakefulness and, to a lesser extent, SWS, REM‐generator neurons in the pons are under strong GABAergic inhibitory tone, presumably to prevent them from firing (Xi et al. 1999 b; Luppi et al. 2006). This hypothesis derives support from the observation that local application of GABAA antagonists in the peri‐LCα of cats (Xi et al. 1999 a, 2001) or in the SLD of rats (Boissard et al. 2002; Pollock & Mistlberger, 2003; Sanford et al. 2003; Fenik & Kubin, 2009) rapidly produces a long‐lasting REM sleep‐like state characterized by desynchronized EEG and postural muscle atonia. Hence disinhibition from GABAergic inputs could be an important synaptic ‘mechanism’ by which SLD REM‐atonia neurons are activated on entry into REM sleep. But what is the synaptic source of SLD‐projecting GABAergic REM‐Off neurons? Studies combining retrograde tracers, immunolabelling for c‐Fos and glutamic acid decarboxylase (GAD) or in situ hybridization for GAD67 or GAD65 mRNAs have identified three potential primary sources of this input, which include the ventrolateral periaqueductal grey matter (vlPAG), the lateral pontine tegmentum (LPT) – also known as deep mesencephalic reticular nucleus (DpMe) – and the oralis pontine (PnO) region, which includes the SLD itself (Fig. 2, Pathway 7, REM‐Off; Boissard et al. 2003; Lu et al. 2006; Sapin et al. 2009).
With respect to these potential sources of GABAergic input, a dense projection from the vlPAG and LPT/DpMe to the SLD has been confirmed in different species (reviewed in Boissard et al. 2003) and, importantly, lesions of both the vlPAG and LPT/DpMe as well as pharmacological inactivation increase REM sleep and can generate a cataplexy‐like state (Sastre et al. 1996; Crochet et al. 2006; Lu et al. 2006; Vanini et al. 2007; Kaur et al. 2009; Sapin et al. 2009). Taken together the foregoing findings strongly suggest that GABAergic projections from the tegmental area may provide critical inhibitory control over the REM generator, including REM‐atonia neurons. Another potential source of SLD‐projecting GABAergic REM‐Off neurons is the PnO (Fig. 2, Pathway 7, REM‐Off), which includes local SLD GABAergic neurons (not represented in Fig. 2). The rostral PnO not only projects to the SLD (Lai et al. 1993; Semba, 1993; Boissard et al. 2003) but contains GABAergic REM‐Off neurons (Maloney et al. 2000). Moreover, antisense disruption of GABA synthesis within the SLD region of the PnO decreases wakefulness and increases REM sleep, suggesting the interesting possibility that local GABAergic SLD neurons may disinhibit glutamatergic SLD REM‐atonia neurons during REM sleep, and inhibit their activity during wakefulness (Xi et al. 1999 a).
In addition to their projections to glutamatergic SLD REM‐atonia neurons (Fig. 2, Pathway 7, REM‐Off), GABAergic REM‐Off neurons of the vlPAG and LPT/DpMe are reciprocally connected with GABAergic REM‐On neurons in the SLD region (Fig. 2, Pathway 6, REM‐On and Pathway 8, REM‐Off). This mutual inhibition has been proposed to form a ‘flip–flop’ switch that would sharpen state transitions, which are typical of the rapid switching from SWS to REM and vice versa (Lu et al. 2006; Fuller et al. 2007). This model has been challenged by the recent finding that disruption of the GABA/glycine transmission in the vlPAG and LPT/DpMe did not produce the predicted increase in REM sleep (Krenzer et al. 2011). Yet a more recent optogenetic study found that inhibition of vlPAG GABA neurons did, in fact, potently promote REM sleep (Weber et al. 2015). Interestingly, Weber et al. used GAD2‐cre mice, while Krenzer et al. used vGat‐cre mice, potentially highlighting alternative mechanisms of GABA transport that differentiate key REM‐inhibiting populations within the vlPAG. Another challenge to this model is the reported lack of REM‐active GABAergic neurons in the SLD region (Sapin et al. 2009), which conflicts with reports from two other groups who found REM‐On GABAergic neurons in the SLD (Maloney et al. 1999; Lu et al. 2006). If the former finding is correct, i.e. REM active GABAergic neurons locate to the vlPAG, not SLD, this might help explain the absence of a REM effect following disruption of GABA/glycine transmission in the vlPAG in the study by Krenzer et al. (2011).
Collectively the available experimental evidence, with minor exceptions, continues to support the flip–flop switch model for REM sleep regulation. In this circuit arrangement, two descending arms of the circuit (from vlPAG and LPT/DpMe REM‐Off GABAergic neurons) provides inhibitory control over SLD REM‐atonia neurons (Fig. 2, Pathway 7, REM‐Off) and SLD GABAergic REM‐Off neurons (Fig. 2, Pathway 8, REM‐Off) during wakefulness, whereas the ascending arm (from SLD GABAergic REM‐On neurons) inhibits REM‐Off vlPAG and LPT/DpMe neurons, and possibly the extra‐SLD PnO, (Fig. 2, Pathway 6, REM‐On) during REM sleep. By its design, this circuit would prevent the inappropriate activation of REM‐atonia neurons during wakefulness, which of course would result in cataplexy, and at the same time ensure rapid reconstitution of muscle tone on awaking. Determining the precise source(s) of GABAergic REM‐Off input to the SLD REM‐atonia neurons remains a clear experimental priority.
Glutamatergic regulation
In addition to disinhibition from monoaminergic and GABAergic inputs, there is emerging experimental evidence that a glutamatergic REM‐On input directly activates SLD REM‐atonia neurons during REM sleep (Fig. 2, Pathway 9, REM‐On; Luppi et al. 2012). For example, local injections of glutamate receptor agonists into the peri‐LCα of cats or into the SLD region of rats induce a REM‐like state with continuous muscle atonia (Lai & Siegel, 1991; Onoe & Sakai, 1995; Hajnik et al. 2000; Boissard et al. 2002). More importantly, local activation of glutamate receptor antagonists rapidly reverses muscle atonia induced by bicuculline application in rats, but does not alter the cortical REM‐like state, suggesting that glutamatergic input to the SLD is required to generate atonia during REM sleep but perhaps is not necessary for REM EEG desynchronization (Boissard et al. 2002). Hence, it remains possible that SLD REM‐atonia neurons are either tonically excited by glutamate during all sleep–waking states – but that this excitation increases further at the onset of REM sleep – or that there are glutamatergic REM‐On neurons that activate the SLD during REM (Luppi et al. 2012, 2013 b). Indeed data on the effects of glutamatergic antagonists in the SLD region across all sleep–wake states, and particularly during naturally occurring REM sleep, are eagerly awaited as they would shed considerable light on this important question.
An important technical consideration for the foregoing is that the methods to definitively identify glutamatergic neurons vis a vis vesicular transporters are relatively new. Previous studies on glutamatergic inputs to the SLD used glutamate antibodies, which lacked both sensitivity and specificity. Hence, the source of glutamatergic input to the SLD remains unknown. For example, the lateral hypothalamus, vlPAG and LPT/DpMe, and ventrolateral medulla all contain glutamatergic neurons and project to the SLD (Fig. 2, Pathway 9, REM‐On; Shammah‐Lagnado et al. 1987; Lai et al. 1993; Semba, 1993; Boissard et al. 2003), but whether or not the projections to the SLD from these regions are glutamatergic remains unknown. In addition, a small number of cells in the contralateral SLD region, and a larger number of cells in the ipsi‐ and contralateral pontine reticular formation, project to the SLD (Lai et al. 1993; Boissard et al. 2003), but their neurochemical phenotype, too, remains unknown. Defining which of these inputs to the SLD are both bona fide glutamatergic and contribute to the development of atonia should be achievable using newer technical approaches, including optogenetics and conditional retrograde tracing systems.
Orexin regulation
Orexin (or hypocretin) neurons are a discrete cluster of neurons in the posterior lateral hypothalamus (de Lecea et al. 1998; Sakurai et al. 1998). Orexin neurons have widespread projections (Peyron et al. 1998) and their receptors (Ox1R and Ox2R) are expressed in virtually all of the brain's major arousal centres, suggesting an important contribution of orexin signalling to the generation and maintenance of wakefulness (Sakurai et al. 2010). Indeed, optogenetic activation of orexin neurons triggers awakening from sleep (Adamantidis et al. 2007), although this effect is attributed to secondary activation of other arousal centres (Carter et al. 2010). Loss of orexin neurons, which is the neuropathological basis of narcolepsy, produces excessive daytime sleepiness, fragmented sleep and cataplexy (Peyron et al. 2000; Thannickal et al. 2000; Crocker et al. 2005; Burgess & Scammell, 2012). Narcoleptic patients also have REM sleep abnormalities including shortened REM sleep onset latency, vivid dreaming and a greater than expected occurrence of RBD, although the mechanism of the motor disturbances during REM sleep remains to be clarified (Frauscher et al. 2011).
There are now several animal models (naturally occurring mutations or genetically engineered) that reproduce human narcolepsy symptoms (sleepiness and cataplexy). All of these models exhibit disruptions in orexin signalling (Chen et al. 2009; Scammell et al. 2009). Disruption of Ox2Rs in dogs results in a phenotype characterized by sleepiness and severe cataplexy (Lin et al. 1999). Yet, interestingly, these dogs have normal cerebrospinal fluid orexin levels (John et al. 2004 b), suggesting that disruption of Ox2Rs is sufficient, at least in this species, to produce severe narcolepsy. In mice, however, fragmented wakefulness and cataplexy are only observed following disruption of the orexin peptide (Chemelli et al. 1999; Mochizuki et al. 2004), the orexin neurons themselves (Hara et al. 2001) or when both Ox1Rs and Ox2Rs are knocked out (Sakurai, 2007; Hasegawa et al. 2014). Mice lacking Ox1Rs or Ox2Rs, but not both concurrently, have milder sleep phenotypes and cataplexy (Willie et al. 2003; Mieda et al. 2011; Mochizuki et al. 2011 and reviewed in Sakurai, 2007).
Physiologically, orexin neurons are active during wakefulness and fire in association with movement. A decrease in firing is observed during quiet waking and these neurons fall silent during SWS and REM sleep, although transient bursts of action potentials do occur during REM sleep (Lee et al. 2005; Mileykovskiy et al. 2005). Of note, extracellular orexin levels in the basal forebrain and lateral hypothalamus are high during wakefulness, low during SWS sleep, and rise again during REM sleep (Kiyashchenko et al. 2002), which is an apparent paradox given that orexin neurons are generally quiescent during REM sleep.
With respect to the regulation of REM sleep, orexin neurons innervate REM sleep nodes in the pons, including the SLD (Fig. 2, Pathway 10, REM‐Off; Zhang et al. 2004; Torterolo et al. 2013). More specifically, orexin neurons innervate noradrenergic and serotonergic REM‐Off neurons of the LC and DRN (Fig. 2, Pathway 11, REM‐Off; Peyron et al. 1998; Date et al. 1999; Sakurai et al. 2005) as well as REM‐Off vlPAG and LPT/DpMe neurons (Fig. 2, Pathway 12, REM‐Off; Peyron et al. 1998; Boissard et al. 2003; Lu et al. 2006). Electrophysiological data further reveal that orexin directly activates LC and DRN neurons (Ivanov & Aston‐Jones, 2000; Brown et al. 2002; Liu et al. 2002; Murai & Akaike, 2005; Kohlmeier et al. 2008, 2013). It has been proposed that orexin might promote REM sleep by directly activating SLD REM‐On neurons (Xi et al. 2002, 2003). This conclusion is, however, not fully supported by experimental findings; for example, orexin neurons fire only occasionally during REM sleep and direct evidence for orexin release in the pons during REM sleep is lacking. On the other hand, orexin injected directly into the dorsal pons – encompassing the SLD region – prolongs waking in animals that are already awake whereas a prolonged state of REM sleep is induced if orexin is applied during SWS (Xi & Chase, 2010).
To explain the foregoing results, the authors proposed that orexin might act on different components of the REM circuit, and thereby bias wakefulness or REM sleep, depending on the state of activation or inhibition of the different nodes (Xi & Chase, 2010). While an intriguing hypothesis, confirmation will require additional experimental work to elucidate a functional circuit framework, which is currently lacking. An alternative hypothesis is that orexin is released in the pons during wakefulness and potentiates REM‐Off GABAergic inhibition of SLD REM‐atonia neurons (Luppi et al. 2006; Lu et al. 2006). Hence orexin projections to the vlPAG and LPT/DpMe (Boissard et al. 2003; Lu et al. 2006) and to the SLD (Zhang et al. 2004; Torterolo et al. 2013) may activate, respectively, vlPAG/LPT/DpMe REM‐Off GABAergic neurons (Fig. 2, Pathway 12, REM‐Off), as well as their presynaptic terminals to SLD REM‐atonia neurons, to prevent cataplexy (Fig. 2, Pathway 10, REM‐Off).
As indicated, positive emotions are the most common trigger for cataplexy in narcoleptic humans (joking, laughter, being tickled or a pleasant surprise; Anic‐Labat et al. 1999; Overeem et al. 2011) and probably as well for dogs (palatable food or play; Mitler et al. 1974; Nishino & Mignot, 1997; Tonokura et al. 2007; Scammell et al. 2009) and mice (palatable food such as chocolate, running wheels, or group housing; Chemelli et al. 1999; Espana et al. 2007; Clark et al. 2009; Scammell et al. 2009; Oishi et al. 2013). In humans anger is also a trigger (Anic‐Labat et al. 1999; Overeem et al. 2011), whereas aversive situations do not trigger cataplexy in dogs (Blouin et al. 2013), but do in narcoleptic cattle and sheep (Strain et al. 1984; White & de Lahunta, 2001). Strong emotions as a ‘trigger’ thus appear to be a common denominator whereas the positive vs. negative valence of the trigger seems to be species‐specific. The circuit mediating emotion‐driven cataplexy is also incompletely understood. It has been proposed, for example, that strong emotions might activate the central and basolateral nucleus of the amygdala (CeA and BLA; Garavan et al. 2001, Straube et al. 2008), possibly following initial activation of the medial prefrontal cortex (mPFC; Damasio et al. 2000, Sabatinelli et al. 2007, Ponz et al. 2010, Etkin et al. 2011, Oishi et al. 2013). Interestingly both the CeA and BLA contain neurons that are active during cataplexy (Gulyani et al. 2002), and inhibitory (vesicular GABA transporter, vGat positive) neurons of the CEA heavily innervate neurons of the vlPAG/LPT/DpMe (Rizvi et al. 1991, Oka et al. 2008, Burgess et al. 2013), which are possibly GABAergic REM‐Off. Under non‐pathological conditions, inhibitory inputs to the vlPAG/LPT/DpMe are opposed by excitatory orexin inputs (Peyron et al. 1998, Boissard et al. 2003, Lu et al. 2006) and maintain inhibitory tone of vlPAG/LPT/DpMe to the SLD REM‐atonia neurons during strong emotions. In narcoleptic patients, however, loss of orexin inputs may ‘disfacilitate’ vlPAG/LPT/DpMe REM‐Off neurons, which could in turn trigger the inappropriate activation of SLD neurons during wake and ultimately lead to loss of postural muscle tone (Lu et al. 2006; Luppi et al. 2006; Burgess & Scammell, 2012). Additional circuit mapping experiments will be required to confirm this model of emotion‐driven cataplexy, and are eagerly awaited.
MCH regulation
Intermingled with orexin neurons is another cell population containing melanin‐concentrating hormone (MCH) peptide. Orexin and MCH neurons have similar projections but their firing patterns across the sleep–wake cycle are roughly opposite, and they are thought to produce opposite effects on their postsynaptic targets (Adamantidis & de Lecea, 2008; Espana & Scammell, 2011; Jones & Hassani, 2013). They are also reciprocally regulated (Gao, 2009): MCH inhibits orexin neurons by depressing the glutamatergic input to orexin neurons (Rao et al. 2008) whereas orexin neurons inhibit MCH neurons by an orexin‐mediated feed‐forward inhibition (Apergis‐Schoute et al. 2015). While MCH neurons are best known for their regulatory role in energy homeostasis (Pissios et al. 2006), several recent studies have demonstrated that they are specifically active during REM sleep (Verret et al. 2003; Hanriot et al. 2007; Hassani et al. 2009). Even more recent experimental work employing optogenetic activation in behaving animal models has shown that acute activation of MCH neurons promotes REM sleep (Jego et al. 2013; Tsunematsu et al. 2014) or both REM and SWS sleep (Konadhode et al. 2013). Thus MCH neurons appear to be involved in both REM and SWS regulation, with the caveats that (1) their ability to drive SWS may be time of day dependent (Jones & Hassani, 2013; Konadhode et al. 2013) and (2) on the basis of silencing and ablation studies (Jego et al. 2013; Tsunematsu et al. 2014), their role in the induction or maintenance of REM sleep could be minor.
The postsynaptic targets and synaptic mechanisms through which MCH neurons promote REM and SWS remain unclear. MCH is an inhibitory peptide that acts through both presynaptic and postsynaptic mechanisms, although presynaptic action seems to be more common (Gao, 2009; van den Pol, 2012). MCH neurons have also been thought to produce and release GABA (Elias et al. 2001; Harthoorn et al. 2005; Del Cid‐Pellitero & Jones, 2012) and optogenetic activation of MCH terminals elicits GABA release within the tuberomammillary nucleus (Jego et al. 2013). On the other hand, other recent studies have reported that MCH neurons do not contain the vesicular GABA transporter (vGat), and hence may not be capable of releasing synaptic GABA (Chee et al. 2015). If MCH neurons indeed release synaptic GABA, they must rely on another, as yet unidentified vesicle transporter, not vGat, to package GABA.
It has been proposed that MCH neurons may contribute to the suppression of the activity of REM‐Off neurons (LC, DRN, and vlPAG and LPT/DpMe neurons) during REM sleep (Fig. 2, Pathways 13 and 14, REM‐On; Monti et al. 2013; Luppi et al. 2013 a) through the release of GABA and MCH. In general support of this model are the findings that MCH neurons send dense projections to all of these regions (Bittencourt et al. 1992; Clement et al. 2012; Del Cid‐Pellitero & Jones, 2012; Yoon & Lee, 2013), that MCH microinjected in the LC or DRN increases the number of REM sleep episodes (Lagos et al. 2009; Monti et al. 2015), and that MCH inhibits the firing of DRN neurons (Devera et al. 2015). If MCH neurons do not indeed package and release synaptic GABA, they may alternatively inhibit REM‐Off neurons directly through MCH release or indirectly through glutamate‐mediated feedforward inhibition (Chee et al. 2015).
MCH neurons also project to REM‐On neurons in the PnO region, which, again, is a pontine region that includes the SLD (Torterolo et al. 2013). Therefore MCH neurons might influence SLD control of muscle atonia during REM sleep via this input. Microinjections of MCH into PnO/SLD significantly increase REM sleep and decrease latency to REM sleep onset, supporting the hypothesis that the MCH system contributes to the generation of both EEG and EMG aspects of REM sleep through pontine circuits (Torterolo et al. 2009; Fig. 2, Pathway 15, REM‐On). How MCH acts on SLD REM‐On neurons is not fully understood, although it is possible that MCH neurons directly activate REM‐On neurons in the region, including SLD REM‐atonia neurons, vis a vis glutamate release. Confirmation of this intriguing possibility will require additional experimental work.
Outputs: descending SLD circuits
SLD REM‐atonia neurons are largely, if not exclusively, glutamatergic. During both REM sleep and REM sleep rebound most c‐Fos positive SLD neurons co‐localize the vesicular glutamate transporter 2 (vGlut2), which is a specific marker for glutamatergic neurons (Lu et al. 2006; Clement et al. 2011). Focal disruption of glutamatergic transmission by SLD neurons in mice produces REM sleep without atonia, which is phenotypically very similar to that observed in human RBD cases (Krenzer et al. 2011). And while is it generally accepted as fact that glutamatergic SLD neurons play a key role in generating postural atonia during REM sleep, the descending pathway by which they do so remains a subject of debate. Specifically, the respective contributions of the direct (SLD→spinal ventral horn; Fig. 2, Pathways 17 and 21 REM‐On) versus indirect (i.e. SLD→ventromedial medulla→spinal ventral horn; Fig. 2, Pathways 16 and 20, REM‐On) projection systems in mediating REM atonia (Fuller et al. 2007; Brown et al. 2012; Chase, 2013; Luppi et al. 2013 b) remain to be clarified, although these two synaptic pathways are likely to provide synergistic control of REM sleep atonia.
The first, ‘direct’ synaptic model proposes that SLD REM‐atonia neurons send axons directly to the spinal cord (Fig. 2, Pathways 17 and 21, REM‐On), forming appositions with parvalbumin‐immunoreactive neurons in lamina VIII (Lu et al. 2006; Fuller et al. 2007), most of which belong to the class V1 interneurons that are known to project to spinal motor neurons of layer IX and to contain glycine, GABA or both (Taal & Holstege, 1994; Alvarez et al. 2005). Focal disruption of glycinergic/GABAergic transmission in the spinal ventral horn produces phasic movements during REM sleep (Krenzer et al. 2011). One challenge to this model is the finding of a previous single unit recording study in cats that reported that spinally projecting neurons in the peri‐LCα were inactive during REM sleep (Sakai et al. 1981). However, only a relatively small number of neurons were sampled and, moreover, many of the recording neurons may have been noradrenergic neurons, which are known to be silent during REM sleep (Aston‐Jones & Bloom, 1981; Bruinstroop et al. 2012). And it is the case that in cats, which is the species used by Sakai et al., glutamatergic SLD neurons intermingle with noradrenergic neurons of the LC complex. Additional studies are therefore required to clarify the circuitry through which SLD neurons directly generate the descending signal for REM muscle atonia.
The second ‘indirect’ synaptic model proposes that SLD glutamatergic REM‐atonia neurons send projections to glycinergic neurons in the ventromedial medulla (VMM), including the ventral gigantocellular reticular nucleus (GiV) and α‐gigantocellular reticular nuclei (GiA) and the adjacent lateral paragigantocellular (LPGi) group (Figs 1 and 2, Pathways 16 and 20, REM‐On; Boissard et al. 2002; Morales et al. 2006; Sapin et al. 2009). Even before the discovery of REM sleep, the medulla was thought to play a key role in the regulation of the overall tone of an animal's spinal motor systems. In 1898, Sherrington described a chronic rigidity resulting from the removal of the cerebral cortex (Sherrington, 1898). Transections through the rostral pons, on the other hand, produced apparent atonia (Keller, 1945). These results seem conflicting: on the one hand, loss of cortical inputs produced hypertonia or rigidity while on the other hand, brainstem transections produced hypotonia/atonia. One potential explanation is the presence of inhibitory brainstem nodes that project to and actively suppress spinal motor function. And, in fact, these putative inhibitory neurons are the likely target of SLD glutamatergic REM‐atonia neurons.
Medullary control of REM sleep and postural atonia
From a historical perspective, atonia was first thought to arise from a ‘tonic tonus‐inhibiting circuit’ in the brainstem that normally receives an antagonistic input (Keller, 1945). In this model, rostral pontine transections released this antagonistic input, allowing the overriding tonus inhibition to win out and suppress spinal muscle tone. Magoun & Rhines (1946) provided critical evidence for the medulla's role in this tonus‐inhibiting circuit. Using electrode stimulation of the medulla, they found inhibition of muscle reflexes and other forms of spinal motor excitation. The effective stimulation points were found to be along the VMM, spanning the rostral–caudal axis of the medulla and including neurons in the GiV and GiA nuclei. This inhibition was due to inhibitory postsynaptic potentials (IPSPs) on spinal motor neurons, which could be produced by stimulation of the medulla (Llinas & Terzuolo, 1964) in the same location of the medullary inhibiting region described by Magoun and colleagues (Jankowska et al. 1968). Given the similarities between this experimental inhibition and REM sleep atonia (Gassel et al. 1964, 1965; Morrison & Pompeiano, 1965; Kubota & Kidokoro, 1966), investigation focused on the VMM and its role in generating postural motor atonia (Pompeiano, 1967; Steriade & Hobson, 1976). It is the case that differences in experimental procedures (e.g. lesion vs. stimulation) and the brainstem anatomy of the experimental models (rats or mice vs. cats) has complicated the identification and characterization of the delimited medullary region mediating REM atonia (Fig. 1). Yet, despite these complications, common neuroanatomical features have emerged: a medullary REM sleep atonia zone near the rostral tip of the inferior olive, centred on the sagittal midline.
VMM neurons, including the more medial GiV and GiA groups and the lateral adjacent LPGi group, are maximally active during REM sleep (Siegel et al. 1979; Kanamori et al. 1980; Boissard et al. 2002; Morales et al. 2006; Sapin et al. 2009) and cataplexy (Siegel et al. 1991). Unit recording shows that these VMM neurons discharge tonically across the sleep–wake cycle, with a progressive increase in firing rate as the animal progresses from active wake to SWS, a dramatic increase in firing rate during the transition into REM, and an equally dramatic slowing of firing upon awaking (Kanamori et al. 1980; Chase et al. 1984). Medullary lesions also produce REM sleep without atonia or other abnormal behaviours during REM sleep (Schenkel & Siegel, 1989; Holmes & Jones, 1994; Lai & Siegel, 1997; Hajnik et al. 2000; Vetrivelan et al. 2009). These ‘effective’ lesions were focused on a specific rostral–caudal level of the medulla, near Magoun's inhibitory centre (Fig. 1). For example, large lesions of GiV and GiA neurons near the rostral pole of the medulla, i.e. near the pontomedullary junction, do not alter postural muscle tone during REM sleep (Sastre et al. 1981; Lu et al. 2006), whereas lesions at a more caudal level, i.e. at the level of the inferior olive, lead to exaggerated muscle twitches during REM sleep (Schenkel & Siegel, 1989; Holmes & Jones, 1994; Lai & Siegel, 1997; Hajnik et al. 2000; Vetrivelan et al. 2009). Both lesion (Schenkel & Siegel, 1989) and stimulation (Takakusaki et al. 2001; Habaguchi et al. 2002) studies indicated that the zone of atonia may extend dorsally above the area adjacent to the inferior olive, although the involvement of these regions may reflect the use of electrical stimulation/lesions that disrupt pontomedullary dialogue critical for REM sleep atonia. Lesion, recording and stimulation experiments suggest that this medullary inhibitory zone is present and functional at birth (Karlsson & Blumberg, 2005).
VMM neurons in the medullary inhibitory region are a mix of glutamatergic, GABA/glycinergic, cholinergic and serotonergic neurons (Reichling & Basbaum, 1990; Jones et al. 1991; Holmes & Jones, 1994; Hossaini et al. 2012). More caudally and laterally, adrenergic REM‐On neurons may play a role in sympathetic activity during REM sleep (Stettner et al. 2013). In generating the postural motor atonia of REM sleep, the indirect model posits that glutamatergic SLD neurons project to inhibitory medullary neurons, which are likely to be GABA/glycinergic. The GiV and GiA neurons of this relay project to spinal and brainstem motor neurons (Fig. 2, Pathway 20, REM‐On; Chase et al. 1984, 1986; Holstege & Bongers, 1991; Castillo et al. 1991 a; Kato et al. 2006) and when stimulated (electrically or pharmacologically; Chase et al. 1984, 1986; Soja et al. 1987 b; Lai & Siegel, 1988; Castillo et al. 1991 a,b; Holstege & Bongers, 1991; Kodama et al. 2003; Kato et al. 2006; Lai et al. 2010) produce short latency glycinergic IPSPs in postsynaptic motor neurons and evoke release of glycine – and possibly GABA – in the spinal ventral horn (Chase et al. 1986; Soja et al. 1987 b; Lai & Siegel, 1988; Castillo et al. 1991 b; Kodama et al. 2003; Lai et al. 2010). Consistent with this model, glutamate release within the ventromedial medulla increases as animals enter REM sleep (Kodama et al. 1998). In addition, endogenous blockade of glutamatergic signalling onto these neurons reverses spinal postural muscle atonia during carbachol‐induced REM sleep (Lai & Siegel, 1988), suggesting that glutamatergic input, likely to be from the SLD, is required for the ventromedial medulla pathway to produce muscle atonia.
One challenge to this model is the finding that elimination of glutamate but not GABA/glycine transmission from medullary neurons at the level of the inferior olive or supraolivary medulla (see Fig. 2, Pathways 18 and 22, REM‐On) results in muscle twitches during REM sleep atonia (Vetrivelan et al. 2009). These findings raise the possibility of an additional medullary relay, excitatory rather than inhibitory, which the authors hypothesized may project to inhibitory spinal interneurons (Takakusaki et al. 2001, 2003). These results also emphasize the importance of the need for greater anatomical precision in defining the rostral–caudal level of the medulla in REM sleep atonia control. For example, GABA/glycine neurons that project to the ventral spinal horn are relatively sparse at the caudal ventromedial medulla compared to levels rostral to the inferior olive (Hossaini et al. 2012). Hence the putative GABA/glycine REM sleep inhibitory region of the medulla is likely to be more rostral to the level that was previously studied (Vetrivelan et al. 2009), near the rostral inferior olive. This putative GABA/glycine population also receives SLD projections but may directly target and inhibit spinal motor neurons.
Taken together, transection, lesion and recording experiments indicate a medullary inhibitory zone within the VMM near the rostral inferior olive that includes GiA, GiV and LPGi cell groups (Fig. 2, Pathways 16 and 20, REM‐On). This zone contains glycinergic and possibly GABAergic pre‐motor neurons (Boissard et al. 2002; Morales et al. 2006; Sapin et al. 2009). During REM sleep SLD neurons activate these medullary neurons, which then directly inhibit spinal motor neurons to maintain muscle atonia (Siegel, 2011; Luppi et al. 2012; Chase, 2013). Cell groups rostral to this zone are likely to not be involved in REM sleep atonia (Sastre et al. 1981; Lu et al. 2006), as even large cell‐body lesions do not significantly disrupt REM sleep atonia. In contrast, cells caudal to this zone may regulate twitching and other motor movements in REM sleep using glutamatergic signalling (Fig. 2, Pathways 18 and 22, REM‐On; Vetrivelan et al. 2009).
In addition to its role as a relay within a feed‐forward model of atonia, i.e. SLD→VMM→spinal ventral horn, the medulla may play an active role in shaping REM sleep generation and, ultimately, motor control. Moreover, the medulla's inhibitory influence may depend on reciprocal interaction between medullary and pontine REM sleep centres. For example, loss of rostral brainstem innervation of the medulla due to experimental transection prevents the motor suppression evoked from medial medulla stimulation (Siegel et al. 1983). Specifically, pontine inhibition reduces the medulla's ability to suppress muscle activity (Kohyama et al. 1998) and inactivation of the pons blocks medullary‐induced muscle tone suppression in the decerebrate cat, suggesting that medulla activity alone is insufficient to generate REM sleep atonia. While medullary activity may normally depend on pontine activity, these studies raise the additional possibility that VMM control of muscle suppression or REM sleep altogether acts via ascending drive to the SLD and other rostral brainstem areas. In this model, REM sleep and atonia generation is initiated in the medulla, rather than the SLD, and atonia is achieved via ascending projections to the SLD. Several lines of evidence support this alternative model. First, large SLD lesions reduce, but do not eliminate, REM sleep, suggesting a supplementary or alternative circuit capable of generating REM sleep (Lu et al. 2006), perhaps in the medulla (Weber et al. 2015). Second, investigations into the projection from the medial medulla to the spinal cord motoneurons and interneurons have not revealed a robust mechanism for transmission of the inhibitory atonia signal (Takakusaki et al. 1989). Third, numerous imaging studies of humans with RBD have documented lesions mostly in the pontomedullary junction, rather than the ventral medulla (Scherfler et al. 2011; St Louis et al. 2014, McCarter et al. 2015). While none of these lines of evidence is conclusive, they raise the possibility of an alternative to the linear, feed‐forward model. Further investigations that identify the critical neurons, their neurochemistry and their interactions with pontine REM sleep‐promoting structures will help define the respective roles of the medulla and pons in REM sleep and atonia generation.
REM atonia at the level of the motor neuron
A series of studies in the 1960s were the first to implicate inhibitory supraspinal inputs in spinal cord motor neuron inhibition (Giaquinto et al. 1964 b; Gassel et al. 1965). Later in the 1970s and 1980s a series of seminal intracellular recording studies – which informed the development of the REM‐On glycinergic pre‐motor neuron model – were conducted that measured the membrane potential of motor neurons during naturally occurring REM sleep (Giaquinto et al. 1964 a; Morales & Chase, 1978; Nakamura et al. 1978; Chase et al. 1980; Glenn & Dement, 1981 b). These studies found that, at the onset of REM sleep, spinal postural and cranial orofacial motor neurons receive large‐amplitude glycinergic IPSPs that hyperpolarize their membrane potential (∼ −10 mV). This hyperpolarized state is maintained for the entirety of the REM periods whereas the membrane of the motor neurons repolarizes upon awakening (Fig. 2, Pathways 20 and 21, REM‐On). Juxtacellular microiontophoretic application of strychnine, but not GABAA antagonists, blocks these large‐amplitude REM‐On IPSPs and prevents REM membrane hyperpolarization of spinal motor neurons (Glenn & Dement, 1981 a; Morales & Chase, 1982; Morales et al. 1987; Chase et al. 1989; Soja et al. 1991). These same glycinergic IPSPs were also observed during pharmacologically induced REM sleep (i.e. injections of carbachol or bicucculine in the PnO/peri‐LCα) as well as following electrical stimulation of the ventromedial medulla (Chase et al. 1986; Soja et al. 1987 b, Kohlmeier et al. 1996; Xi et al. 2001). And, as indicated, focal disruption of spinal ventral horn glycinergic/GABAergic transmission results in aberrant phasic activity during REM sleep (Krenzer et al. 2011). Taken together, these findings provide strong support for a motor‐inhibition model in which postsynaptic inhibitory drive to spinal postural and possibly brainstem motor neurons, be that during natural or pharmacologically induced REM sleep, is primarily mediated by glycine synaptic transmission. However an important consideration, which may link to technical limitations for the spinal postural system, is that direct evidence that these glycine‐mediated IPSPs are responsible for motor atonia is lacking.
To the extent that the ‘glycinergic’ model has provided an important framework for understanding the pathophysiology of a wide range of REM‐based disorders, there have been two notable challenges to this model (Soja et al. 1987 a; Brooks & Peever, 2008 a). Soja et al. found that strychnine applied onto the trigeminal motor nucleus has only a small effect in reactivating the masseteric reflex during REM sleep. In the case of Brooks and Peever, reverse microdialysis was used to apply glycinergic and GABAA antagonists onto trigeminal motor neurons during wakefulness, SWS and REM sleep, revealing a tonic glycinergic/GABAergic drive during wakefulness and SWS that was, surprisingly, absent during REM sleep. This finding was unexpected and essentially refuted two decades of results obtained with intracellular recordings of spinal and brainstem motor neurons (Chase, 2013). A lively and energetic discussion ensued (Berger, 2008; Chase, 2008, 2009; Funk, 2008; Kubin, 2008; Lydic, 2008; Soja, 2008; Brooks & Peever, 2008 b). More recently Brooks and Peever revised their initial model to incorporate multiple receptors, including GABAA, GABAB, glycine, glutamate and noradrenaline, as being involved in the inhibition and disfacilitation of cranial orofacial motor neurons to trigger REM sleep muscle atonia (Brooks & Peever, 2012; Schwarz et al. 2014).
In addition to direct inhibition, a disfaciliation mechanism has been proposed to explain REM‐related suppression of motor neuron activity. The disfacilitatory mechanism was first described in cranial respiratory muscles (Kubin et al. 1992, 1998), and invoked the silencing of brainstem noradrenergic and serotoninergic neurons during REM sleep in the reduction of excitatory drive to motor neurons. In 1993, Kubin et al. proposed that cranial respiratory and spinal postural motor activity may be differently regulated during REM sleep. More specifically, Kubin et al. postulated that inactivation of monoaminergic systems was responsible for the suppression of cranial respiratory muscles (disfacilitation) whereas activation of glycinergic input was responsible for suppression of spinal motor neurons (direct inhibition; Kubin et al. 1993). In 2001 this hypothesis was challenged by the finding that a significant and similar reduction of noradrenaline and serotonin release occurs in both the hypoglossal nucleus and spinal cord when motor atonia is induced, suggesting that disfacilitation contributes to muscle atonia in both systems (cranial respiratory and postural muscles; Lai et al. 2001). In general support of a contributing disfacilitatory mechanism for REM sleep atonia of spinal postural muscles, noradrenaline and serotonin excite spinal motor neurons through α1 and 5HT2 receptors (White et al. 1991, 1996). The observed silencing of LC neurons and reduction in firing of DRN neurons during cataplexy (Wu et al. 1999; John et al. 2004 a) also suggests an important contribution of monoaminergic excitation of motor neurons for the maintenance of postural tone; however, monoaminergic involvement in preventing cataplexy may still be mediated through other nodes rather than through direct spinal control. For example, recent studies have shown that disfacilitation of brainstem motor neurons from excitatory noradrenergic drive is insufficient to trigger atonia in postural muscles during REM sleep (Schwarz et al. 2014).
Also, as indicated, the medullary brainstem contains cholinergic neurons (Armstrong et al. 1983) that may be REM‐On (Holmes & Jones, 1994; Volgin et al. 2008; Grace et al. 2013). These neurons, in fact, directly innervate the facial, trigeminal and hypoglossal motor neurons (Fort et al. 1989, 1990; Travers et al. 2005) and therefore they could inhibit the pharyngeal motor neurons directly (Liu et al. 2005; Grace et al. 2013), although this has not been definitely demonstrated. In summary, the action of multiple neurotransmitters (i.e. glycine, GABA, monoamines, acetylcholine) may be required for complete REM sleep atonia, with direct inhibition of motor neurons being the most important synaptic mechanism mediating REM atonia of the postural muscles.
Cranial respiratory muscles also undergo REM sleep suppression of activity, with the genioglossus (GG) muscle of the tongue arguably showing the most dramatic suppression of activity (Horner, 2009). Work by several groups employing drug delivery via microdialysis while measuring GG activity has concluded that, unlike postural muscles, suppression of glycinergic and GABAA transmission contributes minimally to suppression of GG activity during REM sleep (Kubin et al. 1993; Morrison et al. 2003 a). And further upstream, the reduction of activity in hypoglossal neurons controlling GG, and perhaps all cranial respiratory motor neurons, is thought to be mediated by noradrenergic and glutamatergic disfacilitation (Lai et al. 2001; Fenik et al. 2005 a; Steenland et al. 2008 b). Yet, interestingly, application of an α1‐adrenoceptor agonist onto the hypoglossal motor pool fails to reactivate GG activity (Chan et al. 2006) suggesting that, in addition to a disfacilitation mechanism, a powerful inhibitory mechanism is operative. A competing hypothesis is that hypoglossal motor neurons are directly inhibited during REM sleep by cholinergic, GABAergic and/or glycinergic signals (Brooks & Peever, 2010; Grace et al. 2013; Fung & Chase, 2015). Intracellular recordings have demonstrated that during the transition from SWS to REM, hypoglossal motor neurons start receiving large‐amplitude IPSPs, likely to be mediated by glycine release, and that they become hyperpolarized, as is seen in spinal and brainstem motor neurons (Fung & Chase, 2015). These recording studies therefore suggest an active inhibition vis a vis glycinergic inputs. However, blocking GABAergic and glycinergic transmissions in the hypoglossal motor neurons only partially increases GG activity during REM sleep (Kubin et al. 1993; Morrison et al. 2003 a,b). On the other hand, blocking cholinergic transmission fully restores GG activity to SWS levels (Grace et al. 2013), suggesting that cholinergically mediated inhibition of hypoglossal motor neurons is principally responsible for the suppression of GG activity during REM sleep. Taken together, experimental findings support a coordinated role for disfacilitation and direct inhibition by acetylcholine in the suppression of GG activity during REM sleep.
Across REM sleep, tonic muscle atonia is periodically interrupted by a volley of muscle twitches and jerks, i.e. phasic activity. This myoclonic activity is mediated by a glutamatergic input and by the activation of AMPA postsynaptic glutamate receptors in spinal postural and cranial orofacial motor neurons (Chase & Morales, 1982, 1983; Soja et al. 1995; Burgess et al. 2008). Importantly, blockade of glutamatergic transmission also reduces muscle tone during wakefulness to levels measured during SWS, and has no effect on tonic EEG activity during SWS and REM sleep, indicating that muscle tone is maintained by a glutamatergic drive only during wakefulness. Moreover, application of glutamatergic agonists during REM sleep does not reverse muscle atonia, confirming that REM atonia is not the result of disfacilitation from a glutamatergic excitatory input. The lack of response to glutamate agonists also strongly suggests that motor neurons are actively inhibited during REM sleep (Berger, 2008; Funk, 2008).
Conclusion
Elucidating the synaptic and cellular mechanisms mediating REM sleep atonia continues to be an important experimental pursuit and may have clinical implications reaching far beyond that of treating RBD, cataplexy and sleep disordered breathing. For example, in Parkinson's with RBD, restoration of normal motor control can occur during REM sleep (De Cock et al. 2007). Elucidating the ‘circuit basis’ for this intriguing if puzzling observation may therefore inform a novel therapeutic approach for treating the motoric dysfunction of PD. We moreover propose that newer genetically driven techniques (Adamantidis et al. 2007; Anaclet et al. 2014, 2015; Xu et al. 2015) can and should be employed to fill the existing knowledge gaps. For example, acute inhibition of genetically defined pontine or medullary cell groups or their terminal fields during REM sleep would facilitate a more complete understanding of the roles of specific cell groups in mediating motor atonia, as well as help clarify the synaptic mechanisms, e.g. disfacilitation versus inhibition, by which motor neurons, be they spinal postural, cranial orofacial or cranial respiratory, are themselves suppressed.
Additional information
Competing interests
None declared.
Author contributions
All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was supported by NIH grants: R21NS082854, R01NS073613, R01NS092652 and P01HL095491.
Acknowledgements
We thank Drs Takatoshi Mochizuki and Tana Hoban‐Higgins for helpful comments on the manuscript.
Biographies
Elda Arrigoni is a principal investigator at Harvard Medical School and within the Department of Neurology at Beth Israel Deaconess Medical Center.
Michael C. Chen is a postdoctoral fellow at Harvard Medical School and Beth Israel Deaconess Medical Center.
Patrick M. Fuller is a principal investigator at Harvard Medical School and within the Department of Neurology at Beth Israel Deaconess Medical Centre. They share a common interest in understanding the neuroanatomical, cellular and synaptic bases by which the brain regulates sleep and wakeful consciousness. It is their expectation that this work will enable a greater understanding of the neuropathology underlying a wide range of sleep–wake, neurological and psychiatric disorders. Their experiments employ a wide range of methodologies including morphological methods, genetic engineering techniques in mice and rats, in vivo and in vitro electrophysiology, in vivo imaging, optogenetics, chemogenetics, viral reagents and conditional mapping systems.

And his eyes have all the seeming of a demon's that is dreaming – Edgar Allan Poe, The Raven
Contributor Information
Elda Arrigoni, Email: earrigon@bidmc.harvard.edu.
Patrick M. Fuller, Email: pfuller@bidmc.harvard.edu
References
- Adamantidis A & de Lecea L (2008). Physiological arousal: a role for hypothalamic systems. Cell Mol Life Sci 65, 1475–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K & de Lecea L (2007). Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature 450, 420–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez FJ, Jonas PC, Sapir T, Hartley R, Berrocal MC, Geiman EJ, Todd AJ & Goulding M (2005). Postnatal phenotype and localization of spinal cord V1 derived interneurons. J Comp Neurol 493, 177–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anaclet C, Ferrari L, Arrigoni E, Bass CE, Saper CB, Lu J & Fuller PM (2014). The GABAergic parafacial zone is a medullary slow wave sleep‐promoting centre. Nat Neurosci 17, 1217–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anaclet C, Pedersen NP, Ferrari LL, Venner A, Bass CE, Arrigoni E & Fuller PM (2015). Basal forebrain control of wakefulness and cortical rhythms. Nat Commun 6, 8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anic‐Labat S, Guilleminault C, Kraemer HC, Meehan J, Arrigoni J & Mignot E (1999). Validation of a cataplexy questionnaire in 983 sleep‐disorders patients. Sleep 22, 77–87. [PubMed] [Google Scholar]
- Apergis‐Schoute J, Iordanidou P, Faure C, Jego S, Schone C, Aitta‐Aho T, Adamantidis A & Burdakov D (2015). Optogenetic evidence for inhibitory signaling from orexin to MCH neurons via local microcircuits. J Neurosci 35, 5435–5441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong DM, Saper CB, Levey AI, Wainer BH & Terry RD (1983). Distribution of cholinergic neurons in rat brain, demonstrated by the immunocytochemical localization of choline acetyltransferase. J Comp Neurol 216, 53–68. [DOI] [PubMed] [Google Scholar]
- Aston‐Jones G & Bloom FE (1981). Activity of norepinephrine‐containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep‐waking cycle. J Neurosci 1, 876–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baghdoyan HA, Rodrigo‐Angulo ML, McCarley RW & Hobson JA (1987). A neuroanatomical gradient in the pontine tegmentum for the cholinoceptive induction of desynchronized sleep signs. Brain Res 414, 245–261. [DOI] [PubMed] [Google Scholar]
- Beersma DG, Dijk DJ, Blok CG & Everhardus I (1990). REM sleep deprivation during 5 hours leads to an immediate REM sleep rebound and to suppression of non‐REM sleep intensity. Electroencephalogr Clin Neurophysiol 76, 114–122. [DOI] [PubMed] [Google Scholar]
- Berger AJ (2008). What causes muscle atonia in REM? Sleep 31, 1477–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bittencourt JC, Presse F, Arias C, Peto C, Vaughan J, Nahon JL, Vale W & Sawchenko PE (1992). The melanin‐concentrating hormone system of the rat brain: an immuno‐ and hybridization histochemical characterization. J Comp Neurol 319, 218–245. [DOI] [PubMed] [Google Scholar]
- Blouin AM, Fried I, Wilson CL, Staba RJ, Behnke EJ, Lam HA, Maidment NT, Karlsson KAE, Lapierre JL & Siegel JM (2013). Human hypocretin and melanin‐concentrating hormone levels are linked to emotion and social interaction. Nat Commun 4, 1547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blouin AM, Thannickal TC, Worley PF, Baraban JM, Reti IM & Siegel JM (2005). Narp immunostaining of human hypocretin (orexin) neurons: loss in narcolepsy. Neurology 65, 1189–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeve BF (2013). Idiopathic REM sleep behaviour disorder in the development of Parkinson's disease. Lancet Neurol 12, 469–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeve BF, Silber MH, Saper CB, Ferman TJ, Dickson DW, Parisi JE, Benarroch EE, Ahlskog JE, Smith GE, Caselli RC, Tippman‐Peikert M, Olson EJ, Lin SC, Young T, Wszolek Z, Schenck CH, Mahowald MW, Castillo PR, Del Tredici K & Braak H (2007). Pathophysiology of REM sleep behaviour disorder and relevance to neurodegenerative disease. Brain 130, 2770–2788. [DOI] [PubMed] [Google Scholar]
- Boissard R, Fort P, Gervasoni D, Barbagli B & Luppi PH (2003). Localization of the GABAergic and non‐GABAergic neurons projecting to the sublaterodorsal nucleus and potentially gating paradoxical sleep onset. Eur J Neurosci 18, 1627–1639. [DOI] [PubMed] [Google Scholar]
- Boissard R, Gervasoni D, Schmidt MH, Barbagli B, Fort P & Luppi PH (2002). The rat ponto‐medullary network responsible for paradoxical sleep onset and maintenance: a combined microinjection and functional neuroanatomical study. Eur J Neurosci 16, 1959–1973. [DOI] [PubMed] [Google Scholar]
- Boucetta S, Cisse Y, Mainville L, Morales M & Jones BE (2014). Discharge profiles across the sleep‐waking cycle of identified cholinergic, GABAergic, and glutamatergic neurons in the pontomesencephalic tegmentum of the rat. J Neurosci 34, 4708–4727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PL & Peever JH (2008. a). Glycinergic and GABAA‐mediated inhibition of somatic motoneurons does not mediate rapid eye movement sleep motor atonia. J Neurosci 28, 3535–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PL & Peever JH (2008. b). Unraveling the mechanisms of REM sleep atonia. Sleep 31, 1492–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks PL & Peever JH (2010). GABAergic and glycinergic control of upper airway motoneurons in rapid eye movement sleep. Adv Exp Med Biol 669, 259–262. [DOI] [PubMed] [Google Scholar]
- Brooks PL & Peever JH (2012). Identification of the transmitter and receptor mechanisms responsible for REM sleep paralysis. J Neurosci 32, 9785–9795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Basheer R, McKenna JT, Strecker RE & McCarley RW (2012). Control of sleep and wakefulness. Physiol Rev 92, 1087–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Sergeeva OA, Eriksson KS & Haas HL (2002). Convergent excitation of dorsal raphe serotonin neurons by multiple arousal systems (orexin/hypocretin, histamine and noradrenaline). J Neurosci 22, 8850–8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown RE, Winston S, Basheer R, Thakkar MM & McCarley RW (2006). Electrophysiological characterization of neurons in the dorsolateral pontine rapid‐eye‐movement sleep induction zone of the rat: Intrinsic membrane properties and responses to carbachol and orexins. Neuroscience 143, 739–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruinstroop E, Cano G, Vanderhorst VG, Cavalcante JC, Wirth J, Sena‐Esteves M & Saper CB (2012). Spinal projections of the A5, A6 (locus coeruleus), and A7 noradrenergic cell groups in rats. J Comp Neurol 520, 1985–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess C, Lai D, Siegel J & Peever J (2008). An endogenous glutamatergic drive onto somatic motoneurons contributes to the stereotypical pattern of muscle tone across the sleep‐wake cycle. J Neurosci 28, 4649–4660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess CR, Oishi Y, Mochizuki T, Peever JH & Scammell TE (2013). Amygdala lesions reduce cataplexy in orexin knock‐out mice. J Neurosci 33, 9734–9742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgess CR & Scammell TE (2012). Narcolepsy: neural mechanisms of sleepiness and cataplexy. J Neurosci 32, 12305–12311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter ME, Yizhar O, Chikahisa S, Nguyen H, Adamantidis A, Nishino S, Deisseroth K & de Lecea L (2010). Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat Neurosci 13, 1526–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo P, Pedroarena C, Chase MH & Morales FR (1991. a). A medullary inhibitory region for trigeminal motoneurons in the cat. Brain Res 549, 346–349. [DOI] [PubMed] [Google Scholar]
- Castillo P, Pedroarena C, Chase MH & Morales FR (1991. b). Strychnine blockade of the non‐reciprocal inhibition of trigeminal motoneurons induced by stimulation of the parvocellular reticular formation. Brain Res 567, 346–349. [DOI] [PubMed] [Google Scholar]
- Chan E, Steenland HW, Liu H & Horner RL (2006). Endogenous excitatory drive modulating respiratory muscle activity across sleep‐wake states. Am J Respir Crit Care Med 174, 1264–1273. [DOI] [PubMed] [Google Scholar]
- Chase MH (2008). Confirmation of the consensus that glycinergic postsynaptic inhibition is responsible for the atonia of REM sleep. Sleep 31, 1487–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase MH (2009). Factual errors in Brooks and Peever's rebuttal to critiques. Sleep 32, 845–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chase MH (2013). Motor control during sleep and wakefulness: Clarifying controversies and resolving paradoxes. Sleep Med Rev 17, 299–312. [DOI] [PubMed] [Google Scholar]
- Chase MH, Chandler SH & Nakamura Y (1980). Intracellular determination of membrane potential of trigeminal motoneurons during sleep and wakefulness. J Neurophysiol 44, 349–358. [DOI] [PubMed] [Google Scholar]
- Chase MH, Enomoto S, Hiraba K, Katoh M, Nakamura Y, Sahara Y & Taira M (1984). Role of medullary reticular neurons in the inhibition of trigeminal motoneurons during active sleep. Exp Neurol 84, 364–373. [DOI] [PubMed] [Google Scholar]
- Chase MH & Morales FR (1982). Phasic changes in motoneuron membrane potential during REM periods of active sleep. Neurosci Lett 34, 177–182. [DOI] [PubMed] [Google Scholar]
- Chase MH & Morales FR (1983). Subthreshold excitatory activity and motoneuron discharge during REM periods of active sleep. Science 221, 1195–1198. [DOI] [PubMed] [Google Scholar]
- Chase MH, Morales FR, Boxer PA, Fung SJ & Soja PJ (1986). Effect of stimulation of the nucleus reticularis gigantocellularis on the membrane potential of cat lumbar motoneurons during sleep and wakefulness. Brain Res 386, 237–244. [DOI] [PubMed] [Google Scholar]
- Chase MH, Soja PJ & Morales FR (1989). Evidence that glycine mediates the postsynaptic potentials that inhibit lumbar motoneurons during the atonia of active sleep. J Neurosci 9, 743–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee MJ, Arrigoni E & Maratos‐Flier E (2015). Melanin‐concentrating hormone neurons release glutamate for feedforward inhibition of the lateral septum. J Neurosci 35, 3644–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chemelli RM, Willie JT, Sinton CM, Elmquist JK, Scammell T, Lee C, Richardson JA, Williams SC, Xiong Y, Kisanuki Y, Fitch TE, Nakazato M, Hammer RE, Saper CB & Yanagisawa M (1999). Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell 98, 437–451. [DOI] [PubMed] [Google Scholar]
- Chen L, Brown RE, McKenna JT & McCarley RW (2009). Animal models of narcolepsy. CNS Neurol Disord Drug Targets 8, 296–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark EL, Baumann CR, Cano G, Scammell TE & Mochizuki T (2009). Feeding‐elicited cataplexy in orexin knockout mice. Neuroscience 161, 970–977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement O, Sapin E, Berod A, Fort P & Luppi PH (2011). Evidence that neurons of the sublaterodorsal tegmental nucleus triggering paradoxical (REM) sleep are glutamatergic. Sleep 34, 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement O, Sapin E, Libourel PA, Arthaud S, Brischoux F, Fort P & Luppi PH (2012). The lateral hypothalamic area controls paradoxical (REM) sleep by means of descending projections to brainstem GABAergic neurons. J Neurosci 32, 16763–16774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement O, Valencia Garcia S, Libourel PA, Arthaud S, Fort P & Luppi PH (2014). The inhibition of the dorsal paragigantocellular reticular nucleus induces waking and the activation of all adrenergic and noradrenergic neurons: a combined pharmacological and functional neuroanatomical study. PLoS One 9, e96851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crochet S, Onoe H & Sakai K (2006). A potent non‐monoaminergic paradoxical sleep inhibitory system: a reverse microdialysis and single‐unit recording study. Eur J Neurosci 24, 1404–1412. [DOI] [PubMed] [Google Scholar]
- Crochet S & Sakai K (1999. a). Alpha‐2 adrenoceptor mediated paradoxical (REM) sleep inhibition in the cat. Neuroreport 10, 2199–2204. [DOI] [PubMed] [Google Scholar]
- Crochet S & Sakai K (1999. b). Effects of microdialysis application of monoamines on the EEG and behavioural states in the cat mesopontine tegmentum. Eur J Neurosci 11, 3738–3752. [DOI] [PubMed] [Google Scholar]
- Crocker A, Espana RA, Papadopoulou M, Saper CB, Faraco J, Sakurai T, Honda M, Mignot E & Scammell TE (2005). Concomitant loss of dynorphin, NARP, and orexin in narcolepsy. Neurology 65, 1184–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damasio AR, Grabowski TJ, Bechara A, Damasio H, Ponto LL, Parvizi J & Hichwa RD (2000). Subcortical and cortical brain activity during the feeling of self‐generated emotions. Nat Neurosci 3, 1049–1056. [DOI] [PubMed] [Google Scholar]
- Date Y, Ueta Y, Yamashita H, Yamaguchi H, Matsukura S, Kangawa K, Sakurai T, Yanagisawa M & Nakazato M (1999). Orexins, orexigenic hypothalamic peptides, interact with autonomic, neuroendocrine and neuroregulatory systems. Proc Natl Acad Sci USA 96, 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Cock VC, Vidailhet M, Leu S, Texeira A, Apartis E, Elbaz A, Roze E, Willer JC, Derenne JP, Agid Y & Arnulf I (2007). Restoration of normal motor control in Parkinson's disease during REM sleep. Brain 130, 450–456. [DOI] [PubMed] [Google Scholar]
- de Lecea L, Kilduff TS, Peyron C, Gao X, Foye PE, Danielson PE, Fukuhara C, Battenberg EL, Gautvik VT, Bartlett FS 2nd, Frankel WN, van den Pol AN, Bloom FE, Gautvik KM & Sutcliffe JG (1998). The hypocretins: hypothalamus‐specific peptides with neuroexcitatory activity. Proc Natl Acad Sci USA 95, 322–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Cid‐Pellitero E & Jones BE (2012). Immunohistochemical evidence for synaptic release of GABA from melanin‐concentrating hormone containing varicosities in the locus coeruleus. Neuroscience 223, 269–276. [DOI] [PubMed] [Google Scholar]
- Devera A, Pascovich C, Lagos P, Falconi A, Sampogna S, Chase MH & Torterolo P (2015). Melanin‐concentrating hormone (MCH) modulates the activity of dorsal raphe neurons. Brain Res 1598, 114–128. [DOI] [PubMed] [Google Scholar]
- Egan TM & North RA (1985). Acetylcholine acts on m2‐muscarinic receptors to excite rat locus coeruleus neurones. Br J Pharmacol 85, 733–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias CF, Lee CE, Kelly JF, Ahima RS, Kuhar M, Saper CB & Elmquist JK (2001). Characterization of CART neurons in the rat and human hypothalamus. J Comp Neurol 432, 1–19. [DOI] [PubMed] [Google Scholar]
- Espana RA, McCormack SL, Mochizuki T & Scammell TE (2007). Running promotes wakefulness and increases cataplexy in orexin knockout mice. Sleep 30, 1417–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espana RA & Scammell TE (2011). Sleep neurobiology from a clinical perspective. Sleep 34, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkin A, Egner T & Kalisch R (2011). Emotional processing in anterior cingulate and medial prefrontal cortex. Trends Cogn Sci 15, 85–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenik VB, Davies RO & Kubin L (2005. a). Noradrenergic, serotonergic and GABAergic antagonists injected together into the XII nucleus abolish the REM sleep‐like depression of hypoglossal motoneuronal activity. J Sleep Res 14, 419–429. [DOI] [PubMed] [Google Scholar]
- Fenik VB, Davies RO & Kubin L (2005. b). REM sleep‐like atonia of hypoglossal (XII) motoneurons is caused by loss of noradrenergic and serotonergic inputs. Am J Respir Crit Care Med 172, 1322–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenik VB & Kubin L (2009). Differential localization of carbachol‐ and bicuculline‐sensitive pontine sites for eliciting REM sleep‐like effects in anaesthetized rats. J Sleep Res 18, 99–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fort P, Luppi PH, Sakai K, Salvert D & Jouvet M (1990). Nuclei of origin of monoaminergic, peptidergic, and cholinergic afferents to the cat trigeminal motor nucleus: a double‐labelling study with cholera‐toxin as a retrograde tracer. J Comp Neurol 301, 262–275. [DOI] [PubMed] [Google Scholar]
- Fort P, Sakai K, Luppi PH, Salvert D & Jouvet M (1989). Monoaminergic, peptidergic, and cholinergic afferents to the cat facial nucleus as evidenced by a double immunostaining method with unconjugated cholera toxin as a retrograde tracer. J Comp Neurol 283, 285–302. [DOI] [PubMed] [Google Scholar]
- Frauscher B, Gschliesser V, Brandauer E, Schonwald SV, Falkenstetter T, Ehrmann L, Tokmak I, Poewe W & Hogl B (2011). Motor disturbances during non‐REM and REM sleep in narcolepsy‐cataplexy: a video‐polysomnographic analysis. J Sleep Res 20, 514–521. [DOI] [PubMed] [Google Scholar]
- Fuller PM, Saper CB & Lu J (2007). The pontine REM switch: past and present. J Physiol 584, 735–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller PM, Sherman D, Pedersen NP, Saper CB & Lu J (2011). Reassessment of the structural basis of the ascending arousal system. J Comp Neurol 519, 933–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fung SJ & Chase MH (2015). Postsynaptic inhibition of hypoglossal motoneurons produces atonia of the genioglossal muscle during rapid eye movement sleep. Sleep 38, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Funk GD (2008). Are all motoneurons created equal in the eyes of REM sleep and the mechanisms of muscle atonia? Sleep 31, 1479–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao XB (2009). Electrophysiological effects of MCH on neurons in the hypothalamus. Peptides 30, 2025–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garavan H, Pendergrass JC, Ross TJ, Stein EA & Risinger RC (2001). Amygdala response to both positively and negatively valenced stimuli. Neuroreport 12, 2779–2783. [DOI] [PubMed] [Google Scholar]
- Gassel MM, Marchiafava PL & Pompeiano O (1964). Tonic and phasic inhibition of spinal reflexes during deep, desynchronized sleep in unrestrained cats. Arch Ital Biol 102, 471–479. [PubMed] [Google Scholar]
- Gassel MM, Marchiafava PL & Pompeiano O (1965). An analysis of the supraspinal influences acting on motoneurons during sleep in the unrestrained cat. Modification of the recurrent discharge of the alpha motoneurons during sleep. Arch Ital Biol 103, 25–44. [PubMed] [Google Scholar]
- Gervasoni D, Peyron C, Rampon C, Barbagli B, Chouvet G, Urbain N, Fort P & Luppi PH (2000). Role and origin of the GABAergic innervation of dorsal raphe serotonergic neurons. J Neurosci 20, 4217–4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaquinto S, Pompeiano O & Somogyi I (1964. a). Descending inhibitory influences on spinal reflexes during natural sleep. Arch Ital Biol 102, 282–307. [PubMed] [Google Scholar]
- Giaquinto S, Pompeiano O & Somogyi I (1964. b). Supraspinal modulation of heteronymous monosynaptic and of polysynaptic reflexes during natural sleep and wakefulness. Arch Ital Biol 102, 245–281. [PubMed] [Google Scholar]
- Glenn LL & Dement WC (1981. a). Membrane potential and input resistance of cat spinal motoneurons in wakefulness and sleep. Behav Brain Res 2, 231–236. [DOI] [PubMed] [Google Scholar]
- Glenn LL & Dement WC (1981. b). Membrane potential, synaptic activity, and excitability of hindlimb motoneurons during wakefulness and sleep. J Neurophysiol 46, 839–854. [DOI] [PubMed] [Google Scholar]
- Goutagny R, Luppi PH, Salvert D, Lapray D, Gervasoni D & Fort P (2008). Role of the dorsal paragigantocellular reticular nucleus in paradoxical (rapid eye movement) sleep generation: a combined electrophysiological and anatomical study in the rat. Neuroscience 152, 849–857. [DOI] [PubMed] [Google Scholar]
- Gowda CR & Lundt LP (2014). Mechanism of action of narcolepsy medications. CNS Spectr 19 Suppl 1, 25–33; quiz 25–27, 34. [DOI] [PubMed] [Google Scholar]
- Grace KP & Horner RL (2015). Evaluating the evidence surrounding pontine cholinergic involvement in REM sleep generation. Front Neurol 6, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace KP, Hughes SW & Horner RL (2013). Identification of the mechanism mediating genioglossus muscle suppression in REM sleep. Am J Respir Crit Care Med 187, 311–319. [DOI] [PubMed] [Google Scholar]
- Grace KP, Vanstone LE & Horner RL (2014). Endogenous cholinergic input to the pontine REM sleep generator is not required for REM sleep to occur. J Neurosci 34, 14198–14209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulyani S, Wu MF, Nienhuis R, John J & Siegel JM (2002). Cataplexy‐related neurons in the amygdala of the narcoleptic dog. Neuroscience 112, 355–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habaguchi T, Takakusaki K, Saitoh K, Sugimoto J & Sakamoto T (2002). Medullary reticulospinal tract mediating the generalized motor inhibition in cats: II. Functional organization within the medullary reticular formation with respect to postsynaptic inhibition of forelimb and hindlimb motoneurons. Neuroscience 113, 65–77. [DOI] [PubMed] [Google Scholar]
- Hajnik T, Lai YY & Siegel JM (2000). Atonia‐related regions in the rodent pons and medulla. J Neurophysiol 84, 1942–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanriot L, Camargo N, Courau AC, Leger L, Luppi PH & Peyron C (2007). Characterization of the melanin‐concentrating hormone neurons activated during paradoxical sleep hypersomnia in rats. J Comp Neurol 505, 147–157. [DOI] [PubMed] [Google Scholar]
- Hara J, Beuckmann CT, Nambu T, Willie JT, Chemelli RM, Sinton CM, Sugiyama F, Yagami K, Goto K, Yanagisawa M & Sakurai T (2001). Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron 30, 345–354. [DOI] [PubMed] [Google Scholar]
- Harthoorn LF, Sane A, Nethe M & Van Heerikhuize JJ (2005). Multi‐transcriptional profiling of melanin‐concentrating hormone and orexin‐containing neurons. Cell Mol Neurobiol 25, 1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa E, Yanagisawa M, Sakurai T & Mieda M (2014). Orexin neurons suppress narcolepsy via 2 distinct efferent pathways. J Clin Invest 124, 604–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassani OK, Lee MG & Jones BE (2009). Melanin‐concentrating hormone neurons discharge in a reciprocal manner to orexin neurons across the sleep‐wake cycle. Proc Natl Acad Sci USA 106, 2418–2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendricks JC, Morrison AR & Mann GL (1982). Different behaviors during paradoxical sleep without atonia depend on pontine lesion site. Brain Res 239, 81–105. [DOI] [PubMed] [Google Scholar]
- Henley K & Morrison AR (1974). A re‐evaluation of the effects of lesions of the pontine tegmentum and locus coeruleus on phenomena of paradoxical sleep in the cat. Acta Neurobiol Exp (Wars) 34, 215–232. [PubMed] [Google Scholar]
- Hobson JA, Datta S, Calvo JM & Quattrochi J (1993). Acetylcholine as a brain state modulator: triggering and long‐term regulation of REM sleep. Prog Brain Res 98, 389–404. [DOI] [PubMed] [Google Scholar]
- Hobson JA, McCarley RW & Wyzinski PW (1975). Sleep cycle oscillation: reciprocal discharge by two brainstem neuronal groups. Science 189, 55–58. [DOI] [PubMed] [Google Scholar]
- Holmes CJ & Jones BE (1994). Importance of cholinergic, GABAergic, serotonergic and other neurons in the medial medullary reticular formation for sleep‐wake states studied by cytotoxic lesions in the cat. Neuroscience 62, 1179–1200. [DOI] [PubMed] [Google Scholar]
- Holmes CJ, Mainville LS & Jones BE (1994). Distribution of cholinergic, GABAergic and serotonergic neurons in the medial medullary reticular formation and their projections studied by cytotoxic lesions in the cat. Neuroscience 62, 1155–1178. [DOI] [PubMed] [Google Scholar]
- Holstege JC & Bongers CM (1991). A glycinergic projection from the ventromedial lower brainstem to spinal motoneurons. An ultrastructural double labelling study in rat. Brain Res 566, 308–315. [DOI] [PubMed] [Google Scholar]
- Honda T & Semba K (1994). Serotonergic synaptic input to cholinergic neurons in the rat mesopontine tegmentum. Brain Res 647, 299–306. [DOI] [PubMed] [Google Scholar]
- Horner RL (2009). Emerging principles and neural substrates underlying tonic sleep‐state‐dependent influences on respiratory motor activity. Philos Trans R Soc Lond B Biol Sci 364, 2553–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hossaini M, Goos JA, Kohli SK & Holstege JC (2012). Distribution of glycine/GABA neurons in the ventromedial medulla with descending spinal projections and evidence for an ascending glycine/GABA projection. PLoS One 7, e35293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov A & Aston‐Jones G (2000). Hypocretin/orexin depolarizes and decreases potassium conductance in locus coeruleus neurons. Neuroreport 11, 1755–1758. [DOI] [PubMed] [Google Scholar]
- Jankowska E, Lund S, Lundberg A & Pompeiano O (1968). Inhibitory effects evoked through ventral reticulospinal pathways. Arch Ital Biol 106, 124–140. [PubMed] [Google Scholar]
- Jego S, Glasgow SD, Herrera CG, Ekstrand M, Reed SJ, Boyce R, Friedman J, Burdakov D & Adamantidis AR (2013). Optogenetic identification of a rapid eye movement sleep modulatory circuit in the hypothalamus. Nat Neurosci 16, 1637–1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John J, Wu MF, Boehmer LN & Siegel JM (2004. a). Cataplexy‐active neurons in the hypothalamus: implications for the role of histamine in sleep and waking behavior. Neuron 42, 619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John J, Wu MF, Maidment NT, Lam HA, Boehmer LN, Patton M & Siegel JM (2004. b). Developmental changes in CSF hypocretin‐1 (orexin‐A) levels in normal and genetically narcoleptic Doberman pinschers. J Physiol 560, 587–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BE (1990). Immunohistochemical study of choline acetyltransferase‐immunoreactive processes and cells innervating the pontomedullary reticular formation in the rat. J Comp Neurol 295, 485–514. [DOI] [PubMed] [Google Scholar]
- Jones BE (1991. a). Paradoxical sleep and its chemical/structural substrates in the brain. Neuroscience 40, 637–656. [DOI] [PubMed] [Google Scholar]
- Jones BE (1991. b). The role of noradrenergic locus coeruleus neurons and neighboring cholinergic neurons of the pontomesencephalic tegmentum in sleep‐wake states. Prog Brain Res 88, 533–543. [DOI] [PubMed] [Google Scholar]
- Jones BE (1993). The organization of central cholinergic systems and their functional importance in sleep‐waking states. Prog Brain Res 98, 61–71. [DOI] [PubMed] [Google Scholar]
- Jones BE & Hassani OK (2013). The role of Hcrt/Orx and MCH neurons in sleep‐wake state regulation. Sleep 36, 1769–1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BE, Holmes CJ, Rodriguez‐Veiga E & Mainville L (1991). GABA‐synthesizing neurons in the medulla: their relationship to serotonin‐containing and spinally projecting neurons in the rat. J Comp Neurol 313, 349–367. [DOI] [PubMed] [Google Scholar]
- Jouvet M (1972). The role of monoamines and acetylcholine‐containing neurons in the regulation of the sleep‐waking cycle. Ergeb Physiol 64, 166–307. [DOI] [PubMed] [Google Scholar]
- Jouvet M & Michel F (1960). [New research on the structures responsible for the “paradoxical phase” of sleep.] J Physiol Paris 52, 130–131. [PubMed] [Google Scholar]
- Kanamori N, Sakai K & Jouvet M (1980). Neuronal activity specific to paradoxical sleep in the ventromedial medullary reticular formation of unrestrained cats. Brain Res 189, 251–255. [DOI] [PubMed] [Google Scholar]
- Karlsson KA & Blumberg MS (2005). Active medullary control of atonia in week‐old rats. Neuroscience 130, 275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato G, Yasaka T, Katafuchi T, Furue H, Mizuno M, Iwamoto Y & Yoshimura M (2006). Direct GABAergic and glycinergic inhibition of the substantia gelatinosa from the rostral ventromedial medulla revealed by in vivo patch‐clamp analysis in rats. J Neurosci 26, 1787–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur S, Thankachan S, Begum S, Liu M, Blanco‐Centurion C & Shiromani PJ (2009). Hypocretin‐2 saporin lesions of the ventrolateral periaquaductal grey (vlPAG) increase REM sleep in hypocretin knockout mice. PLoS One 4, e6346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller AD (1945). Generalized atonia and profound dysreflexia following transection of the brain stem through the cephalic pons. J Neurophysiol 8, 275–288. [Google Scholar]
- Khoury J & Doghramji K (2015). Primary sleep disorders. Psychiatr Clin North Am 38, 683–704. [DOI] [PubMed] [Google Scholar]
- Kiyashchenko LI, Mileykovskiy BY, Maidment N, Lam HA, Wu MF, John J, Peever J & Siegel JM (2002). Release of hypocretin (orexin) during waking and sleep states. J Neurosci 22, 5282–5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kodama T, Lai YY & Siegel JM (1998). Enhanced glutamate release during REM sleep in the rostromedial medulla as measured by in vivo microdialysis. Brain Res 780, 176–179. [PubMed] [Google Scholar]
- Kodama T, Lai YY & Siegel JM (2003). Changes in inhibitory amino acid release linked to pontine‐induced atonia: an in vivo microdialysis study. J Neurosci 23, 1548–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmeier KA, Lopez‐Rodriguez F, Liu RH, Morales FR & Chase MH (1996). State‐dependent phenomena in cat masseter motoneurons. Brain Res 722, 30–38. [DOI] [PubMed] [Google Scholar]
- Kohlmeier KA, Tyler CJ, Kalogiannis M, Ishibashi M, Kristensen MP, Gumenchuk I, Chemelli RM, Kisanuki YY, Yanagisawa M & Leonard CS (2013). Differential actions of orexin receptors in brainstem cholinergic and monoaminergic neurons revealed by receptor knockouts: implications for orexinergic signalling in arousal and narcolepsy. Front Neurosci 7, 246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmeier KA, Watanabe S, Tyler CJ, Burlet S & Leonard CS (2008). Dual orexin actions on dorsal raphe and laterodorsal tegmentum neurons: noisy cation current activation and selective enhancement of Ca2+ transients mediated by L‐type calcium channels. J Neurophysiol 100, 2265–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohyama J, Lai YY & Siegel JM (1998). Inactivation of the pons blocks medullary‐induced muscle tone suppression in the decerebrate cat. Sleep 21, 695–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konadhode RR, Pelluru D, Blanco‐Centurion C, Zayachkivsky A, Liu M, Uhde T, Glen WB Jr, van den Pol AN, Mulholland PJ & Shiromani PJ (2013). Optogenetic stimulation of MCH neurons increases sleep. J Neurosci 33, 10257–10263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenzer M, Anaclet C, Vetrivelan R, Wang N, Vong L, Lowell BB, Fuller PM & Lu J (2011). Brainstem and spinal cord circuitry regulating REM sleep and muscle atonia. PLoS One 6, e24998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubin L (2001). Carbachol models of REM sleep: recent developments and new directions. Arch Ital Biol 139, 147–168. [PubMed] [Google Scholar]
- Kubin L (2008). Adventures and tribulations in the search for the mechanisms of the atonia of REM sleep. Sleep 31, 1473–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubin L, Davies RO & Pack AI (1998). Control of upper airway motoneurons during REM sleep. News Physiol Sci 13, 91–97. [DOI] [PubMed] [Google Scholar]
- Kubin L, Kimura H, Tojima H, Davies RO & Pack AI (1993). Suppression of hypoglossal motoneurons during the carbachol‐induced atonia of REM sleep is not caused by fast synaptic inhibition. Brain Res 611, 300–312. [DOI] [PubMed] [Google Scholar]
- Kubin L, Tojima H, Davies RO & Pack AI (1992). Serotonergic excitatory drive to hypoglossal motoneurons in the decerebrate cat. Neurosci Lett 139, 243–248. [DOI] [PubMed] [Google Scholar]
- Kubota K & Kidokoro Y (1966). Excitability of the membrane of lumber motor neurons and natural sleep in the cat. Jpn J Physiol 16, 217–226. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Leung E & Vincent SR (1992). Ultrastructure of cholinergic neurons in the laterodorsal tegmental nucleus of the rat: interaction with catecholamine fibres. Brain Res Bull 29, 479–491. [DOI] [PubMed] [Google Scholar]
- Kushida CA, Bergmann BM & Rechtschaffen A (1989). Sleep deprivation in the rat: IV. Paradoxical sleep deprivation. Sleep 12, 22–30. [DOI] [PubMed] [Google Scholar]
- Lagos P, Torterolo P, Jantos H, Chase MH & Monti JM (2009). Effects on sleep of melanin‐concentrating hormone (MCH) microinjections into the dorsal raphe nucleus. Brain Res 1265, 103–110. [DOI] [PubMed] [Google Scholar]
- Lai YY, Clements JR & Siegel JM (1993). Glutamatergic and cholinergic projections to the pontine inhibitory area identified with horseradish peroxidase retrograde transport and immunohistochemistry. J Comp Neurol 336, 321–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY, Kodama T, Schenkel E & Siegel JM (2010). Behavioural response and transmitter release during atonia elicited by medial medullary stimulation. J Neurophysiol 104, 2024–2033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY, Kodama T & Siegel JM (2001). Changes in monoamine release in the ventral horn and hypoglossal nucleus linked to pontine inhibition of muscle tone: an in vivo microdialysis study. J Neurosci 21, 7384–7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY & Siegel JM (1988). Medullary regions mediating atonia. J Neurosci 8, 4790–4796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY & Siegel JM (1991). Pontomedullary glutamate receptors mediating locomotion and muscle tone suppression. J Neurosci 11, 2931–2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai YY & Siegel JM (1997). Brainstem‐mediated locomotion and myoclonic jerks. I. Neural substrates. Brain Res 745, 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Hassani OK & Jones BE (2005). Discharge of identified orexin/hypocretin neurons across the sleep‐waking cycle. J Neurosci 25, 6716–6720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard TO & Lydic R (1997). Pontine nitric oxide modulates acetylcholine release, rapid eye movement sleep generation, and respiratory rate. J Neurosci 17, 774–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin L, Faraco J, Li R, Kadotani H, Rogers W, Lin X, Qiu X, de Jong PJ, Nishino S & Mignot E (1999). The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 98, 365–376. [DOI] [PubMed] [Google Scholar]
- Liu RJ, van den Pol AN & Aghajanian GK (2002). Hypocretins (orexins) regulate serotonin neurons in the dorsal raphe nucleus by excitatory direct and inhibitory indirect actions. J Neurosci 22, 9453–9464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Sood S, Liu H & Horner RL (2005). Opposing muscarinic and nicotinic modulation of hypoglossal motor output to genioglossus muscle in rats in vivo . J Physiol 565, 965–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinas R & Terzuolo CA (1964). Mechanisms of supraspinal actions upon spinal cord activities. Reticular inhibitory mechanisms on alpha‐extensor motoneurons. J Neurophysiol 27, 579–591. [DOI] [PubMed] [Google Scholar]
- Lu J, Sherman D, Devor M & Saper CB (2006). A putative flip‐flop switch for control of REM sleep. Nature 441, 589–594. [DOI] [PubMed] [Google Scholar]
- Luebke JI, Greene RW, Semba K, Kamondi A, McCarley RW & Reiner PB (1992). Serotonin hyperpolarizes cholinergic low‐threshold burst neurons in the rat laterodorsal tegmental nucleus in vitro. Proc Natl Acad Sci USA 89, 743–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luppi PH, Aston‐Jones G, Akaoka H, Chouvet G & Jouvet M (1995). Afferent projections to the rat locus coeruleus demonstrated by retrograde and anterograde tracing with cholera‐toxin B subunit and Phaseolus vulgaris leucoagglutinin. Neuroscience 65, 119–160. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Clement O & Fort P (2013. a). Paradoxical (REM) sleep genesis by the brainstem is under hypothalamic control. Curr Opin Neurobiol 23, 786–792. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Clement O, Sapin E, Gervasoni D, Peyron C, Leger L, Salvert D & Fort P (2011). The neuronal network responsible for paradoxical sleep and its dysfunctions causing narcolepsy and rapid eye movement (REM) behaviour disorder. Sleep Med Rev 15, 153–163. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Clement O, Sapin E, Peyron C, Gervasoni D, Leger L & Fort P (2012). Brainstem mechanisms of paradoxical (REM) sleep generation. Pflugers Arch 463, 43–52. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Clement O, Valencia Garcia S, Brischoux F & Fort P (2013. b). New aspects in the pathophysiology of rapid eye movement sleep behavior disorder: the potential role of glutamate, gamma‐aminobutyric acid, and glycine. Sleep Med 14, 714–718. [DOI] [PubMed] [Google Scholar]
- Luppi PH, Gervasoni D, Verret L, Goutagny R, Peyron C, Salvert D, Leger L & Fort P (2006). Paradoxical (REM) sleep genesis: the switch from an aminergic‐cholinergic to a GABAergic‐glutamatergic hypothesis. J Physiol Paris 100, 271–283. [DOI] [PubMed] [Google Scholar]
- Lydic R (2008). The motor atonia of REM sleep: A critical topics forum. Sleep 31, 1471–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magoun HW & Rhines R (1946). An inhibitory mechanism in the bulbar reticular formation. J Neurophysiol 9, 165–171. [DOI] [PubMed] [Google Scholar]
- Maloney KJ, Mainville L & Jones BE (1999). Differential c‐Fos expression in cholinergic, monoaminergic, and GABAergic cell groups of the pontomesencephalic tegmentum after paradoxical sleep deprivation and recovery. J Neurosci 19, 3057–3072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloney KJ, Mainville L & Jones BE (2000). c‐Fos expression in GABAergic, serotonergic, and other neurons of the pontomedullary reticular formation and raphe after paradoxical sleep deprivation and recovery. J Neurosci 20, 4669–4679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathis J, Hess CW & Bassetti C (2007). Isolated mediotegmental lesion causing narcolepsy and rapid eye movement sleep behaviour disorder: a case evidencing a common pathway in narcolepsy and rapid eye movement sleep behaviour disorder. J Neurol Neurosurg Psychiatry 78, 427–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarley RW (2007). Neurobiology of REM and NREM sleep. Sleep Med 8, 302–330. [DOI] [PubMed] [Google Scholar]
- McCarley RW & Hobson JA (1975). Neuronal excitability modulation over the sleep cycle: a structural and mathematical model. Science 189, 58–60. [DOI] [PubMed] [Google Scholar]
- McCarter SJ, Tippmann‐Peikert M, Sandness DJ, Flanagan EP, Kantarci K, Boeve BF, Silber MH & St Louis EK (2015). Neuroimaging‐evident lesional pathology associated with REM sleep behaviour disorder. Sleep Med 16, 1502–1510. [DOI] [PubMed] [Google Scholar]
- McGinty DJ & Harper RM (1976). Dorsal raphe neurons: depression of firing during sleep in cats. Brain Res 101, 569–575. [DOI] [PubMed] [Google Scholar]
- Mieda M, Hasegawa E, Kisanuki YY, Sinton CM, Yanagisawa M & Sakurai T (2011). Differential roles of orexin receptor‐1 and ‐2 in the regulation of non‐REM and REM sleep. J Neurosci 31, 6518–6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mignot E, Lammers GJ, Ripley B, Okun M, Nevsimalova S, Overeem S, Vankova J, Black J, Harsh J, Bassetti C, Schrader H & Nishino S (2002). The role of cerebrospinal fluid hypocretin measurement in the diagnosis of narcolepsy and other hypersomnias. Arch Neurol 59, 1553–1562. [DOI] [PubMed] [Google Scholar]
- Mileykovskiy BY, Kiyashchenko LI & Siegel JM (2002). Muscle tone facilitation and inhibition after orexin‐a (hypocretin‐1) microinjections into the medial medulla. J Neurophysiol 87, 2480–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mileykovskiy BY, Kiyashchenko LI & Siegel JM (2005). Behavioural correlates of activity in identified hypocretin/orexin neurons. Neuron 46, 787–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitler MM, Boysen BG, Campbell L & Dement WC (1974). Narcolepsy‐cataplexy in a female dog. Exp Neurol 45, 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki T, Arrigoni E, Marcus JN, Clark EL, Yamamoto M, Honer M, Borroni E, Lowell BB, Elmquist JK & Scammell TE (2011). Orexin receptor 2 expression in the posterior hypothalamus rescues sleepiness in narcoleptic mice. Proc Natl Acad Sci USA 108, 4471–4476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki T, Crocker A, McCormack S, Yanagisawa M, Sakurai T & Scammell TE (2004). Behavioural state instability in orexin knock‐out mice. J Neurosci 24, 6291–6300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monti JM, Lagos P, Jantos H & Torterolo P (2015). Increased REM sleep after intra‐locus coeruleus nucleus microinjection of melanin‐concentrating hormone (MCH) in the rat. Prog Neuropsychopharmacol Biol Psychiatry 56, 185–188. [DOI] [PubMed] [Google Scholar]
- Monti JM, Torterolo P & Lagos P (2013). Melanin‐concentrating hormone control of sleep‐wake behavior. Sleep Med Rev 17, 293–298. [DOI] [PubMed] [Google Scholar]
- Morales FR, Boxer P & Chase MH (1987). Behavioral state‐specific inhibitory postsynaptic potentials impinge on cat lumbar motoneurons during active sleep. Exp Neurol 98, 418–435. [DOI] [PubMed] [Google Scholar]
- Morales FR & Chase MH (1978). Intracellular recording of lumbar motoneuron membrane potential during sleep and wakefulness. Exp Neurol 62, 821–827. [DOI] [PubMed] [Google Scholar]
- Morales FR & Chase MH (1982). Repetitive synaptic potentials responsible for inhibition of spinal cord motoneurons during active sleep. Exp Neurol 78, 471–476. [DOI] [PubMed] [Google Scholar]
- Morales FR, Sampogna S, Rampon C, Luppi PH & Chase MH (2006). Brainstem glycinergic neurons and their activation during active (rapid eye movement) sleep in the cat. Neuroscience 142, 37–47. [DOI] [PubMed] [Google Scholar]
- Morrison AR (1988). Paradoxical sleep without atonia. Arch Ital Biol 126, 275–289. [PubMed] [Google Scholar]
- Morrison AR & Pompeiano O (1965). An analysis of the supraspinal influences acting on motoneurons during sleep in the unrestrained cat: responses of the alpha motoneurons to direct electrical stimulation during sleep. Arch Ital Biol 103, 497–516. [PubMed] [Google Scholar]
- Morrison JL, Sood S, Liu H, Park E, Liu X, Nolan P & Horner RL (2003. a). Role of inhibitory amino acids in control of hypoglossal motor outflow to genioglossus muscle in naturally sleeping rats. J Physiol 552, 975–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JL, Sood S, Liu H, Park E, Nolan P & Horner RL (2003. b). GABAA receptor antagonism at the hypoglossal motor nucleus increases genioglossus muscle activity in NREM but not REM sleep. J Physiol 548, 569–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouret J, Delorme F & Jouvet M (1967). [Lesions of the pontine tegmentum and sleep in rats]. C R Seances Soc Biol Fil 161, 1603–1606. [PubMed] [Google Scholar]
- Murai Y & Akaike T (2005). Orexins cause depolarization via nonselective cationic and K+ channels in isolated locus coeruleus neurons. Neurosci Res 51, 55–65. [DOI] [PubMed] [Google Scholar]
- Nakamura Y, Goldberg LJ, Chandler SH & Chase MH (1978). Intracellular analysis of trigeminal motoneuron activity during sleep in the cat. Science 199, 204–207. [DOI] [PubMed] [Google Scholar]
- Nishino S & Mignot E (1997). Pharmacological aspects of human and canine narcolepsy. Prog Neurobiol 52, 27–78. [DOI] [PubMed] [Google Scholar]
- Nishino S, Ripley B, Overeem S, Lammers GJ & Mignot E (2000). Hypocretin (orexin) deficiency in human narcolepsy. Lancet 355, 39–40. [DOI] [PubMed] [Google Scholar]
- Nitz D & Siegel J (1997. a). GABA release in the dorsal raphe nucleus: role in the control of REM sleep. Am J Physiol Regul Integr Comp Physiol 273, R451–R455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitz D & Siegel JM (1997. b). GABA release in the locus coeruleus as a function of sleep/wake state. Neuroscience 78, 795–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi Y, Williams RH, Agostinelli L, Arrigoni E, Fuller PM, Mochizuki T, Saper CB & Scammell TE (2013). Role of the medial prefrontal cortex in cataplexy. J Neurosci 33, 9743–9751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka T, Tsumori T, Yokota S & Yasui Y (2008). Neuroanatomical and neurochemical organization of projections from the central amygdaloid nucleus to the nucleus retroambiguus via the periaqueductal grey in the rat. Neurosci Res 62, 286–298. [DOI] [PubMed] [Google Scholar]
- Onoe H & Sakai K (1995). Kainate receptors: a novel mechanism in paradoxical (REM) sleep generation. Neuroreport 6, 353–356. [PubMed] [Google Scholar]
- Overeem S, van Nues SJ, van der Zande WL, Donjacour CE, van Mierlo P & Lammers GJ (2011). The clinical features of cataplexy: a questionnaire study in narcolepsy patients with and without hypocretin‐1 deficiency. Sleep Med 12, 12–18. [DOI] [PubMed] [Google Scholar]
- Pace‐Schott EF & Hobson JA (2002). The neurobiology of sleep: genetics, cellular physiology and subcortical networks. Nat Rev Neurosci 3, 591–605. [DOI] [PubMed] [Google Scholar]
- Peever J, Luppi PH & Montplaisir J (2014). Breakdown in REM sleep circuitry underlies REM sleep behavior disorder. Trends Neurosci 37, 279–288. [DOI] [PubMed] [Google Scholar]
- Peyron C, Faraco J, Rogers W, Ripley B, Overeem S, Charnay Y, Nevsimalova S, Aldrich M, Reynolds D, Albin R, Li R, Hungs M, Pedrazzoli M, Padigaru M, Kucherlapati M, Fan J, Maki R, Lammers GJ, Bouras C, Kucherlapati R, Nishino S & Mignot E (2000). A mutation in a case of early onset narcolepsy and a generalized absence of hypocretin peptides in human narcoleptic brains. Nat Med 6, 991–997. [DOI] [PubMed] [Google Scholar]
- Peyron C, Tighe DK, van den Pol AN, de Lecea L, Heller HC, Sutcliffe JG & Kilduff TS (1998). Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci 18, 9996–10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pissios P, Bradley RL & Maratos‐Flier E (2006). Expanding the scales: The multiple roles of MCH in regulating energy balance and other biological functions. Endocr Rev 27, 606–620. [DOI] [PubMed] [Google Scholar]
- Pollock MS & Mistlberger RE (2003). Rapid eye movement sleep induction by microinjection of the GABA‐A antagonist bicuculline into the dorsal subcoeruleus area of the rat. Brain Res 962, 68–77. [DOI] [PubMed] [Google Scholar]
- Pompeiano O (1967). The neurophysiological mechanisms of the postrual and motor events during desynchronized sleep. Res Publ Assoc Res Nerv Ment Dis 45, 351–423. [PubMed] [Google Scholar]
- Ponz A, Khatami R, Poryazova R, Werth E, Boesiger P, Bassetti CL & Schwartz S (2010). Abnormal activity in reward brain circuits in human narcolepsy with cataplexy. Ann Neurol 67, 190–200. [DOI] [PubMed] [Google Scholar]
- Quattrochi JJ, Mamelak AN, Madison RD, Macklis JD & Hobson JA (1989). Mapping neuronal inputs to REM sleep induction sites with carbachol‐fluorescent microspheres. Science 245, 984–986. [DOI] [PubMed] [Google Scholar]
- Rao Y, Lu M, Ge F, Marsh DJ, Qian S, Wang AH, Picciotto MR & Gao XB (2008). Regulation of synaptic efficacy in hypocretin/orexin‐containing neurons by melanin concentrating hormone in the lateral hypothalamus. J Neurosci 28, 9101–9110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichling DB & Basbaum AI (1990). Contribution of brainstem GABAergic circuitry to descending antinociceptive controls: I. GABA‐immunoreactive projection neurons in the periaqueductal grey and nucleus raphe magnus. J Comp Neurol 302, 370–377. [DOI] [PubMed] [Google Scholar]
- Rizvi TA, Ennis M, Behbehani MM & Shipley MT (1991). Connections between the central nucleus of the amygdala and the midbrain periaqueductal grey: topography and reciprocity. J Comp Neurol 303, 121–131. [DOI] [PubMed] [Google Scholar]
- Sabatinelli D, Bradley MM, Lang PJ, Costa VD & Versace F (2007). Pleasure rather than salience activates human nucleus accumbens and medial prefrontal cortex. J Neurophysiol 98, 1374–1379. [DOI] [PubMed] [Google Scholar]
- Sakai K, Crochet S & Onoe H (2001). Pontine structures and mechanisms involved in the generation of paradoxical (REM) sleep. Arch Ital Biol 139, 93–107. [PubMed] [Google Scholar]
- Sakai K & Koyama Y (1996). Are there cholinergic and non‐cholinergic paradoxical sleep‐on neurones in the pons? Neuroreport 7, 2449–2453. [DOI] [PubMed] [Google Scholar]
- Sakai K, Sastre JP, Kanamori N & Jouvet M (1981). State‐specific neurones in the ponto‐medullary reticular formation with specific reference to the postural atonia during paradoxical sleep in cat In Brain Mechanisms of Perceptual Awareness and Purposeful Behaviour, ed. Pompeiano M & Aimone Marsan C, pp. 405–429. Raven Press, New York. [Google Scholar]
- Sakai K, Sastre JP, Salvert D, Touret M, Tohyama M & Jouvet M (1979). Tegmentoreticular projections with special reference to the muscular atonia during paradoxical sleep in the cat: an HRP study. Brain Res 176, 233–254. [DOI] [PubMed] [Google Scholar]
- Sakurai T (2007). The neural circuit of orexin (hypocretin): maintaining sleep and wakefulness. Nat Rev Neurosci 8, 171–181. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Amemiya A, Ishii M, Matsuzaki I, Chemelli RM, Tanaka H, Williams SC, Richardson JA, Kozlowski GP, Wilson S, Arch JR, Buckingham RE, Haynes AC, Carr SA, Annan RS, McNulty DE, Liu WS, Terrett JA, Elshourbagy NA, Bergsma DJ & Yanagisawa M (1998). Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein‐coupled receptors that regulate feeding behaviour. Cell 92, 573–585. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Mieda M & Tsujino N (2010). The orexin system: roles in sleep/wake regulation. Ann N Y Acad Sci 1200, 149–161. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Nagata R, Yamanaka A, Kawamura H, Tsujino N, Muraki Y, Kageyama H, Kunita S, Takahashi S, Goto K, Koyama Y, Shioda S & Yanagisawa M (2005). Input of orexin/hypocretin neurons revealed by a genetically encoded tracer in mice. Neuron 46, 297–308. [DOI] [PubMed] [Google Scholar]
- Sanford LD, Cheng CS, Silvestri AJ, Tang X, Mann GL, Ross RJ & Morrison AR (2001). Sleep behaviour in rats with pontine lesions producing REM without atonia. Sleep Res Online 4, 1–5. [Google Scholar]
- Sanford LD, Tang X, Xiao J, Ross RJ & Morrison AR (2003). GABAergic regulation of REM sleep in reticularis pontis oralis and caudalis in rats. J Neurophysiol 90, 938–945. [DOI] [PubMed] [Google Scholar]
- Sapin E, Lapray D, Berod A, Goutagny R, Leger L, Ravassard P, Clement O, Hanriot L, Fort P & Luppi PH (2009). Localization of the brainstem GABAergic neurons controlling paradoxical (REM) sleep. PLoS One 4, e4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastre JP, Buda C, Kitahama K & Jouvet M (1996). Importance of the ventrolateral region of the periaqueductal grey and adjacent tegmentum in the control of paradoxical sleep as studied by muscimol microinjections in the cat. Neuroscience 74, 415–426. [DOI] [PubMed] [Google Scholar]
- Sastre JP, Sakai K & Jouvet M (1981). Are the gigantocellular tegmental field neurons responsible for paradoxical sleep? Brain Res 229, 147–161. [DOI] [PubMed] [Google Scholar]
- Satoh K & Fibiger HC (1986). Cholinergic neurons of the laterodorsal tegmental nucleus: efferent and afferent connections. J Comp Neurol 253, 277–302. [DOI] [PubMed] [Google Scholar]
- Scammell TE, Willie JT, Guilleminault C & Siegel JM; International Working Group on Rodent Models of Narcolepsy (2009). A consensus definition of cataplexy in mouse models of narcolepsy. Sleep 32, 111–116. [PMC free article] [PubMed] [Google Scholar]
- Schachter M & Parkes JD (1980). Fluvoxamine and clomipramine in the treatment of cataplexy. J Neurol Neurosurg Psychiatry 43, 171–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenck CH, Bundlie SR, Ettinger MG & Mahowald MW (1986). Chronic behavioral disorders of human REM sleep: a new category of parasomnia. Sleep 9, 293–308. [DOI] [PubMed] [Google Scholar]
- Schenck CH, Montplaisir JY, Frauscher B, Hogl B, Gagnon JF, Postuma R, Sonka K, Jennum P, Partinen M, Arnulf I, Cochen de Cock V, Dauvilliers Y, Luppi PH, Heidbreder A, Mayer G, Sixel‐Doring F, Trenkwalder C, Unger M, Young P, Wing YK, Ferini‐Strambi L, Ferri R, Plazzi G, Zucconi M, Inoue Y, Iranzo A, Santamaria J, Bassetti C, Moller JC, Boeve BF, Lai YY, Pavlova M, Saper C, Schmidt P, Siegel JM, Singer C, St Louis E, Videnovic A & Oertel W (2013). Rapid eye movement sleep behavior disorder: devising controlled active treatment studies for symptomatic and neuroprotective therapy–a consensus statement from the International Rapid Eye Movement Sleep Behavior Disorder Study Group. Sleep Med 14, 795–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel E & Siegel JM (1989). REM sleep without atonia after lesions of the medial medulla. Neurosci Lett 98, 159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherfler C, Frauscher B, Schocke M, Iranzo A, Gschliesser V, Seppi K, Santamaria J, Tolosa E, Hogl B, Poewe W & Group S (2011). White and grey matter abnormalities in idiopathic rapid eye movement sleep behavior disorder: a diffusion‐tensor imaging and voxel‐based morphometry study. Ann Neurol 69, 400–407. [DOI] [PubMed] [Google Scholar]
- Schwarz PB, Mir S & Peever JH (2014). Noradrenergic modulation of masseter muscle activity during natural rapid eye movement sleep requires glutamatergic signalling at the trigeminal motor nucleus. J Physiol 592, 3597–3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semba K (1993). Aminergic and cholinergic afferents to REM sleep induction regions of the pontine reticular formation in the rat. J Comp Neurol 330, 543–556. [DOI] [PubMed] [Google Scholar]
- Semba K & Fibiger HC (1992). Afferent connections of the laterodorsal and the pedunculopontine tegmental nuclei in the rat: a retro‐ and antero‐grade transport and immunohistochemical study. J Comp Neurol 323, 387–410. [DOI] [PubMed] [Google Scholar]
- Shammah‐Lagnado SJ, Negrao N, Silva BA & Ricardo JA (1987). Afferent connections of the nuclei reticularis pontis oralis and caudalis: a horseradish peroxidase study in the rat. Neuroscience 20, 961–989. [DOI] [PubMed] [Google Scholar]
- Sherrington CS (1898). Decerebrate rigidity, and reflex coordination of movements. J Physiol 22, 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM (2011). REM sleep: a biological and psychological paradox. Sleep Med Rev 15, 139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Nienhuis R, Fahringer HM, Paul R, Shiromani P, Dement WC, Mignot E & Chiu C (1991). Neuronal activity in narcolepsy: identification of cataplexy‐related cells in the medial medulla. Science 252, 1315–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Nienhuis R & Tomaszewski KS (1983). Rostral brainstem contributes to medullary inhibition of muscle tone. Brain Res 268, 344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel JM, Wheeler RL & McGinty DJ (1979). Activity of medullary reticular formation neurons in the unrestrained cat during waking and sleep. Brain Res 179, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soja PJ (2008). Glycine‐mediated postsynaptic inhibition is responsible for REM sleep atonia. Sleep 31, 1483–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soja PJ, Finch DM & Chase MH (1987. a). Effect of inhibitory amino acid antagonists on masseteric reflex suppression during active sleep. Exp Neurol 96, 178–193. [DOI] [PubMed] [Google Scholar]
- Soja PJ, Lopez‐Rodriguez F, Morales FR & Chase MH (1991). The postsynaptic inhibitory control of lumbar motoneurons during the atonia of active sleep: effect of strychnine on motoneuron properties. J Neurosci 11, 2804–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soja PJ, Lopez‐Rodriguez F, Morales FR & Chase MH (1995). Effects of excitatory amino acid antagonists on the phasic depolarizing events that occur in lumbar motoneurons during REM periods of active sleep. J Neurosci 15, 4068–4076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soja PJ, Morales FR, Baranyi A & Chase MH (1987. b). Effect of inhibitory amino acid antagonists on IPSPs induced in lumbar motoneurons upon stimulation of the nucleus reticularis gigantocellularis during active sleep. Brain Res 423, 353–358. [DOI] [PubMed] [Google Scholar]
- St Louis EK, McCarter SJ, Boeve BF, Silber MH, Kantarci K, Benarroch EE, Rando A, Tippmann‐Peikert M, Olson EJ & Mauermann ML (2014). Lesional REM sleep behaviour disorder localizes to the dorsomedial pons. Neurology 83, 1871–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenland HW, Liu H & Horner RL (2008). Endogenous glutamatergic control of rhythmically active mammalian respiratory motoneurons in vivo. J Neurosci 28, 6826–6835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M (2004). Acetylcholine systems and rhythmic activities during the waking–sleep cycle. Prog Brain Res 145, 179–196. [DOI] [PubMed] [Google Scholar]
- Steriade M & Hobson J (1976). Neuronal activity during the sleep‐waking cycle. Prog Neurobiol 6, 155–376. [PubMed] [Google Scholar]
- Stettner GM, Lei Y, Benincasa Herr K & Kubin L (2013). Evidence that adrenergic ventrolateral medullary cells are activated whereas precerebellar lateral reticular nucleus neurons are suppressed during REM sleep. PLoS One 8, e62410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strain GM, Olcott BM, Archer RM & McClintock BK (1984). Narcolepsy in a Brahman bull. J Am Vet Med Assoc 185, 538–541. [PubMed] [Google Scholar]
- Straube T, Pohlack S, Mentzel HJ & Miltner WH (2008). Differential amygdala activation to negative and positive emotional pictures during an indirect task. Behav Brain Res 191, 285–288. [DOI] [PubMed] [Google Scholar]
- Svensson TH & Engberg G (1980). Effect of nicotine on single cell activity in the noradrenergic nucleus locus coeruleus. Acta Physiol Scand Suppl 479, 31–34. [PubMed] [Google Scholar]
- Taal W & Holstege JC (1994). GABA and glycine frequently colocalize in terminals on cat spinal motoneurons. Neuroreport 5, 2225–2228. [DOI] [PubMed] [Google Scholar]
- Takakusaki K, Kohyama J & Matsuyama K (2003). Medullary reticulospinal tract mediating a generalized motor inhibition in cats: III. Functional organization of spinal interneurons in the lower lumbar segments. Neuroscience 121, 731–746. [DOI] [PubMed] [Google Scholar]
- Takakusaki K, Kohyama J, Matsuyama K & Mori S (2001). Medullary reticulospinal tract mediating the generalized motor inhibition in cats: parallel inhibitory mechanisms acting on motoneurons and on interneuronal transmission in reflex pathways. Neuroscience 103, 511–527. [DOI] [PubMed] [Google Scholar]
- Takakusaki K, Ohta Y & Mori S (1989). Single medullary reticulospinal neurons exert postsynaptic inhibitory effects via inhibitory interneurons upon alpha‐motoneurons innervating cat hindlimb muscles. Exp Brain Res 74, 11–23. [DOI] [PubMed] [Google Scholar]
- Thannickal TC, Moore RY, Nienhuis R, Ramanathan L, Gulyani S, Aldrich M, Cornford M & Siegel JM (2000). Reduced number of hypocretin neurons in human narcolepsy. Neuron 27, 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonokura M, Fujita K & Nishino S (2007). Review of pathophysiology and clinical management of narcolepsy in dogs. Vet Rec 161, 375–380. [DOI] [PubMed] [Google Scholar]
- Tononi G, Pompeiano M & Cirelli C (1991). Suppression of desynchronized sleep through microinjection of the alpha 2‐adrenergic agonist clonidine in the dorsal pontine tegmentum of the cat. Pflugers Arch 418, 512–518. [DOI] [PubMed] [Google Scholar]
- Torterolo P, Sampogna S & Chase MH (2009). MCHergic projections to the nucleus pontis oralis participate in the control of active (REM) sleep. Brain Res 1268, 76–87. [DOI] [PubMed] [Google Scholar]
- Torterolo P, Sampogna S & Chase MH (2013). Hypocretinergic and non‐hypocretinergic projections from the hypothalamus to the REM sleep executive area of the pons. Brain Res 1491, 68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travers JB, Yoo JE, Chandran R, Herman K & Travers SP (2005). Neurotransmitter phenotypes of intermediate zone reticular formation projections to the motor trigeminal and hypoglossal nuclei in the rat. J Comp Neurol 488, 28–47. [DOI] [PubMed] [Google Scholar]
- Trulson ME & Jacobs BL (1979). Raphe unit activity in freely moving cats: correlation with level of behavioural arousal. Brain Res 163, 135–150. [DOI] [PubMed] [Google Scholar]
- Tsunematsu T, Ueno T, Tabuchi S, Inutsuka A, Tanaka KF, Hasuwa H, Kilduff TS, Terao A & Yamanaka A (2014). Optogenetic manipulation of activity and temporally controlled cell‐specific ablation reveal a role for MCH neurons in sleep/wake regulation. J Neurosci 34, 6896–6909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN (2012). Neuropeptide transmission in brain circuits. Neuron 76, 98–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dort CJ, Zachs DP, Kenny JD, Zheng S, Goldblum RR, Gelwan NA, Ramos DM, Nolan MA, Wang K, Weng FJ, Lin Y, Wilson MA & Brown EN (2015). Optogenetic activation of cholinergic neurons in the PPT or LDT induces REM sleep. Proc Natl Acad Sci USA 112, 584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanini G, Torterolo P, McGregor R, Chase MH & Morales FR (2007). GABAergic processes in the mesencephalic tegmentum modulate the occurrence of active (rapid eye movement) sleep in guinea pigs. Neuroscience 145, 1157–1167. [DOI] [PubMed] [Google Scholar]
- Verret L, Fort P, Gervasoni D, Leger L & Luppi PH (2006). Localization of the neurons active during paradoxical (REM) sleep and projecting to the locus coeruleus noradrenergic neurons in the rat. J Comp Neurol 495, 573–586. [DOI] [PubMed] [Google Scholar]
- Verret L, Goutagny R, Fort P, Cagnon L, Salvert D, Leger L, Boissard R, Salin P, Peyron C & Luppi PH (2003). A role of melanin‐concentrating hormone producing neurons in the central regulation of paradoxical sleep. BMC Neurosci 4, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verret L, Leger L, Fort P & Luppi PH (2005). Cholinergic and noncholinergic brainstem neurons expressing Fos after paradoxical (REM) sleep deprivation and recovery. Eur J Neurosci 21, 2488–2504. [DOI] [PubMed] [Google Scholar]
- Vetrivelan R, Fuller PM, Tong Q & Lu J (2009). Medullary circuitry regulating rapid eye movement sleep and motor atonia. J Neurosci 29, 9361–9369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volgin DV, Rukhadze I & Kubin L (2008). Hypoglossal premotor neurons of the intermediate medullary reticular region express cholinergic markers. J Appl Physiol 105, 1576–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber F, Chung S, Beier KT, Xu M, Luo L, Dan Y (2015). Control of REM sleep by ventral medulla GABAergic neurons. Nature 526, 435–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng FJ, Williams RH, Hawryluk JM, Lu J, Scammell TE, Saper CB & Arrigoni E (2014). Carbachol excites sublaterodorsal nucleus neurons projecting to the spinal cord. J Physiol 592, 1601–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White EC & de Lahunta A (2001). Narcolepsy in a ram lamb. Vet Rec 149, 156–157. [DOI] [PubMed] [Google Scholar]
- White SR, Fung SJ & Barnes CD (1991). Norepinephrine effects on spinal motoneurons. Prog Brain Res 88, 343–350. [DOI] [PubMed] [Google Scholar]
- White SR, Fung SJ, Jackson DA & Imel KM (1996). Serotonin, norepinephrine and associated neuropeptides: effects on somatic motoneuron excitability. Prog Brain Res 107, 183–199. [DOI] [PubMed] [Google Scholar]
- William RH, Iqbal SZ, Saper CB & Arrigoni E (2012). The effects of carbachol, norepinephrine, and serotonin on spinally projecting glutamatergic neurons of the sublaterodorsal nucleus. Neuroscience 2012 Abstracts, Program No. 799.714. Society for Neuroscience, Washington, DC. [Google Scholar]
- Williams JA & Reiner PB (1993). Noradrenaline hyperpolarizes identified rat mesopontine cholinergic neurons in vitro. J Neurosci 13, 3878–3883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willie JT, Chemelli RM, Sinton CM, Tokita S, Williams SC, Kisanuki YY, Marcus JN, Lee C, Elmquist JK, Kohlmeier KA, Leonard CS, Richardson JA, Hammer RE & Yanagisawa M (2003). Distinct narcolepsy syndromes in Orexin receptor‐2 and Orexin null mice: molecular genetic dissection of non‐REM and REM sleep regulatory processes. Neuron 38, 715–730. [DOI] [PubMed] [Google Scholar]
- Wu MF, Gulyani SA, Yau E, Mignot E, Phan B & Siegel JM (1999). Locus coeruleus neurons: cessation of activity during cataplexy. Neuroscience 91, 1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu MF, John J, Boehmer LN, Yau D, Nguyen GB & Siegel JM (2004). Activity of dorsal raphe cells across the sleep‐waking cycle and during cataplexy in narcoleptic dogs. J Physiol 554, 202–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi M & Chase MH (2010). The injection of hypocretin‐1 into the nucleus pontis oralis induces either active sleep or wakefulness depending on the behavioral state when it is administered. Sleep 33, 1236–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi MC, Fung SJ, Yamuy J, Morales FR & Chase MH (2002). Induction of active (REM) sleep and motor inhibition by hypocretin in the nucleus pontis oralis of the cat. J Neurophysiol 87, 2880–2888. [DOI] [PubMed] [Google Scholar]
- Xi MC, Fung SJ, Yamuy J, Morales FR & Chase MH (2003). Hypocretinergic facilitation of synaptic activity of neurons in the nucleus pontis oralis of the cat. Brain Res 976, 253–258. [DOI] [PubMed] [Google Scholar]
- Xi MC, Morales FR & Chase MH (1999. a). Evidence that wakefulness and REM sleep are controlled by a GABAergic pontine mechanism. J Neurophysiol 82, 2015–2019. [DOI] [PubMed] [Google Scholar]
- Xi MC, Morales FR & Chase MH (1999. b). A GABAergic pontine reticular system is involved in the control of wakefulness and sleep. Sleep Res Online 2, 43–48. [PubMed] [Google Scholar]
- Xi MC, Morales FR & Chase MH (2001). The motor inhibitory system operating during active sleep is tonically suppressed by GABAergic mechanisms during other states. J Neurophysiol 86, 1908–1915. [DOI] [PubMed] [Google Scholar]
- Xi Z & Luning W (2009). REM sleep behavior disorder in a patient with pontine stroke. Sleep Med 10, 143–146. [DOI] [PubMed] [Google Scholar]
- Xu M, Chung S, Zhang S, Zhong P, Ma C, Chang WC, Weissbourd B, Sakai N, Luo L, Nishino S & Dan Y (2015). Basal forebrain circuit for sleep‐wake control. Nat Neurosci 18, 1641–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon YS & Lee HS (2013). Projections from melanin‐concentrating hormone (MCH) neurons to the dorsal raphe or the nuclear core of the locus coeruleus in the rat. Brain Res 1490, 72–82. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Sampogna S, Morales FR & Chase MH (2004). Distribution of hypocretin (orexin) immunoreactivity in the feline pons and medulla. Brain Res 995, 205–217. [DOI] [PubMed] [Google Scholar]