

Abstract
MicroRNAs belonging to the miR-302 family are emerging as key players in the control of cell growth, and maintaining pluripotency during cell fate determination and differentiation in embryonic stem cells. However, the mechanisms whereby ephA2/ephirnA1 signaling regulates miR-302b expression and attenuates malignant pleural mesothelioma (MPM) cell growth are not known. Our study identified a novel mechanism of ephrin-A1 mediated anti-oncogenic signaling in MPM. Ephrin-A1 treatment up regulates miR-302b expression in MPM cells and attenuates cell proliferation and tumorsphere formation via repression of myeloid cell leukemia-1 (Mcl-1). The expression of miR-302b was analyzed by qPCR, the expression of Mcl-1 was analyzed by RT-PCR, immuno-blotting and Immunofluorescence staining. To confirm that ephrin-A1 regulates the expression of Mcl-1 mRNA through miR-302b up regulation, cells were transfected with and without miR-302b and miR-302b inhibitor prior to ephrinA1 treatment. The cell proliferation and tumorsphere formation was measured by WST-1 and matrigel assays respectively. In addition, to confirm the binding of miR-302b to the 3’UTR of Mcl-1 Luciferase assay was performed. Ephrin-A1 treatment induced several fold increases of miR-302b expression in MM cells. In ephrin-A1 treated MM cells, Mcl-1 expression was significantly down regulated when compared to control. Moreover, ephrin-A1 activation significantly inhibited MM cell proliferation and tumorsphere growth. Furthermore, ephrinA1 and miR-302b induced apoptosis in MM cells. The present data suggests that ephrin-A1 induces the expression of miR-302b in MM cells which targets Mcl-1 thereby inhibits MM tumorsphere growth by inducing apoptosis.
Keywords: Malignant pleural mesothelioma, EphrinA1, EphA2, miR-302b, Mcl-1
Introduction
Malignant pleural Mesothelioma (MPM) is the most aggressive tumor of the mesothelium and predominantly develops from previous exposure to asbestos [1]. In the United States, MPM occurs in approximately 3,000 people per year, with nearly 200 individuals diagnosed in Florida annually, and 19% are women [2]. Worldwide, the incidence is increasing and it is expected to peak [3]. Although asbestos exposure is the main risk factor in the development of mesothelioma, the molecular mechanisms remain unclear. MPM is often resistant to chemotherapy [4] and radiation [5]. For most of the patients, disease is diagnosed at late stages and average survival ranges from a few months to less than 2 years [6,7]. Several clinical problems regarding the diagnosis and treatment of MPM remain unsolved [8].
Emerging investigations reveal that microRNAs (miRs) deregulation is mainly associated with various pathologies including development of cancer [9]. miRs are small non-coding RNAs of approximately 21-25 nucleotide long that regulates posttranscriptional gene expression [10]. MiRs bind to complementary sequences on target messenger RNA transcripts (mRNAs), that results in translational repression or target degradation and gene silencing [11]. Some miRs regulate specific individual gene targets; others can be master regulators of a silencing process. miRs regulate the expression of hundreds of genes simultaneously, and some types of miRs act cooperatively [12]. MiRs have been proposed to contribute to oncogenesis because they can function either as tumor suppressors or oncogenes [13]. In earlier studies we reported that ephrinA1 treatment of MPM cells induced Let-7a miR expression and attenuated cellular proliferation via repression of Ras gene [14]. Here in this report we demonstrated the role of miR-302b in regulation of MPM cells proliferation and apoptosis.
Mcl-1 (myeloid cell leukaemia-1) is an anti-apoptotic protein which is the member of the Bcl-2 family protein [15]. It has been suggested that Mcl-1 may act as an anti-apoptotic factor by sequestering Bak, on the outer mitochondrial membrane and preventing its oligomerization and cytochrome-c release from mitochondria [16,17]. It has been shown to be expressed in multiple cell lineage and renowned as a crucial member of apoptosis control [18]. Although Mcl-1 is one of the necessary anti-apoptotic proteins in normal cells, failure of signaling pathways which regulate Mcl-1 expression often leads to it’s over expression, which contributes to several human malignancies. Mcl-1 expression, regulation can occur at multiple levels as transcriptional regulation, post-transcriptional regulation, translational regulation which can consist of miRs binding in the 3’-UTR of Mcl-1 mRNA and also post translational regulation [19]. On one hand, Mcl-1 has a short half-life and is a highly regulated protein and its rapid regulation suggests that it has a critical role in apoptosis under rapidly changing conditions [19], on the other hand, insufficient Mcl-1 level can cause inappropriate cell death [20]. As Mcl-1 is widely expressed in various human malignant cells, it suggest that Mcl-1 might be a predictive marker and also a target for new anti-cancer therapies [21]. In mesothelioma Mcl-1 has been found to be over expressed in most of the MPM tumor tissues [22,23]. This observation suggests that the resistance to apoptosis in MPM could be related to Mcl-1 protein.
Malignancy of solid tumors such as mesothelioma is a complex process that includes the activation of oncogenic signaling and down regulation of tumor suppressor pathways. Oncogenic conversion, amplification, or overexpression of proto-oncogenes, such as those encoding cell surface receptor tyrosine kinases (RTKs), are frequently observed in variety of human cancers and contribute to most of the malignancies [24,25]. Eph receptors forming the largest sub-family of RTKs of which their extracellular domain interacts with ephrin ligands [26]. The family is subdivided into class A and class B based on binding affinity for 2 distinct types of membrane-anchored ephrin ligands. Class B receptors generally bind to class B ephrins that are attached to the cell membrane by a transmembrane-spanning domain, while A class receptors normally interact with glycosyl-phosphatidylinositol-linked class A ephrins, although interclass binding does occur among certain family members [27]. Emerging evidence indicate that some members of this receptor family, including ephrin A2 (EphA2) receptor, have been linked to tumor progression [28]. EphA2 is the most frequently affected Eph kinases in human cancer and its over expression has been observed in variety of tumor types including pancreatic adenocarcinoma [29], prostate, kidney, lung cancer, ovarian cancer [30,31] and MM [32]. Ephrin-A1, a ligand for the Eph receptor tyrosine kinase, is involved in vascular development, tissue border formation, cell migration, axon guidance, synaptic plasticity, and adult neovascularization [33].
We reported that binding of receptor EphA2 by the ligand ephrin-A1 induces marked reduction in the anchorage-independent growth of MM tumor [34]. Furthermore, vector-based over expression of ephrin-A1 in MM cells demonstrated similar results and also confirmed the tumor-suppressive properties [35]. However, the mechanisms by which ephrin-A1 suppresses the tumor growth in MM are not clear.
For the present study, we identified a novel mechanism of ephrin-A1-mediated tumor growth inhibition in MM. Herein; we demonstrate that ephrin-A1 treatment leads to miR-302b expression in MM cells. In addition; miR-302b functionally targets the pro-survival molecule, Mcl-1 which results in its repression and induces apoptosis in MMCs. Together these findings suggest that ephrin-A1 may have a distinct role in controlling tumor growth pathways and may be a promising candidate in the development of novel therapeutic strategies against MM.
Materials and methods
MMC culture
The CRL-2081 and CRL 5830, two MM cell lines were obtained from American Type Culture Collection (ATCC, Manassas, VA) and maintained in 10 mm dishes in RPMI 1640 (Sigma) containing 10% fetal bovine serum, penicillin (100 U/ml) and streptomycin (100 µg/ml), as reported earlier [14,34]. MM cells were incubated at 37°C in 5% CO2 and 95% air and were cultured to 80% confluence between 3 and 4 days and sub cultured in 60 mm cell culture dishes (5 × 105 cells/dish) and used for various assays as needed.
MMC treatment with recombinant Ephrin-A1
MM Cells in near confluence culture were treated with or without 3.5 µg/ml rm-ephrin-A1/FC Chimera (R and D Systems, Minneapolis, MN) in serum free RPMI 1640, for the indicated times and then processed for immunoblotting and quantitative Real Time PCR.
MicroRNA target prediction
Computer-based programs were used to predict microRNAs that potentially bind Mcl-1. Using “Mcl-1” as a search term, we queried EMBL-EBI, MicroCosm targets (http://www.ebi.ac.uk/). We confirmed the miR-302b target binding by performing Luciferase assay.
Luciferase reporter assay
To construct the luciferase reporter vectors, the whole 3’UTR of Mcl-1 was inserted into the pEZX-MT01 vector (GeneCopiaTM) immediate downstream of the firefly luciferase gene under SV40 enhancer followed by renilla luciferase gene under CMV promoter. Another construct, which is empty, was also constructed as a negative control vector as reported earlier [36]. In brief, to express miR-302b OmicsLinkTM miRNA expression clone (GeneCopiaTM) was constructed with pEZX-MT01 vector with miR-302b stem-loop was inserted into the vector immediate after GFP reporter gene under CMV promoter and also a negative control vector. Each luciferase reporter construct, including Luc+miR302b, Luc+Mcl-1 3’UTR and negative control vectors was co-transfected into CRL-2081 and CRL-5830 cells in 6-well plate using Lipofectamin-2000 (Invitrogen, CA). After incubation for 18 hours, the cells were transferred into 96-well plate. After 24 hours, Firefly and renilla luciferase activities, as indicated by relative luminescence activity (RLA) were determined using Luc-PairTM miR Luciferase Assay kits (GeneCopiaTM) according to the manufacturer’s instructions.
MMCs transfection with miR-302b
To investigate the role of miR-302b in down regulation of Mcl-1 protein, MMCs were transfected with miR-302b mimic (GeneCopia, Inc. Rockville, MD) and miR-302b synthetic inhibitor (GeneCopeia, Inc. Rockville, MD). The MM cells were each plated on 60mm-dishes in density of 500,000 cells/dish and were pre-incubated overnight then miR-302b mimic and miR-302b synthetic inhibitor were introduced into the cells by using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s directions. A negative control was introduced into the cells using the same protocol.
Real time quantitative polymerase chain reaction
Total RNA from treated and untreated MMCs was extracted by applying 375 µl of TRIzol (Invitrogen, Frederick, MD) directly onto the cells by following the manufacturer’s instructions. RNA concentration and purity were determined spectrophotometrically by measuring absorbance at 260/280 nm. Total RNA (1 µg) was reverse transcribed into cDNA and Real Time Q-PCR was performed using an Applied Biosystems 7500 Real Time PCR System and SYBER Green JumpstartTM Taq Ready MixTM (Sigma) as reported earlier [14]. The primers: h18S (sense; 5’-AAACGGCTACCACATCCAAG), (antisense; 5’-TAACGAGGATCCATTGGAGG) as endogenous control and Mcl-1 (forward 5’-CGGTAATCGGACTCAACCTC-3’), (reverse 5’-CCTCCTTCTCCGTAGCCAA-3’) to quantify the expression level of the Mcl-1 gene and hsa-miR-302b primers were purchased from QIAGEN. Expression levels of the genes were based on the amount of the target message relative to the h18S control, to normalize the initial input of total RNA.
Western blot analysis
Total cell lysates were prepared and protein concentration was measured using Pierce’s BCATM Protein Assay Kit (PIERCE, Rockford, IL) following the manufacturer’s instructions as reported earlier [14]. In brief, total proteins (20-30 µg) were separated on 15% SDS-PAGE (Bio-Rad). After electrophoretic transfer onto a nitrocellulose membrane (Bio-Rad), membranes were incubated for 1 hour in phosphate-buffered saline containing 5% nonfat dry milk and 0.1% Tween 20 and then incubated for overnight with Mcl-1 antibody (Cell Signaling, Danvers, MA) at 4°C. After three washes with 0.1% Tween 20 in phosphate-buffered saline, the membranes were incubated for 1 hour with a horseradish peroxidase-conjugated secondary antibody (either anti-Rabbit or anti-mouse IgG; dilution 1:5,000). Finally, the membranes were washed three times, and the bound antibodies were visualized by enhanced Immun-StarTM HRP Chemiluminescent Kit (BIO-RAD).
Immunofluorescence
MM cells were cultured in 4 chamber polystyrene vessel (BD FalconeTM) in density of 25 × 103 per chamber and activated with or without 3.5 µg/ml rm-ephrin-A1/Fc chimera for 24 hours as reported earlier [14,34]. In brief, MM cells were fixed with cold 4% paraformaldehyde and incubated with monoclonal Mcl-1antibody (dilution 1:1000), after washing with PBS, sections were incubated with a goat anti-Rabbit IgG with a green fluorescent label (Invitrogen, Carlsbad, CA) and nuclear stain DAPI (4’,6-diamidino-2-phenylindole) for 1 hour respectively. After removal of antibodies, cells were rinsed with PBS and mounted onto slides using Anti-fade reagents (Slow fade anti-fade kit, Invitrogen) and fluorescence was immediately observed using Immunofluorescence microscope (Nikon).
MMC proliferation assay
In brief, MM cells were cultured in 96-well plates in triplicate at a density of 1 × 104 cells/well as reported earlier [14,34]. The cells were activated with or without 3.5 µg/ml ephrin-A1/Fc chimera in serum free RPMI 1640, transfected with or without 50 nM miR-302b, 100 nM of miR-302b synthetic inhibitor or 100 nM of scrambled sequence in addition to ephrin-A1 for 48 hours. Proliferation was evaluated by incubating cells with tetrazolium salt (WST-1 reagent from Roche) for 4 hours and the absorbance of the formazan generated by the activity of dehydrogenases was measured in wavelength 450 nm and then cell number was also determined by cell counting.
Tumorsphere formation assay
MM cells were plated in 6 well-plates (CELLSTAR) in density of 4 × 104 in 10% FBS RPMI 1640 were activated with or without 3.5 µg/ml ephrin-A1 chimera, and transfected with 50 nM miR-302b, or 100 nM of miR-302b synthetic inhibitor or scrambled sequence 100 nM for 24 hours. After 24 hours, the cells were placed in a 24-well culture plate coated with 200 µl of matrigel per well, followed by polymerization for 1 hour at 37°C. Media were changed every three days and the number of tumorspheres formed was recorded after 10 days of incubation. Four to six randomly chosen fields (10 ×) from the sample were photographed.
Determination of Apoptosis by Annexin V labeling
Approximately 5 × 105 MM cells were plated in 60-mm dishes and incubated overnight at 37°C. Cells were rinsed with Opti-MEM I medium, transfected with or without 50 nM miR-302b, or 100 nM miR-302b synthetic inhibitor and Mcl-1 siRNA prior to treatment with ephrin-A1, and continued to grow for 24 hours. Inactivated MM cells without oligonucleotide treatment were used as control. Cells were harvested by Accutase (SigmaAldrich), and apoptotic cells were assayed with an annexin V-FITC apoptosis detection kit (BD Pharmingen. San Diego, CA) as reported earlier [34,37]. Briefly, 5 × 105 cells after washing twice with PBS were incubated with 100 µl of 1 × of medium-binding reagent, 5 µl of Annexin V-FITC and 5 µl of Propidium Iodide staining solution for 15 min at room temperature in the dark. Then 400 µl of 1 × of medium-binding reagent were added immediately and the flow cytometry analysis was performed by using FACScan (Becton Dickinson, Franklin Lake, NJ).
Statistical analysis
The statistical significance was calculated using Sigma Stat 3.5 statistical software program. Analysis of variance was used to compare the experimental groups from the control groups. The Student Neuman-Keul’s procedure was used for multiple pairwise comparisons. Differences at p values < 0.05 were considered statistically significant.
Results
miR-302b expression is increased in ephrin-A1 treated MMCs
Previously, we have shown that, treatment of MMC with ephrin-A1 suppresses proliferation. If ephrin-A1 is a potent regulator of miR-302b, then miR-302b should be up regulated in activated MM cells. To assess if the treatment of MMCs with ephrin-A1 affect transcriptional regulation of miR-302b, qPCR was performed for MMC1 (CRL-2081) and MMC2 (CRL-5830). We noticed that MMCs treatment with ephrin-A1 for 3, 6, 9 and 12 hours leads to up regulation of miR-302b in a time dependent manner. Treatment with ephrin-A1 at concentration of 3.5 µg for 9 and 12 hours significantly increased miR-302b expression level when compared to 3 and 6 hours of treatment in both MMC1 and MMC2 (Figure 1A, 1B). Ephrin-A1 at the lower concentration was ineffective (< 2 µg). In addition, ephrin-A1 activation down regulates Mcl-1 mRNA and protein levels in MMCs. Mcl-1 is over expressed in both MMC1 and MMC2 cell lines. It was observed that stimulation of EphA2 receptor with its ligand ephrin-A1, negatively regulate the expression of Mcl-1 protein in a time dependent manner in both MM cell lines. MMCs’ total RNAs and lysates were subjected to qPCR and Western blot analysis and β-actin levels were measured to show sample loading equality. Ephrin-A1 treatment down regulated Mcl-1 mRNA and protein levels in MMCs (Figure 1C-F).
Figure 1.
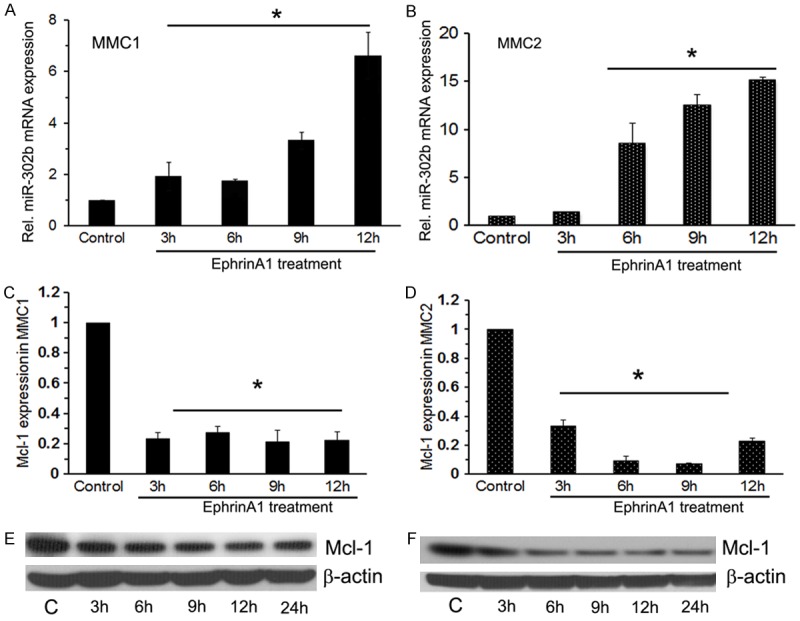
Ephrin-A1 treatment induced miR-302b expression in MM cells in vitro. A and B. Represents miR-302b expression in MMC1 and MMC2 respectively for indicated time points. Data presented as relative expression values using control (Resting MMCs in medium without ephrin-A1 treatment) as the baseline value. Presented data are the mean of three separate observations. To normalize the expression of miR302b, we used 18S RNA as endogenous control. Ephrin-A1 treatment down regulates Mcl-1 mRNA and protein level in MM cells. C and D. Mcl-1 mRNA expression in MMC1 and MMC2. E and F. Western blot analysis of MMC1 and MMC2 subjected to ephrin-A1 treatment at different time points as indicated for determination of Mcl-1 expression. The β-actin was detected to demonstrate equal sample loading. All experiments were performed 3 times separately and data is represented as ± SEM, and values *P < 0.05 as compared to control.
miR-302b down regulates Mcl-1 mRNA and protein expression in MMCs
To determine the effect of ephrin-A1 on Mcl-1 gene expression and to investigate the role of miR-302b in repression of Mcl-1 in MMCs, cultured cells were transfected with or without miR-302b mimic and miR-302b inhibitor before ephrinA1 treatment. Mcl-1 mRNA and protein levels were analysis. The transfection of miR-302b in MMCs significantly inhibited the Mcl-1 mRNA level in MMCs Figure 2A and 2B; whereas transfection of MMCs with miR-302b inhibitor prior to ephrinA1 treatment doesn’t show any effect on Mcl-1 expression as compare to the resting cells Figure 2A and 2B; untreated MMCs showed strong protein expression of Mcl-1, whereas ephrin-A1 treatment and transfection with miR-302b mimic, a decreased expression of Mcl-1 protein was noted as compared to both control and scrambled sequence. In addition, Immunofluorescence analysis also confirmed that treatment of MMCs with ephrin-A1 at a concentration of 3.5 µg for 12 hours significantly decreased Mcl-1 protein levels when compared to other time points (Figure 2E and 2F). MMCs transfected with miR-302b showed decreased expression of Mcl-1 as evidenced by green stain. Taken together these results suggest that miR-302b negatively regulates Mcl-1 expression in MMCs.
Figure 2.
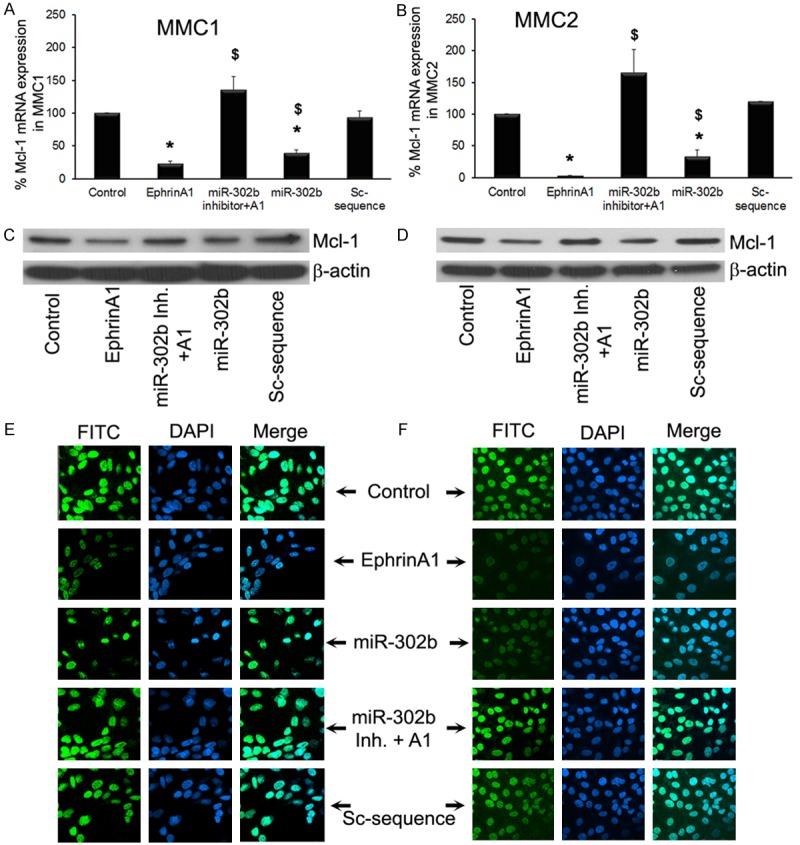
miR-302b synthetic inhibitor blocked ephrin-A1-mediated inhibition of Mcl-1 expression in MMCs. MMCs were transfected with or without miR-302b and miR-302b inhibitor and subsequently treated with ephrin-A1 as described earlier. Mcl-1 mRNA expression (A and B), and protein expression was determined in MMCs subjected to various treatments. The β-actin was detected to demonstrate equal sample loading (C and D). Cellular distribution of Mcl-1 in indicated treatments was analyzed by immunofluorescence microscopy (E and F). The green color represents Mcl-1 expression as stained by FITC, and DAPI was used as nuclear stain (blue color). All experiments were performed 3 times separately and data is represented as ± SEM, and values *P < 0.05 as compared to control; and $P < 0.05 as compared to Sc-sequence.
miR-302b targets Mcl-1 at 3’UTR
To confirm that the miR-302b binds to Mcl-1 in 3’UTR region, we performed a luciferase reporter assay. The alignment of miR-302b with the 3’UTR inserts shown in Figure 3A. Before testing Mcl-1, we confirmed miR-302b transfection efficiency with GFP monitoring in transfected cells with confocal microscopy. When the miR-302b target site from the Mcl-1 3’UTR is inserted into the luciferase construct, expression of luciferase is strongly decreased when co transfected with miR-302b. In contrast, suppression of luciferase activity was almost abolished when the MM cells were co transfected with miRNA negative control vector and Luc+3’UTR Mcl-1 vector Figure 3B and 3C. These data indicate that the 3’UTR Mcl-1 is critical for the direct and specific binding of miR-302b to Mcl-1 mRNA and, miR-302b directly inhibits expression of Mcl-1 by binding to its target sequence.
Figure 3.
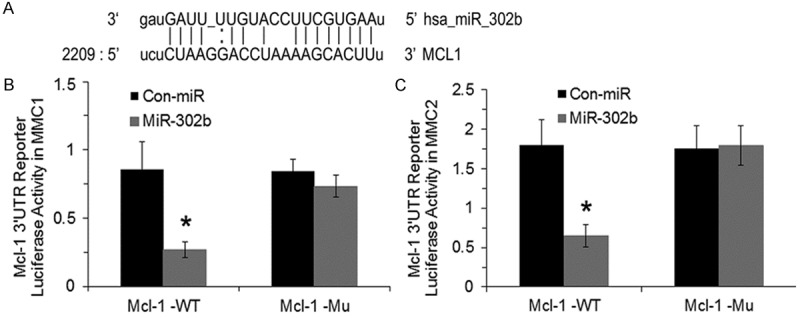
miR-302b inhibits Mcl-1 gene expression via directly binding at the Mcl-1 3’UTR. Alignment of miR-302b with the insert derived from the Mcl-1 3’UTR (A). Cells (1 × 105) were transfected with a Mcl-1 luciferase reporter vector and miR-302b GFP-expression vector. Both firefly luciferase and Renilla luciferase activities were measured as described in the procedure. Firefly luciferase activity was then normalized with Renilla luciferase activities in the same well in MMC1 (B) and MMC2 (C) respectively. The experiments were performed 3 times separately in triplicates and data is represented as ± SEM, and values *P < 0.05 as compared to control-miR vector.
Repression of Mcl-1 inhibited MMC cell proliferation and tumorsphere formation
To assess the role of miR-302b in regulation of MM cell proliferation, we transfected MMCs with miR-302b mimic and inhibitor. The cells were treated in triplicate with ephrin-A1 for 24 hours. As we expected both MM cell lines that generally have high proliferation rate; upon treatment with ephrin-A1 a significant reduction in proliferation rate of MMCs was noticed. In contrast ephrinA1 treatment did not affect the proliferative response when we blocked miR-302b activity through miR-302b synthetic inhibitor and the response was comparable with resting MM cells (Figure 4A). MMCs demonstrated the lowest growth proliferative response after transfection with miR-302b and treatment with ephrin-A1 (Figure 4A). These data suggests that the MMCs proliferation is modulated with ephrin-A1 through the miR-302b expression in MMCs. In addition, to understand the role Mcl-1 in mediation of MMCs proliferation, we have transfected the MMCs with Mcl-1-siRNA or con-siRNA and in some cultures a combination of Mcl-1-siRNA+miR-302b transfection was performed. Mcl-1-siRNA alone inhibited the MMC cell proliferation, whereas the combination treatment showed insignificant inhibition of MMC cell proliferation when compared to only miR-302b or Mcl-1-siRNA. Furthermore, a reduction in proliferation was noted in MMCs transfected with Mcl-1-siRNA+ephrinA1, the inhibition was not significant.
Figure 4.
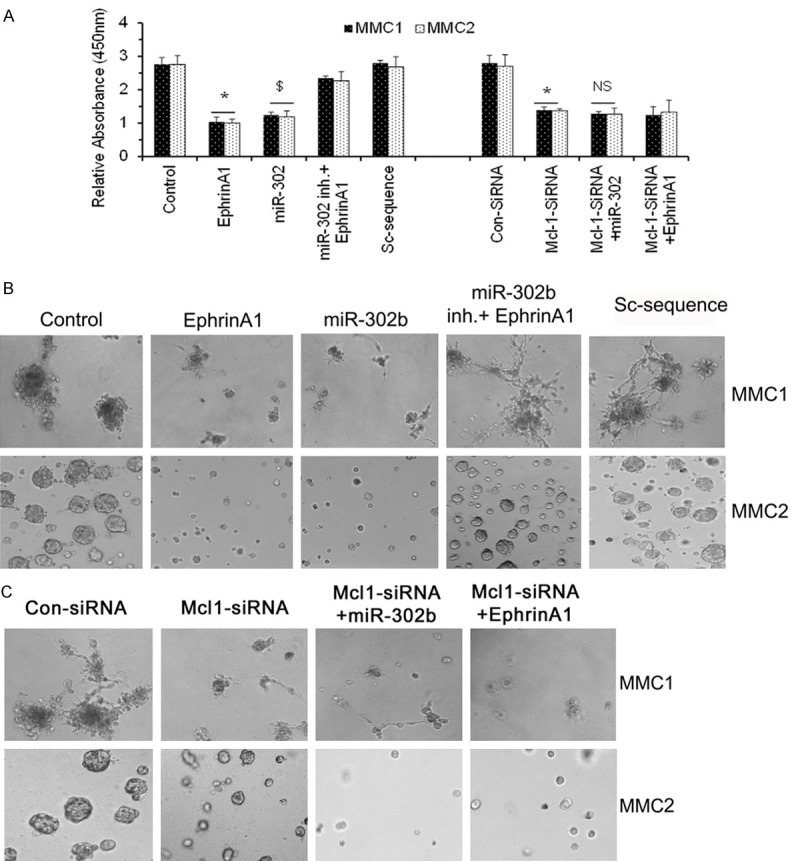
miR-302b inhibitor restored ephrin-A1-mediated proliferation inhibition in MMCs. MMC proliferation as determined by WST-1 assay, wherein control = untreated MMCs and Sc-control = MMC transfected with miR-302b scrambled miRNA (A). miR-302b inhibitor restored ephrin-A1-mediated attenuation of tumorsphere growth in MMC. MMCs were transfected with or without miR-302b and miR-302b inhibitor or Mcl-1 siRNA plus miR-302b as indicated and activated with or without ephrin-A1 and tumorsphere formation was determined (B, C). Data presented is the representatives of three independent observations performed at different times. All experiments were performed 3 times separately and data is represented as ± SEM, and values *p < 0.05 as compared to control; and $p < 0.05 as compared to Sc-sequence; **p < 0.05 as compared to con-siRNA; and NS = not significant when compared to only miR-302b and ephrinA1 alone.
The findings of miR-302b role in cancer progression attenuation lead us to investigate the possibility of using miR-302b as a tumor suppressor. To test this hypothesis MMCs were activated with and without ephrin-A1 and transfected with and without miR-302b mimic and inhibitor or mismatch sequence and cells were plated on 3-D matrigel to determine the tumorsphere growth. The tumorspheres growth was examined in MMCs for 10 days. Light microscopic examination revealed that MMCs transfected with miR-302b mimic showed attenuation in clonal growth as compared to the control cells and both ephrin-A1 treatment and transfection with miR-302b led to suppression of tumorsphere growth. In contrast, ephrin-A1 treatment in the presence of miR-302 inhibitor did not affect tumor colonies in MMCs (Figure 4B). In addition, MMCs tumorsphere formation was also analyzed to understand the role Mcl-1 in tumorsphere development in matrigel. We have transfected the MMCs with Mcl-1-siRNA or con-siRNA or a combination of Mcl-1-siRNA+miR-302b transfection was performed. Mcl-1-siRNA alone inhibited the MMC tumorsphere formation, whereas the combination treatment showed inhibition of MMC tumorsphere growth but when compared to only miR-302b or Mcl-1-siRNA it was not significant. Furthermore, an insignificant reduction in tumorsphere growth was noted in MMCs transfected with Mcl-1-siRNA+ephrinA1 when compared with miR-302b or ephrinA1 alone. Taken together these data suggests that in MMCs miR-302b mediates the rate of cell proliferation/tumorsphere growth via regulation of Mcl-1 (Figure 4C).
miR-302b induces apoptosis in MMCs
To further verify if miR-302b induced apoptotic cell death, we treated MMCs with ephrin-A1 and transfected with miR-302b. We analyzed MMC1 and MMC2 cell lines, 24 hours after transfection with or without miR-302b mimic, miR-302 synthetic inhibitor, and Mcl-1 siRNA prior to ephrin-A1 treatment. To determine the apoptotic effects in MMCs Annexin V-FITC staining was performed. The results showed that ephrin-A1 treatment, and transfection with miR-302b induced significantly greater apoptotic cell death in the MM cell lines as compared with the respective control samples. Flow cytometric cell cycle analysis found no noticeable apoptotic induction in the transfected groups with miR-302b inhibitor and Mcl-1 siRNA prior to ephrin-A1 activation when compared to controls (Figure 5A). In addition, to investigate if Mcl-1 mediated any effect on miR-302b and regulated MMCs cell death, we have tranfected the MMCs with Mcl-1-siRNA or con-siRNA and in some cultures a combination of Mcl-1-siRNA+miR-302b transfection was performed. Mcl-1-siRNA alone induced the MMC apoptosis, whereas the combination treatment showed induction of apoptosis in MMC (26.98 ± 4.56; 20.92 ± 4.20) but when compared to only miR-302b or Mcl-1-siRNA it was not significant. Furthermore, in MMCs tranfected with Mcl-1-siRNA+ephrinA1 an induction of apoptosis was noted but was not significant when compared with ephrinA1 or miR-302b (Figure 5B). Taken together these data suggests that in MMCs miR-302b mediates the rate of apoptosis via regulation of Mcl-1. Interestingly, silencing Mcl-1 in combination with miR-302b did not show any enhanced effect on induction of apoptosis in MMCs.
Figure 5.
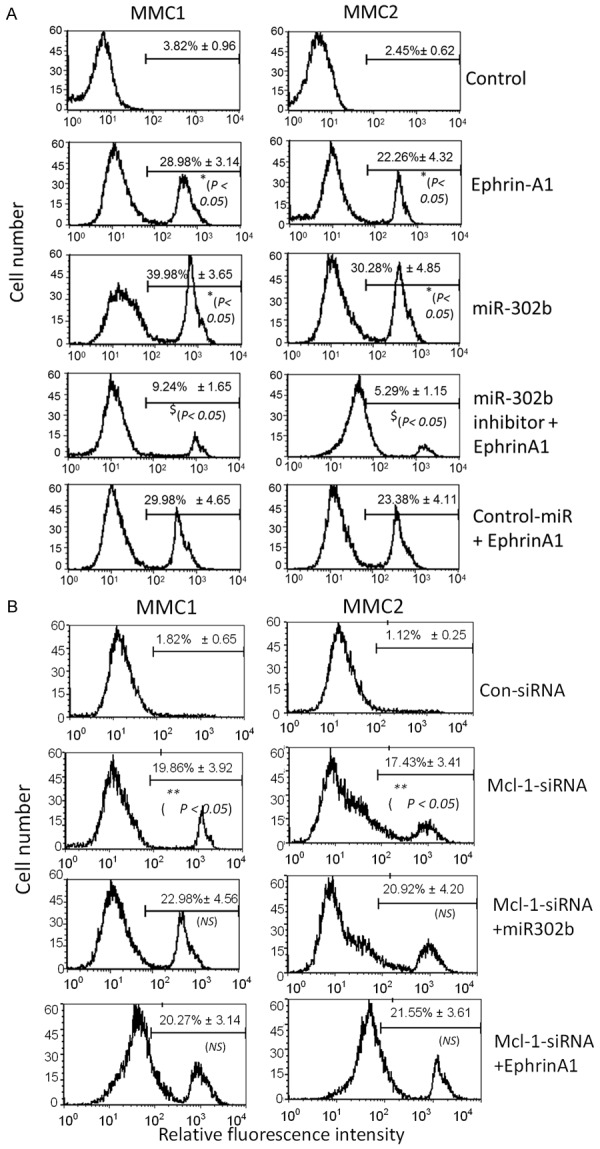
miR-302b inhibitor restored ephrin-A1-mediated cell death in MMCs. MMC apoptosis was determined by Annexin V apoptosis detection assay, wherein Control is untreated MMCs and Sc-control is MMC transfected with scrambled miRNA. miR-302b inhibitor restored ephrin-A1-mediated apoptosis in MMC. MMCs were transfected with or without miR-302b and miR-302b inhibitor and treated with or without ephrin-A1 and apoptosis was determined (A). MMCs were transfected with or without Mcl-1 siRNA and miR-302b and treated with or without ephrin-A1 as shown and apoptosis was determined (B). Date presented is the representatives of three independent observations performed at different times. The values *p < 0.05 as compared to control; and $p < 0.05 as compared to miR-302b alone; **p < 0.05 as compared to con-siRNA; and NS = not significant when compared to Mcl-1-siRNA alone.
Discussion
MPM is one of the most chemoresistant cancers in the world. MPM is usually associated with chronic asbestos exposure. Since, the extremely long latency from time of initial asbestos exposure to tumor development and the lack of effective modes of therapy are barriers to eradicating the MPM [38], and no curative treatment is yet available [39]. Increased activity often is observed in tyrosine kinases functioning which play an essential role in the cell signaling pathways in carcinogenesis [40]. Since the cell-membrane-bound EphA2 receptor, a member of the tyrosine kinases receptor family, has generated great interest in the last few years, and earlier we have shown that EphA2 receptor activation by its ligands down regulates tumor proliferation and tumorsphere growth in MMCs [35]. However, the underlying mechanisms are not clear.
The most widely studied class of non-coding RNAs are microRNAs, which are small RNAs of ~22 nucleotides which mediate post-transcriptional gene silencing by controlling the translation of mRNA into proteins [12,41]. They are involved in regulation of many critical processes in the cells, including proliferation, differentiation, development, and apoptosis [42]. The involvement of miRs in apoptosis was first reported in 2003 with miR-14 and then several miRs were discussed to be involved in cell death [43]. The miR-302 gene encodes a cluster of eight miRs that target cell cycle regulators in the 3’UTR of target mRNAs [44-46]. Evidence shows that miR-302 inhibits tumor proliferation and tumorigenecity and induces apoptosis in pluripotent stem cells [47]. These findings of cell cycle attenuation and cancer cell apoptosis mediated by miR-302 lead us to investigate the possibility of using miR-302 and ephrin-A1 mediated anti-oncogenic effects in MPM. Several observations suggest that EphA2 receptor activation with its ligand, ephrin-A1 regulates MM cell proliferation and tumor formation [32,48].
MM cell lines have been shown to be more resistant to apoptosis than normal mesothelial cells. Unlike many other types of cancer cells which undergo apoptosis as a result of a mutant p53 gene, most mesothelioma cells contain wild-type p53 [49]. Therefore, the mechanism for resistance to apoptosis is yet to be elucidated. The Bcl family of proteins play a role in the regulation of apoptosis may contribute to the resistance of mesothelioma [49,50]. Mcl-1 is an anti-apoptotic member of the Bcl-2 family which has functional and sequence similarity to Bcl-2 [51]. Although Mcl-1 and Bcl-2 both can promote cell survival, there is evidence showing that these protein’s expression is regulated independently [52]. Deregulation of Mcl-1 expression often results in it’s over expression, contributes to human malignancies such as MPM [53]. Even though the ability of ephrin-A1 to regulate the MM cell proliferation is not fully understood yet. In this report we show that, EphA2 receptor binding by ligand ephrin-A1 leads to a significant over expression of miR-302b and results in a down regulation of Mcl-1 an anti-apoptotic protein in MMCs. Transfection of MMCs with miR-302b synthetic inhibitor, before ligand treatment, significantly restored the Mcl-1 expression and proliferation. Furthermore, in MMCs the miR-302b over expression resulted in a significant suppression of Mcl-1, an inhibition of MMC proliferation and tumorsphere growth and induction of apoptosis in vitro. Here for the first time we demonstrate that both mRNA and protein level of Mcl-1 are down regulated in MM cell lines activated with ephrin-A1 suggesting ephrin-A1 may play an important role in attenuating apoptosis resistance and tumor cell proliferation in MM. In mesothelioma Mcl-1 has been found to be over expressed the same as some other solid tumors such as hepatocellular carcinoma [54-56], which demonstrated that Mcl-1 controlled tumor cell viability via suppression of apoptotic pathways [57]. The ability of miR-302b to regulate Mcl-1 protein expression is likely direct as it binds to the 3’UTR region of Mcl-1 messenger RNA. We noticed increased expression of miR-302b in ephrin-A1 treated MMCs leads to down regulation of Mcl-1 mRNA via degradation and also inhibition of Mcl-1 protein translation. Increasing evidence suggests that miRs alteration actively contributes to cancer development and play a role in carcinogenesis by deregulating transcriptional levels of oncogenes and tumor suppressor genes [58]. Our results confirm these concepts by suggesting that lack of miR-302b expression helps MM cells evade cell death, a primordial feature for malignant cells. The increase expression of miR-302b results in deregulation of Mcl-1 expression can explain the up regulation of Mcl-1 in this malignancy. In addition, interestingly silencing Mcl-1 expression and transfection with miR-320b did not show any significant increase in the induction of cell death in MMCs suggesting a compensatory role of other bcl-2 family members such as bcl-xL [59,61]. However, this needs to be further investigated by the application of specific mimetic agents of BH3 that targets only Bcl-2 or bcl-xL but not Mcl-1 in MMCs.
In summary, our findings show a novel mechanism of ephrin-A1-mediated tumor suppressive and apoptosis induction signaling in MMCs which indicates that Mcl-1 mRNA and protein level can be regulated by ephrin-A1 via up regulation of miR-302b. These experiments extend our knowledge of ephrin-A1-mediated apoptotic signaling pathway in MMCs. This is the first report which speaks about apoptosis-inductive properties of ephrin-A1 which induces miR-302b that targets Mcl-1 expression in MMCs. However, further work needs to be performed to learn more about the ephrin-A1-mediated anti-tumorogenesis mechanisms in MMCs.
Acknowledgements
This work was supported by Research grant (4BB02) from Florida Department of Health (Nasreen N); VA Merit Review (Mohammed, KA).
Disclosure of conflict of interest
None.
References
- 1.Lechner JF, Tokiwa T, LaVeck M, Benedict WF, Banks-Schlegel S, Yeager H Jr, Banerjee A, Harris CC. Asbestos-associated chromosomal changes in human mesothelial cells. Proc Natl Acad Sci U S A. 1985;82:3884–3888. doi: 10.1073/pnas.82.11.3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ismail-Khan R, Robinson LA, Williams CC Jr, Garrett CR, Bepler G, Simon GR. Malignant pleural mesothelioma: a comprehensive review. Cancer Control. 2006;13:255–263. doi: 10.1177/107327480601300402. [DOI] [PubMed] [Google Scholar]
- 3.Wagner JC, Sleggs CA, Marchand P. Diffuse pleural mesothelioma and asbestos exposure in the North Western Cape Province. Br J Ind Med. 1960;17:260–271. doi: 10.1136/oem.17.4.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang Y, Zhao R, Chattopadhyay S, Goldman ID. A novel folate transport activity in hu man mesothelioma cell lines with high affinity and specificity for the new-generation antifolate, pemetrexed. Cancer Res. 2002;62:6434–6437. [PubMed] [Google Scholar]
- 5.Narasimhan SR, Yang L, Gerwin BI, Broaddus VC. Resistance of pleural mesothelioma cell lines to apoptosis: relation to expression of Bcl-2 and Bax. Am J Physiol. 1998;275:L165–171. doi: 10.1152/ajplung.1998.275.1.L165. [DOI] [PubMed] [Google Scholar]
- 6.Tsao AS, Wistuba I, Roth JA, Kindler HL. Malignant pleural mesothelioma. J. Clin. Oncol. 2009;27:2081–2090. doi: 10.1200/JCO.2008.19.8523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Steele JP, Klabatsa A, Fennell DA, Pallaska A, Sheaff MT, Evans MT, Shamash J, Rudd RM. Prognostic factors in mesothelioma. Lung Cancer. 2005;49(Suppl 1):S49–52. doi: 10.1016/j.lungcan.2005.03.009. [DOI] [PubMed] [Google Scholar]
- 8.Singhal S, Wiewrodt R, Malden LD, Amin KM, Matzie K, Friedberg J, Kucharczuk JC, Litzky LA, Johnson SW, Kaiser LR, Albelda SM. Gene expression profiling of malignant mesothelioma. Clin Cancer Res. 2003;9:3080–3097. [PubMed] [Google Scholar]
- 9.Calin GA, Croce CM. MicroRNA signatures in human cancers. Nat Rev Cancer. 2006;6:857–866. doi: 10.1038/nrc1997. [DOI] [PubMed] [Google Scholar]
- 10.Di Leva G, Calin GA, Croce CM. MicroRNAs: fundamental facts and involvement in human diseases. Birth Defects Res C Embryo Today. 2006;78:180–189. doi: 10.1002/bdrc.20073. [DOI] [PubMed] [Google Scholar]
- 11.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 13.Calin GA, Croce CM. MicroRNA-cancer connection: the beginning of a new tale. Cancer Res. 2006;66:7390–7394. doi: 10.1158/0008-5472.CAN-06-0800. [DOI] [PubMed] [Google Scholar]
- 14.Khodayari N, Mohammed KA, Goldberg EP, Nasreen N. EphrinA1 inhibits malignant mesothelioma tumor growth via let-7 microRNA-mediated repression of the RAS oncogene. Cancer Gene Ther. 2011;18:806–816. doi: 10.1038/cgt.2011.50. [DOI] [PubMed] [Google Scholar]
- 15.Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, Huang DC. Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell. 2005;17:393–403. doi: 10.1016/j.molcel.2004.12.030. [DOI] [PubMed] [Google Scholar]
- 16.Shimazu T, Degenhardt K, Nur EKA, Zhang J, Yoshida T, Zhang Y, Mathew R, White E, Inouye M. NBK/BIK antagonizes MCL-1 and BCL-XL and activates BAK-mediated apoptosis in response to protein synthesis inhibition. Genes Dev. 2007;21:929–941. doi: 10.1101/gad.1522007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Willis SN, Chen L, Dewson G, Wei A, Naik E, Fletcher JI, Adams JM, Huang DC. Proapoptotic Bak is sequestered by Mcl-1 and Bcl-xL, but not Bcl-2, until displaced by BH3-only proteins. Genes Dev. 2005;19:1294–1305. doi: 10.1101/gad.1304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Michels J, Johnson PW, Packham G. Mcl-1. Int J Biochem Cell Biol. 2005;37:267–271. doi: 10.1016/j.biocel.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 19.Akgul C. Mcl-1 is a potential therapeutic target in multiple types of cancer. Cell Mol Life Sci. 2009;66:1326–1336. doi: 10.1007/s00018-008-8637-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature. 2003;426:671–676. doi: 10.1038/nature02067. [DOI] [PubMed] [Google Scholar]
- 21.Osford SM, Dallman CL, Johnson PW, Ganesan A, Packham G. Current strategies to target the anti-apoptotic Bcl-2 protein in cancer cells. Curr Med Chem. 2004;11:1031–1039. doi: 10.2174/0929867043455486. [DOI] [PubMed] [Google Scholar]
- 22.Soini Y, Kinnula V, Kaarteenaho-Wiik R, Kurttila E, Linnainmaa K, Paakko P. Apoptosis and expression of apoptosis regulating proteins bcl-2, mcl-1, bcl-X, and bax in malignant mesothelioma. Clin Cancer Res. 1999;5:3508–3515. [PubMed] [Google Scholar]
- 23.O’Kane SL, Pound RJ, Campbell A, Chaudhuri N, Lind MJ, Cawkwell L. Expression of bcl-2 family members in malignant pleural mesothelioma. Acta Oncol. 2006;45:449–453. doi: 10.1080/02841860500468927. [DOI] [PubMed] [Google Scholar]
- 24.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 25.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 26.Murai KK, Pasquale EB. ‘Eph’ective signaling: forward, reverse and crosstalk. J Cell Sci. 2003;116:2823–2832. doi: 10.1242/jcs.00625. [DOI] [PubMed] [Google Scholar]
- 27.Brantley-Sieders DM, Chen J. Eph receptor tyrosine kinases in angiogenesis: from development to disease. Angiogenesis. 2004;7:17–28. doi: 10.1023/B:AGEN.0000037340.33788.87. [DOI] [PubMed] [Google Scholar]
- 28.Brantley-Sieders D, Schmidt S, Parker M, Chen J. Eph receptor tyrosine kinases in tumor and tumor microenvironment. Curr Pharm Des. 2004;10:3431–3442. doi: 10.2174/1381612043383160. [DOI] [PubMed] [Google Scholar]
- 29.Duxbury MS, Ito H, Zinner MJ, Ashley SW, Whang EE. Ligation of EphA2 by Ephrin A1-Fc inhibits pancreatic adenocarcinoma cellular invasiveness. Biochem Biophys Res Commun. 2004;320:1096–1102. doi: 10.1016/j.bbrc.2004.06.054. [DOI] [PubMed] [Google Scholar]
- 30.Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, Sloan AE, Cohen ML, Wang B. EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt. Cancer Cell. 2009;16:9–20. doi: 10.1016/j.ccr.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Landen CN, Kinch MS, Sood AK. EphA2 as a target for ovarian cancer therapy. Expert Opin Ther Targets. 2005;9:1179–1187. doi: 10.1517/14728222.9.6.1179. [DOI] [PubMed] [Google Scholar]
- 32.Khodayari N, Mohammed KA, Goldberg EP, Jantz MA, Nasreen N. Abstract 188: EphrinA1 induces microRNA-302b expression in malignant mesothelioma cells and suppresses tumor growth via repression of Mcl-1 gene. Cancer Research. 2012;72(Supplement 1) [Google Scholar]
- 33.Iida H, Honda M, Kawai HF, Yamashita T, Shirota Y, Wang BC, Miao H, Kaneko S. Ephrin-A1 expression contributes to the malignant characteristics of {alpha}-fetoprotein producing hepatocellular carcinoma. Gut. 2005;54:843–851. doi: 10.1136/gut.2004.049486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nasreen N, Mohammed KA, Antony VB. Silencing the receptor EphA2 suppresses the growth and haptotaxis of malignant mesothelioma cells. Cancer. 2006;107:2425–2435. doi: 10.1002/cncr.22254. [DOI] [PubMed] [Google Scholar]
- 35.Lai YM, Mohammed KA, Nasreen N, Baumuratov A, Bellew BF, Antony VB. Induction of cell cycle arrest and apoptosis by BCG infection in cultured human bronchial airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;293:L393–401. doi: 10.1152/ajplung.00392.2006. [DOI] [PubMed] [Google Scholar]
- 36.Nasreen N, Johnson JA, Sadikot RT, Sahn SA, Mohammed KA. MicroRNA-26a promotes epithelial mesenchymal transition in pleural mesothelial cells. Int J Clin Exp Pathol. 2016;9:xxx–xxx. [Google Scholar]
- 37.Mohammed KA, Wang X, Goldberg EP, Antony VB, Nasreen N. Silencing receptor EphA2 induces apoptosis and attenuates tumor growth in malignant mesothelioma. Am J Cancer Res. 2011;1:419–431. [PMC free article] [PubMed] [Google Scholar]
- 38.Barreiro TJ, Katzman PJ. Malignant mesothelioma: a case presentation and review. J Am Osteopath Assoc. 2006;106:699–704. [PubMed] [Google Scholar]
- 39.Ramalingam SS, Belani CP. Recent advances in the treatment of malignant pleural mesothelioma. J Thorac Oncol. 2008;3:1056–1064. doi: 10.1097/JTO.0b013e3181834f66. [DOI] [PubMed] [Google Scholar]
- 40.Tandon M, Vemula SV, Mittal SK. Emerging strategies for EphA2 receptor targeting for cancer therapeutics. Expert Opin Ther Targets. 2011;15:31–51. doi: 10.1517/14728222.2011.538682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mendell JT. MicroRNAs: critical regulators of development, cellular physiology and malignancy. Cell Cycle. 2005;4:1179–1184. doi: 10.4161/cc.4.9.2032. [DOI] [PubMed] [Google Scholar]
- 42.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 43.Vecchione A, Croce CM. Apoptomirs: small molecules have gained the license to kill. Endocr Relat Cancer. 2010;17:F37–50. doi: 10.1677/ERC-09-0163. [DOI] [PubMed] [Google Scholar]
- 44.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 45.Suh MR, Lee Y, Kim JY, Kim SK, Moon SH, Lee JY, Cha KY, Chung HM, Yoon HS, Moon SY, Kim VN, Kim KS. Human embryonic stem cells express a unique set of microRNAs. Dev Biol. 2004;270:488–498. doi: 10.1016/j.ydbio.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 46.Card DA, Hebbar PB, Li L, Trotter KW, Komatsu Y, Mishina Y, Archer TK. Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Mol Cell Biol. 2008;28:6426–6438. doi: 10.1128/MCB.00359-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin SL, Chang DC, Ying SY, Leu D, Wu DT. MicroRNA miR-302 inhibits the tumorigenecity of human pluripotent stem cells by coordinate suppression of the CDK2 and CDK4/6 cell cycle pathways. Cancer Res. 2010;70:9473–9482. doi: 10.1158/0008-5472.CAN-10-2746. [DOI] [PubMed] [Google Scholar]
- 48.Nasreen N, Mohammed KA, Lai Y, Antony VB. Receptor EphA2 activation with ephrinA1 suppresses growth of malignant mesothelioma (MM) Cancer Lett. 2007;258:215–222. doi: 10.1016/j.canlet.2007.09.005. [DOI] [PubMed] [Google Scholar]
- 49.Leard LE, Broaddus VC. Mesothelial cell proliferation and apoptosis. Respirology. 2004;9:292–299. doi: 10.1111/j.1440-1843.2004.00602.x. [DOI] [PubMed] [Google Scholar]
- 50.Metcalf RA, Welsh JA, Bennett WP, Seddon MB, Lehman TA, Pelin K, Linnainmaa K, Tammilehto L, Mattson K, Gerwin BI, et al. p53 and Kirsten-ras mutations in human mesothelioma cell lines. Cancer Res. 1992;52:2610–2615. [PubMed] [Google Scholar]
- 51.Kozopas KM, Yang T, Buchan HL, Zhou P, Craig RW. MCL1, a gene expressed in programmed myeloid cell differentiation, has sequence similarity to BCL2. Proc Natl Acad Sci U S A. 1993;90:3516–3520. doi: 10.1073/pnas.90.8.3516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Krajewski S, Bodrug S, Krajewska M, Shabaik A, Gascoyne R, Berean K, Reed JC. Immunohistochemical analysis of Mcl-1 protein in human tissues. Differential regulation of Mcl-1 and Bcl-2 protein production suggests a unique role for Mcl-1 in control of programmed cell death in vivo. Am J Pathol. 1995;146:1309–1319. [PMC free article] [PubMed] [Google Scholar]
- 53.Varin E, Denoyelle C, Brotin E, Meryet-Figuiere M, Giffard F, Abeilard E, Goux D, Gauduchon P, Icard P, Poulain L. Downregulation of Bcl-xL and Mcl-1 is sufficient to induce cell death in mesothelioma cells highly refractory to conventional chemotherapy. Carcinogenesis. 2010;31:984–993. doi: 10.1093/carcin/bgq026. [DOI] [PubMed] [Google Scholar]
- 54.Derenne S, Monia B, Dean NM, Taylor JK, Rapp MJ, Harousseau JL, Bataille R, Amiot M. Antisense strategy shows that Mcl-1 rather than Bcl-2 or Bcl-x(L) is an essential survival protein of human myeloma cells. Blood. 2002;100:194–199. doi: 10.1182/blood.v100.1.194. [DOI] [PubMed] [Google Scholar]
- 55.Cho-Vega JH, Rassidakis GZ, Admirand JH, Oyarzo M, Ramalingam P, Paraguya A, McDonnell TJ, Amin HM, Medeiros LJ. MCL-1 expression in B-cell non-Hodgkin’s lymphomas. Hum Pathol. 2004;35:1095–1100. doi: 10.1016/j.humpath.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 56.Sieghart W, Losert D, Strommer S, Cejka D, Schmid K, Rasoul-Rockenschaub S, Bodingbauer M, Crevenna R, Monia BP, Peck-Radosavljevic M, Wacheck V. Mcl-1 overexpression in hepatocellular carcinoma: a potential target for antisense therapy. J Hepatol. 2006;44:151–157. doi: 10.1016/j.jhep.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 57.Adams KW, Cooper GM. Rapid turnover of mcl-1 couples translation to cell survival and apoptosis. J Biol Chem. 2007;282:6192–6200. doi: 10.1074/jbc.M610643200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McManus MT. MicroRNAs and cancer. Semin Cancer Biol. 2003;13:253–258. doi: 10.1016/s1044-579x(03)00038-5. [DOI] [PubMed] [Google Scholar]
- 59.Llambi F, Green DR. Apoptosis and oncogenesis: give and take in the BCL-2 family. Curr Opin Genet Dev. 2011;21:12–20. doi: 10.1016/j.gde.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thomas LW, Lam C, Edwards SW. Mcl-1; the molecular regulation of protein function. FEBS Lett. 2010;584:2981–2989. doi: 10.1016/j.febslet.2010.05.061. [DOI] [PubMed] [Google Scholar]
- 61.Kazi A, Sun J, Doi K, Sung SS, Takahashi Y, Yin H, Rodriguez JM, Becerril J, Berndt N, Hamilton AD, Wang HG, Sebti SM. The BH3 alpha-helical mimic BH3-M6 disrupts Bcl-X(L), Bcl-2, and MCL-1 protein-protein interactions with Bax, Bak, Bad, or Bim and induces apoptosis in a Bax-and Bim-dependent manner. J Biol Chem. 2011;286:9382–9392. doi: 10.1074/jbc.M110.203638. [DOI] [PMC free article] [PubMed] [Google Scholar]