

Abstract
Among the various postsynthesis treatments of colloidal nanocrystals that have been developed to date, transformations by cation exchange have recently emerged as an extremely versatile tool that has given access to a wide variety of materials and nanostructures. One notable example in this direction is represented by partial cation exchange, by which preformed nanocrystals can be either transformed to alloy nanocrystals or to various types of nanoheterostructures possessing core/shell, segmented, or striped architectures. In this review, we provide an up to date overview of the complex colloidal nanostructures that could be prepared so far by cation exchange. At the same time, the review gives an account of the fundamental thermodynamic and kinetic parameters governing these types of reactions, as they are currently understood, and outlines the main open issues and possible future developments in the field.
1. Introduction
Colloidal nanocrystals (NCs) have played a pivotal role in both fundamental research and in technological applications over the last 30 years.1−14 Today, a rational synthetic approach to NC is of utmost importance due to the growing demand for nanomaterials having compositional diversity and that need to be engineered in shape, morphology, and surface functionality, such that they will possess well-defined optical, electronic, magnetic, and catalytic features, for use in the most disparate fields of science and technology. Over the years, the synthesis of colloidal NCs has evolved to a point that a fine level of control over size, shape, structural, and compositional parameters has finally become possible for many materials systems.10,15−18 As an example, a huge variety of nanocrystal heterostructures, that is, NCs combining different domains of various chemical compositions, have been reported over the last two decades.19−27 Among these types of nanoheterostructures (NHCs), perhaps semiconductor–semiconductor NHCs are those that have been exploited the most. Here, the fine-tuning of the size, shape, spatial orientation, composition, and crystalline structure of the component materials enables a careful adjustment of the relative band alignments of both semiconductors and, thus, allows for a tight control over the physical and optical properties of the hybrid nanostructures. In type I core|shell NHCs, for instance, the band alignment of the semiconductors results in the confinement of the charges in the core material. By choosing a fluorescent semiconductor as core material, core|shell NHCs of this type can display enhanced photoluminescence, which is required for lighting, lasing, and sensing applications. On the other hand, in NHCs with a type II band alignment the charges can be spatially separated after photoexcitation, which makes these systems interesting for photovoltaic and photocatalytic applications. Even alloy NCs have gained increasing attention in recent years as their band gap, and in some cases their magnetic properties, can be tweaked by controlling the NCs composition, which makes them suitable for applications in displays, biomedical imaging and sensing, lab-on-a-chip, solid-state lighting, and photovoltaic devices.
While on the one hand a consolidated body of knowledge has been established around the colloidal synthesis of NCs, many materials or combinations of them remain poorly accessible in nanocrystalline form. Therefore, new postsynthetic strategies are emerging that compensate for the current limits imposed by direct reaction routes and that expand the library of possible structures that can be fabricated. It still remains a challenge, for example, to directly synthesize NHCs with simultaneous control over the size, shape, spatial orientation, composition, and crystalline structure of the component materials. Even the direct synthesis of alloyed semiconductor NCs, with regulation of the composition as well as the size and shape, is often not straightforward as the control over the reaction kinetics of multiple species in a single reaction step is hardly achievable. In part with the aim to overcome the yet unsolved issues related to direct colloidal synthesis and in part driven by the fundamental interest in chemical and structural transformations of materials at the nanoscale, a new area of research has emerged in the past decade in the field of colloidal NCs. This area encompasses the transformations of nanomaterials driven by cation exchange (CE).28−35 At the nanoscale, CE has been applied and studied on a variety of materials, above all those belonging to the II–VI, I–III–VI, and IV–VI classes of semiconductors. This simple process consists, basically, in the replacement of cations of a starting NC with new cations, with preservation of the original anion sublattice, which in some cases undergoes a certain degree of structural reorganization. CE takes place instead of anion exchange because the diffusion rate of the cations is generally much higher than that of the anions. This can be explained by the fact that, in a crystal, cations are generally smaller than anions, and therefore, one can visualize the cations as diffusing within a comparatively rigid lattice of anions. In most cases, a CE reaction is a topotactic transformation in which the anion framework remains virtually intact and thus the morphology of the overall structure is typically retained.
Reports on CE in NCs have been growing steadily since the first landmark works in the 1990s and especially after important breakthroughs from 2004 on.28,36,37 However, despite CE at the nanoscale having being studied for more than one decade, there is a fundamental knowledge gap for what concerns the detailed mechanisms and forces governing CE at this size regime, and the main reason is that CE had been used primarily as a means of synthesizing nanomaterials. Such a knowledge gap is slowly closing over the years, as recent works are finally addressing in detail the various issues. Examples are the studies that explore the role of the most effective complexing agents in stabilizing the entering/exiting cations in the solution phase, the relevance of various structural parameters in promoting exchange reactions, as well as the importance of the size and valency of cations, which dictates the type of coordination that they can adopt in the lattice and ultimately their diffusivity and whether this occurs primarily via vacancies or via interstitials. All these studies used advanced analysis tools, including many electron microscopy and spectroscopy techniques, and time-resolved X-ray experiments.38−43 A few recent papers have also attempted to model many aspects of CE, at least for the simplest cases. Also, many seminal review articles have been published in recent years by various leading groups on CE.28,32−35
In the present article, while giving ample credit to all those previous review articles on CE, we will present an updated snapshot of the field. To this aim, we will first provide an overview of the well-established thermodynamic concepts that are necessary for predicting the feasibility and the outcome of a CE reaction. This will be followed by a section dedicated to the progress that has been made so far in understanding the various possible mechanisms involved in CE, with particular emphasis on the aspects related to cation diffusion and replacement in NCs. At the same time, we will give an up-to-date overview of the astonishing complex nanostructures that are now accessible via CE (see Scheme 1). These systems, ranging from core|shell, to segmented, and to alloyed NCs, nanorods (NRs), nanoplatelets (NPLs), nanowires (NWs), etc., can be now prepared by extracting just a fraction of the original cations from the starting nanomaterials and replacing them with new types of cations (also called partial CE). Without downplaying the importance of total CE transformations that have started the field and have first aroused the interest of the community in this technique, our review will focus mainly on heterostructured types of nanomaterials, as they represent one of the most fascinating and intriguing developments of CE. It is our conviction that a deeper comprehension of the mechanisms underlying ions interdiffusion and replacement in CE reactions, coupled with a rational choice of semiconductor materials, will generate, in the future, a wide range of sophisticated nanostructures that can be exploited in many disparate applications.
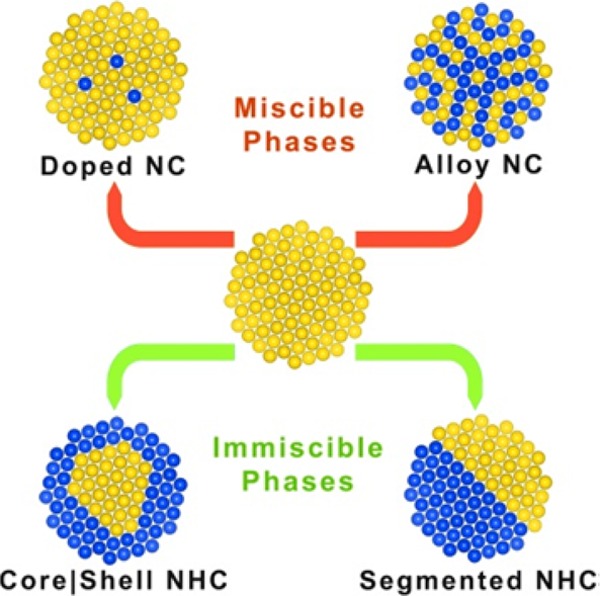
Scheme 1. Schematic Representation of Nanostructures Accessible via Cation Exchange Transformations of a Preformed Colloidal NC.
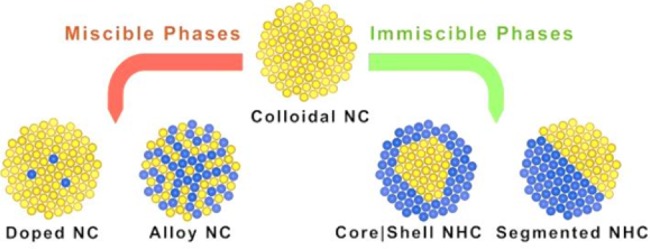
The original cations (yellow spheres) are replaced with new guest cations (blue spheres) with minimal distortion (or no distortion at all) of the anion framework.
Before proceeding further, the question arises on why anion exchange has received much less attention to date.44−52 One important reason, as mentioned earlier, is that at least in binary compounds the comparatively larger anions are generally much less mobile than the cations. Therefore, harsh conditions would be required to trigger transformations that involve the exchange of anions. However, the concept of cation sublattice preservation, and with that the integrity of the overall NCs, is seriously questionable, and one would wonder whether such transformations should be indeed categorized as occurring via ion exchange or if they should rather be seen as recrystallization processes: here, the initial NC seems to act as some sort of “precursor” for the nucleation/recrystallization of a new material on it, with often poor size/morphology retention or no retention at all. Obviously, when the degree of structural complexity and diversity in a crystal lattice increases, this statement shakes considerably. Indeed, in analogy with the well-known high-temperature diffusivity of oxygen ions in various oxide materials, many works (some recent, some dating back to the 1980s) on ternary halide perovskites have demonstrated that the halide ions have a remarkably high diffusivity in such materials.53−55 This was immediately translated in the possibility to exchange, at least in lead halide perovskites, the halide ions (Cl-, Br-, and I-), while the two types of cations involved in the structure remain in place, which enabled tuning of the band gap by means of a simple (and fast) postsynthesis anion exchange at room temperature.56−59 Such materials are receiving impressive attention nowadays due to their many fascinating physical properties and their great promise in photovoltaics, lighting, and lasing applications. Triggered by such surge, we are now witnessing a general revival in the study of materials involving halides in their composition, again both in their bulk form and as nanocrystalline domains. It is therefore plausible to expect that much more knowledge will be generated in the coming years in exchange reactions involving anions. For the time being, our focus here will be on exchange reactions involving cations.
We start this review with a series of basics considerations on the thermodynamic and kinetic of CE in colloidal nanostructures and on how their understanding has evolved over the past years. This section is then followed by an extensive section on selected examples from the literature, with emphasis on those cases in which the degree of structural and compositional complexity arises as a peculiar feature of CE.
2. Thermodynamics of CE
A CE reaction, in which the A+ cations of an AX ionic NC are exposed to a solution of B+ cations to give the BX structure, can be written as
| 1 |
For the sake of simplicity we will consider, in the following discussion, only monovalent exchanging cations, even if typical CE reactions can occur between heterovalent ions. In a first approximation, we will assume that the specific crystallographic phases of the initial and final NC do not play any major role in the overall energy balance. Following the same treatment proposed by Rivest et al., the process can be divided in four ideal steps: A-X dissociation, B-X association, B+ desolvation, and A+ solvation.33 In order to predict a priori the spontaneity of a CE reaction it is necessary to know the energies involved in these four steps. The association and dissociation processes can be defined in terms of lattice and surface energy of the AX and BX crystals, while the determination of the energies involved in the solvation and desolvation requires a precise knowledge of the affinity of the exchanging cations for both the solvent and the possible ligands used in the CE process. No entropic variation is associated with the process as for each A+ ion extracted from the host crystal a B+ cation is desolvated. Furthermore, as will be discussed in more detail in section 2.2, even when working with heterovalent cations, the entropic contribution to the overall free energy can be neglected, as it plays a minor role.
2.1. Association and Dissociation Energies
The lattice energy (ΔHlatt) of a ionic crystal, also called lattice enthalpy, is commonly defined as the energy required to break the crystal apart into isolated ions at absolute zero temperature. This energy gives a measure of the strength of the chemical bonding among ions constituting the ionic solid and, thus, the higher is the lattice energy, the more stable is the crystal. The lattice energy takes into account the Coulomb attraction and repulsion among the ions in the lattice, and it is influenced not only by the charge and the radius of the cations and anions but also by their ordering, i.e. the crystal structure of the solid, as follows:
| 2 |
In the expression above, N is Avogadro’s number, zi and zj are the integral charges on the ions (in units of e), e is the electron charge, r+ and r- are the ionic radii of the cations and anions, respectively, n is the Born exponent (that takes into account the ionic repulsion), and M is the Madelung constant. The latter term is intimately related to the spatial position of the ions, and it is, therefore, dependent on the crystal structure of the material under analysis. It follows that, when working with a given material, its allotropes can have significantly different lattice energies. The lattice energies of a wide range of materials are available in the literature as they have been experimentally calculated through the so-called “Born–Haber cycle”.
Most of the CE reactions performed on NCs deal with metal chalcogenides and, unfortunately, it is not always easy to find the tabulated ΔHlatt values of many of those (see Table 1), especially when they have allotropes. This is particularly true when considering that, in some cases, these reactions can lead to the formation of metastable crystal structures that obviously cannot be predicted a priori (see also Section 4). If the structure of the materials under study is known, then following eq 2 it is possible to make some qualitative conclusions about lattice energies. As ΔHlatt strongly depends on the distance between cations and anions (r), then given a certain cation M+ and by increasing the size of the A– anion (keeping the ionic charges and the crystal structure fixed) a decrease in ΔHlatt is expected. Therefore, in general, the ΔHlatt of metal chalcogenides decreases in absolute value as the ionic radius of the chalcogen increases: ΔHlatt(MxSy) > ΔHlatt(MxSey) > ΔHlatt(MxTey). As shown in Table 1, ΔHlatt values of MxTey are lower than the corresponding MxSey, and the same trend is seen by comparing MxSey with MxSy.
Table 1. Lattice Energies (ΔHlatt) and Bond Dissociation Energies (BDE) of Some Metal Chalcogenides (M-Y) Expressed in kJ/mol (from refs (60−62)).
| sulfides | ΔHlatt | BDE (M-S) | selenides | ΔHlatt | BDE (M-Se) | tellurides | ΔHlatt | BDE (M-Te) |
|---|---|---|---|---|---|---|---|---|
| Ag2S | 2677 | 216.7 ± 14.6 | Ag2Se | 2686 | 210.0 ± 14.6 | Ag2Te | 2600 | 195.8 ± 14.6 |
| CdS | 3460 | 208.5 ± 20.9 | CdSe | 3310 | 127.6 ± 25.1 | CdTe | 100.0 ± 15.1 | |
| Cu2S | 2865 | 274.5 ± 14.6 | Cu2Se | 2936 | 255.2 ± 14.6 | Cu2Te | 2683 | 230.5 ± 14.6 |
| HgS | 3573 | 217.3 ± 22.2 | HgSe | 144.3 ± 30.1 | HgTe | <142 | ||
| PbS | 3161 | 398 | PbSe | 3144 | 302.9 ± 4.2 | PbTe | 3039 | 249.8 ± 10.5 |
| ZnS | 3674 | 224.8 ± 12.6 | ZnSe | 170.7 ± 25.9 | ZnTe | 117.6 ± 18.0 |
When values of ΔHlatt are not known, then an alternative qualitative approach to estimate the relative stabilities of reactants and products, and thus to evaluate the thermodynamics of the CE reaction, is to consider the bond dissociation energies (BDEs).42,63−65 The BDE of a generic AX compound is defined as the standard enthalpy of the following process: A–X → A + X. This energy, being calculated for diatomic molecules, does not take into account any ion ordering and it is independent from the crystal structure of the compound under analysis. As it possible to see in Table 1, using tabulated values of BDE the predicted relative stabilities of many compounds are in agreement with the trend in ΔHlatt. On the other hand, even if the lattice and the solvation/desolvation energies of the species involved in the CE reaction are known, the calculation of the overall energy of the process is still not trivial. The association and dissociation terms in eq 1 require, indeed, a precise knowledge of the surface energy of the NCs under analysis. One of the most common properties of nanomaterials is their large surface-to-volume ratio and thus a large fraction of the atoms are localized on their surface. Therefore, if compared to bulk materials, the high surface-to-volume ratio of NCs is thermodynamically mirrored by a large surface energy. Unfortunately, it is extremely challenging to calculate or even to give a rough estimate of the surface energy contribution in a CE reaction, as this depends on many variables, such as the types of stabilizing molecules bound at the surface of NCs and the chemical and structural nature of the facets by which the NCs are terminated (each of the facets being passivated by the various ligand molecules with different binding strengths).
2.2. Solvation and Desolvation Energies
Let us consider a CE reaction in which monovalent A+ ions from a preformed A2X NC are replaced with divalent B2+ ions to form a BX NC. Such transformation is entropically favored, since for each B2+ cation desolvated and incorporated into the host NC two A+ ions are solvated, with the consequent increase of the overall entropy. If the product BX NC is more stable than the parent A2X NC (that is |ΔHlattBX| > |ΔHlattA2X|), then such CE reaction should be favored by both lattice enthalpy and entropy. The experimental evidence shows that, in many cases, such reaction does not take place if no proper solvents or ligands are used. A typical example is the CE transformation from Cu2X or Ag2X to CdX NCs. The reaction should be promoted by both the increase of entropy, as it proceeds with the solvation of two Cu+ or Ag+ ions for each Cd2+ incorporated in the host NC, and by lattice enthalpy, as the product CdX material is more stable than the starting Cu2X or Ag2X (see Table 1). Nevertheless, unless soft Lewis bases (such as alkyl phospines) are used, no exchange is observed (see next sections for further details). The solvation (and the desolvation) energy, in this example, as in vast majority of CE reactions involving NCs, represents indeed the main driving force of the transformation. Often this energy term can be easily predicted and controlled. To this purpose, a careful choice of solvents and/or ligands can generally increase the solubility (and thus the efficiency of extraction) of the outgoing cations and to promote the entry of the desired host cations.31,66
2.2.1. Hardness
Pearson’s hard and soft acids and bases (HSAB) theory is a qualitative concept that is of utmost importance in CE, as it helps to predict the affinity of metal ions to ligands/solvents and, thus, to finely tune the solubility of cations involved in a given CE reaction. This theory relies on the concept of chemical hardness and softness of Lewis acids (A) and bases (B).67 Using specific reference bases, and depending on the stability of the resulting AB complexes, the Lewis acids are divided in two categories. Similarly, Lewis bases are divided in two categories according to the characteristics of their donor atom. These two types are called hard and soft, where the meaning of the word “hardness” is, in simple terms, the resistance to deformation or change. Soft bases are characterized by a donor atom of high polarizability and low electronegativity and that is easily oxidized; hard bases have a donor atom of low polarizability and high electronegativity that is hard to oxidize. On the other hand, in soft acids the acceptor atom is of low positive charge and large size, while in hard acids the acceptor atom is of high positive charge and small size. According to HSAB theory, hard acids prefer to bind to hard bases forming ionic complexes, whereas soft acids prefer to bind to soft bases yielding covalent complexes.
In order to compare acids and bases in terms of hardness, in 1983 Parr and Pearson introduced the concept of absolute hardness (η). Table 2 reports the absolute hardness of many cations, ligands and solvents typically employed in CE reactions. These values are a guideline for comparing the acidity of cations (Lewis acids) and the basicity of ligands or solvents (Lewis bases) in order to optimize a desired CE reaction. Among the bases, the most significant feature that can be extrapolated from the listed η values is that molecules in which the donor atom is F, O, or N have a strong hardness as a result of the negative values of their electron affinity. For similar molecules in which the donor atom is Cl, S, or P, there is always a large drop in hardness. Following this theory, as it will be discussed in the next sessions, it is easy to understand how many CE reactions are promoted by specific solvents/ligands: NCs in which the cations are hard Lewis acids (e.g., Zn2+ and Cd2+ cations) can be easily cation exchanged with cations acting as weaker acids (e.g., Cu+, Pb2+, and Ag+ ions) when a hard base is used (such has water or alcohols); cations acting as weak acids, instead, are spontaneously exchanged with cations behaving as harder acids, if soft bases are employed (such as alkyl phosphines).
Table 2. Experimental Absolute Hardness, η, of Typical Cations and Ligands or Bases That Can Be Used in CE Reactions (from ref (68)).
| acid | η | base | η |
|---|---|---|---|
| Cu+ | 6.28 | C6H5NH2 | 4.4 |
| Pd2+ | 6.75 | C6H5SH | 4.6 |
| Ag+ | 6.96 | C6H5OH | 4.8 |
| Fe2+ | 7.24 | C5H5N | 5 |
| Hg2+ | 7.7 | CH3COCH3 | 5.6 |
| Sn2+ | 7.94 | CH3CHO | 5.7 |
| Pt2+ | 8 | DMF | 5.8 |
| Co2+ | 8.22 | (CH3)3P | 5.9 |
| Cu2+ | 8.27 | PH3 | 6 |
| Au3+ | 8.4 | (CH3)2S | 6 |
| Pb2+ | 8.46 | CH2O | 6.2 |
| Co3+ | 8.9 | HCONH2 | 6.2 |
| Mn2+ | 9.02 | (CH3)3N | 6.3 |
| Ge2+ | 9.15 | HCO2CH3 | 6.4 |
| Cd2+ | 10.29 | CH3CN | 7.5 |
| Zn2+ | 10.88 | CH3Cl | 7.5 |
| Fe3+ | 12.08 | (CH3)2O | 8 |
| In3+ | 13 | NH3 | 8.2 |
| Ga3+ | 17 | CH3F | 9.4 |
| Al3+ | 45.77 | H2O | 9.5 |
2.2.2. Solubility Product Constants
When working with water as a solvent, the solubility product constants of the starting NCs and the expected products can be used to predict the thermodynamic feasibility of a CE process.31 The solubility product constant (KSP) is a useful parameter for calculating the aqueous solubility of soluble ionic crystals under various conditions. It may be determined by direct measurements or it can be calculated from the standard Gibbs energies of formation ΔfG° of the species involved at their standard states. Let us define KSP = [M+] [A–] as the equilibrium constant for the reaction:
| 3 |
where MA is the soluble ionic crystal and M+ and A– are the ions produced in solution by the dissociation of MA. The Gibbs energy change can be written as
| 2 |
The solubility product constant is calculated from the equation:
| 3 |
It follows that an ionic solid with a relatively high KSP spontaneously transforms trough CE to another ionic solid with a comparatively lower KSP. The solubility product constants of some metal chalcogenides of interest are reported in Table 3.
Table 3. Solubility Product Constants (KSP) at 25°C of Different Metal Chalcogenides (from refs (31 and 69)).
| compound | E=S | E=Se | E=Te |
|---|---|---|---|
| Ag2E | 6.3 × 10–50 | 3 × 10–54 | N.A. |
| Bi2E3 | 1 × 10–97 | 1 × 10–130 | N.A. |
| CdE | 8 × 10–27 | 4 × 10–35 | 1 × 10–42 |
| CuE | 6.3 × 10–36 | 2 × 10–40 | N.A. |
| Cu2E | 2.5 × 10–48 | N.A. | N.A. |
| HgE | 1.6 × 10–52 | 4 × 10–59 | N.A. |
| In2E3 | 5.7 × 10–74 | N.A. | N.A. |
| NiE | 3.2 × 10–19 | 2 × 10–26 | N.A. |
| PbE | 8 × 10–28 | 1 × 10–37 | N.A. |
| PtE | 9.9 × 10–74 | N.A. | N.A. |
| SbE | 2 × 10–26 | N.A. | N.A. |
| SnE | 1 × 10–25 | 5 × 10–34 | N.A. |
| ZnE | 1.6 × 10–24 | 3.6 × 10–26 | N.A. |
Even if the solubility of ionic crystals in many common polar solvents (such as methanol or ethanol) is not available, a rough estimate of the thermodynamic feasibility of a specific CE reaction in these solvents can be made by a glance at the KSP of that material in water.32 With these considerations in mind, it is possible to explain why many CE reactions spontaneously take place in water or in polar solvents. For example, CE reactions involving CdS or CdSe NCs and Pb2+, Ag+, Cu+, Cu2+, or Hg2+ ions are thermodynamically favored as the solubilities of cadmium chalcogenides are higher than those of the expected metal chalcogenides.36,37,70−81 Similarly, the spontaneous exchange of Zn2+ ions in ZnS NCs with softer Lewis acids (such as Ag+, Pb2+, Cu2+, Cd2+, Hg2+, and Bi3+ ions) in water (or polar solvents) can be explained by considering the solubility product constants of these compounds.49,82−86 The same argument applies to many other transformations taking place rapidly at room temperature in water (or in polar solvents): CuS → Ag2S, CdSe → Ag2Se or HgSe, PbS → Cu2S, and ZnSe → CdSe or Ag2Se.28,63,76,79,87−90 Interestingly, when, in a polar solvent, two materials are exposed to an amount of guest cations that enables competition between the two, the solubility product constants can help to predict the outcome of the CE. In a recent work of ours, we demonstrated, for example, that the exposure of Cu2-xSe/Cu2-xS core/shell NCs to a low amount of Hg2+ or Ag+ ions led to a selective exchange of the selenide core, a transformation that can be explained by considering that metal selenides have lower KSP than the corresponding sulfides.65
As it is possible to notice from Table 3, the solubility data of many ionic crystals, such as metal selenides, most of the metal tellurides, ternary chalcogenides (i.e., CuInS2 and CuInSe2) and their possible allotropes are hard to find in literature or simply they are not known.91 In these cases, as pointed out by Moon et al., a qualitative estimate of KSP for a generic MA compound can be made from its molar enthalpy of solvation (ΔHsolv), according to the following reaction:31
ΔHlatt[MA(s)] the lattice energy (or enthalpy) (see also the above discussion in section 2.1), is instead given by
ΔHhyd[M+(g)] and ΔHhyd[A–(g)] are the hydration enthalpies of the M+ cations and A– anions, respectively:
Then, the overall reaction solvation process can be written as
| 4 |
By defining r+ as the radius of the M+ cation and r– as that of the A– anion, ΔHlatt and ΔHhyd are both negative values and are proportional to (1/(r+ + r–)) (see eq 2) and (1/r+ + 1/r–), respectively. If the radius r+ of the metal cation is held fixed and the radius r– of the anion is increased, then the ΔHhyd term decreases more than ΔHlatt term. Considering that rS2– < rSe2– < rTe2–, and that ΔHlatt(MxSy) > ΔHlatt(MxSey) > ΔHlatt(MxTey), it is possible to conclude that metal sulfides are more soluble than the corresponding selenides, as confirmed by the experimental values reported in Table 3.
Following the same line of reasoning, one can infer that metal tellurides have lower solubility product constants than the corresponding selenides (and sulfides). These considerations can be used, for example, to explain the well-known spontaneous CE reaction between CdSe NCs and Cu+ ions in polar solvents.80,92,93 While the KSP of CdSe is known (KSP = 4 × 10–35), no KSP data is apparently available for Cu2Se, but we expect its solubility to be lower than that of the corresponding Cu2S (KSP = 2.5 × 10–48). For this reason, it is possible to realize why the formation of Cu2Se is favored in the CE exchange reaction involving CdSe NCs and Cu+ ions. Using the same argument, one can rationalize why CdTe NCs (KSP = 1 × 10–42) undergo CE with Hg2+, Ag+, and Cu+ ions in polar solvents.94−97 Indeed, even if the solubility product constants of HgTe, Ag2Te, and Cu2Te are not available in the literature, one should expect them to be lower than those of HgSe (KSP = 4 × 10–59), Ag2Se (KSP = 3 × 10–54), and Cu2S (KSP = 2.5 × 10–48), respectively, and thus much lower than that of CdTe.
2.2.3. Ligands in CE
Water and alcohols are particularly suitable for CE reactions in which the acidity of the in-going cations is lower than the one of the out-going cations. On the other hand, also nonpolar solvents have been shown to work in many different CE transformations, if coupled with proper Lewis bases in the form of ligands. In these reactions, a fine-tuning of the affinity between the outgoing cations and the ligands is required. Unfortunately, the hardness values can be used only as a guideline and every specific CE process has to be studied and optimized by choosing proper ligands.
As a general trend, soft acids as Cu+, Ag+, and Pb2+ have been extracted and replaced by harder ones, such as Cd2+, Zn2+, In3+, Sn2+, and Sn4+ in metal chalcogenides and pnictides using either tri-n-octylphosphine (TOP), tri-n-butylphosphine (TBP)29,30,64,72,79,80,92,93,98−115 or carboxylates41,88,116−118 as soft bases. Since phosphines (R3P) have been widely exploited in most of these CE reactions, Gui et al. dedicated a study on their activity in CE reactions between N2E (E = chalcogen and N = Ag or Cu) NCs and M2+ ions in solution, which can generalized by the following reaction scheme:114
| 5 |
These compounds are able bind metal ions by donating their P lone pair and forming a monodentate terminal metal–PR3 group (σ-bonding, see Figure 1a). Differently from amine ligands, capable only of σ-bonding, phosphines can be also good π acceptors, and therefore, they can coordinate metal cations through π-backbonding, to an extent that depends on the nature of the R groups. For these reasons, phosphines can be very high field ligands and can form strong metal–ligand (M–L) bonds. The particular interest in R3P arises from the fact that their affinity to metal cations can be easily modulated in a systematic and predictable way over a wide range by tuning the electron-donor power and steric effect of R groups.119 Moreover, as spectator ligands, R3P could be also used to stabilize metal–ligand (M-L) units as they are able to form (R3P)n Metal–Ligand complexes.120 Using the Ag2S → CdS CE reaction as a case study, Gui et al. could rank the activity of many different phosphines: triethyl phosphine > trimethyl phosphite > triethyl phosphite > tributyl phosphine > triphenyl phosphine > trioctyl phosphine > tri-p-tolylphosphine > tris(o-methoxyphenyl) phosphine. Thus, by varying the phosphine molecules used in the CE transformation, Gui et al. could prove a good control over the residual amount of Ag+ ions inside the product CdS NCs.
Figure 1.

(A) Thermodynamic scheme of a CE reaction initiated by different phosphines or phosphites (R3P). Reproduced with permission from ref (114). Copyright 2015 John Wiley and Sons. (B) A small collection of multidentate ligands, commonly employed in organometallic chemistry. Each molecule has a different binding preference to metal cations, which may allow, in principle, for the modulation of the solvation energy of cations involved in CE reactions. Adapted with permission from ref (120). Copyright 2014 John Wiley and Sons.
In addition to tertiary phosphines, also amines and carboxilates have been shown to be fundamental ligands in many CE reactions as they behave as strong and weak Lewis bases, respectively. For example, the exchange between CdSe and Pb2+ ions was found to be triggered by the presence of oleylamine (OA) that promotes the extraction of Cd2+ ions by forming a stable Cd-OA complex.121 This can be easily understood by considering that Cd2+ is a harder Lewis acid if compared to Pb2+ and OA is a hard base, so that the Cd-OA coupling results in a more stable acid–base combination than Pb-OA. On the other hand, CE reactions involving PbSe or PbS NCs with Cd2+ ions, when performed in a nonpolar solvent, are made possible by the presence of an oleate moiety that, being a weak base, promotes the extraction of Pb2+ cations.41,122−127 Amines with long alkyl chains (such as OA or octadecylamine) have also been shown to trigger CE between ZnSe or ZnTe NCs and Cd2+, Mn2+, and Pb2+ ions. This is due to the fact that such amines bind preferentially Zn2+ ions which are slightly harder than Cd2+, Mn2+, and Pb2+ ones (see Table 2).42,64,100,128−130 Similarly, OA has been successfully employed to drive CE reactions involving TiS2 nanodiscs and Cu+, Ag+, Mn2+, and Cd2+ ions to form the corresponding metal sulfides.131 In analogy with phosphines, the activity of amines can be tuned over a relatively broad range depending on the groups bound to the N atom and on their steric hindrance. As it is possible to see in Table 2, primary (RNH2), secondary (R2NH), ternary (R3N), and aromatic amines have quite different values of hardness, ranging from 4.4 of aniline (C6H5NH2) up to 6.3 of trimethyl amine ((CH3)3N).
A logical consequence of the discussion above is that the “library” of ligands that can effectively work in CE reactions is likely to expand in the near future, and this will be beneficial for improving the control over such reactions and/or to access new ones. For example, multidentate ligands, which have never been tested in CE reactions, could be used for finely tuning CE reactions in organic media, allowing, in principle, to increase the solvation of exiting cations.120 Bidentate diamine ligands, such as ethylenediamine, indeed, have been widely employed in metal–organic chemistry as they can coordinate transition-metal ions in a chelating mode to form stable complexes.132−134 A small collection of such ligands is shown in Figure 1b.
Thiols, thioethers and disulfides (R2S2) might also turn out to be valuable ligands in CE reactions. Coordination complexes of transition metal ions with sulfur-containing ligands are ubiquitous in nature. It is also well-known that many organometallic compounds (containing heavy metals) owe their toxicity to a strong affinity for sulfur, which causes them to bind to biologically important thiol groups. Logically, then, molecules containing thiol groups have been used in antidotes to poisoning by such organometals.135 Moreover, the use of thiols in colloidal synthesis is widespread. As it is possible to see in Table 2, thiols are extremely soft bases, while thioethers, for example (CH3)2S, exhibit typical hardness values that are similar to those of phosphines. These sulfur based ligands could be tested as complementary soft Lewis bases, being, in principle, able to promote CE reactions in which soft acids have to be extracted. This could be particularly beneficial in those CE reactions in which phosphines are not desirable as they might etch metal chalcogenide NCs by extracting either Se2– or S2– anions out of them.136,137
3. Mechanisms and Kinetics of CE
Even if a CE reaction is thermodynamically favored, kinetic factors, such as activation energy barriers and/or ions diffusivity, play a key role in determining its feasibility and the nature of the final products. At present, several aspects related to CE reactions in NCs are debated. When a host NC is exposed to a solution of guest cations, the latter can diffuse inside the NCs lattice, either via an interstitial or via a vacancy-assisted mechanism. Depending on their diffusivity, the guest ions can either probe the whole host NCs or they can only access the outer surface layer. At a later stage, the fast replacement of cations that leads to the nucleation of the product phase occurs at the sites that are energetically favored/allowed. During this step, host cations are expelled from the NCs through an out-diffusion or kick-out mechanism and become solvated by the ligand molecules present in the solution.
The region(s) where the CE take place, also called the “reaction zone”, can include the whole surface of the NC or a fraction of it. When the transformation front includes the whole NC surface, the intermediate steps of a CE reaction are represented by core–shell or graded core–shell NHCs, depending on the solubility of reactant and product materials (see section 5.3.2). When the nucleation of the product occurs at specific sites of the host NC surface (such as the tips of a NR or the corners of a polyhedral NC) the intermediate steps observed in CE are represented by multi domain heterostructures (such as Janus-like, striped or segmented heterostructures). All these structures will be discussed in detail in section 5.3. Once a reaction zone is formed, the CE reaction proceeds then toward the remaining unexchanged part of the NC trough solid state ions diffusion. This step, in most cases, is the rate-limiting one and it strongly influences the outcome of the CE reaction.42,94,138 Many factors influence the rate at which the solid state diffusion can take place: the possibility of cations to diffuse through interstitial sites, the presence of vacancies (even on the surface), dislocations, stacking faults, and possibly grain boundaries inside the NC.40,88,104,137,139
3.1. Solid-State Diffusion via Vacancies
In a recent work, Groenveld et al. proposed a detailed description of the solid state diffusion process taking place during a CE reaction.42 Their suggested model, specifically conceived for the transformation of ZnSe to CdSe NCs, can be generally extended to many of the known CE transformations. It is noteworthy to specify that ZnSe and CdSe structures are completely miscible, so that CE, in this case, occurs trough the formation of CdxZn1–xSe solid solutions, meaning that both Zn2+ and Cd2+ ions can coexist in the same crystal domain (this will be discussed more extensively in section 5.1). Once a reaction zone has formed, two solid-state diffusion fluxes allow the reaction to proceed: inward diffusion of Cd2+ and outward diffusion of Zn2+. In bulk ionic crystals, solid state diffusion processes are promoted by point defects that are able to move through the crystal and to act as “vehicles for diffusion” of atoms.139
Of particular interest for CE reactions are diffusion-relevant point defects such as Frenkel pairs. A Frenkel pair forms when a cation moves from its lattice position to an interstitial site, creating a cation vacancy and a self-interstitial (VZn and Zni in the present discussion, respectively), guaranteeing global charge neutrality (see Figure 2). In the specific case of ZnSe, Frenkel defects have been shown to promote self-diffusion of zinc ions in the bulk. The formation of these defects is strongly temperature-dependent as they minimize the Gibbs free energy of the crystal itself.139 At a given temperature, the concentration of Frenkel pairs (nFP) can be expressed as
where ΔHFP is the formation enthalpy of a Frenkel defect, k is the Boltzmann constant, and T is the temperature. Once formed, both Zni and VZn can move through the solid by hopping from site to site, which requires further activation energy.
Figure 2.
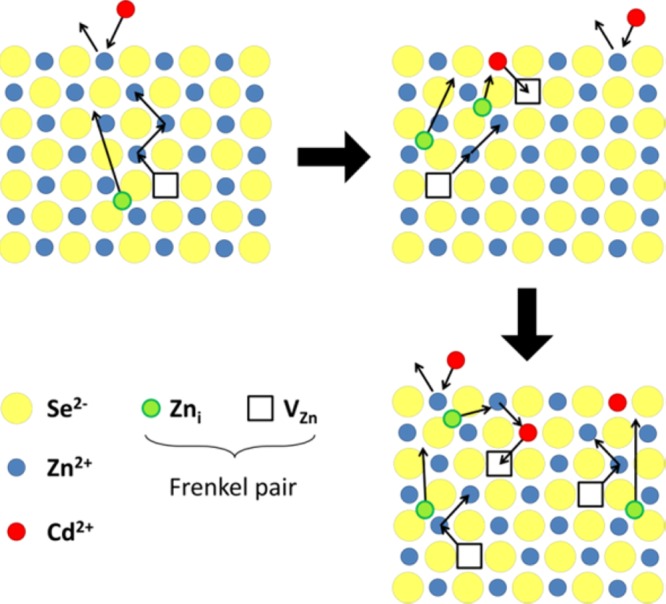
Sketch of the mechanism proposed by Groenveld et al.42 for CE of ZnSe NCs with Cd2+ ions. Fast CE takes place at the NC surface and it is followed by the relatively slower thermally activated solid-state diffusion. The ion diffusion is driven by Frenkel pairs (Zni-VZn), so that the outward diffusion flux consists of interstitial zinc cations (Zni) moving toward the NC surface, while the incoming Cd2+ cations diffuse inwardly by hopping into the VZn.
Depending on the temperature at which the CE is performed, different scenarios can be distinguished: at low temperature (for which nFP is negligible) both inward and outward cation diffusions are precluded, therefore CE either cannot take place or it is limited to the outer surface layer of the NCs. In the specific case of ZnSe NCs exchanged with Cd2+ ions, this is observed at 150 °C and NHCs form that consist of a ZnSe core coated with a thin CdSe shell (one monolayer). At higher temperatures, nFP is sufficiently high to allow solid-state diffusion processes to occur. The host cations migrate to the surface as interstitials (Zni) and are exchanged by Cd2+ guest cations, which hop to the closest available vacancy (VZn) and thus diffuse inward. Once CE takes place at the surface, the Frenkel pair is annihilated, shifting the equilibrium toward the formation of additional Zni-VZn pairs. At the same time, a concentration gradient establishes driving the Cd2+ flux inward (via VZn) and the Zn2+ flux outward (as Zni). In Figure 2 a schematic representation of the solid-state diffusion is depicted. In some cases, this gradient has been shown to keep on driving the ions interdiffusion even after the removal of host NCs from the guest cations solution.94 When the concentration of Frenkel defects is sufficiently high (at high T), the Zn2+ for Cd2+ exchange at the surface occurs simultaneously with solid-state diffusion processes within the NC.
The generation of Frenkel pairs is not the only process through which vacancies can form. Wang et al., in a more recent study, highlighted the relatively low energy required to create a cationic vacancy in ionic NCs.39 For example, the calculated Pb2+-vacancy formation energy in a PbS NC (passivated with Cl– ions) can be as high as 0.96 eV (93.6 kJ/mol) on the edge of a {111} facet, and surprisingly low, that is 0.09 eV (8.7 kJ/mol), on the edge of a {100} facet. The removal of a surface cation and the consequent creation of a surface vacancy obviously require less energy than the formation of a Frenkel defect. It is well-known, indeed, that adatoms, corner atoms, and atoms at terrace steps of a crystal surface possess high energy since they are less tightly bound. The presence of ligands, as Cl– species in the model of Zherebetskyy et al.,39 can further reduce the energy required to extract such surface atoms. Motivated by similar considerations, Casavola et al. studied the chemical transformation of PbSe to CdSe and proposed a surface-vacancy assisted mechanism.41,140,141 The PbSe-CdSe system is different from the ZnSe-CdSe one since PbSe and CdSe are not miscible, meaning that Cd2+ and Pb2+ cannot coexist in the same crystal domain. After the initial surface exchange and the formation of a first CdSe layer, a surface vacancy is allegedly formed and then it diffuses through the CdSe shell to the PbSe/CdSe interface. This happens via multiple moves of Cd2+ ions to the closest vacant sites (see Figure 3a,b). Next, a Pb atom can jump into the vacancy, thereby leaving the PbSe core. Then, through multiple vacancy-assisted hops the Pb atom can reach the surface of the CdSe shell, after which it is exchanged for a Cd atom (see Figure 3c–f).
Figure 3.

Schematic of a plausible mechanism of the CE reaction between PbX NCs and Cd2+ ions. The sketch represents a two-dimensional projection of the PbSe-CdSe interface with Pb (orange) and Cd (yellow) atom positions. Light green dots represent surface Cd atoms; Se anions are not shown. (a) The formation of a cation vacancy at the surface is followed by (b) diffusion of the vacancy to the PbSe-CdSe interface. (c–e) Next, the Pb atom can diffuse to the interface by means of vacancy-assisted migration, whereby many (Cd-associated) vacancy jumps are required to enable (f) diffusion of Pb to the surface, where it undergoes a ligand-enabled Cd-for-Pb exchange. (b–g) This process continues until (h) the Pb layer is completely replaced by Cd atoms, after which (i) the next Pb layer is replaced. (b, d, e, g) only one of many possible vacancy diffusion paths is shown. Reproduced from [Casavola, M.; van Huis, M. A.; Bals, S.; Lambert, K.; Hens, Z.; Vanmaekelbergh, D. Anisotropic Cation Exchange in PbSe/CdSe Core/Shell Nanocrystals of Different Geometry Chem. Mater.2012, 24, 294–302]. Copyright 2012 American Chemical Society.
Differently from the Zn-to-Cd exchange, here the “immiscibility rule” forces the vacancy to travel from the NC surface to the PbSe-CdSe interface and then back, in order to enable CE. HAADF-STEM tomography on these systems confirmed that Pb/Cd intermixing occurs only at the PbSe/CdSe interfacial layer. Interestingly, Lechner et al. could experimentally detect, through SAXS analysis, the presence of a submonolayer of Pb atoms bound on top of the Cd based shell.38 This finding directly supports the proposed exchange mechanism: the Pb2+ ions have to diffuse toward the particle surface, where they are subsequently exchanged by the Cd2+ ions within the solution. The process, in this specific system, proceeds in a layer-by-layer fashion (see Figure 3g–i). The interfacial “corner” Pb atoms in the incomplete Pb/Cd atomic plane are in an energetically unfavorable position: consequently, the removal of such Pb atoms requires much less energy than the removal of a Pb atom from a continuous atomic plane. This is believed to occur because the rock-salt PbSe and the zinc-blend CdSe structures have different cation coordinations (octahedral and tetrahedral, respectively). The same explanation might apply to many CE reactions in which the intermediate structures consist of heterostructures having two domains of different materials sharing a flat interface.
The surface vacancy-assisted mechanism has been also directly demonstrated by Justo et al. in studying the PbS-CdS system.40 They exposed PbS NCs with either a full Pb coverage or surface Pb vacancies (i.e., having a partial Pb-surface coverage) to Cd2+ ions. While no exchange was observed at room temperature for PbS NCs with full excess Pb coverage, the NCs with partial surface coverage exhibited a self-limiting adsorption of Cd, forming PbS|CdS core|shell structures with restricted shell thickness. At higher temperatures, they observed a similar limiting shell thickness for both NCs when using an excess of cadmium. Moreover, in the case of PbS NCs with partial surface coverage, a complete exchange occurred only when working with reduced amount of Cd. This suggests that the CE reaction can run to completion only at low Cd loading, that is, when the surface vacant sites on PbS NCs are not “poisoned” by an excess of Cd2+ ions adsorbed on the NCs surface. These findings support the hypothesis that surface vacancies not only facilitate the adsorption of guest cations from the solution phase, but they also enable the exchange of ions between the inner lattice sites and the surface. This, in turn, demonstrates that surface reactions can indeed limit the rate of a CE reaction and they can play a fundamental role in the kinetics of the overall transformation.
It must be emphasized, however, that thermally activated CE processes are hardly driven by surface vacancies or by Frenkel defects alone but most likely by both types of defects at the same time. In these reactions, the temperature can be used as a sensitive parameter to tune the CE kinetics and consequently to tailor both the composition and the elemental distribution profile of the product.42,100 On the other hand, elevated temperatures may alter the materials stability or trigger secondary processes, such as red-ox reactions, etching, sintering, etc. Equally importantly, at high temperature, the exchange might proceed close to thermodynamic equilibrium, preventing kinetically controlled access to metastable or nonequilibrium structures, which instead represent an interesting feature of CE as a synthetic tool (see section 4).
3.1.1. CE in Systems with High Density of Vacancies
Vacancies are often present in compound nanostructures that are synthesized under wet chemical conditions, as their composition is never perfectly stoichiometric. Aside from this, there are classes of materials which, already in the bulk, can exist in a wide range of stoichiometries of the component ions. This is the case of the entire family of copper chalcogenides, namely, copper sulfides (Cu2-xS), selenides (Cu2-xSe), and tellurides (Cu2-xTe). Colloidal NCs of these materials can be easily synthesized with control over the shape and size with the additional possibility of manipulating the density of copper vacancies,97,142,143 and have been tested in many CE reactions. Our group recently studied these NCs, with particular focus on Cu2-xSe and Cu2-xS ones, to explore the role of vacancies in CE reactions. In a first work, we investigated CE in copper selenide NCs with Zn2+ and Cd2+ ions by comparing the reactivity of close-to-stoichiometric (that is, Cu2Se) NCs with that of substoichiometric (Cu2–xSe) ones.137 Under identical reaction conditions, CE in Cu2Se NCs was found to proceed much slower than in the corresponding Cu2-xSe NCs (see Figure 4a,b). Moreover a small excess of guest cations was found to be sufficient to achieve full exchange in Cu2-xSe NCs, while under the same CE conditions, the Cu2Se NCs still contained a considerable fraction of Cu ions. The evident conclusion of the work was that Cu vacancies in copper chalcogenide NCs accelerate the exchange process, most likely by offering to the exchanging ions several alternative pathways for diffusion, even at low temperature.
Figure 4.
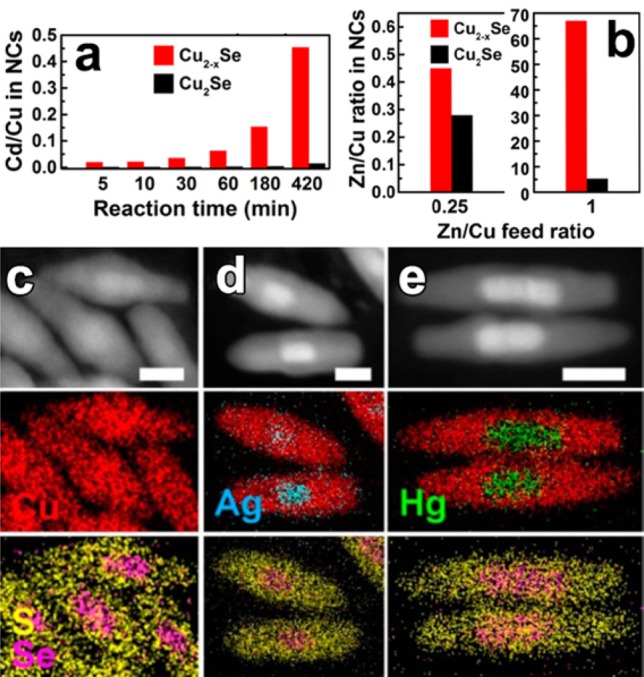
(a) Diagrams displaying the evolution of the Cd/Cu ratio in the heavily substoichiometric (Cu2–xSe, red) and close to stoichiometric (Cu2Se, black) NCs over the time of the Cu+ → Cd2+ CE reaction at RT using a Cd/Cu feed ratio of 0.5. (b) Diagrams displaying the dependence of the Zn/Cu ratio in the vacant (red) and stoichiometric (black) NCs on the Zn/Cu feed ratio in overnight CE reactions at RT. Reproduced from [Lesnyak, V.; Brescia, R.; Messina, G. C.; Manna, L. Cu Vacancies Boost Cation Exchange Reactions in Copper Selenide Nanocrystals J. Am. Chem. Soc.2015, 137, 9315–9323]. Copyright 2015 American Chemical Society. (c–e) CE in Cu2–xSe/Cu2–xS NRs with Ag+ and Hg2+ ions. HAADF-STEM images of groups of (c) pristine, (d) Ag+, and (e) Hg2+ partially exchanged Cu2–xSe/Cu2–xS NRs with the corresponding STEM-EDS elemental maps. Scale bars are 20 nm. Reproduced from [Miszta, K.; Gariano, G.; Brescia, R.; Marras, S.; De Donato, F.; Ghosh, S.; De Trizio, L.; Manna, L. Selective Cation Exchange in the Core Region of Cu2-xSe/Cu2-xS Core/Shell Nanocrystals J. Am. Chem. Soc. 2015, 137, 12195–12198]. Copyright 2015 American Chemical Society.
In another work, we studied CE reactions between Cu3-xP NCs and In3+ ions in order to synthesize InP NCs.104 Copper phosphide NCs are naturally substoichiometric in copper, and therefore, in analogy with copper chalcogenides, they are characterized by a high density of Cu vacancies. Our findings were that Cu3-xP NCs can undergo total CE, to form InP NCs, at mild conditions and by employing only a small excess of In3+ ions. On the other hand, CE reactions to prepare III–V semiconductor NCs have been shown to require much higher temperatures and a high excess of guest cations (for the Cd3P2 → InP CE reaction, for example, a In/Cd feed ratio of 100 was required to get full exchange) since the diffusivity of ions in covalent semiconductors can be orders of magnitude lower than that in ionic systems.144 This brought us to conclude that CE reactions in Cu3-xP NCs are strongly favored and assisted by Cu vacancies.
In a recent work, we demonstrated that that Cu vacancies in copper chalcogenide NCs not only boost CE reactions, but also can provide sufficient mobility for the guest cations to initiate the exchange in the preferred zone of the host crystal.65 To prove this point, we used core|shell Cu2–xSe|Cu2–xS nanorods as starting seeds, Ag+ or Hg2+ ions as guest cations and methanol as polar solvent. At the early stages of the CE transformation, the guest cations diffused through the Cu2-xS shell and selectively replaced Cu+ ions in the Cu2-xSe core region (see Figure 4c–e). The preferential exchange in the core is driven by thermodynamic factors, which favor the formation of metal selenides over the corresponding sulfides (see section 2.2.2). Nearly identical CE transformations were performed on CdSe|CdS core|shell NRs with Cu+ or Ag+ ions. In such reactions, the thermodynamic driving force should likewise promote the initial selective replacement of the selenide core region. In this case, however, CE starts with a partial replacement of cadmium ions from the sulfide shell, while the core region remains unaltered. These control experiments suggests that, most likely, it is the high ions diffusivity in copper chalcogenide NCs, promoted by Cu vacancies, that enables guest cations to probe the whole host structure, such that they initiate the exchange where it is mostly thermodynamically favored. On the contrary, in cadmium chalcogenide NHCs this is not possible. This work suggests that in CE reactions involving nanoscale systems with high ionic diffusivity, the reaction zone that is formed at the beginning of the transformation might not be necessarily limited to the surface of a NC, but it could involve any region of the host material.
3.2. Solid-State Diffusion via Interstitials
Bothe et al., in a recent study, proposed a model in which the ion diffusion and exchange processes are exclusively mediated by interstitial lattice sites.145 This conclusion was reached by studying two model CE reactions that involve the exposure of PbSe or ZnSe NCs to Cd2+ ions. Both CE processes, as seen in the previous section, had been previously explained as mediated by vacancies.40−42 In this model, instead, the relatively small Cd2+ ions (ionic radius 78 pm) are assumed to be diffusing in both ZnSe and PbSe structures by occupying tetrahedral interstitial sites. Moreover, in the PbSe → CdSe CE transformation the interstitial sites of the rock-salt PbSe lattice represent the lattice sites of the developing CdSe crystal, while in the ZnSe → CdSe case the exchange has to take place on the existing tetrahedral sites of the zb-ZnSe lattice. By monitoring the kinetics and the temperature-dependent behavior of these CE reactions, Bothe et al. extrapolated activation energy values in the range of 30–40 kJ/mol, which are in good agreement with an ion exchange transformation occurring without the creation of vacancies. A vacancy-mediated CE in bulk materials would require higher energies, of the order of 102 kJ/mol. Motivated also by the fact that these ions have short diffusion pathways and that the concentration of vacancies in these NCs is rather low at RT, the authors proposed an interstitial-mediated mechanism to rationalize such CE reactions (see Figure 5). In the PbSe → CdSe reaction, Cd2+ ions start occupying tetrahedral interstitial sites in the PbSe lattice, while at the same time the Pb2+ ions out-diffuse by hopping through the octahedral interstitial sites. Clearly, this process is completely controlled by diffusion and the activation energies involved are, thus, the ones for the diffusion through the interstitial sites. In the ZnSe → CdSe transformation, Cd2+ ions can diffuse through both octahedral and unoccupied tetrahedral sites by replacing Zn2+ ions in their respective lattice positions by a kick-out mechanism. Zinc ions, now occupying interstitials, will then leave the crystal by diffusion through interstitials. For these reactions, it was proposed that the rate-determining step is not the diffusion but the ion exchange. In this case, in fact, the exchange has small activation energy as it is believed to proceed through a kick-out mechanism, that is, without any vacancy creation.
Figure 5.

Exchange mechanism for the CE with cadmium in (a–c) PbSe and (d–f) ZnSe NCs.(g) The lattice structures involved in these CE experiments. Adapted with permission from ref (145). Copyright 2015 John Wiley and Sons.
3.3. Cooperativity in CE Transformations in NCs
CE in many cases was assumed to be a diffusion-limited transformation. In some systems, however, as pointed out by White et al., sharp and instantaneous CE transformations, evidencing some sort of cooperativity, are observed.43,146,147 A typical feature of a cooperative system is the “all-or-nothing” behavior: rather than progressively transforming through intermediate steps, the system makes a sudden sharp transition from the initial to the final state in response to a specific input. The CE transformation of CdSe to Ag2Se (or to Cu2Se) NCs seems to follow such behavior. At the early stages of the process, the incorporation of guest cations (Ag+ or Cu+) into a NC lattice has a relatively low probability of occurrence. However, once this initial step does take place, it activates the NC for further doping. Each doping event increases the affinity of the same NC for the subsequent guest ions incorporation, until a critical doping concentration is reached. Then, the NC is ready to transform by taking up every available Cu+ or Ag+ ion, which results in a cascade of guest ion incorporation events and in an abrupt phase transition from a doped CdSe NC to Cu2Se or Ag2Se. Thus, at the intermediate steps of the CE reaction, each NC has either a CdSe composition (with some Ag+ or Cu+ dopants) or it has transitioned to a fully exchanged Ag2Se or Cu2Se NC, thus exhibiting the typical “all-or-nothing” behavior of a cooperative system. Optical monitoring such reaction with single NC resolution revealed that indeed each CdSe NC waits a specific amount of time, of the order of few hundred milliseconds, before making a sudden transition to the Ag2Se phase.43,147
Ott et al. recently proposed a detailed microscopic model (based on density-functional theory calculations and dynamical simulations of CE) that is in line with the co-operative behavior observed by Jain and co-workers.115,148 The model, specifically built for the CdSe → Ag2Se transformation, considers as first step the entrance of Ag+ ions in the CdSe lattice as interstitials (Ag+int), followed by their rapid diffusion, still through interstitial sites, toward the central region of the lattice (see Figure 6a). As the concentration of Ag+int ions increases, their mutual repulsion becomes dominant and drives the ions toward the surface. At this stage, to compensate electrically for the positive charge accumulating near the surface, a “kick-out reaction” occurs, in which an Ag+int ion moves onto a Cd site forming a Ag–Cd. The Cd2+ ion can immediately leave the NC by out-diffusing from the NC to the surrounding solution. At the same time, the Ag–Cd attracts a mobile Ag+int, generating a Ag2 pair around a Se anion, that is a Ag2Se nucleus (see Figure 6b). The formation of such Ag2 pair triggers a cascade of CE steps through Coulomb interactions, as the formation of contiguous regions of electrostatically bound Ag2 pairs is strongly favored. The model explains why initial Ag+int doping of a NC strongly enhances further silver doping within the same NC, causing eventually a sharp transition from a lightly doped CdSe NC to the Ag2Se phase (a typical example of co-operative behavior).
Figure 6.
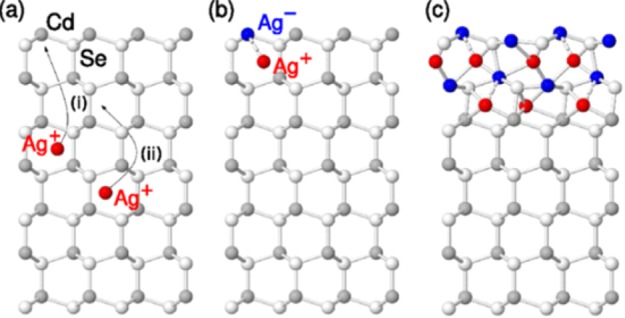
Microscopic model of Ag doping and CE of CdSe NCs. (a) At low concentration, the Ag+ ions diffuse through interstitial sites (Ag+int). As their concentration in the lattice increases, their mutual repulsion pushes them toward the NC surface, where a Cd kick-out reaction can occur [step (i)]. The substitutional Ag–Cd created by the kick-out reaction attracts the remaining Ag+int [step (ii)]. (b) An electrostatically bound complex is then formed by two Ag at the surface, generating a nucleus of Ag2Se. (c) At even higher Ag concentrations, all interstitial and Cd sites at the surface become occupied with Ag. This fully substituted domain closely resembles the naumannite crystal structure of Ag2Se. Reproduced with permission from ref (148). Copyright 2014 American Physical Society.
A common feature of the transformations from CdSe to Ag2Se or to Cu2Se and those involving CE of Cu2-xSe/Cu2-xS NRs with Ag+ or Hg2+ ions (discussed in section 3.1.1) is that the guest ions appear to be able to probe the whole host NC and to start the CE in a “preferred” region. While in CdSe this is possible through the high diffusivity of Ag+ and Cu+ ions via interstitials, in copper chalcogenides the fast ion diffusion is favored by the high density of vacancies. These examples support the idea that the reaction zone, which is established at the beginning of a CE transformation, might not be necessarily limited to the surface of a NC, as commonly believed.
3.4. Influence of Crystal Structure on CE Reactions
In the simplified discussion of section 2, the overall CE reaction was reduced to a model that consists of the dissolution of a starting AX crystal to form A+ cations and X– anions which, upon reaction with B+ cations, generate a BX crystal. In an actual CE reaction, the host AX crystal is not dissolved and the X– anion framework can be thought as a rigid skeleton with a limited flexibility that sustains the whole reaction, while the cations diffuse in and out. The activation barrier for the diffusion and the exchange of cations depends on many factors, including structural differences between the reactant and the product phases. It is therefore important, before performing a CE reaction, to compare the starting AX and the final BX crystal structures. If the anion framework of the expected BX crystal structure can be accessed only by a severe deformation of the starting AX lattice, the AX → BX conversion might not take place or it might result in deformed BX NCs.66 For example, the CE involving Cu2-xSe NCs and Sn4+ ions cannot proceed to the expected SnSe2 phase, but it reaches a composition limit of Cu2SnSe3.149 This can be rationalized by considering that the initial Cu2-XSe NCs have a fcc anion sublattice, while SnSe2 has a trigonal crystal structure characterized by a stack of SnSe2 layers bound together by weak Se–Se van der Waals bonds. Therefore, even if the exchange between Cu+ and Sn4+ cations is promoted by a proper choice of ligands, complete CE in this case would entail a pervasive reorganization of the anion framework, which is not possible under the mild reaction condition at which the exchange is carried out. Harsher conditions on the other hand, were found to trigger redox reactions.149
Similarly, van der Stam et al. demonstrated that Cu2-xS NCs with hexagonal crystal phase (low-chalcocite) can undergo CE with In3+ ions up to a CuInS2 stoichiometry.103 The resulting NCs exhibit a wurtzite crystal structure whose anion sublattice, in line with that of the starting low-chalcocite Cu2-xS NCs, has a hexagonal close-packed arrangement. On the other hand, In2S3 can exist in three different crystal structures, denoted as α, β, and γ, all of them characterized by an fcc stacking of S2– anions and differing from each other only in the ordering of the In3+ cations. Hence, the anion sublattice is intrinsically different from that of hexagonal Cu2–xS and of wurtzite CuInS2, preventing the full conversion from Cu2–xS to In2S3 (see Figure 7A,B).
Figure 7.

(A and B) Comparison of the anionic sublattice of low-chalcocite Cu2–xS with wurtzite CuInS2, evidencing that the anionic sublattice of low chalcocite Cu2–xS (blue spheres) is compatible with that of wurtzite CuInS2 (red spheres) since both sublattices have an hcp arrangement (A). Comparison of low-chalcocite unit cell (blue spheres) with In2S3 (green spheres). The spinel In2S3 structure has a fcc anionic sublattice, where the C layer has to dislocate by 58% of a S–S distance in order to fit in the low-chalcocite lattice (B). Reproduced from [van der Stam, W.; Berends, A. C.; Rabouw, F. T.; Willhammar, T.; Ke, X.; Meeldijk, J. D.; Bals, S.; de Mello Donega, C. Luminescent CuInS2 Quantum Dots by Partial Cation Exchange in Cu2–xS Nanocrystals Chem. Mater. 2015, 27, 621–628]. Copyright 2015 American Chemical Society. (C) Description of the In–Se layer shown in [0–1–10] projection of the In2Se3 structure (bigger dark circle, Se; smaller light circle, In). (D) [110] projection of CuInSe2 structure (smaller dark circle: Cu). Adapted with permission from ref (91). Copyright 2011 IOP Publishing. All rights reserved.
Gupta et al. demonstrated that the exchange between wurtzite CdSe nanorods and Hg2+ ions in water, even if thermodynamically favored (see Table 3 and section 2.2.2) on the basis of the much lower solubility product of HgSe relative to that of CdSe, was strongly limited.150 This was attributed to the differences between the two end point structures of the transformation: wurtzite CdSe on the starting side and cubic HgSe on the end side. In order for the full exchange to take place, the Se2– framework would have to be deformed, creating a high-strain field in the NCs. Eventually the entering Hg2+ ions would experience repulsive elastic forces from the host CdSe lattice, and this explains why their inclusion was limited up to a composition corresponding to Cd0.9Hg0.1Se. There are nonetheless cases of CE transformations in which a significant reorganization of the crystal structures takes place. Yuho et al. reported, for example, a CE transformation from trigonal In2Se3 NCs to tetragonal CuInSe2 NCs at room temperature in water.91 Similarly to SnSe2, In2Se3 has a layered structure consisting of alternating sheets (Se–In–Se–In–Se) held together by van der Waals bonds (see Figure 7C,D). During the CE process, Cu+ ions both substitute In3+ cations and occupy “vacant sites” within the In2Se3 layers (see the dashed line in Figure 7C). The reorganization of the lattice upon inclusion of Cu+ ions produces a small volume expansion of the unit cell (∼5%) and the cleavage of weak Se–Se van der Waals bonds. It is remarkable that, in this specific system, these requirements are fulfilled even at RT.
Along the same lines, in a recent work of ours, we observed that the CE transformation from covellite CuS to cinnabar HgS, which requires a marked reorganization of the crystal lattice of the starting NCs, can take place already at room temperature.151 The covellite (CuS) structure is formed by Cu–S layers stitched together by S–S covalent bonds. Every Cu–S layer, in turn, is composed by a trilayer motif made of a layer of triangular CuS3 units between two layers of CuS4 tetrahedra. Even if the transformation from CuS to HgS should be promoted by a polar solvent (see section 2.2.2), no exchange was observed by exposing the CuS NCs to Hg2+ ions in methanol. The formation of a HgS phase requires, in fact, not only a simple replacement of Cu+ with Hg2+ ions, but also a chemical reduction of a fraction of the sulfur anions (2/3) in order to break the covalent S–S bonds. The transformation from covellite to hexagonal cinnabar HgS phase (although with a low yield) was made possible at RT in the presence of a mild reducing agent (i.e., ascorbic acid) that provided the required electrons to reduce the disulfide bonds. In another work, we observed that hexagonal Cu2Te disk-shaped NCs could be converted into wurtzite CdTe ones.109 This CE reaction, interestingly, preserved the NCs morphology, even if a substantial reconstruction of the anion sublattice took place. As it possible to notice in the sketches of Figure 8A–C, a reorientation of the c-axis must take place when going from Cu2Te to CdTe nanodisks. This is accomplished by a displacements of Te anions, mainly along the z direction, resulting in a splitting of a pristine (001) Te-plane of Cu2Te into the (110) Te “bilayer” of CdTe. The temperature, in this particular CE reactions, is believed to play a fundamental role in supplying the system with enough energy for the reorganization of the lattice and, consequently, for the cation replacement. Indeed, while above 150 °C the reactions required only 5 min to reach completion, almost no exchange was observed when working at 50 °C, even after 24h.
Figure 8.

(A) Sketches of the Cu2Te and CdTe disks, with arrows indicating the respective crystal orientations. (B and C) Schematic views of slabs of the Cu2Te and CdTe structures, evidencing a few atomic layers of the nanodisks, according to the respective phases and orientations determined by HRTEM. Each Te anion (larger yellow spheres) in the (001) plane of Cu2Te forms an hexagonal lattice (cell indicated by blue dashed lines) with the three neighboring anions (magenta) on the same plane. The (110) anionic sublattice in wurtzite CdTe still resembles a distorted hexagon in projection (red dashed lines). However, the Te atoms of the original Cu2Te structure have to move considerably in the z-direction to accommodate for the Cd atoms and form the two (110) layers in wurtzite CdTe. The volume occupied by the same number of Te atoms is included within the blue and red frames in Cu2Te and CdTe, respectively. Reproduced from [Li, H.; Brescia, R.; Povia, M.; Prato, M.; Bertoni, G.; Manna, L.; Moreels, I. Synthesis of Uniform Disk-Shaped Copper Telluride Nanocrystals and Cation Exchange to Cadmium Telluride Quantum Disks with Stable Red Emission J. Am. Chem. Soc. 2013, 135, 12270–12278]. Copyright 2013 American Chemical Society. (D) Schematic of the CE transformation from Metallic|Ag2X core|amorphous-shell nanoparticles to Au|MX core|monocrystalline-shell HNCs promoted by TBP molecules. (Bottom) Series of high-resolution TEM images highlighting different synthetic stages (indicated with (i), (ii), and (iii)) in the CE reaction from Au|Ag2S to Au|CdS NCs. The scale bar is 5 nm in all the images. Red and yellow dashed lines are guides for the eye, distinguishing the core and shell boundaries, respectively. Adapted with permission from ref (106). Copyright 2010 The American Association for the Advancement of Science.
As shown in the previous examples, the crystal phase resulting from a CE reaction can be sometimes hard to predict. This is especially true in some systems in which during CE there is a transition from an amorphous to a monocrystalline structure: Zhang et al. studied CE reactions taking place on an amorphous Ag2X (X=S, Se, Te) shell grown onto preformed metallic NC. By exchanging the Ag+ ions of the shell with M2+ ions (M= Cd, Zn, Pb), the resulting MX shell was monocrystalline (see Figure 8D, top panel).106 The alkyl phosphine used in the synthesis, TBP, was believed to make the CE reaction possible (by selectively solvating the Ag+ ions) and to promote the reorganization of the MX crystalline lattice to form a monocrystalline domain, even at low temperature (i.e., 50 °C). As it is possible to appreciate from HRTEM micrographs, reported in Figure 8D, the CE takes place from a certain site on the Ag2X surface and then gradually transforms the amorphous domain into a monocrystalline MX one.
3.5. Effects of Volume Change
In the previous section we have shown various examples of CE reactions in which the morphology of the host NCs is retained, even if a strong deformation of their anion framework occurs. There are also reported examples of CE transformations in which the crystal structure is retained, albeit with a large change in the volume of the structure. The morphology retention of NCs, in these cases, may not be assured anymore as the volume change (ΔV/V) can have various deleterious effects on the product NCs. Depending on the degree of lattice stress developed in the exchanged NCs, the formation of voids or a fragmentation can be observed. This was seen, for example, by Wark et al. when studying CE reactions between CdE (E=S, Se and Te) NCs and Pd2+ or Pt2+ ions.66 The volume change in almost all these transformations is approximately −30%. As shown in Figure 9, the CdSe → PdSe (Figure 9 c → d) and CdTe → PtTe (Figure 9 a → b) reactions yielded NCs characterized by voids, especially when starting from larger NCs. This was attributed to the release of stress accumulated in the NCs lattice during the CE reaction, as a consequence of the large volume change. In the CdS → PtS (Figure 9 e → f) and CdSe → PtSe (Figure 9 g → h) cases, fragmentation of the NCs occurred. The fragmentation was tentatively explained by considering a continuous pealing-off of the ion-exchanged parts from the main body of the reacting NC, due to the excessive lattice mismatch at the heterointerface. Similarly, Moon et al. showed that the exposure of CdTe nanowires to Pt4+ ions leads to the formation of PtTe2 nanotubes (see Figure 9 i → l).31 Also in this case, the void formation was not attributed to the Kirkendall effect (see section 4.1) but to the release of the mechanical stress accumulated during the reaction.
Figure 9.

TEM images of the reactant CdE (E = S, Se, and Te) and fully exchanged product NCs. In the Figure, each reactant-product pair is indicated by an arrow connecting the two panels: (a → b) CdTe-PtTe, (c → d) CdSe-PdSe, (e → f) CdS-PtS, (g → h) CdSe-PtSe, (i → l) CdTe-PtTe2. Scale bars in a–h are 10 nm. (a–h) Reproduced from [Wark, S. E.; Hsia, C.-H.; Son, D. H. Effects of Ion Solvation and Volume Change of Reaction on the Equilibrium and Morphology in Cation-Exchange Reaction of Nanocrystals J. Am. Chem. Soc. 2008, 130, 9550–9555]. Copyright 2008 American Chemical Society. (i–l) Reproduced from [Moon, G. D.; Ko, S.; Xia, Y.; Jeong, U. Chemical Transformations in Ultrathin Chalcogenide Nanowires ACS Nano2010, 4, 2307–2319]. Copyright 2010 American Chemical Society.
4. Materials
As it is possible to see from Table 4, many materials are now accessible via CE reactions on NCs, emphasizing how such a synthetic tool has been refined over the last years. As regarding binary compounds, most of the work has been done on metal chalcogenide NCs, with most efforts on metal sulfides and selenides. Few CE reactions, on the other hand, have been reported on metal telluride NCs. This might be due to the weak M-Te bonds (i.e., low lattice energy, see section 2) that make the whole anion lattice less rigid and thus more prone to deformation or dissolution during a CE reaction. Also, much less has been reported on metal oxides. This can be tentatively explained considering that, in general, the M-O bonds are much shorter than the M-X (X = S, Se, and Te) bonds, hindering, in many cases, the diffusion of cations in oxides NCs.152 CE reactions are even more challenging in pnictides: in these systems, the bonds are much more covalent than those in metal chalcogenides. In order to promote a CE reaction in such materials high temperatures are required, but such harsh conditions may lead to the dissolution of the starting NCs.
Table 4. List of Reactant and Product Materials of Known CE Reactions in NCs.
| class | reactant | shape | product |
|---|---|---|---|
| oxides | CuO | sphere | NiO, CoO, Fe2O3, Mn3O4153 |
| selenides | CdSe | rod, sphere, wire, sheet | Cu2Se,75,92,154 Ag2Se28,76,78,155 PdSe, PtSe,66 PbSe118,121,156 |
| PbSe | rods, cubes | CdSe41,145 | |
| Cu2-xSe | rod, sphere, cube | ZnSe,92 Ag2Se,157 SnSe149 | |
| SnSe | sphere | PbSe158 | |
| ZnSe | sphere | Ag2Se,159 PbSe,89,130 CdSe145 | |
| Ag2Se | wire, sphere, rod | CdSe,28,29,99 Cd1-xZnxSe111 | |
| sulfides | CdS | sphere, rod, wire, platelet | CuS,77 Cu2S,88,117,160−163 PdS, PtS,66 PbS118,164 |
| CuS | sphere | Ag2S87 | |
| ZnS | rod, sphere | Cu2S,49 PbS118 | |
| Ag2S | sphere | CdS114 | |
| Cu2S | Sphere, rod, platelet, wire | ZnS107,160 CdS, PbS,88,117,160 Au2S,165 Ag2S162 | |
| PbS | sphere | CdS40 | |
| selenide-sulfide | Cu2-x(SySe1-y) | sphere, platelet | Cd(SySe1-y)105,166 Zn(SySe1-y)166 |
| CdSe|CdS | core|shell | Cu2Se|Cu2S73,79,80,93,160,167,168 | |
| Cu2Se|Cu2S | core|shell | PbSe|PbS,79,160 ZnSe|ZnS,93,160 CdSe|CdS,80 | |
| tellurides | CdTe | wire, sphere, tetrapod | PbTe,118 Ag2Te,28 PtTe2,31 PdTe, PtTe,66 Cu2Te97 |
| Cu2Te | platelet | CdTe109 | |
| pnictides | Cd3As2 | sphere | InAs, GaAs144 |
| Cd3P2 | sphere | InP, GaP144 | |
| Cu3P | platelet | InP104 | |
| lantanide fluorides | GdF3, EuF3 | sphere | LaF3169 |
| NaYF4 | sphere | LaF3169 | |
| Ln:CaF2 (Ln = Yb, Ho, Er, Tm) | cube, sphere | Ln:NaGdF4170 | |
| hybrid metal-chalchogenide | Se|Ag2Se | core|shell sphere | Se|ZnSe, Se|CdSe, Se|PbSe30,98 |
| Au|Ag2S | core|shell rod, sphere | Au|CdS112,171 | |
| Au|Ag2X (X = S, Se, Te) | core|shell sphere | Au|CdSe, Au|CdTe, Au|PbS, Au|ZnS, Au|Cd(SaSe1-a)106 |
CE reactions have been exploited even in complex structures, such as the so-called nano heterostructures (NHCs) (see Table 4). In these cases, the anion frameworks of the different sections of the NHCs remain unaltered while all the original cations are replaced with new desired ones. As a few examples, spherical, dot-in-rod and platelet-like CdSe|CdS core|shell NHCs have been transformed into the corresponding Cu2Se|Cu2S NHCs and subsequently to PbSe|PbS, ZnSe|ZnS, or back to CdSe|CdS NHCs, with complete morphology preservation.79,80,93,160,167,168 This is assured by the topotactic nature of CE transformations that, in some cases, also leads to unexpected results, such as metastable crystal structures. For example, the CE reaction of wurtzite (wz) CdSe NCs with Cu+ ions produces an unknown hexagonal (hex) Cu2Se phase.92 Similarly, metastable hex-CdTe phase formed when starting from hex-Cu2Te NCs, and wz-InP NCs resulted from the CE transformation of hex-Cu3P NCs by exchanging Cu+ with In3+ ions.104,109 As highlighted in the introduction, important recent reviews on CE reactions have already discussed most of the above-mentioned cases and have given a comprehensive overview on this topic.28,32−34 This second part of the review, with the aim of being complementary to those works, will focus only on those complex structures that were made accessible by CE transformations. More specifically, we will provide an overview of the nanostructures that can be prepared with partial replacement of the original cations, with a special attention on the morphology of the resulting products (see section 5). Also, we decided to include those CE transformations that do not proceed topotactically, resulting, consequently, in NCs with complex morphologies, such as hollow nanostructures (see next Section).
4.1. Kirkendall Effect in CE Reactions: Hollow NCs
CE reactions naturally involve the diffusive motion of two different cation species, which, in general, have different intrinsic diffusion coefficients. Two ions fluxes take place during such transformations: one flux corresponds to that of the outgoing host cations, while the other refers to ingoing guest ions. If one cation species diffuses much faster than the other, a net mass flow accompanies the interdiffusion process, which is balanced by an opposite flow of vacancies. In bulk materials, these vacancies can condense forming voids, or they can annihilate at dislocations. This phenomenon, discovered by Kirkendall and co-workers in the 1940s studying a copper-brass diffusion couple, is indeed known as the Kirkendall effect.139 If the outgoing cations of a NC diffuse much faster than the ingoing cations, a significant flow of vacancies can be established, favored by the high surface-to-volume ratio of the NC.172 Within the small volume of a transforming NC, the supersaturated vacancy cloud is likely to coalesce into a single void, with the consequent formation of a hollow structure. The Kirkendall effect has been demonstrated in some CE reactions and has led to exotic nanostructures such as toroids, hollow tubes and ring-like morphologies.
4.1.1. TiS2 → Cu2S, CdS
Jeong et al. studied the CE reactions between 2D layered TiS2 nanodiscs (NDs) and copper(I) ions.131 TiS2 consists of S–Ti–S layers stacked in the (001) direction and offering preferential nanometer sized channels for the ion diffusion. Upon exposure to excess of copper ions at relatively low temperature (100 °C), TiS2 NDs transform into Cu2S toroidal nanostructures. The transformation starts at the edges of the 2D NDs with the nucleation of the Cu2S phase. While the CE process proceeds inward, the sulfur ions most likely migrate outward, through nanometer-sized channels of the TiS2 structure, eventually producing a hole in the middle of the original NDs and forming a toroid (see Figure 10B,C). The same process was observed to take place using Cd2+ ions as guest cations, with the consequent formation of CdS toroidal NCs. This final morphology is unconventional, since usually the hollow nanostructures obtained with shape-transformative reactions, for example of spherical NCs, result in concavo-convex curvature (see Figure 10A). This unique shape obtained through CE reaction is made possible as a consequence of the different diffusivities of the in-going and out-going cations coupled with anion migration through the layered structure of TiS2. It is noteworthy that the whole ion diffusion produces, upon CE, a monocrystalline material (hexagonal Cu2S) whose crystal structure is templated by the starting TiS2.
Figure 10.

(A) Concavo-convex curvature formed as a consequence of a transformation from a sphere to a hollow sphere. (B) Double-convex curvature from a layer-structured disc to a toroid. (C) Schematic illustration of 2D layered nanocrystals, regioselectively reacting with incoming ions, to form a toroid as the final product. Reproduced from [Jeong, S.; Han, J. H.; Jang, J.-t.; Seo, J.-w.; Kim, J.-G.; Cheon, J. Transformative Two-Dimensional Layered Nanocrystals J. Am. Chem. Soc. 2011, 133, 14500–14503]. Copyright 2011 American Chemical Society. (D) Schematic representation of the Kirkendall Effect occurring in the transformation of Cu2-xS NDs into hollow CuInS2 NDs. Schematic representation TEM images of (E) the template Cu2-xS NCs and (F) CuInS2 nanocrystals resulting from ion exchange with InI3 (150 °C), revealing large central holes and related thickness variations. Reproduced from [Mu, L.; Wang, F.; Sadtler, B.; Loomis, R. A.; Buhro, W. E. Influence of the Nanoscale Kirkendall Effect on the Morphology of Copper Indium Disulfide Nanoplatelets Synthesized by Ion Exchange ACS Nano2015, 9, 7419–7428]. Copyright 2015 American Chemical Society.
4.1.2. Cu2-xS → CuInS2
Mu et al. prepared CuInS2 hollow NDs starting from Cu2-xS NDs and In3+ ions.110 The CE in this system takes place from the edges of the NDs and proceeds toward the core. The formation of an internal void is not directly correlated to intrinsic ions diffusion rates, but it is governed by the availability of In3+ ions at the NCs surface. More specifically, a high reaction barrier for In3+ entry at the NCs edges decreases the effective rate of indium incorporation, allowing for the out-diffusion of Cu+ (and S2–) to outcompete the in-diffusion of In3+. Hollow-centered CuInS2 nanorings form under these conditions (see Figure 10D–F). In contrast, low reaction barriers for In3+ entry at the NDs edges provide higher rate of indium incorporation in the lattice, balancing the fluxes of ingoing and outgoing ions. In this case, no formation of voids is observed and the NDs preserve their morphology. Remarkably, Mu et al. observed that this reaction barrier can be modulated experimentally by varying the temperature and the nature of the In-complex used in the CE reaction. Therefore, the variation of these two synthetic parameters could either promote or suppress the nanoscale Kirkendall effect, effectively regulating the final NCs morphology.
4.1.3. Co3O4 → ZnxCo3-xO4
Tian et al. reported the formation of hollow single crystal ZnxCo1–xCo2O4 nanocubes as a result of the in situ CE reaction between Co3O4 nanocubes and Zn2+ ions (see Figure 11).173 The transformation represents actually the last step of a hydrothermal synthetic route in which, at the beginning, Co3O4 nanocubes with a cubic spinel like crystal lattice form. The Zn2+ ions in the reaction solution, then, start replacing the Co2+ ions in tetrahedral positions and progressively transform the host crystal lattice to ZnxCo1–xCo2O4. The zinc content in the NCs slowly varies with time, up to a maximum value of x = 0.75, with the corresponding formation of a void in the center of each NC (see Figure 11). The formation of hollow NCs, comes, most likely, as a consequence of the fast out-diffusion of Co2+ and a less efficient in-diffusion of Zn2+ which produce a net mass transport between the inner part and the more external part of the Zn–Co–O spinel nanocubes. The end products of this process (i.e., Kirkendal effect) are hollow nanocubes that are surprisingly monocrystalline, as shown by HRTEM images of Figure 11a-c.
Figure 11.

High-magnification (A) dark and (B) bright-field TEM images of ZnxCo1–xCo2O4 hollow nanocubes. (C) HRTEM image of a single hollow nanocube along with its FFT (inset). (D) Illustration of the nanoscale differential diffusion (CE) and the hollowing process. Reproduced from [Tian, L.; Yang, X.; Lu, P.; Williams, I. D.; Wang, C.; Ou, S.; Liang, C.; Wu, M. Hollow Single-Crystal Spinel Nanocubes: The Case of Zinc Cobalt Oxide Grown by a Unique Kirkendall Effect Inorg. Chem. 2008, 47, 5522–5524]. Copyright 2008 American Chemical Society.
5. Partial CE
The outcome of partial exchange experiments depends mostly on the miscibility of reactant and product materials. In the case of miscible materials, either alloy or doped NCs can be produced, otherwise multi domain NHCs, such as core|shell or segmented NCs, are observed. All these structures are listed in Table 5. Thus, the phase diagrams of reactant-product solid solutions have to be carefully consulted in order to predict a desired nano architecture.37
Table 5. List of Nanostructures Accessible via Partial CE Transformations.
| class | reactant | shape | product | morphology |
|---|---|---|---|---|
| oxides | Co3O4 | cube | ZnxCo1-xCo2O4173 | alloy |
| Fe3O4 | sphere | Co–Fe–O174 | ||
| FeO|CoFe2O4 | core|shell | Fe1-xCoxO4|CoFe2O4174 | ||
| selenides | CdSe | sphere, rod | CdxHg1-xSe90,96,150 | |
| Cu2-xSe | sphere | Cu2–4ySnySe149 | ||
| ZnSe | sphere | ZnxCd1-xSe,42,63,64,100,145 | ||
| In2Se3 | sphere | CuInSe291 | ||
| sulfides | In2S3 | sphere | CuxInS2175 | |
| CdS | sphere, wire | ZnxCd1-xS,176,177 HgxCd1-xS96 | ||
| Cu2S | sphere | CuInS2,102,103,110 Cu–In–Zn-S102 | ||
| AgInS2 | sphere | Ag–In–Zn-S178,179 | ||
| Cu1-xInS2 | sphere | Cu–In–Zn-S113 | ||
| Cu2SnS3 | sphere | Cu-M–Sn-S (M = Zn, Fe, Co, Ni, Cd)180 | ||
| selenide-sulfide | Cu2-x(SeyS1-y) | platelet | Cu–Zn–Se-S | |
| Cu–Sn–Se-S | ||||
| Cu–Zn–Sn–Se-S108 | ||||
| Cu2Se|Cu2S | core|shell | CuInSe2|CuInS2168 | ||
| tellurides | CdTe | sphere, rod | CdxHg1-xTe96,181,182 | |
| ZnTe | wire | CdxZn1-xTe129 | ||
| lantanide fluorides | NdF3 | sphere | LaxNd1-xF3 | |
| GdF3 | EuxGd1-xF3169 | |||
| KLnF4 (Ln=Y, Yb, Er,Gd) | rod | NaLnF4183 | ||
| lantanide fluorides | NaYF4:Yb,Tm | sphere | NaYF4:Yb,Tm-NaGdF4184 | core|shell |
| selenides | CdSe | sphere | CdSe|PdSe66 | |
| PbSe | sphere, rod, cubes, star-shaped | PbSe|CdSe41,122−124,126,145,185,186 | ||
| selenide-sulfide | CuInSexS2-x | sphere | CuInSexS2-x|CdSexS2-x187 | |
| sulfides | PbS | sphere, rod | PbS|CdS38,40,88,116,122,126,188,189 | |
| CdS | sphere, wire | CdS|PbS,37 CdS|HgS,36 CdS|Cu2-XS161,162 | ||
| CuInS2 | sphere | CuInS2|ZnS190 | ||
| Cu2S | sphere | Cu2S|Au2S165 | ||
| tellurides | PbTe | sphere | PbTe|CdTe127 | |
| selenides | CdSe | sphere, rod | CdSe/PbSe118,121 | segmented |
| Cu2-xSe | sphere | Cu2-xSe/SnSe,149 Cu2-xSe/CdSe, Cu2-xSe/ZnSe137 | ||
| sulfides | Cu2S | sphere, wire | Cu2S/ZnS, Cu2S/CdS,107 Cu2S/Ag2S162 | |
| CdS | rod, sphere, wire | CdS/PbS,72,118 CdS/Ag2S,71 CdS/Cu2S,70,162 CdS/PdS, CdS/PtS,66 CdS/Pd4S191 | ||
| TiS2 | disc | TiS2/Cu2S131 | ||
| tellurides | CdTe | rod | CdTe/Cu2-xTe192 | |
| oxides | YVO4 | sphere | Eu3+:YVO4193 | doping |
| selenides | CdSe | sphere | Ag:CdSe115 | |
| selenides | ZnSe | sphere | Ag:ZnxCd1-xSe89 | |
| Cu:ZnxCd1-xSe89,100 | ||||
| selenides | ZnSe | sphere | Cu:ZnSe159 | |
| selenide-sulfide | CdSe|ZnS | sphere | 64Cu:CdSe|ZnS194 | |
| tellurides | ZnTe | cluster | Mn:ZnTe128 |
5.1. Partial CE with Miscible Phases
The direct synthesis of alloy NCs requires a deep understanding of the specific reactivities of the precursors employed, together with the correct choice of ligands for the size and shape control of the resulting NCs. It is easy to understand that, in many cases, the simultaneous control over the composition, size distribution and morphology of alloy NCs, especially when quaternary or even quinary systems need to be prepared, can be extremely challenging. A useful method to sidestep these problems has recently emerged; it consists of first growing binary/ternary compound NCs using well-established synthesis procedures, and then using such NCs as templates for CE reactions in order to include other cations species. A wide range of alloyed metal chalcogenide NCs realized by CE have been demonstrated (see Table 5). Starting from Cu-based chalcogenides, for example, ternary, quaternary and even quinary systems, such as Cu–Zn–Sn–Se-S, have been produced with a fine control over the composition. The alloying, in almost all the reported systems, is exclusively exploited to achieve control over the optical properties of the corresponding NCs, as it allows, for example, to tune the absorbance or, in specific cases, the photoluminescence of the products.
5.1.1. Cd-Based Chalcogenides
ZnSe NCs and ZnTe magic size clusters, both in the zinc-blende (zb) crystal structure, have been alloyed with Cd2+ to get ternary NCs.42,63,64,100,129 As this CE transformation is thermally activated (see section 3.1), the process is particularly sensible to the reaction temperature that can be used to govern the final NCs composition. Sheng et al., working with ZnSe NCs in water (that is at 95 °C), could tune the PL of the resulting CdxZn1–xSe NCs from 375 to 480 nm.63 By performing the reaction in organic media at higher temperatures (up to 250 °C-300 °C), the exchange could be pushed further, achieving eventually the total conversion of ZnSe to CdSe: the PL of the alloy NCs could be shifted up to almost 600 nm (see Figure 12B).42,64,100 Analogously, the reverse CE (that is, from CdS to ZnS) enabled the fabrication of alloy ZnxCd1–xS structures. If the reaction was performed at relatively low temperature (100 °C), the Zn2+ ions started replacing Cd2+ ions gradually from the surface of the NCs, yielding composition-gradient ZnxCd1–xS NCs.177 These NCs showed enhanced PL emission, as a consequence of the fact that the composition-gradient Cd–Zn–S shell optimally alleviates the lattice strain caused by the lattice mismatches between the CdS core and the Zn-based shell.
Figure 12.
Normalized UV–vis (left panels) and PL spectra (right panels) of: (A and B) ∼3 nm ZnSe and cation-exchanged CdxZn1–xSe NCs; (C and D) 4 nm CdTe and cation-exchanged HgxCd1–xTe NCs. (A and B) Reproduced with permission from ref (64). Copyright 2007 IOP Publishing. All rights reserved. (C and D) Reproduced from [Smith, A. M.; Nie, S. Bright and Compact Alloyed Quantum Dots with Broadly Tunable Near-Infrared Absorption and Fluorescence Spectra through Mercury Cation Exchange J. Am. Chem. Soc. 2011, 133, 24–26]. Copyright 2011 American Chemical Society.
Hg2+ ions can be easily inserted in zb-CdTe NCs through partial CE at RT to form the corresponding ternary Cd–Hg–Te alloy NCs.96,181,182 As CdTe and HgTe are completely miscible and have nearly identical lattice constants (aCdTe = 6.48 Å, aHgTe = 6.46 Å) the amount of Hg in HgxCd1–xTe NCs can be easily tuned from x = 0 to x = 1. The Cd2+ by Hg2+ CE in CdTe NCs does not change the lattice parameters or the particle size but greatly alters the band gap energy of the corresponding alloy NCs, since the two end points of this transformation (CdTe and HgTe) have very different band gap energies (the band gap energy of CdTe is 1.5 eV whereas that of HgTe is −0.15 eV). Consequently the PL emission of such NCs can be tuned from 600 nm to almost 1000 nm depending on the amount of Hg incorporated (see Figure 12c,d).
The transformation of binary CdSe to ternary alloy CdxHg1–xSe NCs via Cd by Hg CE is strongly influenced by the crystal structure of the starting NCs.90,96,150 This is due to the fact that HgSe crystallizes only in a zinc-blende phase, while CdSe can exist either in cubic zinc-blende or wurtzite structures. Consequently, zb-CdSe NCs can be easily converted at RT to isostructural alloy CdxHg1–xSe NCs with precise control over the composition.90,96 On the other hand, wz-CdSe NCs have been shown to incorporate a limited amount of Hg2+ ions, requiring at the same time longer reaction times.90,150 Gupta et al. showed, for example, that in wz-CdSe NRs the inclusion of Hg2+ ions is limited to a composition of Cd0.9Hg0.1Se (see also Section 3.4) with the resulting NCs exhibiting many stacking faults.150 This was attributed to the poor miscibility of the two end point structures, as the wurtzite Cd–Hg–Se bulk alloy structure can incorporate at maximum 20% of Hg.195 By increasing the amount of Hg2+, the bulk alloy adopts a zinc-blende structure. Similarly, Prudnikauet al. observed that upon exposure of wz-CdSe NRs to Hg2+ ions, defective nonluminescent alloy structures formed at the early stage of the CE reaction.90 Notably, at longer reaction times (i.e., 24h at RT) these structures were found to gradually undergo structural reorganization forming eventually zb-CdxHg1–xSe NCs (as predicted by the corresponding phase diagram195) with PL emission in the NIR (up to 850 nm).
5.1.2. Cu-Based Chalcogenides
Numerous recent works have shown that Cu-based chalcogenide NCs are able to incorporate many different guest cations forming quaternary or even quinary alloy systems with finely tunable optical properties. It is worth to note that, even if the possibility of different materials to form alloys is known (such as Cu2Se and SnSe2), not all the compositions allowed by CE experiments can be predicted by phase diagrams.149 Indeed, NCs with metastable compositions and with hitherto unexplored optical proprieties have been recently shown in various reports on CE, as will be shortly discussed below.
Different groups demonstrated that Cu2S NCs or NPLs with hexagonal crystal lattice can be partially exchanged to form ternary Cu–In–S NCs with wurtzite structure.102,103,110 In turn, the ternary alloy NCs, having either hexagonal or tetragonal crystal structure, can be further transformed into quaternary Cu–In–Zn-S or Cu–In–Zn-S|ZnS core|shell-like NCs with precise control over the composition and with enhanced PL emission.102,113 Remarkably, the direct transformation of Cu2S NCs into quaternary Cu–In–Zn-S NCs can be achieved in one-step CE by exposing the starting NCs to both In3+ and Zn2+ ions (see Figure 13A).102 Being AgInS2 and CuInS2 both chalcopyrite ternary solids belonging to I–III–VI2 class of semiconductors, the CE reactions working on the latter have been shown to work also on the former yielding quaternary Ag–In–Zn-S NCs.178,179 Tang et al., for example, showed that Ag–In–Zn-S NCs can be prepared by CE from AgInS2 NCs with a fine control over the Zn concentration, from 36.4 to 56.5%.179
Figure 13.
(A) Transformations of spherical Cu2S NCs into ternary and quaternary alloy NCs. The sequential introduction of In3+ and Zn2+ ions leads to the formation of Cu–In–S196 NCs and core|shell-like Cu–In–Zn-S (CIZS) NCs, respectively. The simultaneous introduction of indium and zinc produces homogeneous CIZS NCs (left). Reproduced from [Akkerman, Q. A.; Genovese, A.; George, C.; Prato, M.; Moreels, I.; Casu, A.; Marras, S.; Curcio, A.; Scarpellini, A.; Pellegrino, T.et al. From Binary Cu2S to Ternary Cu–In–S and Quaternary Cu–In–Zn–S Nanocrystals with Tunable Composition via Partial Cation Exchange ACS Nano2015, 9, 521–531]. Copyright 2015 American Chemical Society. (B) Schematic representation of the lattice conservation during CE reactions between zb- or wz-Cu2SnS3 NCs and Zn2+ ions. Reproduced from [Wang, Y.-X.; Wei, M.; Fan, F.-J.; Zhuang, T.-T.; Wu, L.; Yu, S.-H.; Zhu, C.-F. Phase-Selective Synthesis of Cu2ZnSnS4 Nanocrystals through Cation Exchange for Photovoltaic Devices Chem. Mater. 2014, 26, 5492–5498]. Copyright 2014 American Chemical Society.
In a similar manner, in a recent work from our group, we demonstrated that Cu2-xSeyS1–y nanoplatelets (NPLs) with hexagonal chalcocite-like structure can be partially exchanged with either Sn4+ or Zn2+ ions and form ternary alloys.108 Also, if the tin and zinc precursors are used together in the partial CE reaction, it is possible to insert both ions at the same time to get quinary Cu–Sn–Zn-Se(S) NCs. The bandgap of the alloy NPLs can be tuned from 1.0 to 2.1 eV depending on the composition. As an example, Cu0.26Zn0.25Sn0.06Se0.3S0.13 NPLs, having the highest Zn content and the lowest Sn content, are characterized by a wide band gap (2.1 eV), which narrows down by reducing Zn and increasing Sn amount (1.3 eV for Cu0.29Zn0.14Sn0.12Se0.33S0.12). The incorporation of only tin ions into Cu2-xSeyS1–y NPLs produces a further narrowing of their band gap from 1.5 to 1.0 eV. An analogous behavior was observed by us in spherical Cu2-xSe NCs with a cubic structure (berzelianite), which can accommodate Sn4+ ions and form ternary Cu–Sn–Se systems when partially exchanged.149 Interestingly, the same NCs, if exposed to Zn2+ ions, undergo CE, but in this case NHCs are formed (see section 5.3.1.6).137 The crystal structure of the host NCs, evidently, has a major role in accommodating the guest ions and in dictating the outcome of the CE. On the other hand, Wang et al. demonstrated that both zinc-blende and wurtzite Cu2SnS3 NCs can be partially exchanged with numerous guest ions, such as Zn2+, Fe3+, Co2+, Ni2+, and Cd2+, forming, independently from the initial crystal structure, single crystal alloy NCs (see Figure 13B).180
5.1.3. Iron Oxides
The formation of alloy NCs prepared by partial CE has almost exclusively concerned metal chalcogenide systems (see Table 5). The only example of alloying in oxide NCs was reported by Heiss et al. when exposing Fe3O4 NCs to Co2+ cations to give Co–Fe–O alloy NCs.174 The alloying is promoted by the structural similarity between Fe3O4 (more precisely written as Fe2+Fe3+2O4) and metal-doped spinel-type ferrites, M2+Fe3+2O4 (M=Co, Ni or Mn). Ideally, a complete replacement of Fe2+ ions, occupying the octahedral sites of the inverse spinel structure of Fe3O4, with Co2+ cations would lead to the CoFe2O4 structure. Heiss et al. observed indeed that partial Co2+-for-Fe2+ CE of Fe3O4 NCs resulted in homogeneous alloy NCs with size and shape preservation (see Figure 14a,b). The Co alloying, on the other hand, had a strong influence on the magnetic properties of the resulting NCs due to strong spin–orbit couplings at the Co2+ sites and the concomitant increase of the magnetic anisotropy (see Figure 14c,d).
Figure 14.
TEM images of small Fe3O4 NCs with a mean diameter of 9 nm before (a) and after (b) partial CE with Co2+ (performed at 200 °C for 40 min). The size, shape and size distribution of the parental Fe3O4 NCs are preserved after the exchange process. (c) The blocking temperature TB increases from 130 to 210 K. (d) The opening of the hysteresis loop observed a 5 K increase from 836.6 to 4674.8 Oe. Reproduced from [Sytnyk, M.; Kirchschlager, R.; Bodnarchuk, M. I.; Primetzhofer, D.; Kriegner, D.; Enser, H.; Stangl, J.; Bauer, P.; Voith, M.; Hassel, A. W. et al. Tuning the Magnetic Properties of Metal Oxide Nanocrystal Heterostructures by Cation Exchange Nano Lett. 2013, 13, 586–593]. Copyright 2013 American Chemical Society.
5.2. Doping with Partial CE
When partial CE reactions are performed using a severe substoichiometric amount of guest ions, few “impurities” can be added to each host NC. This process is commonly defined as doping, even if, compared to doped bulk semiconductors, the concentration of dopants is enormously higher. Doping in direct synthesis requires a fine balance or precursor reactivity, as the impurities have to be incorporated in a controlled way during the growth of the NCs. At the same time, a precise control over the size and shape of the NCs is required in order to have a homogeneous sample. One additional challenge in the synthesis of doped NCs is the so-called “self-purification” process, in which the host matrix tends to expel the dopant ions to the NC surface during the synthesis, especially when working at high temperatures.197 One key advantage of the CE approach is that it enables the temporal separation of the growth stage of the NCs from that of the inclusion of impurities. The original sample, eventually, can be compared with that after the doping process, allowing a more direct study of the doping effects. That would be otherwise hard to produce through a direct approach.
Sahu et al. prepared lightly doped Ag:CdSe NCs by adapting a standard CE procedure normally used to convert CdSe into Ag2Se NCs.115 Ag can be added as electronically active impurity in CdSe NCs, which led to a strong enhancement of their PL emission, with the maximum intensity peaking around 2 Ag per NC (see Figure 15A). Additional Ag ions were found to decrease the PL intensity from this maximum. A further increase in Ag incorporation eventually led to the nucleation of a Ag2Se phase. Acharya et al. observed instead the formation of Cu-doped CdxZn1–xSe NCs by exchanging simultaneously Cu+ and Cd2+ ions with Zn2+ ones in preformed ZnSe NCs (see Figure 15B,C).100 Such doping procedure is quite challenging as a fine-tuning of copper incorporation can be achieved only through the proper choice of the relative concentration of Cu- and Cd-precursors, as well as the reaction temperature. This is due not only to the fact that Cu+ and Cd2+ ions have different substitution rates in ZnSe NCs, but also because Cu+ ions can be easily inserted in ZnSe, but not in CdSe. Consequently, at the early stage of the CE, both Cd2+ and Cu+ ions are exchanged with zinc. As long as the CE proceeds with the further inclusion of cadmium, copper ions are progressively ejected from the NCs (see Figure 15C). The Cu:CdxZn1–xSe tunability covers the range from 500 to 750 nm, accompanied by the composition tunable spectral broadening which arises due to tailoring of the band gap of the host (see Figure 15B).
Figure 15.
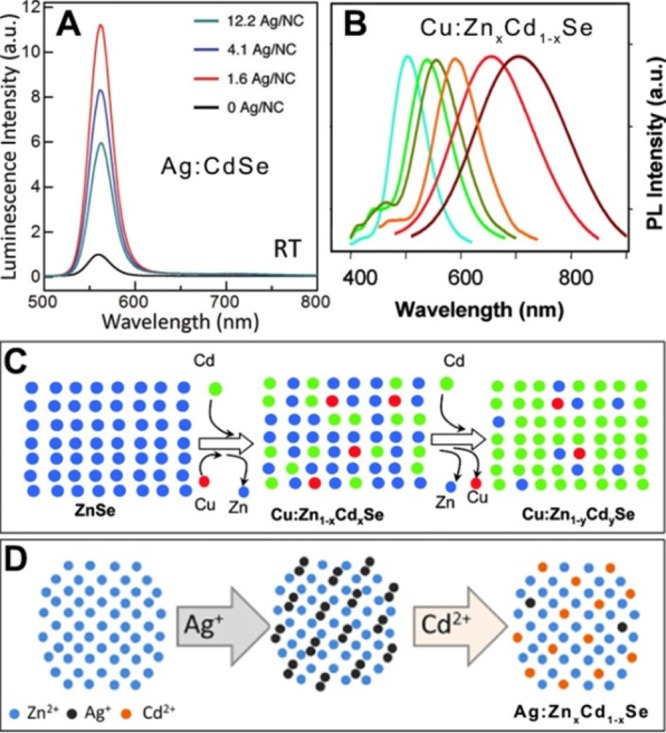
(A) Room-temperature PL emission spectra of 3.1 nm-diameter CdSe NCs dispersed in hexane with no Ag (black), 1.6 Ag/NC (0.3% Ag, red), 4.1 Ag/NC (0.74% Ag, blue), and 12.2 Ag/NC (2.22% Ag, green). The excitation wavelength was 350 nm. (B) PL spectra (normalized) of Cu-doped alloyed CdxZn1–xSe NCs. Cu precursors are incorporated along with the Cd precursor. The excitation wavelength is 350 nm. Reproduced from [Sahu, A.; Kang, M. S.; Kompch, A.; Notthoff, C.; Wills, A. W.; Deng, D.; Winterer, M.; Frisbie, C. D.; Norris, D. J. Electronic Impurity Doping in CdSe Nanocrystals Nano Lett. 2012, 12, 2587–2594]. Copyright 2012 American Chemical Society. (C) Sketch of the formation of Cu: CdxZn1–xSe NCs trough the one-pot CE reaction used by Acharya et al. Dopants are incorporated together with Cd ions, and ejected when the amount of Cd increases with the progress of the reaction. In the alloy composition, x is greater than y. Reproduced from [Acharya, S.; Pradhan, N. Insertion/Ejection of Dopant Ions in Composition Tunable Semiconductor Nanocrystals J. Phys. Chem. C2011, 115, 19513–19519]. Copyright 2011 American Chemical Society. (D) Schematic of the sequential CE approach used by Haibaoet al. to synthesize Ag (or Cu)-doped CdxZn1–xSe NCs. In both (C and D) the arrangement of the colored balls represents the placement of cations in the crystal lattice. Adapted with permission from ref (89). Copyright 2014 IOP Publishing. All rights reserved.
Direct and sequential hydrazine-promoted CE approaches have been demonstrated by Haibao et al. to produce Cu:ZnSe NCs and Cu- or Ag-doped Cd1–xZnxSe NCs.89 In the first step, ZnSe NCs were exposed to Ag+ (or Cu+) ions to get alloyed Zn–Ag–Se (or Zn–Cu–Se) NCs. This was followed by a second CE reaction with Cd2+ ions that resulted in Ag- or Cu-doped Cd1–xZnxSe NCs (see Figure 15D). The final amount of dopants concentration could be controlled by tuning the concentration of Ag+ or Cu+ ions inserted in ZnSe NCs during the first step, while keeping fixed the amount of Cd2+ ions used in the second step. Starting from 3.3 nm ZnSe NCs, the PL emission of the resulting Ag-doped ZnCdSe NCs could be tuned from 515 to 660 nm, while Cu-doped ZnCdSe ternary NCs exhibited a much broader tunable spectrum range, covering emission from 550 to 790 nm.
The potentialities of CE reactions have been also exploited to prepare more elaborate doped systems. Sun et al. for example synthesized 64Cu-doped CdSe|ZnS NCs by inserting positron-emitting 64Cu2+ radionuclides in core|shell CdSe|ZnS NCs through CE.194 Meijerink et al., on the other hand, observed that CE reactions can even work for doping extremely small (less than 2 nm) ZnTe nanoclusters.128 The exposure of ZnTe nanoclusters to Mn2+ ions resulted, indeed, in doped Mn:ZnTe systems whose PL spectra showed a red 620 nm emission characteristic of manganese impurities in bulk ZnTe. As a last example, Du et al. exploited CE reactions to dope YVO4 NCs (10–15 nm in size) with Eu3+ ions, forming Eu3+:YVO4 NCs.193 All these complex systems, obviously, give credit to CE reactions as a powerful tool to prepare nanostructures that would be otherwise extremely hard to synthesize through a direct approach.
5.3. Partial CE with Immiscible Phases
The systems discussed in this section deal with materials that are known to be immiscible in the bulk. Partial CE, in these cases, leads to the formation of NHCs made of two (or more) different domains sharing, usually, a sharp epitaxial interface. The spatial arrangement of the two materials within each NC strongly depends on kinetic and thermodynamic factors and it is hardy predictable a priori. The final architecture is mainly governed by the relative activation barriers for CE to initiate at different facets of the NC and by the energetics of the interface(s) between the two materials, as the reaction front proceeds through the NC. The diffusion rates of the exchanging ions can play a dominant role in determining the morphology of the intermediate CE structures which, in turn, can be dictated by kinetic rather than by thermodynamic factors. The observed morphologies can be divided in two main groups: core|shell and segmented NHCs. As a general trend, core|shell NHCs are formed when CE proceeds following a isotropic-like cations substitution, without the generation of a significant strain between the two materials (see section 5.3.2). On the other hand, segmented NHCs, such as Janus-like, striped and sandwich-like ones are characterized by the presence of a preferred interface between the two materials that minimizes both the interface energy and the strain of the different domains (see section 5.3.1). It is easily understood that the shape of the starting NCs plays a pivotal role in determining the morphology of the intermediate NHCs, since different types of surfaces, and thus, different entry points for the entering cations, are exposed to the reaction solution. Wark et al. showed, for example, that wz-CdSe NCs and NRs, when partially exchanged with Pd2+ ions, produce core|shell or segmented NHCs, respectively.66 Also, it is important to underline that the NHCs formed upon CE do not always exhibit morphologies that can be explained by simple considerations based on interfacial energy, strain or ions diffusivities. The presence of point, line or planar defects in the host NCs, in fact, may lead to unpredicted structures: these defects can act as preferential nucleation points for the new phase, or as preferential entry/diffusion channel for the guest cations.70,88,162 These cases are discussed in section 5.3.3.
5.3.1. Segmented Heterostructures
The synthesis of NHCs, consisting of two or more components within each particle, has been for long exploited to create multifunctional materials and to control the electronic coupling between nanoscale units. Typically the synthesis of such NHCs implies the nucleation and growth of a secondary material on specific facets of seed NCs. While this seeded-growth methodology has been applied to a wide range of material combinations, one of the major drawbacks is that that the desired heterogeneous nucleation on the existing NCs surface often competes with homogeneous nucleation of separate NCs of the secondary material. CE reactions have been exploited as an alternative reaction tool for synthesizing NHCs. Thanks to this approach, a portion of the seed NCs can be transformed into a new composition or structural phase, therefore circumventing any separate homonucleation. Moreover, as the CE process in most of the cases is topotactic, the different materials share a common anion sublattice and have a sharp epitaxial interface. This, of course, makes such NHCs particularly suitable for applications.
Segmented NHCs seem to form in CE reactions as a result of the presence of a specific interface between the reactant and the product materials that minimizes both the interface energy and the strain. In all the systems in which segmented structures are formed upon partial CE, the ion mobility is usually high enough to favor the arrangements of the resulting domains in the “lowest energy” configuration. Typically, once the product material nucleates at preferred sites on the surface of the host NC, the CE proceeds further only from there. The ion exchange tends to favor specific crystallographic directions, with the consequent formation of NHCs whose domains exhibit a preferential low energy interface. As a general trend, when the host NCs have a rod-like shape, the CE tends to initiate at the tip regions of the NRs,37,70,191 while in case of NDs the nucleation of the product phase has a stronger tendency to take place at the lateral edges.104,131,151 In case of spherical NCs the CE can start from just one site, generating Janus-like nanoarchitectures,118,137,149 from two diametrically opposite sites, forming sandwich-like NHCs,107 or even from multiple random points, forming polycrystalline NCs.149
5.3.1.1. CdX/Cu2X (X = S, Te) Systems
CdS/Cu2S is among the most studied binary systems prepared with CE reactions. As no miscibility is allowed in the whole compositional range, Cu- partial exchange of CdS (or vice versa) produces binary NHCs. Sadtler et al. provided a detailed study of partial CE reactions involving CdS NRs and Cu+ ions.70 In these reactions, Cu+ ions start replacing cadmium preferentially at one or both ends of each NR, so that one or two orthorhombic Cu2S domains grow inward starting from the tip regions. The result of this process is the formation of segmented Cu2S/CdS NHCs in which the two materials share flat interfaces perpendicular to the elongation direction of the NRs (see Figure 16). As the hexagonal sulfur sublattices of wurtzite CdS and orthorhombic Cu2S are almost identical and the lattice volume contracts just by 8% when going from CdS to Cu2S, the morphology and size of NRs is preserved upon such conversion.
Figure 16.
(A) Color-composite EFTEM image of CdS/Cu2S binary NRs, where the orange regions correspond to the Cd energy-filtered mapping and blue regions correspond to the Cu mapping. (B) high-resolution TEM image of a CdS/Cu2S NR, (C) Sketch representing the development of the morphology of binary NRs produced by CE for increasing amounts of Cu+ added to CdS NRs. Reproduced from [Sadtler, B.; Demchenko, D. O.; Zheng, H.; Hughes, S. M.; Merkle, M. G.; Dahmen, U.; Wang, L.-W.; Alivisatos, A. P. Selective facet reactivity during cation exchange in cadmium sulfide nanorods J. Am. Chem. Soc. 2009, 131, 5285–5293]. Copyright 2009 American Chemical Society.
The elastic contributions to the interface formation energies are small in this system and the relative values of the chemical formation energies determine the stability of the different CdS-Cu2S interfaces. The S sublattices of the two different crystal structures are perfectly aligned along the [001] axis of the orthorhombic Cu2S cell that corresponds to the [0001] axis of the hexagonal CdS lattice. The interfaces with the lowest energy of formation are perpendicular to the elongation direction of the NR (c axis). This explains why the CE selectively starts at the tips of the rod and why Cu2S/CdS interfaces parallel to the cross section of the NRs are observed most often by TEM. Moreover, the probability of nucleation of Cu2S at the (000–1) end facet is slightly lower if compared to the (0001) facet, which justifies why in some NRs the CE takes places at only one tip, while in some other cases asymmetric Cu-CE is observed.
Analogous NHCs were recently observed by Kriegel et al. when exposing zb-CdTe NRs to Cu+ ions.192 In such CE reactions the exchange took place preferentially from just one side of the parent CdTe NRs, yielding CdTe/Cu2-xTe NHCs. Also in this case, even if CE in most NRs started from only one of the NRs’ tips, in some other NRs CE was observed to start simultaneously from both tips.
5.3.1.2. CdS/Ag2S System
Another important combination of materials, represented by the Ag2S–CdS binary system, has been deeply investigated in colloidal NCs using CE. Robinson et al., for example, found that CdS NRs transform to Ag2S/CdS striped superstructures (n-n junctions) after partial CE with Ag+ ions.71,198 Differently from the Cu2S/CdS case, the addition of low amounts of Ag+, rather than leading to a preferential exchange at the tips, produces instead small Ag2S regions that are dispersed randomly over the surface of the starting CdS NR. This is due to the negative chemical formation energies for each of the CdS/Ag2S attachments that favor, initially, the creation of Cd–S–Ag interfacial bonds on both ends and sides of the CdS NRs. By increasing the amount of silver ions, the Ag2S domains coalesce and form regular segments spanning the diameter of the NR, with flat interfaces parallel to the NR cross section (see Figure 17). As the Ag2S domains grow further, the elastic strain originating at the Ag2S/CdS interface becomes a more important contribution to the total formation energy, driving the ripening of the Ag2S regions in order to reduce the interfacial area. Surprisingly, the system does not tend to the lowest energy structure, that would consist in a full phase segregation of the CdS and Ag2S regions to the opposite ends of the NR (as in the case of Cu2S/CdS NRs), but the Ag2S segments remain separated from each other. This separation is stabilized by the large interfacial strain that leads to a repulsive elastic interaction between segments that decreases with increasing separation between them. Differently from the CdS-Cu2S case, thus, here the factor that governs the morphology of the resulting NHCs is the interfacial strain, rather than the interface energy.
Figure 17.
(A) TEM image of Ag2S-CdS superlattices formed through partial CE. (Inset) Histogram of Ag2S segment spacing (center-to-center). The average spacing is 13.8 ± 3.8 nm. Adapted with permission from ref (71). Copyright 2007 The American Association for the Advancement of Science. (b) Sketch representing the development of the morphology of binary nanorods produced by CE at increasing amounts of Ag+ added to CdS NRs. Reproduced from [Sadtler, B.; Demchenko, D. O.; Zheng, H.; Hughes, S. M.; Merkle, M. G.; Dahmen, U.; Wang, L.-W.; Alivisatos, A. P. Selective facet reactivity during cation exchange in cadmium sulfide nanorods J. Am. Chem. Soc. 2009, 131, 5285–5293]. Copyright 2009 American Chemical Society. (C) Color maps of the z-component of strain in the interatomic bonds, for a 4.8 nm diameter Ag2S/CdS striped NR. The segment separations are 6, 8, and 10 nm. The color map limits indicate a strain ranging from −2% (compression, blue) to 2% (tension, red). The atoms are pulled in opposite directions (indicated by the black lines at the center of the second rod), which leads to an increased interaction energy when the segments separation decreases. Reproduced from [Demchenko, D. O.; Robinson, R. D.; Sadtler, B.; Erdonmez, C. K.; Alivisatos, A. P.; Wang, L.-W. Formation Mechanism and Properties of CdS-Ag2S Nanorod Superlattices ACS Nano2008, 2, 627–636]. Copyright 2008 American Chemical Society.
5.3.1.3. CdX/PbX (X = S, Se, Te) Systems
The CdX/PbX system (X = S, Se, Te) represents one of the most studied systems in CE reactions (see also section 5.3.2.1). As PbX and CdX materials are immiscible, partial exchange of Cd2+ with Pb2+ ions results in segmented CdX/PbX NHCs. In a recent work, Lee et al. showed that the transformation of CdSe NRs, upon exposure to Pb2+ ions, leads to rock-salt (rs) PbSe NRs with intermediate structures represented by wz-CdSe/rs-PbSe heterojunctions.121 The replacement of Cd2+ ions was found to take place anisotropically from the tips of the NRs and forming interfaces parallel to the (0001) plane of wz-CdSe NRs, exactly as in the CdS/Cu2S case (see Figure 18A,B).70 These interfaces were proven to minimize the interfacial energy between the two materials as the {111} facets of rs-PbSe form nearly strain-free interface with {0001} facets of wz-CdSe. The lattice mismatch at the {111}PbSe/{0001}CdSe interface is estimated around 0.74%, implying the high stability of this particular epitaxial interface. Interestingly, as in the case of CdS-Cu2S system, the CE process initiates at both tips of CdSe NRs, and then it proceeds faster along the (000–1) planes. In the case of Cd-to-Cu conversion in CdS NRs this asymmetry was explained as resulting from the lack of inversion symmetry about the c axis in the wz-CdS lattice; namely, the densities of dangling bonds and their geometries are not equal at the (0001) and (000–1) end facets of the NRs, hence the two facets have different energies. Analogously, as the (000–1) facet of CdSe NRs is more reactive than the (0001) one, the Cd-to-Pb exchange appears to be facet-selective.
Figure 18.

(A,B) HAADF images of w-CdSe/rs-PbSe NRs obtained with Cd-to-Pb partial conversion of CdSe NRs. The NHC have either only one or both tip converted into PbSe, in the latter case with different extents of conversion. The resulting clear PbSe/CdSe interfaces are parallel to the (0001) plane of w-CdSe. Reproduced from [Lee, D.; Kim, W. D.; Lee, S.; Bae, W. K.; Lee, S.; Lee, D. C. Direct Cd-to-Pb Exchange of CdSe Nanorods into PbSe/CdSe Axial Heterojunction Nanorods Chem. Mater. 2015, 27, 5295–5304]. Copyright 2015 American Chemical Society. (C) Janus-like spherical NC where approximately half of each QD is made PbS and half is CdS. The resulting c PbS/CdS interface is parallel to the (111) plane of both cubic structures. Reproduced from [Zhang, J.; Chernomordik, B. D.; Crisp, R. W.; Kroupa, D. M.; Luther, J. M.; Miller, E. M.; Gao, J.; Beard, M. C. Preparation of Cd/Pb Chalcogenide Heterostructured Janus Particles via Controllable Cation Exchange ACS Nano 2015, 9, 7151–7163]. Copyright 2015 American Chemical Society. (D–G) CdS/Pd4S hybrid nanostructures: (D) tip growth of Pd4S on 5.9 nm diameter nanorods; (E) more extensive growth of Pd4S on a sample of CdS nanorods with a broad size distribution; (F) close-up of the sample shown in (D); (G) HRTEM of a single CdS/Pd4S NR from the sample shown in (E). Adapted with permission from ref (191). Copyright 2010 John Wiley and Sons.
Using similar reaction conditions, Zhang et al. prepared zb-CdS/rs-PbS and zb-CdSe/rs-PbSe Janus-like NHCs upon partial CE.118 In this case too, even if starting from cubic CdX NCs, the CE proceeds anisotropically: it is initiated at specific sites or planes and then forms sharp (111)PbE/(111)CdE interfaces (see Figure 18c). The presence of only one interface in all the Janus-like NHCs, again, proves that the process tends to proceed by minimizing both strain and interface energy. The specific (111)PbE/(111)CdE interface suits this requirements as it is composed by a Se atomic layer sandwiched by a Pb layer on one side and a Cd layer at the other side along the <111> direction.
5.3.1.4. CdS/Pd4S System
Shemeshet al. reported the synthesis of CdS/Pd4S segmented NRs by exposing CdS NRs to Pd2+ ions in the presence of oleylamine at 200 °C.191 Surprisingly, while the direct exchange of Cd2+ with Pd2+ ions would produce a PdS phase, in this particular work the formation of a Pd4S phase, in which Pd is present both in the +2 and 0 oxidation states, was observed. The process itself, consequently, can be thought as a combination of a high-temperature reduction of Pd2+ to Pd0 (driven by the oleylamine) and the simultaneous replacement of Cd2+ ions with both palladium species. Interestingly, the formation of Pd4S, as in the case of CdS/Cu2S and CdX/PbX systems, starts either at one or at both tips of the initial CdS NRs (see Figure 18D–G). The growth of the new phase produces segmented CdS/Pd4S structures in which the two domains share flat interfaces perpendicular to the elongation direction of the NRs, most likely to ensure a minimal interface energy and strain. As the reactivity of the crystallographically anisotropic ends of the wz-CdS NRs is different, also in this case the formation of the Pd4S domains was not symmetrical on both sides of the NRs.
5.3.1.5. Cu2-xS/ZnS and Cu2-xS/CdS Systems
Ha et al. reported the formation of dual-interface NHCs by exposing roxybite Cu2-xS NCs to Zn2+ ions.107 The CE starts with the symmetrical nucleation of two ZnS grains on opposite sides of each spherical copper sulfide NC (see Figure 19). As ZnS domains grow, the copper sulfide region in the center of the NC eventually becomes a disk-like 2D layer (see Figure 19c–e), whose thickness can be tuned by controlling the extent of CE. In the presence of an excess of Zn2+ the CE leads to fully converted ZnS NCs preserving the spherical shape and size of the initial copper sulfide NCs. As revealed by HAADF and HRTEM characterizations, the Cu2-xS/ZnS NHCs exhibit atomically sharp interfaces along the <100> direction of Cu2-xS and <001> direction of ZnS, with no intermixing of zinc and copper ions. These particular epitaxial interfaces minimize the interface energy as the sulfur sublattices shared by Cu1.81S roxybite (triclinic) and wz-ZnS are well-matched. Indeed, even if roxbyite copper sulfide has a low symmetry crystal structure, its sulfur sublattice along the (100) or (−100) planes is hexagonal, with maximum deviations of ±0.35 Å from the sulfur sublattice spacing in wz-ZnS along the (001) or (00–1) planes (see Figure 19f–g). Along the other planes, a poor match between the two crystal structures was found. Additionally, the dual interface formation in this specific systems can be explained by considering that the atomic arrangements of the (100) and (−100) planes of the roxbyite phase are extremely similar, providing a comparable chemical environment for the CE, and, thus, similar interface energies with the ZnS domains. The presence of such favorable epitaxial matching with only a few atomic planes is likely the reason why Cu1.81S/ZnS NHCs do not form a core–shell structure. The strain in Cu1.81S/ZnS NHCs however plays a minor role in determining the arrangements of the different domains, since the lattice constant of the sulfur sublattice in ZnS (001/00–1) is only 1.1% smaller than that of the sulfur sublattice in roxbyite (100/–100). This lattice mismatch results in the formation of curved interfaces between copper sulfide and ZnS (see Figure 19h) and the strain relaxation occurs only by the development of atomic steps along the interfaces. It is interesting to notice that the same CE procedure was shown to work with Cd2+ ions and led again to the formation of dual-interface Cu1.81S/CdS NHCs. As the CdS sulfur sublattice spacing is 6.8% larger than that of roxbyite copper sulfide, the stress at the interface is reversed with respect to the Cu1.81S/ZnS NHCs case; therefore, an opposite curvature develops (see Figure 19i).
Figure 19.

CE transformation of spherical Cu2-xS NCs into dual-interface NHCs with ZnS caps. Schematic (top panel) and TEM images (a–e) show the composition change from the initial copper sulfide NC (a) into a ZnS with a residual copper sulfide thin disk (e). The copper sulfide disk thickness (t) is indicated. (f,g) Atomic models of the Cu1.81S roxbyite (100) plane (f) and wz-ZnS (001) plane (g). The sulfur atoms are shown in yellow. HRTEM images of Cu2-xS/ZnS (h) and Cu2-xS/CdS (i) dual-interface NHCs showing the convex curvature of ZnS caps (h) and concave curvature of CdS caps (i) on the copper sulfide central layer. The opposite curvature is due to the reversal of the interfacial strain for the ZnS (copper sulfide is in compression) versus the CdS (copper sulfide is in tension) caps. Reproduced from [Ha, D.-H.; Caldwell, A. H.; Ward, M. J.; Honrao, S.; Mathew, K.; Hovden, R.; Koker, M. K. A.; Muller, D. A.; Hennig, R. G.; Robinson, R. D. Solid–Solid Phase Transformations Induced through Cation Exchange and Strain in 2D Heterostructured Copper Sulfide Nanocrystals Nano Lett. 2014, 14, 7090–7099]. Copyright 2014 American Chemical Society.
5.3.1.6. Cu2-xSe/MSe (M=Cd,Sn,Zn) Systems
The replacement of Cu+ ions in berzelianite Cu2-xSe spherical NCs with Zn2+, Cd2+, and Sn2+ produces, as intermediates, Janus-like Cu2-xSe/MSe NHCs.137,149 This indicates that the MSe phase nucleates at one location of the NCs and grows from there at the expenses of the residual Cu2-xSe domain, forming a single epitaxial interface. In all the cases, the appearance of a Janus-like architecture suggests that once a MSe nucleus is formed on a given location on the host NC surface, the CE reaction can proceed only from there and creates a single interface between the two different materials. The possibility of a simultaneous formation of multiple MSe nuclei on the surface of a single NC, with the consequent increase of the overall interface energy, is rarely observed, but is not statistically impossible, and it leads to the formation of polycrystalline NHCs, as shown in Figure 20F.149 The Cd2+-for-Cu+ partial CE in Cu2-xSe spherical NCs produces Cu2-xSe/CdSe Janus-like NCs with a clear distinction between the sphalerite CdSe (a = 6.1 Å) and the berzelianite Cu2Se (a = 5.8 Å) domains. In these structures, interestingly, the HRTEM analysis enables for the detection of a 4.5 (±0.9)% mean dilation in the CdSe domain relative to the Cu2Se one (see Figure 20A,B).137 The CE between Cu2-xSe NCs and Zn2+ ions, on the other hand, produces Cu2Se/ZnSe NHCs whose morphological attribution is more complex as neither HRTEM nor XRD analyses can directly prove that pure Cu2Se and ZnSe domains form.137 The mismatch between the berzelianite Cu2Se and sphalerite ZnSe (a = 5.7 Å) is indeed so low that HRTEM analysis of a single NHC cannot detect any variation in the lattice parameter (see Figure 20C). On the other hand, STEM-EDS mapping of such NCs indicates the presence of two distinct domains, one Cu-rich and the other Zn-rich (see Figure 20D). As these two materials are completely immiscible in the bulk, these NHCs are most likely Cu2Se/ZnSe Janus-like NCs (see Figure 20D).
Figure 20.
HRTEM images of (A) Cu2Se/CdSe, (C) Cu2Se/ZnSe, and (E) Cu2Se/SnSe Janus-like NCs. (B) Mean dilation map as obtained by geometric phase analysis of the Cu2Se/CdSe NC in panel A. (D) STEM-EDS map of the Cu2Se/ZnSe NC shown in panel C. (F) HRTEM image a polycrystalline Cu2-xSe/SnSe NC exhibiting different domains (Cu2-xSe in green, SnSe in red and light blue). Scale bars in all the HRTEM images are 5 nm. (A–D) Reproduced from [Lesnyak, V.; Brescia, R.; Messina, G. C.; Manna, L. Cu Vacancies Boost Cation Exchange Reactions in Copper Selenide Nanocrystals J. Am. Chem. Soc. 2015, 137, 9315–9323] Copyright 2015 American Chemical Society. (E and F) Reproduced from [De Trizio, L.; Li, H.; Casu, A.; Genovese, A.; Sathya, A.; Messina, G. C.; Manna, L. Sn Cation Valency Dependence in Cation Exchange Reactions Involving Cu2-xSe Nanocrystals J. Am. Chem. Soc. 2014, 136, 16277–16284]. Copyright 2014 American Chemical Society.
Differently from the systems discussed above, the substitution of Cu+ ions with Sn2+ ions in Cu2-xSe NCs produces a significant distortion of the Se2– anion sublattice, leading to the formation of an orthorhombic SnSe phase.149 This distortion is responsible, in the case of partial CE, for the formation of Cu2-xSe/SnSe Janus-like NCs characterized by clean interfaces, but exhibiting an imperfect epitaxial relationship: as can be seen from Figure 20E, there is a visible angular distortion between the (220) planes of Cu2-xSe and the (−511) planes of SnSe, which is also affected by a lattice mismatch of the order of 10%. The formation of Janus-like NHCs in this system can be attributed, most likely, to the high energy required for the nucleation of the SnSe phase. A local distortion of Cu2-xSe the anion sublattice is indeed necessary to enable the large Sn2+ ions to form the orthorhombic SnSe phase.
5.3.1.7. TiS2/Cu2S System
Jeong et al. showed that layered TiS2 nanoplatelets can undergo CE with Cu+ ions, leading to unconventional intermediate structures: TiS2/Cu2S core-crown-like NCs.131 The formation of the Cu2S phase starts and propagates from the edges of the TiS2 NDs in a heteroepitaxial manner. The interface between the two materials is not atomically flat, but the two lattices are precisely aligned, as found by HRTEM analysis (see Figure 21). On the edge of TiS2 NDs, the (002) planes of Cu2S are indeed parallel to the (001) planes of TiS2. Clearly, the presence of a preferential interface is lacking in this system and the reactive peripheral edges of TiS2 offer nonselective nucleation and growth points for the product material. The role of the strain in TiS2/Cu2S NHCs is hardly computable as the morphology of the starting Ti2S NCs is not retained during the CE process. Indeed, in this case even a reorganization of the anion framework accompanies the overall transformation. The initial disc-like shape transforms into a toroid; a highly symmetric, double-convex geometrical structure with a hole in its center (see section 4.1.1). The very same process seems to apply even when working with other guest cations such as Ag+, Mn2+ and Cd2+, which consistently resulted in similar intermediate nanostructures of TiS2/Ag2S, TiS2/MnS and a fully converted toroid of CdS, respectively.
Figure 21.
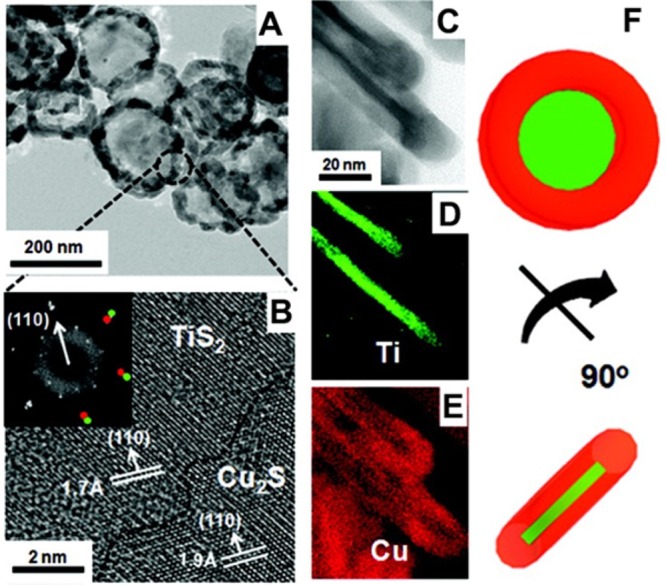
(A) Large-area TEM image of TiS2/Cu2S core-crown-like NHCs. (B) High-magnification image of the interface between TiS2 and Cu2S showing that the (110) planes of Cu2S with a 1.9 Å lattice fringe are oriented in the same direction as the (110) planes of TiS2 with a 1.7 Å lattice fringe. The FFT pattern (inset) shows green and red dots corresponding to TiS2 and Cu2S, respectively. (C–E) Side-view TEM image (C) and EELS elemental analysis (D and E) of a core-crown-like NHCs. Ti is shown in green and Cu in red. (F) Schematic rotation of a TiS2/Cu2S NHC by 90°, showing both planar and side-view images. Reproduced from [Jeong, S.; Han, J. H.; Jang, J.-t.; Seo, J.-w.; Kim, J.-G.; Cheon, J. Transformative Two-Dimensional Layered Nanocrystals J. Am. Chem. Soc.2011, 133, 14500–14503]. Copyright 2011 American Chemical Society.
5.3.2. Core|Shell Heterostructures
The synthesis of core|shell structures typically involves the epitaxial growth of a second semiconductor onto seed NCs, either in a single-step or in multistep reactions. In wet chemical approaches, the inorganic shell growth requires, in general, relatively high temperatures, i.e. above 150 °C, at which undesirable homogeneous nucleation of the shell material NCs or degradation of the starting seeds can occur: for example, lead chalcogenides NCs might undergo Ostwald ripening. Also, the growth of a monocrystalline shell might be hindered once the shell thickness becomes larger than the critical layer thickness (about two monolayers) due to the existence of strain-induced defects.199 This is particularly true when growing a shell onto quasi-spherical core NCs, as they are characterized by highly curved surfaces and they expose many different crystallographic facets.
Pioneering works, dating back to the nineties, demonstrated that CE reactions could lead, in some cases, to core|shell structures (for example exposing CdS QDs either to Pb2+ or Hg2+ ions) with tunable layer thickness.36,37 Recently, CE reactions have emerged as an alternative strategy to synthesize many core|shell NHCs of compounds such as PbX|CdX, CdX|PbX (where X = S, Se, or Te), and CuInS2|ZnS NCs (see Table 5). CE reactions can be used to overcome the problems connected to the traditional seeded-growth approach, as they can be performed at relatively low temperature, with consequent preservation of the starting NC seeds.122 Also, as the cation replacement proceeds in most cases topotactically, strain-induced effects can be minimized and in principle monocrystalline shells with tunable thickness can be prepared.
Differently from the segmented NHCs, the core|shell NCs formed upon CE seem to be metastable (i.e., kinetically driven), and under annealing or beam irradiation they transform into bidomain NHCs sharing one single interface that minimizes both interface energy and strain (see the discussion below). This suggests that this kind of architecture forms when the diffusivity of the ions involved in the CE is sufficiently slow to allow, statistically, the nucleation of the product material unselectively on the whole surface of the parent NCs. This is also favored by the absence of a specific interface that has an overwhelming lower energy with respect to the other interfaces. The growth of the product phase, therefore, proceeds from the surface to the inner core of each NC, with the consequent increment of the shell thickness and of the interface energy. Even if the shell growth can be thought as the result of an isotropic replacement of host cations with new ones, it has been demonstrated that this process is rather anisotropic. There is always, in fact, a favored interface, having the lowest energy, that preferentially forms, with consequent heterogeneity in shell thickness and in core position, size and shape.122,127,186
5.3.2.1. PbX-CdX System
Cd-exchanged PbX NCs are among the most studied core|shell systems that can be prepared by CE. As shown in section 5.3.1.3 the intermediate structures formed upon Cd2+ for Pb2+ exchange are typically segmented CdX/PbX heterojunctions, while the opposite reaction, that is, the replacement of Pb2+ ions with Cd2+ in PbX NCs, leads, surprisingly, to PbX|CdX core|shell NCs (see Figure 22a–c). This suggests that the two processes have different dynamics, i.e. the ions have different mobilities, which leads to different CE pathways. It is also important to consider that, while CE from CdX to PbX entails the reorganization of the overall NC lattice, from either wz-CdX or zb-CdX to rs-PbX, the inverse reaction (discussed in this section) always implies the transformation of a rs-PbX structure into a zb-CdX one. In this last case PbX and CdX materials have both a cubic crystal structure, with almost no lattice mismatch between them, and no relevant strain is generated at their interface.41,122,127 No preferential interfaces are thermodynamically predominant so that CE can proceed from each surface of the NC toward the inner part, generating core|shell architectures.127
Figure 22.
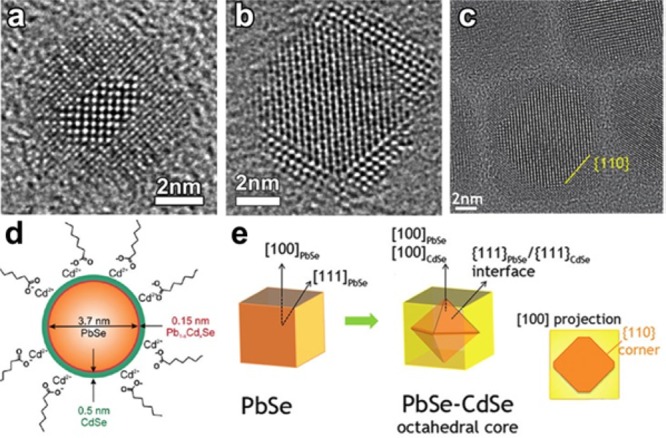
HR-TEM images of (a) spherical PbTe|CdTe, (b) spherical and (c) cubic PbSe|CdSe core|shell NCs observed in the (a,c) [100] and (b) [100] directions. (d) Schematic representation of a typical PbSe|CdSe core|shell QD. (e) Sketch of the structural transformation from pure PbSe cube-shaped NCs to core|shell PbSe|CdSe NHCs with a nearly octahedron PbSe core. The PbSe is indicated in orange, CdSe in yellow. (a,b) Reproduced from [Lambert, K.; Geyter, B. D.; Moreels, I.; Hens, Z. PbTe|CdTe Core|Shell Particles by Cation Exchange, a HR-TEM study Chem. Mater. 2009, 21, 778–780]. Copyright 2009 American Chemical Society. (c,e) Reproduced from [Casavola, M.; van Huis, M. A.; Bals, S.; Lambert, K.; Hens, Z.; Vanmaekelbergh, D. Anisotropic Cation Exchange in PbSe/CdSe Core/Shell Nanocrystals of Different Geometry Chem. Mater. 2012, 24, 294–302]. Copyright 2012 American Chemical Society. (d) Reproduced from [Abel, K. A.; FitzGerald, P. A.; Wang, T.-Y.; Regier, T. Z.; Raudsepp, M.; Ringer, S. P.; Warr, G. G.; van Veggel, F. C. J. M. Probing the Structure of Colloidal Core/Shell Quantum Dots Formed by Cation Exchange J. Phys. Chem. C2012, 116, 3968–3978]. Copyright 2012 American Chemical Society.
Pietryga et al. were the first group to exploit CE reactions in order to achieve inorganic passivation of PbSe NCs with a CdSe shell in 2008.122 They purposely employed a relatively slow-reacting cadmium precursor allowing for a controlled ion substitution at the surface and, in turn, resulting in a slow, self-limiting growth of a CdSe shell at the expenses of the starting PbSe NCs. Additional works followed this synthetic route and some of them demonstrated that PbS and PbTe, when exposed to Cd2+, form similar core|shell structures.40,41,88,116,123,124,126,127,185,186,188 As a general trend, the formation of PbX|CdX core|shell NCs leads to improved photostability and higher PL quantum efficiencies with respect to the “bare” starting PbX NCs.38,40,116,122,188 Moreover, when the shell is grown thick enough, the NHCs exhibit also the characteristic absorption and emission features of the CdX material.40,126,188
As the direct observation of such core|shell structure was initially challenging, detailed structural characterizations of CdX and PbX domains were published in later works.38,127,188 For what concerns the exact chemical composition of PbX|CdX NHCs, Abel et al., by combining advanced HRTEM with synchrotron XPS as well as X-ray and neutron scattering methods, demonstrated that the exchange of PbSe NCs with Cd2+ ions leads, indeed, to core|shell structures with a purely PbSe core, an outer shell made of pure CdSe (with a Cd-oleate capping layer), and a sharp interface of (strained) Pb1–xCdxSe material (see Figure 22d).185 No hints of the presence of alloyed structures emerged in any analysis.
In the PbS-CdS case, Lechner et al. showed that during the first stages of CE the shell grows at the expenses of the PbS core, preserving its original crystal structure and, thus, originating a metastable rocksalt CdS phase.38 As the shell gets thicker, the CdS shell transforms into the more stable zinc-blende structure. The growth of the shell, surprisingly, is not isotropic, as expected, but rather anisotropic, as proven by Lambert et al.127 This finding came as a comparison of ab initio calculations of interfacial energies and HRTEM observations of PbTe|CdTe NCs. While similar interface energies, of around 0.20 J/m2, were calculated for the {111}, {110}, and {100} facets, mainly {111} interfaces were observed by HRTEM.125 This suggests that CE in that system is not determined by thermodynamics, but rather by an anisotropic growth mechanism, since preferential interfaces are formed. The same mechanism was found to apply to PbSe|CdSe NHCs.41,127 More specifically, Casavola et al. showed that, when starting with rs-PbSe NRs or nano cubes, the CE reaction with Cd2+ leads to sharp {111}-faceted quantum dot cores of rs-PbSe embedded in a zb-CdSe shell (see Figure 22). The anisotropy of the exchange was tentatively explained by considering that the peculiarity of the {111} interface, with respect to the other interfaces (such as {001} and {110}), is that it consists entirely of either cations or anions. Thus, at the boundary the interfacial Se{111} atomic layer is sandwiched by a Pb{111} layer at one side, and by a Cd{111} layer at the other side providing a stress-free interface.41,141
The intrinsic anisotropy of the exchange process leads to a strong increase in the heterogeneity of the cores formed, not only in terms of core size and shell thickness but also at the level of the shape and position of the core. The initial size dispersion of the PbTe cores, for example, increases from 6.6% to 25%. At the same time, the almost spherical PbTe NCs transform into cores with an aspect ratio of about 1:2. The position of these cores can be either at the center or completely at one side of the final NHC. Clearly, this lack of control represents a drawback of an otherwise straightforward core|shell synthesis technique.
PbSe|CdSe core|shell NHCs prepared by CE have been shown to be metastable and to evolve into Janus-like systems upon annealing at temperatures as low as 150 °C under vacuum.125 Prior to annealing, the spherical NHCs are characterized by a rs-PbSe core with an approximately octahedral morphology (showing eight {111} facets), and a CdSe shell with a zinc-blende crystal structure (see Figure 23A,B). Upon in situ annealing, the NHCs preserve their original nearly spherical shape, but they undergo a drastic reconstruction whereby the total number of atoms stays constant. Both CdSe and PbSe domains reconstruct to form a Janus-like architecture with a single preferential rs-PbSe{111}/zb-CdSe{111} heterointerface. The mechanism of the thermally induced structural reconstruction is schematically depicted in Figure 23. The formation of perfect bihemispherical NCs requires that the starting volumes of PbSe and CdSe domains must be equal. Obviously, depending on the initial core/shell volume ratios, different shapes of NHCs can be formed after thermal annealing, with consequent different heterointerfaces, as shown in Figure 23C. These annealing experiments prove that the core|shell NHCs that form upon Cd-for-Pb partial CE are most likely kinetically accessed, and that these systems thermodynamically evolve to Janus-like architectures, with the consequent minimization of the CdSe/PbSe interface, if enough energy is provided.
Figure 23.
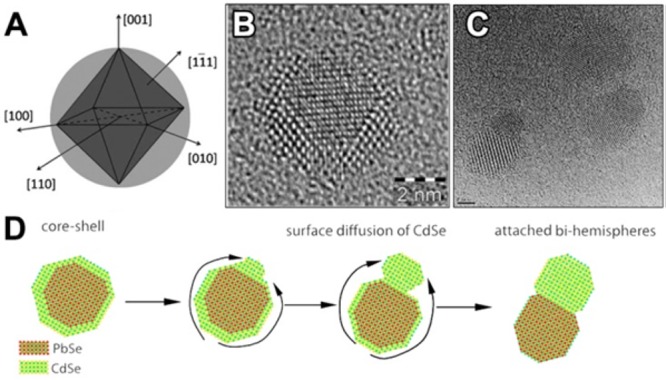
Model of the PbSe|CdSe core|shell NC structure evidencing the octahedral shape of the PbSe core (A). HR-TEM images of a PbSe|CdSe core|shell NC (B) and PbSe/CdSe Janus-like NCs (C). The latter are obtained after thermal annealing at 200 °C of initial core|shell NCs. The bottom-left particle clearly shows that the volume of the CdSe shell was significantly larger than that of the PbSe core, consequently the PbSe/CdSe heterointerface is considerably different from that observed for equal volume bihemispheres, in the sense that it is no longer a flat interface, as can be clearly observed for the two top-right NHCs. (D) The mechanism of the thermally induced structural transformation from core|shell to Janus-like NHC is schematically depicted. Reproduced from [Grodzinska, D.; Pietra, F.; van Huis, M. A.; Vanmaekelbergh, D.; de Mello Donega, C. Thermally induced atomic reconstruction of PbSe/CdSe core/shell quantum dots into PbSe/CdSe bihemisphere heteronanocrystals J. Mater. Chem. 2011, 21, 11556–11565]. Copyright 2011 American Chemical Society.
5.3.2.2. Cu2-xS|Au2S System
Wang et al. studed the CE reaction between Cu2-xS NCs and gold ions and found that the replacement of copper ions took place gradually from the surface to the core of NCs, yielding eventually Au2S NCs (see Figure 24).165 This process leads initially to the formation of Au2S islands decorating the host NCs followed by their growth to form a cubic Au2S shell covering the inner hexagonal Cu2-xS core. In analogy to the CdS-Ag2S system, the preliminary formation of Au2S islands suggests that the interface energy is sufficiently low to allow a nonspecific nucleation of the new phase along the Cu2-xS NCs surface.71 The growth of the Au2S phase then proceeded from there under kinetic control and led to a core|shell architecture that was found to be metastable: upon exposure to the electron beam of the HR-TEM, such NHCs initiated a transformation to a more stable configuration that resembled that of Janus-like structures (see Figure 24). Hence, when enough energy is provided the system tends to the most stable morphology, in which a single interface is shared between the two materials.
Figure 24.
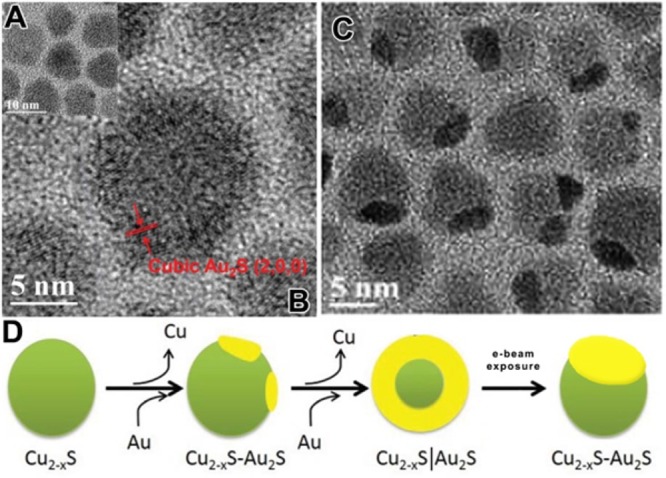
HRTEM images of (A) NCs synthesized exposing Cu2-xS NCs to Au ions using (A) nominal 1:4 Au:Cu ratio, and (B) nominal 1:1 Au:Cu ratio (Cu2-xS|Au2S NHCs). (C) HRTEM image of Cu2–xS–Au2S structures achieved after exposure of Cu2-xS|Au2S NHCs to electron beam irradiation. (D) schematic illustration of the CE process and the subsequent evolution of the core|shell structures upon electron beam irradiation. Adapted with permission from ref (165). Copyright 2014 Royal Society of Chemistry.
5.3.2.3. CuInS2|MS (M = Cd, Zn) Systems
In some CE reactions the exact morphology of the resulting structures cannot be defined easily. For example, the exposure of CuInS2 (CIS) QDs to Zn2+ or Cd2+ ions leads to NCs with a blue-shifted and improved PL emission.113,187,190 More specifically, in both cases the CE process completely eliminates the fast PL component of the parent CuInS2, associated with the surface defects, resulting in uniform single-exponential decay across the whole emission profile. As the size and morphology of the starting NCs is retained, the observed optical improvements have been explained in terms of a reduction of the CuInS2 domain size and the consequent formation of a ZnS or CdS shell. Unfortunately, CIS roquesite, ZnS sphalerite and CdS hawleyite involved in these CE reactions are miscible and have very similar lattice parameters. Therefore, XRD and HRTEM analyses are of little help in ruling out the formation of alloyed NCs, and in univocally demonstrating the presence of core|shell NCs.113,190,200 Moreover, as the size of the QDs is extremely small (in the order or 3 nm), any precise element distribution, trough STEM EDX for example, has not been reported yet. Our group, supported mainly by XPS analyses, has proposed that alloyed Cu–In–Zn-S (CIZS) QDS rather than core|shell structures are most likely formed upon CE.102,113
5.3.2.4. CdS|Cu2S System
The CE reactions between CdS NRs and Cu+ ions, as it was discussed in section 5.3.1.1, tend to produce segmented CdS-Cu2S NHCs in which the two materials share a low energy interface.70 On the other hand, CE experiments performed on wz-CdS NWs with Cu+ ions were found to lead to core|shell architectures.161,162 As an example, Zhang et al. observed CdS|Cu2S core|shell structures upon exposure of 30–40 nm thick and 10 μm long CdS NWs to copper ions (see Figure 25). Both CdS NRs and NWs under analysis are elongated along the [001] direction, exposing, consequently, the same crystallographic planes to the reaction solution. In the case of NWs, CE takes place preferentially at the tips (as in the case of NRs), but with a non-negligible nucleation of Cu2S islands along the side facets of the wires (see Figure 25D,E). The chemical formation energy of this process was calculated to be approximately 7 times greater than the energy of nucleation of Cu2S of at the CdS NRs tips.70 In NWs instead, being the lateral surface markedly higher than that of NRs, such nucleation becomes statistically possible. Once the exchange takes place at the tips and at the surface of the NWs, it proceeds from there to the inner part of the crystals through solid state diffusion and eventually produces core|shell structures.
Figure 25.

(A–D) EDS mapping for Cd–L (in green) and Cu–K (in red) and corresponding STEM images of CdS–Cu2–xS NWs. (A,B) Conversion at the side facets. (C,D) Conversion at the tips of the NWs. (E) HRTEM image of CdS–Cu2–xS core–shell NW and. Scale bars in (A–D) are 10 nm. Reproduced from [Zhang, D.; Wong, A. B.; Yu, Y.; Brittman, S.; Sun, J.; Fu, A.; Beberwyck, B.; Alivisatos, A. P.; Yang, P. Phase-Selective Cation-Exchange Chemistry in Sulfide Nanowire Systems J. Am. Chem. Soc. 2014, 136, 17430–17433]. Copyright 2014 American Chemical Society.
5.3.2.5. ZnSe|CdSe System
The ions diffusion can determine, in some particular circumstances, the formation of core|shell structures, even in case of miscible reactant and product materials. Groeneveld et al., in a recent work, showed that the ZnSe → CdSe CE transformation is a thermally activated isotropic process, and, thus, the reaction temperature can be used to precisely govern both the composition and morphology of the products.42 More specifically, at 150 °C ZnSe|CdSe core|shell NCs are obtained, while higher temperatures produce either gradient or homogeneous (Zn1–xCdx)Se alloys. This is quite interesting if one considers that ZnSe and CdSe materials are fully miscible, enabling, in principle, the formation of alloy NCs of any arbitrary composition. At relatively low temperature, i.e., 150 °C, the CE process is most likely limited to the surface of the NCs, as shown by EELS elemental mapping (see Figure 26) leading, unexpectedly, to core|shell structures with a CdSe shell thickness up to 0.5 nm. These structures, synthesized in a kinetic regime were found to be metastable. Upon thermal treatment at 250 °C, the core|shell NHCs could be converted to the expected alloy Zn1–xCdxSe NCs (see Figure 26F).
Figure 26.
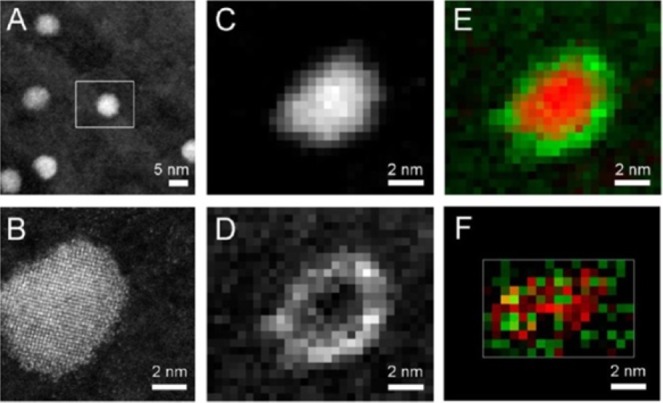
(A) STEM-HAADF images of ZnSe|CdSe core|shell NCs obtained from 5.6 nm ZnSe NCs by Zn2+ for Cd2+ exchange. The square indicates the region selected for EELS analysis. (B) HR-STEM image of a representative ZnSe|CdSe core|shell NC. Chemical maps for Zn (C) and Cd (D) of the ZnSe|CdSe core|shell NC selected in (A). These maps are combined in (E) to produce a chemical distribution profile of the NC (Zn in red, Cd in green). Panel (F) gives the chemical distribution profile map for a sample obtained by heating the ZnSe/CdSe core|shell NHCs, shown in (A–E) at 250 °C for 20 min. Reproduced from [Groeneveld, E.; Witteman, L.; Lefferts, M.; Ke, X.; Bals, S.; Van Tendeloo, G.; de Mello Donega, C. Tailoring ZnSe–CdSe Colloidal Quantum Dots via Cation Exchange: From Core/Shell to Alloy Nanocrystals ACS Nano2013, 7, 7913–7930]. Copyright 2013 American Chemical Society.
5.3.2.6. NaYF4:Yb,Tm|NaGdF4 System
Much less work has been done to date on CE reactions involving lantanide-based NCs. Core|shell NCs were reported by van Dong et al. in CE experiments with water-dispersible spherical NaYF4:Yb,Tm NCs and Gd3+ ions.184 The result of the CE reaction is represented by NaYF4:Yb,Tm|NaGdF4 core|shell NCs with a thin, tunable, and uniform NaGdF4 shell (see Figure 27). The presence of a shell in the product NCs could be visualized directly from HAADF images, where the signal difference between the shell and the center gave rise to a contrast, with the shell being brighter (see Figure 27B). Direct proof of the presence of a Gd3+ rich shell came from EELS 2D elemental maps in which a stronger Gd3+ signal was detected at the edge of each NC (see Figure 27C). In this system too, the interdiffusion of ions appears to govern the shell thickness, so that, by varying the CE reaction temperature from 25 to 100 °C, the shell thickness could be tuned from 0.3 to 1.9 nm. Neither in this case was the shell found to be uniform, most likely because preferred interfaces made the overall process slightly anisotropic.
Figure 27.
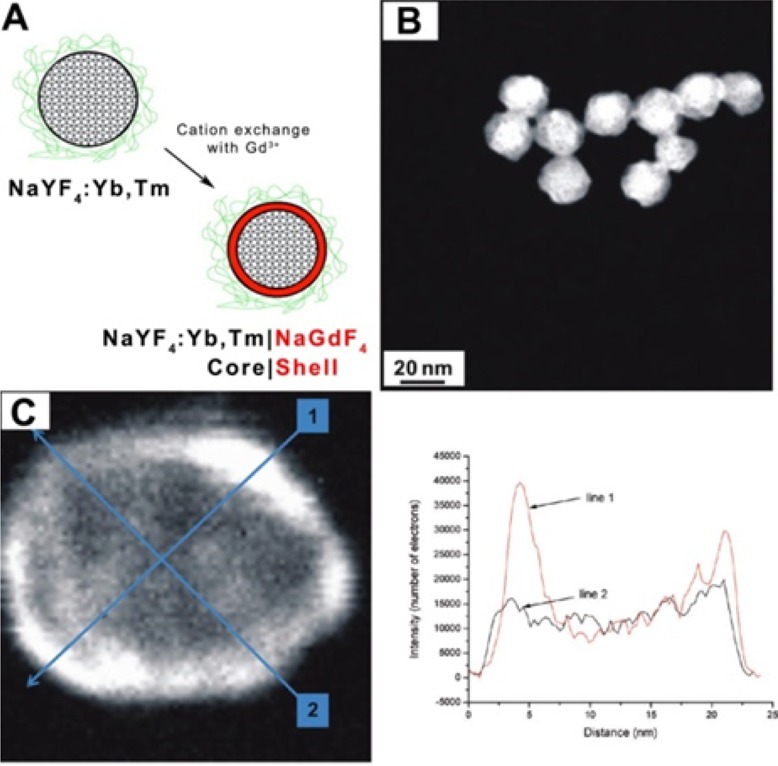
(A) Sketch representing the CE transformation of NaYF4:Yb,Tm NCs exposed to Gd3+ ions. (B) HAADF image, and (C) EELS 2D elemental maps and line profiles of Gd3+ in NaYF4:Yb,Tm|NaGdF4 core|shell NCs prepared by CE of NaYF4:Yb,Tm nanoparticles with 10-fold Gd3+ ions at 75 °C. Reproduced from [Dong, C.; Korinek, A.; Blasiak, B.; Tomanek, B.; van Veggel, F. C. J. M. Cation Exchange: A Facile Method To Make NaYF4:Yb,Tm-NaGdF4 Core–Shell Nanoparticles with a Thin, Tunable, and Uniform Shell Chem. Mater. 2012, 24, 1297–1305]. Copyright 2012 American Chemical Society.
5.3.3. Defects Driven Heterostructures
The presence of crystal defects, as already anticipated, can alter the outcome of the CE reactions. Dislocations, stacking faults and grain boundaries in host NCs can, indeed, greatly alter the ions diffusion pathways and the energetics involved. Consequently, the morphology of the NHCs, resulting from CE experiments on such NCs, can be substantially different from what expected. Justo et al., for example, observed that the partial CE of polycrystalline PbS NRs with Cd2+ ions leads to multiple dot-in-rod PbS/CdS NHCs (see Figure 28A–C).88 Similarly Casavola et al., when studying the CE transformation of rs-PbSe NRs into zb-CdSe, observed that, in some cases, single CdSe NRs embedding multiple PbSe dots were produced (see Figure 28).41 In both cases the final NHCs can be seen as NRs made of PbX|CdX core–shell NCs (see Figure 28). These unique nanostructures are believed to form because CE proceeds faster along the grain boundaries that are present in the original PbS NRs. Moreover, the grain boundaries act as preferential nucleation sites for the product CdX phase.
Figure 28.
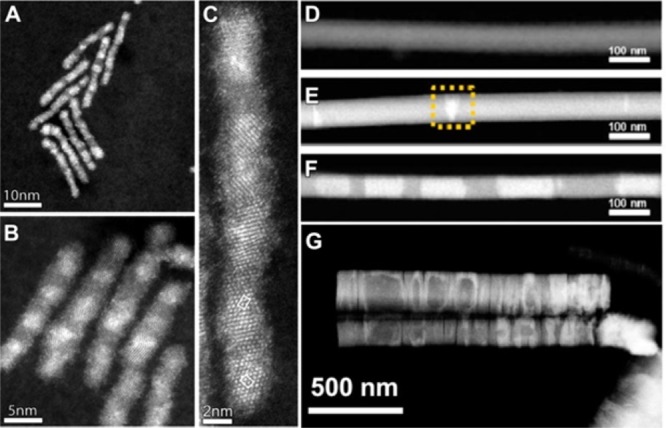
(A–C) STEM-HAADF images of PbS/CdS rods showing multiple PbS QDs inside the rods. The white rectangles in (C) indicate the ⟨110⟩ unit cell. STEM high-angle annular dark-field (HAADF) images of Cu2S NWs (D) and Cu2S–Ag2S superlattice NWs (E–G) formed after Ag-partial CE of (E,F) fcc or (G) monoclinic Cu2S NWs. The bright stripe regions are assigned to Ag2S through EDS chemical mapping, while the darker regions are assigned to Cu2S. (A–C) Reproduced from [Justo, Y.; Goris, B.; Kamal, J. S.; Geiregat, P.; Bals, S.; Hens, Z. Multiple Dot-in-Rod PbS/CdS Heterostructures with High Photoluminescence Quantum Yield in the Near-Infrared J. Am. Chem. Soc. 2012, 134, 5484–5487]. Copyright 2012 American Chemical Society. (D–G) Reproduced from [Tan, C.-S.; Hsiao, C.-H.; Wang, S.-C.; Liu, P.-H.; Lu, M.-Y.; Huang, M. H.; Ouyang, H.; Chen, L.-J. Sequential Cation Exchange Generated Superlattice Nanowires Forming Multiple p–n Heterojunctions ACS Nano2014, 8, 9422–9426]. Copyright 2014 American Chemical Society.
Tan et al. prepared Cu2S NWs bearing multiple twins along the wire and exposed them to Ag+ cations, which led to the formation of p-n Cu2S–Ag2S striped heterojunctions (see Figure 28D–G).162 This process was proven not to be driven by any interfacial energy or strain minimization, as in the case of the CdS-Ag2S system. Conversely, it was argued that the twin boundaries in the Cu2S NWs acted as preferred sites for the nucleation of Ag2S segments. Notably, the orientation of the twin planes was shown to have a fundamental role in driving the formation of regular striped Cu2S–Ag2S NHCs. Indeed, while regular Ag2S segments could be grown starting from fcc Cu2S NWs with twin planes perpendicular to the growth direction (see Figure 28E,F), irregular patterning was observed when starting from monoclinic Cu2S NWs having twin planes parallel to the growth direction (see Figure 28G). The presence of stacking faults, on the other hand, induced the degradation of CdSe NRs when exposed to Pb2+ ions, as observed by Lee et al.121 These defects were believed to provide high energy sites (such as corners, edges, or kinks) on side-walls of CdSe NRs which became more prone to CE. Once the exchange started from those sites, multiple unstable wz-CdSe/rs-PbSe were formed that, ultimately, caused the dismantling of the original NRs.
5.4. Engineering Heterostructures through Sequential CE
As shown in the previous sections, partial CE can lead to many different NHCs with specific morphologies. These NHCs, in turn, can be further used in CE reactions in which only one of the two components is converted. Thus, the spatial arrangement of the components of the NHC can be controlled via the first exchange reaction, while the final composition is determined by the second one. By a proper choice of starting NCs seeds and type of CE reaction, it is now possible to finely engineer complex NHCs that are difficult or currently impossible to synthesize through a classical seeded growth approach.
Luther et al., for example, adopted this strategy to prepare CdS-PbS NHCs with different topologies.117 Partial replacement of Cu-for-Cd ions in CdS NRs was used to synthesize Cu2S-CdS segmented NHCs in which the Cu2S region is located at one or both ends of the NRs (see section 5.3.1.1). These structures, in a second step, were exposed to Pb2+ ions to yield PbS-CdS NRs in which one or both tips are made of PbS (see Figure 29A). Starting with the same parent CdS NRs, completely different NHCs can be achieved instead by using Ag+ ions in the first partial CE step. In this case, the first the partial CE leads to the formation of small Ag2S regions distributed along the walls of the CdS NRs and, under careful conditions, to striped NHCs (see Figure 29B and section 5.3.1.2). Then the selective replacement of Ag+ with Pb2+ ions leads to the corresponding PbS-CdS NHCs in which PbS is embedded in CdS (see Figure 29C,D). The same strategy has been proven to work on wz-CdS NWs to create 1D CdS/PbS heterostructured NWs.72
Figure 29.

NHCs with different topologies in chemical composition prepared from wz-CdS NRs. (A) HRTEM image showing epitaxy between rock salt PbS and wurtzite CdS in a segmented CdS/PbS NHC produced using Cu+ ions in the first partial CE step. TEM images of striped CdS/Ag2S NHCs (B) that, after exposure to Pb2+ ions, are transformed into CdS/PbS NHCs (C). The contrast between Pb and Cd containing regions is less obvious than the contrast between Ag and Cd. (D) HRTEM and FFT images of various segments of striped CdS/PbS NHC clearly demonstrate that PbS sections exist throughout the CdS NRs. The lower FFT image evidence a rock salt structure, while the FFT image of the upper side of the rod is compatible with a wurtzite structure. It is clear how the distribution of the CdS and PbS domains depends on the first partial CE step. Reproduced from [Luther, J. M.; Zheng, H.; Sadtler, B.; Alivisatos, a. P. Synthesis of PbS nanorods and other ionic nanocrystals of complex morphology by sequential cation exchange reactions J. Am. Chem. Soc. 2009, 131, 16851–16857]. Copyright 2009 American Chemical Society. .
Adel et al. synthesized CdSe|CdS/ZnS NRs built in a segment like manner, that is, a CdSe core which is embedded in a CdS rod, which then epitaxially ends with ZnS tips (see Figure 30).101 This elaborate NHC is realized by exploiting a sequential CE procedure in which first CdSe|CdS core|shell NRs are exposed to Cu+ ions to get CdSe|CdS/Cu2-xS NHCs. In a second step, the Cu2-xS domains are converted into ZnS. A simple variation of the relative concentration of Cu+ ions in the first CE step allows for a fine control over the position of the final boundary between CdS and ZnS domains. On the other hand, it is important to underline that a direct Zn-for-Cd CE reaction would have led to NHCs with a completely different topology, most likely to CdSe|Cd1–xZnxS core|graded-shell NRs, as suggested by other works.176,177
Figure 30.
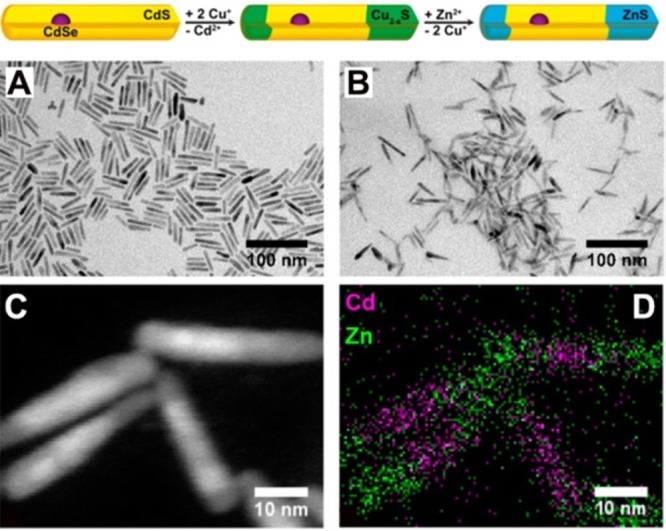
Schematic representation of the sequential CE reactions through which CdSe|CdS NRs are transformed first into CdSe|CdS/Cu2-xS and then into CdSe|CdS/ZnS segment-like NHCs. The TEM images of the original CdSe|CdS (A) and the CdSe|CdS/ZnS (B) samples show that neither the length nor the diameter of the NRs have changed significantly during the sequential ion exchange steps. (C) STEM-HAADF image of several NRs from a sample which has undergone a 50% ion exchange, along with the corresponding EDX mapping (D) showing the spatial distribution of Zn and Cd. The distinct CdS and ZnS sections can be clearly detected. Reproduced from [Adel, P.; Wolf, A.; Kodanek, T.; Dorfs, D. Segmented CdSe@CdS/ZnS Nanorods Synthesized via a Partial Ion Exchange Sequence Chem. Mater. 2014, 26, 3121–3127]. Copyright 2014 American Chemical Society.
As discussed in section 3.4, Zhang et al. demonstrated that CE reactions can be exploited to prepare NHCs consisting of a metal core and a monocrystalline semiconductor shell with substantial lattice mismatches between them, which cannot be obtained by conventional epitaxial techniques.106 Their strategy relied on exposing preformed metallic spherical NCs (such as Au, FePt and Pt) covered with an amorphous Ag2X (X=S, Se and Te) shell to M2+ (M = Cd, Pb, Zn) ions in the presence of TBP. As a result, a monocrystalline shell forms around the metallic core, even if the lattice mismatch between the core and the shell materials can be as high as 40%, as in the case of Au|CdS NHCs. In conventional epitaxial techniques it is well-known that some energy is required to accommodate for an epitaxial layer of lattice-mismatched material, since a certain amount of elastic strain accumulates during its growth. This energy, which depends on both the shell thickness and the extent of lattice mismatch, can be accommodated by the shell material, without the formation of defects, only up to a certain thickness (typically referred to as “critical layer thickness”). Consequently, in conventional epitaxial synthesis the shell material can be grown up to a limited thickness, otherwise strain-induced defects can form, negatively affecting the properties of the resulting heterostructure. Surprisingly, in the procedure reported by Zhang et al. a total absence of a critical layer thickness was observed. The same authors demonstrated that this strategy, if coupled with sequential partial CE reactions, can be used to prepare even more complex NHCs with precise structural and compositional tailoring. For example, as illustrated in Figure 31, Au|(CdS+PbS) core|shell NHCs can be realized by converting the first section of the amorphous Ag2S shell into monocrystalline CdS, followed by the sequential Pb-for-Ag CE that leads to the growth of PbS (see Figure 31).
Figure 31.
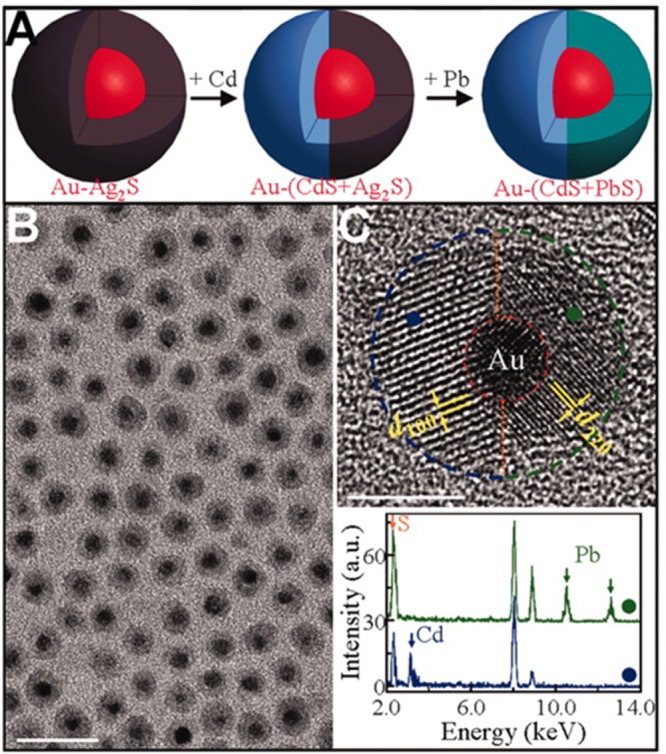
Growth of complex hybrid core|shell NHCs with tailored structures and compositions of the monocrystalline shells. (A–C) Control of the monocrystalline cation species within the shell: the case of Au|(CdS+PbS). (A) Sketch of the growth procedure. (B) Low-resolution TEM image. Scale bar, 20 nm. (C) (top) High-resolution TEM image. Blue and green dashed arc curves highlight the monocrystalline CdS and PbS domains, respectively. CdS and PbS manifest distinct lattice planes that can be assigned to (100) and (220), respectively. Scale bar, 5 nm. (bottom) Single-particle EDS measurements in the CdS and PbS domains. Peaks from Cd, Pb, and S elements are highlighted. Adapted with permission from ref (106). Copyright 2010 The American Association for the Advancement of Science.
6. Conclusions and Perspectives
Research on CE reactions in NCs has gone through astonishing developments in recent years, both in understanding the mechanisms and forces involved in these types of reactions and in the complexity in the topology of composition and structure that can be achieved, as was highlighted in this review. With the progress in experimental techniques, for example in high resolution electron microscopy, X-ray photoelectron spectroscopy (using a monochromatic synchrotron beam), X-ray and neutron scattering experiments,38,185 but also in computational tools, more in depth characterization and modeling of the process will be possible in the near future. Clearly, there are many questions that remain unanswered or poorly addressed, and they are briefly outlined below.
For example, the role of surface ligands during CE is relatively unexplored. They obviously need to attach and detach dynamically in order to allow for the inflow and outflow of cations, and in many cases there are strong indications that some of the ligands are lost during the process, as the NCs can aggregate. To what extent, for example, they promote or hinder CE? Some ligands coating in the form of polymer shell have been found to block CE altogether.201 The degradation of the shell by an ionizing radiation such that of an electron beam or an X-ray beam can cause degradation of the ligands to a point that can block CE and even anion exchange on a film of NCs deposited on a substrate, as recently shown by us.167,ref202 Then, a natural development in this direction would be the use of various libraries of photopolymerizable or photodegradable ligands able to block CE on specific regions/domains of a NC, in order to create segmented or striped heterostructures. Also, as discussed in this review, there is a wide range of ligands that need to be studied in CE reactions (for example multidentate ligands) in order to improve the solvation of exiting cations.120,132−134 This, in turn, could not only expand the range of accessible materials, but also optimize the actual synthetic CE conditions, allowing for a better control over the purity of resulting NCs.
Another major concern in reactions involving CE, somehow connected with the previous discussion, is that lattice defects can form upon exchange, and in addition is in not always clear whether such reactions truly reach completion, or if a fraction of the initial cations is still present in the lattice, compromising the optical/electronic properties of the final NCs. Recent works have started addressing these issues in fluorescent semiconductor NCs.80,93 Then the logical question is to what extent CE can be truly regarded as an alternative/complementary tool to the direct synthesis of a desired nanostructure, if the exchange can be plagued by the formation of defects and the persistence of residual cations from the parent NC? Are there ways of performing CE such that these defects are eliminated? Addressing these points will require extensive research in the near future.
The capability of CE in accessing NCs in metastable phases, which cannot be prepared otherwise, has already been exploited to a certain extent. However, reports on new metastable materials are continuously reported,202 and we believe that many unexplored materials, perhaps with novel and/or exotic properties, are yet to be synthesized, and is likely that some of them will only be accessible via CE. Another exciting research direction could be the study of “dry” solid state CE transformations in NHCs. Some systems, like Cu3P/CdSe NHCs, have been show to undergo CE if annealed under vacuum, by transforming to Cu2Se NCs through concomitant sublimation of Cd and P.203 Also, in principle, “sacrificial” NCs can be used as a source of cations to be exchanged with cations of desired host NCs. The possibility of triggering such CE reaction under specific external inputs, such as heat or e-beam irradiation, might represent an alternative route in those cases in which the host NCs are prone to degradation by chemical agents while being processed in solution.196
Acknowledgments
We acknowledge the 7th European Community Framework Programme under Grant Agreement No. 614897 (ERC Consolidator Grant “TRANS-NANO”).
Biographies
Liberato Manna received his Ph.D. in Chemistry in 2001 from the University of Bari (Italy) and worked at UC Berkeley (USA) as a Visiting Student and subsequently at the Lawrence Berkeley Lab (USA) as a Postdoc until 2003. He was then Scientist at the National Nanotechnology Lab in Lecce (Italy) and he moved to the Istituto Italiano di Tecnologia (IIT), Genova (Italy) in 2009 as Director of the Nanochemistry Department. Since 2010, he has also been part-time Professor at TU Delft (The Netherlands). Currently, he is also Deputy Director of IIT for the Materials and Nanotechnology programs. His research interests include the synthesis and assembly of colloidal nanocrystals, the study of structural, chemical and surface transformations in nanoscale materials, and their applications in energy, photonics, electronics, and biology.
Luca De Trizio graduated in Materials Science in 2008 from the University of Milano Bicocca and obtained his Ph.D. in Nanostructures and Nanotechnology from the same institution in 2013. During his Ph.D., in 2010-2011, he worked as collaborator at the Italian Institute of Technology in Genova with Liberato Manna and, in 2012, at the Molecular Foundry in the Lawrence Berkeley National Laboratory with Delia Milliron. From 2013 to now, he has been working as a postdoc researcher at the Nanochemistry Department in the Italian Institute of Technology (IIT) in Genova (Italy). His research interests include the synthesis of transparent conductive oxides, luminescent semiconductors, and the study of postsynthetic chemical transformations of nanocrystals.
The authors declare no competing financial interest.
References
- Yin Y.; Alivisatos A. P. Colloidal nanocrystal synthesis and the organic-inorganic interface. Nature 2005, 437, 664–670. 10.1038/nature04165. [DOI] [PubMed] [Google Scholar]
- Nurmikko A. What future for quantum dot-based light emitters?. Nat. Nanotechnol. 2015, 10, 1001–1004. 10.1038/nnano.2015.288. [DOI] [PubMed] [Google Scholar]
- Clifford J. P.; Konstantatos G.; Johnston K. W.; Hoogland S.; Levina L.; Sargent E. H. Fast, sensitive and spectrally tuneable colloidal-quantum-dot photodetectors. Nat. Nanotechnol. 2009, 4, 40–44. 10.1038/nnano.2008.313. [DOI] [PubMed] [Google Scholar]
- Alivisatos A. P.; Gu W. W.; Larabell C.. Annual Review of Biomedical Engineering; Annual Reviews: Palo Alto, 2005; Vol. 7. [DOI] [PubMed] [Google Scholar]
- Li Z.-B.; Cai W.; Chen X. Semiconductor Quantum Dots for In Vivo Imaging. J. Nanosci. Nanotechnol. 2007, 7, 2567–2581. 10.1166/jnn.2007.628. [DOI] [PubMed] [Google Scholar]
- Jain P.; Huang X.; El-Sayed I.; El-Sayed M. Review of Some Interesting Surface Plasmon Resonance-enhanced Properties of Noble Metal Nanoparticles and Their Applications to Biosystems. Plasmonics 2007, 2, 107–118. 10.1007/s11468-007-9031-1. [DOI] [Google Scholar]
- Klimov V. I.; Mikhailovsky A. A.; Xu S.; Malko A.; Hollingsworth J. A.; Leatherdale C. A.; Eisler H.-J.; Bawendi M. G. Optical Gain and Stimulated Emission in Nanocrystal Quantum Dots. Science 2000, 290, 314–317. 10.1126/science.290.5490.314. [DOI] [PubMed] [Google Scholar]
- Bruchez M.; Moronne M.; Gin P.; Weiss S.; Alivisatos A. P. Semiconductor Nanocrystals as Fluorescent Biological Labels. Science 1998, 281, 2013–2016. 10.1126/science.281.5385.2013. [DOI] [PubMed] [Google Scholar]
- Brus L. Electronic wave functions in semiconductor clusters: experiment and theory. J. Phys. Chem. 1986, 90, 2555–2560. 10.1021/j100403a003. [DOI] [Google Scholar]
- Talapin D. V.; Lee J.-S.; Kovalenko M. V.; Shevchenko E. V. Prospects of Colloidal Nanocrystals for Electronic and Optoelectronic Applications. Chem. Rev. 2010, 110, 389–458. 10.1021/cr900137k. [DOI] [PubMed] [Google Scholar]
- Efros A. L.; Efros A. Interband absorption of light in semiconductor sphere. Semiconductors 1982, 16, 772–775. [Google Scholar]
- Hergt R.; Dutz S.; Müller R.; Zeisberger M. Magnetic particle hyperthermia: nanoparticle magnetism and materials development for cancer therapy. J. Phys.: Condens. Matter 2006, 18, S2919–S2934. 10.1088/0953-8984/18/38/S26. [DOI] [Google Scholar]
- Kovalenko M. V. Opportunities and challenges for quantum dot photovoltaics. Nat. Nanotechnol. 2015, 10, 994–997. 10.1038/nnano.2015.284. [DOI] [PubMed] [Google Scholar]
- Tian N.; Zhou Z.-Y.; Sun S.-G.; Ding Y.; Wang Z. L. Synthesis of Tetrahexahedral Platinum Nanocrystals with High-Index Facets and High Electro-Oxidation Activity. Science 2007, 316, 732–735. 10.1126/science.1140484. [DOI] [PubMed] [Google Scholar]
- Pang X.; Zhao L.; Han W.; Xin X.; Lin Z. A general and robust strategy for the synthesis of nearly monodisperse colloidal nanocrystals. Nat. Nanotechnol. 2013, 8, 426–431. 10.1038/nnano.2013.85. [DOI] [PubMed] [Google Scholar]
- Milliron D. J.; Hughes S. M.; Cui Y.; Manna L.; Li J.; Wang L.-W.; Paul Alivisatos A. Colloidal nanocrystal heterostructures with linear and branched topology. Nature 2004, 430, 190–195. 10.1038/nature02695. [DOI] [PubMed] [Google Scholar]
- Oh N.; Nam S.; Zhai Y.; Deshpande K.; Trefonas P.; Shim M. Double-heterojunction nanorods. Nat. Commun. 2014, 5, 3642. 10.1038/ncomms4642. [DOI] [PubMed] [Google Scholar]
- Kovalenko M. V.; Heiss W.; Shevchenko E. V.; Lee J.-S.; Schwinghammer H.; Alivisatos A. P.; Talapin D. V. SnTe Nanocrystals: A New Example of Narrow-Gap Semiconductor Quantum Dots. J. Am. Chem. Soc. 2007, 129, 11354–11355. 10.1021/ja074481z. [DOI] [PubMed] [Google Scholar]
- Mauser C.; Limmer T.; Da Como E.; Becker K.; Rogach A. L.; Feldmann J.; Talapin D. V. Anisotropic optical emission of single CdSe/CdS tetrapod heterostructures: Evidence for a wavefunction symmetry breaking. Phys. Rev. B: Condens. Matter Mater. Phys. 2008, 77, 153303. 10.1103/PhysRevB.77.153303. [DOI] [Google Scholar]
- Müller J.; Lupton J. M.; Lagoudakis P. G.; Schindler F.; Koeppe R.; Rogach A. L.; Feldmann J.; Talapin D. V.; Weller H. Wave Function Engineering in Elongated Semiconductor Nanocrystals with Heterogeneous Carrier Confinement. Nano Lett. 2005, 5, 2044–2049. 10.1021/nl051596x. [DOI] [PubMed] [Google Scholar]
- Talapin D. V.; Koeppe R.; Götzinger S.; Kornowski A.; Lupton J. M.; Rogach A. L.; Benson O.; Feldmann J.; Weller H. Highly Emissive Colloidal CdSe/CdS Heterostructures of Mixed Dimensionality. Nano Lett. 2003, 3, 1677–1681. 10.1021/nl034815s. [DOI] [Google Scholar]
- Scotognella F.; Miszta K.; Dorfs D.; Zavelani-Rossi M.; Brescia R.; Marras S.; Manna L.; Lanzani G.; Tassone F. Ultrafast Exciton Dynamics in Colloidal CdSe/CdS Octapod Shaped Nanocrystals. J. Phys. Chem. C 2011, 115, 9005–9011. 10.1021/jp203000n. [DOI] [Google Scholar]
- Pal B. N.; Ghosh Y.; Brovelli S.; Laocharoensuk R.; Klimov V. I.; Hollingsworth J. A.; Htoon H. ‘Giant’ CdSe/CdS Core/Shell Nanocrystal Quantum Dots As Efficient Electroluminescent Materials: Strong Influence of Shell Thickness on Light-Emitting Diode Performance. Nano Lett. 2012, 12, 331–336. 10.1021/nl203620f. [DOI] [PubMed] [Google Scholar]
- Ivanov S. A.; Nanda J.; Piryatinski A.; Achermann M.; Balet L. P.; Bezel I. V.; Anikeeva P. O.; Tretiak S.; Klimov V. I. Light Amplification Using Inverted Core/Shell Nanocrystals: Towards Lasing in the Single-Exciton Regime. J. Phys. Chem. B 2004, 108, 10625–10630. 10.1021/jp0483371. [DOI] [Google Scholar]
- Kudera S.; Carbone L.; Casula M. F.; Cingolani R.; Falqui A.; Snoeck E.; Parak W. J.; Manna L. Selective Growth of PbSe on One or Both Tips of Colloidal Semiconductor Nanorods. Nano Lett. 2005, 5, 445–449. 10.1021/nl048060g. [DOI] [PubMed] [Google Scholar]
- Costi R.; Saunders A. E.; Banin U. Colloidal Hybrid Nanostructures: A New Type of Functional Materials. Angew. Chem., Int. Ed. 2010, 49, 4878–4897. 10.1002/anie.200906010. [DOI] [PubMed] [Google Scholar]
- Vaneski A.; Susha A. S.; Rodríguez-Fernández J.; Berr M.; Jäckel F.; Feldmann J.; Rogach A. L. Hybrid Colloidal Heterostructures of Anisotropic Semiconductor Nanocrystals Decorated with Noble Metals: Synthesis and Function. Adv. Funct. Mater. 2011, 21, 1547–1556. 10.1002/adfm.201002444. [DOI] [Google Scholar]
- Son D. H.; Hughes S. M.; Yin Y.; Alivisatos A. P. Cation Exchange Reactions in Ionic Nanocrystals. Science 2004, 306, 1009–1012. 10.1126/science.1103755. [DOI] [PubMed] [Google Scholar]
- Jeong U.; Camargo P. H. C.; Lee Y. H.; Xia Y. Chemical transformation: a powerful route to metal chalcogenide nanowires. J. Mater. Chem. 2006, 16, 3893–3897. 10.1039/b606682h. [DOI] [Google Scholar]
- Camargo P. H. C.; Lee Y. H.; Jeong U.; Zou Z.; Xia Y. Cation Exchange: A Simple and Versatile Route to Inorganic Colloidal Spheres with the Same Size but Different Compositions and Properties. Langmuir 2007, 23, 2985–2992. 10.1021/la0632070. [DOI] [PubMed] [Google Scholar]
- Moon G. D.; Ko S.; Xia Y.; Jeong U. Chemical Transformations in Ultrathin Chalcogenide Nanowires. ACS Nano 2010, 4, 2307–2319. 10.1021/nn9018575. [DOI] [PubMed] [Google Scholar]
- Moon G. D.; Ko S.; Min Y.; Zeng J.; Xia Y.; Jeong U. Chemical transformations of nanostructured materials. Nano Today 2011, 6, 186–203. 10.1016/j.nantod.2011.02.006. [DOI] [Google Scholar]
- Rivest J. B.; Jain P. K. Cation exchange on the nanoscale: an emerging technique for new material synthesis, device fabrication, and chemical sensing. Chem. Soc. Rev. 2013, 42, 89–96. 10.1039/C2CS35241A. [DOI] [PubMed] [Google Scholar]
- Fayette M.; Robinson R. D. Chemical transformations of nanomaterials for energy applications. J. Mater. Chem. A 2014, 2, 5965–5965. 10.1039/C3TA13982D. [DOI] [Google Scholar]
- Gupta S.; Kershaw S. V.; Rogach A. L. 25th Anniversary Article: Ion Exchange in Colloidal Nanocrystals. Adv. Mater. 2013, 25, 6923–6944. 10.1002/adma.201302400. [DOI] [PubMed] [Google Scholar]
- Mews A.; Eychmüller A.; Giersig M.; Schooss D.; Weller H. Preparation, characterization, and photophysics of the quantum dot quantum well system CdS/HgS/CdS. J. Phys. Chem. 1994, 98, 934–941. 10.1021/j100054a032. [DOI] [Google Scholar]
- Zhou H. S.; Sasahara H.; Honma I.; Komiyama H.; Haus J. W. Coated Semiconductor Nanoparticles: The CdS/PbS System’s Photoluminescence Properties. Chem. Mater. 1994, 6, 1534–1541. 10.1021/cm00045a010. [DOI] [Google Scholar]
- Lechner R. T.; Fritz-Popovski G.; Yarema M.; Heiss W.; Hoell A.; Schülli T. U.; Primetzhofer D.; Eibelhuber M.; Paris O. Crystal Phase Transitions in the Shell of PbS/CdS Core/Shell Nanocrystals Influences Photoluminescence Intensity. Chem. Mater. 2014, 26, 5914–5922. 10.1021/cm502521q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zherebetskyy D.; Zhang Y.; Salmeron M.; Wang L.-W. Tolerance of Intrinsic Defects in PbS Quantum Dots. J. Phys. Chem. Lett. 2015, 6, 4711–4716. 10.1021/acs.jpclett.5b02202. [DOI] [PubMed] [Google Scholar]
- Justo Y.; Sagar L. K.; Flamee S.; Zhao Q.; Vantomme A.; Hens Z. Less Is More. Cation Exchange and the Chemistry of the Nanocrystal Surface. ACS Nano 2014, 8, 7948–7957. 10.1021/nn5037812. [DOI] [PubMed] [Google Scholar]
- Casavola M.; van Huis M. A.; Bals S.; Lambert K.; Hens Z.; Vanmaekelbergh D. Anisotropic Cation Exchange in PbSe/CdSe Core/Shell Nanocrystals of Different Geometry. Chem. Mater. 2012, 24, 294–302. 10.1021/cm202796s. [DOI] [Google Scholar]
- Groeneveld E.; Witteman L.; Lefferts M.; Ke X.; Bals S.; Van Tendeloo G.; de Mello Donega C. Tailoring ZnSe–CdSe Colloidal Quantum Dots via Cation Exchange: From Core/Shell to Alloy Nanocrystals. ACS Nano 2013, 7, 7913–7930. 10.1021/nn402931y. [DOI] [PubMed] [Google Scholar]
- Routzahn A. L.; Jain P. K. Single-Nanocrystal Reaction Trajectories Reveal Sharp Cooperative Transitions. Nano Lett. 2014, 14, 987–992. 10.1021/nl4044289. [DOI] [PubMed] [Google Scholar]
- Cao H.; Qian X.; Wang C.; Ma X.; Yin J.; Zhu Z. High Symmetric 18-Facet Polyhedron Nanocrystals of Cu7S4 with a Hollow Nanocage. J. Am. Chem. Soc. 2005, 127, 16024–16025. 10.1021/ja055265y. [DOI] [PubMed] [Google Scholar]
- Geng J.; Liu B.; Xu L.; Hu F.-N.; Zhu J.-J. Facile Route to Zn-Based II–VI Semiconductor Spheres, Hollow Spheres, and Core/Shell Nanocrystals and Their Optical Properties. Langmuir 2007, 23, 10286–10293. 10.1021/la701299w. [DOI] [PubMed] [Google Scholar]
- Saruyama M.; So Y.-G.; Kimoto K.; Taguchi S.; Kanemitsu Y.; Teranishi T. Spontaneous Formation of Wurzite-CdS/Zinc Blende-CdTe Heterodimers through a Partial Anion Exchange Reaction. J. Am. Chem. Soc. 2011, 133, 17598–17601. 10.1021/ja2078224. [DOI] [PubMed] [Google Scholar]
- Park J.; Zheng H.; Jun Y.-w.; Alivisatos A. P. Hetero-Epitaxial Anion Exchange Yields Single-Crystalline Hollow Nanoparticles. J. Am. Chem. Soc. 2009, 131, 13943–13945. 10.1021/ja905732q. [DOI] [PubMed] [Google Scholar]
- Zhang F.; Shi Y.; Sun X.; Zhao D.; Stucky G. D. Formation of Hollow Upconversion Rare-Earth Fluoride Nanospheres: Nanoscale Kirkendall Effect During Ion Exchange. Chem. Mater. 2009, 21, 5237–5243. 10.1021/cm902231s. [DOI] [Google Scholar]
- Dloczik L.; Könenkamp R. Nanostructure Transfer in Semiconductors by Ion Exchange. Nano Lett. 2003, 3, 651–653. 10.1021/nl0340879. [DOI] [Google Scholar]
- Dawood F.; Schaak R. E. ZnO-Templated Synthesis of Wurtzite-Type ZnS and ZnSe Nanoparticles. J. Am. Chem. Soc. 2009, 131, 424–425. 10.1021/ja808455u. [DOI] [PubMed] [Google Scholar]
- Wang X.; Zhan S.; Wang Y.; Wang P.; Yu H.; Yu J.; Hu C. Facile synthesis and enhanced visible-light photocatalytic activity of Ag2S nanocrystal-sensitized Ag8W4O16 nanorods. J. Colloid Interface Sci. 2014, 422, 30–37. 10.1016/j.jcis.2014.02.009. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Gao L. Synthesis of Nanocrystalline Aluminum Nitride by Nitridation of δ-Al2O3 Nanoparticles in Flowing Ammonia. J. Am. Ceram. Soc. 2006, 89, 415–421. 10.1111/j.1551-2916.2005.00715.x. [DOI] [Google Scholar]
- Mizusaki J.; Arai K.; Fueki K. Ionic conduction of the perovskite-type halides. Solid State Ionics 1983, 11, 203–211. 10.1016/0167-2738(83)90025-5. [DOI] [Google Scholar]
- Tress W.; Marinova N.; Moehl T.; Zakeeruddin S. M.; Nazeeruddin M. K.; Gratzel M. Understanding the rate-dependent J-V hysteresis, slow time component, and aging in CH3NH3PbI3 perovskite solar cells: the role of a compensated electric field. Energy Environ. Sci. 2015, 8, 995–1004. 10.1039/C4EE03664F. [DOI] [Google Scholar]
- Stranks S. D.; Snaith H. J. Metal-halide perovskites for photovoltaic and light-emitting devices. Nat. Nanotechnol. 2015, 10, 391–402. 10.1038/nnano.2015.90. [DOI] [PubMed] [Google Scholar]
- Akkerman Q. A.; D’Innocenzo V.; Accornero S.; Scarpellini A.; Petrozza A.; Prato M.; Manna L. Tuning the Optical Properties of Cesium Lead Halide Perovskite Nanocrystals by Anion Exchange Reactions. J. Am. Chem. Soc. 2015, 137, 10276–10281. 10.1021/jacs.5b05602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedelcu G.; Protesescu L.; Yakunin S.; Bodnarchuk M. I.; Grotevent M. J.; Kovalenko M. V. Fast Anion-Exchange in Highly Luminescent Nanocrystals of Cesium Lead Halide Perovskites (CsPbX3, X = Cl, Br, I). Nano Lett. 2015, 15, 5635–5640. 10.1021/acs.nanolett.5b02404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellet N.; Teuscher J.; Maier J.; Grätzel M. Transforming Hybrid Organic Inorganic Perovskites by Rapid Halide Exchange. Chem. Mater. 2015, 27, 2181–2188. 10.1021/acs.chemmater.5b00281. [DOI] [Google Scholar]
- Solis-Ibarra D.; Smith I. C.; Karunadasa H. I. Post-synthetic halide conversion and selective halogen capture in hybrid perovskites. Chem. Sci. 2015, 6, 4054–4059. 10.1039/C5SC01135C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y. R.Comprehensive Handbook of Chemical Bond Energies; CRC Press: Boca Raton, FL, 2007. [Google Scholar]
- Lide D. R.CRC Handbook of Chemistry and Physics, 84th ed.; Taylor & Francis: London, 2003. [Google Scholar]
- Mu L.; Feng C.; He H. Topological research on lattice energies for inorganic compounds. MATCH Commun. Math. Comput. Chem. 2006, 56, 97–111. [Google Scholar]
- Sheng Y.; Wei J.; Liu B.; Peng L. A facile route to synthesize CdZnSe core–shell-like alloyed quantum dots via cation exchange reaction in aqueous system. Mater. Res. Bull. 2014, 57, 67–71. 10.1016/j.materresbull.2014.05.033. [DOI] [Google Scholar]
- Xinhua Z.; Yaoyu F.; Yuliang Z.; Zhenyu G.; Lei Z. A facile route to violet- to orange-emitting CdxZn1–xSe alloy nanocrystals via cation exchange reaction. Nanotechnology 2007, 18, 385606. 10.1088/0957-4484/18/38/385606. [DOI] [Google Scholar]
- Miszta K.; Gariano G.; Brescia R.; Marras S.; De Donato F.; Ghosh S.; De Trizio L.; Manna L. Selective Cation Exchange in the Core Region of Cu2-xSe/Cu2-xS Core/Shell Nanocrystals. J. Am. Chem. Soc. 2015, 137, 12195–12198. 10.1021/jacs.5b06379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wark S. E.; Hsia C.-H.; Son D. H. Effects of Ion Solvation and Volume Change of Reaction on the Equilibrium and Morphology in Cation-Exchange Reaction of Nanocrystals. J. Am. Chem. Soc. 2008, 130, 9550–9555. 10.1021/ja802187c. [DOI] [PubMed] [Google Scholar]
- Parr R. G.; Pearson R. G. Absolute hardness: companion parameter to absolute electronegativity. J. Am. Chem. Soc. 1983, 105, 7512–7516. 10.1021/ja00364a005. [DOI] [Google Scholar]
- Pearson R. G. Absolute electronegativity and hardness: application to inorganic chemistry. Inorg. Chem. 1988, 27, 734–740. 10.1021/ic00277a030. [DOI] [Google Scholar]
- Lange N. A.; Dean J. A.. Lange’s Handbook of chemistry; McGraw-Hill: New York, 1979. [Google Scholar]
- Sadtler B.; Demchenko D. O.; Zheng H.; Hughes S. M.; Merkle M. G.; Dahmen U.; Wang L.-W.; Alivisatos A. P. Selective facet reactivity during cation exchange in cadmium sulfide nanorods. J. Am. Chem. Soc. 2009, 131, 5285–5293. 10.1021/ja809854q. [DOI] [PubMed] [Google Scholar]
- Robinson R. D.; Sadtler B.; Demchenko D. O.; Erdonmez C. K.; Wang L.-W.; Alivisatos A. P. Spontaneous superlattice formation in nanorods through partial cation exchange. Science 2007, 317, 355–358. 10.1126/science.1142593. [DOI] [PubMed] [Google Scholar]
- Mukherjee B.; Peterson A.; Subramanian V. 1D CdS/PbS heterostructured nanowire synthesis using cation exchange. Chem. Commun. 2012, 48, 2415–2417. 10.1039/c2cc16254g. [DOI] [PubMed] [Google Scholar]
- Miszta K.; Dorfs D.; Genovese A.; Kim M. R.; Manna L. Cation exchange reactions in colloidal branched nanocrystals. ACS Nano 2011, 5, 7176–7183. 10.1021/nn201988w. [DOI] [PubMed] [Google Scholar]
- Wong A. B.; Brittman S.; Yu Y.; Dasgupta N. P.; Yang P. Core–Shell CdS–Cu2S Nanorod Array Solar Cells. Nano Lett. 2015, 15, 4096–4101. 10.1021/acs.nanolett.5b01203. [DOI] [PubMed] [Google Scholar]
- Yao Q.; Arachchige I. U.; Brock S. L. Expanding the Repertoire of Chalcogenide Nanocrystal Networks: Ag2Se Gels and Aerogels by Cation Exchange Reactions. J. Am. Chem. Soc. 2009, 131, 2800–2801. 10.1021/ja900042y. [DOI] [PubMed] [Google Scholar]
- Li J.; Zhang T.; Ge J.; Yin Y.; Zhong W. Fluorescence Signal Amplification by Cation Exchange in Ionic Nanocrystals. Angew. Chem., Int. Ed. 2009, 48, 1588–1591. 10.1002/anie.200805710. [DOI] [PubMed] [Google Scholar]
- Lubeck C. R.; Han T. Y. J.; Gash A. E.; Satcher J. H.; Doyle F. M. Synthesis of Mesostructured Copper Sulfide by Cation Exchange and Liquid-Crystal Templating. Adv. Mater. 2006, 18, 781–784. 10.1002/adma.200501653. [DOI] [Google Scholar]
- Dorn A.; Allen P. M.; Harris D. K.; Bawendi M. G. In Situ Electrical Monitoring of Cation Exchange in Nanowires. Nano Lett. 2010, 10, 3948–3951. 10.1021/nl102560b. [DOI] [PubMed] [Google Scholar]
- Jain P. K.; Amirav L.; Aloni S.; Alivisatos A. P. Nanoheterostructure Cation Exchange: Anionic Framework Conservation. J. Am. Chem. Soc. 2010, 132, 9997–9999. 10.1021/ja104126u. [DOI] [PubMed] [Google Scholar]
- Jain P. K.; Beberwyck B. J.; Fong L.-K.; Polking M. J.; Alivisatos A. P. Highly Luminescent Nanocrystals From Removal of Impurity Atoms Residual From Ion-Exchange Synthesis. Angew. Chem., Int. Ed. 2012, 51, 2387–2390. 10.1002/anie.201107452. [DOI] [PubMed] [Google Scholar]
- Rivest J. B.; Buonsanti R.; Pick T. E.; Zhu L.; Lim E.; Clavero C.; Schaible E.; Helms B. A.; Milliron D. J. Evolution of Ordered Metal Chalcogenide Architectures through Chemical Transformations. J. Am. Chem. Soc. 2013, 135, 7446–7449. 10.1021/ja403071w. [DOI] [PubMed] [Google Scholar]
- Yao J.; Han X.; Zeng S.; Zhong W. Detection of Femtomolar Proteins by Nonfluorescent ZnS Nanocrystal Clusters. Anal. Chem. 2012, 84, 1645–1652. 10.1021/ac202910y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X.; Xue H.; Xu J.; Huang X.; Zhang J.; Tang Y. B.; Ng T. W.; Kwong H. L.; Meng X. M.; Lee C. S. Synthesis of porous ZnS:Ag2S nanosheets by ion exchange for photocatalytic H2 generation. ACS Appl. Mater. Interfaces 2014, 6, 9078–9084. 10.1021/am5020953. [DOI] [PubMed] [Google Scholar]
- Qu H.; Cao L.; Su G.; Liu W.; Gao R.; Xia C.; Qin J. Silica-coated ZnS quantum dots as fluorescent probes for the sensitive detection of Pb2+ ions. J. Nanopart. Res. 2014, 16, 1–12. 10.1007/s11051-014-2762-y. [DOI] [Google Scholar]
- Shi Y.; Chen J.; Shen P. ZnS micro-spheres and flowers: Chemically controlled synthesis and template use in fabricating MS(shell)/ZnS(core) and MS (M = Pb, Cu) hollow microspheres. J. Alloys Compd. 2007, 441, 337–343. 10.1016/j.jallcom.2006.09.104. [DOI] [Google Scholar]
- Qu Z.; Yan L.; Li L.; Xu J.; Liu M.; Li Z.; Yan N. Ultraeffective ZnS Nanocrystals Sorbent for Mercury(II) Removal Based on Size-Dependent Cation Exchange. ACS Appl. Mater. Interfaces 2014, 6, 18026–18032. 10.1021/am504896w. [DOI] [PubMed] [Google Scholar]
- Pang M.; Hu J.; Zeng H. C. Synthesis, Morphological Control, and Antibacterial Properties of Hollow/Solid Ag2S/Ag Heterodimers. J. Am. Chem. Soc. 2010, 132, 10771–10785. 10.1021/ja102105q. [DOI] [PubMed] [Google Scholar]
- Justo Y.; Goris B.; Kamal J. S.; Geiregat P.; Bals S.; Hens Z. Multiple Dot-in-Rod PbS/CdS Heterostructures with High Photoluminescence Quantum Yield in the Near-Infrared. J. Am. Chem. Soc. 2012, 134, 5484–5487. 10.1021/ja300337d. [DOI] [PubMed] [Google Scholar]
- Haibao S.; Chunlei W.; Shuhong X.; Yuan J.; Yujie S.; Fan B.; Zhuyuan W.; Yiping C. Hydrazine-promoted sequential cation exchange: a novel synthesis method for doped ternary semiconductor nanocrystals with tunable emission. Nanotechnology 2014, 25, 025603. 10.1088/0957-4484/25/2/025603. [DOI] [PubMed] [Google Scholar]
- Prudnikau A.; Artemyev M.; Molinari M.; Troyon M.; Sukhanova A.; Nabiev I.; Baranov A. V.; Cherevkov S. a.; Fedorov A. V. Chemical substitution of Cd ions by Hg in CdSe nanorods and nanodots: Spectroscopic and structural examination. Mater. Sci. Eng., B 2012, 177, 744–749. 10.1016/j.mseb.2011.12.038. [DOI] [Google Scholar]
- Yuho M.; Geon Dae M.; Jaeyoon P.; Minwoo P.; Unyong J. Surfactant-free CuInSe2 nanocrystals transformed from In2Se3 nanoparticles and their application for a flexible UV photodetector. Nanotechnology 2011, 22, 465604. 10.1088/0957-4484/22/46/465604. [DOI] [PubMed] [Google Scholar]
- Li H.; Zanella M.; Genovese A.; Povia M.; Falqui A.; Giannini C.; Manna L. Sequential Cation Exchange in Nanocrystals: Preservation of Crystal Phase and Formation of Metastable Phases. Nano Lett. 2011, 11, 4964–4970. 10.1021/nl202927a. [DOI] [PubMed] [Google Scholar]
- Li H.; Brescia R.; Krahne R.; Bertoni G.; Alcocer M. J. P.; D’Andrea C.; Scotognella F.; Tassone F.; Zanella M.; De Giorgi M.; et al. Blue-UV-Emitting ZnSe(Dot)/ZnS(Rod) Core/Shell Nanocrystals Prepared from CdSe/CdS Nanocrystals by Sequential Cation Exchange. ACS Nano 2012, 6, 1637–1647. 10.1021/nn204601n. [DOI] [PubMed] [Google Scholar]
- Gupta S.; Zhovtiuk O.; Vaneski A.; Lin Y.-C.; Chou W.-C.; Kershaw S. V.; Rogach A. L. CdxHg(1–x)Te Alloy Colloidal Quantum Dots: Tuning Optical Properties from the Visible to Near-Infrared by Ion Exchange. Part. Part. Syst. Charact. 2013, 30, 346–354. 10.1002/ppsc.201200139. [DOI] [Google Scholar]
- Cao Y.; Zhang A.; Ma Q.; Liu N.; Yang P. Application of hybrid SiO2-coated CdTe nanocrystals for sensitive sensing of Cu2+ and Ag+ ions. Luminescence 2013, 28, 287–293. 10.1002/bio.2379. [DOI] [PubMed] [Google Scholar]
- Smith A. M.; Nie S. Bright and Compact Alloyed Quantum Dots with Broadly Tunable Near-Infrared Absorption and Fluorescence Spectra through Mercury Cation Exchange. J. Am. Chem. Soc. 2011, 133, 24–26. 10.1021/ja108482a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriegel I.; Rodríguez-Fernández J.; Wisnet A.; Zhang H.; Waurisch C.; Eychmüller A.; Dubavik A.; Govorov A. O.; Feldmann J. Shedding Light on Vacancy-Doped Copper Chalcogenides: Shape-Controlled Synthesis, Optical Properties, and Modeling of Copper Telluride Nanocrystals with Near-Infrared Plasmon Resonances. ACS Nano 2013, 7, 4367–4377. 10.1021/nn400894d. [DOI] [PubMed] [Google Scholar]
- Jeong U.; Kim J.-U.; Xia Y.; Li Z.-Y. Monodispersed Spherical Colloids of Se@CdSe: Synthesis and Use as Building Blocks in Fabricating Photonic Crystals. Nano Lett. 2005, 5, 937–942. 10.1021/nl050482i. [DOI] [PubMed] [Google Scholar]
- Jeong U.; Xia Y.; Yin Y. Large-scale synthesis of single-crystal CdSe nanowires through a cation-exchange route. Chem. Phys. Lett. 2005, 416, 246–250. 10.1016/j.cplett.2005.09.106. [DOI] [Google Scholar]
- Acharya S.; Pradhan N. Insertion/Ejection of Dopant Ions in Composition Tunable Semiconductor Nanocrystals. J. Phys. Chem. C 2011, 115, 19513–19519. 10.1021/jp2052147. [DOI] [Google Scholar]
- Adel P.; Wolf A.; Kodanek T.; Dorfs D. Segmented CdSe@CdS/ZnS Nanorods Synthesized via a Partial Ion Exchange Sequence. Chem. Mater. 2014, 26, 3121–3127. 10.1021/cm500431m. [DOI] [Google Scholar]
- Akkerman Q. A.; Genovese A.; George C.; Prato M.; Moreels I.; Casu A.; Marras S.; Curcio A.; Scarpellini A.; Pellegrino T.; et al. From Binary Cu2S to Ternary Cu–In–S and Quaternary Cu–In–Zn–S Nanocrystals with Tunable Composition via Partial Cation Exchange. ACS Nano 2015, 9, 521–531. 10.1021/nn505786d. [DOI] [PubMed] [Google Scholar]
- van der Stam W.; Berends A. C.; Rabouw F. T.; Willhammar T.; Ke X.; Meeldijk J. D.; Bals S.; de Mello Donega C. Luminescent CuInS2 Quantum Dots by Partial Cation Exchange in Cu2–xS Nanocrystals. Chem. Mater. 2015, 27, 621–628. 10.1021/cm504340h. [DOI] [Google Scholar]
- De Trizio L.; Gaspari R.; Bertoni G.; Kriegel I.; Moretti L.; Scotognella F.; Maserati L.; Zhang Y.; Messina G. C.; Prato M.; et al. Cu3-xP Nanocrystals as a Material Platform for Near-Infrared Plasmonics and Cation Exchange Reactions. Chem. Mater. 2015, 27, 1120–1128. 10.1021/cm5044792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dilena E.; Dorfs D.; George C.; Miszta K.; Povia M.; Genovese A.; Casu A.; Prato M.; Manna L. Colloidal Cu2-x(SySe1-y) alloy nanocrystals with controllable crystal phase: synthesis, plasmonic properties, cation exchange and electrochemical lithiation. J. Mater. Chem. 2012, 22, 13023–13031. 10.1039/c2jm30788j. [DOI] [Google Scholar]
- Zhang J.; Tang Y.; Lee K.; Ouyang M. Nonepitaxial Growth of Hybrid Core-Shell Nanostructures with Large Lattice Mismatches. Science 2010, 327, 1634–1638. 10.1126/science.1184769. [DOI] [PubMed] [Google Scholar]
- Ha D.-H.; Caldwell A. H.; Ward M. J.; Honrao S.; Mathew K.; Hovden R.; Koker M. K. A.; Muller D. A.; Hennig R. G.; Robinson R. D. Solid–Solid Phase Transformations Induced through Cation Exchange and Strain in 2D Heterostructured Copper Sulfide Nanocrystals. Nano Lett. 2014, 14, 7090–7099. 10.1021/nl5035607. [DOI] [PubMed] [Google Scholar]
- Lesnyak V.; George C.; Genovese A.; Prato M.; Casu A.; Ayyappan S.; Scarpellini A.; Manna L. Alloyed Copper Chalcogenide Nanoplatelets via Partial Cation Exchange Reactions. ACS Nano 2014, 8, 8407–8418. 10.1021/nn502906z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Brescia R.; Povia M.; Prato M.; Bertoni G.; Manna L.; Moreels I. Synthesis of Uniform Disk-Shaped Copper Telluride Nanocrystals and Cation Exchange to Cadmium Telluride Quantum Disks with Stable Red Emission. J. Am. Chem. Soc. 2013, 135, 12270–12278. 10.1021/ja404694k. [DOI] [PubMed] [Google Scholar]
- Mu L.; Wang F.; Sadtler B.; Loomis R. A.; Buhro W. E. Influence of the Nanoscale Kirkendall Effect on the Morphology of Copper Indium Disulfide Nanoplatelets Synthesized by Ion Exchange. ACS Nano 2015, 9, 7419–7428. 10.1021/acsnano.5b02427. [DOI] [PubMed] [Google Scholar]
- Nguyen A. T.; Lin W.-H.; Lu Y.-H.; Chiou Y.-D.; Hsu Y.-J. First demonstration of rainbow photocatalysts using ternary Cd1-xZnxSe nanorods of varying compositions. Appl. Catal., A 2014, 476, 140–147. 10.1016/j.apcata.2014.02.023. [DOI] [Google Scholar]
- Lambright S.; Butaeva E.; Razgoniaeva N.; Hopkins T.; Smith B.; Perera D.; Corbin J.; Khon E.; Thomas R.; Moroz P.; et al. Enhanced Lifetime of Excitons in Nonepitaxial Au/CdS Core/Shell Nanocrystals. ACS Nano 2014, 8, 352–361. 10.1021/nn404264w. [DOI] [PubMed] [Google Scholar]
- De Trizio L.; Prato M.; Genovese A.; Casu A.; Povia M.; Simonutti R.; Alcocer M. J. P.; D’Andrea C.; Tassone F.; Manna L. Strongly Fluorescent Quaternary Cu–In–Zn–S Nanocrystals Prepared from Cu1-xInS2 Nanocrystals by Partial Cation Exchange. Chem. Mater. 2012, 24, 2400–2406. 10.1021/cm301211e. [DOI] [Google Scholar]
- Gui J.; Ji M.; Liu J.; Xu M.; Zhang J.; Zhu H. Phosphine-Initiated Cation Exchange for Precisely Tailoring Composition and Properties of Semiconductor Nanostructures: Old Concept, New Applications. Angew. Chem., Int. Ed. 2015, 54, 3683–3687. 10.1002/anie.201410053. [DOI] [PubMed] [Google Scholar]
- Sahu A.; Kang M. S.; Kompch A.; Notthoff C.; Wills A. W.; Deng D.; Winterer M.; Frisbie C. D.; Norris D. J. Electronic Impurity Doping in CdSe Nanocrystals. Nano Lett. 2012, 12, 2587–2594. 10.1021/nl300880g. [DOI] [PubMed] [Google Scholar]
- Kovalenko M. V.; Schaller R. D.; Jarzab D.; Loi M. A.; Talapin D. V. Inorganically Functionalized PbS–CdS Colloidal Nanocrystals: Integration into Amorphous Chalcogenide Glass and Luminescent Properties. J. Am. Chem. Soc. 2012, 134, 2457–2460. 10.1021/ja2087689. [DOI] [PubMed] [Google Scholar]
- Luther J. M.; Zheng H.; Sadtler B.; Alivisatos a. P. Synthesis of PbS nanorods and other ionic nanocrystals of complex morphology by sequential cation exchange reactions. J. Am. Chem. Soc. 2009, 131, 16851–16857. 10.1021/ja906503w. [DOI] [PubMed] [Google Scholar]
- Zhang J.; Chernomordik B. D.; Crisp R. W.; Kroupa D. M.; Luther J. M.; Miller E. M.; Gao J.; Beard M. C. Preparation of Cd/Pb Chalcogenide Heterostructured Janus Particles via Controllable Cation Exchange. ACS Nano 2015, 9, 7151–7163. 10.1021/acsnano.5b01859. [DOI] [PubMed] [Google Scholar]
- Tolman C. A. Steric effects of phosphorus ligands in organometallic chemistry and homogeneous catalysis. Chem. Rev. 1977, 77, 313–348. 10.1021/cr60307a002. [DOI] [Google Scholar]
- Crabtree R. H.The Organometallic Chemistry of the Transition Metals; Wiley: New York, 2014. [Google Scholar]
- Lee D.; Kim W. D.; Lee S.; Bae W. K.; Lee S.; Lee D. C. Direct Cd-to-Pb Exchange of CdSe Nanorods into PbSe/CdSe Axial Heterojunction Nanorods. Chem. Mater. 2015, 27, 5295–5304. 10.1021/acs.chemmater.5b01548. [DOI] [Google Scholar]
- Pietryga J. M.; Werder D. J.; Williams D. J.; Casson J. L.; Schaller R. D.; Klimov V. I.; Hollingsworth J. a. Utilizing the lability of lead selenide to produce heterostructured nanocrystals with bright, stable infrared emission. J. Am. Chem. Soc. 2008, 130, 4879–4885. 10.1021/ja710437r. [DOI] [PubMed] [Google Scholar]
- De Geyter B.; Justo Y.; Moreels I.; Lambert K.; Smet P. F.; Van Thourhout D.; Houtepen A. J.; Grodzinska D.; de Mello Donega C.; Meijerink A.; et al. The Different Nature of Band Edge Absorption and Emission in Colloidal PbSe/CdSe Core/Shell Quantum Dots. ACS Nano 2011, 5, 58–66. 10.1021/nn102980e. [DOI] [PubMed] [Google Scholar]
- Grodzińska D.; Evers W. H.; Dorland R.; van Rijssel J.; van Huis M. A.; Meijerink A.; de Mello Donegá C.; Vanmaekelbergh D. Two-Fold Emission From the S-Shell of PbSe/CdSe Core/Shell Quantum Dots. Small 2011, 7, 3493–3501. 10.1002/smll.201101819. [DOI] [PubMed] [Google Scholar]
- Grodzinska D.; Pietra F.; van Huis M. A.; Vanmaekelbergh D.; de Mello Donega C. Thermally induced atomic reconstruction of PbSe/CdSe core/shell quantum dots into PbSe/CdSe bi-hemisphere hetero-nanocrystals. J. Mater. Chem. 2011, 21, 11556–11565. 10.1039/c0jm04458j. [DOI] [Google Scholar]
- Lin Q.; Makarov N. S.; Koh W.-k.; Velizhanin K. A.; Cirloganu C. M.; Luo H.; Klimov V. I.; Pietryga J. M. Design and Synthesis of Heterostructured Quantum Dots with Dual Emission in the Visible and Infrared. ACS Nano 2015, 9, 539–547. 10.1021/nn505793y. [DOI] [PubMed] [Google Scholar]
- Lambert K.; Geyter B. D.; Moreels I.; Hens Z. PbTe|CdTe Core|Shell Particles by Cation Exchange, a HR-TEM study. Chem. Mater. 2009, 21, 778–780. 10.1021/cm8029399. [DOI] [Google Scholar]
- Eilers J.; Groeneveld E.; de Mello Donegá C.; Meijerink A. Optical Properties of Mn-Doped ZnTe Magic Size Nanocrystals. J. Phys. Chem. Lett. 2012, 3, 1663–1667. 10.1021/jz300300g. [DOI] [PubMed] [Google Scholar]
- Groeneveld E.; van Berkum S.; van Schooneveld M. M.; Gloter A.; Meeldijk J. D.; van den Heuvel D. J.; Gerritsen H. C.; de Mello Donega C. Highly Luminescent (Zn,Cd)Te/CdSe Colloidal Heteronanowires with Tunable Electron–Hole Overlap. Nano Lett. 2012, 12, 749–757. 10.1021/nl203695m. [DOI] [PubMed] [Google Scholar]
- Kim S.; Marshall A. R.; Kroupa D. M.; Miller E. M.; Luther J. M.; Jeong S.; Beard M. C. Air-Stable and Efficient PbSe Quantum-Dot Solar Cells Based upon ZnSe to PbSe Cation-Exchanged Quantum Dots. ACS Nano 2015, 9, 8157–8164. 10.1021/acsnano.5b02326. [DOI] [PubMed] [Google Scholar]
- Jeong S.; Han J. H.; Jang J.-t.; Seo J.-w.; Kim J.-G.; Cheon J. Transformative Two-Dimensional Layered Nanocrystals. J. Am. Chem. Soc. 2011, 133, 14500–14503. 10.1021/ja2049594. [DOI] [PubMed] [Google Scholar]
- Narayanan B.; Bhadbhade M. M. X-Ray Structure Of Thermochromic Bis(N,N-Diethylethylenediamine)-Copper(II)Tetrafluoroborate. J. Coord. Chem. 1998, 46, 115–123. 10.1080/00958979808047201. [DOI] [Google Scholar]
- Fabbrizzi L.; Micheloni M.; Paoletti P. Continuous and discontinuous thermochromism of copper(II) and nickel(II) complexes with N,N-diethylethylenediamine. Inorg. Chem. 1974, 13, 3019–3021. 10.1021/ic50142a049. [DOI] [Google Scholar]
- Grenthe I.; Paoletti P.; Sandstroem M.; Glikberg S. Thermochromism in copper(II) complexes. Structures of the red and blue-violet forms of bis(N,N-diethylethylenediamine)copper(II) perchlorate and the nonthermochromic violet bis(N-ethylethylenediamine)copper(II) perchlorate. Inorg. Chem. 1979, 18, 2687–2692. 10.1021/ic50200a010. [DOI] [Google Scholar]
- Thayer J.Organometallic Compounds and Living Organisms; Elsevier Science: Amsterdam, 2012. [Google Scholar]
- Sines I. T.; Schaak R. E. Phase-Selective Chemical Extraction of Selenium and Sulfur from Nanoscale Metal Chalcogenides: A General Strategy for Synthesis, Purification, and Phase Targeting. J. Am. Chem. Soc. 2011, 133, 1294–1297. 10.1021/ja110374d. [DOI] [PubMed] [Google Scholar]
- Lesnyak V.; Brescia R.; Messina G. C.; Manna L. Cu Vacancies Boost Cation Exchange Reactions in Copper Selenide Nanocrystals. J. Am. Chem. Soc. 2015, 137, 9315–9323. 10.1021/jacs.5b03868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ethayaraja M.; Bandyopadhyaya R. Model for Core–Shell Nanoparticle Formation by Ion-Exchange Mechanism. Ind. Eng. Chem. Res. 2008, 47, 5982–5985. 10.1021/ie0715106. [DOI] [Google Scholar]
- Mehrer H.Diffusion in Solids: Fundamentals, Methods, Materials, Diffusion-Controlled Processes; Springer: Berlin, 2007. [Google Scholar]
- Yalcin A. O.; Fan Z.; Goris B.; Li W.-F.; Koster R. S.; Fang C.-M.; van Blaaderen A.; Casavola M.; Tichelaar F. D.; Bals S.; et al. Atomic Resolution Monitoring of Cation Exchange in CdSe-PbSe Heteronanocrystals during Epitaxial Solid–Solid–Vapor Growth. Nano Lett. 2014, 14, 3661–3667. 10.1021/nl501441w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bals S.; Casavola M.; van Huis M. A.; Van Aert S.; Batenburg K. J.; Van Tendeloo G.; Vanmaekelbergh D. Three-Dimensional Atomic Imaging of Colloidal Core–Shell Nanocrystals. Nano Lett. 2011, 11, 3420–3424. 10.1021/nl201826e. [DOI] [PubMed] [Google Scholar]
- Dorfs D.; Härtling T.; Miszta K.; Bigall N. C.; Kim M. R.; Genovese A.; Falqui A.; Povia M.; Manna L. Reversible Tunability of the Near-Infrared Valence Band Plasmon Resonance in Cu2–xSe Nanocrystals. J. Am. Chem. Soc. 2011, 133, 11175–11180. 10.1021/ja2016284. [DOI] [PubMed] [Google Scholar]
- Luther J. M.; Jain P. K.; Ewers T.; Alivisatos A. P. Localized surface plasmon resonances arising from free carriers in doped quantum dots. Nat. Mater. 2011, 10, 361–366. 10.1038/nmat3004. [DOI] [PubMed] [Google Scholar]
- Beberwyck B. J.; Alivisatos A. P. Ion Exchange Synthesis of III–V Nanocrystals. J. Am. Chem. Soc. 2012, 134, 19977–19980. 10.1021/ja309416c. [DOI] [PubMed] [Google Scholar]
- Bothe C.; Kornowski A.; Tornatzky H.; Schmidtke C.; Lange H.; Maultzsch J.; Weller H. Solid-State Chemistry on the Nanoscale: Ion Transport through Interstitial Sites or Vacancies?. Angew. Chem., Int. Ed. 2015, 54, 14183–14186. 10.1002/anie.201507263. [DOI] [PubMed] [Google Scholar]
- White S. L.; Smith J. G.; Behl M.; Jain P. K. Co-operativity in a nanocrystalline solid-state transition. Nat. Commun. 2013, 4, 2933. 10.1038/ncomms3933. [DOI] [PubMed] [Google Scholar]
- Routzahn A. L.; Jain P. K. Luminescence Blinking of a Reacting Quantum Dot. Nano Lett. 2015, 15, 2504–2509. 10.1021/acs.nanolett.5b00068. [DOI] [PubMed] [Google Scholar]
- Ott F. D.; Spiegel L. L.; Norris D. J.; Erwin S. C. Microscopic Theory of Cation Exchange in CdSe Nanocrystals. Phys. Rev. Lett. 2014, 113, 156803. 10.1103/PhysRevLett.113.156803. [DOI] [PubMed] [Google Scholar]
- De Trizio L.; Li H.; Casu A.; Genovese A.; Sathya A.; Messina G. C.; Manna L. Sn Cation Valency Dependence in Cation Exchange Reactions Involving Cu2-xSe Nanocrystals. J. Am. Chem. Soc. 2014, 136, 16277–16284. 10.1021/ja508161c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S.; Kershaw S. V.; Susha A. S.; Wong T. L.; Higashimine K.; Maenosono S.; Rogach A. L. Near-Infrared-Emitting CdxHg1–xSe Nanorods Fabricated by Ion Exchange in an Aqueous Medium. ChemPhysChem 2013, 14, 2853–2858. 10.1002/cphc.201300084. [DOI] [PubMed] [Google Scholar]
- Xie Y.; Bertoni G.; Riedinger A.; Sathya A.; Prato M.; Marras S.; Tu R.; Pellegrino T.; Manna L. Nanoscale Transformations in Covellite (CuS) Nanocrystals in the Presence of Divalent Metal Cations in a Mild Reducing Environment. Chem. Mater. 2015, 27, 7531–7537. 10.1021/acs.chemmater.5b03892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batsanov S. S.; Batsanov A. S.. Introduction to Structural Chemistry; Springer: The Netherlands, 2012. [Google Scholar]
- Ning J.; Xiao G.; Wang L.; Zou B.; Liu B.; Zou G. Facile synthesis of magnetic metal (Mn, Fe, Co, and Ni) oxides nanocrystals via a cation-exchange reaction. Nanoscale 2011, 3, 741–745. 10.1039/C0NR00684J. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Zhukovskyi M.; Tongying P.; Tian Y.; Kuno M. Synthesis of Ultrathin and Thickness-Controlled Cu2–xSe Nanosheets via Cation Exchange. J. Phys. Chem. Lett. 2014, 5, 3608–3613. 10.1021/jz5019288. [DOI] [PubMed] [Google Scholar]
- Li X.; Chen K.; Huang L.; Lu D.; Liang J.; Han H. Sensitive immunoassay for porcine pseudorabies antibody based on fluorescence signal amplification induced by cation exchange in CdSe nanocrystals. Microchim. Acta 2013, 180, 303–310. 10.1007/s00604-012-0934-y. [DOI] [Google Scholar]
- Zhang J.; Gao J.; Church C. P.; Miller E. M.; Luther J. M.; Klimov V. I.; Beard M. C. PbSe Quantum Dot Solar Cells with More than 6% Efficiency Fabricated in Ambient Atmosphere. Nano Lett. 2014, 14, 6010–6015. 10.1021/nl503085v. [DOI] [PubMed] [Google Scholar]
- Li W.; Zamani R.; Ibáñez M.; Cadavid D.; Shavel A.; Morante J. R.; Arbiol J.; Cabot A. Metal Ions To Control the Morphology of Semiconductor Nanoparticles: Copper Selenide Nanocubes. J. Am. Chem. Soc. 2013, 135, 4664–4667. 10.1021/ja400472m. [DOI] [PubMed] [Google Scholar]
- Kovalenko M. V.; Talapin D. V.; Loi M. A.; Cordella F.; Hesser G.; Bodnarchuk M. I.; Heiss W. Quasi-Seeded Growth of Ligand-Tailored PbSe Nanocrystals through Cation-Exchange-Mediated Nucleation. Angew. Chem., Int. Ed. 2008, 47, 3029–3033. 10.1002/anie.200705604. [DOI] [PubMed] [Google Scholar]
- Shao H.; Wang C.; Xu S.; Wang Z.; Yin H.; Cui Y. Fast Doping of Cu into ZnSe NCs by Hydrazine Promoted Cation Exchange in Aqueous Solution at Room Temperature. J. Fluoresc. 2015, 25, 305–310. 10.1007/s10895-015-1509-1. [DOI] [PubMed] [Google Scholar]
- Bouet C.; Laufer D.; Mahler B.; Nadal B.; Heuclin H.; Pedetti S.; Patriarche G.; Dubertret B. Synthesis of Zinc and Lead Chalcogenide Core and Core/Shell Nanoplatelets Using Sequential Cation Exchange Reactions. Chem. Mater. 2014, 26, 3002–3008. 10.1021/cm5008608. [DOI] [Google Scholar]
- Zhang D.; Wong A. B.; Yu Y.; Brittman S.; Sun J.; Fu A.; Beberwyck B.; Alivisatos A. P.; Yang P. Phase-Selective Cation-Exchange Chemistry in Sulfide Nanowire Systems. J. Am. Chem. Soc. 2014, 136, 17430–17433. 10.1021/ja511010q. [DOI] [PubMed] [Google Scholar]
- Tan C.-S.; Hsiao C.-H.; Wang S.-C.; Liu P.-H.; Lu M.-Y.; Huang M. H.; Ouyang H.; Chen L.-J. Sequential Cation Exchange Generated Superlattice Nanowires Forming Multiple p–n Heterojunctions. ACS Nano 2014, 8, 9422–9426. 10.1021/nn5035247. [DOI] [PubMed] [Google Scholar]
- Manzi A.; Simon T.; Sonnleitner C.; Döblinger M.; Wyrwich R.; Stern O.; Stolarczyk J. K.; Feldmann J. Light-induced cation exchange for copper sulfide based CO2 reduction. J. Am. Chem. Soc. 2015, 137, 14007–14010. 10.1021/jacs.5b06778. [DOI] [PubMed] [Google Scholar]
- Jang S. Y.; Song Y. M.; Kim H. S.; Cho Y. J.; Seo Y. S.; Jung G. B.; Lee C.-W.; Park J.; Jung M.; Kim J.; et al. Three Synthetic Routes to Single-Crystalline PbS Nanowires with Controlled Growth Direction and Their Electrical Transport Properties. ACS Nano 2010, 4, 2391–2401. 10.1021/nn100163k. [DOI] [PubMed] [Google Scholar]
- Wang X.; Liu X.; Zhu D.; Swihart M. T. Controllable conversion of plasmonic Cu2-xS nanoparticles to Au2S by cation exchange and electron beam induced transformation of Cu2-xS-Au2S core/shell nanostructures. Nanoscale 2014, 6, 8852–8857. 10.1039/C4NR02114B. [DOI] [PubMed] [Google Scholar]
- Han S.-K.; Gong M.; Wang Z.-M.; Gu C.; Yu S.-H. Colloidal Synthesis of Cu2SxSe1–x Hexagonal Nanoplates and Their Transformation to CdSxSe1–x and ZnSxSe1–x by the Cation-Exchange Reaction. Part. Part. Syst. Charact. 2013, 30, 1024–1029. 10.1002/ppsc.201300166. [DOI] [Google Scholar]
- Miszta K.; Greullet F.; Marras S.; Prato M.; Toma A.; Arciniegas M.; Manna L.; Krahne R. Nanocrystal Film Patterning by Inhibiting Cation Exchange via Electron-Beam or X-ray Lithography. Nano Lett. 2014, 14, 2116–2122. 10.1021/nl500349j. [DOI] [PubMed] [Google Scholar]
- van der Stam W.; Bladt E.; Rabouw F. T.; Bals S.; de Mello Donega C. Near-Infrared Emitting CuInSe2/CuInS2 Dot Core/Rod Shell Heteronanorods by Sequential Cation Exchange. ACS Nano 2015, 9, 11430–11438. 10.1021/acsnano.5b05496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C.; van Veggel F. C. J. M. Cation Exchange in Lanthanide Fluoride Nanoparticles. ACS Nano 2009, 3, 123–130. 10.1021/nn8004747. [DOI] [PubMed] [Google Scholar]
- Lei L.; Chen D.; Yu Y.; Zhang R.; Ling H.; Xu J.; Huang F.; Wang Y. Growth of hexagonal NaGdF4 nanocrystals based on cubic Ln3+: CaF2 precursors and the multi-color upconversion emissions. J. Alloys Compd. 2014, 591, 370–376. 10.1016/j.jallcom.2013.12.238. [DOI] [Google Scholar]
- Li M.; Yu X.-F.; Liang S.; Peng X.-N.; Yang Z.-J.; Wang Y.-L.; Wang Q.-Q. Synthesis of Au–CdS Core–Shell Hetero-Nanorods with Efficient Exciton–Plasmon Interactions. Adv. Funct. Mater. 2011, 21, 1788–1794. 10.1002/adfm.201002233. [DOI] [Google Scholar]
- Yin Y.; Rioux R. M.; Erdonmez C. K.; Hughes S.; Somorjai G. A.; Alivisatos A. P. Formation of Hollow Nanocrystals Through the Nanoscale Kirkendall Effect. Science 2004, 304, 711–714. 10.1126/science.1096566. [DOI] [PubMed] [Google Scholar]
- Tian L.; Yang X.; Lu P.; Williams I. D.; Wang C.; Ou S.; Liang C.; Wu M. Hollow Single-Crystal Spinel Nanocubes: The Case of Zinc Cobalt Oxide Grown by a Unique Kirkendall Effect. Inorg. Chem. 2008, 47, 5522–5524. 10.1021/ic702457b. [DOI] [PubMed] [Google Scholar]
- Sytnyk M.; Kirchschlager R.; Bodnarchuk M. I.; Primetzhofer D.; Kriegner D.; Enser H.; Stangl J.; Bauer P.; Voith M.; Hassel A. W.; et al. Tuning the Magnetic Properties of Metal Oxide Nanocrystal Heterostructures by Cation Exchange. Nano Lett. 2013, 13, 586–593. 10.1021/nl304115r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B.; Chang S.; Li D.; Chen L.; Wang Y.; Chen T.; Zou B.; Zhong H.; Rogach A. L. Template Synthesis of CuInS2 Nanocrystals from In2S3 Nanoplates and Their Application as Counter Electrodes in Dye-Sensitized Solar Cells. Chem. Mater. 2015, 27, 5949–5956. 10.1021/acs.chemmater.5b01971. [DOI] [Google Scholar]
- Zhang B.; Jung Y.; Chung H.-S.; Vugt L. V.; Agarwal R. Nanowire Transformation by Size-Dependent Cation Exchange Reactions. Nano Lett. 2010, 10, 149–155. 10.1021/nl903059c. [DOI] [PubMed] [Google Scholar]
- Choi D.; Pyo J.-Y.; Kim Y.; Jang D.-J. Facile synthesis of composition-gradient Cd1-xZnxS quantum dots by cation exchange for controlled optical properties. J. Mater. Chem. C 2015, 3, 3286–3293. 10.1039/C4TC02795G. [DOI] [Google Scholar]
- Mao B.; Chuang C.-H.; Lu F.; Sang L.; Zhu J.; Burda C. Study of the Partial Ag-to-Zn Cation Exchange in AgInS2/ZnS Nanocrystals. J. Phys. Chem. C 2013, 117, 648–656. 10.1021/jp309202g. [DOI] [Google Scholar]
- Tang X.; Ho W. B. A.; Xue J. M. Synthesis of Zn-Doped AgInS2 Nanocrystals and Their Fluorescence Properties. J. Phys. Chem. C 2012, 116, 9769–9773. 10.1021/jp207711p. [DOI] [Google Scholar]
- Wang Y.-X.; Wei M.; Fan F.-J.; Zhuang T.-T.; Wu L.; Yu S.-H.; Zhu C.-F. Phase-Selective Synthesis of Cu2ZnSnS4 Nanocrystals through Cation Exchange for Photovoltaic Devices. Chem. Mater. 2014, 26, 5492–5498. 10.1021/cm501424n. [DOI] [Google Scholar]
- Tang B.; Yang F.; Lin Y.; Zhuo L.; Ge J.; Cao L. Synthesis and Characterization of Wavelength-Tunable, Water-Soluble, and Near-Infrared-Emitting CdHgTe Nanorods. Chem. Mater. 2007, 19, 1212–1214. 10.1021/cm062805x. [DOI] [Google Scholar]
- Wang H.; Lou S.; Tang Z.; Xu W.; Shang H.; Shen H.; Li L. S. Thiolate-assisted cation exchange reaction for the synthesis of near-infrared photoluminescent HgxCd1-xTe nanocrystals. Dalton Trans. 2012, 41, 12726–12732. 10.1039/c2dt31602a. [DOI] [PubMed] [Google Scholar]
- Yang L. W.; Li Y.; Li Y. C.; Li J. J.; Hao J. H.; Zhong J. X.; Chu P. K. Quasi-seeded growth, phase transformation, and size tuning of multifunctional hexagonal NaLnF4 (Ln = Y, Gd, Yb) nanocrystalsvia in situ cation-exchange reaction. J. Mater. Chem. 2012, 22, 2254–2262. 10.1039/C1JM14425A. [DOI] [Google Scholar]
- Dong C.; Korinek A.; Blasiak B.; Tomanek B.; van Veggel F. C. J. M. Cation Exchange: A Facile Method To Make NaYF4:Yb,Tm-NaGdF4 Core–Shell Nanoparticles with a Thin, Tunable, and Uniform Shell. Chem. Mater. 2012, 24, 1297–1305. 10.1021/cm2036844. [DOI] [Google Scholar]
- Abel K. A.; FitzGerald P. A.; Wang T.-Y.; Regier T. Z.; Raudsepp M.; Ringer S. P.; Warr G. G.; van Veggel F. C. J. M. Probing the Structure of Colloidal Core/Shell Quantum Dots Formed by Cation Exchange. J. Phys. Chem. C 2012, 116, 3968–3978. 10.1021/jp2112928. [DOI] [Google Scholar]
- Abel K. A.; Qiao H.; Young J. F.; van Veggel F. C. J. M. Four-Fold Enhancement of the Activation Energy for Nonradiative Decay of Excitons in PbSe/CdSe Core/Shell versus PbSe Colloidal Quantum Dots. J. Phys. Chem. Lett. 2010, 1, 2334–2338. 10.1021/jz1007565. [DOI] [Google Scholar]
- McDaniel H.; Fuke N.; Pietryga J. M.; Klimov V. I. Engineered CuInSexS2–x Quantum Dots for Sensitized Solar Cells. J. Phys. Chem. Lett. 2013, 4, 355–361. 10.1021/jz302067r. [DOI] [PubMed] [Google Scholar]
- Zhao H.; Chaker M.; Wu N.; Ma D. Towards controlled synthesis and better understanding of highly luminescent PbS/CdS core/shell quantum dots. J. Mater. Chem. 2011, 21, 8898–8904. 10.1039/c1jm11205h. [DOI] [Google Scholar]
- Zhao H.; Wang D.; Zhang T.; Chaker M.; Ma D. Two-step synthesis of high-quality water-soluble near-infrared emitting quantum dots via amphiphilic polymers. Chem. Commun. 2010, 46, 5301–5303. 10.1039/c0cc00067a. [DOI] [PubMed] [Google Scholar]
- Park J.; Kim S.-W. CuInS2/ZnS core/shell quantum dots by cation exchange and their blue-shifted photoluminescence. J. Mater. Chem. 2011, 21, 3745–3750. 10.1039/c0jm03194a. [DOI] [Google Scholar]
- Shemesh Y.; Macdonald J. E.; Menagen G.; Banin U. Synthesis and Photocatalytic Properties of a Family of CdS-PdX Hybrid Nanoparticles. Angew. Chem., Int. Ed. 2011, 50, 1185–1189. 10.1002/anie.201006407. [DOI] [PubMed] [Google Scholar]
- Kriegel I.; Wisnet A.; Srimath Kandada A. R.; Scotognella F.; Tassone F.; Scheu C.; Zhang H.; Govorov A. O.; Rodriguez-Fernandez J.; Feldmann J. Cation exchange synthesis and optoelectronic properties of type II CdTe-Cu2-xTe nano-heterostructures. J. Mater. Chem. C 2014, 2, 3189–3198. 10.1039/c3tc32049a. [DOI] [Google Scholar]
- Tang L.; Gui W.; Ding K.; Chen N.; Du G. Ion exchanged YVO4:Eu3+ nanocrystals and their strong luminescence enhanced by energy transfer of thenoyltrifluoroacetone ligands. J. Alloys Compd. 2014, 590, 277–282. 10.1016/j.jallcom.2013.12.112. [DOI] [Google Scholar]
- Sun X.; Huang X.; Guo J.; Zhu W.; Ding Y.; Niu G.; Wang A.; Kiesewetter D. O.; Wang Z. L.; Sun S.; et al. Self-Illuminating 64Cu-Doped CdSe/ZnS Nanocrystals for in Vivo Tumor Imaging. J. Am. Chem. Soc. 2014, 136, 1706–1709. 10.1021/ja410438n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalb A.; Leute V. The miscibility gap of the system CdSe-HgSe. Phys. Status Solidi A 1971, 5, K199–K201. 10.1002/pssa.2210050352. [DOI] [Google Scholar]
- Casu A.; Genovese A.; Manna L.; Longo P.; Buha J.; Botton G. A.; Lazar S.; Kahaly M. U.; Schwingenschloegl U.; Prato M.; et al. Cu2Se and Cu Nanocrystals as Local Sources of Copper in Thermally Activated In Situ Cation Exchange. ACS Nano 2016, 10.1021/acsnano.5b07219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalpian G. M.; Chelikowsky J. R. Self-Purification in Semiconductor Nanocrystals. Phys. Rev. Lett. 2006, 96, 226802. 10.1103/PhysRevLett.96.226802. [DOI] [PubMed] [Google Scholar]
- Demchenko D. O.; Robinson R. D.; Sadtler B.; Erdonmez C. K.; Alivisatos A. P.; Wang L.-W. Formation Mechanism and Properties of CdS-Ag2S Nanorod Superlattices. ACS Nano 2008, 2, 627–636. 10.1021/nn700381y. [DOI] [PubMed] [Google Scholar]
- Chen X.; Lou Y.; Samia A. C.; Burda C. Coherency Strain Effects on the Optical Response of Core/Shell Heteronanostructures. Nano Lett. 2003, 3, 799–803. 10.1021/nl034243b. [DOI] [Google Scholar]
- Li L.; Pandey A.; Werder D. J.; Khanal B. P.; Pietryga J. M.; Klimov V. I. Efficient Synthesis of Highly Luminescent Copper Indium Sulfide-Based Core/Shell Nanocrystals with Surprisingly Long-Lived Emission. J. Am. Chem. Soc. 2011, 133, 1176–1179. 10.1021/ja108261h. [DOI] [PubMed] [Google Scholar]
- Ostermann J.; Merkl J.-P.; Flessau S.; Wolter C.; Kornowksi A.; Schmidtke C.; Pietsch A.; Kloust H.; Feld A.; Weller H. Controlling the Physical and Biological Properties of Highly Fluorescent Aqueous Quantum Dots Using Block Copolymers of Different Size and Shape. ACS Nano 2013, 7, 9156–9167. 10.1021/nn4037859. [DOI] [PubMed] [Google Scholar]
- Palazon F.; Akkerman Q. A.; Prato M.; Manna L. X-ray Lithography on Perovskite Nanocrystals Films: From Patterning with Anion-Exchange Reactions to Enhanced Stability in Air and Water. ACS Nano 2016, 10, 1224–1230. 10.1021/acsnano.5b06536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell A. E.; Hodges J. M.; Schaak R. E. Preserving Both Anion and Cation Sublattice Features during a Nanocrystal Cation-Exchange Reaction: Synthesis of Metastable Wurtzite-Type CoS and MnS. J. Am. Chem. Soc. 2016, 138, 471–474. 10.1021/jacs.5b10624. [DOI] [PubMed] [Google Scholar]
- De Trizio L.; De Donato F.; Casu A.; Genovese A.; Falqui A.; Povia M.; Manna L. Colloidal CdSe/Cu3P/CdSe Nanocrystal Heterostructures and Their Evolution upon Thermal Annealing. ACS Nano 2013, 7, 3997–4005. 10.1021/nn3060219. [DOI] [PubMed] [Google Scholar]