

Abstract
Poly(ADP-ribosyl)ation (PARylation) is a widespread and highly conserved post-translational modification catalysed by a large family of enzymes called poly(ADP-ribose) polymerases (PARPs). PARylation plays an essential role in various cardinal processes of cellular physiology and recent approvals and breakthrough therapy designations for PARP inhibitors in cancer therapy have sparked great interest in pharmacological targeting of PARP proteins. Although, many PARP inhibitors have been developed, existing compounds display promiscuous inhibition across the PARP superfamily which could lead to unwanted off-target effects. Thus the prospect of isoform-selective inhibition is being increasingly explored and research is now focusing on understanding specific roles of PARP family members. PARP-2, alongside PARP-1 and PARP-3 are the only known DNA damage-dependent PARPs and play critical roles in the DNA damage response, DNA metabolism and chromatin architecture. However, growing evidence shows that PARP-2 plays specific and diverse regulatory roles in cellular physiology, ranging from genomic stability and epigenetics to proliferative signalling and inflammation. The emerging network of PARP-2 target proteins has uncovered wide-ranging functions of the molecule in many cellular processes commonly dysregulated in carcinogenesis. Here, we review novel PARP-2-specific functions in the hallmarks of cancer and consider the implications for the development of isoform-selective inhibitors in chemotherapy. By considering the roles of PARP-2 through the lens of tumorigenesis, we propose PARP-2-selective inhibition as a potentially multipronged attack on cancer physiology.
Keywords: Poly(ADP-ribose) polymerases, poly(ADP-ribosyl)ation, cancer, isoform-selective inhibition
Introduction
Poly(ADP-ribose) polymerase-2 (PARP-2) belongs to a family of seventeen ADP-ribosyltransferases, termed poly(ADP-ribose) polymerases (PARPs) [1]. While some PARPs are enzymatically inactive (PARP-9 and PARP-13) or catalyse only mono(ADP-ribosyl)ation of targets, PARP-2 alongside PARP-1, PARP-5A and PARP-5B are the only known PARPs which possess poly(ADP-ribosyl)ation activity (PARylation) [1]. PARylation involves the homopolymerisation of the ADP-ribose unit of β-NAD+ on specific target amino acids and the resulting poly(ADP-ribose) (PAR) polymers vary in size and branching, conferring diverse functional and structural effects on target proteins [1-3]. The modification itself is short-lived as the PAR polymer is rapidly degraded by poly(ADP-ribose) glycohydrolase (PARG) [4] and poly(ADP-ribose) hydrolase 3 (ARH3) [5]. PARP-2, together with PARP-1 and PARP-3 are activated by DNA strand breaks and are the only known DNA damage-dependent PARPs [6-8]. They target a plethora of proteins, largely involved in the DNA damage response, DNA metabolism and chromatin architecture but are also capable of automodification [1] (Figure 1). Recently, advances in proteomic screens have allowed identification of PARylation targets of PARP-1, PARP-2 and PARP-3, revealing new and specific functions of these modifications in cellular processes [20-22] including cell fate, genomic integrity, replication and metabolism [17,23-26] and thus PARP inhibition may offer wide-ranging therapeutic utility (Figure 1).
Figure 1.
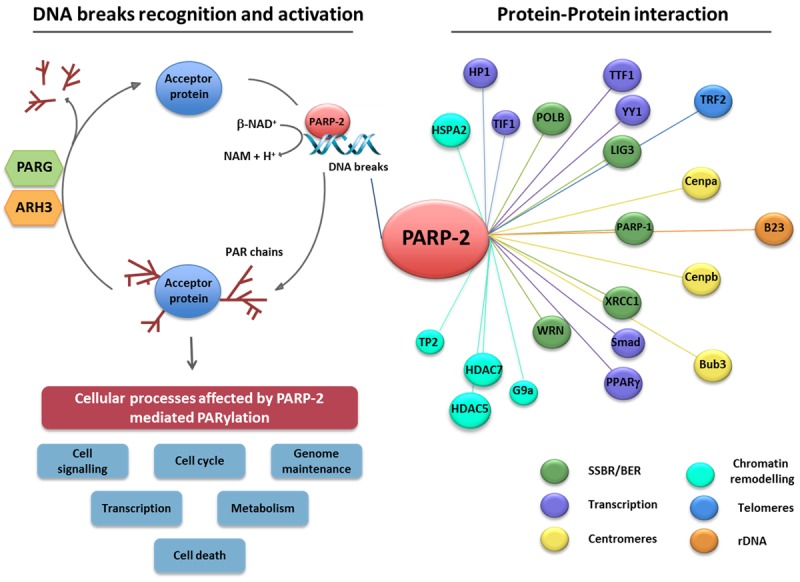
Poly(ADP-ribose) polymerase-2 (PARP-2) executes its biological functions via physical interaction with or PARylation of target proteins. PARP-2 is rapidly recruited to sites of DNA breakage leading to its enzymatic activation (left panel). Activated PARP-2, as well as other bona fide PARPs, hydrolyse NAD+ to nicotinamide (Nam) and one proton (H+), in doing so transferring an ADP-ribose moiety onto specific residues of protein substrates. The subsequent homopolymerisation of ADP-ribose molecules forms variable, long and branched chains of ADP-ribose polymer (PAR). Acceptor proteins subject to PARP-2-dependent PARylation are implicated in a wide range of biological processes, including genome maintenance, transcription, cell cycle regulation, cell death, cell signalling, and metabolism. The reverse reaction involves the rapid degradation of the PAR polymer by poly(ADP-ribose) glycohydrolase (PARG) and poly(ADP-ribose) hydrolase-3 (ARH3) which hydrolyse poly(ADP-ribose) into ADP-ribose units. Biochemical approaches and proteomics show that PARP-2 also physically associates with various protein partners (right panel) involved in different biological processes [9-19].
PARP-1 and PARP-2 activity are intricately interwoven and double knockout Parp-1-/- Parp-2-/- mice exhibit early embryonic lethality [27]. Although at present, PARP inhibitors display promiscuous inhibition among the PARP superfamily [28,29], it is becoming increasingly clear that PARP-2 regulates an overlapping but distinct target set to that of PARP-1 [20-22,30]. New PARP-2 specific roles are being uncovered in diverse functions ranging from DNA repair [17] to telomeric integrity [15] and PARP-2, through physical interaction with or PARylation of partner proteins, impinges on various cellular processes dysregulated in tumorigenesis (Figure 1). Here, we review emerging roles of PARP-2 in many of the hallmarks of cancer and how this may impact the design and therapeutic potential of PARP-2-selective inhibitors in cancer treatment.
Parp-2 gene and protein organization
The Parp-2 gene, mapping to position 14q11.2 and 14C1 [6,31] in the human and murine genome respectively, consists of a sequence of around 13 kb comprising 16 exons and 15 introns (Figure 2A). Eleven transcripts, generated by alternative splicing, have been described (www.ensembl.org), of which some encode PARP-2 protein isoforms (Figure 2B). However, the biological significance of these protein variants is largely unknown. The 62 kDa PARP-2 protein is composed of a modular structure, conserved across the DNA damage-dependent PARPs, comprising an N-terminal region (NTR), a central WGR (Trp-Gly-Arg) domain and a C-terminal catalytic (CAT) domain composed of a helical subdomain (HD) and the ADP-ribosyltransferase (ART) subdomain, which allow coupling of catalysis to DNA break detection [32] (Figure 2C). The PARP-2 NTR is natively unstructured [32] and bears homology with the SAP domain of other nuclear proteins involved in DNA repair and chromosomal structure such as Ku70 and APE-1 [7,32]. Residues 1-65 contain a highly basic DNA-binding domain (DBD) with lysine or arginine residues constituting 27% of its sequence [6], a bipartite nuclear localisation signal (NLS) and a nucleolar localisation signal (NoLS) [3,19]. In contrast, the PARP-1 DBD contains three zinc finger DNA-binding motifs and a BRCA C-terminus (BRCT) domain [33]. These architectural differences between the PARP-1 and the PARP-2 DBDs translate into disparate DNA structure recognition. Although PARP-1 and PARP-2 exhibit similar binding affinity for nicked DNA, PARP-1, unlike PARP-2, also binds strongly to double-strand breaks (DSBs) and to a lesser extent, undamaged DNA, revealing greater specificity of PARP-2 for single-strand break (SSB) recognition [34]. Post-translational modifications on the NLS by the nuclear histone acetyltransferases P/CAF and GCN5L acetylating K36 and K37 serve to decrease DNA binding and enzymatic activity in vitro by an unknown mechanism [35]. Recent mutational analysis has shown that the PARP-2 NTR is, unexpectedly, not required for DNA-binding but is critical for PARP-2 activation on SSBs. Indeed, the WGR and CAT domain display cooperative binding to DNA damage substrates [32]. Interdomain contacts between the WGR and CAT domains appear to be particularly important for this interaction, with mutational disruption thereof reducing DNA-binding activity and abrogating PARP-2 DNA-dependent allosteric activation [32]. Furthermore, while the NTR confers nuclear and nucleolar localisation, it is not necessary for recruitment of PARP-2 to sites of DNA damage, a function mediated instead by a combination of the WGR and CAT domains [32].
Figure 2.
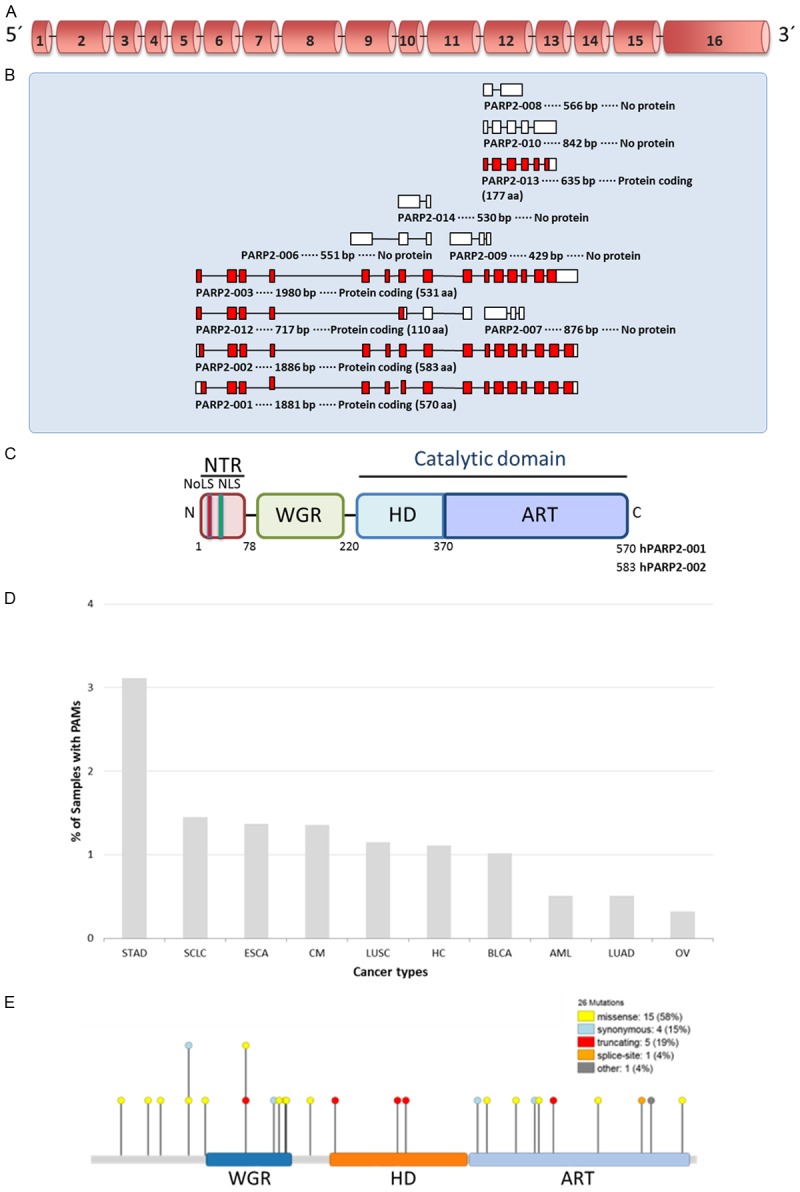
Gene and protein organization of PARP-2. (A) Schematic representation of the genomic DNA organization of the human PARP-2 gene showing exons numbered 1-16 (B) Schematic representation of the transcript variants of PARP-2 (www.ensembl.org). (C) Schematic representation of the domain structure of the human PARP-2 protein. NTR, N-terminal region; WGR, Trp-Gly-Arg domain; HD, helical domain; ART, ADP-ribosyltransferase domain; NoLS, Nucleolar localization signal; NLS, Nuclear localization signal. (D) Graph showing the frequency of PARP-2 mutations in different cancers (www.intogen.org). PAMs, Protein affecting mutations; STAD, stomach adenocarcinoma; SCLC, small cell lung carcinoma; ESCA, esophageal carcinoma; CM, cutaneous melanoma; LUSC, lung squamous cell carcinoma; HC, hepatocarcinoma; BLCA, bladder carcinoma; AML, acute myeloid leukemia; LUAD, lung adenocarcinoma; OV, serous ovarian adenocarcinoma. (E) Distribution of mutations along the PARP-2 protein identified in different types of tumour (www.intogen.org).
The PARP-2 WGR domain is homologous in structure to that of PARP-1 but mediates homodimerisation, DNA damage-dependent autoPARylation and protein-protein interactions-functions attributed to the BRCT motif-bearing domain D of PARP-1 [17]. The WGR domain of PARP-2 is interposed between a caspase-3 cleavage site 58-DNRD-61, and a caspase-8 cleavage site 183-LQMD-186 just proximal to the border with the C-terminal catalytic domain. The effect of caspase-8 cleavage is to inactivate PARP-2 but both PARP-1 and PARP-2 are also cleaved by caspase-3, which may enhance DNA damage and allow for swift chromatin degradation in apoptosis by inhibition of PARylation [36]. Moreover, apoptosis is ATP-dependent [37] and PARP inactivation prevents ATP depletion from NAD+ salvage reactions, thus also ensuring the seamless execution of cell suicide [36].
The C-terminal catalytic domain of PARP-2 bears 69% homology with PARP-1 [38] and operates by a similar mechanism of PARylation. Some structural differences, which may be of importance to the design of isoform-selective inhibitors, are evident from structural comparisons of the catalytic domains of human PARP-1 and PARP-2 in complex with the PARP-1/2 inhibitor BMN 673 [39]. Crystallographic analysis of these PARP-inhibitor complexes reveal similar modes of binding of BMN 673 to the PARP-1 and PARP-2 nicotinamide-binding pocket, mediated by a conserved network of hydrogen bonding and π-stacking forces [39]. In contrast, residues at the periphery of the NAD+ recognition site exhibit least conservation between the catalytic domains. Unique interactions between the PARP active site N-terminal helical bundle and D-loop with the di-branched scaffold of the inhibitor suggest new targets for the design of PARP-2-selective inhibitors [39].
New evidence is emerging revealing PARP-2 polymorphisms and mutations (www.intogen.org) associated with different cancers (Figure 2D and 2E), suggesting a role of PARP-2 in tumorigenesis (Table 1).
Table 1.
PARP-2 polymorphisms in human cancer
| Reference cluster ID (rs#) | Chr Pos* | Nucleotide change | Consequence | Amino acid change | Cancer association | Ref. |
|---|---|---|---|---|---|---|
| rs200603922 | 14:20811843 | c.43A>G | Missense | R15G | Prostate† | [40] |
| rs3093926 | 14:20354893 | c.848G>A | Missense | R283Q | Prostate | [41] |
| Ovarian (BRCA2 mutation carriers) | [42] | |||||
| rs878157 | 14:20356854 | c.1329+165C>T | Intron variant | - | Breast | [43] |
| rs1713413 | 14:19891955 | c.601-129C>G | Intron variant | - | Bladder | [44] |
| rs3093938 | 14:20356935 | c.1330-116A>G | Intron variant | - | Colorectal adenoma | [45] |
| rs878156 | 14:20356700 | c.1329+11T>C | Intron variant | - | Breast‡ | [46] |
Chromosome position.
Partial segregation with prostate cancer in 5 individuals lacking mutations in BRCA1/2.
Carriers of rs878156 with breast cancer exhibit differential prognosis dependent on type of chemotherapy with improved survival with anthracycline-based chemotherapy.
PARP-2 and the hallmarks of cancer
Carcinogenesis is a multistage process which involves the stepwise accumulation of various mutations, some of which confer tumour cells a survival advantage. Many of the resulting phenotypes acquired are shared between different tumours and are considered ‘hallmarks of cancer’ (Figure 3) [47]. Among these hallmarks are genomic instability, sustained growth signalling, dysregulated cellular metabolism, angiogenesis, inflammation, tumour invasion and immune evasion [47]. As we continue to uncover specific biological functions of PARP-2, it is becoming increasingly clear that PARP-2 is variously involved in many of these hallmarks of cancer (Figure 3), thus raising the possibility of targeting specific PARP-isoforms in cancer therapy.
Figure 3.
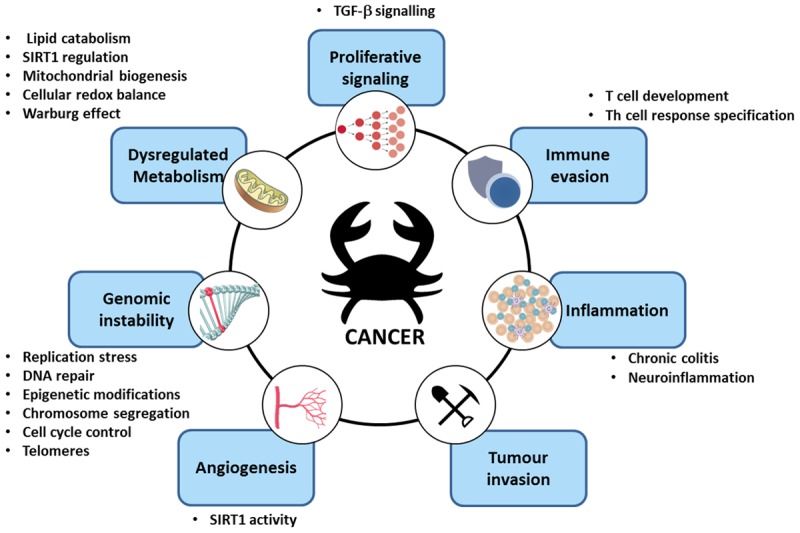
PARP-2 in the hallmarks of cancer. Schematic representation of the involvment of PARP-2 in biological processes commonly dysregulated in tumorigenesis.
Role of PARP-2 in maintaining genomic stability
The genome is vulnerable to a multitude of genotoxic insults, both exogenous (ionising radiation, mutagenic chemicals) and endogenous (reactive oxygen species, recombination intermediates, replicative errors). Thus, mechanisms exist in cells, globally termed the DNA damage response (DDR), to detect damage, signal its presence to effectors and repair the lesion, coordinated with apoptotic or cell cycle arrest responses. Defects in these processes result in genomic instability, which is a major factor in tumorigenesis [48]. High-fidelity replication, intact DNA repair pathways, accurate chromosome segregation in cell division and proper coupling of these processes to cell cycle progression are the major factors driving genome surveillance and maintenance [48]. Genome instability is thus found to arise from various factors including replication stress, aberrant DNA repair, disruption of chromatin architecture, chromosome segregation defects, cell cycle dysregulation and telomere dysfunction [49]. New data are increasingly revealing specific functions of PARP-2 in these biological processes, information critical to the rational development and use of PARP-2-specific inhibitors.
The first indication of PARP-2 involvement in genomic stability came from the observation that Parp-2-/- mice are highly sensitive to ionising radiation (IR) and alkylating agents, although to a different extent to PARP-1 deficiency, with PARP-2 but not PARP-1 deficiency also producing hyperradiosensitivity to low-dose IR (<2 Gy) [50]. Parp-2-/- murine bone marrow cells irradiated with 2 Gy IR exhibit increase chromosome breakage, particularly in centromeres and treatment with the alkylating agent, N-methyl-N-nitrosourea (MNU), results in increased sister chromatid exchange events [27]. While PARP-2 deficiency is not alone tumorigenic, if combined with loss of p53 (Table 2), the rate of spontaneous tumour formation is accelerated [51] and this coupled with the aforementioned sensitivities to genotoxicity and known DNA-damage induced DNA-binding of PARP-2 [6], point towards a role of the molecule in the DDR. Mouse models deficient in both PARP-2 and other DDR proteins (Table 2) are increasingly revealing specific functional interactions of PARP-2, information both critical to understanding its role in the maintenance of genomic stability and the development and use of PARP-2-specific inhibitors.
Table 2.
PARP-2-deficient mouse models
| Genotype | Developmental defects | Fertility defects | Spontaneous tumorigenesis | Ref. |
|---|---|---|---|---|
| Parp-2-/- | Impaired thymopoiesis, adipogenesis, spermatogenesis, fetal liver erythropoiesis | Hypofertility, testicular hypoplasia, oligospermia | None | [13,23-25,51] |
| Parp-2-/-Parp-1-/- | Early embryonic lethality | NA | NA | [27] |
| Parp-2-/-Atm-/- | Early embryonic lethality | NA | NA | [7] |
| Parp-2-/-p53-/- | Partial embryonic lethality | None | Early onset of T cell lymphoma | [51] |
| Parp-2-/-p21-/- | Newborn lethality due to impaired fetal liver erythropoiesis | NA | NA | [25] |
| Parp-2-/-Xrcc5-/- | Partial newborn lethality and growth retardation | NA | NA | [52] |
| Parp-2-/-Prkdc-/- | Shortened lifespan and accumulation of telomere fusions similar to Parp-2+/-Prkdc-/- | Unknown | 50% thymic lymphoma similar to Parp-2+/-Prkdc-/- | [52] |
| Parp-2-/-H2afx-/- | Early embryonic lethality | NA | NA | [52] |
NA, not applicable.
Role of PARP-2 in replication stress
DNA replication must be tightly regulated to prevent genomic instability and therefore carcinogenesis [53]. Indeed, impaired replication fork progression and increased replication-dependent DNA damage have been observed in early stages of tumour development [54,55]. Furthermore, oncogene activation has been found to induce replication stress by various mechanisms [53]. Perturbations in replication can give rise to mutagenesis, genomic rearrangements and chromosome segregation defects [53]. Thus, a variety of checkpoints and DDR pathways act to detect and respond to replicative abnormalities [53,56]. Replication stress is prevalent among cancers and it has been proposed as an additional hallmark of carcinogenesis, driving genomic instability and evasion of apoptosis [57].
While PARP-1 is known to protect stalled replication forks against degradation and acts to recruit the DNA repair machinery required for fork restart [58-61], there is growing evidence for a role of PARP-2 therein. PARP1-/- and/or PARP-2-/- in SW480SN.3 human cells, abolishes homologous recombination (HR)-mediated restart of hydroxyurea-stalled replication forks [58]. Combined with a role of PARP-1 in Mre11 recruitment to stalled forks, it has been proposed that PARP-1 and PARP-2 cooperate in stalled fork detection and restart via Mre11-dependent initiation of HR [58].
Recent work in mouse erythroblasts reveals a specific role of PARP-2 in the replication stress response in erythropoiesis. Parp-2-/-, but not Parp-1-/-, mice display chronic anaemia in steady-state conditions and Parp-2-/- erythroblasts exhibit various biomarkers of replication stress, including micronuclei formation, elevated γH2AX formation in S-phase, increased CHK1 phosphorylation and RPA phosphorylation and newborn lethality with Parp-2/p21 double-knockout (Table 2) [25]. PARP-2 appears to play tissue-specific roles in processes characterised by high proliferation rates, similar to patterns observed in other mouse models defective in the replication stress response, such as ATR hypomorphism [62]. Indeed, PARP-2-specific roles have been uncovered in spermatogenesis [23], thymopoiesis [24,51] and haematopoietic stress responses [50] and PARP-2 was recently identified in a genome-wide RNAi screen for replication stress response genes [63]. Interestingly, PARP-2 plays an important role in hydroxyurea-induced replication stress in plants [64]. Together these recent findings point towards specific functions of PARP-2 in the replication stress response.
Role of PARP-2 in DNA repair
With the panoply of intrinsic and extrinsic sources of genotoxicity faced by cells everyday comes a similarly diverse gamut of lesions, ranging from base modifications and conversion to single- and double-strand breakage. In turn, cells have evolved a large range of DNA repair mechanisms, broadly classed as single-stranded DNA repair or double-stranded break repair [65]. Mutagenesis is critical for carcinogenesis and dysfunction of DNA repair mechanisms is an important source of genomic instability in cancer [66]. Conversely, these pathways also act to repair damage from chemo- and radiotherapy and thus cancer therapies have been developed to target cancer DNA repair defects, halt proliferation and induce apoptosis via DDR activation or potentiate existing treatments [67].
Single-stranded DNA repair is largely mediated by three major pathways: base excision repair (BER)/single-strand break repair (SSBR), nucleotide excision repair (NER) and mismatch repair (MMR). BER involves the recognition and excision of a damaged base by a DNA glycosylase to produce an apurinic/apymidinic (AP) site, followed by AP endonuclease 1 (APE1) mediated strand incision to produce a single-strand break (SSB). Pol β then catalyses gap filling and the nick is sealed by a ligase, either ligase I or the DNA ligase III/XRCC1 complex [68]. Although both PARP-1 and PARP-2 are considered to contribute in SSBR/BER, they exhibit specific interactions with the BER pathway and their roles appear to be spatiotemporally distinct. While both PARPs can bind to AP sites, DNA binding of PARP-2 is not regulated by autoPARylation [69]. Parp-2-/- MEFs exhibit significantly slower repair of MNU-induced DNA strand breaks, similar to PARP-1-deficient cells suggesting a non-redundant overlapping function of PARP-2 in BER [17]. In contrast, human A549 cells deficient in PARP-2 displayed only a slight reduction in initial SSBR rate upon hydrogen peroxide exposure, compared to a significant delay observed with PARP-1 deficiency, calling into question the role of PARP-2 in SSBR/BER [70]. While PARP-1 is recruited early and transiently to laser-microirradiated DNA, PARP-2 exhibits delayed persistent recruitment in a PARP-1 and PARylation-dependent manner [71]. Recent single-molecule AFM imaging studies of PARP-2 DNA binding have confirmed preferential recruitment to SSBs over DSBs and undamaged DNA [34] and Kd values measured for a range of DNA structures have shown that PARP-2 has highest affinity for flap-containing DNA [72]. In addition PARP-2, like PARP-3, is preferentially activated by 5’ phosphorylated DNA breaks [73,74]. Together, these findings argue for a role of PARP-2 in the recognition of later repair intermediates in SSBR.
PARP-1 and -2 have been found to differentially interact with multiple SSBR/BER factors (Figure 1) including the non-enzymatic scaffold protein XRCC1 [75]. XRCC1 is recruited to microirradiated DNA in a PARP-1, but not PARP-2 dependent manner [71,76], acting to inhibit the dissociative automodification activities of both molecules and being PARylated itself on its BRCT1/2 domains by PARP-2 [17]. PARP-1 and 2 also interact with Pol β, FEN1 and DNA Ligase III (Figure 1) [17,72], inhibiting the DNA synthetic and endonuclease activities of the former two proteins respectively. This inhibition can be relieved by PARP-1 but not PARP-2 PARylation but PARP-2 acts to abrogate the restorative polymerisation of PARP-1 [72]. Together these results suggest spatiotemporal segregation but mutually modifiable functions of PARP-1 and PARP-2 in SSBR/BER.
While recent data are uncovering a role of PARP-1 in NER and MMR [77-79], the role of PARP-2 therein remains unknown.
Double-strand break repair (DSBR) proceeds via two main pathways, non-homologous end-joining (NHEJ) and homologous recombination (HR). The presence of DSBs is signalled by the protein Ataxia telangiectasia mutated (ATM), a serine/threonine kinase, phosphorylating targets such as p53, CHK2, NBS1 and BRCA1 to initiate repair and cell cycle responses [80]. Double knockout mouse models of PARP-1 or PARP-2 with ATM (Parp-1-/-Atm-/- and Parp-2-/-Atm-/-) both result in early embryonic lethality (Table 2) [7,81]. This may be due to the conversion of unrepaired spontaneous SSBs in hyperproliferative embryonic cells, into DSBs during replication and again going unrepaired due to ATM deficiency. GST-pulldowns of GST-ATM fusions have, however, revealed a physical interaction with PAR molecules and PARylated PARP-1 via putative PAR-binding domains on the ATM N-terminus (amino acid residues 99-120) and overlapping with the C-terminal PI3K domain [82]. The N-terminal PAR-binding domain contains a protein interaction domain critical for interaction with and phosphorylation of p53 [83] which suggests coupled functions of PARP-1 and downstream effectors of DSBR. Within the HR pathway, PARP-1 has been shown to interact with MRN complex components Mre11 and NBS1 [84] and combined with the aforementioned interactions of PARP-1, PARP-2 and Mre11 in replication stress, this could implicate PARP-2 in a new role in HR [58].
PARP-1 is known to act in conjunction with the ligation activity of the DNA ligase III/XRCC4 complex [85], in an alternative Ku-independent end-joining pathway, complementary to classical NHEJ, with competition between Ku and PARP-1 for DNA binding thought to mediate the switch between pathways [86]. Furthermore, class-switching recombination (CSR) of immunoglobulin genes has been demonstrated to involve activation-induced cytidine deaminase (AID)-induced PARP-1 activity. Here, PARP-1 is thought to promote joining of switch regions in a non-classical microhomology-mediated pathway [87], favoured in the absence of NHEJ proteins e.g. XRCC4 and DNA ligase IV [88]. AID-induced DSBs are a critical component of CSR, without which short-range recombination is abnormally frequent and chromosomal translocations with proto-oncogenes such as c-myc may arise [89]. Monitoring the frequency of IgH/c-myc translocations in Parp-1-/- or Parp-2-/- murine B-lymphocytes showed suppression of translocations by PARP-2 but not PARP-1 [87]. However, a mechanistic involvement of PARP-2 in NHEJ is at present unclear.
The recent development of new double-knockout mouse models has allowed the identification of interconnections between PARP-1 and PARP-2 with the DSBR machinery (Table 2). The histone variant H2AX serves an important role in DSB detection and signalling and neither Parp-1-/-H2AX-/- nor Parp-2-/-H2AX-/- mice are viable [52,90,91]. In contrast, unlike PARP-1, PARP-2 deficiency combined with loss of the NHEJ factors Ku80 or DNA-PKcs, does not reveal any considerable exacerbation of phenotype [52].
PARP-2 in chromatin architecture and epigenetic modifications
Chromatin remodelling is actively involved in the DDR via modulation of accessibility of damaged sites to signalling and repair machinery. This has obvious implications for genome integrity as well as cellular senescence and cancer which involve DDR activation and alterations in chromatin architecture [92,93]. Furthermore chromatin architectural changes, such as by histone modifications, form part of the nominal ‘epigenetic landscape’, the dysregulation of which is a hallmark of cancer [94]. Indeed, epigenetic therapy targeting reversible epigenetic marks, as opposed to irreversible tumorigenic mutations, is increasingly becoming a potential therapeutic strategy against cancer [95].
Early experiments found that nucleosomal PARylation resulted in chromatin decondensation, pointing towards a role of PARPs in chromatin architecture [96,97]. PARP-1 and PARP-2 have both been shown to differentially PARylate the nucleosome with PARP-1 preferentially targeting the linker histone H1 while core histones H2A/H2B serve as a preferential substrate of PARP-2 [35,98]. PARP-2-dependent PARylation also appears to have a role in facultative heterochromatin maintenance such as that in meiotic sex chromosome inactivation in male germ cells. Indeed, Parp-2-/- murine pachytene spermatocytes display histone H4 hypoacetylation and H3K9 hypomethylation throughout the nucleus compared to wild-type cells [23].
PARP-2 has also been shown to be involved in heterochromatin reorganisation in endodermal differentiation via interactions with non-histone proteins heterochromatin protein 1 (HP1) proteins and transcriptional intermediary factor 1β (TIF1β) [9]. The HP1 family of proteins is involved in chromatin packing and heterochromatinisation-mediated gene repression [99]. TIF1β is a co-repressor for the KRAB-transcriptional repression domain-containing family of Zn-finger proteins and promotes transcriptional silencing involving HP1 binding and histone deacetylation [100-103]. PARP-2 has been shown to bind directly to HP1α and HP1β and with high affinity both directly to TIF1β and indirectly via HP1α. In comparison, PARP-1 demonstrates only weak binding to TIF1β and HP1β. Both PARP-1 and PARP-2 PARylate HP1α but shRNA knockdowns demonstrate distinct roles in endodermal differentiation [9].
Together, these results point towards specific roles of PARP-2 in chromatin structure and function and establishment of the histone code and subcodes, distinct from PARP-1. Further studies are needed to elucidate the precise role of PARP-2 in these processes, which will allow the evaluation of PARP-2 as a potential target of epigenetic therapy for cancer.
PARP-2 in chromosome segregation
Centromeric and kinetochore integrity are crucial to faithful chromosome segregation in mitosis [104] and aneuploidy secondary to chromosomal instability and segregation defects is a common feature of cancers [105]. Both PARP-1 and PARP-2 exhibit cell cycle-dependent association with mammalian centromeres, albeit with different localisation. While PARP-1 occupies a diffuse centromeric distribution extending to the surrounding pericentric region, PARP-2 demonstrates sequence-independent accumulation at the centromere periphery, which increases with microtubule inhibitor treatment [14,106]. Both PARP-1 and PARP-2 co-immunoprecipitate with the constitutive centromere proteins Cenpa and Cenpb and the spindle assembly checkpoint protein Bub3 (Figure 1), which are PARylated with DNA damage [14,107]. Furthermore, Parp-2-/- MEFs treated with the alkylating agent MNU exhibit G2/M arrest marked by polyploidy and chromosome mis-segregation, indicative of centromeric and kinetochore dysfuntion [23,27]. These findings argue for a role of PARP-2 distinct from but overlapping with that of PARP-1 in centromeric structure and function as well as checkpoint activation.
In addition to the aforementioned effects on mitotic cells, PARP-2-deficiency in mice also produces chromosome segregation defects at meiotic metaphase I in male gametogenesis [23]. The fidelity of meiotic chromosome segregation is dependent on the integrity of chiasma formation between homologous chromosomes [108]. Concurrently, the accumulation of univalents in Parp-2-/- metaphase I spermatocytes and in turn, aneuploid metaphase II cells was found against a backdrop of a 59% reduction in Mlh1 foci formation, a marker of chiasmata sites, compared to wild-type controls. While mechanistically unclear, this segregation failure could be ascribed to PARP-2 deficiency marring centromeric heterochromatin integrity [27], as in somatic cells, or to putative roles of PARP-2 in resolution of recombination intermediates or meiotic spindle assembly [23].
PARP-2 in cell cycle regulation
The coupling of the aforementioned processes involved in the maintenance of genomic integrity with cell cycle progression and checkpoint controls, ensures the timely execution of events in cellular proliferation. Cell cycle dysregulation gives rise to the uncontrolled proliferation observed in cancers and checkpoint dysfunction can lead to inheritance of damaged DNA and chromosome segregation defects, thus producing genomic instability [109].
New data are beginning to shed light on potential roles of PARP-2 in the eukaryotic cell cycle. Loss of PARP-2 in mouse erythroid progenitors results in cell cycle arrest at the G2/M transition [25] and PARP-2 overexpression in human HEK293 cells delays G1 exit [10]. PARP-2 is found to cause PARylation-independent transcriptional repression of the promoters of various cell cycle-related genes including p21, RB, E2F1 and c-MYC and this co-repressor activity involves recruitment of the histone deacetylases HDAC5 and HDAC7 and the H3K9 trimethyltransferase G9a [10]. p21, a cyclin-dependent kinase inhibitor, is an important cell cycle regulator, acting at both G1/S and G2/M transitions [110] and double knockout of PARP-2 and p21 causes newborn lethality in mice suggesting a functional interaction between the two molecules [25]. Together with the interaction of PARP-2 with the spindle assembly checkpoint protein Bub3 [14], these findings point towards a role of PARP-2 in the coordination of cell cycle progression and checkpoint regulation. Further studies are necessary to elucidate the precise action of PARP-2 in these processes.
PARP-2 in telomere maintenance
Telomeres are double-stranded DNA tracts of short tandem repeats at chromosome termini and are maintained by telomerase [111]. In mammals, the repeat sequence is TTAGGG [112] and forms a length of around 10-15 kb at birth with a single-stranded 3’ overhang of 50-150 nucleotides which may invade the telomeric duplex to form a lariat structure called the T-loop as well as a locally displaced single-stranded DNA tract called the D-loop [113]. The TTAGGG repeats associate with the multisubunit shelterin complex which protects the telomere from recognition as a DNA strand break and is also involved in maintaining the integrity thereof [114]. TRF2 is a core component of the shelterin complex and is involved in T-loop formation [115] as well as recruitment of proteins involved in telomeric protection and maintenance [111]. PARP-2 has been shown to physically interact with TRF2 (Figure 1) and inhibits TRF2 DNA-binding by both PARylation of its dimerisation domain and non-covalent competitive binding of PAR polymers to the TRF2 DBD [15]. Such regulation may serve to open the T-loop lariat to ease access of DNA repair machinery and/or modulate TRF2 binding partner interactions, pointing to a role of PARP-2 in telomeric integrity [15]. Indeed, PARP-2-deficient MEFs display hallmarks of telomeric dysfunction including increased frequency of metaphase chromosomal and chromatid breaks and TTAGGG-free ends despite normal telomere length [15].
Lagging strand replication occurs by a series of short DNA Okazaki fragments downstream of RNA primers, the most terminal of which is not replicated - the nominal ‘end-replication problem’ and thus with repeated rounds of replication, a cell’s telomeres will gradually shorten [116] until a certain critical threshold is reached and a potent DDR is induced by the uncapped chromosomes which results in cellular senescence, thus setting a cap on the replicative potential of a cell [93,117,118]. However, some cells with dysfunctional checkpoints, such as inactivated Rb or p53 pathways in cancer cells, may continue to divide with marked genomic instability and accumulation of rearranged DNA, largely resulting in cell death in a period termed ‘cellular crisis’ [119]. A subset of these cells may reactivate telomere maintenance mechanisms via telomerase expression [120] or ALT pathway activation [121], effectively immortalising the cell, an important hallmark of cancer [47]. Through its role in telomere integrity, PARP-2 may be an important therapeutic target in cancer.
Role of PARP-2 in proliferative signalling
The TGFβ signalling pathway is an important ‘tumour suppressor’ pathway with roles in embryonic development and differentiation, morphogenesis and tissue homeostasis [122]. Regulation of downstream Smad signal transducers is an important point of control in the pathway. Proximity ligation assays and immunoprecipitation experiments revealed the induction of PARP-1/Smad3 and PARP-2/Smad2/3 complexes upon TGFβ signalling [11]. Activation of the TGFβ pathway was found to promote Smad3 PARylation and PARylated Smad3 and Smad4 co-precipitate with both PARP-1 and PARP-2 (Figure 1) [11]. PARP-2 overexpression has been found to repress the Smad3/4 promoter and PARP-2 siRNA knockdown results in increased expression of TGFβ gene targets fibronectin and Smad7, concurrent with an inhibitory role of PARP-2 in the pathway [11]. Through regulation of TGFβ signalling, these results point towards a role of PARP-2 in the control of proliferation and cell fate, but further studies are required to elucidate the functional role of PARP-2 in TGFβ-dependent cell behaviour and tumorigenesis.
Role of PARP-2 in metabolism
The role of PARP-2, as of PARP-1, in metabolic regulation is becoming increasingly apparent. Both PARPs mediate diverse regulation of oxidative metabolism and mitochondrial biogenesis via the NAD+-dependent deacetylase SIRT1 of the Sirtuin family of proteins. SIRT1 regulates a range of transcription factors and cofactors including PPARγ, p53, PGC-1α and the FOXO family of transcription factors to regulate metabolic homeostasis in response to changing NAD+ levels in states of energetic stress such as fasting and exercise [123]. While loss of either PARP produces increased SIRT1 activity, PARP-1 and PARP-2 regulate SIRT1 via different mechanisms. PARP-1 deletion acts to relieve competition for intracellular NAD+, while shRNA-mediated depletion of PARP-2 produces little effect on NAD+ balance [124]. Instead, PARP-2 is found to bind directly to the SIRT1 proximal promoter where it acts to negatively regulate SIRT1 expression [26]. Due to SIRT1 derepression, Parp-2-/- mice exhibit elevated SIRT1 expression with decreased acetylation of SIRT1 targets FOXO1 and PGC-1α which are transcriptional regulators of mitochondrial bioenergetics and biogenesis [26]. Indeed Parp-2 deletion in mice produces increased mitochondrial biogenesis with a shift towards oxidative metabolism and upregulation of genes involved in mitochondrial function and lipid oxidation such as tpn1, SDH, UCP2, MCAD and PGC-1α [26]. Concurrently, PARP-2 knockout mice display a leaner phenotype marked by a predominance of lipid catabolism as an energy source and interestingly, are more protected against diet-induced obesity and insulin resistance with a high-fat diet [26]. This protection is at least in part due to increased energy expenditure but may also be due to a direct effect of PARP-2 on white adipose tissue (WAT). Like PARP-1, PARP-2 acts to positively regulate PPARγ-dependent promoters to promote adipogenesis [13,125]. Parp-2-/- mice exhibit impaired adipocyte differentiation with reduced adipocyte size and decreased WAT deposition concomitant with downregulation of PPARγ target genes [13]. PARP-2 mRNA is additionally regulated by the miR-149, producing upregulation of SIRT1 activity to promote mitochondrial function and biogenesis via PGC-1α activation [126]. Related to the role of PARP-2 in mitochondrial bioenergetics, cellular redox balance also appears to be regulated by PARP-2. Syngeneic lung adenocarcinomas from Parp-1-/- or Parp2-/- mice are smaller and exhibit reduced proliferation rates imputed to an observed increase in autophagy and apoptosis secondary to increased oxidative stress. Indeed loss of either PARP produces downregulation of key antioxidant enzymes such as catalase [127].
PARP-2 has been to found to regulate both hepatic and systemic lipid homeostasis and negatively regulates the promoter of SREBP1, an important transcriptional regulator of sterol biosynthesis [128]. Parp-2-/- mice exhibit increased hepatic cholesterol levels and Parp-2 shRNA knockdown in HepG2 cells shows upregulation of SREBP1-dependent genes [128]. Parp-2 deletion also produces reduced serum HDL (high-density lipoprotein) levels, a known risk factor for cardiovascular disease [128,129]. This may be ascribed to an observed decrease in expression of the ATP-binding cassette transporter ABCA1, which is involved in reverse cholesterol transport from peripheral cells to the liver [128,130].
Cancers are marked by a characteristic metabolic reprogramming producing a switch from oxidative bioenergetics to a regime of aerobic glycolysis (Warburg effect) which balances energy requirements with biogenesis [131]. Tumour metabolism is also marked by extensive lipid anabolism and redox balance changes to regulate the generation of tumorigenic reactive oxygen species (ROS) [131]. From the above it is clear that PARP-2 has a net antagonistic action on mitochondrial function and thus pharmacological PARP-2 inhibition may produce a shift away from glycolytic tumour metabolism towards an oxidative programme. Furthermore, increased lipid catabolism and oxidative stress-induced apoptosis and autophagy may intensify the putative antitumorigenic effect of PARP-2 loss.
While Parp-1-/- and Parp-2-/- mice display similar metabolic changes, unlike Parp-1 deletion, Parp-2 knockout mice exhibit increased glucose intolerance due to pancreatic dysfunction marked by reduced islet size and fasting blood insulin [26]. Parp-2-/- mice have a substantial loss of expression of the pancreatic transcription factor pdx1, imputed to upregulated pancreatic SIRT1 expression and consequent FOXO1 activation [26]. Thus, any consideration of selective PARP-2 inhibition will require better understanding of the exact mechanisms of PARP2-dependent metabolic regulation.
Role of PARP-2 in angiogenesis
Neovascularisation of tissues is a physiological response to tissue damage but is also an important process in tumour expansion, serving to adequately oxygenate and nourish the tumour, remove waste and to function as a route for metastasis [132]. The angiogenic effect of PARP-1 is well established [133-135]. Moreover, PARP inhibition using the PARP-1/PARP-2 inhibitor GPI 15427 [133] results in impaired endothelial cell proliferation, migration, adhesion and tube-formation in response to angiogenic growth factor stimulation by vascular endothelial growth factor (VEGF) [136] and placental growth factor (PIGF) [133,137] suggesting that some of the antiangiogenic effect of inhibition was ascribable to loss of PARP-2. The role of PARP-2 in the regulation of proangiogenic genes such as Pecam-1, OPN and Hif-1α, known to be upregulated by PARP-1 [134,138], has not as yet been investigated.
However, as aforementioned, loss of PARP-2 produces upregulation of SIRT1 activity in mice [26]. Aside from known metabolic roles, the NAD+-dependent deacetylase exerts a proangiogenic effect via multiple mechanisms including repression of the antiangiogenic transcription factor FOXO1 [139] and activation of the proangiogenic transcriptional regulator PGC-1α [140,141]. SIRT1 also regulates the Notch signalling pathway, an important controller of blood vessel patterning and growth [142], both by deacetylation and inactivation of signalling intermediates [140] and epigenetic changes in Notch target genes [143]. Further investigations will be necessary to characterise the effects of PARP2-selective inhibition in neovascularisation through its role in SIRT1 modulation.
Role of PARP-2 in inflammation
The tumorigenic role of the proinflammatory microenvironment of a cancer is being increasingly understood and is an important feature of cancer development [47]. A role of PARP-1 in the inflammatory response has been established for a range of disease models [144,145], imputed to either a functional interaction with NF-κB, an important transcriptional regulator of inflammation, or NAD+ depletion secondary to PARP overactivation [146,147]. In contrast, far less is understood of the potential roles of PARP-2 therein and mechanisms thereof.
Current evidence demonstrates differential importance of PARP-2 in different inflammatory disease models. In secretagogue-induced murine models of acute pancreatitis, PARP-1 but not PARP-2 deficiency attenuates disease severity [145], whereas Parp-2 knockdown, like non-specific PARP inhibition, reduces inflammation in IL-10-deficient mouse models of chronic colitis [148,149]. More recently, it was shown that Parp-2 deletion, like non-selective PARP-1/2 inhibition, abates neuroinflammation and neurological dysfunction in mouse models of multiple sclerosis [150,151]. Further studies are required to elucidate roles of PARP-2 in other inflammatory diseases and the mechanisms thereof.
Role of PARP-2 in immunomodulation and immune evasion
Despite considerable enthusiasm regarding the prospect of PARP inhibition in cancer therapy, little attention has been paid to the role of these proteins in the modulation of the immune response to neoplasia. In addition to tumour cells, the tumour microenvironment is composed of stromal cells and tumor-infiltrating immune cells of both innate and adaptive lineages [152]. These diverse cells communicate with one another by means of direct contact and/or indirect paracrine signaling mediated by soluble factors such as cytokines and chemokines. This intercellular signaling can drive immune responses which may either hinder or facilitate tumour growth [153]. Interestingly, PARP-1 and PARP-2 have been shown to play a role in both innate and adaptive immune responses [154-156]. One of the major players in the immune response to cancer is the T cell. Although peripheral T cell homeostasis appears unaffected by either PARP-1 or PARP-2-deficiency in mice [24], PARP-1 is implicated in the regulation of NFAT [157] and Foxp3 [158,159] expression, transcription factors important in T cell development and function. In contrast, the role of PARP-2 in such immunoregulation is as yet unexplored. PARP-1-deficiency is also found to promote Th1 cell responses [156], whereas PARP-2 deletion in EAE mouse models reduces Th1 and Th17 cell infiltration in the central nervous system suggesting non-redundant roles of PARP-2 in modulating T cell responses [150]. While PARP-1 is dispensable for thymocyte development, PARP-2-deficiency is found to halve CD4+/CD8+ (double positive; DP) thymocyte cellularity, and produce impaired DP survival accompanied by a marked increase in DP sensitivity to apoptosis [24].
Recent studies of tumour transplantation in a BALB/c syngeneic tumour model demonstrate a reduction in lung adenocarcinoma growth in Parp-1-/- host mice, and to a lesser extent in Parp-2-/- host mice compared to wild-type [127,160]. This effect may be mediated, at least in part, by modulation of the immune response against the tumour in the absence of either PARP-1 or PARP-2. Further studies are required to understand the specific function of PARP-2 in immunomodulation and its role in tumour destruction and immune evasion. Although biological strategies directing the immune system against tumour cells are under extensive study, small-molecule approaches targeting intracellular signalling pathways [161] such as those mediated by PARP proteins, may represent a potential therapeutic approach to fighting cancer.
Future prospects for PARP-2-selective inhibition in cancer therapy
PARP inhibition for cancer therapy has classically focused on either one of two antitumorigenic mechanisms of action, either the potentiation of DNA-damaging chemo- or radiotherapy [162-165] or synthetic lethality in DSBR-deficient tumours which are thus dependent on SSBR/BER and PARP [166-168]. However, most clinically trialled ‘PARP inhibitors’ show weak or no selectivity among the PARP superfamily and even major clinical candidates such as veliparib and olaparib show no discrimination between PARP-1 and PARP-2 [169]. The activity of such agents should thus be considered as the effect of either pan-PARP inhibition or in many cases, PARP-1/2 blockade. The search for selective inhibitors is important both for the specific targeting of the non-redundant functions of PARPs but also to reduce off-target effects.
The above account of the pleiotropic influence of PARP-2 in the hallmarks of cancer reveals a wide-ranging molecular network regulated by this ADP-ribosyltransferase. The critical role of PARP-2 in DNA repair and the results of double-knockout mouse models immediately suggests the potential utility of PARP-2-selective inhibition in radio- or chemopotentiation or synthetic lethality approaches to cancer therapy. In addition, the broadening roles of PARP-2 in genomic stability and the bias of its action for highly proliferative cells suggest that PARP-2 inhibition could also serve to resist tumour growth via exacerbation of replication stress, induction of chromosome mis-segregation and dysregulation of cancer epigenetics, cell cycle progression and telomere integrity. Furthermore, emerging roles of PARP-2 in regulation of the tumour suppressor TGFβ signalling cascade may provide another means of halting tumour proliferation. Of great interest is also the function of PARP-2 in metabolism; the aforementioned biochemical, cellular and organismal effects of PARP-2 loss on metabolism point towards a shift in metabolic profile away from the neoplastic Warburg effect. It may be that in addition to multilateral genomic destabilisation, PARP-2 inhibition could additionally hinder tumour growth by establishment of a metabolic programme non-permissive for carcinogenesis. We speculate that the immunomodulatory and seemingly tissue-specific anti-inflammatory effect of PARP-2 deletion in mice combined with putative roles in angiogenesis could reveal that PARP-2 inhibition further impairs tumour development through deprivation of essential oxygenation, nutrition, immunoprotection and growth signals. In addition, functions of PARP-2 in tumour invasion need to be explored. From these heterogeneous effects in the hallmarks of cancer, we propose that pharmacological inhibition of PARP-2 may provide a multipronged attack on tumour physiology on a genomic, cellular and microenvironmental level. Further studies will be needed to characterise the expanding regulatory network of PARP-2 to inform the design of PARP-2-selective inhibitors and evaluate the therapeutic utility thereof. Greater specificity may even be achieved by pharmacological targeting of PARP-2 protein substrates, such as blockade of epitopes subject to PARP-2-mediated PARylation, permitting ‘fine-tuning’ of target molecule functionality. There is thus great unexplored potential for the selective inhibition of PARP-2 or its targets and future work will be needed to guide the development of novel therapeutics.
Acknowledgements
The JY laboratory is supported by grants from Spanish Ministerio de Economía y Competitividad (SAF2014-53467-R), Fundació La Marató de TV3 (20134130), and CIBERehd.
Disclosure of conflict of interest
None.
References
- 1.Hottiger MO. Nuclear ADP-ribosylation and its role in chromatin plasticity, cell differentiation, and epigenetics. Ann Rev Biochem. 2015;84:227–263. doi: 10.1146/annurev-biochem-060614-034506. [DOI] [PubMed] [Google Scholar]
- 2.Schreiber V, Dantzer F, Ame JC, De Murcia G. Poly(ADP-ribose): novel functions for an old molecule. Nat Rev Mol Cell Biol. 2006;7:517–528. doi: 10.1038/nrm1963. [DOI] [PubMed] [Google Scholar]
- 3.Yélamos J, Schreiber V, Dantzer F. Toward specific functions of poly(ADP-ribose) polymerase-2. Trends Mol Med. 2008;14:169–178. doi: 10.1016/j.molmed.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 4.Slade D, Dunstan MS, Barkauskaite E, Weston R, Lafite P, Dixon N, Ahel M, Leys D, Ahel I. The structure and catalytic mechanism of a poly(ADP-ribose) glycohydrolase. Nature. 2011;477:616–620. doi: 10.1038/nature10404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oka S, Kato J, Moss J. Identification and characterization of a mammalian 39-kDa poly(ADP-ribose) glycohydrolase. J Biol Chem. 2006;281:705–713. doi: 10.1074/jbc.M510290200. [DOI] [PubMed] [Google Scholar]
- 6.Amé JC, Rolli V, Schreiber V, Niedergang C, Apiou F, Decker P, Muller S, Höger T, Ménissier-de Murcia J, de Murcia G. PARP-2, A novel mammalian DNA damage-dependent poly (ADP-ribose) polymerase. J Biol Chem. 1999;274:17860–17868. doi: 10.1074/jbc.274.25.17860. [DOI] [PubMed] [Google Scholar]
- 7.Huber A, Bai P, de Murcia JM, de Murcia G. PARP-1, PARP-2 and ATM in the DNA damage response: functional synergy in mouse development. DNA Repair (Amst) 2004;3:1103–1108. doi: 10.1016/j.dnarep.2004.06.002. [DOI] [PubMed] [Google Scholar]
- 8.Boehler C, Dantzer F. PARP-3, a DNA-dependent PARP with emerging roles in double-strand break repair and mitotic progression. Cell Cycle. 2011;10:1023–1024. doi: 10.4161/cc.10.7.15169. [DOI] [PubMed] [Google Scholar]
- 9.Quénet D, Gasser V, Fouillen L, Cammas F, Sanglier-Cianferani S, Losson R, Dantzer F. The histone subcode: poly(ADP-ribose) polymerase-1 (Parp-1) and Parp-2 control cell differentiation by regulating the transcriptional intermediary factor TIF1β and the heterochromatin protein HP1α. FASEB J. 2008;22:3853–3865. doi: 10.1096/fj.08-113464. [DOI] [PubMed] [Google Scholar]
- 10.Liang YC, Hsu CY, Yao YL, Yang WM. PARP-2 regulates cell cycle-related genes through histone deacetylation and methylation independently of Poly(ADP-ribosyl)ation. Biochem Biophys Res Commun. 2013;431:58–64. doi: 10.1016/j.bbrc.2012.12.092. [DOI] [PubMed] [Google Scholar]
- 11.Dahl M, Maturi V, Lönn P, Papoutsoglou P, Zieba A, Vanlandewijck M, van der Heide LP, Watanabe Y, Söderberg O, Hottiger MO. Fine-Tuning of Smad Protein Function by Poly(ADP-Ribose) Polymerases and Poly(ADP-ribose) Glycohydrolase during Transforming Growth Factor β Signaling. PLoS One. 2014;9:e103651–e103651. doi: 10.1371/journal.pone.0103651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quénet D, Mark M, Govin J, Van Dorsselear A, Schreiber V, Khochbin S, Dantzer F. Parp2 is required for the differentiation of post-meiotic germ cells: identification of a spermatid-specific complex containing Parp1, Parp2, TP2 and HSPA2. Exp Cell Res. 2009;315:2824–2834. doi: 10.1016/j.yexcr.2009.07.003. [DOI] [PubMed] [Google Scholar]
- 13.Bai P, Houten SM, Huber A, Schreiber V, Watanabe M, Kiss B, De Murcia G, Auwerx J, Ménissier-de Murcia J. Peroxisome proliferator-activated receptor (PPAR)-2 controls adipocyte differentiation and adipose tissue function through the regulation of the activity of the retinoid X receptor/PPARγ heterodimer. J Biol Chem. 2007;282:37738–37746. doi: 10.1074/jbc.M701021200. [DOI] [PubMed] [Google Scholar]
- 14.Saxena A, Wong LH, Kalitsis P, Earle E, Shaffer LG, Choo KH. Poly(ADP-ribose) polymerase 2 localizes to mammalian active centromeres and interacts with PARP-1, Cenpa, Cenpb and Bub3, but not Cenpc. Hum Mol Genet. 2002;11:2319–2329. doi: 10.1093/hmg/11.19.2319. [DOI] [PubMed] [Google Scholar]
- 15.Dantzer F, Giraud-Panis MJ, Jaco I, Amé JC, Schultz I, Blasco M, Koering CE, Gilson E, Ménissier-de Murcia J, de Murcia G. Functional interaction between poly(ADP-ribose) polymerase 2 (PARP-2) and TRF2: PARP activity negatively regulates TRF2. Mol Cell Biol. 2004;24:1595–1607. doi: 10.1128/MCB.24.4.1595-1607.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Maeda Y, Hunter TC, Loudy DE, Davé V, Schreiber V, Whitsett JA. PARP-2 interacts with TTF-1 and regulates expression of surfactant protein-B. J Biol Chem. 2006;281:9600–9606. doi: 10.1074/jbc.M510435200. [DOI] [PubMed] [Google Scholar]
- 17.Schreiber V, Amé JC, Dollé P, Schultz I, Rinaldi B, Fraulob V, Ménissier-de Murcia J, de Murcia G. Poly(ADP-ribose) polymerase-2 (PARP-2) is required for efficient base excision DNA repair in association with PARP-1 and XRCC1. J Biol Chem. 2002;277:23028–23036. doi: 10.1074/jbc.M202390200. [DOI] [PubMed] [Google Scholar]
- 18.Von Kobbe C, Harrigan JA, Schreiber V, Stiegler P, Piotrowski J, Dawut L, Bohr VA. Poly(ADP-ribose) polymerase 1 regulates both the exonuclease and helicase activities of the Werner syndrome protein. Nucleic Acids Res. 2004;32:4003–4014. doi: 10.1093/nar/gkh721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Meder VS, Boeglin M, De Murcia G, Schreiber V. PARP-1 and PARP-2 interact with nucleophosmin/B23 and accumulate in transcriptionally active nucleoli. J Cell Sci. 2005;118:211–222. doi: 10.1242/jcs.01606. [DOI] [PubMed] [Google Scholar]
- 20.Gibson BA, Zhang Y, Jiang H, Hussey KM, Shrimp JH, Lin H, Schwede F, Yu Y, Kraus WL. Chemical genetic discovery of PARP targets reveals a role for PARP-1 in transcription elongation. Science. 2016;353:45–50. doi: 10.1126/science.aaf7865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Y, Wang J, Ding M, Yu Y. Site-specific characterization of the Asp-and Glu-ADP-ribosylated proteome. Nat Methods. 2013;10:981–984. doi: 10.1038/nmeth.2603. [DOI] [PubMed] [Google Scholar]
- 22.Jungmichel S, Rosenthal F, Altmeyer M, Lukas J, Hottiger MO, Nielsen ML. Proteome-wide identification of Poly(ADP-ribosyl)ation targets in different genotoxic stress responses. Mol Cell. 2013;52:272–285. doi: 10.1016/j.molcel.2013.08.026. [DOI] [PubMed] [Google Scholar]
- 23.Dantzer F, Mark M, Quenet D, Scherthan H, Huber A, Liebe B, Monaco L, Chicheportiche A, Sassone-Corsi P, De Murcia G. Poly(ADP-ribose) polymerase-2 contributes to the fidelity of male meiosis I and spermiogenesis. Proc Natl Acad Sci U S A. 2006;103:14854–14859. doi: 10.1073/pnas.0604252103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yélamos J, Monreal Y, Saenz L, Aguado E, Schreiber V, Mota R, Fuente T, Minguela A, Parrilla P, De Murcia G. PARP-2 deficiency affects the survival of CD4+ CD8+ double-positive thymocytes. EMBO J. 2006;25:4350–4360. doi: 10.1038/sj.emboj.7601301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farrés J, Llacuna L, Martin-Caballero J, Martínez C, Lozano JJ, Ampurdanés C, López-Contreras AJ, Florensa L, Navarro J, Ottina E, Dantzer F, Schreiber V, Villunger A, Fernández-Capetillo O, Yélamos J. PARP-2 sustains erythropoiesis in mice by limiting replicative stress in erythroid progenitors. Cell Death Differ. 2015;22:1144–57. doi: 10.1038/cdd.2014.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bai P, Canto C, Brunyánszki A, Huber A, Szántó M, Cen Y, Yamamoto H, Houten SM, Kiss B, Oudart H. PARP-2 regulates SIRT1 expression and whole-body energy expenditure. Cell Metab. 2011;13:450–460. doi: 10.1016/j.cmet.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Murcia JM, Ricoul M, Tartier L, Niedergang C, Huber A, Dantzer F, Schreiber V, Amé JC, Dierich A, LeMeur M. Functional interaction between PARP-1 and PARP-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003;22:2255–2263. doi: 10.1093/emboj/cdg206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sonnenblick A, De Azambuja E, Azim HA Jr, Piccart M. An update on PARP inhibitors - moving to the adjuvant setting. Nat Rev Clin Oncol. 2015;12:27–41. doi: 10.1038/nrclinonc.2014.163. [DOI] [PubMed] [Google Scholar]
- 29.O’Connor MJ. Targeting the DNA damage response in cancer. Mol Cell. 2015;60:547–560. doi: 10.1016/j.molcel.2015.10.040. [DOI] [PubMed] [Google Scholar]
- 30.Daniels CM, Ong SE, Leung AK. The promise of proteomics for the study of ADP-ribosylation. Mol Cell. 2015;58:911–924. doi: 10.1016/j.molcel.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heilig R, Eckenberg R, Petit JL, Fonknechten N, Da Silva C, Cattolico L, Levy M, Barbe V, de Berardinis V, Ureta-Vidal A. The DNA sequence and analysis of human chromosome 14. Nature. 2003;421:601–607. doi: 10.1038/nature01348. [DOI] [PubMed] [Google Scholar]
- 32.Riccio AA, Cingolani G, Pascal JM. PARP-2 domain requirements for DNA damage-dependent activation and localization to sites of DNA damage. Nucleic Acids Res. 2016;44:1691–702. doi: 10.1093/nar/gkv1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Langelier MF, Servent KM, Rogers EE, Pascal JM. A third zinc-binding domain of human poly(ADP-ribose) polymerase-1 coordinates DNA-dependent enzyme activation. J Biol Chem. 2008;283:4105–4114. doi: 10.1074/jbc.M708558200. [DOI] [PubMed] [Google Scholar]
- 34.Sukhanova MV, Abrakhi S, Joshi V, Pastre D, Kutuzov MM, Anarbaev RO, Curmi PA, Hamon L, Lavrik OI. Single molecule detection of PARP1 and PARP2 interaction with DNA strand breaks and their Poly(ADP-ribosyl)ation using high-resolution AFM imaging. Nucleic Acids Res. 2015;44:e60. doi: 10.1093/nar/gkv1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Haenni SS, Hassa PO, Altmeyer M, Fey M, Imhof R, Hottiger MO. Identification of lysines 36 and 37 of PARP-2 as targets for acetylation and auto-ADP-ribosylation. Int J Biochem Cell Biol. 2008;40:2274–2283. doi: 10.1016/j.biocel.2008.03.008. [DOI] [PubMed] [Google Scholar]
- 36.Benchoua A, Couriaud C, Guégan C, Tartier L, Couvert P, Friocourt G, Chelly J, Ménissier-de Murcia J, Onténiente B. Active caspase-8 translocates into the nucleus of apoptotic cells to inactivate poly(ADP-ribose) polymerase-2. J Biol Chem. 2002;277:34217–34222. doi: 10.1074/jbc.M203941200. [DOI] [PubMed] [Google Scholar]
- 37.Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer research. 1997;57:1835–1840. [PubMed] [Google Scholar]
- 38.Amé JC, Spenlehauer C, de Murcia G. The PARP superfamily. Bioessays. 2004;26:882–893. doi: 10.1002/bies.20085. [DOI] [PubMed] [Google Scholar]
- 39.Aoyagi-Scharber M, Gardberg AS, Yip BK, Wang B, Shen Y, Fitzpatrick PA. Structural basis for the inhibition of poly(ADP-ribose) polymerases 1 and 2 by BMN 673, a potent inhibitor derived from dihydropyridophthalazinone. Acta Crystallogr F Struct Biol Commun. 2014;70:1143–1149. doi: 10.1107/S2053230X14015088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hunter SM, Rowley SM, Clouston D, Li J, Lupat R, Krishnananthan N, Risbridger G, Taylor R, Bolton D, Campbell IG. Searching for candidate genes in familial BRCAX mutation carriers with prostate cancer. Urol Oncol. 2016;34:120, e9–16. doi: 10.1016/j.urolonc.2015.10.009. [DOI] [PubMed] [Google Scholar]
- 41.Johnson AM, Zuhlke KA, Plotts C, McDonnell SK, Middha S, Riska SM, Schaid DJ, Thibodeau SN, Douglas JA, Cooney KA. Mutational landscape of candidate genes in familial prostate cancer. Prostate. 2014;74:1371–1378. doi: 10.1002/pros.22849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osorio A, Milne RL, Kuchenbaecker K, Vaclová T, Pita G, Alonso R, Peterlongo P, Blanco I, De la Hoya M, Duran M. DNA glycosylases involved in base excision repair may be associated with cancer risk in BRCA1 and BRCA2 mutation carriers. PLoS Genet. 2014;10:e1004256–e1004256. doi: 10.1371/journal.pgen.1004256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Popanda O, Seibold P, Nikolov I, Oakes CC, Burwinkel B, Hausmann S, Flesch-Janys D, Plass C, Chang-Claude J, Schmezer P. Germline variants of base excision repair genes and breast cancer: A polymorphism in DNA polymerase gamma modifies gene expression and breast cancer risk. Int J Cancer. 2013;132:55–62. doi: 10.1002/ijc.27665. [DOI] [PubMed] [Google Scholar]
- 44.Xie H, Gong Y, Dai J, Wu X, Gu J. Genetic variations in base excision repair pathway and risk of bladder cancer: A case-control study in the United States. Mol Carcinog. 2015;54:50–57. doi: 10.1002/mc.22073. [DOI] [PubMed] [Google Scholar]
- 45.Gao Y, Hayes RB, Huang WY, Caporaso NE, Burdette L, Yeager M, Chanock SJ, Berndt SI. DNA repair gene polymorphisms and tobacco smoking in the risk for colorectal adenomas. Carcinogenesis. 2011;32:882–7. doi: 10.1093/carcin/bgr071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Seibold P, Schmezer P, Behrens S, Michailidou K, Bolla MK, Wang Q, Flesch-Janys D, Nevanlinna H, Fagerholm R, Aittomäki K. A polymorphism in the base excision repair gene PARP2 is associated with differential prognosis by chemotherapy among postmenopausal breast cancer patients. BMC cancer. 2015;15:1–1. doi: 10.1186/s12885-015-1957-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 48.Shen Z. Genomic instability and cancer: an introduction. J Mol Cell Biol. 2011;3:1–3. doi: 10.1093/jmcb/mjq057. [DOI] [PubMed] [Google Scholar]
- 49.Aguilera A, García-Muse T. Causes of genome instability. Ann Rev Genet. 2013;47:1–32. doi: 10.1146/annurev-genet-111212-133232. [DOI] [PubMed] [Google Scholar]
- 50.Farrés J, Martín-Caballero J, Martínez C, Lozano JJ, Llacuna L, Ampurdanés C, Ruiz-Herguido C, Dantzer F, Schreiber V, Villunger A. Parp-2 is required to maintain hematopoiesis following sublethal γ-irradiation in mice. Blood. 2013;122:44–54. doi: 10.1182/blood-2012-12-472845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nicolas L, Martinez C, Baro C, Rodriguez M, Baroja-Mazo A, Sole F, Flores JM, Ampurdanes C, Dantzer F, Martin-Caballero J. Loss of poly(ADP-ribose) polymerase-2 leads to rapid development of spontaneous T-cell lymphomas in p53-deficient mice. Oncogene. 2010;29:2877–2883. doi: 10.1038/onc.2010.11. [DOI] [PubMed] [Google Scholar]
- 52.Ghosh R, Roy S, Kamyab J, Dantzer F, Franco S. Common and unique genetic interactions of the poly(ADP-ribose) polymerases PARP1 and PARP2 with DNA double-strand break repair pathways. DNA Repair (Amst) 2016;45:56–62. doi: 10.1016/j.dnarep.2016.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gaillard H, García-Muse T, Aguilera A. Replication stress and cancer. Nat Rev Cancer. 2015;15:276–289. doi: 10.1038/nrc3916. [DOI] [PubMed] [Google Scholar]
- 54.Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. doi: 10.1038/nature05268. [DOI] [PubMed] [Google Scholar]
- 55.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Nuciforo PG, Bensimon A, Maestro R. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006;444:638–642. doi: 10.1038/nature05327. [DOI] [PubMed] [Google Scholar]
- 56.Mazouzi A, Velimezi G, Loizou JI. DNA replication stress: causes, resolution and disease. Exp Cell Res. 2014;329:85–93. doi: 10.1016/j.yexcr.2014.09.030. [DOI] [PubMed] [Google Scholar]
- 57.Macheret M, Halazonetis TD. DNA replication stress as a hallmark of cancer. Ann Rev Pathol. 2015;10:425–448. doi: 10.1146/annurev-pathol-012414-040424. [DOI] [PubMed] [Google Scholar]
- 58.Bryant HE, Petermann E, Schultz N, Jemth AS, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T. PARP is activated at stalled forks to mediate Mre11-dependent replication restart and recombination. EMBO J. 2009;28:2601–2615. doi: 10.1038/emboj.2009.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ying S, Chen Z, Medhurst AL, Neal JA, Bao Z, Mortusewicz O, McGouran J, Song X, Shen H, Hamdy FC. DNA-PKcs and PARP1 bind to unresected stalled DNA replication forks where they recruit XRCC1 to mediate repair. Cancer Res. 2016;76:1078–1088. doi: 10.1158/0008-5472.CAN-15-0608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ying S, Hamdy FC, Helleday T. Mre11-dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res. 2012;72:2814–2821. doi: 10.1158/0008-5472.CAN-11-3417. [DOI] [PubMed] [Google Scholar]
- 61.Chaudhuri AR, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ. Replication fork stability confers chemoresistance in BRCA-deficient cells. Nature. 2016;535:382–387. doi: 10.1038/nature18325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Murga M, Bunting S, Montaña MF, Soria R, Mulero F, Cañamero M, Lee Y, McKinnon PJ, Nussenzweig A, Fernandez-Capetillo O. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat Genet. 2009;41:891–898. doi: 10.1038/ng.420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kavanaugh G, Ye F, Mohni KN, Luzwick JW, Glick G, Cortez D. A whole genome RNAi screen identifies replication stress response genes. DNA Repair (Amst) 2015;35:55–62. doi: 10.1016/j.dnarep.2015.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rybaczek D. Hydroxyurea-induced replication stress causes poly(ADP-ribose) polymerase-2 accumulation and changes its intranuclear location in root meristems of Vicia faba. J Plant Physiol. 2016;198:89–102. doi: 10.1016/j.jplph.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 65.Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell. 2010;40:179–204. doi: 10.1016/j.molcel.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jeggo PA, Pearl LH, Carr AM. DNA repair, genome stability and cancer: a historical perspective. Nat Rev Cancer. 2016;16:35–42. doi: 10.1038/nrc.2015.4. [DOI] [PubMed] [Google Scholar]
- 67.Gavande NS, VanderVere-Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS, Turchi JJ. DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol Ther. 2016;160:65–83. doi: 10.1016/j.pharmthera.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kim YJ, Wilson Iii DM. Overview of base excision repair biochemistry. Curr Mol Pharmacol. 2012;5:3–3. doi: 10.2174/1874467211205010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kutuzov MM, Khodyreva SN, Ilina ES, Sukhanova MV, Ame JC, Lavrik OI. Interaction of PARP-2 with AP site containing DNA. Biochimie. 2015;112:10–19. doi: 10.1016/j.biochi.2015.02.010. [DOI] [PubMed] [Google Scholar]
- 70.Fisher AE, Hochegger H, Takeda S, Caldecott KW. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol Cell Biol. 2007;27:5597–5605. doi: 10.1128/MCB.02248-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mortusewicz O, Amé JC, Schreiber V, Leonhardt H. Feedback-regulated Poly(ADP-ribosyl)ation by PARP-1 is required for rapid response to DNA damage in living cells. Nucleic Acids Res. 2007;35:7665–7675. doi: 10.1093/nar/gkm933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kutuzov MM, Khodyreva SN, Amé JC, Ilina ES, Sukhanova MV, Schreiber V, Lavrik OI. Interaction of PARP-2 with DNA structures mimicking DNA repair intermediates and consequences on activity of base excision repair proteins. Biochimie. 2013;95:1208–1215. doi: 10.1016/j.biochi.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 73.Langelier MF, Riccio AA, Pascal JM. PARP-2 and PARP-3 are selectively activated by 5’ phosphorylated DNA breaks through an allosteric regulatory mechanism shared with PARP-1. Nucleic Acids Res. 2014;42:7762–7775. doi: 10.1093/nar/gku474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Talhaoui I, Lebedeva NA, Zarkovic G, Saint-Pierre C, Kutuzov MM, Sukhanova MV, Matkarimov BT, Gasparutto D, Saparbaev MK, Lavrik OI. Poly(ADP-ribose) polymerases covalently modify strand break termini in DNA fragments in vitro. Nucleic Acids Res. 2016 doi: 10.1093/nar/gkw675. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Caldecott KW. XRCC1 and DNA strand break repair. DNA Repair. 2003;2:955–969. doi: 10.1016/s1568-7864(03)00118-6. [DOI] [PubMed] [Google Scholar]
- 76.El-Khamisy SF, Masutani M, Suzuki H, Caldecott KW. A requirement for PARP-1 for the assembly or stability of XRCC1 nuclear foci at sites of oxidative DNA damage. Nucleic Acids Res. 2003;31:5526–5533. doi: 10.1093/nar/gkg761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Liu Y, Kadyrov FA, Modrich P. PARP-1 enhances the mismatch-dependence of 5’-directed excision in human mismatch repair in vitro. DNA Repair (Amst) 2011;10:1145–1153. doi: 10.1016/j.dnarep.2011.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Pines A, Vrouwe MG, Marteijn JA, Typas D, Luijsterburg MS, Cansoy M, Hensbergen P, Deelder A, de Groot A, Matsumoto S. PARP1 promotes nucleotide excision repair through DDB2 stabilization and recruitment of ALC1. J Cell Biol. 2012;199:235–249. doi: 10.1083/jcb.201112132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Robu M, Shah RG, Petitclerc N, Brind’Amour J, Kandan-Kulangara F, Shah GM. Role of poly(ADP-ribose) polymerase-1 in the removal of UV-induced DNA lesions by nucleotide excision repair. Proc Natl Acad Sci U S A. 2013;110:1658–1663. doi: 10.1073/pnas.1209507110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hickson I, Zhao Y, Richardson CJ, Green SJ, Martin NM, Orr AI, Reaper PM, Jackson SP, Curtin NJ, Smith GC. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152–9159. doi: 10.1158/0008-5472.CAN-04-2727. [DOI] [PubMed] [Google Scholar]
- 81.Ménissier-de Murcia J, Mark M, Wendling O, Wynshaw-Boris A, de Murcia G. Early Embryonic Lethality in PARP-1 AtmDouble-Mutant Mice Suggests a Functional Synergy in Cell Proliferation during Development. Mol Cell Biol. 2001;21:1828–1832. doi: 10.1128/MCB.21.5.1828-1832.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Haince JF, Kozlov S, Dawson VL, Dawson TM, Hendzel MJ, Lavin MF, Poirier GG. Ataxia telangiectasia mutated (ATM) signaling network is modulated by a novel poly(ADP-ribose)-dependent pathway in the early response to DNA-damaging agents. J Biol Chem. 2007;282:16441–16453. doi: 10.1074/jbc.M608406200. [DOI] [PubMed] [Google Scholar]
- 83.Fernandes N, Sun Y, Chen S, Paul P, Shaw RJ, Cantley LC, Price BD. DNA damage-induced association of ATM with its target proteins requires a protein interaction domain in the N terminus of ATM. J Biol Chem. 2005;280:15158–15164. doi: 10.1074/jbc.M412065200. [DOI] [PubMed] [Google Scholar]
- 84.Haince JF, McDonald D, Rodrigue A, Déry U, Masson JY, Hendzel MJ, Poirier GG. PARP1-dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem. 2008;283:1197–1208. doi: 10.1074/jbc.M706734200. [DOI] [PubMed] [Google Scholar]
- 85.Audebert M, Salles B, Calsou P. Involvement of poly(ADP-ribose) polymerase-1 and XRCC1/DNA ligase III in an alternative route for DNA double-strand breaks rejoining. J Biol Chem. 2004;279:55117–55126. doi: 10.1074/jbc.M404524200. [DOI] [PubMed] [Google Scholar]
- 86.Wang M, Wu W, Wu W, Rosidi B, Zhang L, Wang H, Iliakis G. PARP-1 and Ku compete for repair of DNA double strand breaks by distinct NHEJ pathways. Nucleic Acids Res. 2006;34:6170–6182. doi: 10.1093/nar/gkl840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Robert I, Dantzer F, Reina-San-Martin B. Parp1 facilitates alternative NHEJ, whereas Parp2 suppresses IgH/c-myc translocations during immunoglobulin class switch recombination. J Exp Med. 2009;206:1047–1056. doi: 10.1084/jem.20082468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yan CT, Boboila C, Souza EK, Franco S, Hickernell TR, Murphy M, Gumaste S, Geyer M, Zarrin AA, Manis JP. IgH class switching and translocations use a robust non-classical end-joining pathway. Nature. 2007;449:478–482. doi: 10.1038/nature06020. [DOI] [PubMed] [Google Scholar]
- 89.Robbiani DF, Nussenzweig MC. Chromosome translocation, B cell lymphoma, and activation-induced cytidine deaminase. Ann Rev Pathol. 2013;8:79–103. doi: 10.1146/annurev-pathol-020712-164004. [DOI] [PubMed] [Google Scholar]
- 90.Kinner A, Wu W, Staudt C, Iliakis G. γ-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008;36:5678–5694. doi: 10.1093/nar/gkn550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Orsburn B, Escudero B, Prakash M, Gesheva S, Liu G, Huso DL, Franco S. Differential requirement for H2AX and 53BP1 in organismal development and genome maintenance in the absence of poly (ADP) ribosyl polymerase 1. Mol Cell Biol. 2010;30:2341–2352. doi: 10.1128/MCB.00091-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sulli G, Di Micco R, d’Adda di Fagagna F. Crosstalk between chromatin state and DNA damage response in cellular senescence and cancer. Nat Rev Cancer. 2012;12:709–720. doi: 10.1038/nrc3344. [DOI] [PubMed] [Google Scholar]
- 93.d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- 94.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ahuja N, Sharma AR, Baylin SB. Epigenetic therapeutics: a new weapon in the war against cancer. Ann Rev Med. 2016;67:73–89. doi: 10.1146/annurev-med-111314-035900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Poirier GG, De Murcia G, Jongstra-Bilen J, Niedergang C, Mandel P. Poly(ADP-ribosyl)ation of polynucleosomes causes relaxation of chromatin structure. Proc Natl Acad Sci U S A. 1982;79:3423–3427. doi: 10.1073/pnas.79.11.3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Huletsky A, De Murcia G, Muller S, Hengartner M, Menard L, Lamarre D, Poirier GG. The effect of poly(ADP-ribosyl)ation on native and H1-depleted chromatin. A role of poly(ADP-ribosyl) ation on core nucleosome structure. J Biol Chem. 1989;264:8878–8886. [PubMed] [Google Scholar]
- 98.Bürkle A. Poly(ADP-Ribosyl)ation. US: Springer; 2008. [Google Scholar]
- 99.Nielsen AL, Oulad-Abdelghani M, Ortiz JA, Remboutsika E, Chambon P, Losson R. Heterochromatin formation in mammalian cells: interaction between histones and HP1 proteins. Mol Cell. 2001;7:729–739. doi: 10.1016/s1097-2765(01)00218-0. [DOI] [PubMed] [Google Scholar]
- 100.Nielsen AL, Ortiz JA, You J, Oulad-Abdelghani M, Khechumian R, Gansmuller A, Chambon P, Losson RG. Interaction with members of the heterochromatin protein 1 (HP1) family and histone deacetylation are differentially involved in transcriptional silencing by members of the TIF1 family. EMBO J. 1999;18:6385–6395. doi: 10.1093/emboj/18.22.6385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Le Douarin B, Nielsen AL, Garnier JM, Ichinose H, Jeanmougin F, Losson R, Chambon P. A possible involvement of TIF1 alpha and TIF1 beta in the epigenetic control of transcription by nuclear receptors. EMBO J. 1996;15:6701–6701. [PMC free article] [PubMed] [Google Scholar]
- 102.Friedman JR, Fredericks WJ, Jensen DE, Speicher DW, Huang XP, Neilson EG, Rauscher FJ. KAP-1, a novel corepressor for the highly conserved KRAB repression domain. Genes Dev. 1996;10:2067–2078. doi: 10.1101/gad.10.16.2067. [DOI] [PubMed] [Google Scholar]
- 103.Cammas F, Herzog M, Lerouge T, Chambon P, Losson R. Association of the transcriptional corepressor TIF1β with heterochromatin protein 1 (HP1): an essential role for progression through differentiation. Genes Dev. 2004;18:2147–2160. doi: 10.1101/gad.302904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Westhorpe FG, Straight AF. Functions of the centromere and kinetochore in chromosome segregation. Curr Opin Cell Biol. 2013;25:334–340. doi: 10.1016/j.ceb.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;432:338–341. doi: 10.1038/nature03099. [DOI] [PubMed] [Google Scholar]
- 106.Earle E, Saxena A, MacDonald A, Hudson DF, Shaffer LG, Saffery R, Cancilla MR, Cutts SM, Howman E, Choo KH. Poly(ADP-ribose) polymerase at active centromeres and neocentromeres at metaphase. Hum Mol Genet. 2000;9:187–194. doi: 10.1093/hmg/9.2.187. [DOI] [PubMed] [Google Scholar]
- 107.Saxena A, Saffery R, Wong LH, Kalitsis P, Choo KH. Centromere proteins Cenpa, Cenpb, and Bub3 interact with poly(ADP-ribose) polymerase-1 protein and are poly (ADP-ribosyl) ated. J Biol Chem. 2002;277:26921–26926. doi: 10.1074/jbc.M200620200. [DOI] [PubMed] [Google Scholar]
- 108.Vogt E, Kirsch-Volders M, Parry J, Eichenlaub-Ritter U. Spindle formation, chromosome segregation and the spindle checkpoint in mammalian oocytes and susceptibility to meiotic error. Mutat Res. 2008;651:14–29. doi: 10.1016/j.mrgentox.2007.10.015. [DOI] [PubMed] [Google Scholar]
- 109.Diaz-Moralli S, Tarrado-Castellarnau M, Miranda A, Cascante M. Targeting cell cycle regulation in cancer therapy. Pharmacol Ther. 2013;138:255–271. doi: 10.1016/j.pharmthera.2013.01.011. [DOI] [PubMed] [Google Scholar]
- 110.Niculescu AB, Chen X, Smeets M, Hengst L, Prives C, Reed SI. Effects of p21Cip1/Waf1 at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol. 1998;18:629–643. doi: 10.1128/mcb.18.1.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Palm W, de Lange T. How shelterin protects mammalian telomeres. Ann Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 112.Morin GB. The human telomere terminal transferase enzyme is a ribonucleoprotein that synthesizes TTAGGG repeats. Cell. 1989;59:521–529. doi: 10.1016/0092-8674(89)90035-4. [DOI] [PubMed] [Google Scholar]
- 113.Griffith JD, Comeau L, Rosenfield S, Stansel RM, Bianchi A, Moss H, De Lange T. Mammalian telomeres end in a large duplex loop. Cell. 1999;97:503–514. doi: 10.1016/s0092-8674(00)80760-6. [DOI] [PubMed] [Google Scholar]
- 114.Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336:593–597. doi: 10.1126/science.1218498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Stansel RM, de Lange T, Griffith JD. T-loop assembly in vitro involves binding of TRF2 near the 3’ telomeric overhang. EMBO J. 2001;20:5532–5540. doi: 10.1093/emboj/20.19.5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ohki R, Tsurimoto T, Ishikawa F. In vitro reconstitution of the end replication problem. Mol Cell Biol. 2001;21:5753–5766. doi: 10.1128/MCB.21.17.5753-5766.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 118.Herbig U, Jobling WA, Chen BP, Chen DJ, Sedivy JM. Telomere shortening triggers senescence of human cells through a pathway involving ATM, p53, and p21 CIP1, but not p16 INK4a. Mol Cell. 2004;14:501–513. doi: 10.1016/s1097-2765(04)00256-4. [DOI] [PubMed] [Google Scholar]
- 119.Chang S, Khoo CM, Naylor ML, Maser RS, DePinho RA. Telomere-based crisis: functional differences between telomerase activation and ALT in tumor progression. Genes Dev. 2003;17:88–100. doi: 10.1101/gad.1029903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Counter CM, Avilion AA, LeFeuvre CE, Stewart NG, Greider CW, Harley CB, Bacchetti S. Telomere shortening associated with chromosome instability is arrested in immortal cells which express telomerase activity. EMBO J. 1992;11:1921–1921. doi: 10.1002/j.1460-2075.1992.tb05245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Bollmann FM. Targeting ALT: the role of alternative lengthening of telomeres in pathogenesis and prevention of cancer. Cancer Treat Rev. 2007;33:704–709. doi: 10.1016/j.ctrv.2007.08.006. [DOI] [PubMed] [Google Scholar]
- 122.Weiss A, Attisano L. The TGFbeta superfamily signaling pathway. Wiley Interdiscip Rev Dev Biol. 2013;2:47–63. doi: 10.1002/wdev.86. [DOI] [PubMed] [Google Scholar]
- 123.Houtkooper RH, Cantó C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocr Rev. 2010;31:194–223. doi: 10.1210/er.2009-0026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bai P, Cantó C, Oudart H, Brunyánszki A, Cen Y, Thomas C, Yamamoto H, Huber A, Kiss B, Houtkooper RH. PARP-1 inhibition increases mitochondrial metabolism through SIRT1 activation. Cell Metab. 2011;13:461–468. doi: 10.1016/j.cmet.2011.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Erener S, Hesse M, Kostadinova R, Hottiger MO. Poly(ADP-ribose) polymerase-1 (PARP1) controls adipogenic gene expression and adipocyte function. Mol Endocrinol. 2011;26:79–86. doi: 10.1210/me.2011-1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mohamed JS, Hajira A, Pardo PS, Boriek AM. MicroRNA-149 inhibits PARP-2 and promotes mitochondrial biogenesis via SIRT-1/PGC-1α network in skeletal muscle. Diabetes. 2014;63:1546–1559. doi: 10.2337/db13-1364. [DOI] [PubMed] [Google Scholar]
- 127.Mateu-Jiménez M, Cucarull-Martínez B, Yelamos J, Barreiro E. Reduced tumor burden through increased oxidative stress in lung adenocarcinoma cells of PARP-1 and PARP-2 knockout mice. Biochimie. 2016;121:278–286. doi: 10.1016/j.biochi.2015.11.030. [DOI] [PubMed] [Google Scholar]
- 128.Szántó M, Brunyánszki A, Márton J, Vámosi G, Nagy L, Fodor T, Kiss B, Virág L, Gergely P, Bai P. Deletion of PARP-2 induces hepatic cholesterol accumulation and decrease in HDL levels. Biochim Biophys Acta. 2014;1842:594–602. doi: 10.1016/j.bbadis.2013.12.006. [DOI] [PubMed] [Google Scholar]
- 129.Ali KM, Wonnerth A, Huber K, Wojta J. Cardiovascular disease risk reduction by raising HDL cholesterol-current therapies and future opportunities. Br J Pharmacol. 2012;167:1177–1194. doi: 10.1111/j.1476-5381.2012.02081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Oram JF, Lawn RM. ABCA1: the gatekeeper for eliminating excess tissue cholesterol. J Lipid Res. 2001;42:1173–1179. [PubMed] [Google Scholar]
- 131.Wallace DC. Mitochondria and cancer. Nat Rev Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nishida N. Angiogenesis in cancer. Vasc Health Risk Manag. 2006;2:213–219. doi: 10.2147/vhrm.2006.2.3.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tentori L, Lacal PM, Muzi A, Dorio AS, Leonetti C, Scarsella M, Ruffini F, Xu W, Min W, Stoppacciaro A. Poly(ADP-ribose) polymerase (PARP) inhibition or PARP-1 gene deletion reduces angiogenesis. Eur J Cancer. 2007;43:2124–2133. doi: 10.1016/j.ejca.2007.07.010. [DOI] [PubMed] [Google Scholar]
- 134.Tentori L, Muzi A, Dorio AS, Bultrini S, Mazzon E, Lacal PM, Shah GM, Zhang J, Navarra P, Nocentini G. Stable depletion of poly(ADP-ribose) polymerase-1 reduces in vivo melanoma growth and increases chemosensitivity. Eur J Cancer. 2008;44:1302–1314. doi: 10.1016/j.ejca.2008.03.019. [DOI] [PubMed] [Google Scholar]
- 135.Mao H, Lockyer P, Townley-Tilson WH, Xie L, Pi X. LRP1 regulates retinal angiogenesis by inhibiting PARP-1 activity and endothelial cell proliferation. Arterioscler Thromb Vasc Biol. 2016;36:350–360. doi: 10.1161/ATVBAHA.115.306713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ferrara N. Role of vascular endothelial growth factor in the regulation of angiogenesis. Kidney Int. 1999;56:794–814. doi: 10.1046/j.1523-1755.1999.00610.x. [DOI] [PubMed] [Google Scholar]
- 137.Ziche M, Maglione D, Ribatti D, Morbidelli L, Lago CT, Battisti M, Paoletti I, Barra A, Tucci M, Parise G. Placenta growth factor-1 is chemotactic, mitogenic, and angiogenic. Lab Invest. 1997;76:517–531. [PubMed] [Google Scholar]
- 138.Elser M, Borsig L, Hassa PO, Erener S, Messner S, Valovka T, Keller S, Gassmann M, Hottiger MO. Poly(ADP-Ribose) Polymerase 1 Promotes Tumor Cell Survival by Coactivating Hypoxia-Inducible Factor-1-Dependent Gene Expression. Mol Cancer Res. 2008;6:282–290. doi: 10.1158/1541-7786.MCR-07-0377. [DOI] [PubMed] [Google Scholar]
- 139.Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, Haendeler J, Mione M, Dejana E, Alt FW. SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev. 2007;21:2644–2658. doi: 10.1101/gad.435107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Guarani V, Deflorian G, Franco CA, Krüger M, Phng LK, Bentley K, Toussaint L, Dequiedt F, Mostoslavsky R, Schmidt MH. Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature. 2011;473:234–238. doi: 10.1038/nature09917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Arany Z, Foo SY, Ma Y, Ruas JL, Bommi-Reddy A, Girnun G, Cooper M, Laznik D, Chinsomboon J, Rangwala SM. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 142.Phng LK, Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell. 2009;16:196–208. doi: 10.1016/j.devcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 143.Mulligan P, Yang F, Di Stefano L, Ji JY, Ouyang J, Nishikawa JL, Toiber D, Kulkarni M, Wang Q, Najafi-Shoushtari SH. A SIRT1-LSD1 corepressor complex regulates Notch target gene expression and development. Mol Cell. 2011;42:689–699. doi: 10.1016/j.molcel.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Di Paola R, Mazzon E, Xu W, Genovese T, Ferrraris D, Muià C, Crisafulli C, Zhang J, Cuzzocrea S. Treatment with PARP-1 inhibitors, GPI 15427 or GPI 16539, ameliorates intestinal damage in rat models of colitis and shock. Eur J Pharmacol. 2005;527:163–171. doi: 10.1016/j.ejphar.2005.09.055. [DOI] [PubMed] [Google Scholar]
- 145.Mota RA, Sánchez-Bueno F, Saenz L, Hernández-Espinosa D, Jimeno J, Tornel PL, Martínez-Torrano A, Ramírez P, Parrilla P, Yélamos J. Inhibition of poly(ADP-ribose) polymerase attenuates the severity of acute pancreatitis and associated lung injury. Lab Invest. 2005;85:1250–1262. doi: 10.1038/labinvest.3700326. [DOI] [PubMed] [Google Scholar]
- 146.Oliver FJ, Ménissier-de Murcia J, Nacci C, Decker P, Andriantsitohaina R, Muller S, de la Rubia G, Stoclet JC, de Murcia G. Resistance to endotoxic shock as a consequence of defective NF-κB activation in poly (ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999;18:4446–4454. doi: 10.1093/emboj/18.16.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Giansanti V, Donà F, Tillhon M, Scovassi AI. PARP inhibitors: new tools to protect from inflammation. Biochem Pharmacol. 2010;80:1869–1877. doi: 10.1016/j.bcp.2010.04.022. [DOI] [PubMed] [Google Scholar]
- 148.Popoff I, Jijon H, Monia B, Tavernini M, Ma M, McKay R, Madsen K. Antisense oligonucleotides to poly(ADP-ribose) polymerase-2 ameliorate colitis in interleukin-10-deficient mice. J Pharmacol Exp Ther. 2002;303:1145–1154. doi: 10.1124/jpet.102.039768. [DOI] [PubMed] [Google Scholar]
- 149.Jijon HB, Churchill T, Malfair D, Wessler A, Jewell LD, Parsons HG, Madsen KL. Inhibition of poly(ADP-ribose) polymerase attenuates inflammation in a model of chronic colitis. Am J Physiol Gastrointest Liver Physiol. 2000;279:G641–G651. doi: 10.1152/ajpgi.2000.279.3.G641. [DOI] [PubMed] [Google Scholar]
- 150.Kamboj A, Lu P, Cossoy MB, Stobart JL, Dolhun BA, Kauppinen TM, de Murcia G, Anderson CM. Poly(ADP-ribose) polymerase 2 contributes to neuroinflammation and neurological dysfunction in mouse experimental autoimmune encephalomyelitis. J Neuroinflammation. 2013;10:49. doi: 10.1186/1742-2094-10-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Scott GS, Kean RB, Mikheeva T, Fabis MJ, Mabley JG, Szabó C, Hooper DC. The therapeutic effects of PJ34 [N-(6-oxo-5, 6-dihydrophenanthridin-2-yl)-N, N-dimethylacetamide. HCl] , a selective inhibitor of poly(ADP-ribose) polymerase, in experimental allergic encephalomyelitis are associated with immunomodulation. J Pharmacol Exp Ther. 2004;310:1053–1061. doi: 10.1124/jpet.103.063214. [DOI] [PubMed] [Google Scholar]
- 152.Chen F, Zhuang X, Lin L, Yu P, Wang Y, Shi Y, Hu G, Sun Y. New horizons in tumor microenvironment biology: challenges and opportunities. BMC Med. 2015;13:45. doi: 10.1186/s12916-015-0278-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory Pathways in Immunotherapy for Cancer. Ann Rev Immunol. 2016;34:539–573. doi: 10.1146/annurev-immunol-032414-112049. [DOI] [PubMed] [Google Scholar]
- 154.Hareendran S, Ramakrishna B, Jayandharan GR. Synergistic inhibition of PARP-1 and NF-κB signaling downregulates immune response against recombinant AAV2 vectors during hepatic gene therapy. Eur J Immunol. 2016;46:154–166. doi: 10.1002/eji.201545867. [DOI] [PubMed] [Google Scholar]
- 155.Rosado MM, Bennici E, Novelli F, Pioli C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology. 2013;139:428–437. doi: 10.1111/imm.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Saenz L, Lozano JJ, Valdor R, Baroja-Mazo A, Ramirez P, Parrilla P, Aparicio P, Sumoy L, Yélamos J. Transcriptional regulation by poly (ADP-ribose) polymerase-1 during T cell activation. BMC Genomics. 2008;9:171. doi: 10.1186/1471-2164-9-171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Valdor R, Schreiber V, Saenz L, Martínez T, Munoz-Suano A, Dominguez-Villar M, Ramírez P, Parrilla P, Aguado E, García-Cózar F. Regulation of NFAT by poly(ADP-ribose) polymerase activity in T cells. Mol Immunol. 2008;45:1863–1871. doi: 10.1016/j.molimm.2007.10.044. [DOI] [PubMed] [Google Scholar]
- 158.Luo X, Nie J, Wang S, Chen Z, Chen W, Li D, Hu H, Li B. Poly(ADP-ribosyl)ation of FOXP3 Protein Mediated by PARP-1 Protein Regulates the Function of Regulatory T Cells. J Biol Chem. 2015;290:28675–28682. doi: 10.1074/jbc.M115.661611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhang P, Maruyama T, Konkel JE, Abbatiello B, Zamarron B, Wang ZQ, Chen W. PARP-1 controls immunosuppressive function of regulatory T cells by destabilizing Foxp3. PLoS One. 2013;8:e71590–e71590. doi: 10.1371/journal.pone.0071590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Chacon-Cabrera A, Fermoselle C, Salmela I, Yelamos J, Barreiro E. MicroRNA expression and protein acetylation pattern in respiratory and limb muscles of Parp-1-/- and Parp-2-/- mice with lung cancer cachexia. Biochim Biophys Acta. 2015;1850:2530–2543. doi: 10.1016/j.bbagen.2015.09.020. [DOI] [PubMed] [Google Scholar]
- 161.Adams JL, Smothers J, Srinivasan R, Hoos A. Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Discov. 2015;14:603–622. doi: 10.1038/nrd4596. [DOI] [PubMed] [Google Scholar]
- 162.Somnay YR, Lubner S, Kunnimalaiyaan M, Chen H. Velaparib (ABT-888), a Novel Inhibitor of Poly-Adp Ribosyl Polymerase (PARP), Synergizes With 5-Fluorouracil in Pancreatic Carcinoid Cells. J Surg Res. 2013;179:311–311. [Google Scholar]
- 163.Donawho CK, Luo Y, Luo Y, Penning TD, Bauch JL, Bouska JJ, Bontcheva-Diaz VD, Cox BF, DeWeese TL, Dillehay LE. ABT-888, an orally active poly(ADP-ribose) polymerase inhibitor that potentiates DNA-damaging agents in preclinical tumor models. Clin Cancer Res. 2007;13:2728–2737. doi: 10.1158/1078-0432.CCR-06-3039. [DOI] [PubMed] [Google Scholar]
- 164.Muñoz-Gámez JA, Martín-Oliva D, Aguilar-Quesada R, Cañuelo A, Nunez MI, Valenzuela MT, de Almodovar JM, de Murcia G, Oliver FJ. PARP inhibition sensitizes p53-deficient breast cancer cells to doxorubicin-induced apoptosis. Biochem J. 2005;386:119–125. doi: 10.1042/BJ20040776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Delaney CA, Wang LZ, Kyle S, White AW, Calvert AH, Curtin NJ, Durkacz BW, Hostomsky Z, Newell DR. Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly (adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Clin Cancer Res. 2000;6:2860–2867. [PubMed] [Google Scholar]
- 166.Tutt A, Robson M, Garber JE, Domchek SM, Audeh MW, Weitzel JN, Friedlander M, Arun B, Loman N, Schmutzler RK. Oral poly(ADP-ribose) polymerase inhibitor olaparib in patients with BRCA1 or BRCA2 mutations and advanced breast cancer: a proof-of-concept trial. Lancet. 2010;376:235–244. doi: 10.1016/S0140-6736(10)60892-6. [DOI] [PubMed] [Google Scholar]
- 167.Mendes-Pereira AM, Martin SA, Brough R, McCarthy A, Taylor JR, Kim JS, Waldman T, Lord CJ, Ashworth A. Synthetic lethal targeting of PTEN mutant cells with PARP inhibitors. EMBO Mol Med. 2009;1:315–322. doi: 10.1002/emmm.200900041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ashworth A. A synthetic lethal therapeutic approach: poly (ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. 2008;26:3785–3790. doi: 10.1200/JCO.2008.16.0812. [DOI] [PubMed] [Google Scholar]
- 169.Wahlberg E, Karlberg T, Kouznetsova E, Markova N, Macchiarulo A, Thorsell AG, Pol E, Frostell Å, Ekblad T, Öncü D. Family-wide chemical profiling and structural analysis of PARP and tankyrase inhibitors. Nat Biotechnol. 2012;30:283–288. doi: 10.1038/nbt.2121. [DOI] [PubMed] [Google Scholar]