

Abstract
Peters Plus syndrome (PPS), a congenital disorder of glycosylation, results from recessive mutations affecting the glucosyltransferase B3GLCT, leading to congenital corneal opacity and diverse extra-ocular manifestations. Together with the fucosyltransferase POFUT2, B3GLCT adds Glucoseβ1-3Fucose disaccharide to a consensus sequence in thrombospondin type 1 repeats (TSRs) of several proteins. Which of these target proteins is functionally compromised in PPS is unknown. We report here that haploinsufficiency of murine Adamts9, encoding a secreted metalloproteinase with 15 TSRs, leads to congenital corneal opacity and Peters anomaly (persistent lens-cornea adhesion), which is a hallmark of PPS. Mass spectrometry of recombinant ADAMTS9 showed that 9 of 12 TSRs with the O-fucosylation consensus sequence carried the Glucoseβ1-3Fucose disaccharide and B3GLCT knockdown reduced ADAMTS9 secretion in HEK293F cells. Together, the genetic and biochemical findings imply a dosage-dependent role for ADAMTS9 in ocular morphogenesis. Reduced secretion of ADAMTS9 in the absence of B3GLCT is proposed as a mechanism of Peters anomaly in PPS. The functional link between ADAMTS9 and B3GLCT established here also provides credence to their recently reported association with age-related macular degeneration.
Anterior segment dysgenesis (ASD) encompasses a group of congenital eye disorders which are an important cause of severe visual loss in children. Typically, one or more ocular anterior segment structures, such as the cornea, lens, iris, ciliary body, and/or aqueous humor drainage apparatus are malformed. The observed congenital anomalies can include corneal opacity, iris hypoplasia, developmental cataract, and adhesions between the iris, cornea and lens, occurring alone or in combination1. Mild ASD with abnormalities in the aqueous humor drainage structures can result in glaucoma and subsequent visual impairment2. FOXC1, PITX2, PITX3, FOXE3, PAX6, CYP1B1, and COL4A1 mutations are known causes of ASD3,4,5,6, and these genes also participate in ocular morphogenesis in mice7,8,9,10,11,12,13, but they do not account for all genetic forms of ASD.
A canonical subtype of ASD, named Peters anomaly, constituting corneal opacity, defects in the posterior layers of the cornea, and lenticulo-corneal and/or irido-corneal adhesions14, is the most common cause of congenital corneal opacity15. It is thought to arise from defective separation of the lens from the surface ectoderm during early eye development. Peters Plus Syndrome (PPS, MIM261540) combines ASD, primarily Peters anomaly, with short stature and brachydactyly, as well as a variable incidence of cleft palate, intellectual disability, genitourinary anomalies and heart defects16,17. Classic PPS (obligate triad of Peters anomaly, short stature and brachydactyly) is due to mutations in B3GLCT18,19, previously known as B3GALTL, which encodes a β1,3-glucosyltransferase, and is thus a congenital disorder of glycosylation. B3GLCT is an exquisitely specific enzyme, which appends glucose to O-linked fucose (O-fucose) added by protein O-fucosyltransferase 2 (POFUT2) only to thrombospondin type 1 repeats (TSRs). Together, these two enzymes generate a Ser/Thr-linked Glucoseβ1-3Fucose disaccharide20. POFUT2 and B3GLCT act in the endoplasmic reticulum and exclusively modify correctly folded TSRs, providing a quality control mechanism regulating the secretion of TSR-containing proteins21,22,23. The specificity of B3GLCT is pre-determined by POFUT2, which modifies Ser/Thr residues (underlined) within the defined consensus sequence CXX(S/T)CXXG in TSRs, which is found in 49 proteins encoded by the human and mouse genomes24. Although POFUT2 is related to POFUT1, which adds fucose to epidermal growth-factor-like repeats and is involved in Notch signaling, it has exquisite specificity for TSRs and is not involved in Notch signaling. ASD in PPS likely results from impaired secretion, and thus, a functional loss of one or more TSR-containing proteins indispensable for proper ocular morphogenesis, but which of these proteins are essential in this context is presently undetermined. Furthermore, modification by B3GLCT has only been investigated in a few TSR-containing proteins21. Of the 49 TSR-containing proteins having the requisite consensus sequence, ADAMTS (a disintegrin-like and metalloproteinase domain with thrombospondin type 1 motif) proteins are the most numerous, with 26 predicted targets, including 19 secreted proteinases and 7 ADAMTS-like (ADAMTSL) proteins that are not proteinases25. ADAMTS proteins have been established to have diverse and crucial roles in development and human disease via analysis of human and animal genetic disorders and engineered mutations26.
ADAMTS9 and ADAMTS20 are highly homologous and evolutionarily conserved, reflecting duplication of an ancestral gene27. Adamts9LacZ/LacZ embryos and embryos arising from germline Cre deletion of an Adamts9 floxed allele (Adamts9del/del) die prior to eye development28,29. Adamts20 Belted (bt/bt) mutant mice are normal but for a white spotting defect and delayed closure of the secondary palate that leads to a low incidence of cleft palate28,30. Adamts9 haploinsufficient (Adamts9del/+ or Adamts9LacZ/+) mice survive and are fertile, although they have cardiovascular abnormalities31. Adamts20bt/bt; Adamts9LacZ/+ and Adamts20bt/bt; Adamts9del/+ mice die at birth as a result of cleft palate28. Here, we demonstrate using Adamts9del/+ mice, that the corneal opacity originally noted postnatally in Adamts9LacZ/+ eyes32 is of developmental origin, a result of ASD that includes Peters anomaly and lens abnormalities. Therefore, we investigated O-fucosylation of ADAMTS9 to ask whether its deficiency could be a possible mechanism for ASD in PPS patients. We show that ADAMTS9 modification by B3GLCT is required for its secretion, which together with Peters anomaly observed in Adamts9+/− mice, and its previously defined roles in cardiac and palate development elicited in combination with ADAMTS20, potentially link ADAMTS9 mechanistically to PPS.
Results
Adamts9 haploinsufficiency leads to anterior segment dysgenesis (ASD)
An Adamts9 germline mutant (Adamts9del) was generated using a previously described floxed Adamts9 allele and maintained in the C57BL/6 background29. As previously seen in Adamts9LacZ/+ mice32, Adamts9del/+ mice had corneal opacities with high penetrance (Fig. 1a and Supplementary Fig. S1a), which were discernible as soon as their eyelids opened (~2 weeks of age). Opacities were bilateral in about half the affected mice, and unilateral in the other half with preferential involvement of the right eye (Supplementary Fig. S1a). The corneal opacities were of variable severity, ranging from faint central clouding to opacity of the entire cornea (Fig. 1a). A central corneal contour anomaly was visible externally in some eyes with corneal opacity (Fig. 1a, central panels). Moreover, Adamts9del/+ eyes were significantly smaller than Adamts9+/+ eyes (Fig. 1b). Optical coherence tomography (OCT) at 3 weeks of age showed a flattened cornea and a shallow anterior chamber in Adamts9del/+ mice compared to the wild-type littermates, comprising little more than a potential space in the most severely affected eyes (Fig. 1c,d). Although the anterior chamber volume was reduced in most Adamts9del/+ eyes, comparison with wild-type chamber volumes did not reach statistical significance owing to the considerable variability of ASD and the presence of buphthalmos (enlarged eye) in at least one Adamts9del/+ eye (Fig. 1e). Peters anomaly, never seen in the wild-type eyes, was identified by OCT in 3-week-old Adamts9del/+ eyes having the central corneal contour anomaly and confirmed histologically by observation of lens-cornea adhesion with disrupted posterior corneal layers, and iridocorneal adhesions (anterior synechiae) (Fig. 1c,f, Table 1). Adamts9del/+ eyes had a smaller lens (Fig. 1g) and some eyes showed ciliary body hypoplasia/dysplasia, signs of vacuolar cataract and persistence of lens fiber nuclei in the posterior lens, none of which were seen in wild-type littermates (Table 1, Supplementary Fig. S1b–d). No histological anomalies of the retina, RPE and choroid were evident in Adamts9del/+ eyes.
Figure 1. Adamts9del/+ mice have a highly penetrant congenital corneal opacity resulting from ASD.

(a–g) Corneal opacity in Adamts9del/+ eyes is associated with Peters anomaly (a), a smaller eye (b), shallow anterior chamber (c–e), iridocorneal and iridolenticular adhesions (f) and a smaller lens (g). 3 week-old Adamts9+/+ and Adamts9del/+ enucleated eyes were analyzed by stereomicroscopy (a), OCT (c,d) and H&E staining of paraffin sections (f). The arrowheads and arrow indicate, respectively, the corneal opacity and the Peters anomaly. Adamts9del/+ eyes had a significantly smaller diameter than Adamts9+/+ eyes (b). Anterior chamber volume was determined from OCT data using the Amira segmentation tool (d). Anterior chamber volumes were highly variable in Adamts9del/+ eyes while quite constant in Adamts9+/+ eyes (e). Lens area was measured from H&E stained sections and was significantly smaller in Adamts9del/+ eyes as compared to Adamts9+/+ eyes (g). The images are representative of 6 Adamts9+/+ and 12 Adamts9del/+ eyes analyzed by OCT. Scale bar = 1 mm. Significance was determined using a 2-tailed student’s t test (*p < 0.05).
Table 1. Summary of eye phenotypes in Adamts9del/+ mice.
| Phenotype | Smaller lens | Peters anomaly | Posterior lens extrusions | Disorganized corneal stroma | Lens-cornea adhesion | Iris-cornea adhesion | ciliary body dysplasia | Vacuolar cataract | Persistent nuclei in posterior lens |
|---|---|---|---|---|---|---|---|---|---|
| Newborn | 15/17 | 1/17 | 16/17 | 13/17 | N/A | N/A | N/A | 3/17 | 1/17 |
| 10 day-old | 4/6 | 0/6 | 6/6 | 5/6 | 4/6 | 5/6 | 3/6 | 0/6 | 2/6 |
| 3 week-old | 8/10 | 2/10 | 10/10 | 7/10 | 7/10 | 7/10 | 7/10 | 0/10 | 2/10 |
These data are based on observations from H&E stained sections (see also Supplementary Fig. S1b–d). N/A, not available. Note that the fused eyelids in the newborn push the cornea toward the lens, precluding proper visualization of the anterior chamber. Anterior chamber development is not complete until 2–3 weeks, e.g., the ciliary body is not yet developed in the newborn.
Adamts9 is expressed at specific sites during mouse eye development
To determine the role of Adamts9 in these anomalies, we defined its spatio-temporal expression pattern in the eye during development (Fig. 2a). At gestational age 10.5 days (E10.5), i.e., shortly after lens vesicle formation, Adamts9 (red signal) was expressed strongly in the optic cup, preponderantly at its anterior pole, with weak Adamts9 expression in the invaginating lens vesicle. E11.5 eyes showed the strongest Adamts9 expression of all developmental stages analyzed, localizing mRNA mostly in the anterior pole of the developing retina/optic cup and more faintly, in the hyaloid vascular plexus and the lens vesicle. At E12.5 and E13.5, strong Adamts9 expression persisted in the anterior pole of the retina. After E12.5, Adamts9 mRNA was no longer detected in the lens, but was present in endothelial cells of the tunica vasculosa lentis and vasa hyaloidea propria, and in the choroid and sclera. In newborn eyes, Adamts9 mRNA was most strongly expressed in the ciliary margin zone and prospective ciliary body, sclera, corneal keratocytes and choroidal vasculature (Fig. 2a). This mRNA distribution pattern was similar to β-galactosidase staining elicited from the intragenic lacZ reporter in Adamts9Lacz/+ mice (Supplementary Fig. S2).
Figure 2. Adamts9 mRNA is expressed during murine eye development consistent with a developmental sequence of ASD in Adamts9del/+ eyes.

(a) In situ hybridization using an Adamts9 probe was performed on E10.5 through newborn (P0) Adamts9+/+ eyes. Adamts9 mRNA expression is visualized as red dots overlying cells, against a purple hematoxylin counterstain. Adamts9 was expressed throughout this period in the anterior pole of the retina (arrows) and hyaloid vasculature (single asterisk). Adamts9 expression was also observed in the lens and the lens epithelium (arrowheads) and in mesenchymal cells within the developing cornea (double asterisk). L = lens, R = optic cup/retina, I = iris, C = cornea. The boxed areas in the upper panels are shown at higher magnification (lower black boxed panels). Scale bar = 50 μm; blue scale bar in P0 images = 250 μm. (b,c) Sections from Adamts9+/+ and Adamts9del/+ eyes from E14.5 to P0 were stained by H&E and the lens area was measured. At E14.5 Adamts9+/+ and Adamts9del/+ eyes were of comparable size. Posterior to the lens, abnormal material (indicated by an arrow) was observed in the E16.5 and P0 Adamts9del/+ eyes shown here (b) but never in Adamts9+/+ eyes. At E16.5 and P0, the Adamts9del/+ lens was significantly smaller than the Adamts9+/+ lens (c). Images are representative of at least 3 eyes analyzed at each time point. L = lens. Scale bar = 100 μm. Significance was determined using a 2-tailed Student’s t test (*p < 0.05).
Adamts9 haploinsufficiency affects lens capsule integrity, its composition, and lens growth
To determine the pathogenic sequence of ASD, and because of continuous expression of Adamts9 during eye development, Adamts9del/+ and Adamts9+/+ eyes were compared histologically at different developmental stages. H&E staining of E14.5 and older Adamts9del/+ eyes revealed eosinophilic globules adjacent to the posterior aspect of the lens (Fig. 2b, Supplementary Fig. S1e). In contrast, no differences in lens morphology were evident from E10.5–E12.5 (Supplementary Fig. S1f). From E15.5, Adamts9del/+ eyes were smaller than wild-type eyes with a disproportionately smaller lens (Fig. 2b,c and Supplementary Fig. S3a,b).
The retro-lenticular globules noted from E14.5 were identified as arising from extruded lens fibers using anti-γ-crystallin antibody, suggesting impaired lens capsule integrity (Fig. 3a). Therefore, we characterized the composition and structure of the lens capsule. Periodic acid-Schiff (PAS) staining, an indicator of lens capsule glycoprotein content, was subtly reduced and diffuse in the E16.5 Adamts9del/+ lens capsule (Fig. 3b, insets in left-hand column), although relatively normal in newborn (post-natal (P) 0) and 3-week old (P21) Adamts9del/+ eyes (Fig. 3b, center and right-hand columns). At E16.5, collagen IV immunostaining (Fig. 3c) and laminin immunostaining (Fig. 3d) were conspicuously reduced throughout the circumference of the lens capsule in Adamts9del/+ eyes. At P0, the reduction in collagen IV and laminin immunostaining in the Adamts9del/+ lens capsule was less pronounced than at E16.5 (Fig. 3c,d, center panels). At P21, the lens capsule showed regions where collagen IV and laminin immunostaining of the lens capsule were interrupted (Fig. 3c,d, right-hand panels). In contrast, collagen IV and laminin staining in capillaries comprising the tunica vasculosa lentis, the vascular network adjacent to the embryonic lens, were not altered in Adamts9del/+ eyes (Fig. 3c,d).
Figure 3. Adamts9 haploinsufficiency leads to posterior lens extrusions owing to a defective lens capsule.

(a) E12.5 to E16.5 Adamts9del/+ and Adamts9+/+ eyes were stained using an antibody against γ-crystallin which identified the extrusions lying posterior to the lens as extruded lens fibers (arrows). Images are representative of at least 3 eyes analyzed for each time point. L = lens. Scale bar = 25 μm. (b–d) E16.5, P0 and P21 Adamts9del/+ and Adamts9+/+ eyes were stained by the periodic acid Schiff (b, PAS stain, magenta, indicative of glycoproteins) or using antibodies directed against collagen IV (c, red) or laminin (d, red). Collagen IV and laminin immunostaining of the lens capsule (arrow) had reduced intensity in Adamts9del/+ eyes as compared to Adamts9+/+ eyes, most evident in E16.5 embryos. However, the capillary basement membrane of vessels comprising the tunica vasculosa lentis (asterisk) had similar staining in both genotypes. At higher magnification (framed images), segmentally weaker or discontinuous lens capsule immunostaining (arrowheads) suggestive of fenestrae, was observed in mutant eyes. The images are representative of at least 3 eyes analyzed at each time point. L = lens. Scale bar = 50 μm.
Versican, fibrillin-2 and fibronectin, which are major components of embryonic ECM, were analyzed by immunofluorescence (Supplementary Fig. S4). Although versican is a known ADAMTS9 substrate, no difference in staining intensity or distribution of versican was detected between Adamts9del/+ and Adamts9+/+ eyes at E12.5 and E14.5 (Supplementary Fig. S4). Both fibrillin-2 and fibronectin were present around the lens at E12.5, especially, in the hyaloid tissue (Supplementary Fig. S4). Fibrillin-2 immunostaining was more intense at E12.5 and E14.5 in Adamts9del/+ eyes while fibronectin immunostaining was increased in Adamts9del/+ eyes at E14.5, but not at E12.5 (Supplementary Fig. S4). In contrast, a consistent difference in fibrillin-2 or fibronectin immunostaining between Adamts9del/+ and Adamts9+/+ eyes older than E14.5 was not observed, indicating a transient alteration of hyaloid ECM from E12.5 to E14.5 (Supplementary Fig. S4).
Despite the marked differences in lens capsule immunostaining between Adamts9del/+ and Adamts9+/+ eyes at E16.5, transmission electron microscopy (TEM) at this age showed comparable lens capsule appearance, including thickness, layering and electron density over almost the entire extent of the lens capsule in Adamts9del/+ and Adamts9+/+ eyes (Fig. 4a,b). Notwithstanding this apparent normalcy, discontinuities, i.e., fenestrations in the lens capsule were also detected by TEM in Adamts9del/+ eyes (Fig. 4c,d). Posteriorly, Adamts9del/+ eyes contained extruded material continuous with lens fibers (Fig. 4c) and anteriorly, TEM revealed that lens epithelium cells had translocated through lens capsule fenestrae into the anterior chamber (Fig. 4d). The translocated cells were surrounded by an ECM with a similar ultrastructural appearance as the lens capsule, and a duplicated lens capsule was observed anterior to the ectopic cell nests (Fig. 4d). In one eye analyzed by TEM, we observed continuity of the lens fibers with corneal stroma, along with an interrupted corneal endothelium and Descemet’s membrane, together satisfying identification of Peters anomaly (Fig. 4e).
Figure 4. Transmission electron microscopy (TEM) identifies lens capsule fenestrae in E16.5 Adamts9del/+ eyes.

(a) The anterior and posterior lens capsule were of comparable appearance in Adamts9del/+ and Adamts9+/+ eyes through most of their extent. Arrowheads indicate endothelial cells of microvessels in the pupillary membrane (anterior) or the tunica vasculosa lentis (posterior). Panels are representative of 4 eyes of each genotype. Scale bar = 500 nm. (b) Anterior, equatorial and posterior lens capsule thickness was similar in Adamts9del/+ and Adamts9+/+eyes. (c) A defect in the posterior lens capsule associated with a lens fiber extrusion in an Adamts9del/+ eye (left-hand panel). The right-hand panel shows that the extruded material lying in the vitreous is membrane-bound. Scale bar = 2 μm. (d) In Adamts9del/+ eyes, lens epithelial cells traversed the discontinuous anterior lens capsule (white arrowhead) and formed nests of cells surrounded by basement membrane similar to the lens capsule. Higher magnifications of the indicated regions are shown as framed panels on the right. The asterisk in the upper panel shows the extraneous anterior layer of lens capsule formed by the ectopic cells, the center panel shows a local duplication or folding of the lens capsule, and the lower panel shows a lens epithelial cell (arrowhead) penetrating the fenestrated lens capsule. Scale bar = 2 μm. (e) In an Adamts9del/+ eye, Peters anomaly was identified by continuity of the lens with surface ectoderm across a discontinuous cornea (left-hand panel). The right-hand panel shows the resulting corneal edge at high magnification to indicate the interrupted corneal endothelium. Scale bar = 2 μm. L = lens, C = cornea, Lc = lens capsule, Le = lens extrusion, Lf = lens fibers, Ce = corneal endothelium. Significance was determined using a 2-tailed student’s t test.
Adamts9 del/+ lens epithelium undergoes epithelium to mesenchyme transition
The lens epithelium normally comprises a single layer of cells arranged in a columnar format reflecting strict apical-basal polarity, as was seen in Adamts9+/+ embryos (Fig. 5a,b). In contrast, up to 3 layers of lens epithelial cells were present in E16.5 and P0 Adamts9del/+ eyes (Fig. 5a,b). In P21 Adamts9del/+ eyes, lens epithelium cells were embedded in the corneal stroma (Fig. 5a,b). Combined with staining for laminin, DAPI stained nuclei indicated that the cells adopted aberrant orientations in mutant E16.5 eyes, indicating disruption of cell polarity (Fig. 5b, upper panel). At birth, we observed aberrant nests of cells anterior to the lens capsule and within the cornea that were morphologically distinct from corneal keratocytes, which have a flattened nucleus and an abundant cytoplasm (Fig. 5b, center panels). ECM surrounding these ectopic cells stained positive for laminin and collagen IV, indicative of ectopic basement membrane (Fig. 5b, center panel), consistent with the TEM observations of a multilayered lens epithelium and duplicated anterior lens capsule (Fig. 4d). At 21 days of age, the cell nests were more prominent than at birth, and were embedded in an abundant collagen IV and laminin (Fig. 5b, lower panel, inset). We concluded that the aberrant cell nests in the anterior chamber and corneal stroma were ectopic lens epithelium cells that continued to deposit lens capsule after E16.5. In addition, the keratocyte density was greater in the P21 Adamts9del/+ corneal stroma than the Adamts9+/+ littermates (Supplementary Fig. S5). However, cell density in the corneal stroma and corneal thickness were not significantly altered in embryonic or newborn Adamts9del/+ eyes as compared to Adamts9+/+ eyes (Supplementary Fig. S5), indicative of post-natal corneal changes.
Figure 5. Adamts9del/+ eyes have aberrant and intra-corneal nests of cells arising from epithelium-mesenchyme transformation of lens epithelium.

(a,b) E16.5, P0 and P21 Adamts9+/+ and Adamts9del/+ eyes were stained with H&E (a) or an anti-collagen IV antibody (red) and the nuclei were counterstained with DAPI (white) (b). At E16.5, a disorganized lens epithelium (arrowhead) was associated with weaker collagen IV staining in Adamts9del/+ eyes. At P0 and P21, aberrant nests of cells surrounded by collagen IV-positive ECM were observed only in the Adamts9del/+ eyes (arrow). Higher magnification of these cells in P21 Adamts9del/+ eyes is shown (inset). These images represent at least 3 eyes analyzed at each time point. L = lens, C = cornea. Scale bar = 25 μm. (c) P0 and P21 Adamts9del/+ and Adamts9+/+ eyes were stained using antibodies directed against α-smooth muscle actin (α-SMA) (red), E-cadherin (red) or N-cadherin (red) and nuclei were stained by DAPI (nucleus: white). At P0 and P21, the ectopic cells in Adamts9del/+ corneas stained positive for α-SMA, E-cadherin and N-cadherin (arrows) whereas no α-SMA expression was observed in the lens or the cornea of Adamts9+/+ mice, and E-cadherin was distinctly expressed by the lens and corneal epithelium. The corneal endothelium of Adamts9+/+ mice strongly expressed N-cadherin, but also low levels of E-cadherin. C = cornea, E = corneal endothelium, L = lens. (d) Adamts9del/+ and Adamts9+/+ mice were bred with mT/mG; Wnt1-Cre mice for lineage tracing of neural crest-derived cells (green). In adult Adamts9del/+; mT/mG; Wnt1-Cre eyes (lower panel), ectopic cells are red (arrow) and thus, unlike corneal keratocytes and corneal endothelium (green, see mT/mG; Wnt1-Cre cornea in upper panel), they do not arise from the neural crest. Scale bar = 25 μm.
Corneal invasion by lens epithelium suggested occurrence of epithelium to mesenchyme transition (EMT) of lens epithelium. During EMT, epithelial cells lose or reduce expression of epithelial markers, such as E-cadherin, and acquire mesenchymal markers, such as N-cadherin and α-smooth muscle actin (α-SMA), as well as altered polarity. In Adamts9del/+ newborn eyes, the anteriorly extruded lens epithelium cells expressed the mesenchymal markers α-SMA and N-cadherin, as well as the epithelial marker, E-cadherin (Fig. 5c). In Adamts9+/+ eyes, α-SMA staining was absent in corneal stroma and lens epithelium, whereas E-cadherin was expressed only by the lens and the corneal epithelium, and N-cadherin by the lens epithelium and lens fibers (Fig. 5c). At P21, ectopic lens epithelium in Adamts9del/+ eyes still expressed the three markers, although α-SMA immunostaining was stronger than at P0, and E-cadherin expression was reduced (Fig. 5c). In P21 Adamts9+/+ eyes, α-SMA staining was not detectable in the cornea or the lens, and E-cadherin and N-cadherin were only expressed, as expected, by lens epithelium and corneal endothelium, respectively. These changes are indicative of age-related progression of EMT of Adamts9del/+ ectopic lens epithelial cells, while retaining some characteristics of the lens epithelium. Since lens epithelium does not express Adamts9 (Fig. 2a, Supplementary Fig. S2), EMT may be secondary to lens capsule alteration and loss of appropriate lens epithelium basal contacts with the lens capsule.
To further establish the origin of the ectopic cell nests, Adamts9del/+ and Adamts9+/+ mice were crossed with Wnt1-Cre mice carrying a dual fluorescent reporter (mT/mG). In Adamts9+/+; mT/mG; Wnt1-Cre eyes, neural crest-derived cells (e.g., keratocytes in the cornea and corneal endothelium) were marked by green fluorescence, reflecting excision of the Td Tomato reporter (red) by Cre and a switch to GFP expression, whereas corneal epithelium (Fig. 5d, top panel) and lens epithelium displayed constitutive red fluorescence. In Adamts9del/+; mT/mG; Wnt1-Cre eyes, the aberrant cell nests in the anterior chamber exhibited red fluorescence, demonstrating that in contrast to corneal stroma (green), these cells were not neural crest-derived (Fig. 5d, lower panel). Taken together with the presence of lens capsule components detected by immunostaining around these cells, and lens epithelium-like morphology in TEM (Fig. 4) we suggest that the ectopic cells in mutant cornea arise from lens epithelium by EMT.
POFUT2 and B3GLCT modify ADAMTS9 to regulate its secretion
ADAMTS9 contains 15 TSRs, of which 12 contain the O-fucosylation consensus motif, CXX(S/T)CXXG (Fig. 6a). Full-length ADAMTS9 is secreted into the medium of transfected cells at levels too low to permit efficient recombinant protein purification, and natural sources of this molecule are unavailable. Therefore, to determine whether ADAMTS9 TSRs are modified by POFUT2 and B3GLCT, we generated two recombinant human ADAMTS9 constructs (Fig. 6a), that, between them, include all its TSRs, for analysis by mass spectral glycoproteomic methods22,23. These constructs, containing the first 8 TSRs (hADAMTS9-N-L2) or TSR9-15 (hADAMTS9 TSR9-15) (Fig. 6a), were purified from the conditioned medium of stably transfected HEK cells and were subjected to tryptic digestion. Mass spectral analysis of the peptides confirmed modification of TSR5-9, TSR11-13 and TSR15 by POFUT2 and B3GLCT (Fig. 6b, Supplementary Table S1 and Supplementary Fig. S6). Semi-quantitative analysis by extracted ion chromatography (EIC) showed the fully modified, Glucoseβ1-3Fucose disaccharide forms of the peptides to be the most abundant (Fig. 6b). Since POFUT2 and B3GLCT are the only known enzymes capable of adding these two sugars to TSRs33,34,35,36,37, these data strongly support the conclusion that ADAMTS9 is modified by both POFUT2 and B3GLCT. Whether or not O-fucosylation governed quality control of ADAMTS9 for secretion was determined by knockdown of POFUT2 or B3GLCT mRNA using specific siRNAs and a scrambled siRNA as a control in HEK cells. The secreted ADAMTS9-N-L2 levels in the medium of POFUT2, B3GLCT or control siRNA transfected cells were compared relative to the impact on secretion of a co-transfected plasmid expressing IgG (Fig. 7a, complete gel is shown in Supplementary Fig. S7). As previously seen, ADAMTS9-N-L2 migrated more rapidly in the medium of transfected cells than in the cell lysate (Fig. 7a, Supplementary Fig. S7) because of furin-mediated excision of the N-terminal propeptide (Fig. 7a) which reduces molecular mass by ∼25kDa38. Knockdown of POFUT2 led to a nearly complete loss of ADAMTS9-N-L2 secretion compared to the scrambled siRNA (Fig. 7a,b, Supplementary Fig. S7). Knockdown of B3GLCT also caused a statistically significant reduction of ADAMTS9 secretion (Fig. 7b). In contrast, knockdown of POFUT2 or B3GLCT had no effect on secretion of transfected IgG chain, which contains no TSRs (Fig. 7a,b, Supplementary Fig. S7). Thus, modification of ADAMTS9 with the Glucoseβ1-3Fucose disaccharide was required for its optimal secretion. Although ADAMTS9 carrying only O-fucose was also secreted after B3GLCT knockdown, it was at much lower levels than in the control cells (Fig. 7a,b).
Figure 6. Specific ADAMTS9 TSRs are O-fucosylated.

(a) Domain structure of ADAMTS9 and of the ADAMTS9 constructs used for mass spectrometry (hADAMTS9-N-L2 and hADAMTS9 TSR9-15). TSRs that carry a consensus sequence for O-fucosylation are shown in red. (b) Extracted ion chromatograms showing the relative amounts of the unmodified form (black line), O-fucose monosaccharide form (red line, fucose indicated by red triangle), and Glucoseβ1-3Fucose disaccharide form (blue line, glucose indicated by blue circle) of peptides from TSRs 5–9, TSRs11-13 and TSR15 of ADAMTS9 generated using the corresponding masses listed in Appendix Table S1. Note that lack of data for TSRs 1, 4, and 14 does not indicate that they are unmodified, but that the ions corresponding to those sites were not detected. (*=contaminating peptide).
Figure 7. POFUT2 is indispensable for ADAMTS9 secretion whereas B3GLCT is required for optimal secretion.
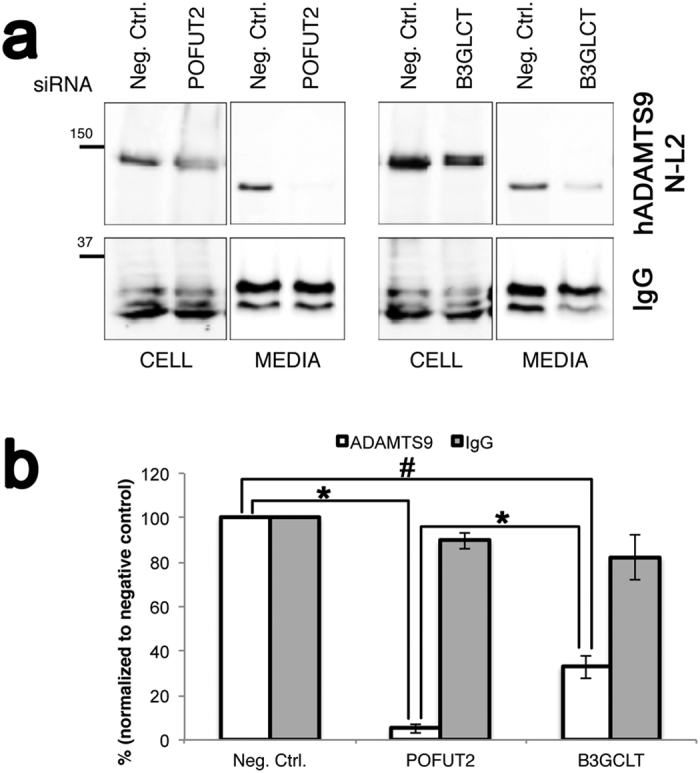
(a) HEK293T cells were co-transfected with siRNA targeting POFUT2, or B3GLCT, or a non-targeting control (Neg. Ctrl.), and hADAMTS9-N-L2 or human IgG heavy chain. Media and lysates were analyzed by western blotting with anti-myc (hADAMTS9-N-L2) or anti-human IgG. ADAMTS-N-L2 in medium migrates more rapidly compared to the cell lysate because the N-terminal propeptide (~25 kDa) is excised by furin at the cell-surface. The western blot images are cropped from gels which were provided for review as Supplementary information (Supplementary Fig. S7). (b) Quantitation of data from the western blot is shown relative to the control siRNA (Neg. Ctrl.). Data are mean+/− standard deviation (n = 3). Significance was determined using a 2-tailed Student’s t test (*p < 0.005, #p < 0.001).
Although the present studies clearly demonstrated ADAMTS9 post-translational modification by POFUT2 and B3GLCT, and B3GLCT has long been known as the causative gene in PPS, there is little known about the spatial and temporal regulation of expression of these transferases during eye developement. Because this information is crucial for providing a physiological context for the action of these enzymes on ADAMTS9, we determined their mRNA expression pattern using ISH. As shown in Supplementary Fig. S8, both Pofut2 and B3glct were expressed in the developing eye from E11.5 to birth. Their mRNAs were widely expressed, being evident in the optic cup/retina, lens epithelium, cornea and peri-ocular mesenchyme. They not only overlap entirely with Adamts9 mRNA distribution (Fig. 2a, Supplementary Fig. S2), but show a broader expression than Adamts9 mRNA consistent with their having 48 other potential targets, which may each have very different expression patterns. We conclude that Pofut2 and B3glct mRNAs are expressed in the very same cells that express Adamts9 and are therefore, physiologically relevant to ADAMTS9 post-translational modification and secretion in the eye.
Discussion
We demonstrate here that congenital corneal opacity and Peters anomaly results from Adamts9 haploinsufficiency in mice. Because enzyme deficiencies are typically recessive in their manifestation, the hemizygous impact of Adamts9 is remarkable and implies a major role for this gene in ocular morphogenesis. Corneal opacity occurs in two distinct Adamts9 alleles, Adamts9del/+ and Adamts9LacZ/+, but not in mice hemizigous for a hypomorphic gene trap allele Adamts9GT, which has ADAMTS9 function intermediate between the wild-type and haploinsufficient39. Together, these observations indicate that a critical dosage threshold of ADAMTS9 is involved in eye development. Adamts9LacZ/+ and Adamts9del/+ alleles differed in the timing of detection of the ocular defect. In Adamts9LacZ/+ mice, corneal opacity was evident in only 20% after 5 weeks of age and in 80% after 25 weeks of age32, whereas in Adamts9del/+ mice, the corneal opacity was obvious as early as 2 weeks of age and present in nearly all mice by 3 weeks. Although Peters anomaly was not observed in Adamts9LacZ/+ mice, anterior synechiae (lens-iris-cornea adhesions) and lens extrusions similar to those detected in Adamts9del/+ mice were present. These differences could result from different targeting strategies employed in the two alleles or slightly different genetic backgrounds of the two strains. The Adamts9LacZ/+ allele was generated in a hybrid genetic background (129Sv X C57Bl/6) by targeting exon 3 (encoding the propeptide), and was backcrossed for 10 generations into the C57Bl/6 background32, whereas the Adamts9del/+ allele was generated and subsequently maintained in the C57Bl/6 background, and it was engineered to lack exons 5-8 (encoding the catalytic domain)29. Each targeted allele results in a frame-shift and the Adamts9 mRNA, if stable, would generate only the N-terminal propeptide in the mutants, to which no innate activity has been ascribed in any ADAMTS protease. Consistent with a possible role for the genetic background, the ocular phenotype was not initially apparent in 129Sv X C57Bl/6 Adamts9LacZ/+ mice, and was only noted after six to eight crosses into the C57Bl/6 strain32. Although it was previously reported that eye abnormalities occur spontaneously at low frequency in C57Bl/6 mice, and have a strong predilection for the right side40,41, we did not notice eye anomalies in wild type littermates and the preponderance of anomalies in the right eye of Adamts9del/+ mice is presently unexplained.
Although Adamts9del/+ eyes do not exhibit morphologic anomalies prior to E12.5, the presence of Peters anomaly indicates that lens separation, which occurs at E11, is compromised in a significant proportion of eyes. After the lens separation period, Adamts9del/+ eyes consistently showed impaired lens growth and loss of structural integrity of the lens capsule resulting in lens fiber extrusion posteriorly, and migration of the lens epithelium into the cornea anteriorly through an EMT-like process. The lens capsule is a specialized and exceptionally thick basement membrane composed mainly of collagen IV, laminin, entactin/nidogen and perlecan42. It is deposited by lens epithelium anteriorly and lens fibers posteriorly as successive basement membrane layers during development. PAS stain and immunostaining demonstrated that the lens capsule composition was transiently altered during development of Adamst9del/+ eyes. Other mouse mutants reported to have lens extrusions and/or outgrowth of lens epithelium had defined defects in the lens capsule components, i.e., perlecan43 and laminin44. Col4a1 mutant mice develop ASD, although lens fiber extrusions through the lens capsule were not described13,45. We conclude that ADAMTS9 may participate in stabilization of basement membrane or in lens capsule remodeling during lens growth. Such a role, if compromised, could also have an adverse effect on “pinching off” of the lens and result in Peters anomaly. Interestingly, mice deficient in peroxidasin, an enzyme responsible for collagen IV crosslinking through formation of sulfilimine bonds46, developed ASD associated with a small lens, loss of lens capsule integrity and posterior and anterior lens extrusions47. These mice also have white spotting closely resembling the pigmentation defect in Adamts20bt/bt mice48 which is exacerbated in Adamts20bt/bt;Adamts9del/+ newborns28. We speculate that these findings could suggest that ADAMTS9 and peroxidasin may work in the same developmental pathway, a potential future direction for this work. Adamts9 haploinsufficiency may affect assembly of collagen IV or laminin subunits expressed in the embryonic period, such as laminin α1, which is essential for embryonic lens capsule development, and collagen IV assemblies with chain composition α1α1α2:α1α1α2 and α1α1α2:α5α5α6, which are expressed in the embryonic lens capsule and replaced at birth by collagen IV with the chain composition α3α4α5:α3α4α542. The ADAMTS9 substrate versican is present in the hyaloid space from E10-E14, but its staining in the mutant eyes was not consistently different from the wild-type.
A proper interaction of the lens epithelial cells with the lens capsule at their basal aspect maintains their polarity, as demonstrated in integrin and integrin-linked kinase knockout mice49,50,51. Our findings indicate that lens epithelium cells transitioned to an intermediate EMT state between P0 and P21, since they deposited lens capsule-like basement membrane, identified by immunostaining and TEM analysis and retained E-cadherin expression, yet lost their polarity and acquired α-SMA and N-cadherin expression. In addition to Peters anomaly, corneal invasion by lens epithelium, lens capsule deposition in the corneal stroma, as well as increased keratocyte density are together likely to be significant contributors to corneal opacity. We propose that the consecutive changes we observed comprise a pathogenic sequence arising from a flawed lens capsule.
Of 49 known B3GLCT targets, ADAMTS9 is the first to be associated with Peters anomaly. The similarities between the ocular phenotype of Adamts9del/+ mice and the ocular phenotype of PPS in humans suggests ADAMTS9 as the first B3GLCT substrate whose impairment potentially explains the ocular anomalies of PPS. In strong support of this possibility, we have further shown, 1. That several ADAMTS9 TSRs are O-fucosylated, 2. That B3GLCT or POFUT2 knockdown, i.e. defective O-fucosylation, is essential for ADAMTS9 secretion and 3. That Adamts9, B3glct and Pofut2 have overlapping expression in the eye throughout development. POFUT2 and B3GLCT likely affect ADAMTS9 secretion by a quality control process occurring in the ER that was previously established by analysis of ADAMTSL1, ADAMTSL2 and ADAMTS13, which showed that the modifying enzymes recognized properly folded TSRs and stabilized them by glycosylation21. Because TSR modification by B3GLCT is dependent on prior attachment of O-linked fucose, POFUT2 knockdown inevitably affects both monosaccharide and disaccharide forms of O-fucosylation. Consistent with the severe reduction of ADAMTS9 secretion upon POFUT2 knockdown, Pofut2 null embryos, like Adamts9 nulls embryos, die in early development with essentially identical phenotypes37,52, although Pofut2+/− mice do not have eye anomalies. In contrast, knockdown of B3GLCT reduced, but did not eliminate ADAMTS9 secretion in vitro, predicting a milder outcome than loss of POFUT2. Although B3glct deficient mice are currently unavailable for comparison with Pofut2 and Adamts9-deficient mice, survival of humans with PPS supports the milder-than-expected outcome of B3GLCT than POFUT2 deficiency. ADAMTS9, in addition to the ASD reported here, has additional relevance to PPS. Notably, Adamts9 haploinsufficiency resulted in cardiovascular defects and, in combination with Adamts20 deficiency, cleft palate, which are anomalies seen frequently in PPS28.
Since Adamts9del/+ mice were of normal size and limb-specific Adamts9 conditional deletion did not affect limb length29, impairment of ADAMTS9 does not explain short stature in PPS. PPS is predicted to be a composite phenotype resulting from reduced secretion (and therefore functional loss) of a subset of developmentally crucial B3GLCT substrates from among 49 predicted targets. Of these, not all are likely to be subjected to quality control by B3GLCT, so the number of functionally impaired targets in PPS is likely to be even smaller. Which are some of the other likely participants in PPS from among the 26 target ADAMTS proteins? Geleophysic dysplasia, which is caused by recessive ADAMTSL2 mutations, results in short stature, and brachydactyly, which are two major defining characteristics of PPS, and affected individuals have similar facial features to PPS17,53. Appropriately, B3GLCT knockdown abolished ADAMTSL2 secretion21. ADAMTS10 mutations lead to short stature and brachydactyly in Weill-Marchesani syndrome54, whereas ADAMTS17 mutations lead to short stature55. Thus, ADAMTS10 and ADAMTS17 are potentially relevant to PPS, but the effect of B3GLCT on their secretion is presently unknown. On the other hand, some potential targets could be excluded. ADAMTS2 and ADAMTS13 are required for skin integrity and hemostasis respectively, but neither function is impaired in PPS, and, indeed, ADAMTS13 is not subject to B3GLCT quality control21. From these observations, it seems reasonable to conclude that impairment of ADAMTS9 (in the eye, palate and heart) and ADAMTSL2 (in skeletal growth) contributes to the PPS phenotype, but that other relevant targets remain to be evaluated.
Intriguingly, both ADAMTS9 and B3GLCT were recently identified in genome-wide association studies as loci linked to age-related macular degeneration56,57,58. The direct functional link between their protein products demonstrated here for the first time, expression of Adamts9 in the microvasculature32, and its role in eye development strengthens this bi-allelic association. Future functional analysis of ADAMTS9 and B3GLCT in age-related macular degeneration is thus warranted. In addition, the mouse eye phenotype reported here suggests consideration of ADAMTS9 itself as a candidate gene for ASD. Although Adamts9 mRNA was strongly expressed in the anterior pole of the optic cup, especially in E11.5 eyes, the current study demonstrated severe ASD, and not retinal anomalies. Because the eye develops from crosstalk between the optic cup and lens ectoderm, this suggest that ADAMTS9, a cell-surface and secreted protease produced mainly by the optic cup and hyaloid plexus, may well act in trans on the lens vesicle. Future experiments using Adamts9 conditional mutagenesis in specific ocular structures or embryonic lineages contributing to the eye will be insightful in this regard.
Materials and Methods
Mouse alleles and genotyping
Mice with targeted germline inactivation of Adamts9 (referred to as Adamts9del/+) using the Adamts9 floxed allele (RRID: JAX_026103) and their genotyping were described previously29. Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo mice59 (RRID: MGI_3722405), referred to as mT/mG, were from Jackson Laboratories (Bar Harbor, ME). Detection of the mT/mG transgene was done using PCR primers recommended by Jackson Laboratories (http://jaxmice.jax.org/). Wnt1-cre mice (B6.Cg-Tg(Wnt1-cre)11Rth Tg(Wnt1-GAL4)11Rth/J; Jackson Laboratories, Bar Harbor, ME; RRID: IMSR_JAX:009107) were bred with mT/mG mice and Adamts9del/+ mice to obtain Adamts9del/+ and Adamts9+/+ mice with conditional deletion of the membrane-bound TdTomato reporter and expression of GFP in neural crest cell-derived tissues. Wnt1-Cre mice were genotyped using the following primers: Wnt1 forward = 5′-TAAGAGGCCTATAAGAGGCGG-3′; Wnt1 reverse = 5′-CGCATAACCAGTGAAACAGCATTGC-3′. For timed pregnancies, the date of the vaginal plug was designated E (embryonic age) 0.5. All analyses of Adamts9del/+ eyes used wild-type littermates as controls.
Gross Morphology and Optical Coherence Tomography
We evaluated adult mouse eyes for the presence of leukoma (corneal opacity) using a Leica MZ6 stereomicroscope coupled to an Insight Spot2 FireWire camera (Diagnostic Instruments, Inc, Sterling Heights, MI) immediately after dissection and immersion in PBS. E12.5 and E15.5 mouse embryos were fixed in 4% paraformaldehyde (PFA) and their eyes were photographed. The eye surface and eye diameter was measured using ImageJ® software (NIH, Bethesda, MD). At E15.5, the lens surface area, rendered opaque by 4% PFA fixation, was also measured using ImageJ® software.
Optical Coherence Tomography (OCT) was performed on enucleated P21 mouse eyes fixed overnight in 4% PFA. The eyes were imaged using a custom-built Fourier domain OCT system with a quasi-telecentric scanner, linear-in-wavenumber spectrometer and a line-scan camera with a line rate of 47 kHz40,41. The axial resolution as well as the lateral resolution is approximately 10 μm in tissue. Custom MATLAB programs (MathWorks; Natick, MA) were utilized to create OCT images from the raw data. Amira software (FEI Visualization Sciences Group; Burlingtong, MA) was used to visualize OCT data and quantify anterior chamber volume. All mouse work was performed under a protocol approved by the Cleveland Clinic Institutional Animal Care and Use Committee. Animal husbandry and euthanasia was done in accordance with guidelines established by the American Veterinary Medical Association.
Histology, immunohistochemistry (IHC) and electron microscopy
Mouse embryos or enucleated eyes were fixed overnight at 4 °C in 4% PFA prior to paraffin embedding, and 5 μm thick sections were taken for hematoxylin and eosin (H&E) stain, periodic acid-Schiff stain, oxytalan fiber staining, or immunohistochemistry/immunofluorescence. Cornea cell density and thickness were quantified on DAPI stained sections using ImageJ® software.
For immunofluorescence, sections were subjected to microwave heating in 10 mM sodium citrate-EDTA pH 6.2 for 4 × 1.5 min, followed by gradual cooling to ambient temperature or by incubation with 5 μg/ml proteinase K (Invitrogen, Carlsbad, CA) for 3 min at room temperature (for anti-laminin and anti-collagen IV immunostaining). The sections were then incubated with the following antibodies: anti-γ-crystallin rabbit polyclonal antibody (dilution:1/200; catalog number FL-175, sc-227346, Santa Cruz Biotechnology, Santa Cruz, CA), anti-collagen IV rabbit polyclonal antibody (dilution 1/500; catalog number 600-401-106S, Rockland, Gilbertsville, PA), anti-laminin rabbit polyclonal antibody (dilution: 1/400; catalog number L9393, Sigma-Aldrich, St-Louis, MO), anti-N-cadherin mouse monoclonal antibody (dilution:1/100; catalog number 610920, BD Transduction Laboratories, San Jose, CA), anti-E-cadherin mouse monoclonal antibody (dilution:1/100; catalog number 610181, BD Transduction Laboratories), anti-α-smooth muscle actin Cy3 coupled mouse monoclonal antibody (dilution:1/200; catalog number C6198, Sigma-Aldrich), anti-fibrillin-2 rabbit polyclonal antibody (dilution:1/500; kindly provided by Robert Mecham60), anti-fibronectin rabbit polyclonal antibody (dilution:1/500; catalog number AB2033, Millipore, Billerica, MA), anti-versican GAGβ rabbit polyclonal antibody (dilution: 1/500, catalog number AB1033, Millipore), anti-DPEAAE rabbit polyclonal antibody (dilution:1/500; catalog number PA1-1748A, Thermo-Fisher Scientific, Waltham, MA), anti-Ki67 (dilution:1/100; clone SP6, Thermo-Fisher Scientific). Alexa fluor 568-conjugated goat anti-rabbit immunoglobulin and goat anti-mouse immunoglobulin (Molecular Probes, Invitrogen) were used as the second antibodies and the nuclei were counterstained and slides were mounted with ProLong Gold Antifade with 4′, 6-diamidino-2-phenylindole (DAPI) mounting medium (Life Technologies, Grand Island, NY). mT/mG mouse tissues were embedded in OCTR compound and frozen for cryostat sectioning and slides were mounted with ProLong Gold Antifade with DAPI mounting medium (Life Technologies).
For electron microscopy, eyes were fixed with 2.5% glutaraldehyde plus 4% PFA in 0.2 M sodium cacodylate buffer, pH 7.4 prior to processing and embedding in epoxy resin. Thin sections (85 nm) were stained with osmium tetroxide and viewed with a Phillips CM12/STEM transmission electron microscope (FEI Company, Delmont, PA, USA) equipped with a digital 11-megapixel CCD camera (Gatan, Pleasanton, CA, USA). Lens capsule thickness was measured using Image J software.
β-galactosidase (β-gal) histochemistry and RNA in situ hybridization (ISH)
Whole mount β-gal staining of Adamts9lacZ/+ eyes32 was followed by paraffin embedding and 5 μm sections were counterstained with eosin as previously described32,61. For RNA in situ hybridization, (ISH), mouse embryo heads or enucleated eyes were obtained fresh at various ages. Tissues were fixed overnight in 4% PFA/PBS, embedded in paraffin and sectioned (6 μm). ISH was carried out using the RNAscope® technique and custom-designed Adamts9, Pofut2 and B3glct probes and a HybEZ™ Oven (RNAscope® 2.0; Advanced Cell Diagnostics, Hayward, CA) according to the manufacturer’s instructions. Target probe binding was disclosed by alkaline phosphatase-staining with Fast Red as substrate and Gill’s hematoxylin as a counterstain. As a negative control, some sections were hybridized with target probe against DapB, a bacterial gene encoding dihydrodipicolinate reductase. A target probe directed against ubiquitously expressed Polr2a served as a positive control.
Expression plasmids, cell culture, western blotting and protein purification
hADAMTS9-N-L2 expression plasmid was previously described38. hADAMTS9 TSR9-15 was generated by PCR amplification of the encoding cDNA and cloning into pSecTagA (Life Technologies) for in-frame expression with an N-terminal signal peptide and C-terminal myc-His6 tag in mammalian cells. PCR was performed using full-length ADAMTS9 cDNA27 as the template and primers 5′-AAGGCGCGCCCTCGGTGGAAACCAGTGGAGAACT-3′ and 5′-AACTCGAGTGCAATTCTGGGGTAACTCACAGTT-3′ (restriction enzyme recognition sequences employed for cloning are underlined). Plasmids were transfected into CHO cells (ATCC, Manassas, VA) using Fugene® 6 (Promega, Madison, WI) and stably transfected clones were selected using 500 μg/ml Zeocin (hADAMTS9 TSR9-15, Life Technologies) or 300 μg/ml G418 (hADAMTS9-N-L2). Clones were expanded in DMEM supplemented with 10% fetal bovine serum; conditioned media were tested by western blotting using anti-myc mouse monoclonal antibody (clone 9E10, Life Technologies). At confluence, medium was removed and replaced with serum-free medium. After 48 h, the conditioned media were collected and affinity-purified by Ni-NTA chromatography (Qiagen, Germantown, MD) using an AKTA FPLC instrument (GE Healthcare).
Mass spectral glycoproteomic analysis
The hADAMTS9 constructs purified as described above were reduced, alkylated, and subjected to digestion with Trypsin or Chymotrypsin (Promega). The resulting peptides were analyzed on an Agilent 6340 HPLC-Chip Cube nano LC-Ion trap mass spectrometer as described previously62.
siRNA knockdown experiments
HEK293T cells were purchased (ATCC, Manassas, VA) and co-transfected with negative control (non-targeting siRNA), POFUT2 or B3GLCT siRNA21 and plasmid encoding hADAMTS9-N-L2 using Lipofectamine 2000 (Life Technologies) according to manufacturer’s protocol. A plasmid encoding the human IgG heavy chain was co-transfected with siRNAs as a control and IgG analysis in medium and cells was used for normalization of ADAMTS9 levels. 48 hours post-transfection, the cell and media fractions were collected and analyzed by quantitative western blotting using mouse anti-myc (9E10) or anti-human IgG (Rockland Immunochemicals, Limerick, PA) on an Odyssey 9120 infrared imaging system (LI-COR, Lincoln, NE).
Statistics
All data are reported as the mean ± SD. Statistical differences between two groups were analyzed with a 2-tailed Student’s t test, assuming a normal distribution. A p value of less than 0.05 was considered statistically significant.
Additional Information
How to cite this article: Dubail, J. et al. Impaired ADAMTS9 secretion: A potential mechanism for eye defects in Peters Plus Syndrome. Sci. Rep. 6, 33974; doi: 10.1038/srep33974 (2016).
Supplementary Material
Acknowledgments
We are grateful to Elias Traboulsi for helpful discussion and Timothy Mead for valuable comments on the manuscript. We thank Andrew Rollins for the OCT system, and Megumi Takeuchi, Joshua Kantharia, and Beth Harvey for assistance in the mass spectral analysis of ADAMTS9 protein. This work was supported by Pediatric Ophthalmology Career-Starter Research and Competitive renewal grants from the Knight Templar Eye Foundation (to J.D.), and awards from the National Eye Institute (EY024943 to S.A.) and the National Institute of General Medical Sciences (GM061126 to R.H.). S.E. was funded by NIH award T32EY024236.
Footnotes
Author Contributions J.D.: designed research studies, undertook mouse husbandry, acquired histology, in situ hybridization and immunostaining data, generated protein constructs, acquired data, analyzed data, wrote the manuscript; D.V.: conducted experiments, acquired and analyzed data for mass spectral glycoproteomic analysis and siRNA knock-down experiments; L.W.W.: purified protein for mass spectral glycoproteomic analysis, S.E.E.: undertook mouse husbandry, acquired in situ hybridization data; M.W.J.: conducted experiments, acquired and analyzed data for optical coherence tomography; R.S.H.: analyzed data and wrote the section for spectral glycoproteomic analysis and siRNA knock-down experiments; S.S.A.: designed research studies, analyzed data, wrote the manuscript.
References
- Sowden J. C. Molecular and developmental mechanisms of anterior segment dysgenesis. Eye (Lond) 21, 1310–1318, doi: 10.1038/sj.eye.6702852 (2007). [DOI] [PubMed] [Google Scholar]
- Gould D. B. & John S. W. Anterior segment dysgenesis and the developmental glaucomas are complex traits. Hum Mol Genet 11, 1185–1193 (2002). [DOI] [PubMed] [Google Scholar]
- Weh E. et al. Whole exome sequence analysis of Peters anomaly. Hum Genet 133, 1497–1511, doi: 10.1007/s00439-014-1481-x (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mataftsi A., Islam L., Kelberman D., Sowden J. C. & Nischal K. K. Chromosome abnormalities and the genetics of congenital corneal opacification. Mol Vis 17, 1624–1640 (2011). [PMC free article] [PubMed] [Google Scholar]
- Reis L. M. & Semina E. V. Genetics of anterior segment dysgenesis disorders. Curr Opin Ophthalmol 22, 314–324, doi: 10.1097/ICU.0b013e328349412b (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito Y. A. & Walter M. A. Genomics and anterior segment dysgenesis: a review. Clin Experiment Ophthalmol 42, 13–24, doi: 10.1111/ceo.12152 (2014). [DOI] [PubMed] [Google Scholar]
- Ormestad M. et al. Foxe3 haploinsufficiency in mice: a model for Peters’ anomaly. Invest Ophthalmol Vis Sci 43, 1350–1357 (2002). [PubMed] [Google Scholar]
- Collinson J. M. et al. Primary defects in the lens underlie complex anterior segment abnormalities of the Pax6 heterozygous eye. Proc Natl Acad Sci USA 98, 9688–9693, doi: 10.1073/pnas.161144098 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidson S. H., Kume T., Deng K., Winfrey V. & Hogan B. L. The forkhead/winged-helix gene, Mf1, is necessary for the normal development of the cornea and formation of the anterior chamber in the mouse eye. Developmental biology 211, 306–322, doi: 10.1006/dbio.1999.9314 (1999). [DOI] [PubMed] [Google Scholar]
- Evans A. L. & Gage P. J. Expression of the homeobox gene Pitx2 in neural crest is required for optic stalk and ocular anterior segment development. Hum Mol Genet 14, 3347–3359, doi: 10.1093/hmg/ddi365 (2005). [DOI] [PubMed] [Google Scholar]
- Semina E. V., Murray J. C., Reiter R., Hrstka R. F. & Graw J. Deletion in the promoter region and altered expression of Pitx3 homeobox gene in aphakia mice. Hum Mol Genet 9, 1575–1585 (2000). [DOI] [PubMed] [Google Scholar]
- Libby R. T. et al. Modification of ocular defects in mouse developmental glaucoma models by tyrosinase. Science 299, 1578–1581, doi: 10.1126/science.1080095 (2003). [DOI] [PubMed] [Google Scholar]
- Van Agtmael T. et al. Dominant mutations of Col4a1 result in basement membrane defects which lead to anterior segment dysgenesis and glomerulopathy. Hum Mol Genet 14, 3161–3168, doi: 10.1093/hmg/ddi348 (2005). [DOI] [PubMed] [Google Scholar]
- Shigeyasu C. et al. Clinical features of anterior segment dysgenesis associated with congenital corneal opacities. Cornea 31, 293–298, doi: 10.1097/ICO.0b013e31820cd2ab (2012). [DOI] [PubMed] [Google Scholar]
- Kurilec J. M. & Zaidman G. W. Incidence of Peters anomaly and congenital corneal opacities interfering with vision in the United States. Cornea 33, 848–850, doi: 10.1097/ico.0000000000000182 (2014). [DOI] [PubMed] [Google Scholar]
- Lesnik Oberstein S. A. J., van Belzen M. & Hennekam R. In GeneReviews(R) (eds Pagon R. A. et al.) (University of Washington, Seattle, 1993). [Google Scholar]
- Maillette de Buy Wenniger-Prick L. J. & Hennekam R. C. The Peters’ plus syndrome: a review. Ann Genet 45, 97–103 (2002). [DOI] [PubMed] [Google Scholar]
- Weh E. et al. Novel B3GALTL mutations in classic Peters plus syndrome and lack of mutations in a large cohort of patients with similar phenotypes. Clin Genet 86, 142–148, doi: 10.1111/cge.12241 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesnik Oberstein S. A. et al. Peters Plus syndrome is caused by mutations in B3GALTL, a putative glycosyltransferase. Am J Hum Genet 79, 562–566, doi: 10.1086/507567 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinonen T. Y. & Maki M. Peters’-plus syndrome is a congenital disorder of glycosylation caused by a defect in the beta1,3-glucosyltransferase that modifies thrombospondin type 1 repeats. Ann Med 41, 2–10, doi: 10.1080/07853890802301975 (2009). [DOI] [PubMed] [Google Scholar]
- Vasudevan D., Takeuchi H., Johar S. S., Majerus E. & Haltiwanger R. S. Peters plus syndrome mutations disrupt a noncanonical ER quality-control mechanism. Curr Biol 25, 286–295, doi: 10.1016/j.cub.2014.11.049 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L. W. et al. O-fucosylation of thrombospondin type 1 repeats in ADAMTS-like-1/punctin-1 regulates secretion: implications for the ADAMTS superfamily. J Biol Chem 282, 17024–17031 (2007). [DOI] [PubMed] [Google Scholar]
- Ricketts L. M., Dlugosz M., Luther K. B., Haltiwanger R. S. & Majerus E. M. O-fucosylation is required for ADAMTS13 secretion. J Biol Chem 282, 17014–17023 (2007). [DOI] [PubMed] [Google Scholar]
- Vasudevan D. & Haltiwanger R. S. Novel roles for O-linked glycans in protein folding. Glycoconj J 31, 417–426, doi: 10.1007/s10719-014-9556-4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apte S. S. A disintegrin-like and metalloprotease (reprolysin type) with thrombospondin type 1 motifs: the ADAMTS family. Int J Biochem Cell Biol 36, 981–985 (2004). [DOI] [PubMed] [Google Scholar]
- Dubail J. & Apte S. S. Insights on ADAMTS proteases and ADAMTS-like proteins from mammalian genetics. Matrix Biol, doi: 10.1016/j.matbio.2015.03.001 (2015). [DOI] [PubMed] [Google Scholar]
- Somerville R. P. et al. Characterization of ADAMTS-9 and ADAMTS-20 as a distinct ADAMTS subfamily related to Caenorhabditis elegans GON-1. J Biol Chem 278, 9503–9513 (2003). [DOI] [PubMed] [Google Scholar]
- Enomoto H., Nelson C., Somerville R. P. T., Mielke K., Dixon L., Powell K. & Apte S. S. Cooperation of two ADAMTS metalloproteases in closure of the mouse palate identifies a requirement for versican proteolysis in regulating palatal mesenchyme proliferation. Development 137, 4029–4038 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubail J. et al. A new Adamts9 conditional mouse allele identifies its non-redundant role in interdigital web regression. Genesis 52, 702–712, doi: 10.1002/dvg.22784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao C. et al. A defect in a novel ADAMTS family member is the cause of the belted white-spotting mutation. Development 130, 4665–4672 (2003). [DOI] [PubMed] [Google Scholar]
- Kern C. B. et al. Reduced versican cleavage due to Adamts9 haploinsufficiency is associated with cardiac and aortic anomalies. Matrix Biol 29, 304–316, doi: S0945-053X(10)00009-0 [pii] 10.1016/j.matbio.2010.01.005 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo B. H. et al. ADAMTS9 Is a Cell-Autonomously Acting,Anti-Angiogenic Metalloprotease Expressed by Microvascular Endothelial Cells. Am J Pathol, doi: ajpath.2010.090655 [pii] 10.2353/ajpath.2010.090655 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y., Nita-Lazar A. & Haltiwanger R. S. Two distinct pathways for O-fucosylation of epidermal growth factor-like or thrombospondin type 1 repeats. J Biol Chem 281, 9385–9392 (2006). [DOI] [PubMed] [Google Scholar]
- Luo Y., Koles K., Vorndam W., Haltiwanger R. S. & Panin V. M. Protein O-fucosyltransferase 2 adds O-fucose to thrombospondin type 1 repeats. J Biol Chem 281, 9393–9399 (2006). [DOI] [PubMed] [Google Scholar]
- Kozma K. et al. Identification and characterization of abeta1,3-glucosyltransferase that synthesizes the Glc-beta1,3-Fuc disaccharide on thrombospondin type 1 repeats. J Biol Chem 281, 36742–36751 (2006). [DOI] [PubMed] [Google Scholar]
- Sato T. et al. Molecular cloning and characterization of a novel human beta1,3-glucosyltransferase, which is localized at the endoplasmic reticulum and glucosylates O-linked fucosylglycan on thrombospondin type 1 repeat domain. Glycobiology 16, 1194–1206 (2006). [DOI] [PubMed] [Google Scholar]
- Du J. et al. O-fucosylation of thrombospondin type 1 repeats restricts epithelial to mesenchymal transition (EMT) and maintains epiblast pluripotency during mouse gastrulation. Developmental biology 346, 25–38, doi: 10.1016/j.ydbio.2010.07.008 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koo B. H. et al. Cell-surface processing of pro-ADAMTS9 by furin. J Biol Chem 281, 12485–12494 (2006). [DOI] [PubMed] [Google Scholar]
- Nandadasa S., Nelson C. M. & Apte S. S. ADAMTS9-Mediated Extracellular Matrix Dynamics Regulates Umbilical Cord Vascular Smooth Muscle Differentiation and Rotation. Cell Rep 11, 1519–1528, doi: 10.1016/j.celrep.2015.05.005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Z. & Rollins A. M. Fourier domain optical coherence tomography with a linear-in-wavenumber spectrometer. Opt Lett 32, 3525–3527 (2007). [DOI] [PubMed] [Google Scholar]
- Karunamuni G. et al. Ethanol exposure alters early cardiac function in the looping heart: a mechanism for congenital heart defects? Am J Physiol Heart Circ Physiol 306, H414–H421, doi: 10.1152/ajpheart.00600.2013 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danysh B. P. & Duncan M. K. The lens capsule. Exp Eye Res 88, 151–164, doi: 10.1016/j.exer.2008.08.002 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi M. et al. Heparan sulfate chains of perlecan are indispensable in the lens capsule but not in the kidney. EMBO J 22, 236–245, doi: 10.1093/emboj/cdg019 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pathania M., Semina E. V. & Duncan M. K. Lens extrusion from Laminin alpha 1 mutant zebrafish. ScientificWorldJournal 2014, 524929, doi: 10.1155/2014/524929 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould D. B., Marchant J. K., Savinova O. V., Smith R. S. & John S. W. Col4a1 mutation causes endoplasmic reticulum stress and genetically modifiable ocular dysgenesis. Hum Mol Genet 16, 798–807, doi: 10.1093/hmg/ddm024 (2007). [DOI] [PubMed] [Google Scholar]
- Lazar E. et al. Structure-function analysis of peroxidasin provides insight into the mechanism of collagen IV crosslinking. Free Radic Biol Med 83, 273–282, doi: 10.1016/j.freeradbiomed.2015.02.015 (2015). [DOI] [PubMed] [Google Scholar]
- Yan X. et al. Peroxidasin is essential for eye development in the mouse. Hum Mol Genet 23, 5597–5614, doi: 10.1093/hmg/ddu274 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver D. L., Hou L., Somerville R., Young M. E., Apte S. S. & Pavan W. J. The secreted metalloprotease ADAMTS20 is required for melanoblast survival. PLoS Genet 4, 1–15 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo Z. L. et al. Integrin linked kinase (ILK) is required for lens epithelial cell survival, proliferation and differentiation. Exp Eye Res 121, 130–142, doi: 10.1016/j.exer.2014.01.013 (2014). [DOI] [PubMed] [Google Scholar]
- Wederell E. D. & de Iongh R. U. Extracellular matrix and integrin signaling in lens development and cataract. Semin Cell Dev Biol 17, 759–776, doi: 10.1016/j.semcdb.2006.10.006 (2006). [DOI] [PubMed] [Google Scholar]
- Walker J. & Menko A. S. Integrins in lens development and disease. Exp Eye Res 88, 216–225, doi: 10.1016/j.exer.2008.06.020 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benz B. A. et al. Genetic and biochemical evidence that gastrulation defects in Pofut2 mutants result from defects in ADAMTS9 secretion. Developmental biology 416, 111–122, doi: 10.1016/j.ydbio.2016.05.038 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Goff C. et al. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat Genet 40, 1119–1123 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagoneau N. et al. ADAMTS10 Mutations in Autosomal Recessive Weill-Marchesani Syndrome. Am J Hum Genet 75, 801–806 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales J. et al. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am J Hum Genet 85, 558–568, doi: S0002-9297(09)00407-8 [pii] 10.1016/j.ajhg.2009.09.011 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fritsche L. G. et al. Seven new loci associated with age-related macular degeneration. Nat Genet 45, 433–439, 439e431–432, doi: 10.1038/ng.2578 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helisalmi S. et al. ADAMTS9 locus associates with increased risk of wet AMD. Acta Ophthalmol 92, e410, doi: 10.1111/aos.12341 (2014). [DOI] [PubMed] [Google Scholar]
- de Jong E. K. et al. Chronic central serous chorioretinopathy is associated with genetic variants implicated in age-related macular degeneration. Ophthalmology 122, 562–570, doi: 10.1016/j.ophtha.2014.09.026 (2015). [DOI] [PubMed] [Google Scholar]
- Muzumdar M. D., Tasic B., Miyamichi K., Li L. & Luo L. A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605, doi: 10.1002/dvg.20335 (2007). [DOI] [PubMed] [Google Scholar]
- Trask T. M. et al. Interaction of tropoelastin with the amino-terminal domains of fibrillin-1 and fibrillin-2 suggests a role for the fibrillins in elastic fiber assembly. J Biol Chem 275, 24400–24406, doi: 10.1074/jbc.M003665200 (2000). [DOI] [PubMed] [Google Scholar]
- McCulloch D. R. et al. ADAMTS metalloproteases generate active versican fragments that regulate interdigital web regression. Dev Cell 17, 687–698, doi: S1534-5807(09)00390-6 [pii] 10.1016/j.devcel.2009.09.008 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonhard-Melief C. & Haltiwanger R. S. O-fucosylation of thrombospondin type 1 repeats. Methods Enzymol 480, 401–416, doi: 10.1016/s0076-6879(10)80018-7 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.