

Abstract
Introduction: Based on the amyloid cascade hypothesis of Alzheimer’s disease (AD) pathogenesis, a series of clinical trials involving immunotherapies have been undertaken including infusion with the IgG1 monoclonal anti-Aβ antibody solanezumab directed against the middle of the soluble Aβ peptide. In this report, we give an account of the clinical history, psychometric testing, gross and microscopic neuropathology as well as immunochemical quantitation of soluble and insoluble Aβ peptides and other proteins of interest related to AD pathophysiology in a patient treated with solanezumab. Materials and Methods: The solanezumab-treated AD case (SOLA-AD) was compared to non-demented control (NDC, n = 5) and non-immunized AD (NI-AD, n = 5) subjects. Brain sections were stained with H&E, Thioflavine-S, Campbell-Switzer and Gallyas methods. ELISA and Western blots were used for quantification of proteins of interest. Results: The SOLA-AD subject’s neuropathology and biochemistry differed sharply from the NDC and NI-AD groups. The SOLA-AD case had copious numbers of amyloid laden blood vessels in all areas of the cerebral cortex, from leptomeningeal perforating arteries to arteriolar deposits which attained the cerebral amyloid angiopathy (CAA) maximum score of 12. In contrast, the maximum CAA for the NI-AD cases averaged a total of 3.6, while the NDC cases only reached 0.75. The SOLA-AD subject had 4.4-fold more soluble Aβ40 and 5.6-fold more insoluble Aβ40 in the frontal lobe compared to NI-AD cases. In the temporal lobe of the SOLA-AD case, the soluble Aβ40 was 80-fold increased, and the insoluble Aβ40 was 13-fold more abundant compared to the non-immunized AD cases. Both soluble and insoluble Aβ42 levels were not dramatically different between the SOLA-AD and NI-AD cohort. Discussion: Solanezumab immunotherapy provided no apparent relief in the clinical evolution of dementia in this particular AD patient, since there was a continuous cognitive deterioration and full expression of amyloid deposition and neuropathology.
Keywords: Alzheimer’s disease, immunotherapy, solanezumab, amyloid-beta, amyloid precursor protein, cerebral amyloid angiopathy
Introduction
Alzheimer’s disease (AD) is the most common form of dementia and cause of death among the elderly. AD is characterized by the abundant deposition of amyloid-beta (Aβ) peptides in parenchymal plaques and cerebral blood vessel walls as well as intracellular neurofibrillary tangles (NFT), mainly composed of tau protein. The profuse accumulation of fibrillar Aβ peptides led to the postulation of the amyloid cascade hypothesis [1] for the pathogenesis, pathophysiology and clinical evolution of AD. A series of clinical trials involving immunotherapies targeting Aβ peptides have been implemented including the IgG1 monoclonal anti-Aβ antibody solanezumab directed against the mid-region of the soluble Aβ peptide.
Few postmortem studies of AD subjects who received immunotherapy have been published. We provide an account of the clinical history, psychometric testing results, gross and microscopic neuropathology as well as immunochemical quantitation of Aβ soluble and insoluble peptides, amyloid precursor protein (APP), tumor necrosis factor-α (TNF-α), interleukin-1β (IL-1β), presenilin-1 (PSEN1), β-secretase-1 (BACE1), Tau, α-synuclein, Bcl-2, S100B and apolipoprotein E (ApoE) in a patient who died as the result of AD after receiving solanezumab treatment for a period of 9 months.
Clinical summary
The subject was a 79 year-old man who died with a clinical diagnosis of probable AD dementia. According to family members, symptoms had first been noticed about 12 years earlier. The available private medical records date from six years prior to his death, when he presented with a 5-6 year history of memory dysfunction and loss of ability to live independently. Aside from repeating the same stories and questions frequently, he was often disoriented to time and place, was not able to handle his own finances medications or complete common household tasks. He had been having trouble recognizing people who were familiar to him, including family and friends. His personality had changed in that he was more withdrawn and had a shorter temper. He had been having paranoid delusions and possibly illusions or hallucinations. He had been treated with Aricept for up to 5 years. On examination he had an antalgic gait and hypertonia with variable resistance during passive movement, increased muscle tone without bradykinesia or tremor. He had fluent speech and was able to follow simple commands. He scored 16/30 on the Mini Mental State Examination (MMSE), recalling none of 5 words after a few minutes delay. A metabolic PET brain scan showed hypometabolism in the parietal and temporal lobes as well as the posterior cingulate gyrus. The diagnostic impression was of dementia due to probable AD. He was continued on Aricept and Namenda was added a few months later. Over subsequent years, his cognitive status declined further and he developed physically aggressive behavior; he was treated with anti-psychotic agents. Four years prior to death, he scored 6/30 on the MMSE, developed an unsteady gait and started using a walker. Three years prior to death, he was rated as 3 on the Clinical Dementia Rating Scale.
Other past medical history includes obesity, hypertension, hyperlipidemia, coronary artery disease, myocardial infarction, status post-stent placement, status post-coronary bypass, hepatic steatosis, chronic renal insufficiency, pancreatitis, cholecystectomy, osteoarthritis and lumbar laminectomy. He had had “lung surgery” but no additional details were provided. A prior history of bipolar disorder was indicated, but was not described. He had been treated with phenobarbital for years after a single suspected seizure. He had at least one episode of transient loss of responsiveness two years prior to death, but it is not clear whether this was a seizure or a transient ischemic attack. The family history revealed the subject had a strong family history of dementia; his father and a sister had Alzheimer’s dementia, and his mother had “mental problems” in her old age.
Psychometric testing summary
6 years prior to death (PD): MMSE 16. 5 years PD: ADAS-Cog 46, CDR 1, ADL 50. 4.7 years PD: ADAS-cog 44, ADL 50. 4.5 years PD: MMSE 10, ADAS-cog 52, CDR 2, ADL 46. 4.2 years PD: ADAS-cog 51, ADL 47. 4 years PD: MMSE 8, ADAS-cog discontinued secondary to patient agitation, CDR 2 (with informant feedback only), ADL 40. 3.7 years PD: ADAS-cog 61, ADL 33. 3.3 years PD: MMSE 6, ADAS-cog 72, CDR 2, ADL 43. 3.2 years PD: ADAS-cog 74, ADL 30. 2.9 years PD: MMSE 8, ADAS-cog not completed secondary to degree of impairment, CDR 2 (with informant feedback only), ADL 31. 2.6 years PD: MMSE 4, ADAS-cog not completed secondary to degree of impairment, CDR 2 (with caregiver feedback only), ADL 23.
Review of performance/ratings over 3 years reveals consistent cognitive decline with progressively failing functional status. At the end of study, the participant was no longer testable and required 24-hour care.
Neuropathology summary
Gross description of brain
The brain mass was 1042 grams. The dura mater was normal and the leptomeninges showed patchy mild and moderate fibrosis of the temporal lobes and midsagittal areas, respectively. The convexities were symmetrical and showed moderate to severe gyral atrophy of the anterior frontal lobes, moderate gyral atrophy of the posterior frontal lobes and parietal lobes and mild to moderate gyral atrophy of the occipital lobes. Paracentral regions exhibited mild gyral atrophy of the precentral gyri and mild to moderate gyral atrophy of the postcentral gyri. No focal lesions were present on the convexities or base of the brain. The circle of Willis had moderate patchy atherosclerosis. The mammillary bodies were slightly flattened in appearance demonstrating dusky brown discoloration and atrophy without evidence of hemosiderin deposition or capillary proliferation. The temporal lobes and unci showed moderate to severe atrophy. The cerebellum and brainstem were externally normal. Cerebral slices showed mild to moderate enlargement of the lateral ventricles. The basal ganglia, thalamus and subthalamic nucleus were unremarkable. The amygdala, head and body of the hippocampus and the parahippocampal gyrus all showed severe atrophy. The substantia nigra had severe depigmentation bilaterally. Respective axial and parasagittal slices of the brainstem and cerebellum were normal.
Microscopic description
Paraffin-embedded sections of left-sided brain structures stained with H&E showed marked gliosis of upper neocortical layers; senile plaques were frequent. The amygdala, as well as the entorhinal area, showed marked gliosis. Area CA1 of the hippocampus showed narrowing and marked gliosis with frequent hypertrophic astrocytes and numerous ghost tangles. Sections of the caudate nucleus and putamen showed mild patchy gliosis; occasional larger neurons had NFT. Several mineralized blood vessels were noted in the globus pallidus. The hypothalamus and medial thalamus had marked gliosis otherwise, sections of the thalamus and subthalamic regions were unremarkable. The substantia nigra and locus ceruleus were markedly depleted of pigmented neurons; there were several pale bodies and two Lewy bodies in the former region. Sections of the cerebellum showed cortical sclerosis with frequent corpora amylacea over a region measuring 0.8 x 0.4 cm. The remainder of the cerebellum was normal. Remaining sections of the brainstem and cervical spinal cord were unremarkable. Large cerebral sections stained with H&E showed no infarcts in any of the examined sections. Sections stained immunohistochemically for amyloid, as well as with Campbell-Switzer and Thioflavine-S methods showed frequent senile plaques with both neuritic and diffuse plaques in neocortical areas and fibrillar amyloid bundles in the cerebellar cortex. Plaques were also frequent in the striatum and thalamus, while they were rare to focally frequent in the cerebellar cortex. Amyloid angiopathy was highly prominent in all cerebral cortex regions as well as in the cerebellar and cerebral convexity leptomeninges. Neurofibrillary tangles were assessed by Gallyas staining and phosphorylated tau antibodies and were found to be present at frequent densities in neocortical areas except the primary visual area. Tangles were frequent in the amygdala, hippocampal CA1 region and entorhinal area. Sections stained immunohistochemically for phosphorylated α-synuclein showed frequent neuronal perikaryal inclusions and associated fibers in the olfactory bulb and amygdala while these are rare in the medulla, locus ceruleus and cingulate gyrus, and absent in cerebral neocortex.
Pathology diagnosis
Alzheimer’s disease; microscopic changes of Lewy body disease, insufficient for diagnosis; focal cerebellar cortical sclerosis, left cerebellar superior vermis.
Materials and methods
Human subjects
In order to establish the degree of variation in the levels of proteins of interest between the solanezumab treated AD patient (SOLA-AD) and non-treated AD and control individuals, we selected 5 non-demented controls (NDC) and 5 NI-AD cases for comparison analyses. The demographic and neuropathological assessments are shown in Table 1. All brain tissues were obtained from our Brain and Body Donation Program at BSHRI [2,3]. Written informed consent was obtained for all clinical and autopsy procedures related to the present study and all approvals were obtained by the Banner Health and Western Institutional Review Boards. Verbal informed consent was provided by the donor or the donor’s next of kin. APOE genotypes for all cases were obtained from DNA isolated from cerebellar samples using a modified technique of Hixson and Vernier [4]. Demographic information (age of death, gender and APOE genotype), postmortem interval (PMI), brain weight and neuropathological scoring are presented in Table 1.
Table 1.
Study Subject Demographics and Neuropathology Scores
| NDC | Expired Age (yrs) | Gender | PMI (h) | APOE Genotype | Brain Weight (g) | Total Plaque Score | Total NFT Score | Braak Score | Total WMRScore | Total CAAScore |
|
| ||||||||||
| 1 | 74 | M | 2.50 | 3/3 | 1230 | 0.0 | 3.5 | II | 1 | n/a |
| 2 | 67 | F | 3.50 | 3/4 | 1150 | 9.9 | 4.0 | II | 2 | 0 |
| 3 | 69 | M | 2.00 | 3/3 | 1190 | 0.0 | 0.3 | I | 0 | 0 |
| 4 | 76 | M | 2.33 | 3/4 | 1375 | 5.5 | 0.0 | I | 1 | 3 |
| 5 | 68 | F | 2.60 | 3/3 | 1140 | 0.0 | 3.5 | III | 4 | 0 |
| Mean | 71 | 2.59 | 1217 | 3.1 | 2.3 | II* | 1.6 | 0.75 | ||
|
| ||||||||||
| NI-AD | Expired Age (yrs) | Gender | PMI (h) | APOE Genotype | Brain Weight (g) | Total Plaque Score | Total NFT Score | Braak Score | Total WMR Score | Total CAA Score |
|
| ||||||||||
| 12 | 65 | F | 3.25 | 3/3 | 800 | 15.0 | 15.0 | VI | 2 | 0 |
| 13 | 77 | M | 2.33 | 3/4 | 1080 | 14.0 | 15.0 | VI | 7 | 2 |
| 14 | 68 | F | 3.50 | 3/4 | 915 | 14.5 | 15.0 | VI | 1 | 2 |
| 15 | 79 | F | 5.00 | 3/4 | 1070 | 15.0 | 15.0 | VI | 4 | 3 |
| 16 | 68 | F | 4.16 | 3/3 | 1074 | 15.0 | 15.0 | VI | 4 | 11 |
| Mean | 71 | 3.65 | 988 | 14.7 | 15.0 | VI* | 3.6 | 3.6 | ||
|
| ||||||||||
| Expired Age (yrs) | Gender | PMI(h) | APOE Genotype | Brain Weight (g) | Total Plaque Score | Total NFTScore | Braak Score | Total WMRScore | Total CAAScore | |
|
| ||||||||||
| SOLA-AD | 79 | M | 2.4 | 4/4 | 1042 | 13.5 | 15 | V | 5 | 12 |
NDC, non-demented control; NI-AD, non-immunized Alzheimer’s disease; SOLA-AD, solanezumab Alzheimer’s disease patient; yrs, years; h, hours; APOE, apolipoprotein E; g, grams; NFT, neurofibrillary tangle; WMR, white matter rarefaction; CAA, cerebral amyloid angiopathy; n/a, not available.
Median reported.
Histological examination
Brain coronal sections (~1 cm thick) of the left hemispheres were fixed in formalin and large blocks comprising half coronal sections (80 μm thickness) were sectioned utilizing a frozen microtome. Frontal, parietal, occipital, cerebellum and temporal sections including the amygdala and hippocampus, mid-brain at the level of the substantia nigra, thalamus and striatum, were evaluated. Histological sections were stained using H&E, Campbell-Switzer, Gallyas and Thioflavine-S methods and scored [3]. Coronal sections of the corresponding right hemispheres were immediately frozen in slabs of dry ice, packed separately under vacuum and stored at -86°C. All brains were neuropathologically appraised for amyloid plaques, NFT, white matter rarefaction (WMR) and cerebral amyloid angiopathy (CAA). Frontal, temporal, parietal, hippocampal and entorhinal areas were scored as follows as 0 (none), 1 (sparse), 2 (moderate) and 3 (frequent) for a maximum additive value of 15 for total amyloid plaque and NFT scoring. Braak scores were evaluated following the Braak stage method and range from I-VI [5]. Total CAA scores were appraised in the frontal, temporal, parietal and occipital lobes with maximum total score of 12, and scored as none (0), mild (1), moderate (2) and severe (3). The total WMR score was scored as none (0), mild (less than 25% affected = 1) to moderate (25 to 50% affected = 2) to severe (greater than 50% affected = 3). The results of these neuropathological scores as well as brain weight at autopsy are summarized in Table 1.
Preparation of cortical blood vessels
For assessing the degree of CAA, approximately 1 g of cerebral cortex from the frontal, temporal and occipital cortices of the SOLA-AD case were sectioned into pieces measuring ~5 mm3. The material from each lobe was gently stirred in 300 ml of 5% SDS prepared in 10 mM Tris-HCl buffer, pH 7.5 for 72 h at room temperature. The resulting tufts of cortical blood vessels were washed 3 X with distilled water to remove the SDS and spread on microscopic glass slides, dried in an oven at 60°C for 2 h, fixed with absolute ethanol for 1 h and stained with 1% Thioflavine-S. Excess of unbound Thioflavine-S was removed by multiple rinses in 70% ethanol.
Aβ40 and Aβ42 ELISA
Gray matter tissue from the frontal and temporal lobes (200 mg) were each gently homogenized in 1600 µl of 20 mM Tris, 5 mM EDTA, pH 7.8 supplemented with a complete™ protease inhibitor cocktail (PIC, Roche, Mannheim, Germany) using a Teflon tissue homogenizer. The samples were centrifuged at 435,000 x g for 20 min, 4°C in a Beckman TLA 120.2 rotor (Brea, CA), the supernatant recovered and retained for analyses as the ‘soluble’ fraction. The Tris-insoluble pellet was suspended in 1200 µl of 90% glass-distilled formic acid (GDFA) with an Omni TH tissue grinder (Kennesaw, GA) and centrifuged at 435,000 x g for 20 min, 4°C in a Beckman TLA 120.2 rotor. The supernatant was dialyzed twice against deionized water (1 h each) 3 times with 0.1 M ammonium bicarbonate buffer (1 h each) and lyophilized. The lyophilized proteins were reconstituted in 1000 µl 5 M guanidine-hydrochloride, 50 mM Tris, pH 8.0 with PIC, centrifuged as described above and the supernatant saved as the ‘insoluble’ fraction. Total protein in the soluble and insoluble fractions was determined with a Pierce Micro BCA protein assay kit (Rockford, IL). The following ELISA kits from Life Technologies Corp (Carlsbad, CA), were used to quantify Aβ following the manufacturer’s instructions: Aβ40 (KHB3481; limit of detection <6 pg/ml), Aβ42 (KHB3441; limit of detection = 10 pg/ml) and Aβ42 ultrasensitive (KHB3544; limit of detection = 1 pg/mL).
TNF-α and IL-1β ELISA
Two-hundred mg of frontal and temporal gray matter tissue was homogenized in 2 ml of RIPA buffer (Sigma) containing PIC with an Omni TH tissue grinder and centrifuged at 18,000 x g for 30 min at 4°C in a Beckman 22R centrifuge. The supernatant was collected and submitted to Pierce’s Micro BCA protein assay kit for total protein determination. TNF-α (PK-EL-63707) and IL-1β (PK-EL-61127) were quantified with ELISA kits from PromoKine (Heidelberg, Germany) following the manufacturer’s instructions.
Western blots
Gray matter tissue from the frontal and temporal lobe (200 mg) was homogenized in 2 ml of 5% SDS, 5 mM EDTA, 20 mM Tris, pH 8.0 that included PIC using an Omni TH tissue grinder. The homogenates were centrifuged at 435,000 x g for 20 min, 20°C in a Beckman TLA 120.2 rotor, the supernatant was recovered and total protein determined by a Micro BCA protein assay kit (Pierce). Samples containing 50 µg of total protein were mixed with 2 X NuPage LDS (Life Technology Corp) sample buffer containing 50 mM DTT (Sigma) and heated at 80°C for 10 min. Proteins were electrophoretically separated on 4-12% Bis-Tris gels (Life Technology Corp) using 1 X MES SDS run buffer and NuPage antioxidant (Life Technology Corp). Bio-Rad’s prestained kaleidoscope marker was used as a molecular mass standard (Hercules, CA). Proteins were transferred onto 0.45 µm nitrocellulose membranes with 1 X NuPage transfer buffer (Life Technology Corp) and 20% methanol. After blocking in 5% Quick-Blocker (G-Biosciences, St. Louis, MO), 1 X PBS, 0.5% Tween 20 for 1 h, blots were incubated at 4°C overnight in blocking buffer solution containing the primary antibody (Table 2). The blots were washed 4 times for 5 min in 1 X PBS, 0.5% Tween 20 and incubated 1 h in HRP conjugated secondary antibody (Table 2). After incubation with secondary antibody, washing was repeated. SuperSignal WestPico chemiluminescent substrate (Pierce) was added with CL-Xpose film (Pierce) and a Konica Minolta SRX-101A film processor (Wayne, NJ) used for signal detection. All blots were boiled for 10 min in 1 X PBS to remove antibodies and re-probed with either anti-mouse actin or anti-rabbit actin (Table 2) to provide a total protein loading control. The films were scanned with a GS-800 calibrated densitometer (Bio-Rad) and the band density determined with the trace quantity feature in the Quantity One software (Bio-Rad). Final density values were normalized to actin.
Table 2.
Antibodies Used in Western Blots
| Primary antibody | Antigen specificity or immunogen | Secondary antibody | Company/Catalog # |
|---|---|---|---|
| CT20APP | C1/6.1 clone, last 20 aa of APP | M | Covance/SIG-39152 |
| PSEN1-CTF | D39D1 clone, CTF peptide of PSEN | R | Cell Signaling Technology/5643 |
| BACE1 | 3D5 clone, aa 46-460 | M | Kindly provided by Dr. R. Vassar |
| Tau | HT7 clone, aa 159-163 | M | Pierce/MN1000 |
| α-synuclein | 42/α-synuclein clone, aa 15-123 of rat synuclein-1 | M | BD Transduction Laboratories/610786 |
| ApoE | Recombinant human ApoE | G | Millipore/AB947 |
| Bcl-2 | Bcl-2-100 clone, aa 41-54 of human Bcl-2 | M | Life Technologies Corp./13-8800 |
| S100B | EP1576Y clone, CT peptide of human S100B | R | Abcam/ab52642 |
| Actin | C4 clone, chicken gizzard muscle actin | M | BD Transduction Laboratories/612657 |
| Actin | N-terminus of human α-actin | R | Abcam/ab37063 |
CT, C-terminal; aa, amino acid; APP, amyloid-β precursor protein; PSEN1, preseniln-1; CTF, C-terminal fragment; BACE, β-secretase; ApoE, apolipoprotein E; M, HRP conjugated AffiniPure goat-anti mouse IgG (catalog # 115-035-146, Jackson Laboratory); R, HRP conjugated AffiniPure goat-anti rabbit IgG, (catalog # 111-035-144, Jackson Laboratory); G, HRP conjugated AffiniPure bovine-anti goat IgG (catalog # 805-035-180, Jackson Laboratory).
Results
The neuropathology scores differed substantially between the NDC and AD cases (Table 1). The SOLA-AD subject neuropathology resembled the NI-AD cohort with the exception of total CAA scores (Table 1). The SOLA-AD case had copious numbers of blood vessels loaded with amyloid in all areas of the cerebral cortex (Figures 1D, 1E and 2) from leptomeningeal perforating arteries to arteriolar/capillary deposits which attained the CAA maximum score of 12. Figure 2 shows that the walls of the brain microvessels in the cerebral cortex of the frontal, temporal and occipital lobes were heavily loaded with amyloid deposits as demonstrated by Thioflavine-S. In contrast, the corresponding CAA scores for the 5 NI-AD cases averaged a total of 3.6 while the NDC cases only reached 0.75 (Table 1). In addition, the SOLA-AD patient demonstrated an abundant number of cortical amyloid plaques, most of them showing compact deposits of Aβ, which were similar to the number of plaque deposits observed in the NI-AD cases (total plaque scores: 13.5 and 14.7, respectively), as shown in Table 1 and Figure 1. An analogous trend was observed when total NFT score was compared between the NI-AD cases and the SOLA-AD individual in which there were abundant numbers of NFT (Figure 1G-I and Table 1) in the frontal, parietal and, temporal cortices, and the hippocampus. The SOLA-AD subject’s visual cortex had no NFT and the total Braak score was V. The total WMR score (a postmortem measure of leukoaraiosis) in the NDC and non-immunized NI-AD cases averaged 1.6 and 3.6, respectively, while the SOLA-AD patient scored 5.0. Another interesting feature was the presence of foci of fibrillar amyloid bundles in the cerebellum of the SOLA-AD subject (Figure 1C), which are consistent with advanced amyloid dissemination observed in the final stages of AD.
Figure 1.
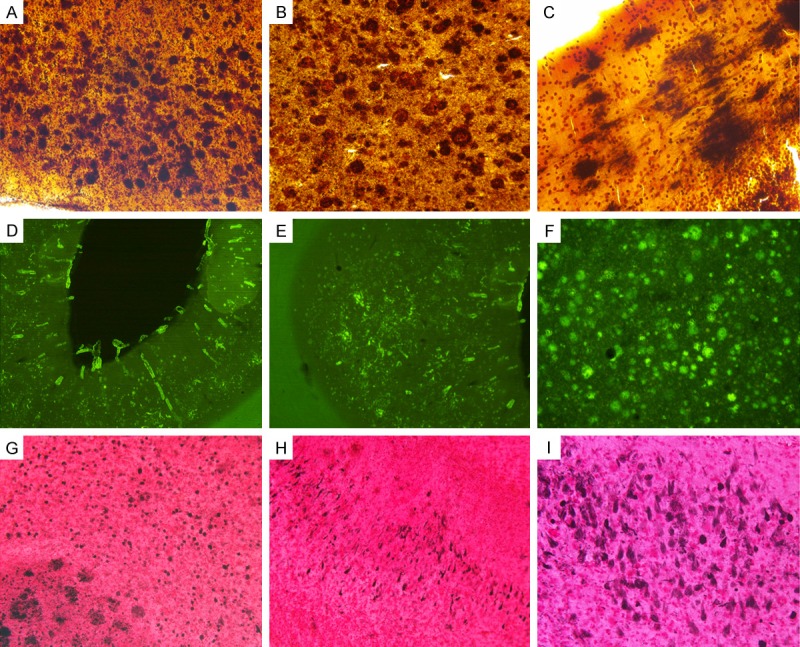
Amyloid plaques, vascular amyloid and neurofibrillary tangles in a case of a solanezumab-immunized AD patient. A. Temporal lobe amyloid plaques stained with Campbell-Switzer, 100 X. B. Frontal lobe amyloid plaques stained with Campbell-Switzer, 100 X. C. Cerebellum amyloid plaques stained with Campbell-Switzer stain, 200 X. D and E. Visual cortex showing rich deposits of vascular amyloid stained with Thioflavine-S, 25 X. F. Parietal cortex amyloid plaques stained with Thioflavine-S, 25 X. G. Temporal cortex amyloid plaques and NFT stained with Gallyas, 100 X. H. Temporal cortex NFT stained with Gallyas, 100 X. I. Temporal cortex NFT stained with Gallyas, 200 X.
Figure 2.
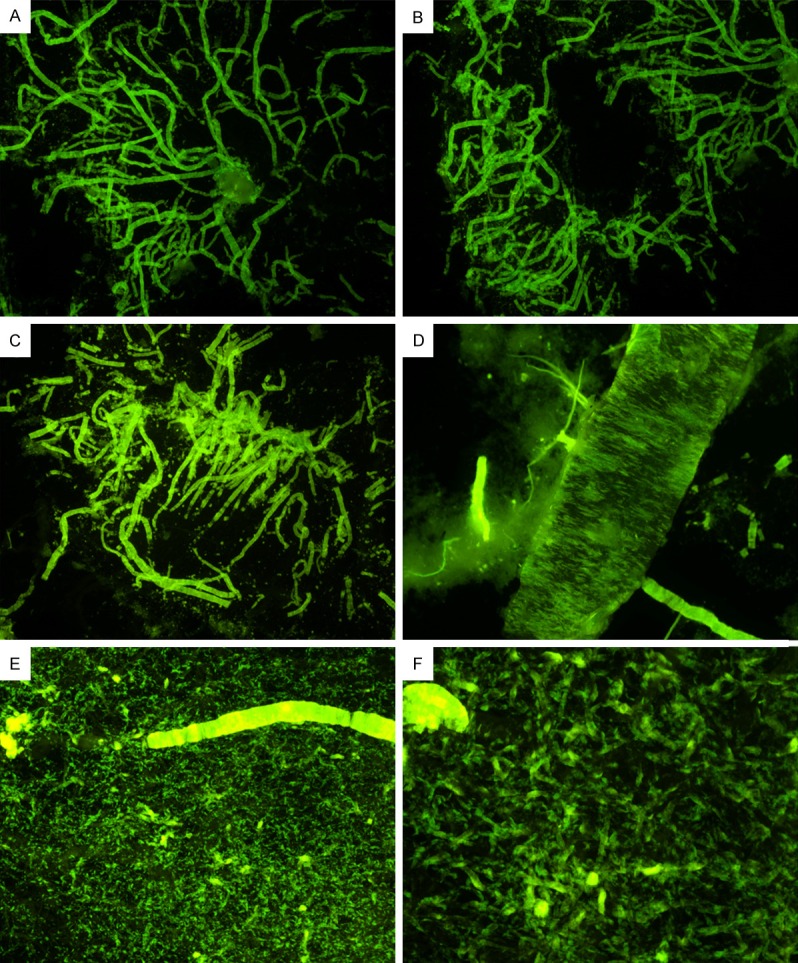
Cerebrovascular amyloidosis in a case of solanezumab-immunized patient stained with Thioflavine-S. (A and B) Frontal lobe cortex, 20 X. (C) Temporal cortex, 20 X. (D) Occipital cortex, 40 X. (E) Occipital cortex, 100 X. Notice that all small arteries and arterioles are loaded with amyloid, which are more evident at 200 X as shown in caption (F).
For biochemical studies, frontal and temporal cortical samples were examined. ELISA quantification of Aβ demonstrated some interesting results shown in Table 3. The amounts of Aβ40 extracted by Tris buffer and GDFA/GHCl treatment were substantially increased in the frontal and temporal lobes of the SOLA-AD case compared to the NI-AD and NDC cases (Table 3). These elevated amounts probably reflect the large number of vessels profusely loaded with Aβ as observed in the histological sections (Figures 1 and 2). In this respect, the difference between our SOLA-AD case and the NI-AD cases (n = 5), in terms of the soluble and insoluble fractions, are large. In the frontal lobe, the Tris-soluble Aβ40 fraction amounted to 13,634 pg/mg total protein vs. 3,067 pg/mg total protein, respectively, and for the GDFA/GHCl-soluble Aβ40 1,711 ng/mg total protein vs. 304 ng/mg total protein, respectively. In the temporal lobe, the Tris-soluble Aβ40 fraction in the SOLA-AD case and the NI-AD group were 13,128 pg/mg total protein vs. 165 pg/mg total protein, respectively. A large difference was also found in the temporal lobe between the SOLA-AD case and the AD NI-AD individuals for the GDFA/GHCL solubilized Aβ40: 838 ng/mg total protein vs. 66 ng/mg total protein, respectively. In this regard, the SOLA-AD had 4.4-fold more soluble Aβ40 and 5.6-fold more insoluble Aβ40 in the frontal lobe compared to NI-AD cases. In the temporal lobe of the SOLA-AD case, the soluble Aβ40 was 80-fold increased, and the insoluble Aβ40 13-fold more abundant than in the NI-AD cases. However, both soluble and insoluble Aβ42 levels in the frontal or temporal lobes were not dramatically different between the SOLA-AD individual and NI-AD cohort (Table 3).
Table 3.
Aβ ELISA quantification
| Frontal | ||||
|
| ||||
| NDC | Tris Aβ40 pg/mg total protein | Tris Aβ42 pg/mg total protein | GHCl Aβ40 ng/mg total protein | GHCl Aβ42 ng/mg total protein |
|
| ||||
| 1 | 56 | 0 | 0 | 0 |
| 2 | 128 | 152 | 8 | 556 |
| 3 | 44 | 0 | 0 | 0 |
| 4 | 82 | 6 | 7 | 81 |
| 5 | 69 | 0 | 0 | 0 |
| Mean | 76 | 32 | 3 | 127 |
|
| ||||
| NI-AD | Tris Aβ40 pg/mg total protein | Tris Aβ42 pg/mg total protein | GHCl Aβ40 ng/mg total protein | GHCl Aβ42 ng/mg total protein |
|
| ||||
| 12 | 98 | 207 | 9 | 491 |
| 13 | 157 | 162 | 23 | 509 |
| 14 | 118 | 370 | 33 | 880 |
| 15 | 71 | 177 | 31 | 1089 |
| 16 | 14892 | 541 | 1425 | 813 |
| Mean | 3,067 | 291 | 304 | 756 |
|
| ||||
| Tris Aβ40 pg/mg total protein | Tris Aβ42 pg/mg total protein | GHCl Aβ40 ng/mg total protein | GHCl Aβ42 ng/mg total protein | |
|
| ||||
| SOLA-AD | 13,634 | 291 | 1711 | 586 |
|
| ||||
| Temporal | ||||
|
| ||||
| NDC | Tris Aβ40 pg/mg total protein | Tris Aβ42 pg/mg total protein | GHCl Aβ40 ng/mg total protein | GHCl Aβ42 ng/mg total protein |
|
| ||||
| 1 | 26 | 0 | 2 | 0 |
| 2 | 74 | 74 | 2 | 144 |
| 3 | 18 | 0 | 0 | 0 |
| 4 | 134 | 530 | 6 | 152 |
| 5 | 50 | 0 | 0 | 0 |
| Mean | 61 | 121 | 2 | 59 |
|
| ||||
| NI-AD | Tris Aβ40 pg/mg total protein | Tris Aβ42 pg/mg total protein | GHCl Aβ40 ng/mg total protein | GHCl Aβ42 ng/mg total protein |
|
| ||||
| 12 | 66 | 102 | 7 | 351 |
| 13 | 95 | 118 | 20 | 336 |
| 14 | 132 | 164 | 6 | 120 |
| 15 | 66 | 99 | 10 | 299 |
| 16 | 465 | 142 | 287 | 360 |
| Mean | 165 | 125 | 66 | 293 |
|
| ||||
| Tris Aβ40 pg/mg total protein | Tris Aβ42 pg/mg total protein | GHCl Aβ40 ng/mg total protein | GHCl Aβ42 ng/mg total protein | |
|
| ||||
| SOLA-AD | 13,128 | 291 | 838 | 425 |
NDC, non-demented control; NI-AD, non-immunized Alzheimer’s disease; SOLA-AD, solanezumab Alzheimer’s disease patient; Tris, 20 mM Tris, 5 mM EDTA, pH 7.8 with PIC; GHCl, 5 M guanidine-hydrochloride, 50 mM Tris, pH 8.0 with PIC after GDFA extraction. Note that the Tris-soluble results are in pg/mg of total protein, while the GHCl-soluble results are in ng/mg total protein.
Regarding pro-inflammatory TNF-α and IL-1β, the levels of these cytokines were very similar between the NI-AD subjects and the SOLA-AD individual (Figure 3).
Figure 3.
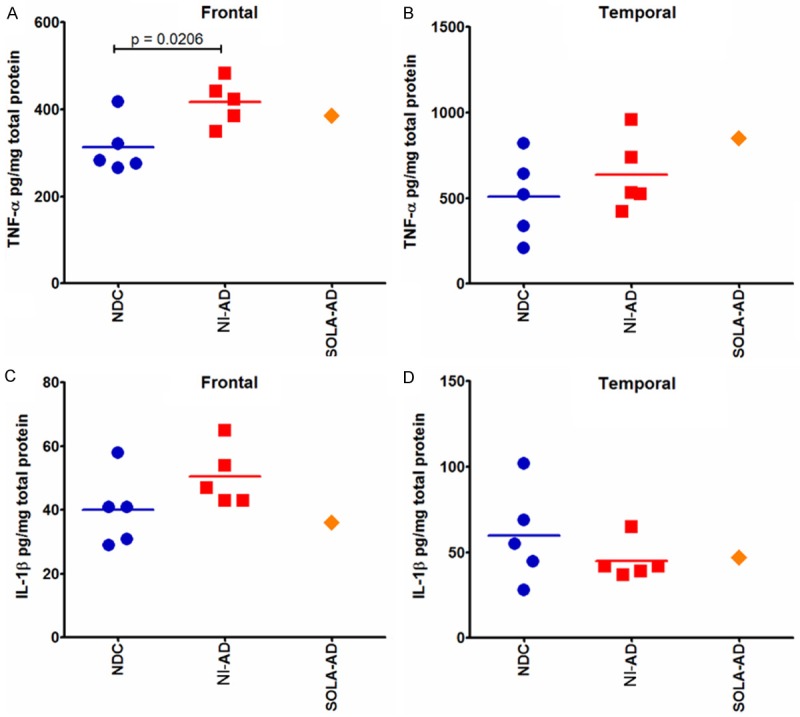
TNF-α and IL-1β ELISA quantification. Frontal (A, C) and temporal (B, D) cerebral cortices were investigated in SOLA-AD, NI-AD and NDC cases. An unpaired, 2-tailed t-test was used to compare NI-AD and NDC cases.
Eight different antibodies were used for semi-quantification of proteins of interest by Western blots. Estimation of the levels in the frontal and temporal lobes of total APP and its C-terminal fragments revealed no substantial deviation in the former (Figure 4A and 4B). However, the CT99 and CT83 fragments of APP were moderately elevated in the SOLA-AD patient, relative to NDC and NI-AD cases (Figure 4A and 4B). The PSEN1-CTF (Figure 4C and 4D), BACE1 (Figure 4E and 4F), α-synuclein (Figure 4G and 4H), ApoE (Figure 4K and 4L), S100B (Figure 4M and 4N) and Bcl-2 (Figure 4O and 4P) showed no major deviations between the SOLA-AD and the NI-AD group. Tau also showed a modest elevation in the frontal lobe of the SOLA-AD case when compared with the rest of the samples (Figure 4I).
Figure 4.
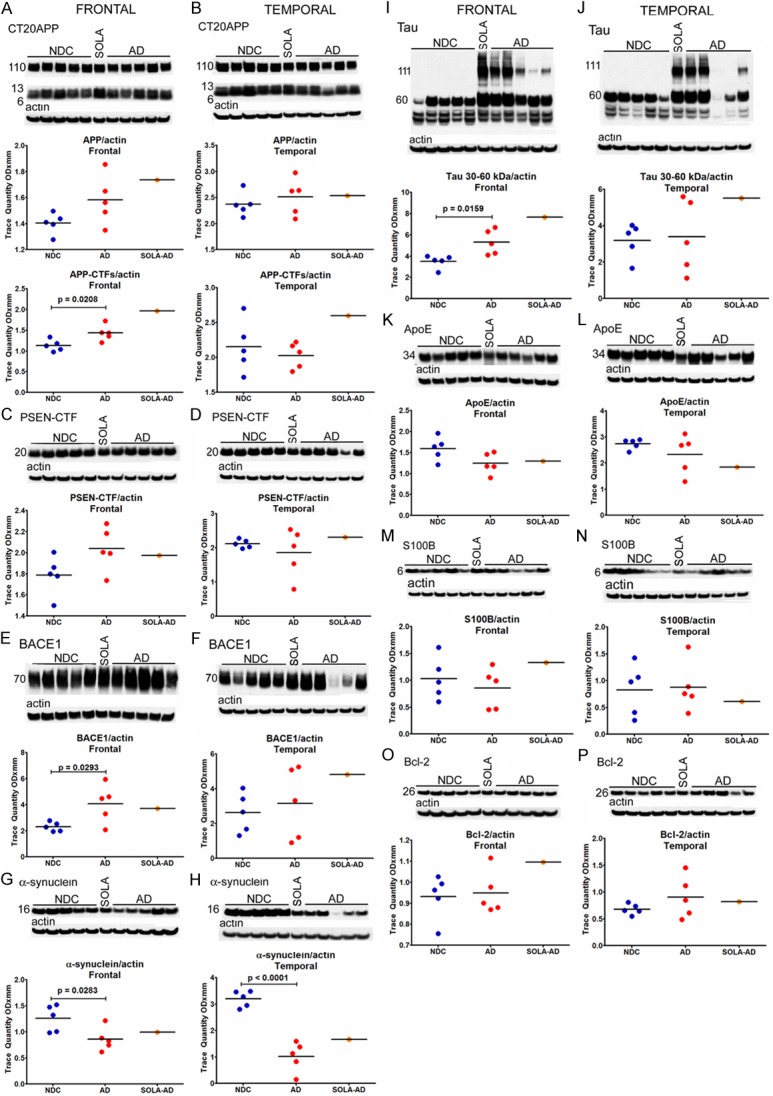
Western blots of: (A, B) amyloid precursor protein (APP) and APP-C-terminal fragments (CTF); (C, D) presenilin C-terminal fragments (PSEN-CTF); (E, F) β-sectretase (BACE-1); (G, H) α-synuclein; (I, J) tau; (K, L) apolipoprotein E (ApoE); (M, N) S100B; (O, P) Bcl-2. Frontal and temporal cerebral cortices were investigated in SOLA-AD, NI-AD and NDC cases. An unpaired, 2-tailed t-test was used to compare NI-AD and NDC cases.
Discussion
A recent investigation examining the possible therapeutic value and risks of solanezumab found some positive results on secondary analyses in patients with mild AD [6]. These observations were made based on ADAS-Cog11, ADAS-Cog14, MMSE and ADCS-iADL which demonstrated some statistical significance [6] and encouraged the continuation of solanezumab clinical trials.
The patient in this case report was enrolled in the initial double blind H8A-MC-LZAM solanezumab study in which subjects received either placebo or solanezumab (400 mg every 4 weeks) for a total of 22 infusions. At the present time there is no information as to whether this patient was in the solanezumab-treated or placebo branch. However, 5 years prior to death, the patient scored MMSE 16, ADAS-Cog 46, CDR 1 and ADL 50. At the time of screening, 5 years prior to death, the patient had a Florbetapir-PET scan, which was repeated at the end of the H8A-MC-LZAM solanezumab study approximately 1.7 years later. Psychometric testing revealed that the patient had MMSE 6, ADAS-cog 72, CDR 2, ADL 43. At this time, the patient was transitioned into the H8A-MC-LZAO open label solanezumab clinical trial. After receiving 9 doses of 400 mg each of solanezumab, the treatment was suspended due to general worsening of the patient complicated by encephalopathy and hyperammonemia. The patient survived an additional 2.6 years after the discontinuation of solanezumab treatment.
Neurochemical characterization of proteins of interest suggested that in general there were small differences between the SOLA-AD patient and the randomly selected NI-AD subjects used for comparison. However, there was a remarkable density of leptomeningeal and cortical amyloid loaded vessels in the immunized individual which was determined by histological examination. In this context, two important caveats need to be considered: the SOLA-AD subject results are the outcome from a single case and this patient had an APOE genotype of ε4/4. APOE ε4/4 individuals with AD harbor extremely high levels of both amyloid plaques and vascular amyloid. Whether the massive vascular amyloid burden observed in our SOLA-AD case represents solubilized Aβ plaques mobilized to the brain vasculature by solanezumab, as speculated to have occurred in AN-1792 vaccinated subjects [7], remains to be established.
Amyloid-β peptides in the AD brain are a structurally diverse family of 3-dimensional molecular conformers dictated by several posttranslational modifications and proteolysis to yield a huge number of shorter and longer altered species [8]. In the AD brain, solanezumab may recognize a limited quantity of Aβ conformers, mostly related to Aβ42 rather than Aβ40, a peptide mostly associated with vascular deposits. The SOLA-AD case examined in this study contained abundant Aβ40. Access of solanezumab to vascular Aβ could had been limited by the extracellular matrix which is intimately enmeshed with the vascular amyloid deposits [8,9].
In recent investigations, we described the neuropathological and neurochemical results from 7 patients who were treated with AN-1792, an active immunization clinical trial which utilized aggregated synthetic Aβ1-42 antigen. The AN-1792 immunized cases showed a large amount of compact and diffuse amyloid plaques as well as an abundant vascular amyloid. Both leptomeningeal and parenchymal amyloid deposits contained profuse Aβ40 [7,10]. In spite of an appreciable patchy clearance of amyloid plaques in the brains of some treated patients, AN-1792 therapy did not significantly enhance cognitive function or halt neurodegeneration. In contrast to AN-1792, postmortem analysis of 3 AD patients immunized with bapineuzumab demonstrated no neuropathological relief. As in the case of AN-1792, there were no significant clinical changes in the development of their dementia when compared to non-treated AD individuals. However, in the bapineuzumab-treated individuals there was an apparent neurochemical change in which the amounts of Aβ42 seemingly decreased and the quantities of Aβ40 increased [11], resulting in altered Aβ42:Aβ40 ratios. These apparent changes were also observed in the AN-1792 treated patients.
The postmortem findings invite speculation as to the disease process and its possible modification by solanezumab treatment in this subject. Conceivably, the extraordinary deposition of leptomeningeal and cortical vascular amyloid had already reached levels where clearance was failing years prior to his death, while neurons were still actively synthesizing Aβ that remained trapped in the brain parenchyma and vessels. The destruction of cortical blood vessels, and specifically of myocytes by amyloid infiltration must have resulted in compromised vasodilation and vasoconstriction rendering the blood vessels rigid. In addition, the provision of oxygen and nutrients as well as the removal of catabolic by-products resulting from cortical vascular devastation may have reduced energy production and accelerated neurodegeneration. The rapid decline complicated by encephalopathy and hyperammonemia probably aggravated vascular compromise. This case also illustrates dementia subjects exhibit a tremendous variability in amyloid burden patterns from extraordinarily profuse amyloid accumulation to those in whom amyloid is apparently absent [12]. On the other hand, there are cognitively intact persons with substantial amyloid accumulation [13].
Solanezumab immunotherapy provided no apparent relief in the clinical course of this AD patient. However, it is possible that the disease severity in this APOE ε4 homozygote patient may have already reached an advanced irreversible stage when solanezumab treatment was initiated. Furthermore, the overwhelming and generalized deposits of vascular amyloid in the present case report suggests a compromised brain perfusion that may have had a negative synergistic impact on the ongoing neurodegenerative processes. Given the quantitative and qualitative heterogeneity of the amyloid load in AD, it is possible that solanezumab treatment may reduce or stabilize amyloid deposition and improve quality of life. The information provided in this case report underscores the importance of neuropathological and neurochemical postmortem studies in the characterization of immunotherapy clinical trials.
Solanezumab may represent an effective treatment for patients in the early stages of AD, as hinted by the EXPEDITION and EXPEDITION 2 clinical trials, a concept which is currently being investigated in the EXPEDITION 3 clinical study [6], or as a preventative intervention for amyloidosis if administered prior to the clinical manifestations of AD, as in the DIAN and A4 clinical trials. The results of these trials will be pivotal in understanding the role of Aβ in AD pathophysiology and on the potential value of solanezumab as an anti-Aβ therapeutic agent.
Acknowledgements
This study was supported by the National Institute on Aging grants R01 AG019795 (AER) and by Midwestern University, Glendale, AZ. The Brain and Body Donation Program at Banner Sun Health Research Institute (TGB) is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026 National Brain and Tissue Resource for Parkinson’s Disease and Related Disorders), the National Institute on Aging (P30 AG19610 Arizona Alzheimer’s Disease Core Center), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research. The funders had no role in study design, data collection, analysis or interpretation of data, decision to publish or preparation of the manuscript.
Disclosure of conflict of interest
None.
Authors’ contribution
Sabbagh MS has clinical support (research or grant) from: Avid, Lilly, Merck, VTV therapeutics, Lundbeck, AC Immune, Roche, Biogen, and advisor to: VTV Therapeutics, Lilly, Biogen, and stock ownership: Muses Labs and Veranum. Beach TG is a consultant for Avid Radiopharmaceuticals and GE Healthcare, has research contracts with Avid Radiopharmaceuticals and Navidea Biopharmaceuticals, and receives grant support from the National Institutes of Health, the Michael J. Fox Foundation for Parkinson’s Research and the State of Arizona.
References
- 1.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 2.Beach TG, Adler CH, Sue LI, Serrano G, Shill HA, Walker DG, Lue L, Roher AE, Dugger BN, Maarouf C, Birdsill AC, Intorcia A, Saxon-Labelle M, Pullen J, Scroggins A, Filon J, Scott S, Hoffman B, Garcia A, Caviness JN, Hentz JG, Driver-Dunckley E, Jacobson SA, Davis KJ, Belden CM, Long KE, Malek-Ahmadi M, Powell JJ, Gale LD, Nicholson LR, Caselli RJ, Woodruff BK, Rapscak SZ, Ahern GL, Shi J, Burke AD, Reiman EM, Sabbagh MN. Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program. Neuropathology. 2015;34:354–389. doi: 10.1111/neup.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Beach TG, Sue LI, Walker DG, Roher AE, Lue L, Vedders L, Connor DJ, Sabbagh MN, Rogers J. The Sun Health Research Institute Brain Donation Program: description and experience, 1987-2007. Cell Tissue Bank. 2008;9:229–245. doi: 10.1007/s10561-008-9067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hixson JE, Vernier DT. Restriction isotyping of human apolipoprotein E by gene amplification and cleavage with HhaI. J Lipid Res. 1990;31:545–548. [PubMed] [Google Scholar]
- 5.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- 6.Siemers ER, Sundell KL, Carlson C, Case M, Sethuraman G, Liu-Seifert H, Dowsett SA, Pontecorvo MJ, Dean RA, Demattos R. Phase 3 solanezumab trials: Secondary outcomes in mild Alzheimer’s disease patients. Alzheimers Dement. 2016;12:110–120. doi: 10.1016/j.jalz.2015.06.1893. [DOI] [PubMed] [Google Scholar]
- 7.Patton RL, Kalback WM, Esh CL, Kokjohn TA, Van Vickle GD, Luehrs DC, Kuo YM, Lopez J, Brune D, Ferrer I, Masliah E, Newel AJ, Beach TG, Castano EM, Roher AE. Amyloid-beta peptide remnants in AN-1792-immunized Alzheimer’s disease patients: a biochemical analysis. Am J Pathol. 2006;169:1048–1063. doi: 10.2353/ajpath.2006.060269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roher AE, Lowenson JD, Clarke S, Wolkow C, Wang R, Cotter RJ, Reardon IM, Zurcher-Neely HA, Heinrikson RL, Ball MJ, Greenberg BD. Structural alterations in the peptide backbone of beta-amyloid core protein may account for its deposition and stability in Alzheimer’s disease. J Biol Chem. 1993;268:3072–3083. [PubMed] [Google Scholar]
- 9.Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, Ball MJ. beta-Amyloid-(1-42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci U S A. 1993;90:10836–10840. doi: 10.1073/pnas.90.22.10836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kokjohn TA, Roher AE. Antibody responses, amyloid-beta peptide remnants and clinical effects of AN-1792 immunization in patients with AD in an interrupted trial. CNS Neurol Disord Drug Targets. 2009;8:88–97. doi: 10.2174/187152709787847315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Roher AE, Cribbs DH, Kim RC, Maarouf CL, Whiteside CM, Kokjohn TA, Daugs ID, Head E, Liebsack C, Serrano G, Belden C, Sabbagh MN, Beach TG. Bapineuzumab alters abeta composition: implications for the amyloid cascade hypothesis and anti-amyloid immunotherapy. PLoS One. 2013;8:e59735. doi: 10.1371/journal.pone.0059735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Monsell SE, Kukull WA, Roher AE, Maarouf CL, Serrano G, Beach TG, Caselli RJ, Montine TJ, Reiman EM. APOE4 carriers and non-carriers with the clinical diagnosis of Alzheimer’s dementia and minimal amyloid plaques. JAMA Neurol. 2015;71:1124–1131. doi: 10.1001/jamaneurol.2015.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maarouf CL, Daugs ID, Kokjohn TA, Walker DG, Hunter JM, Kruchowsky JC, Woltjer R, Kaye J, Castano EM, Sabbagh MN, Beach TG, Roher AE. Alzheimer’s disease and non-demented high pathology control nonagenarians: comparing and contrasting the biochemistry of cognitively successful aging. PLoS One. 2011;6:e27291. doi: 10.1371/journal.pone.0027291. [DOI] [PMC free article] [PubMed] [Google Scholar]