

Abstract
Despite remarkable successes with targeted therapies in the treatment of cancer, resistance can occur which limits the clinical outcome. In this study, we generated and characterized resistant cell clones derived from two different head and neck squamous cell carcinoma (HNSCC) cell lines (Cal27, UD-SCC-5) by long-term exposure to five targeted- and chemotherapeutics (afatinib, MK2206, BEZ235, olaparib and cisplatin). The resistant tumor cell clones showed an increased ERK1/2 expression and an altered expression of the stem-cell markers CD44, ALDH1, Oct4, Sox2, Nanog and Bmi1. None of the single markers alone was predictive for resistance to all five targeted- and chemotherapeutics. Furthermore, long-term exposure of tumor cells to these five drugs resulted in an eightfold increase in the mutational rate compared to untreated cells. Interestingly, targeted- and chemotherapy resistant cell clones remained sensitive to irradiation. Lastly, clones that were resistant to afatinib, MK2206 or BEZ235 showed cross-resistance to further treatment with therapeutics that affect the same signaling pathway, but remained sensitive to those affecting different pathways such as cisplatin and olaparib. In contrast, cell clones which were once resistant to cisplatin or olaparib were found to be multidrug-resistant. These data might indicate that patients with HNSCC benefit more by a first line targeted therapy followed by cisplatin as a second line therapy.
Keywords: HNSCC, resistant cells, hypermutation
Introduction
Squamous cell carcinomas constitute to 90% of all head and neck cancers (HNSCC). HNSCC is the sixth most common neoplasia worldwide, showing rising incidence rates [1]. Mortality due to this cancer remains largely unimproved despite ongoing advancements in tumor surgery, radiotherapy, and chemotherapy. The 5-year survival rate of advanced HNSCC is below 50% [2]. The prognosis of HNSCC is worsened by a poor loco-regional disease control and significant morbidity [3]. In cases of advanced unresectable and metastatic cancer multi-agent chemotherapy or biological therapies are presently investigated [4,5]. These include drugs targeting growth factors and their receptors. The Epidermal Growth Factor Receptor (EGFR) is overexpressed in 90% of HNSCC and thus represents an important target.
Cetuximab received its FDA approval for the therapy of HNSCC in 2006. The human-murine chimeric immunoglobulin cetuximab competitively targeting the EGFR prevents receptor activation by endogenous ligands and results in EGFR downregulation by internalization of the receptor/antibody complex [6]. Tyrosine-kinase inhibitors like erlotinib or gefitinib influence downstream signaling processes. They were used in locally advanced or metastatic NSCLC pancreatic carcinomas and were studied in HNSCC.
NSCLC patients harboring the activating mutation in the EGFR kinase domain respond well to small tyrosine-kinase inhibitors like erlotinib or gefitinib. However, after an initial clinical response, 50% of all tumors develop the EGFR T790M mutation [7] which alters the binding pocket of the receptor and thus prevents binding of the inhibitor [8]. The knowledge of the molecular mechanisms of drug resistance is important for the selection of the patients for further therapy. It was shown, that tyrosine-kinase inhibitors of the third generation like AZD9291 or rociletinib are targets for tumors with the EGFR mutation T790M [9,10].
Different resistance mechanisms such as cell-cycle alterations, mutations, activation of alternate signaling pathways have been elucidated [11]. Cancer stem cells which have been identified in many tumor entities have also been associated with drug resistance [12,13]. Various cell surface markers such as CD44 and ALDH1 have been associated with a stem cell phenotype in HNSCC [14,15]. Sox2, Oct4, Nanog and Bmi1 are transcription factors that play pivotal roles in maintaining the pluripotency of embryonic stem cells [16,17] and are also found in tumors [18-20].
In the presented study, we generated resistant tumor cell clones derived from two different HNSCC cell lines by a long-term incubation to five targeted- and chemotherapeutics (afatinib, MK2206, BEZ235, olaparib and cisplatin). The tumor cell clones were screened for alterations in the EGFR signaling pathway, for the expression of stem-cell markers and for upcoming mutation rates. In addition, therapeutic strategies to overcome resistancy were investigated.
Methods
Selection of inhibitors
We conducted a systematic literature research to identify the most promising targets and inhibitors for HNSCC which had been clinically successful in other solid tumors. Most of our targets were in the EGFR pathway which plays a pivotal role in HNSCC development and progression [21]. In order to generate resistant cells, we selected the EGFR tyrosine-kinase inhibitor afatinib (Selleckchem, Houston, United States of America), the AKT inhibitor MK2206 (Adooq, Irvine, United States of America), the dual mTOR/PI3K inhibitor BEZ235 (Adooq), the PARP inhibitor olaparib (Selleckchem) and cisplatin (Teva, Ulm, Germany), the standard chemotherapy in HNSCC. By PARP inhibition, olaparib is able to enhance pre-existing DNA repair defects leading to the accumulation of unrepaired DNA double strand breaks [22]. Therapeutics can thus be divided into two classes: EGFR pathway inhibitors such as afatinib, MK2206 and BEZ235, and DNA-damaging therapeutics such as olaparib and cisplatin.
Cell culture and reagents
The Cal27 cell line was obtained from DSMZ (Braunschweig, Germany), UD-SCC-5 cells were obtained from the University of Düsseldorf (Clinic for Otolaryngology, Düsseldorf, Germany). The cells were cultured in Dulbecco’s modified Eagle medium (DMEM) (Invitrogen, Darmstadt, Germany) containing 10% fetal calf serum (FBS) (Biochrom, Berlin, Germany), 2 mM glutamine (Biochrom), 100 μg/ml streptomycin (Biochrom), and 100 U/ml penicillin (Biochrom), maintained at 37°C in an atmosphere of 5% CO2, and grown to 70-90% confluence.
IC50 determination
For IC50 determination cells were treated with increasing concentrations of the five targeted- and chemotherapeutics. Cell survival was assessed by applying the crystal-violet assay. The cells were seeded in a 6-well plate (5 × 103 cells/well). After 1 d, the cells were treated with the drug and then cultured for 10 days. Ten days after treatment, the culture medium was aspirated and 500 µl of 4% formaldehyde/1 × PBS were added to each well for 30 minutes. After washing with 0.1% Triton-X-100/1 × PBS and H2O, crystal violet (0.04%) was added to the fixed cells and allowed to act for 30 minutes. Finally, SDS (1%) was added and the optical density was measured at 590 nm using an ELISA reader 1 h later. The IC50 was calculated by nonlinear regression of three independent experiments by using GraphPad Prism 6.0 software (Graph Pad Software Inc., La Jolla, United States of America).
Development of resistant cells
To establish resistant HNSCC cells we started treating the two cell lines with the IC50 concentration of each targeted- and chemotherapeutic (IC50 for Cal27/UD-SCC-5: afatinib 3.45/2.24 nM, MK2206: 0.21/0.18 µM, BEZ235 9.21/5.45 nM, cisplatin: 1.00/1.96 µM, olaparib 8.96/7.06 µM). We increased the dose stepwise for 6 months, and finished by decreasing the concentration slightly to regain normally proliferating cells for the following experiments. We did not perform single-cell cloning, as we think the generated potentially heterogeneous cells are a more realistic clinical model. Tumors in patients have been demonstrated to be heterogeneous as well.
Western blot
The expression of key proteins of the EGFR receptor pathways and stem-cell markers was assessed by western blotting. For that purpose cells were seeded in 6-well-plates (2.5 × 106 per culture dish). After treatment cells were lysed in an appropriate buffer (20 mM Tris/HCl, pH 7.5, 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1 % Triton-X-100, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 μg/ml leupeptin, 1 mM PMSF). Lysates were centrifuged at 10.000 rpm for 15 minutes at 4°C to precipitate insoluble membrane particles. Protein concentrations of the lysates were quantified by using the Bradford assay to ensure equal amounts of protein loaded per lane in SDS-PAGE. The proteins were then transferred to a PVDF membrane (Immobilon-P, Millipore, Germany). Before incubation with primary antibodies, membranes were blocked with 5% non-fat dry milk in Tris-buffered saline containing 0.1% Tween 20 (TTBS) for 1 h at room temperature. After 12 hours of incubation on a shaker at 4°C, the secondary anti-IgG antibodies labelled with peroxidase were added in 5% non-fat dry milk in TTBS. Bands were detected by means of chemiluminescence using the ECL Detection System with the Imager SRX-101° (Konica Minolta, Langenhagen, Germany). Primary antibodies against the following antigens were used (dilution): p-EGFR Tyr1068 (1:2500), p-Akt Ser473 (1:1000), p-ERK 1/2 Thr202/Tyr204 (1:1000), ALDH1 (1:1000), Oct4 (1:1000), Nanog (1:2000), Sox2 (1:2000), Bmi1 (1:1000), Tubulin (1:5000). Antibodies were obtained from Cell Signaling Technology (Danvers MA, USA), US Biological (Marblehead MA, USA).
X-ray irradiation kinetics
For determining the kinetics of cell death after X-ray exposure, cells were seeded in 6-well plates at a density of 5 × 103 cells/well. The irradiation of cells was performed by using an X-ray generator (Gulmay CP-2225; maximum dose rate 3 Gy/min) at the Department of Radiotherapy (Technische Universität München, Germany). The cells were irradiated at room temperature (70 kV, 10 mA) with X-ray doses ranging between 0.5 and 10 Gy 24 h after seeding. Control cells were treated accordingly, however, without irradiation. Cell survival was assessed by crystal-violet assay as described above.
Random mutation capture assay
We followed the random mutation capture assay protocol of Wright et al. [23] with slight variations described below. In order to calculate the DNA copy number per well qPCR was performed using a 10-fold dilution series and the control primer (forward 5’-CACTGACAACCACCCTTAACC-3’, reverse 5’-TCAGCATCTTATCCGAGTGGA-3’). A reaction volume of 25 µl was used: 12.5 µl SYBR Green Master mix (Peqlab Erlangen, Germany), 7.25 µl double deionized, UV-irradiated water (Cayman Chemical, Ann Arbor, United States of America), 0.25 µl 20 pM Primer (MWG Biotech, Ebersberg, Germany), 5 µl DNA. PCR was performed in 96-well format (4titude Frame Star, Wotton, Surrey, United Kingdom) with a C1000 thermal cycler (Biorad, Munich, Germany) machine. The following qPCR protocol was used: 1. 95.0°C for 10:00; 2. 95.0°C for 0:30; 3. 65.0°C for 0:30; 4. 74.0°C for 0:10 + plate read; 5. GOTO 2 for 57 more times; 6. melt Curve 65.0°C to 95.0°C, increment 0.5°C for 0:05 + plate read.
Then TaqαI digestion was performed as described by Wright et al. [23]. The reaction was incubated in an Eppendorf thermomixer (Eppendorf, Hamburg, Germany) for 3 hours, while shaking the reaction mixture at 750 rpm for a total of nine times. Before the third, fifth and seventh addition of TaqαI, precipitated BSA was removed by transferring the sample to a new tube and 8 µl fresh BSA were added. We did not perform the Microcon YM-50 buffer exchange described by Wright, as we experienced a heavy DNA loss when using these concentrator tubes [23].
After that, we tested for TaqαI (New England Biolabs, Ipswich, United Kingdom) digestion efficiency using the mutation-specific primer (forward 5’-CAAGCAGGGGAGGCCTTTT-3’, reverse 5’-TCCTGGCTAACGGTGAAACC-3’) and chose a dilution for screening of mutants [23]. The master mix and the qPCR protocol described above were applied.
Then qPCR plate was performed to screen for mutants. Primer-specific products which had been positive as confirmed by melt curves were then analyzed by means of post-PCR restriction and gel electrophoresis to differentiate between the wild-type and the mutant PCR product. 5 µl of PCR product were digested with 0.25 µl TaqαI, 0.2 µl CutSmart (New England Biolabs) and 1.55 µl double-deionized water [23].
Next, the number of DNA copies per well was determined by standard curve and mutation frequency was calculated [23].
All experiments were performed three times independently.
A total of 777 mutations were detected by gel electrophoresis. A random sample was chosen and 100 mutations were sent to sequencing with the mutation-specific primer.
Proliferation assay
Cell proliferation was assessed by a crystal violet assay. The binding of crystal violet to cellular DNA was used to assess cell proliferation by means of ELISA as described previously [24]. Briefly, 5 × 103 cells/well were seeded in six-well plates 24 h before treatment. On the next day, cells were incubated with the IC50 dose of each drug. Ten days after treatment, culture medium was aspirated and 500 µl of 4% formaldehyde were added to each well for 30 minutes. After washing with 0.1% Triton-X-100/PBS and H2O, crystal violet (0.04%) was added to the fixed cells and allowed to act for 30 minutes. Finally, SDS (1%) was added and the optical density was measured at 590 nm, using an ELISA reader 1 h later.
Statistical analysis
Statistical analysis was performed by using GraphPad Prism 6.0 software (Graph Pad Software Inc.). Assuming a symmetry correlation structure for all experiments, all hypotheses were tested with the One-way ANOVA test. The means of treated cells and untreated controls were compared by applying Student’s t-test. The level of statistical significance was set at p < 0.05.
Results
Establishment of drug-resistant HNSCC cell lines
According to a protocol described by Kim et al., a resistant tumor cell clone which displays the typical T790M mutation could be established after exposure of the NSCLC cell line PC9 with escalating doses of afatinib [25]. In the present study we chose a similar approach and generated HNSCC cell lines which are resistant to five targeted- and chemotherapeutics.
After increasing the drug doses stepwise for 6 months we observed that resistant cell clones could be divided in two groups: Clones resistant to EGFR pathway inhibitors could be dose escalated much more than clones resistant to DNA-damaging therapeutics. The final concentration of Cal27 clones resistant to the pathway inhibitors was at 800 × IC50, 47 × IC50 and 50 × IC50 for afatinib, MK2206, and BEZ235, respectively, whereas the final concentration to DNA-damaging cisplatin was 9 × IC50 and to olaparib 4 × IC50. Similar results were obtained with the cell line UD-SCC-5: afatinib 800 × IC50, MK2206 50 × IC50, BEZ235 16 × IC50, cisplatin and olaparib only 1 × IC50. The development of the resistant clones of both cell lines is shown in Figure 1A, 1B.
Figure 1.

Development on the time axis of the resistant tumor cell clones. Starting with the two cell lines Cal27 and UD-SCC-5 and the IC50 dose of the three EGFR signaling inhibitors afatinib, MK2206 and BEZ235 and the two DNA-damaging substances cisplatin and olaparib, a total of 10 resistant cell lines were generated by increasing the dose of a single durg tepwise over 24 weeks. Each dot represents a cell passages. Cells resistant to EGFR-pathway inhibitors could be dose-escalated far higher (e.g. Cal27 afatinib: 800 x IC50 vs. Cal27 cisplatin: 9x).
Switch to ERK1/2 pathway in resistant cells
Next we assessed changes in the EGFR signaling pathway by applying a western-blot analysis to the expression of p-EGFR Tyr1068, p-AKT Ser473 and p-ERK1/2 Thr202/Tyr204 (Figure 2A, 2B). In untreated UD-SCC-5 cells p-EGFR was phosphorylated at tyrosine 1068, whereas we did not detect any phosphorylation of p-ERK1/2 or p-AKT. In all five drug-resistant UD-SCC-5 cell lines p-EGFR phosphorylation at tyrosine 1068 was lost, whereas ERK1/2 and AKT phosphorylation newly occurred. In untreated Cal27 cells we detected a basal phosphorylation of p-EGFR Tyr1068, p-ERK1/2 Thr202/Tyr204 and p-AKT Ser473. In treated Cal27 cells, ERK1/2 phosphorylation increased generally, whereas there was an inhomogeneous pattern in AKT and EGFR phosphorylation (Figure 2A, 2B). These results indicate an activation of the ERK1/2 in UD-SCC-5-resistant cells and, less pronounced, in resistant Cal27 cell lines.
Figure 2.
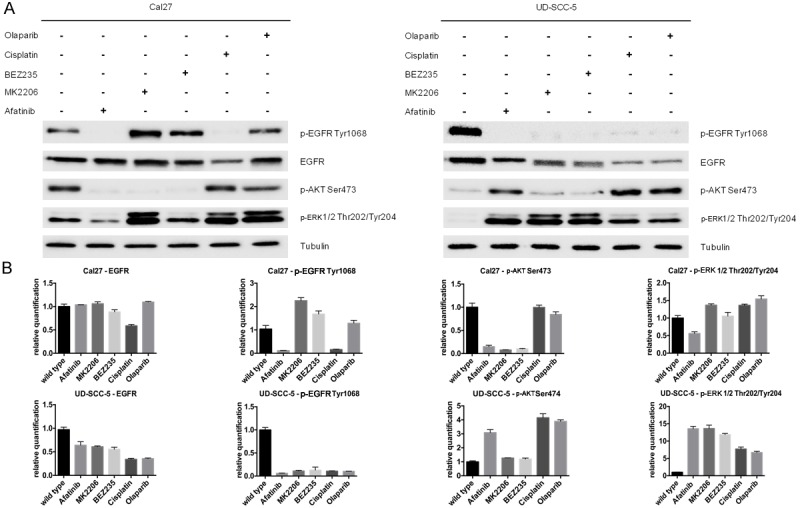
Western blot and concordant relative quantification displaying the changes in signaling of the resistant cells compared to untreated cells. No clear pattern of signaling alternations in EGFR, p-EGFR Tyr1068 and p-AKT Ser473 could be identified. Untreated UD-SCC-5 cells however showed a much weaker phosphorylation of Thr202/Tyr204 at ERK1/2 while all resistant cell lines show a sharp rise in phosphorylation. Cal27 resistant cell lines show an overall increase in ERK1/2 phosphorylation, although this effect is less pronounced than in UD-SCC-5 due to basal level of phosphorylation in untreated cells. Together these findings indicate a switch to ERK1/2 during the development of the resistances.
Heterogeneous alterations in stem cell marker expression
To explore common patterns of stem-cell populations after pathway inhibition or targeted- or chemotherapy, we determined the expression of the stem-cell markers CD44, ALDH1, Oct4, Sox2, Nanog and Bmi1 in untreated and resistant clones. Untreated Cal27 and UD-SCC-5 cells expressed all of the abovementioned stem-cell markers, with the exception of CD44 in wild-type UD-SCC-5 and ALDH1 in wild-type Cal27. UD-SCC-5 clones resistant to the pathway inhibitors expressed CD44 except for ALDH1, whereas UD-SCC-5 clones resistant to DNA-damaging substances expressed ALDH1, with the exception of CD44 (Figure 3A, 3B). Bmi1 was downregulated in all resistant UD-SCC-5 cells. These patterns were not observed in resistant Cal27 cells. We detected heterogeneous alterations in the expression of Oct4, Nanog and Sox2. No clear pattern of change in the expression of these markers was observed in the two cell lines when they were treated with the same inhibitor (Figure 3A, 3B). In conclusion, stem-cell makers had already been expressed differently in the untreated cell lines, and no single marker could be attributed to the resistance to a specific drug.
Figure 3.
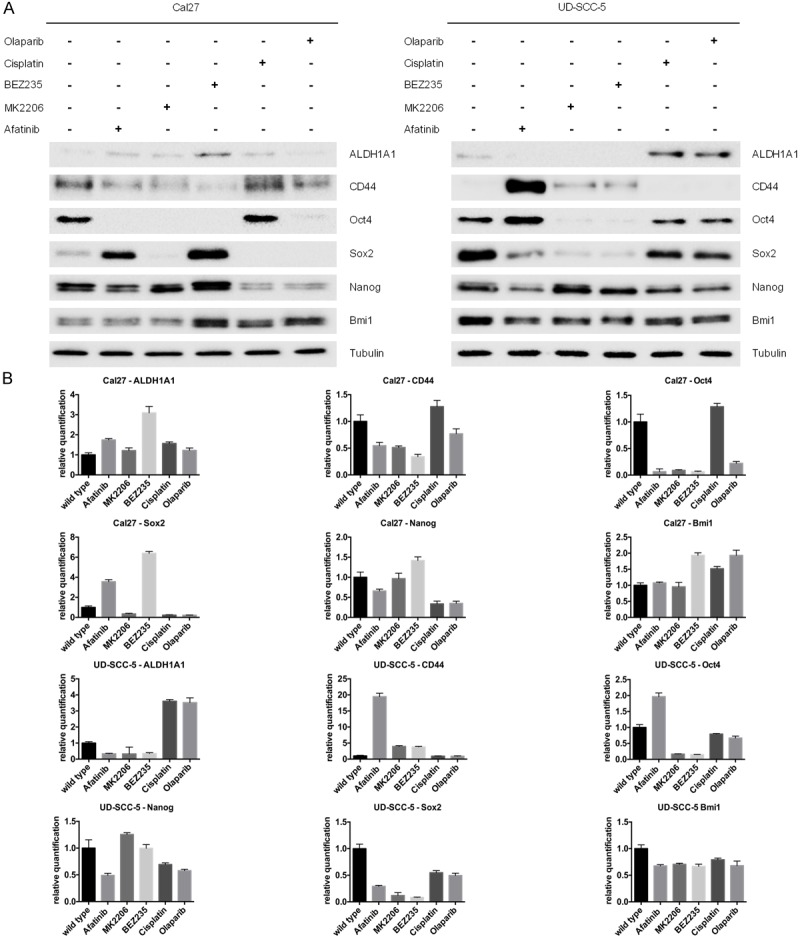
The western blot and the concordant relative quantification showing changes in the stem-cell marker expression of CD44, ALDH1, Oct4, Sox2, Nanog and Bmi1 in untreated and resistant clones. With the exception of CD44 in UD-SCC-5 and ALDH1 in Cal27, all stem-cell markers had already been expressed in the untreated cells. While there are similarities, for example, a downregulation of BMI1 in all resistant UD-SCC-5 cells, overall alternation in stem-cell marker expression was heterogeneous and no single marker could be attributed to the resistance to all therapeutics.
Hypermutation of the cancer-cell genome in drug-resistant cells
Genetic change can be a powerful mechanism of acquiring resistance. To test this hypothesis, we determined the point-mutation frequency by applying the random mutation capture assay [23].
Untreated cancer cells were co-cultivated over the period of 6 months. Untreated Cal27 and UD-SCC-5 cells had a mutation frequency of 7.00 × 10-8 and 1.57 × 10-7, respectively (Figure 4). The average mutation frequency of all resistant Cal27 clones was at 5.78 × 10-7 and thus 8.3 times higher than in the untreated control (p < 0.001). The median mutation frequency of resistant UD-SCC-5 cells was at 1.22 × 10-6 and thus 7.8 times higher compared to the control (p=0.007). The mutation frequency can be transformed into the number of gained mutations in coding regions by assuming 3.3 × 109 bases per genome of which only 1 % are coding. Untreated Cal27 cells gained 2.3 mutations in coding regions, while untreated UD-SCC-5 cells gained 5.2. Resistant Cal27 clones accumulated on average 19.0 mutations, whereas resistant UD-SCC-5 clones accumulated 40.3 mutations. Mutation frequency increases of the various drugs ranged from 4.4-14.8 × in resistant clones (Figure 4). As expected, the mutation frequency incremented most in cells treated with DNA-damaging substances (cisplatin and olaparib). Interestingly, after treatment with the pathway inhibitors (afatinib, MK2206 and BEZ235), the mutation frequency increased as well (Figure 4). In conclusion, long-term targeted- and chemotherapeutical treatment leads to a hypermutation of the cancer-cell genome.
Figure 4.
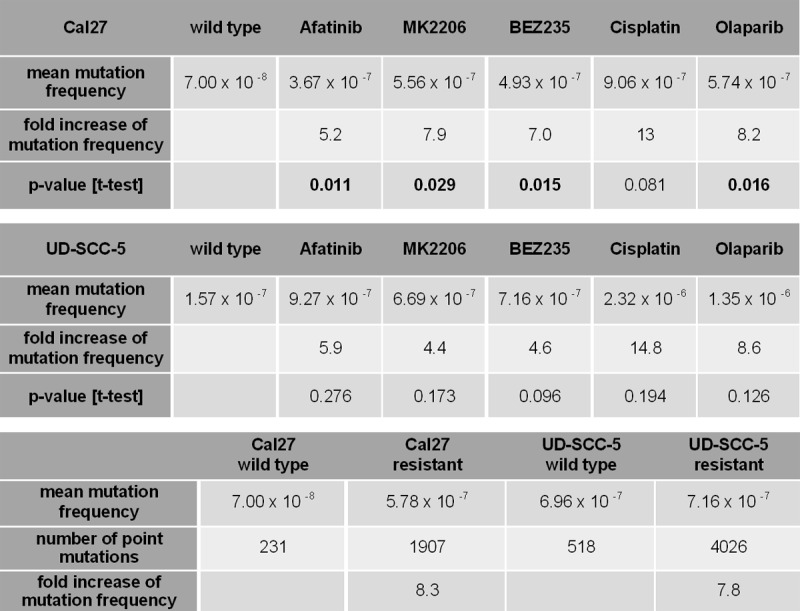
Table showing the point-mutation frequency determined by application of the random mutation capture assay. The overall increase in mutation frequency compared to untreated, co-cultivated cells is 8.3 in cell lines resistant to Cal27, and 7.8 in those resistant to UD-SCC-5, indicating the importance of point mutations in acquiring resistance to all therapeutics. The fold increase in mutation frequency ranged from 4.4 in Cal27 cells resistant to MK2206 to 14.8 in cisplatin-resistant UD-SCC-5 clones, displaying comparability of increases of the mechanistically per se nonmutational tyrosine-kinase inhibitors and DNA-crosslinking cytotoxic cisplatin.
Drug-resistant cells are sensitive to irradiation
To assess the response to radiation therapy we conducted X-ray kinetics with wild-type and resistant cells. As shown in Figure 5, with the exception of Cal27 cells which had been treated with cisplatin and olaparib, changes were rather minimal. The resistant cells are thus as sensitive to irradiation as their untreated wild-type counterparts, indicating that different mechanisms are mediating chemo- and radioresistance.
Figure 5.
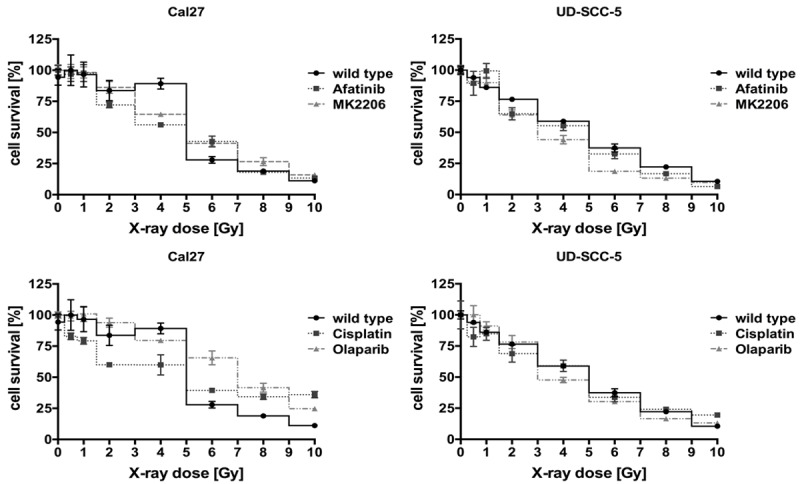
X-ray kinetics of untreated wild-type Cal27 and UD-SCC-5 compared to the resistant clones. With the exception of olaparib-resistant Cal27 cells, which seem slightly more resistant to X-ray exposure, overall alternation in response to irradiation is largely minimal, leaving radiation therapy as an option for a further treatment of chemoresistant clones.
Cells resistant to pathway inhibitors show “cross-resistance”, whereas DNA-damaging substances show “multidrug resistance”
To characterize the change in sensitivity of cell lines resistant to various other forms of targeted- and chemotherapy, we treated each resistant cell line with a single IC50 dose of one of the five drugs. All cells lines resistant to EGFR pathway inhibitors showed a cross-resistance with respect to the other EGFR pathway inhibitors afatinib, MK2206 or BEZ235 and a strong reduction in their proliferative capacity when treated with cisplatin or olaparib.
In contrast, cells resistant to DNA-damaging therapeutics like cisplatin or olaparib showed multidrug resistance in UD-SCC-5 and Cal27 cell lines (Figure 6A, 6B).
Figure 6.
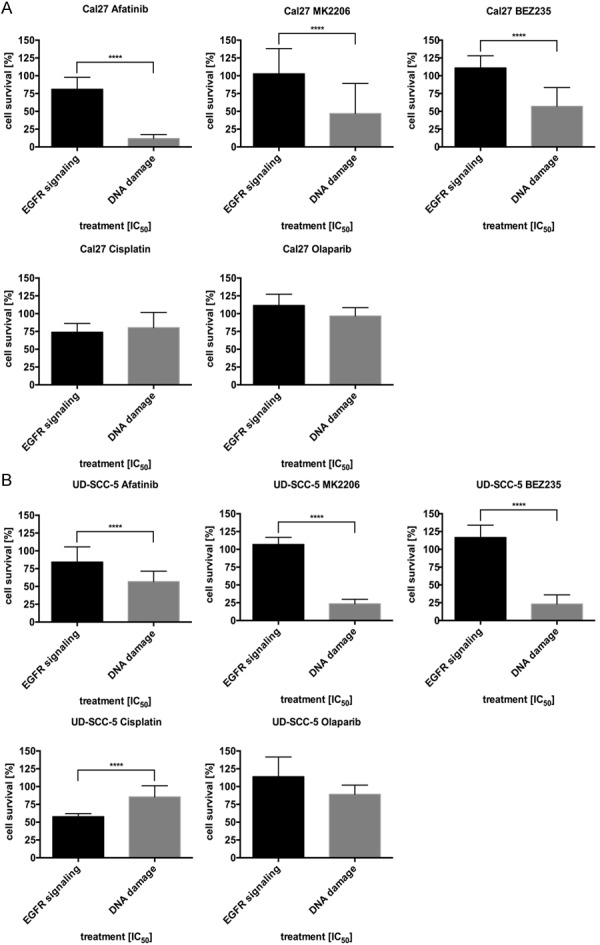
Reduction of proliferation in resistant cells treated with a single IC50 dose of the three EGFR signaling inhibitors: afatinib, MK2206 and BEZ235 and the two DNA-damaging substances cisplatin and olaparib. While clones resistant to EGFR signaling inhibitors were cross-resistant relative to the other EGFR pathway inhibitors, they were still sensitive to the DNA-damaging substances. In contrast, cells resistant to the DNA-damaging substances only showed a minimal response after treatment with all substances, thus indicating multidrug-resistance.
Discussion
Survival in chemotherapeutic treatment in advanced cancer disease is limited. While properly selected patients for targeted therapy respond initially, resistances develop and patients’ tumors relapse. Generation of resistant cell lines by chronic exposure and dose escalation has been widely used as a model to study resistance mechanisms at the molecular level, for example, in lung cancer [25], colon cancer [26], breast cancer [27] and melanoma [28]. While most authors chose one cell line and only one treatment, in this study, we generated cells with an acquired resistance in two HNSCC cell lines and five different drugs to explore whether there is a common pattern of adaption to targeted- or chemotherapy . Our results show that clones resistant to inhibitors of the EGFR signaling pathway, such as afatinib, MK2206 and BEZ235, could be dose-escalated much more than clones resistant to the DNA-damaging substances olaparib and cisplatin. The highest dose escalation was seen in receptor-targeting afatinib, whereas far lower dose increases were possible in downstream targeting MK2206 and BEZ235. We think that this is due to more alternative and downstream pathways available for acquiring resistance to therapy.
Our western-blot analyses show that resistant cancer cells switch towards the ERK1/2 pathway. This is in acccordance with previous findings described in the literature. Rampias et al. reported that oncogenic HRAS promoted the activation of the ERK1/2 pathway when mediating cetuximab resistance in HNSCC [29]. In ovarian cancer, Wang et al. showed that ERK2 promotes phosphorylation of mitogen-activated protein kinase phosphatase-1, leading to increased cisplatin resistance [30]. Upregulation of RAS has been described as a mechanism of acquired resistance to B-RAF (V600E) inhibition in melanoma [31]. Whether cisplatin activates ERK signaling directly or through induction of DNA damage remains unclear [32]. Also further research is needed to clarify whether a switch to the RAS/RAF/ERK pathway alone is sufficient for acquiring resistance to targeted- or chemotherapy.
The cancer stem-cell hypothesis has been used to explain various properties of cancer cells including drug resistance [33-35]. Cancer stem-cell enrichment was reported after treatment with gemcitabine in pancreatic cancer [36], with cyclophosphamide in colon cancer [37], and with irradiation in glioma [38]. We explored the changes in expression of the well described stem-cell markers CD44, ALDH1, Oct4, Sox2, Nanog and Bmi1 [14-17]. All untreated cancer cells expressed Oct4, Sox2, Nanog and Bmi1, only Cal27 expressed CD44 and UD-SCC-5 ALDH1. While we found alterations in expression of stem-cell markers in resistant clones, they were heterogeneous and no clear pattern of enrichment or depletion could be identified. This is in concordance with findings of Liu et al., who reported a distinctive rather than a common pattern of CD44, CD24 and ALDH1 expression in two breast cancer cell lines treated with doxorubicin, docetaxel and radiation [39].
Genetic change is an important mechanism in acquiring resistance [40]. Our data supports this hypothesis. Untreated Cal27 cells accumulated about 2.3 point mutations in coding regions, and untreated UD-SCC-5 accumulated 5 mutations over the six month period. All resistant clones acquired additional point mutations compared with untreated cancer cells. On average, a single resistant Cal27 cell acquired around 19 mutations in coding regions, whereas a resistant UD-SCC-5 cell acquired about 40 mutations. The number of mutations acquired was about eight times higher in resistant cells than in untreated cells. In accordance with our findings, Jia et al. found newly acquired single nucleotide variants by next generation sequencing of an erlotinib-resistant NSCLC cell line in comparison with the parental line 18-91 [41].
In addition, we showed that the response of the targeted- and chemoresistant cell lines to irradiation remains mostly unchanged. These data might imply that mechanisms of targeted- and chemo- and radioresistance are independent from each other and thus irradiation might provide an option to treat targeted- and chemoresistant tumors in the clinical setting. There is no final consent in the literature on chemo- and radio-cross-sensitivity or -resistance. Pauwels et al. described a cisplatin-treated bladder-cell line which showed cross-resistance to chemo- and radiotherapy [42]. On the other hand, Gigante et al. described a cisplatin-resistant and multidrug-resistant colon-cancer cell line, which is radiosensitive [43], and Shi et al. reported that the radioresistant lung-adenocarcinoma cell line A549 is still sensitive to five chemotherapeutic drugs [44].
Furthermore, we showed that clones resistant to one targeted therapy are less sensitive to the further application of a differently targeted therapy involving the same pathway. Interestingly, the “cross-resistant” cancer cells were still sensitive to treatment with cisplatin or olaparib, whereas cell clones resistant to DNA-damaging substances tend to be “multidrug-resistant”. This indicates that there are specific mechanisms of resistance to the pathway inhibitors like mutations or use of alternative pathways, whereas unspecific mechanism exist in DNA-targeting pathways. This is in agreement with previous findings of mutations affecting the binding site of targets [45] or an activation of alternative pathways after treatment with targeted therapies [46]. In contrast, drug efflux [47] or reduced cellular uptake have been suggested as resistance mechanisms effective in conventional chemotherapy [48].
To prevent multidrug resistance in cancers in first-line treatment we hypothesize that it would be more useful to treat patients with a combination of targeted therapies first and then start a second-line treatment with DNA-damaging substances.
Disclosure of conflict of interest
None.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA Cancer J Clin. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- 2.Bernier J, Domenge C, Ozsahin M, Matuszewska K, Lefèbvre JL, Greiner RH, Giralt J, Maingon P, Rolland F, Bolla M, Cognetti F, Bourhis J, Kirkpatrick A, van Glabbeke M European Organization for Research and Treatment of Cancer Trial 22931. Postoperative Irradiation with or without Concomitant Chemotherapy for Locally Advanced Head and Neck Cancer. N Engl J Med. 2004;350:1945–1952. doi: 10.1056/NEJMoa032641. [DOI] [PubMed] [Google Scholar]
- 3.Shirai K, Kaneshiro T, Wada M, Furuya H, Bielawski J, Hannun YA, Obeid LM, Ogretmen B, Kawamori T. A Role of Sphingosine Kinase 1 in Head and Neck Carcinogenesis. Cancer Prev Res (Phila) 2011;4:454–462. doi: 10.1158/1940-6207.CAPR-10-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vermorken JB, Specenier P. Optimal treatment for recurrent/metastatic head and neck cancer. Ann Oncol. 2010;21:vii252–vii261. doi: 10.1093/annonc/mdq453. [DOI] [PubMed] [Google Scholar]
- 5.Kundu SK, Nestor M. Targeted therapy in head and neck cancer. Tumour Biol. 2012;33:707–721. doi: 10.1007/s13277-012-0350-2. [DOI] [PubMed] [Google Scholar]
- 6.Dequanter D, Shahla M, Paulus P, Lothaire PH. The role of EGFR-targeting strategies in the treatment of head and neck cancer. Onco Targets Ther. 2012;5:127–131. doi: 10.2147/OTT.S31863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kosaka T, Yatabe Y, Endoh H, Yoshida K, Hida T, Tsuboi M, Tada H, Kuwano H, Mitsudomi T. Analysis of Epidermal Growth Factor Receptor Gene Mutation in Patients with Non-Small Cell Lung Cancer and Acquired Resistance to Gefitinib. Clin Cancer Res. 2006;12:5764–5769. doi: 10.1158/1078-0432.CCR-06-0714. [DOI] [PubMed] [Google Scholar]
- 8.Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR Mutation and Resistance of Non-Small-Cell Lung Cancer to Gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 9.Jänne PA, Yang JC, Kim DW, Planchard D, Ohe Y, Ramalingam SS, Ahn MJ, Kim SW, Su WC, Horn L, Haggstrom D, Felip E, Kim JH, Frewer P, Cantarini M, Brown KH, Dickinson PA, Ghiorghiu S, Ranson M. AZD9291 in EGFR Inhibitor-Resistant Non-Small-Cell Lung Cancer. N Engl J Med. 2015;372:1689–1699. doi: 10.1056/NEJMoa1411817. [DOI] [PubMed] [Google Scholar]
- 10.Sequist LV, Soria JC, Goldman JW, Wakelee HA, Gadgeel SM, Varga A, Papadimitrakopoulou V, Solomon BJ, Oxnard GR, Dziadziuszko R, Aisner DL, Doebele RC, Galasso C, Garon EB, Heist RS, Logan J, Neal JW, Mendenhall MA, Nichols S, Piotrowska Z, Wozniak AJ, Raponi M, Karlovich CA, Jaw-Tsai S, Isaacson J, Despain D, Matheny SL, Rolfe L, Allen AR, Camidge DR. Rociletinib in EGFR-Mutated Non-Small-Cell Lung Cancer. N Engl J Med. 2015;372:1700–1709. doi: 10.1056/NEJMoa1413654. [DOI] [PubMed] [Google Scholar]
- 11.Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer. 2013;13:714–726. doi: 10.1038/nrc3599. [DOI] [PubMed] [Google Scholar]
- 12.Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011:313–319. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 13.Baccelli I, Trumpp A. The evolving concept of cancer and metastasis stem cells. J Cell Biol. 2012;198:281–293. doi: 10.1083/jcb.201202014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Clay MR, Tabor M, Owen JH, Carey TE, Bradford CR, Wolf GT, Wicha MS, Prince ME. Single-marker identification of head and neck squamous cell carcinoma cancer stem cells with aldehyde dehydrogenase. Head Neck. 2010;32:1195–1201. doi: 10.1002/hed.21315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Prince ME, Sivanandan R, Kaczorowski A, Wolf GT, Kaplan MJ, Dalerba P, Weissman IL, Clarke MF, Ailles LE. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc Natl Acad Sci U S A. 2007;104:973–978. doi: 10.1073/pnas.0610117104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loh YH, Wu Q, Chew JL, Vega VB, Zhang W, Chen X, Bourque G, George J, Leong B, Liu J, Wong KY, Sung KW, Lee CWH, Zhao XD, Chiu KP, Lipovich L, Kuznetsov VA, Robson P, Stanton LW, Wei CL, Ruan Y, Lim B, Ng HH. The Oct4 and Nanog transcription network regulates pluripotency in mouse embryonic stem cells. Nat Genet. 2006;38:431–440. doi: 10.1038/ng1760. [DOI] [PubMed] [Google Scholar]
- 17.Park Ik, Qian D, Kiel M, Becker MW, Pihalja M, Weissman IL, Morrison SJ, Clarke MF. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature. 2003;423:302–305. doi: 10.1038/nature01587. [DOI] [PubMed] [Google Scholar]
- 18.Luo W, Li S, Peng B, Ye Y, Deng X, Yao K. Embryonic Stem Cells Markers SOX2, OCT4 and Nanog Expression and Their Correlations with Epithelial-Mesenchymal Transition in Nasopharyngeal Carcinoma. PLoS One. 2013;8:e56324. doi: 10.1371/journal.pone.0056324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guo Y, Liu S, Wang P, Zhao S, Wang F, Bing L, Zhang Y, Ling EA, Gao J, Hao A. Expression profile of embryonic stem cell-associated genes Oct4, Sox2 and Nanog in human gliomas. Histopathology. 2011;59:763–775. doi: 10.1111/j.1365-2559.2011.03993.x. [DOI] [PubMed] [Google Scholar]
- 20.Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, Dirbas FM, Somlo G, Pera RAR, Lao K, Clarke MF. Downregulation of miRNA-200c Links Breast Cancer Stem Cells with Normal Stem Cells. Cell. 2009;138:592–603. doi: 10.1016/j.cell.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11:9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 22.Weston VJ, Oldreive CE, Skowronska A, Oscier DG, Pratt G, Dyer MJ, Smith G, Powell JE, Rudzki Z, Kearns P, Moss PA, Taylor AM, Stankovic T. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood. 2010;116:4578–4587. doi: 10.1182/blood-2010-01-265769. [DOI] [PubMed] [Google Scholar]
- 23.Wright JH, Modjeski KL, Bielas JH, Preston BD, Fausto N, Loeb LA, Campbell JS. A random mutation capture assay to detect genomic point mutations in mouse tissue. Nucleic Acids Research. 2011;39:e73–e73. doi: 10.1093/nar/gkr142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Noeske K. [The binding of crystal violet on deoxyribonucleic acid. Cytophotometric studies on normal and tumor cell nuclei] . Histochemie. 1966;7:273–287. doi: 10.1007/BF00577848. [DOI] [PubMed] [Google Scholar]
- 25.Kim Y, Ko J, Cui Z, Abolhoda A, Ahn JS, Ou SH, Ahn MJ, Park K. The EGFR T790M Mutation in Acquired Resistance to an Irreversible Second-Generation EGFR Inhibitor. Mol Cancer Ther. 2012;11:784–791. doi: 10.1158/1535-7163.MCT-11-0750. [DOI] [PubMed] [Google Scholar]
- 26.Misale S, Yaeger R, Hobor S, Scala E, Janakiraman M, Liska D, Valtorta E, Schiavo R, Buscarino M, Siravegna G, Bencardino K, Cercek A, Chen CT, Veronese S, Zanon C, Sartore-Bianchi A, Gambacorta M, Gallicchio M, Vakiani E, Boscaro V, Medico E, Weiser M, Siena S, Di Nicolantonio F, Solit D, Bardelli A. Emergence of KRAS mutations and acquired resistance to anti EGFR therapy in colorectal cancer. Nature. 2012;486:532–536. doi: 10.1038/nature11156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ward A, Balwierz A, Zhang JD, Kublbeck M, Pawitan Y, Hielscher T, Wiemann S, Sahin O. Re-expression of microRNA-375 reverses both tamoxifen resistance and accompanying EMT-like properties in breast cancer. Oncogene. 2013;32:1173–1182. doi: 10.1038/onc.2012.128. [DOI] [PubMed] [Google Scholar]
- 28.Villanueva J, Vultur A, Lee JT, Somasundaram R, Fukunaga-Kalabis M, Cipolla AK, Wubbenhorst B, Xu X, Gimotty PA, Kee D, Santiago-Walker AE, Letrero R, D’Andrea K, Pushparajan A, Hayden JE, Brown KD, Laquerre S, McArthur GA, Sosman JA, Nathanson KL, Herlyn M. Acquired resistance to BRAF inhibitors mediated by a RAF kinase switch in melanoma can be overcome by co-targeting MEK and IGF-1R/PI3K. Cancer cell. 2010;18:683–695. doi: 10.1016/j.ccr.2010.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rampias T, Giagini A, Siolos S, Matsuzaki H, Sasaki C, Scorilas A, Psyrri A. RAS/PI3K Crosstalk and Cetuximab Resistance in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res. 2014;20:2933–2946. doi: 10.1158/1078-0432.CCR-13-2721. [DOI] [PubMed] [Google Scholar]
- 30.Wang J, Zhou JY, Wu GS. ERK-Dependent MKP-1-Mediated Cisplatin Resistance in Human Ovarian Cancer Cells. Cancer Res. 2007;67:11933–11941. doi: 10.1158/0008-5472.CAN-07-5185. [DOI] [PubMed] [Google Scholar]
- 31.Nazarian R, Shi H, Wang Q, Kong X, Koya RC, Lee H, Chen Z, Lee MK, Attar N, Sazegar H, Chodon T, Nelson SF, McArthur G, Sosman JA, Ribas A, Lo RS. Melanomas acquire resistance to B-RAF(V600E) inhibition by RTK or N-RAS upregulation. Nature. 2010;468:973–977. doi: 10.1038/nature09626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Woessmann W, Chen X, Borkhardt A. Ras-mediated activation of ERK by cisplatin induces cell death independently of p53 in osteosarcoma and neuroblastoma cell lines. Cancer Chemother Pharmacol. 2002;50:397–404. doi: 10.1007/s00280-002-0502-y. [DOI] [PubMed] [Google Scholar]
- 33.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–284. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 34.Donnenberg VS, Donnenberg AD. Multiple Drug Resistance in Cancer Revisited: The Cancer Stem Cell Hypothesis. J Clin Pharmacol. 2005;45:872–877. doi: 10.1177/0091270005276905. [DOI] [PubMed] [Google Scholar]
- 35.Alison MR, Lin WR, Lim SM, Nicholson LJ. Cancer stem cells: In the line of fire. Cancer Treat Rev. 2012;38:589–598. doi: 10.1016/j.ctrv.2012.03.003. [DOI] [PubMed] [Google Scholar]
- 36.Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct Populations of Cancer Stem Cells Determine Tumor Growth and Metastatic Activity in Human Pancreatic Cancer. Cell Stem Cell. 2007;1:313–323. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 37.Dylla SJ, Beviglia L, Park IK, Chartier C, Raval J, Ngan L, Pickell K, Aguilar J, Lazetic S, Smith-Berdan S, Clarke MF, Hoey T, Lewicki J, Gurney AL. Colorectal Cancer Stem Cells Are Enriched in Xenogeneic Tumors Following Chemotherapy. PLoS One. 2008;3:e2428. doi: 10.1371/journal.pone.0002428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, Dewhirst MW, Bigner DD, Rich JN. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–760. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y, Nenutil R, Appleyard MV, Murray K, Boylan M, Thompson AM, Coates PJ. Lack of correlation of stem cell markers in breast cancer stem cells. Br J Cancer. 2014;110:2063–2071. doi: 10.1038/bjc.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Turner NC, Reis-Filho JS. Genetic heterogeneity and cancer drug resistance. Lancet Oncol. 2012;13:e178–185. doi: 10.1016/S1470-2045(11)70335-7. [DOI] [PubMed] [Google Scholar]
- 41.Jia P, Jin H, Meador CB, Xia J, Ohashi K, Liu L, Pirazzoli V, Dahlman KB, Politi K, Michor F, Zhao Z, Pao W. Next-generation sequencing of paired tyrosine kinase inhibitor-sensitive and -resistant EGFR mutant lung cancer cell lines identifies spectrum of DNA changes associated with drug resistance. Genome Res. 2013;23:1434–1445. doi: 10.1101/gr.152322.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pauwels O, Gozy M, Van Houtte P, Pasteels JL, Atassi G, Kiss R. Cross resistance and collateral sensitivity between cytotoxic drugs and radiation in two human bladder cell lines. Radiother Oncol. 1996;39:81–86. doi: 10.1016/0167-8140(95)01696-1. [DOI] [PubMed] [Google Scholar]
- 43.Gigante M, Toffoli G, Bertola A, Biscontin G, Dassie A, Zanelli GD, Zanin E, Trovo MG, Muzzio PC. Radiosensitivity in multidrug-resistant and cisplatin-resistant human carcinoma cell lines. Am J Clin Oncol. 2003;26:e73–79. doi: 10.1097/01.COC.0000077939.04389.B6. [DOI] [PubMed] [Google Scholar]
- 44.Shi D, Shi G, Huang G, Zhang J, Lartigau E. Chemosensitivity of radioresistant cells in the multicellular spheroids of A549 lung adenocarcinoma. J Exp Clin Cancer Res. 2009;28:72. doi: 10.1186/1756-9966-28-72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oerlemans R, Franke NE, Assaraf YG, Cloos J, van Zantwijk I, Berkers CR, Scheffer GL, Debipersad K, Vojtekova K, Lemos C, van der Heijden JW, Ylstra B, Peters GJ, Kaspers GL, Dijkmans BA, Scheper RJ, Jansen G. Molecular basis of bortezomib resistance: proteasome subunit β5 (PSMB5) gene mutation and overexpression of PSMB5 protein. Blood. 2008;112:2489–2499. doi: 10.1182/blood-2007-08-104950. [DOI] [PubMed] [Google Scholar]
- 46.Wheeler DL, Huang S, Kruser TJ, Nechrebecki MM, Armstrong EA, Benavente S, Gondi V, Hsu KT, Harari PM. Mechanisms of acquired resistance to cetuximab: role of HER (ErbB) family members. Oncogene. 2008;27:3944–3956. doi: 10.1038/onc.2008.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control. 2003;10:159–165. doi: 10.1177/107327480301000207. [DOI] [PubMed] [Google Scholar]
- 48.Arnold CN, Goel A, Boland CR. Role of hMLH1 promoter hypermethylation in drug resistance to 5-fluorouracil in colorectal cancer cell lines. Int J Cancer. 2003;106:66–73. doi: 10.1002/ijc.11176. [DOI] [PubMed] [Google Scholar]