

Abstract

Contributing to the need for new graphene nanoribbon (GNR) structures that can be synthesized with atomic precision, we have designed a reactant that renders chiral (3,1)-GNRs after a multistep reaction including Ullmann coupling and cyclodehydrogenation. The nanoribbon synthesis has been successfully proven on different coinage metals, and the formation process, together with the fingerprints associated with each reaction step, has been studied by combining scanning tunneling microscopy, core-level spectroscopy, and density functional calculations. In addition to the GNR’s chiral edge structure, the substantial GNR lengths achieved and the low processing temperature required to complete the reaction grant this reactant extremely interesting properties for potential applications.
Keywords: on-surface synthesis, Ullmann coupling, cyclodehydrogenation, scanning tunneling microscopy, core-level photoemission, density functional theory
Graphene nanoribbons (GNRs) are drawing enormous interest, partly due to their attractive electronic properties.1,2 Those properties vary dramatically with changes in the nanoribbon’s atomic structure in terms of width,3−5 crystallographic symmetry,6,7 dopant heteroatoms,8−13 and edge termination.14 Moreover, the electronic properties can be modulated even further by the appropriate design of GNR heterostructures.12,15−17 This enormous tunability of electronic properties is thus extremely promising for next-generation nanoelectronic and optoelectronic devices.18,19 However, the high susceptibility of those properties to minimum changes in the GNR structure also indicates the stringent need for atomic precision in GNR synthesis. With the advent of bottom-up synthesis,1,2,20 increasingly high hopes are being placed on this approach, but the field is still in its infancy. Thus, although a large pool of GNRs with different edge orientations, widths, or heteroatoms (and heterostructures thereof) should become available to really allow for the envisioned breakthroughs in nanoelectronics and the development of full GNR-based circuitry; so far, only few GNRs have been successfully synthesized with the required selectivity and precision.1,2,8−10,20−24
To date, the most widely studied nanoribbon is the armchair-oriented GNR with seven dimer lines across its width (7-AGNR) that grows from 10,10′-dibromo-9,9′-bianthracene (reactant 1 in Figure 1a) in a multistep reaction, including dehalogenation, polymerization (also known as Ullmann coupling), and cyclodehydrogenation.20,25,26 The synthesis of 7-AGNR has been shown to work reproducibly on substrates such as Au(111),20 Au(110),27 or Ag(111).20,28 Surprisingly, the same reactant 1 designed to render AGNRs turned out to form chiral (3,1)-GNRs on Cu(111) (Figure 1).29,30 This result has been subject to debate31−34 since the polymerization does not involve the carbon atoms attached to bromines. However, the debate has been recently settled by unambiguous high-resolution imaging of the resulting bonding structure.35 These results mirror a very system-specific reaction mechanism not translatable to other substrates, based on the surface-catalyzed, selective activation of particular C–H bonds. In fact, similar results were obtained from 1 and its nonhalogenated sister molecule, the latter producing slightly longer GNRs.35 This implies an absent or even negative impact of the halogenation of 1 for GNR growth on Cu(111).
Figure 1.

(a) Schemes of the chemical reactions of precursor 1 on various metallic surfaces. On Au(111) and Ag(111), it affords armchair GNRs. On Cu(111), it affords chiral (3,1)-GNRs. (b) Our work (highlighted with the red line) reports the transformation of reactant 2 into chiral GNRs independently of the substrate [Au(111), Ag(111), and Cu(111)]. Associated STM images are shown for poly-2 after initial polymerization by Ullmann coupling (5.6 nm × 2.2 nm, I = 0.09 nA, U = 1.5 V), as well as for the final (3,1)-GNR after cyclodehydrogenation (5.6 nm × 2.2 nm, I = 0.2 nA, U = −650 mV), both on Au(111). Steric hindrance causes poly-2 to be nonplanar. The high parts (circled in yellow) are correspondingly marked in the polymer’s wireframe structure above.
Nevertheless, those results have inspired this work, in which we introduce monomer 2, specifically designed to obtain (3,1)-GNRs in a more efficient manner (Figure 1). In particular, the bromine atoms in positions 10,10′ of reactant 1 have been shifted in reactant 2 to positions 2,2′, which are the positions selectively activated on Cu(111) and through which the polymerization preceding the (3,1)-GNR formation takes place. The same structure can thus be obtained from 2 following a conventional Ullmann coupling/cyclodehydrogenation sequence. This has been shown to work independently of the used substrate, as proven with growth studies on Au(111) and Ag(111). A similar study on Cu(111) is out of the scope of this paper due to the readily proven growth of (3,1)-GNRs from 1 (and even from nonhalogenated precursors) on that substrate. However, also on Cu(111), we show how changing the halogen functionalization site, and thereby changing the polymerization mechanism from a selective C–H bond activation to Ullmann coupling, is still a significant advancement by greatly increasing the resultant GNR length.
Results and Discussion
Key to this work is the synthesis of bianthracene reactants with adequately chosen bromine atom positions. We prepare 2,2′-dibromo-9,9′-bianthracene (2) starting from phthalic anhydride (3), following the four-step synthetic route shown in Figure 2 (see Supporting Information, Scheme S1 and Figures S1 and S2 for details). The key reaction in this protocol is the reductive coupling of bromoanthrone 4 promoted by Zn.36
Figure 2.

Synthetic route to obtain dibromobianthracene 2 from phthalic anhydride 3.
Starting from reactant 2, the reaction pathway associated with the surface-supported synthesis of (3,1)-GNRs is closely related to the well-known transformation of 1 into 7-AGNRs. That is, in a first step, the molecules polymerize by Ullmann coupling into poly-2. This polymer is a highly nonplanar molecular structure in which the steric hindrance between the H atoms in neighboring anthracene units drives their alternating tilting (Figure 1b). As a consequence, the polymer’s imaging by scanning tunneling microscopy (STM) displays a sequence of protruding features that we associate with the up-pointing ends of the anthracene units. This correspondence is highlighted by yellow circles in the wireframe chemical structure and in the STM image. In a following reaction step, cyclodehydrogenation sets in and poly-2 transforms into the planar (3,1)-GNR structure, as can be directly discerned in the high-resolution STM images in Figure 1b and Figure S3.
Figure 3 summarizes, as observed by STM, the growth process of (3,1)-GNRs on top of Au(111) and Ag(111). On either substrate, the images correspond to the same sample at different stages of its growth: after deposition of 2 on substrates held at room temperature, after being annealed to 150 °C, and after being annealed to 205 °C.
Figure 3.

Large-scale (36 nm × 36 nm) and smaller-scale (10 nm × 6.5 nm) STM images on Au(111) (a–f) and Ag(111) (g–l) of the same samples at different growth stages: after deposition on substrates held at room temperature (a,b,g,h), after being annealed to 150 °C (c,d,i,j), and after being annealed to 205 °C (e,f,k,l). STM imaging parameters are (a) I = 0.086 nA, U = 1.4 V; (b) I = 0.086 nA, U = 1.5 V; (c) I = 0.16 nA, U = 1.76 V; (d) I = 0.16 nA, U = 1.76 V; (e) I = 0.16 nA, U = 0.47 V; (f) I = 1.29 nA, U = −0.13 V; (g) I = 0.09 nA, U = 1.5 V; (h) I = 0.06 nA, U = −2.02 V; (i) I = 0.36 nA, U = 1.76 V; (j) I = 0.36 nA, U = −1.6 V; (k) I = 0.42 nA, U = 1.07 V; and (l) I = 0.19 nA, U = −0.13 V.
We first focus on Au(111). After room temperature deposition, the molecules aggregate into islands of linear structures formed by a zig-zag arrangement of protrusions comparable to those expected from poly-2 (Figure 3a,b). Upon annealing to 150 °C, we observe clear changes in the sample’s topology. It is difficult to discern changes in the STM contrast within the linear structures (see Supporting Information for details). However, their overall length substantially increases and the spacing between them (perpendicular to the structure’s long axis) becomes less regular and decreases the minimum distance (Figure 3c,d). Annealing to 205 °C brings about more notorious changes, displaying arrays of planar structures clearly recognizable from the edge topology as (3,1)-GNRs (Figure 3e,f).
In the case of Ag(111), two distinct sections are observed after molecular deposition at room temperature. On the one hand, we find regions of ordered, linear structures packed side by side (Figure 3g). The linear structures are imaged again as zig-zagging protrusions (Figure 3h). Instead, other regions display a disordered arrangement of adsorbates with increased mobility and a much larger apparent height (∼2.7 Å vs ∼1.8 Å). The areal ratio between these two different sections is approximately 1:1 (Figure 3h). Annealing to 150 °C brings about the growth of the ordered, linear structures at the expense of the disappearing disordered regions (Figure 3i,j). As opposed to the findings on Au(111), on Ag(111), the arrangement within the ordered arrays of linear structures remains unchanged after this annealing. Annealing the sample to 205 °C triggers the cyclodehydrogenation and thereby the ultimate formation of (3,1)-GNRs (Figure 3k,l).
Complementary information on the chemical transformation process is obtained from core-level X-ray photoemission spectroscopy (XPS) measurements. As in the STM experiment, molecules were deposited on Au(111) and Ag(111) substrates held at room temperature. The samples were then annealed stepwise while their Br 3p and C 1s core-level spectra were monitored. The data are summarized in Figure 4.
Figure 4.

Photoemission spectra of the C 1s core levels of 2 deposited on (a) Au(111) and (d) Ag(111) held at room temperature and their evolution as a function of sample annealing temperature. Similar measurements of the Br 3p core levels are shown in panels (b) and (e). (c,f) Br 3p spectra, together with their associated fits (blue and green lines correspond to organic and metal-bound Br components, respectively), of two representative temperatures marked with the colored lines in (b) and (e), respectively. The spectra are shifted along the intensity axis for better comparison.
On Au(111), the molecules remain intact upon deposition at room temperature and only start showing chemical changes for substrate temperatures of around 125 °C. As the temperature increases above that threshold, the most evident change in the core-level spectra is a pronounced shift of the Br 3p peaks to ∼2 eV lower binding energies. This effect is well-known from other studies on Ullmann coupling of different precursors and relates to the dehalogenation process and the new chemical environment as Br detaches from the organic molecule and binds to the metallic surface.27,37,38 Concomitantly, the C 1s peak displays a smaller shift (∼0.3 eV) to lower binding energies. Similar C 1s shifts have also been observed in previous studies on Ullmann coupling with different precursors and surfaces, for which a variety of explanations have been given: (i) bond formation between the C atoms hosting the generated radicals and the substrate atoms or adatoms,27,37 (ii) a change in the supramolecular assembly prior to dehalogenation that brings about changes in the interaction with the substrate,38 or (iii) a change of work function caused by the chemisorption of Br to the substrate.37 We discard the first because on Au the formation of organometallic compounds is disfavored and the molecules are known to polymerize as the radicals are formed.39,40 We also discard the second because we observe the C 1s and Br 3p shifts simultaneously in a correlated way. Thus, we ascribe the observed C 1s shift to the change of work function generated by the metal-bound Br atoms. Besides, this is further supported by the changes observed in the core levels as the temperature is increased further: as Br desorption starts to set in, the C 1s level shifts in the opposite direction, toward higher binding energy. A similar shift on closely related systems has also been ascribed to different reaction processes like the transformation from an organometallic to a polymer phase27,41 or cyclodehydrogenation.37 In addition to the chemical change, these reactions also cause an alteration in the molecule–substrate distance that may, in turn, additionally affect the core-hole screening effects and thereby the core-level spectra. However, we can again discard these justifications for our system because we know the polymerization and cyclodehydrogenation to occur at lower temperatures. Altogether, XPS on Au(111) thus shows that the structures observed on Au(111) at room temperature are a noncovalent self-assembled supramolecular arrangement, polymerizing only upon annealing above 125 °C, in line with the overall sample topology changes observed by STM at 150 °C and described above.
On Ag(111), the molecules are readily partially dehalogenated upon deposition at room temperature. This is clearly observed in the Br 3p core-level spectra, which shows the coexistence of organic and metal-linked Br atoms in a 1:1 ratio. As the temperature is increased, the metal-bound Br increases at the expense of the organic Br. From the correlation with the STM observations, we can readily ascribe the pristine precursors to the disordered structures found at room temperature and the dehalogenated molecules to be the building blocks forming the ordered structures. As in Au(111), the dehalogenation brings about a minor shift to lower binding energy in the C 1s spectrum, which shifts back again as the Br is desorbed at higher temperatures. Again, we ascribe the dominating effect behind the C 1s shifts to the changes in work function as Br binds or leaves the metal surface. However, spectroscopy-wise, this leaves the question open as to what is the nature of the linear, ordered structures formed by the dehalogenated precursors. Do those radical species link covalently or via metal–organic coordination, as is commonly the case on Ag at temperatures below ∼150 °C? In the absence of clear spectroscopic fingerprints, the answer will be given based on periodicity analysis along the one-dimensional structures, organometallic structures typically having significantly larger periodicities than polymers.40,42,43
Density functional theory (DFT) calculations for free-standing structures predict periodicities of 9.52 Å for the metal–organic chain (Figure 5c), 8.21 Å for the polymer (Figure 5b), and 8.96 Å for the flat (3,1)-GNR (Figure 5a), the latter in good agreement with the value of 8.89 Å that results from assuming an undisturbed graphene lattice for the GNR. Our STM measurements reveal the periodicity of the GNRs to be 9.0 ± 0.8 Å, excellently fitting the calculations. For the nonplanar structures, we observe the same periodicity at room temperature and after annealing to 150 °C, with an average value of 8.2 ± 0.7 Å. The value is similar to that of poly-2 on Au(111) (8.3 ± 0.6 Å) and in excellent agreement with a polymeric phase, with the error margins remaining clearly under the metal–organic periodicity (Figure 5d). Thus, in spite of the flexibility associated with the nonplanarity of poly-2 and the metal–organic chain, which may introduce minor uncertainties in the calculated periodicities, the results still allow us to conclude that the polymer is directly formed after dehalogenation.
Figure 5.

Relaxed structures for (a) free-standing (3,1)-GNRs, (b) poly-2, and (c) metal–organic chains. (d) Comparison of the periodicities of the calculated structures with those measured experimentally for the GNRs and for the nonplanar structure on Ag(111). The latter fits the polymer period, and its error margin (shown as the standard deviation) is well below the periodicity of the metal–organic chain.
As only reported with a few other systems,44 we thus observe a covalent polymer formation readily at room temperature instead of the metal–organic intermediates reported with most precursors linking through Ullmann coupling on Ag(111) and Cu(111).37,39−42 The reason behind this may be sought in the different coordination geometry imposed by the nonplanar precursors. As observed in most previous Ullmann coupling studies on Ag surfaces, Ag atoms present linear coordination geometry,39,40,42 while the anthracene units hosting the generated radicals upon dehalogenation of 2 display a strong tilt with respect to the substrate plane due to the steric congestion within the organic backbone (Figure 5c). Under these circumstances, and based on our experimental observations, we conclude that the metal–organic intermediate is not sufficiently stable and the reaction directly proceeds toward the polymeric phase.
As noted above, at 205 °C, the GNRs are fully cyclodehydrogenated on both Ag(111) and Au(111). Similarly, low cyclodehydrogenation temperatures (177 °C) are reported for reactant 1 on Ag(111),28 but higher temperatures, similar to those required on Au(111), are needed to really form GNRs (377 °C).20,28 On Cu(111), the required temperature for fully cyclodehydrogenated GNRs to be formed is 250 °C.33,35 Most remarkably, reactant 2 renders fully dehydrogenated GNRs at significantly lower temperatures than 1 even on the low reactivity Au(111) surface. This surprisingly large change relates to the substantially altered strain in the two polymer structures since sterically induced strain is known to weaken the involved C–H bonds and thereby lower the cyclodehydrogenation barriers.25,45 In poly-1, the anthracene units are linked covalently along their short axis by a bond that allows free rotational movement with respect to their neighbors. This freedom results in alternatively tilted anthracene units along the polymer backbone so as to minimize the steric hindrance from opposing H atoms. Instead, the anthracene units within poly-2 are linked covalently to their neighbors both along their long and short axes. Thus, although the anthracene units still display the same alternative tilt to reduce the steric hindrance, the covalent bonds along the long anthracene’s axes limit the structure’s rotational freedom, resulting in a substantially strained geometry. It is this strain opposing the anthracene’s tilting which favors the planarization of the structure and thus reduces the cyclodehydrogenation temperature threshold regardless of the substrate.
Both on Ag(111) and Au(111), the cyclodehydrogenation threshold is between 150 °C, at which no cyclodehydrogenation is observed, and 205 °C, at which the whole sample has readily become fully planar (Figure 3). On Ag(111), where polymerization readily starts at room temperature, there is still a substantial temperature gap before the onset of cyclodehydrogenation. However, on Au(111), the threshold temperatures for polymerization (∼125 °C) and cyclodehydrogenation are in close proximity. This may have an impact on the growth process and the resulting GNRs since liberated H in the cyclodehydrogenation process could quench the available radicals and terminate the polymerization. To shed light on this issue, we have increased the number of sampling temperatures, figuring the cyclodehydrogenation onset on Au(111) to be below 175 °C, at which most of the sample has readily become a planar GNR but some of the polymer units still remain unreacted (inset in Figure 6). A systematic GNR length analysis of samples as a function of the substrate temperature upon reactant deposition is shown in Figure 6, with all samples having coverages of around 0.8 ML. For temperatures below the cyclodehydrogenation threshold, a second annealing step to 205 °C was applied to the sample for GNR formation before performing the length analysis.
Figure 6.

(a) Length distribution of GNRs grown on Au, at coverages close to the full monolayer, for different substrate temperatures upon first deposition. For temperatures below the cyclodehydrogenation threshold (T < 174 °C), a second annealing step to 205 °C was applied to the sample for GNR formation by cyclodehydrogenation. (b) Median length for each substrate temperature. The inset depicts an STM image (7.4 nm × 2.9 nm, I = 0.16 nA, U = 0.47 V) of a sample deposited at 174 °C, revealing a mostly, but not yet fully, cyclodehydrogenated structure. This value has been thus taken as the cyclodehydrogenation threshold temperature.
Representative distributions for selected temperatures are shown in Figure 6a, making immediately obvious that high temperatures narrow the distribution significantly and prevent formation of long GNRs. Because of the asymmetric length distribution, we take the median length as a representative value and plot it versus substrate temperature upon first deposition (Figure 6b). We observe an important drop in the length with increasing temperature once the cyclodehydrogenation threshold is passed. Under this scenario, radical step growth and cyclodehydrogenation take place simultaneously. Thus, radical quenching by liberated H atoms competes with the radical step-growth polymerization. Deposition on Au held at room temperature and subsequent annealing to cyclodehodrogenation temperatures suffers from the same effect because the precursors on the surface remain intact at room temperature, and both polymerization and cyclodehydrogenation occur during the same subsequent annealing process. However, the length-limiting effect is less pronounced, among other reasons, due to the finite heating rate. Longest GNRs are obtained at substrate temperatures that first activate polymerization, only to form the GNRs in a subsequent annealing process. Under these circumstances, GNRs in excess of 30 nm can be easily obtained, well beyond the longest (3,1)-GNRs obtained from 1 on Cu(111).30 Moreover, additional studies to maximize GNR lengths by optimizing surface coverages or heating rates may bring about even further improvements in the future.
Lastly, we have confirmed the suitability of this molecule to form longer (3,1)-GNRs than 1 also on Cu(111). This is shown in Figure 7 and underlines the great advancement provided by this new GNR precursor. In addition to the (3,1)-GNR formation, a concomitant etching of triangular holes into the remaining uncovered Cu(111) surface is observed, lined along their sides by Br atoms (Figure 7). A detailed study and description of this process, however, is beyond the scope of this paper. We also want to note that the GNR length analysis of this sample on Cu(111) (Figure 7c) should not be compared with that on Au(111) (Figure 6) because the growth was performed in a different chamber with different coverage and a different heating rate, two parameters that may play an important role in the length distribution. However, most importantly, we want to remark that, different from what occurs with precursor 1 on Cu(111), the precursor 2 allows on the one hand to grow chiral GNRs on different materials not relying on specific and strong molecule–substrate interactions. On the other hand, on all surfaces studied, it forms long GNRs that easily exceed several tens of nanometers, a great advantage for their implementation in actual device structures.
Figure 7.
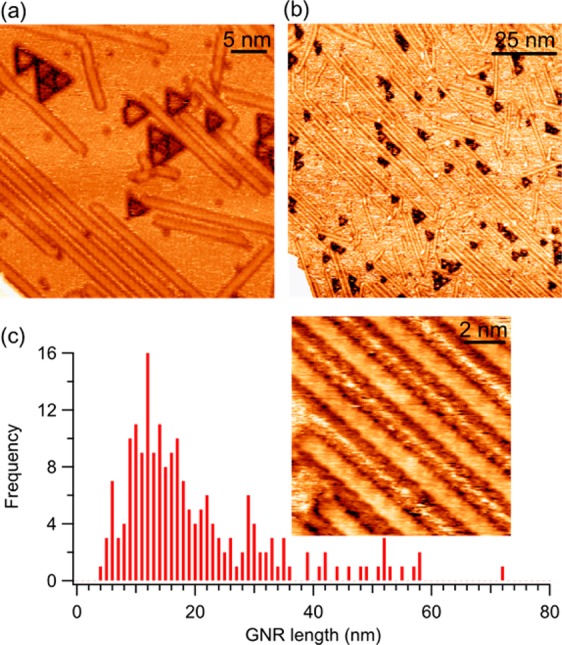
Constant current (a) 36 × 36 nm2 (I = 0.58 nA, U = −400 mV) and (b) 100 × 100 nm2 (I = 0.05 nA, U = −200 mV) STM images of (3,1)-GNRs on Cu(111). (c) GNR length distribution as obtained from large-scale images as in (b), showing a significant portion of GNRs to be longer than 40–50 nm and the average and median length values well above those of GNRs grown from 1 on Cu(111). The inset depicts a 10 × 10 nm2 close-up view of the GNRs (I = 1 nA, U = −150 mV).
Conclusions
Inspired by the previously reported system-specific growth of (3,1)-GNRs on Cu(111) from precursor 1, we have designed an alternative building block 2 that renders the same (3,1)-GNRs but now independently of the substrate material. This has been proven on Au(111), Ag(111), and Cu(111), revealing additional advantages of the use of this monomer in the growth of selective and atomically precise GNRs, as is the substantially increased length of the resultant GNRs and the low processing temperature required for their formation. Furthermore, the growth process has been followed in detail combining core-level spectroscopy, scanning tunneling microscopy, and density functional theory calculations, providing a clear correlation between the spectroscopic fingerprints and the different reaction processes, as well as revealing the unusual absence of a metastable metal–organic intermediate preceding the covalent polymerization in the Ullmann coupling process on Ag(111).
Methods
The various metal (111) surfaces were all prepared by standard sputtering–annealing cycles. Subsequently, the samples were prepared by thermal evaporation of 2 at ∼140 °C onto the substrate. Substrate temperature was controlled by resistive heating, and the calibration for the STM experiments was performed by direct measurement of the substrate surface temperature, as a function of the resistive heating current, with a thermocouple fixed and glued to the surface with silver paint after completion of the experiments (see Supporting Information for more details). STM was measured in a commercial UHV system at room temperature, except for the GNR image in Figure 1b, measured in a commercial UHV system at 5 K. WSxM software was used to process all STM images.47 XPS measurements were performed using a non-monochromatized source. The XPS data were collected by means of a SPECS Phoibos 100 hemispherical electron analyzer, making use of Al Kα X-ray emission.
Ab initio calculations were carried out on free-standing structures using DFT, as implemented in the SIESTA code.48−50 The optB88-vdW functional, which accounts for nonlocal corrections, was adopted for the exchange and correlation potential. We employed a double-ζ plus polarization basis set and a mesh cutoff of 300 Ry for the real-space integrations. A variable-cell relaxation of the periodic systems was performed until residual forces on all atoms were less than 0.01 eV/Å and a Monkhorst–Pack mesh with 101 × 1 × 1 k-point sampling of the three-dimensional Brillouin zone was used.
Details on the four-step synthetic route to obtain dibromobianthracene 2 from phthalic anhydride 3 are given in the Supporting Information.
Acknowledgments
The project leading to this publication has received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (Grant Agreement No. 635919), from ICT-FET EU integrated project PAMS (Agreement No. 610446), from the Spanish Ministry of Science and Competitiveness (MINECO, Grant Nos. MAT2013-46593-C6-01, MAT2013-46593-C6-2-P, MAT2013-46593-C6-4-P, MAT2013-46593-C6-6-P), FEDER, from the Basque Government (Grant Nos. IT-621-13, IT-756-13, and PI2015-042), from the World Premier International Center (WPI) for Materials Nanoarchitectonics (MANA) of the National Institute for Materials Science (NIMS), Tsukuba, Japan, and from JSPS Bilateral Open Partnership Joint Research Project.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.6b05269.
Details on the reactant synthesis, intramolecular resolution STM imaging, description of the temperature measurement method and its calibration, details on the temperature-dependent STM contrast along linear structures (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Talirz L.; Ruffieux P.; Fasel R. On-Surface Synthesis of Atomically Precise Graphene Nanoribbons. Adv. Mater. 2016, 28, 6222–6231. 10.1002/adma.201505738. [DOI] [PubMed] [Google Scholar]
- Narita A.; Wang X.-Y.; Feng X.; Müllen K. New Advances in Nanographene Chemistry. Chem. Soc. Rev. 2015, 44, 6616–6643. 10.1039/C5CS00183H. [DOI] [PubMed] [Google Scholar]
- Son Y.-W.; Cohen M. L.; Louie S. G. Energy Gaps in Graphene Nanoribbons. Phys. Rev. Lett. 2006, 97, 216803. 10.1103/PhysRevLett.97.216803. [DOI] [PubMed] [Google Scholar]
- Yang L.; Park C.-H.; Son Y.-W.; Cohen M. L.; Louie S. G. Quasiparticle Energies and Band Gaps in Graphene Nanoribbons. Phys. Rev. Lett. 2007, 99, 186801. 10.1103/PhysRevLett.99.186801. [DOI] [PubMed] [Google Scholar]
- Jiang Z.; Song Y. Band Gap Oscillation and Novel Transport Property in Ultrathin Chiral Graphene Nanoribbons. Phys. B 2015, 464, 61–67. 10.1016/j.physb.2015.02.003. [DOI] [Google Scholar]
- Yazyev O. V.; Capaz R. B.; Louie S. G. Theory of Magnetic Edge States in Chiral Graphene Nanoribbons. Phys. Rev. B: Condens. Matter Mater. Phys. 2011, 84, 115406. 10.1103/PhysRevB.84.115406. [DOI] [Google Scholar]
- Tao C.; Jiao L.; Yazyev O. V.; Chen Y.-C.; Feng J.; Zhang X.; Capaz R. B.; Tour J. M.; Zettl A.; Louie S. G.; Dai H.; Crommie M. F. Spatially Resolving Edge States of Chiral Graphene Nanoribbons. Nat. Phys. 2011, 7, 616–620. 10.1038/nphys1991. [DOI] [Google Scholar]
- Cloke R. R.; Marangoni T.; Nguyen G. D.; Joshi T.; Rizzo D. J.; Bronner C.; Cao T.; Louie S. G.; Crommie M. F.; Fischer F. R. Site-Specific Substitutional Boron Doping of Semiconducting Armchair Graphene Nanoribbons. J. Am. Chem. Soc. 2015, 137, 8872–8875. 10.1021/jacs.5b02523. [DOI] [PubMed] [Google Scholar]
- Kawai S.; Saito S.; Osumi S.; Yamaguchi S.; Foster A. S.; Spijker P.; Meyer E. Atomically Controlled Substitutional Boron-Doping of Graphene Nanoribbons. Nat. Commun. 2015, 6, 8098. 10.1038/ncomms9098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen G. D.; Toma F. M.; Cao T.; Pedramrazi Z.; Chen C.; Rizzo D. J.; Joshi T.; Bronner C.; Chen Y.-C.; Favaro M.; Louie S. G.; Fischer F. R.; Crommie M. F. Bottom-Up Synthesis of N = 13 Sulfur-Doped Graphene Nanoribbons. J. Phys. Chem. C 2016, 120, 2684–2687. 10.1021/acs.jpcc.5b09986. [DOI] [Google Scholar]
- Basagni A.; Vasseur G.; Pignedoli C. A.; Vilas-Varela M.; Peña D.; Nicolas L.; Vitali L.; Lobo-Checa J.; de Oteyza D. G.; Sedona F.; Casarin M.; Ortega J. E.; Sambi M. Tunable Band Alignment with Unperturbed Carrier Mobility of On-Surface Synthesized Organic Semiconducting Wires. ACS Nano 2016, 10, 2644–2651. 10.1021/acsnano.5b07683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai J.; Pignedoli C. A.; Talirz L.; Ruffieux P.; Söde H.; Liang L.; Meunier V.; Berger R.; Li R.; Feng X.; Müllen K.; Fasel R. Graphene Nanoribbon Heterojunctions. Nat. Nanotechnol. 2014, 9, 896–900. 10.1038/nnano.2014.184. [DOI] [PubMed] [Google Scholar]
- Bronner C.; Stremlau S.; Gille M.; Brauße F.; Haase A.; Hecht S.; Tegeder P. Aligning the Band Gap of Graphene Nanoribbons by Monomer Doping. Angew. Chem., Int. Ed. 2013, 52, 4422–4425. 10.1002/anie.201209735. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Yazyev O. V.; Feng J.; Xie L.; Tao C.; Chen Y.-C.; Jiao L.; Pedramrazi Z.; Zettl A.; Louie S. G.; Dai H.; Crommie M. F. Experimentally Engineering the Edge Termination of Graphene Nanoribbons. ACS Nano 2013, 7, 198–202. 10.1021/nn303730v. [DOI] [PubMed] [Google Scholar]
- Chen Y.-C.; Cao T.; Chen C.; Pedramrazi Z.; Haberer D.; de Oteyza D. G.; Fischer F.; Louie S. G.; Crommie M. F. Molecular Bandgap Engineering of Bottom-Up Synthesized Graphene Nanoribbon Heterojunctions. Nat. Nanotechnol. 2015, 10, 156–160. 10.1038/nnano.2014.307. [DOI] [PubMed] [Google Scholar]
- Sevinçli H.; Topsakal M.; Ciraci S. Superlattice Structures of Graphene-Based Armchair Nanoribbons. Phys. Rev. B: Condens. Matter Mater. Phys. 2008, 78, 245402. 10.1103/PhysRevB.78.245402. [DOI] [Google Scholar]
- Prezzi D.; Varsano D.; Ruini A.; Molinari E. Quantum Dot States and Optical Excitations of Edge-Modulated Graphene Nanoribbons. Phys. Rev. B: Condens. Matter Mater. Phys. 2011, 84, 041401. 10.1103/PhysRevB.84.041401. [DOI] [Google Scholar]
- Schwierz F. Graphene Transistors. Nat. Nanotechnol. 2010, 5, 487–496. 10.1038/nnano.2010.89. [DOI] [PubMed] [Google Scholar]
- Bennett P. B.; Pedramrazi Z.; Madani A.; Chen Y.-C.; de Oteyza D. G.; Chen C.; Fischer F. R.; Crommie M. F.; Bokor J. Bottom-Up Graphene Nanoribbon Field-Effect Transistors. Appl. Phys. Lett. 2013, 103, 253114. 10.1063/1.4855116. [DOI] [Google Scholar]
- Cai J.; Ruffieux P.; Jaafar R.; Bieri M.; Braun T.; Blankenburg S.; Muoth M.; Seitsonen A. P.; Saleh M.; Feng X.; Mullen K.; Fasel R. Atomically Precise Bottom-up Fabrication of Graphene Nanoribbons. Nature 2010, 466, 470–473. 10.1038/nature09211. [DOI] [PubMed] [Google Scholar]
- Chen Y.-C.; de Oteyza D. G.; Pedramrazi Z.; Chen C.; Fischer F. R.; Crommie M. F. Tuning the Band Gap of Graphene Nanoribbons Synthesized from Molecular Precursors. ACS Nano 2013, 7, 6123–6128. 10.1021/nn401948e. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Lin H.; Sun K.; Chen L.; Zagranyarski Y.; Aghdassi N.; Duhm S.; Li Q.; Zhong D.; Li Y.; Mullen K.; Fuchs H.; Chi L. On-Surface Synthesis of Rylene-Type Graphene Nanoribbons. J. Am. Chem. Soc. 2015, 137, 4022–4025. 10.1021/ja511995r. [DOI] [PubMed] [Google Scholar]
- Kimouche A.; Ervasti M. M.; Drost R.; Halonen S.; Harju A.; Joensuu P. M.; Sainio J.; Liljeroth P. Ultra-Narrow Metallic Armchair Graphene Nanoribbons. Nat. Commun. 2015, 6, 10177. 10.1038/ncomms10177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffieux P.; Wang S.; Yang B.; Sánchez-Sánchez C.; Liu J.; Dienel T.; Talirz L.; Shinde P.; Pignedoli C. A.; Passerone D.; Dumslaff T.; Feng X.; Müllen K.; Fasel R. On-Surface Synthesis of Graphene Nanoribbons with Zigzag Edge Topology. Nature 2016, 531, 489–492. 10.1038/nature17151. [DOI] [PubMed] [Google Scholar]
- Blankenburg S.; Cai J.; Ruffieux P.; Jaafar R.; Passerone D.; Feng X.; Mullen K.; Fasel R.; Pignedoli C. A. Intraribbon Heterojunction Formation in Ultranarrow Graphene Nanoribbons. ACS Nano 2012, 6, 2020–2025. 10.1021/nn203129a. [DOI] [PubMed] [Google Scholar]
- Bjork J.; Stafstrom S.; Hanke F. Zipping Up: Cooperativity Drives the Synthesis of Graphene Nanoribbons. J. Am. Chem. Soc. 2011, 133, 14884–14887. 10.1021/ja205857a. [DOI] [PubMed] [Google Scholar]
- Batra A.; Cvetko D.; Kladnik G.; Adak O.; Cardoso C.; Ferretti A.; Prezzi D.; Molinari E.; Morgante A.; Venkataraman L. Probing the Mechanism for Graphene Nanoribbon Formation on Gold Surfaces Through X-Ray Spectroscopy. Chem. Sci. 2014, 5, 4419–4423. 10.1039/C4SC01584C. [DOI] [Google Scholar]
- Huang H.; Wei D.; Sun J.; Wong S. L.; Feng Y. P.; Castro Neto A. H.; Wee A. T. S. Spatially Resolved Electronic Structures of Atomically Precise Armchair Graphene Nanoribbons. Sci. Rep. 2012, 2, 983. 10.1038/srep00983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han P.; Akagi K.; Canova F. F.; Mutoh H.; Shiraki S.; Iwaya K.; Weiss P. S.; Asao N.; Hitosugi T. Bottom-Up Graphene-Nanoribbon Fabrication Reveals Chiral Edges and Enantioselectivity. ACS Nano 2014, 8, 9181–9187. 10.1021/nn5028642. [DOI] [PubMed] [Google Scholar]
- Han P.; Akagi K.; Canova F. F.; Shimizu R.; Oguchi H.; Shiraki S.; Weiss P. S.; Asao N.; Hitosugi T. Self-Assembly Strategy for Fabricating Connected Graphene Nanoribbons. ACS Nano 2015, 9, 12035–12044. 10.1021/acsnano.5b04879. [DOI] [PubMed] [Google Scholar]
- Han P.; Akagi K.; Canova F. F.; Mutoh H.; Shiraki S.; Iwaya K.; Weiss P. S.; Asao N.; Hitosugi T. Reply to “Comment on ’Bottom-Up Graphene-Nanoribbon Fabrication Reveals Chiral Edges and Enantioselectivity’. ACS Nano 2015, 9, 3404–3405. 10.1021/acsnano.5b01687. [DOI] [PubMed] [Google Scholar]
- Simonov K. A.; Vinogradov N. A.; Vinogradov A. S.; Generalov A. V.; Zagrebina E. M.; Martensson N.; Cafolla A. A.; Carpy T.; Cunniffe J. P.; Preobrajenski A. B. Effect of Substrate Chemistry on the Bottom-Up Fabrication of Graphene Nanoribbons: Combined Core-Level Spectroscopy and STM Study. J. Phys. Chem. C 2014, 118, 12532–12540. 10.1021/jp502215m. [DOI] [Google Scholar]
- Simonov K. A.; Vinogradov N. A.; Vinogradov A. S.; Generalov A. V.; Zagrebina E. M.; Martensson N.; Cafolla A. A.; Carpy T.; Cunniffe J. P.; Preobrajenski A. B. Comment on “Bottom-Up Graphene-Nanoribbon Fabrication Reveals Chiral Edges and Enantioselectivity. ACS Nano 2015, 9, 3399–3403. 10.1021/nn506439a. [DOI] [PubMed] [Google Scholar]
- Simonov K. A.; Vinogradov N. A.; Vinogradov A. S.; Generalov A. V.; Zagrebina E. M.; Svirskiy G. I.; Cafolla A. A.; Carpy T.; Cunniffe J. P.; Taketsugu T.; Lyalin A.; Martensson N.; Preobrajenski A. B. From Graphene Nanoribbons on Cu(111) to Nanographene on Cu(110): Critical Role of Substrate Structure in the Bottom-Up Fabrication Strategy. ACS Nano 2015, 9, 8997–9011. 10.1021/acsnano.5b03280. [DOI] [PubMed] [Google Scholar]
- Sánchez-Sánchez C.; Dienel T.; Deniz O.; Ruffieux P.; Berger P.; Feng X.; Müllen K.; Fasel R. Purely Armchair or Partially Chiral: Non-Contact Atomic Force Microscope Characterization of Dibromo-Bianthryl-Based Graphene Nanoribbons Grown on Cu(111). ACS Nano 2016, 10, 8006–8011. 10.1021/acsnano.6b04025. [DOI] [PubMed] [Google Scholar]
- Weiler-Feilchenfeld H.; Bergmann E. D.; Hirschfeld A. The Conformation of 9,9′-bianthryl. Tetrahedron Lett. 1965, 6, 4129–4131. 10.1016/S0040-4039(01)99576-0. [DOI] [Google Scholar]
- Chen M.; Xiao J.; Steinrück H.-P.; Wang S.; Wang W.; Lin N.; Hieringer W.; Gottfried J. M. Combined Photoemission and Scanning Tunneling Microscopy Study of the Surface-Assisted Ullmann Coupling Reaction. J. Phys. Chem. C 2014, 118, 6820–6830. 10.1021/jp4121468. [DOI] [Google Scholar]
- Basagni A.; Sedona F.; Pignedoli C. A.; Cattelan M.; Nicolas L.; Casarin M.; Sambi M. Molecules–Oligomers–Nanowires–Graphene Nanoribbons: A Bottom-Up Stepwise On-Surface Covalent Synthesis Preserving Long-Range Order. J. Am. Chem. Soc. 2015, 137, 1802–1808. 10.1021/ja510292b. [DOI] [PubMed] [Google Scholar]
- Dong L.; Liu P. N.; Lin N. Surface-Activated Coupling Reactions Confined on a Surface. Acc. Chem. Res. 2015, 48, 2765–2774. 10.1021/acs.accounts.5b00160. [DOI] [PubMed] [Google Scholar]
- Shi K. J.; Yuan D. W.; Wang C. X.; Shu C. H.; Li D. Y.; Shi Z. L.; Wu X. Y.; Liu P. N. Ullmann Reaction of Aryl Chlorides on Various Surfaces and the Application in Stepwise Growth of 2D Covalent Organic Frameworks. Org. Lett. 2016, 18, 1282–1285. 10.1021/acs.orglett.6b00172. [DOI] [PubMed] [Google Scholar]
- Di Giovannantonio M.; El Garah M.; Lipton-Duffin J.; Meunier V.; Cardenas L.; Fagot Revurat Y.; Cossaro A.; Verdini A.; Perepichka D. F.; Rosei F.; Contini G. Insight into Organometallic Intermediate and Its Evolution to Covalent Bonding in Surface-Confined Ullmann Polymerization. ACS Nano 2013, 7, 8190–8198. 10.1021/nn4035684. [DOI] [PubMed] [Google Scholar]
- Walch H.; Gutzler R.; Sirtl T.; Eder G.; Lackinger M. Material- and Orientation-Dependent Reactivity for Heterogeneously Catalyzed Carbon-Bromine Bond Homolysis. J. Phys. Chem. C 2010, 114, 12604–12609. 10.1021/jp102704q. [DOI] [Google Scholar]
- Zhang Y. Q.; Kepcija N.; Kleinschrodt M.; Diller K.; Fischer S.; Papageorgiou A. C.; Allegretti F.; Bjork J.; Klyatskaya S.; Klappenberger F.; Ruben M.; Barth J. V. Homo-Coupling of Terminal Alkynes on a Noble Metal Surface. Nat. Commun. 2012, 3, 1286. 10.1038/ncomms2291. [DOI] [PubMed] [Google Scholar]
- Lahrood A. R.; Björk J.; Heckl W. M.; Lackinger M. 1,3-Diiodobenzene on Cu(111) – an Exceptional Case of On-Surface Ullmann Coupling. Chem. Commun. 2015, 51, 13301–13304. 10.1039/C5CC04453G. [DOI] [PubMed] [Google Scholar]
- Treier M.; Pignedoli C. A.; Laino T.; Rieger R.; Müllen K.; Passerone D.; Fasel R. Surface-Assisted Cyclodehydrogenation Provides a Synthetic Route towards Easily Processable and Chemically Tailored Nanographenes. Nat. Chem. 2011, 3, 61–67. 10.1038/nchem.891. [DOI] [PubMed] [Google Scholar]
- Horcas I.; Fernández R.; Gómez-Rodríguez J. M.; Colchero J.; Gómez-Herrero J.; Baro A. M. WSXM: A Software for Scanning Probe Microscopy and a Tool for Nanotechnology. Rev. Sci. Instrum. 2007, 78, 013705. 10.1063/1.2432410. [DOI] [PubMed] [Google Scholar]
- Soler J. M.; Artacho E.; Gale J. D.; Garcia A.; Junquera J.; Ordejón P.; Sanchez-Portal D. The SIESTA Method for Ab Initio Order-N Materials Simulation. J. Phys.: Condens. Matter 2002, 14, 2745. 10.1088/0953-8984/14/11/302. [DOI] [PubMed] [Google Scholar]
- Sánchez-Portal D.; Ordejón P.; Artacho E.; Soler J. M. Density-Functional Method for Very Large Systems with LCAO Basissets. Int. J. Quantum Chem. 1997, 65, 453–461. . [DOI] [Google Scholar]
- Artacho E.; Sánchez-Portal D.; Ordejón P.; Garcia A.; Soler J. M. Linear-Scaling Ab-Initio Calculations for Large and Complex Systems. Phys. Status Solidi B 1999, 215, 809–817. . [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.