

Abstract
Gastric carcinoma is one of the most lethal malignancies of cancers and its prognosis remains dismal due to the paucity of effective therapeutic targets. Herein, we showed that HRAS is markedly up-regulated in gastric carcinoma. Prognostic analysis indicated that HRAS expression might be a prognostic indicator for the survival of patients with gastric carcinoma. Ectopic expression of HRAS in gastric carcinoma cells accelerated proliferation, migration, invasion, angiogenesis, and clone formation ability of gastric carcinoma cells in vitro. Furthermore, HRAS over-expressing significantly promoted the tumorigenicity of gastric carcinoma cells in vivo whereas silencing endogenous HRAS caused opposite outcomes. Moreover, we demonstrated that HRAS enhanced gastric carcinoma aggressiveness by activating VEGFA/PI3K/AKT pathway and Raf-1 signaling. Together, our results provide new evidence that HRAS overexpression promotes the progression of gastric carcinoma and might represent a novel therapeutic target for its treatment.
Keywords: HRAS, gastric carcinoma, growth, angiogenesis
Introduction
Gastric carcinoma remains one of the most common malignant solid cancer and a worldwide major public health concern [1]. Gastric carcinogenesis is a complex phenomenon involving multiple epigenetic and genetic factors; several genetic, environmental and infectious agents interact causing a cumulative effect in the early steps of gastric carcinogenesis [2]. Despite improvement in surgical morbidity and mortality, as well as significant advancement of chemotherapy and radiotherapy options, its incidence and the survival for gastric cancer patient has not significantly improved over the past decades [3]. Therefore, it is of great clinical value to explore the molecular mechanisms in development of gastric carcinoma and identify effective treatment strategies to improve the survival rate for gastric carcinoma patients [4].
There is mounting evidences suggest that poor therapeutic efficacy and the dismal overall survival rate of gastric carcinoma patients are associated with aberrantly activated signaling pathways. RAS proteins (HRAS, KRAS and NRAS) are small GTPases that cycle between inactive guanosine diphosphate (GDP)-bound and active guanosine triphosphate (GTP)-bound conformations [5]. Dysregulation of this pathway is frequently observed in several cancers including gastric cancer. The RAS signaling pathway is activated by several cellular stimuli regulating various physiological functions such as cell growth, cell survival, cell cycle progression, protein translation, and metabolism [6]. RAS activity regulates a complex signaling network including the RAF-MEK-ERK cascade, the phosphatidylinositol 3-kinase (PI3K) pathway and the effector family of exchange factors for the RAL small GTPases. Through the combined action of these signaling pathways, expression of activated mutant RAS is thought to promote several of the characteristics of malignant transformation [7]. The KRAS oncogene is one of the most frequently mutated genes in human cancer, being altered in approximately 20% of all human tumors. Oncogenic forms of mutant KRAS is arguably the most studied oncogene [8]. Therefore, clearly there is a lot known about this molecule. NRAS mutations are found in various malignancies including melanoma (20%), adenocarcinoma of the lung (1%), neuroblastoma (0.83%) and cutaneous T-cell lymphoma (4%) [9]. NRAS mutations sensitize towards inhibition of MEK in cutaneous T-cell lymphoma, lung cancer and neuroblastoma cell lines. Mutations of NRAS are found at typical hotspots including codon 12, 13 and 61 which results in G12C/S, G13R/V and Q61R/L mutations [10]. These mutations block GTPase activity and lock the RAS isoforms in continuous activation in which they signal to downstream effectors such as MEK and ERK. Several studies could show that mutant NRAS activates the PI3K/mechanistic target of rapamycin (mTOR)-signaling cascade in melanoma and lung cancer [11]. Direct targeting of mutant NRAS by farnesylation inhibitors have failed but blocking downstream MEK kinase by MEK kinase inhibitors was successful in a preclinical setting.
Nevertheless, recent observations have shed a new light on the mechanisms involved in HRAS mediated oncogenesis that emphasize the importance of HRAS activity. Among the three RAS genes (H-, K-, and N-RAS), HRAS is commonly mutated in tumors originated from stratified epithelial tissues including squamous cell carcinoma in the skin, head, and neck cancer as well as bladder cancer [12]. Histological subtypes could play a role as a report described a high frequency of HRAS mutations in inverted urothelial papilloma (IUP) -an uncommon neoplasm of the urinary bladder with distinct morphologic features. In addition, HRAS mutations seem to be more frequent in squamous cell cancer of the lung (2.8%) than in adenocarcinoma of the lung (1%) [13]. Experimental and genomic sequencing studies have revealed that the vast majority of RAS mutations are missense, point mutations at amino acid residues glycine 12 (G12), glycine 13 (G13), or glutamine 61 (Q61) [14]. Wild-Type HRAS suppresses the earliest stages of tumorigenesis in a genetically engineered mouse model of pancreatic cancer. These promising results establish a role of HRAS in tumors. HRAS pathway would provide a promising target for cancer therapy is currently under preclinical and early preclinical investigation. Further, little is known about signaling of oncogenic HRAS in gastric carcinoma.
In the present study, we reported that HRAS expression was significantly up-regulated in gastric carcinoma, and was associated with the clinical prognosis of gastric cancer. Overexpression of HRAS promoted the proliferation, metastasis and angiogenesis of gastric carcinoma cells, whereas silencing endogenous HRAS caused an opposite outcome. Our findings suggest that HRAS plays critical oncogenic role in human gastric carcinoma progression and highlights its potential as a target for patient therapy.
Materials and methods
Cell lines
Human gastric carcinoma cell lines BGC-823, SGC7901, MGC-803, MKN-28, and MKN-45 were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). All cancerous cell lines were grown in RPMI-1640 or DMEM (Thermo Scientific, MA, USA) supplemented with 10% FBS (Hyclone, Rockford, IL, USA) and 100 units/ml penicillin and streptomycin. Non-tumorigenic gastric epithelial cells GES-1 used as control were obtained from the Chinese Academy of Sciences Cell Bank of Type Culture Collection (CBTCCCAS, Shanghai, China). All cell lines were maintained in a humidified incubator with 5% CO2 at 37°C.
Vectors, retroviral infection and transfection
A HRAS expression construct was generated by subcloning PCR-amplied full-length human HRAS cDNA into the pMSCV retrovirus plasmid. Stable cell lines expressing HRAS were selected for 10 days with 0.5 μg/ml puromycin 48 h after infection. HRAS expression in MKN-28 cells was knocked down using siRNA. The HRAS and non-targeted RNA were obtained from GenScript (Nanjing, China). The sequence of HRAS siRNA was: 5’-GUAUGAGUAUGACUUUGAAUU-3’. The siRNAs were transfected to cells according to the manufacturer’s instruction. The expression levels of HRAS were examined using a western blot after 48 h. The growth rates were examined daily using an 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay for 3 days. Transfection of siRNA or plasmids was performed using the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA) according to the manufacturer’s instruction.
Oncomine analysis
The expression level of HRAS genes in the selected cancers was analyzed using Oncomine (https://www.oncomine.org). For this, we compared clinical specimens of gastric cancer vs. normal patient datasets. In order to reduce our false discovery rate, we selected P < 0.01 as a threshold [15].
Cell viability assay
Briefly, cells (3 × 104 cells per well) were seeded in 96-well plates. After 48 hours. Cell viability was measured by 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2-H-tetrazolium bromide (MTT) assay and the optical density (OD) was then measured at 450 nm using a Spectra MAX M5 micro-plate spectrophotometer (Molecular Devices, Sunnyvale, CA, USA). Three independent experiments with triplicate were carried out.
Wound healing assay
We examined the migration of cells using a wound-healing assay. Briefly, cells were each grown on 6-well plates. After the growing cell layers had reached confluence, we inflicted a uniform wound in each plate using a pipette tip, and washed the wounded layers with phosphate buffer saline (PBS) to remove all cell debris. We evaluated the closure at 48 h using bright-field microscopy [16]. The relative wound area was obtained by quantitatively analyzing the areas in the scratch overlapped by cells using Image Pro-plus (IPP) software.
Invasion assay
Cells were then seeded on the upper chamber of Boyden (8 μm; BD Biosciences) and allowed to invasive to the lower chamber. After 6 hours incubation, noninvasive cells were removed with cotton swabs, and invasive cells were fixed with cold 4% paraformaldehyde and stained with 1% crystal violet. Images were taken using a ZEISS digital microscope and invading cells were counted by manual counting in random 5 fields [17]. Three independent experiments with triplicate were carried out.
Colony formation assay
Cells were digested in 0.25% trypsin to reconstitute the single-cell suspension at a density of 1 × 105 cells per ml. Cells then were plated onto a 6-well tissue culture plate in complete medium and incubated at 37°C. Cells were allowed to grow in complete medium at 37°C and at an atmosphere of 5% CO2 for 14 days. The supernatants were discarded, and cells were rinsed in PBS for twice and fixed in methanol for 10 min. Then cells were fixed and stained with 1% crystal violet and allowed to air dry at room temperature. The experiments were triplicated and the numbers of colonies containing more than 50 cells were microscopically counted to calculate the colony formation rate as number of colonies.
Chicken chorioallantoic membrane (CAM) assay
CAM assay was performed at day 8 of fertilized chicken eggs using a method previously described. A 1.0-cm diameter window was opened on the egg shell (Hongwei Chicken farm, Luhe District Nanjing, China). The surface of the dermic sheet on the floor of the air sac was removed to expose the CAM. A 0.5-cm diameter filter paper was first placed on top of the CAM, and 100 μl conditioned medium from HRAS over-expression cells or cells HRAS siRNA was added onto the center of the paper. After the window was closed with sterile adhesive tape, the eggs were incubated at 37°C under 80-90% relative humidity for 4 days [18]. Following fixation with stationary solution (methanol:acetone = 1:1) for 15 min, the CAMs were cut and harvested, and gross photos of each CAM were taken with ZEISS digital microscope.
ELISA
Cells (7 × 105) were plated in six-well dishes. Supernatants were collected and ELISA for VEGFA was performed with a Quantikine immunoassay kit (DVE00; R&D Systems) following the manufacturer’s instructions.
Western blotting analysis
Proteins were resolved by SDS-PAGE and transferred to Immobilon-P-membrane (Millipore, Billerica, MA). After blocking, the membrane was incubated with primary antibodies against HRAS (Abcam, USA; 1:1000), p-PI3K p55Tyr199, PI3K, p-AKTSer473, AKT, p-GSK-3βSer9, GSK-3β, p-NF-κB p65Ser536, NF-κB p65, p-Raf-1Ser289, Raf-1, p-ERK1/2Thr202/Tyr204, ERK1/2, p-MEK1/2Ser221, MEK1/2, p-p38 MAPKThr180/Tyr182, p38 MAPK, and VEGFA antibodies (Cell Signaling, Danvers, MA, USA). After washing, the membranes were incubated with HRP-labeled Goat Anti-Rabbit IgG (Abcam, USA; 1:1000) and signals were detected using the ECL Western blot detection system. β-Tublin antibody (Sigma, Saint Louis, MI) was used as the protein loading control.
Real-time PCR
Total RNA was isolated using TRIzol according to the manufacturer’s instructions (Invitrogen, USA) and the concentration of total RNA was detected by spectrophotometry at OD260. Reverse transcription (RT) was carried out using superscript III reverse transcriptase (Invitrogen, USA) as described in the manufacturer’s manual. The real-time PCR was performed on ABI Prism 7500 Sequence detection system (Applied Biosystems, CA) with the KAPA SYBR® qPCR Kit (KAPA Biosystems, USA) according to the manufacturer’s instructions. The primers used were as follow: HRAS sense, 5’-CCCTTGGGTGTCAAAGGTAAA-3’ and antisense, 5’-AAACTGATGCGTGAAGTGCTG-3’; β-actin sense, 5’-GCGAGCACAGAGCCTCGCCTTTG-3’ and antisense, 5’-GATGCCGTGCTCGATGGGGTAC-3’. The target mRNA level of control cells normalized to the level of β-actin mRNA, was defined as 1. Results were obtained from three independent experiments.
Immunocytochemistry
After MKN-28 cells on glass coverslips were treated by indicated agents, they were fixed by pre-cold acetone, and then rinsed three times with PBS. The cells were permeabilized in 0.1% Triton X-100 and incubated with 1% BSA/PBS to block nonspecific binding. Subsequently, the cells were immunostained by incubating with rabbit monoclonal antibody against VEGFA (diluted 1:500, Epitomics) overnight at 4°C. After being washed with PBS, cells were incubated with FITC-conjugated goat anti-rabbit secondary antibody (diluted 1:60, Boster Biotechnology, Wuhan, China). Nuclei were counterstained with Hoechst 33258 (Biotime Biotech, Haimen, China). Images were taken and analyzed using the ZEN 2011 imaging software on a Zeiss invert microscope (CarlZeiss, Hallbergnoos, Germany) under 400-fold magnification.
Cell immunohistochemistry
MKN28 for immunohistochemistry were grown to 60-80% confluence on glass coverslips. Subsequently, cells were washed three times with PBS and permeabilized in 0.1% Triton X-100 for 10 min. Cells were blocked for 1 h in 5% IgG and protease-free BSA (Jackson ImmunoResearch) at room temperature and after another three washes incubated overnight at 4°C in primary antibody supplemented with 0.5% BSA. Unless otherwise stated, VEGFA was stained with primary antibody (1:500, Invitrogen). The next day, cells were rinsed three times in PBS and then incubated with the secondary antibody for 1 h at room temperature before being washed another three times. Images were taken using DAB chromogenic kit and analyzed using the ZEN 2011 imaging software on a Zeiss invert microscope (CarlZeiss, Hallbergnoos, Germany).
Xenografted tumor model, IHC, and H&E staining
BALB/c-nu mice (4-5 weeks of age, 18-20 g) were purchased from the Center of Experimental Animal of Nanjing University of Chinese Medicine. All experimental procedures were approved by the Institutional Animal Care and Use Committee of Nanjing University of Chinese Medicine. The BALB/c nude mice were randomly divided into two groups (n = 6/group). One group of mice was inoculated subcutaneously with MKN28/RNAi-vector cells (5 × 106) in the left dorsal flank and with MKN-28/HRAS-RNAi cells (5 × 106) in the right dorsal flank. Tumors were examined twice weekly; length and width measurements were obtained with calipers and tumor volumes were calculated using the equation (L*W2)/2. On day 25, animals were euthanized, tumors were excised, weighed and paraffin-embedded [19]. Serial 6.0 μm sections were cut and subjected to IHC analyzed using an anti-CD31, anti-VEGFA, anti-PI3K and anti-AKT antibodies.
TUNEL staining
To detect apoptotic cells, TUNEL staining was performed using an In situ Apoptosis Detection Kit (Merck Millipore) according to the manufacturer’s protocol. The mice were sacrificed and tumor samples were collected and sectioned. DNA fragments were determined by terminal deoxynucleotidyl transferase mediated dUTP nick end labeling (TUNEL) assay.
Statistical analysis
Statistical tests for data analysis included Fisher’s exact test, log-rank test, Chi-square test, and Student’s 2-tailed t test. Multivariate statistical analysis was performed using a Cox regression model. Statistical analyses were performed using the SPSS 11.0 statistical software package. Data represent mean ± SD. P < 0.05 was considered statistically significant.
Results
Over-expression of HRAS correlates with gastric cancer poor prognosis
By analyzing the published mRNA expression profiles obtained from 22 gastric carcinoma tissues and 8 normal tissues (NCBI/GEO/GSE2685), we found that HRAS was markedly up-regulated in gastric cancer tissues compared with normal tissues (Figure 1A). Consistently, real-time PCR and western blotting assays revealed that HRAS was significantly overexpressed in gastric cancer cell lines at both protein and mRNA levels, compared with the normal gastric cells GES-1 (Figure 1B and 1C). Oncomine analysis of neoplastic vs. normal tissue from TCGA dataset [20] proved that HRAS was significantly overexpression in gastric cancer (Figure 1D). Furthermore, Kaplan-Meier analysis (http://kmplot.com) revealed that in overall cancer high levels of expression of HRAS correlate with low survival rate (Figure 1E). These studies emphasized the importance the HRAS gene expression during gastric cancer progression.
Figure 1.
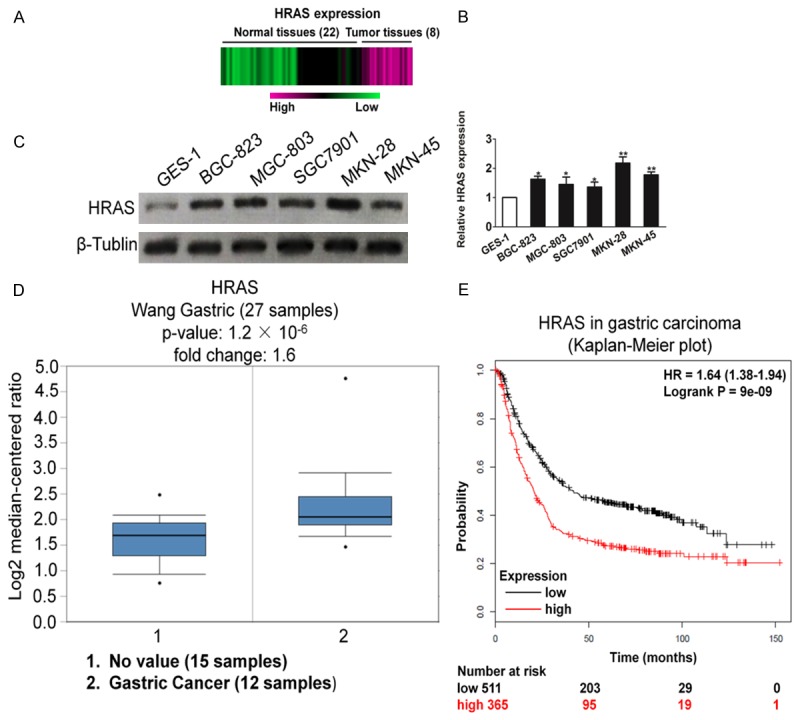
Overexpression of HRAS correlates with poor prognosis of gastric carcinoma. A. Expression profiling of mRNAs showing that HRAS was up-regulated in gastric carcinoma tissues compared to normal tissues (n = 30). B. Real-time PCR analysis of HRAS mRNA in one immortalized cell line and five gastric carcinoma cell lines. C. Western blotting analysis of HRAS expression in one immortalized cell line GSE-1 and five gastric carcinoma cell lines, including BGC-823, SGC7901, MGC-803, MKN-28, and MKN-45. D. Box plots derived from gene expression data in Oncomine comparing expression of a specific HRAS gene in normal (left plot) and gastric carcinoma tissue (right plot). E. Kaplan-Meier plots shown overall survival in gastric carcinoma. In red: patients with expression above the median and in black, patients with expressions below the median. HRAS, P = 9 × 10-9.
Up-regulation of HRAS promotes the aggressiveness of gastric cancer cells
To investigate the biological role of HRAS up-regulation in gastric carcinoma progression, MKN28 cell line that stably expressed HRAS were established. The transfection efficiency was confirmed by the expression of green fluorescence protein (GFP) (Figure 2A). We future confirmed the overexpression of HRAS by western blot with HRAS antibody (Figure 2B) and PCR (Figure 2C). Uncontrolled, unlimited and accelerated multiplication and colony-formation ability are the most fundamental biologic behaviors of cancer cells. We found that ectopic expression of HRAS in gastric cancer cells markedly increased the growth rate (Figure 2D). To investigate what role, if any, HRAS might play in the non-tumorigenic gastric epithelial cells GES-1, the proliferation and clone formation ability in HRAS-expressing GES-1 cells were assessed by MTT and colony assay, respectively. As shown in Supplementary Figure 3B and 3C, overexpression of HRAS in GES-1 cells led to higher proliferation rate and stronger colony forming ability compared with that of control cells, which suggest the tumorigenic of HRAS in non-tumorigenic gastric epithelial cells GES-1. The ability to grow in an anchorage-independent manner is one of the fundamental properties of tumor cells and is a key for metastasis. We found that HRAS overexpression augmented the anchorage-independent growth ability of MKN28 cells (Figure 2E). Furthermore, the migration (Figure 2F) and invasive (Figure 2G) ability of MKN28 cells was significantly increased in the gastric cancer cells that overexpressed HRAS, which indicating its potential role in tumor aggressiveness. PI3K and Raf-1 signaling pathways are involved in cell migration and invasion. After demonstrating the HRAS function in the aggressiveness of gastric cancer cells, we inquired whether PI3K and Raf-1 signaling pathway activity are associated with HRAS over-expression. As shown in Figure 2H, HRAS overexpression indeed facilitated the activation of PI3K and Raf-1 signaling pathway in gastric cancer MKN28 cells. Collectively, these results suggest that HRAS up-regulation promotes the aggressiveness of gastric cancer cells in vitro.
Figure 2.

Up-regulation of HRAS expression promotes cell aggressiveness in vitro. A. HRAS was cloned into vector and transfected into cell. The cells transfected with an empty vector were used as control. The transfection efficiency was evaluated by the expression of GFP. B. MKN28 cells stably expressed HRAS or transfected with vector. After for 10 d post transfections, cells were subjected to western blot for measuring protein level of HRAS. C. Cells were treated as above and then total RNAs were extracted. HRAS mRNA levels were determined by means of quantitative real-time PCR and normalized to the level of β-actin mRNA. The fold changes of mRNA expression of indicated genes were compared as a ratio to the vehicle control. The data are shown as mean ± SD of triplicates experiments. **P < 0.01 compared with the control group. D. Overexpression of HRAS promoted MKN28 cell proliferation. Cells were treated as above and cell viability was determined by MTT assay. E. Representative results of the colony numbers of MKN28 cells. Number of multicellular colonies was increased by HRAS over-expression. Colonies with > 50 cells per colony were counted. The average number of established colonies per field was presented as mean ± SD (n = 5 fields). **P < 0.01, versus control. F. Wound healing assay was performed to determine the metastatic potential of cells overexpression HRAS. The percentage of wound closure was quantified. G. Representative pictures and quantification of invaded cells were analyzed using a Transwell invasion assay. H. Cells were transfected with HRAS and then were subjected to western blot for measuring protein levels of phosphor-PI3K, total-PI3K, phosphor-AKT, total-AKT, phosphor-mTOR, total-mTOR, phosphor-GSK-3β, total-GSK-3β, phosphor-NF-κB p65, total-NF-κB p65, phosphor-Raf-1, total-Raf-1, phosphor-ERK1/2, total-ERK1/2, phosphor-MEK1/2, total-MEK1/2, phosphor-p38 MAPK and total-p38 MAPK respectively.
Over-expression HRAS facilitates tumor angiogenesis and VEGF-A expression
Tumor growth is angiogenesis dependent, and inhibit breast cancer angiogenesis may aid the development of more effective therapeutic strategies for combating tumor. To investigate the effects of HRAS on tumor angiogenesis, chick embryo chorioallantoic membrane (CAM) assay was performed to detect the ability of MKN28 cells that overexpressed HRAS to induced chorioallantoic membrane (CAM) neovascularization (Figure 3A). The conditioned medium from MKN28-HRAS overexpression cells clearly increased new blood vessel formation in the CAM model. Vascular endothelial growth factor (VEGF)-A has been identified as the predominant tumor angiogenesis factor and well-studied molecular factor in the majority of human and experimental murine cancers, acting via VEGF receptor (VEGFR)-1 and VEGFR-2. To further clarify the role of HRAS in the tumor angiogenesis, we set out to determine whether HRAS over-expression was correlated with VEGFA. As indicated in Figure 3B, the expression of VEGFA was greatly increased in MKN28 cells overexpressing HRAS. We also performed western blot and ELISA assays to evaluate the expression of VEGFA (Figure 3C) and its secretion (Figure 3D) in MKN28 cells. Consistently, VEGFA level in MKN28-HRAS overexpression cells was significantly increased compared with control cells. These observations demonstrate that HRAS overexpression effectively promotes the formation of blood vessels in the CAM of the chick embryo models and VEGFA expression in gastric cancer cells.
Figure 3.
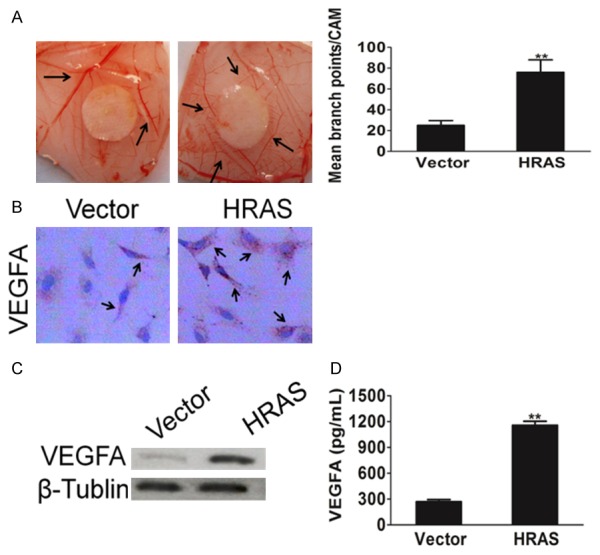
HRAS facilities tumor cells induced angiogenesis. A. Representative Images of CAM blood vessels stimulated with conditioned medium from MKN28 cells. B. A representative cell immunohistochemistry assay shown VEGFA protein expression in control cells and MKN28 cells overexpression HRAS. C. Western blot shown that VEGFA was elevated in cells transfected with vector or HRAS retrovirus plasmid. β-Tublin was used as a loading control. D. MKN28 cells grown to 70-90% confluence were co-transfected with vector or HRAS plasmid. Cell culture supernatants from indicated cells were performed by ELISA assay for assay VEGFA mount. Each bar represents the mean ± SD of three independent experiments. *P < 0.05, **P < 0.01.
Down-regulation of HRAS suppresses the aggressiveness of gastric cancer cells
To confirm the biological role of HRAS in gastric carcinoma progression, MKN28 cells that stably knocked down HRAS were established (Supplementary Figure 1). The down-regulation of HRAS in MKN28 cell line dramatically inhibited the proliferative capacity of gastric cancer cells (Supplementary Figure 2). Additionally, down-regulation of HRAS decreased MKN28 cells colony formation number (Figure 4A), as well as repressed the ability of gastric cancer cells invasion (Figure 4B). Moreover, HRAS siRNA attenuated PI3K and Raf-1 signaling pathway activity in MKN28 cells (Figure 4C), which demonstrated the role of PI3K-AKT and Raf-1 signaling in HRAS-mediated cell aggressiveness. Altogether, our data suggest that down-regulation of HRAS inhibited gastric cancer aggressiveness in vitro. Furthermore, the ability of MKN28 cells to induced CAM neovascularization was dramatically inhibited after MKN28 cells knock-down HRAS (Figure 4D). We also measured VEGFA expression in MKN28 cells after HRAS siRNA by immunofluorescence assay. As shown in Figure 4E, the expression of VEGFA was significantly inhibited in MKN28 cells down-expressing HRAS. Consistently, when HRAS was silenced by siRNA in MKN28 cells, VEGFA protein expression was down regulation (Figure 4F) in MKN28 cells. VEGFA secretion mount were also decreased in MKN28 cells culture medium upon transfection with HRAS siRNA plasima (Figure 4G). Collectively, these results suggest that HRAS down-regulation attenuated the aggressiveness of gastric cancer cells and angiogenesis in vitro.
Figure 4.
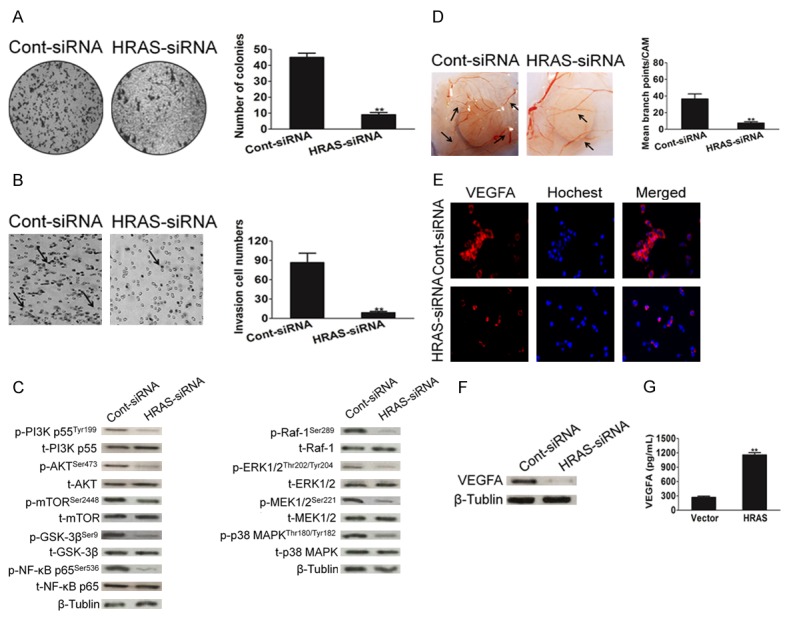
Down-regulation of HRAS suppresses the aggressiveness of MKN28 cells. A. Representative pictures of (left panel) and quantification (right panel) of colony numbers of indicated cells as determined by an anchorage-independent growth assay. All data were expressed as mean ± SD. **P < 0.01, versus control. B. Effect of HRAS on MKN28 cells invasiveness performed by Transwell invasion analysis. All data were expressed as mean ± SD (n = 5 fields). **P < 0.01, versus control. C. MKN28 cells were transfected with control siRNA or HRAS siRNA plasmid. After for 48 h transfections, cells were subjected to western blot for measuring protein level of phosphor-PI3K, total-PI3K, phosphor-AKT, total-AKT, phosphor-mTOR, total-mTOR, phosphor-GSK-3β, total-GSK-3β, phosphor-NF-κB p65, total-NF-κB p65, phosphor-Raf-1, total-Raf-1, phosphor-ERK1/2, total-ERK1/2, phosphor-MEK1/2, total-MEK1/2, phosphor-p38 MAPK and total-p38 MAPK respectively. D. Angiogenesis assay by chorioallantoic membrane (CAM) model as described in Methods. Representative images of CAM blood vessels stimulated with conditioned medium from MKN28 cells. The data represented as mean ± SD of blood vessel numbers were normalized to those of the control. E. MKN28 cells were transfected with control siRNA or HRAS siRNA. Then cells were fixed and incubated with primary antibodies against VEGFA. MKN28 cells were immunostained with anti-rabbit FITC-conjugated secondary antibody and then stained with Hoechst 33258. The specimens were visualized and photographed using a fluorescence microscopeand VEGFA expression was detected by immunofluorescence assay. F. Western blot shown that VEGFA protein expression was inhibited in cells transfected with HRAS siRNA plasmid. G. MKN28 cells were transfected with siRNA control or HRAS siRNA plasmid and VEGFA in culture medium was detected by ELISA. Each bar represents the mean ± SD of three independent experiments. For indicated comparisons, **P < 0.01.
Down-expression of HRAS inhibits gastric cancer progression in vivo
In order to test whether HRAS attenuates progression of gastric cancer in vivo, we engineered MKN28 cells to HRAS down-expression, which were subsequently implanted into immunodeficient mice, and tumor sizes were measured once every three days. At the end of the experiment, the average tumor volume of the HRAS-siRNA group was significantly lower compared with that of the Cont-siRNA cells-injected control group (Figure 5A and 5B). Interestingly, no difference was detected in body weight between the HRAS-siRNA and v Cont-siRNA (Figure 5C). Consist with in vitro studies, the immunohistochemistry results showed that the proliferation of gastric cancer cells was inhibited by HRAS-siRNA as there was less and weak expression for Ki67 in HRAS-siRNA group compared with that in control group (Figure 5D). Furthermore, numbers of TUNEL-positive cells (apoptotic cell death) were greater in the tumor tissues from HRAS-siRNA injected mice than in those of control mice (Figure 5D). To further confirm the role of HRAS in gastric cancer in vivo, MKN28 cells that stably express HRAS were subsequently implanted into immunodeficient mice. As shown in Supplementary Figure 4, tumors formed by HRAS-overexpressing cells exhibited a greater size than tumors formed by the control cells. Additionally, further examination of tumorigenic of up-regulation of HRAS by non-tumorigenic gastric epithelial cells GES-1 showed that HRAS also promoted the tumorigenicity of GES-1 cells in vivo, which further demonstrated HRAS was an oncogenic gene (Supplementary Figure 3D).
Figure 5.
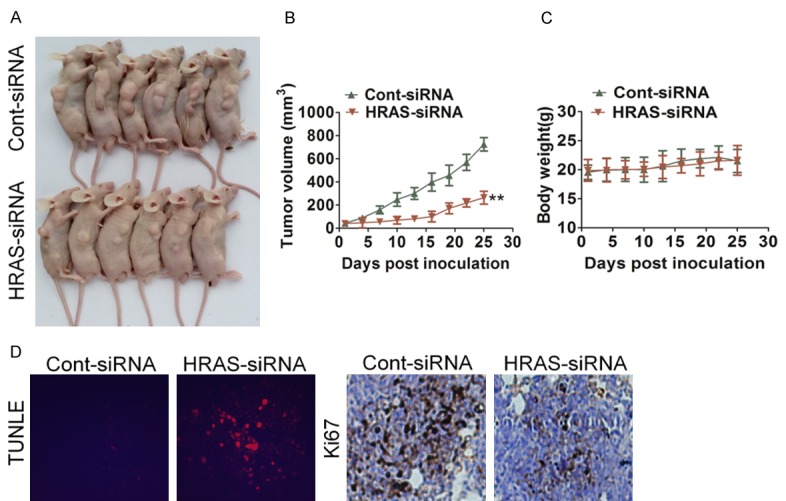
Down-expression of HRAS inhibits gastric cancer progression in vivo. A. Effect of HRAS on the growth of MKN28 cells inoculated into nude mice. BALB/c-nu mice were subcutaneously injected with MKN28/RNAi-vector or MKN28/HRAS-RNAi cells. Tumor volume and weight were monitored over time as indicated, and the tumor was excised after 25 days. HRAS down-expression causes a decrease in tumor volume. B. Tumor growth curve upon implantation. For indicated comparisons, **P < 0.01. C. Body weight changes in mice subcutaneously injected with MKN28/RNAi-vector or MKN28/HRAS-RNAi cells. There was no significant difference in body weight between RNAi-vector and HRAS-RNAi group. D. Tumor sections were analyzed by immunohistochemistry for detection of Ki67 expression in each group of nude mice. Apoptotic cells were examined by TUNEL staining. Each image was representative of six independent mice.
Down-expression of HRAS inhibits gastric cancer VEGFA/PI3K/AKT and Raf-1 signaling pathway in vivo
Tumor angiogenesis is recognized as a process that is critical to tumor growth. To delineate the signaling molecules involved in HRAS regulation of tumor growth and in support of the above results, which HRAS facilitated tumor angiogenesis and regulated VEGFA expression, we examined the effect of HRAS down-expression on the tumor induced-angiogenesis in xenografts by immunohistochemistry analysis. Immunohistochemistry staining revealed that the expression levels of CD31 and VEGFA were significantly repressed by HRAS-siRNA in gastric cancer tissues (Figure 6A). In agreement with in vitro studies, VEGFA downstream pathway proteins, such as p-PI3K and p-AKT were significantly suppressed after HRAS siRNA in gastric cancer tissues (Figure 6A). To further decipher the underlying mechanism of HRAS-siRNA inhibited proliferation and progression in gastric cancer growth in vivo; the HRAS down-regulation signaling pathway was then assayed by western blot assay. As would be expected, the phosphorylation level of PI3K (p-PI3K), AKT (p-AKT), mTOR (p-mTOR), GSK-3β (p-GSK-3β), p-NF-κB p65 (p-NF-κB p65), as well as Raf-1 signaling activity, including phosphor-ERK1/2, total-ERK1/2, phosphor-MEK1/2, total-MEK1/2, phosphor-p38 MAPK and total-p38 MAPK in the HRAS-siRNA-treated group was significantly increased compared to the control group (Figure 6B). Taken together, our findings indicate that HRAS-down-regulation inhibits gastric cancer growth and angiogenesis in vivo through regulation PI3K-AKT and Raf-1 signaling molecules.
Figure 6.
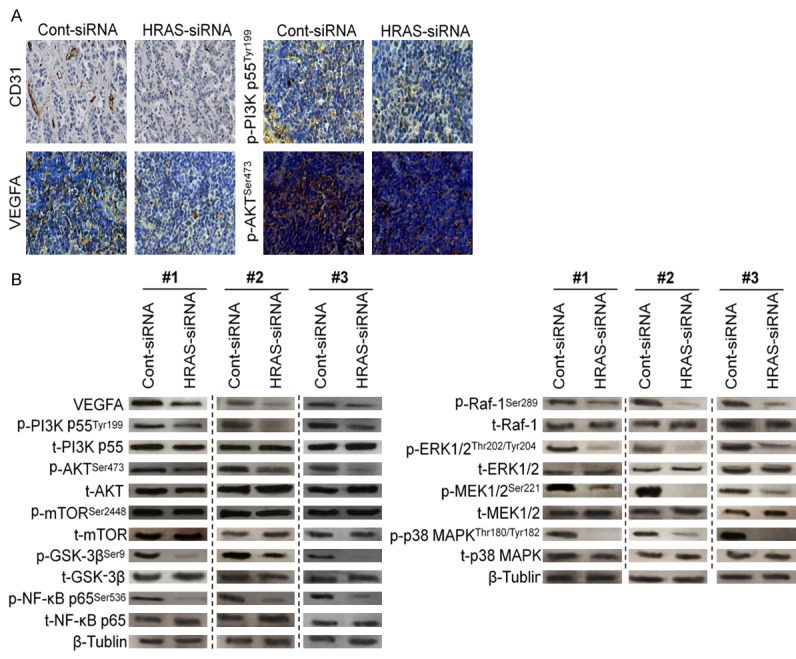
HRAS siRNA inhibits gastric cancer induced angiogenesis in vivo. A. The expression levels of tumor induced angiogenesis markers including CD31 and VEGFA from the tumor tissues of MKN28/HRAS-siRNA group were lower than that of control group by immunohistochemistry assay. B. The proteins were extracted from tumor xenografts and were subjected to western blot for measuring phosphor-PI3K, total-PI3K, phosphor-AKT, total-AKT, phosphor-mTOR, total-mTOR, phosphor-GSK-3β, total-GSK-3β, phosphor-NF-κB p65, total-NF-κB p65, phosphor-Raf-1, total-Raf-1, phosphor-ERK1/2, total-ERK1/2, phosphor-MEK1/2, total-MEK1/2, phosphor-p38 MAPK and total-p38 MAPK, respectively.
Discussion
Gastric cancer is a common disease with limited treatment options and a poor prognosis. Many gastric cancers harbor potentially actionable targets, including over-expression and mutations in tyrosine kinase pathways [21]. Herein, we found that HRAS was up-regulated in gastric cancer and that HRAS overexpression promoted gastric cancer aggressiveness both in vitro and in vivo, which is in agreement with oncogenic-effect of other RAS family members. These findings provide novel insights into the potential roles of HRAS deregulation in promoting carcinogenesis and progression of gastric cancer.
HRAS is a small G protein in the RAS subfamily of the RAS superfamily of small GTPases. Once bound to Guanosine triphosphate, H-RAS will activate a RAF kinase like c-RAF, the next step in the MAPK/ERK pathway [22]. HRAS acts as a molecular on/off switch; once it is turned on it recruits and activates proteins necessary for the propagation of the receptor’s signal, such as c-RAF and PI3-kinase. HRAS binds to GTP in the active state and possesses an intrinsic enzymatic activity that cleaves the terminal phosphate of this nucleotide converting it to GDP. Upon conversion of GTP to GDP, HRAS is turned off [23]. HRAS has been shown to be a proto-oncogene. The altered HRAS protein is permanently activated within the cell. This overactive protein directs the cell to grow and divide in the absence of outside signals, leading to uncontrolled cell division and the formation of a tumor. Recent studies suggest that HRAS deregulations are common in thyroid, salivary duct carcinoma, epithelial-myoepithelial carcinoma, and kidney cancers [24]. The HRAS protein also may be produced at higher levels (overexpressed) in other types of cancer cells. Additionally, NRAS was also significantly up-regulated in human prostate cancers and found to play an important role in prostate cancer progression [25]. Furthermore, mutations in the HRAS gene also have been associated with the progression of bladder cancer and an increased risk of tumor recurrence after treatment. Somatic mutations in the HRAS gene are probably involved in the development of several other types of cancer. These mutations lead to an HRAS protein that is always active and can direct cells to grow and divide without control. Therapeutic targeting of the RAS pathway has been aggressively pursued for the treatment of a wide range of malignant tumor, including melanoma.
The NRAS has been reported to be fundamentally involved in many tumor processes, such as tumor cell proliferation, apoptosis, angiogenesis, and invasion. In general, there is little data on the regulation of HRAS gene expression, and molecular mechanism in gastric carcinoma [26]. Herein, we found that HRAS was over-expressed in several gastric cancer cell lines and furthermore, according to The Cancer Genome Atlas (TCGA; http://cancergenome.nih.gov/), we found that HRAS amplification was positive in 12 of 27 cases, suggesting that the over-expression of HRAS in gastric cancer might be associated with genomic amplification. Our analysis of Kaplan-Meier Plotter also found correlations between HRAS gene deregulation and survival rates. Overall, having higher levels of HRAS gene expression led to lower survival amongst patients with gastric cancer patients. Tumor progression, for instance, metastasis is a complicate process. The cancer cell has first to acquire the ability to invade into the adjacent organs and/or to migrate through bloodstream or lymphatic system to distant sites and to survive. Ectopic expression of HRAS in gastric cancer cells markedly increased the proliferation and anchorage-independent growth ability. Tumor progressions always require increase in number of blood vessels and similarly decrease in number of blood vessels in milieu leads to dormancy of tumor. Angiogenic pathway is a sound target to obstruct the excessive proliferation of cells because the nutrients and growth factors are supplied through blood vessels to the tumor cells [27]. Over-expression of HRAS augmented the anchorage-independent growth and the invasive abilities of gastric cancer cells, provoked their ability to induce CAM neovascularization and enhanced their resistance to apoptosis. RNA interference (RNAi) is a powerful method for gene inactivation and cancer gene therapy [28]. Silencing HRAS significantly both inhibited the malignant behavior of the tumor cells and repressed the expression of VEGFA, PI3K, AKT that are specifically regulate proliferation, apoptosis, metastasis and angiogenesis in gastric carcinoma, suggesting that HRAS could contribute to PI3K-AKT signaling activation and thereby represent a novel target for gastric cancer treatment.
In summary, we reported that HRAS was markedly up-regulated in gastric carcinoma and that a positive correlation existed between HRAS expression and the prognosis of gastric cancer patients. Overexpression of HRAS augmented gastric carcinoma aggressiveness in vitro and in vivo and activated the PI3K-AKT and Raf-1 signaling pathway. Therefore, understanding the biological function of HRAS in gastric carcinoma progression both advance our knowledge of the mechanisms that underlie gastric carcinoma aggressiveness, and establish HRAS as a potential therapeutic target for the treatment of gastric carcinoma.
Acknowledgements
National science foundation of China (81402523 to WXY, 81373990 to WXY, 81201797 to LG), Jiangsu Province “Six talents” high peak plan (2015-WSN-052 to WXY), Jiangsu Province TCM Project (JD201510 to WXY YXQ), Jiangsu Province “Six adults just” high peak (2009-D-63 to LG), Youth Projects of Jiangsu Provincial Health Department (H201065 to LG), the Natural Science Foundation of Jiangsu Province (BK2009445 to LG).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Zhang H, Zhan Z, Cui M, Gao Y, Wang D, Feng Y. Hydroxyflavanone inhibits gastric carcinoma MGC-803 cell proliferation. Int J Clin Exp Med. 2015;8:16955–16959. [PMC free article] [PubMed] [Google Scholar]
- 2.Chang Q, Zhang L, He C, Zhang B, Zhang J, Liu B, Zeng N, Zhu Z. HOXB9 induction of mesenchymal-to-epithelial transition in gastric carcinoma is negatively regulated by its hexapeptide motif. Oncotarget. 2015;6:42838–42853. doi: 10.18632/oncotarget.5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ju J, Wang N, Wang X, Chen F. A novel all-trans retinoic acid derivative inhibits proliferation and induces differentiation of human gastric carcinoma xenografts via up-regulating retinoic acid receptor beta. Am J Transl Res. 2015;7:856–865. [PMC free article] [PubMed] [Google Scholar]
- 4.Tang J, Wang X, Wang T, Chen F, Zhou J. In vivo pharmacokinetics, biodistribution and antitumor effect of amphiphilic poly(L-amino acids) micelles loaded with a novel all-trans retinoic acid derivative. Eur J Pharm Sci. 2014;51:157–164. doi: 10.1016/j.ejps.2013.09.016. [DOI] [PubMed] [Google Scholar]
- 5.Li Z, Li D, Zhang G, Xiong J, Jie Z, Cheng H, Cao Y, Jiang M, Lin L, Le Z, Tan S, Zou W, Gong B, Lin S, Yang K. Methylation-associated silencing of MicroRNA-335 contributes tumor cell invasion and migration by interacting with RASA1 in gastric cancer. Am J Cancer Res. 2014;4:648–662. [PMC free article] [PubMed] [Google Scholar]
- 6.Zhao W, Chan TL, Chu KM, Chan AS, Stratton MR, Yuen ST, Leung SY. Mutations of BRAF and KRAS in gastric cancer and their association with microsatellite instability. Int J Cancer. 2004;108:167–169. doi: 10.1002/ijc.11553. [DOI] [PubMed] [Google Scholar]
- 7.Zandvakili I, Davis AK, Hu G, Zheng Y. Loss of RhoA Exacerbates, Rather Than Dampens, Oncogenic K-Ras Induced Lung Adenoma Formation in Mice. PLoS One. 2015;10:e0127923. doi: 10.1371/journal.pone.0127923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng D, Wang R, Zhang Y, Pan Y, Cheng X, Cheng C, Zheng S, Li H, Gong R, Li Y, Shen X, Sun Y, Chen H. The prevalence and prognostic significance of KRAS mutation subtypes in lung adenocarcinomas from Chinese populations. Onco Targets Ther. 2016;9:833–843. doi: 10.2147/OTT.S96834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang T, Li C, Xia C, Dong Y, Yang D, Geng Y, Cai J, Zhang J, Zhang X, Wang J. Oncogenic NRAS hyper-activates multiple pathways in human cord blood stem/progenitor cells and promotes myelomonocytic proliferation in vivo. Am J Transl Res. 2015;7:1963–1973. [PMC free article] [PubMed] [Google Scholar]
- 10.Vaughn CP, Zobell SD, Furtado LV, Baker CL, Samowitz WS. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosomes Cancer. 2011;50:307–312. doi: 10.1002/gcc.20854. [DOI] [PubMed] [Google Scholar]
- 11.Seth R, Crook S, Ibrahem S, Fadhil W, Jackson D, Ilyas M. Concomitant mutations and splice variants in KRAS and BRAF demonstrate complex perturbation of the Ras/Raf signalling pathway in advanced colorectal cancer. Gut. 2009;58:1234–1241. doi: 10.1136/gut.2008.159137. [DOI] [PubMed] [Google Scholar]
- 12.Miglietta G, Gouda AS, Cogoi S, Pedersen EB, Xodo LE. Nucleic Acid Targeted Therapy: G4 Oligonucleotides Downregulate HRAS in Bladder Cancer Cells through a Decoy Mechanism. ACS Med Chem Lett. 2015;6:1179–1183. doi: 10.1021/acsmedchemlett.5b00315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kiessling MK, Curioni-Fontecedro A, Samaras P, Atrott K, Cosin-Roger J, Lang S, Scharl M, Rogler G. Mutant HRAS as novel target for MEK and mTOR inhibitors. Oncotarget. 2015;6:42183–42196. doi: 10.18632/oncotarget.5619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weyandt JD, Lampson BL, Tang S, Mastrodomenico M, Cardona DM, Counter CM. Wild-Type Hras Suppresses the Earliest Stages of Tumorigenesis in a Genetically Engineered Mouse Model of Pancreatic Cancer. PLoS One. 2015;10:e0140253. doi: 10.1371/journal.pone.0140253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ortega CE, Seidner Y, Dominguez I. Mining CK2 in cancer. PLoS One. 2014;9:e115609. doi: 10.1371/journal.pone.0115609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiao Y, Li X, Wang H, Wen R, He J, Tang J. Epigenetic regulation of miR-129-2 and its effects on the proliferation and invasion in lung cancer cells. J Cell Mol Med. 2015;19:2172–2180. doi: 10.1111/jcmm.12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Huang HL, Jiang Y, Wang YH, Chen T, He HJ, Liu T, Yang T, Yang LW, Chen J, Song ZQ, Yao W, Wu B, Liu G. FBXO31 promotes cell proliferation, metastasis and invasion in lung cancer. Am J Cancer Res. 2015;5:1814–1822. [PMC free article] [PubMed] [Google Scholar]
- 18.Lu Z, Xiao Z, Liu F, Cui M, Li W, Yang Z, Li J, Ye L, Zhang X. Long non-coding RNA HULC promotes tumor angiogenesis in liver cancer by up-regulating sphingosine kinase 1 (SPHK1) Oncotarget. 2016;7:241–254. doi: 10.18632/oncotarget.6280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Min A, Im SA, Kim DK, Song SH, Kim HJ, Lee KH, Kim TY, Han SW, Oh DY, Kim TY, O’Connor MJ, Bang YJ. Histone deacetylase inhibitor, suberoylanilide hydroxamic acid (SAHA), enhances anti-tumor effects of the poly (ADP-ribose) polymerase (PARP) inhibitor olaparib in triple-negative breast cancer cells. Breast Cancer Res. 2015;17:33. doi: 10.1186/s13058-015-0534-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Q, Wen YG, Li DP, Xia J, Zhou CZ, Yan DW, Tang HM, Peng ZH. Upregulated INHBA expression is associated with poor survival in gastric cancer. Med Oncol. 2012;29:77–83. doi: 10.1007/s12032-010-9766-y. [DOI] [PubMed] [Google Scholar]
- 21.Chu SJ, Wang G, Zhang PF, Zhang R, Huang YX, Lu YM, Da W, Sun Q, Zhang J, Zhu JS. MicroRNA-203 suppresses gastric cancer growth by targeting PIBF1/Akt signaling. J Exp Clin Cancer Res. 2016;35:47. doi: 10.1186/s13046-016-0323-1. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Castellano E, Downward J. Role of RAS in the regulation of PI 3-kinase. Curr Top Microbiol Immunol. 2010;346:143–169. doi: 10.1007/82_2010_56. [DOI] [PubMed] [Google Scholar]
- 23.Kompier LC, Lurkin I, van der Aa MN, van Rhijn BW, van der Kwast TH, Zwarthoff EC. FGFR3, HRAS, KRAS, NRAS and PIK3CA mutations in bladder cancer and their potential as biomarkers for surveillance and therapy. PLoS One. 2010;5:e13821. doi: 10.1371/journal.pone.0013821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Engen-van Grunsven AC, van Dijk MC, Ruiter DJ, Klaasen A, Mooi WJ, Blokx WA. HRAS-mutated Spitz tumors: A subtype of Spitz tumors with distinct features. Am J Surg Pathol. 2010;34:1436–1441. doi: 10.1097/PAS.0b013e3181f0a749. [DOI] [PubMed] [Google Scholar]
- 25.Stearns M, Tran J, Francis MK, Zhang H, Sell C. Activated Ras enhances insulin-like growth factor I induction of vascular endothelial growth factor in prostate epithelial cells. Cancer Res. 2005;65:2085–2088. doi: 10.1158/0008-5472.CAN-04-4100. [DOI] [PubMed] [Google Scholar]
- 26.Vaque JP, Navascues J, Shiio Y, Laiho M, Ajenjo N, Mauleon I, Matallanas D, Crespo P, Leon J. Myc antagonizes Ras-mediated growth arrest in leukemia cells through the inhibition of the Ras-ERK-p21Cip1 pathway. J Biol Chem. 2005;280:1112–1122. doi: 10.1074/jbc.M409503200. [DOI] [PubMed] [Google Scholar]
- 27.Qian CN, Tan MH, Yang JP, Cao Y. Revisiting tumor angiogenesis: vessel co-option, vessel remodeling, and cancer cell-derived vasculature formation. Chin J Cancer. 2016;35:10. doi: 10.1186/s40880-015-0070-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di D, Chen L, Wang L, Sun P, Liu Y, Xu Z, Ju J. Downregulation of human intercellular adhesion molecule-1 attenuates the metastatic ability in human breast cancer cell lines. Oncol Rep. 2016;35:1541–1548. doi: 10.3892/or.2016.4543. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.