

Abstract
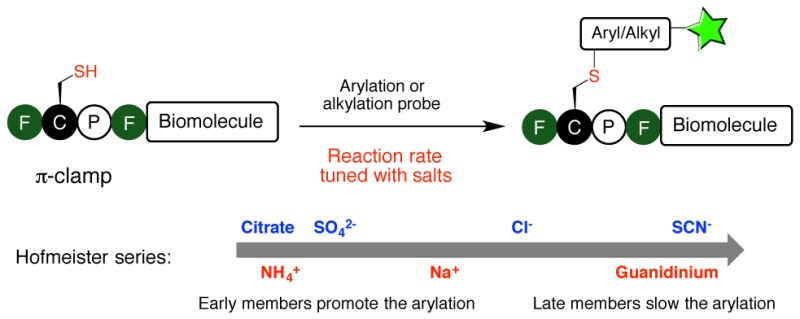
Highly efficient and selective chemical reactions are desired. For small molecule chemistry, the reaction rate can be varied by changing the concentration, temperature, and solvent used. In contrast for large biomolecules, the reaction rate is difficult to modify by adjusting these variables because stringent biocompatible reaction conditions are required. Here we show that adding salts can change the rate constant over 4 orders of magnitude for an arylation bioconjugation reaction between a cysteine residue within a four-residue sequence (π-clamp) and a perfluoroaryl electrophile. Biocompatible ammonium sulfate significantly enhances the reaction rate without influencing the site-specificity of π-clamp mediated arylation, enabling the fast synthesis of two site-specific antibody–drug conjugates that selectively kill HER2-positive breast cancer cells. Computational and structure–reactivity studies indicate that salts may tune the reaction rate through modulating the interactions between the π-clamp hydrophobic side chains and the electrophile. On the basis of this understanding, the salt effect is extended to other bioconjugation chemistry, and a new regioselective alkylation reaction at π-clamp cysteine is developed.
Short abstract
Salt in aqueous solution significantly changed the reaction rate of π-clamp mediated cysteine bioconjugation, enabling fast and site-specific modification of proteins including antibodies. The salt effect on π-clamp mediated arylation follows the Hofmeister series.
Introduction
Salts in aqueous solution influence a wide range of processes by tuning molecular interactions in an ion-specific manner;1,2 examples include enzyme activity,3−5 protein–protein interactions,6,7 gel formation,8 protein crystallization,9 and optical rotation of sugar and amino acids.10 The salt effects on various processes exhibit a recurring empirical trend called the Hofmeister series.11 For instance, ions could be ranked in their ability to decrease or increase protein solubility in water according to the Hofmeister series. Early members in the series decrease protein solubility, while later members increase the solubility. This observation is widely exploited in protein purification to salt out proteins with ammonium sulfate.12
The salt effect on hydrophobic interactions follows the Hofmeister series.1,13,14 Hydrophobic interactions describe the tendency of nonpolar molecules (hydrophobes) to exclude water and associate in aqueous solution. Such interactions are essential for chemical and biological processes including protein folding,15,16 DNA double helix stabilization,17 formation of protein–protein complexes,18−20 and self-assembly of synthetic molecules.21 Early members in Hofmeister series are believed to strengthen the hydrophobic interactions, while later members weaken them. The salt effect was even exploited to tune the reaction rate for the Diels–Alder22−24 and benzoin condensation25 processes in aqueous solution presumably by changing the interaction between the hydrophobic substrates.
Exploring the salt effect on bioconjugation is tempting because it may tune reaction rate while maintaining stringent biocompatible conditions, and there are many known biocompatible salts. However, such efforts are impeded because of the ubiquitous presence of hydrophobic structures and interactions in biomolecules. A unique hydrophobic environment is needed to selectively tune the bioconjugation reaction in the presence of other hydrophobic regions on the biomolecule of interest, but none of the commonly used bioconjugation reactions appear to be modulated by hydrophobicity. Recently we reported the π-clamp mediated conjugation in which the cysteine in the four-residue motif Phe-Cys-Pro-Phe (π-clamp) can be selectively arylated with perfluoroaryl probes.26 Notably, this reaction is regioselective for the cysteine residue within the π-clamp and leaves other cysteine thiols unchanged (Figure 1a). The interactions between the hydrophobic phenylalanine side chains and the hydrophobic perfluoroaryl probes are thought to be one of the main driving forces for the observed reactivity and selectivity of the π-clamp. This hypothesis inspired us to investigate whether salts would affect the reaction rate of the π-clamp mediated arylation. Here we report a 37 000-fold variation in reaction rate of the π-clamp-mediated conjugation in the presence of different salts.
Figure 1.

Salts significantly tuned the reaction rate of the π-clamp mediated arylation on peptides. (a) π-Clamp-mediated site-specific conjugation on proteins. (b) Salts used in this study and their positions in the Hofmeister series. (c) Rate constants of the reactions between π-clamp peptide 1A and perfluoroaryl probe 2 with salts at different concentrations. The black dashed line indicates the rate constant with no additional salt. General reaction conditions for measuring rate constant: 200 mM phosphate, 20 mM TCEP, pH 8.0, 37 °C. (See Figures S1–S15 for detailed conditions of each reaction.) Error was obtained from linear fitting of the kinetics curves for measuring the reaction rate.
Results
Salts Affect the Rate of π-Clamp Mediated Cysteine Arylation in Model Peptides
We observed a concentration-dependent and ion-specific effect on the π-clamp reaction rate that followed the Hofmeister series trend (Figure 1b). The rate constants were tunable by 4 orders of magnitude according to the salt added. We measured the rate constants of the reactions between the π-clamp peptide 1A and perfluoroaryl probe 2 in the presence of different salts (Figure 1c, Figures S1–S15 and Table S2). Salts composed of early member ions in the Hofmeister series accelerated the reaction, while late members decreased the rate. Compared to the reaction rate without additional salts (secondary rate constant k = 0.63 ± 0.02 M–1 s–1), it was significantly enhanced by the addition of ammonium sulfate (3 M, k = 62 ± 3 M–1 s–1) or ammonium citrate (2 M, k = 74 ± 3 M–1 s–1) and decreased by the addition of guanidinium chloride (3 M, k = 0.010 ± 0.001 M–1 s–1) or guanidinium thiocyanate (3 M, k = 0.0020 ± 0.0002 M–1 s–1). Sodium chloride, a middle member in the Hofmeister series, had little effect on the reaction rate.
Ammonium Sulfate Enhanced the Efficiency of the π-Clamp Mediated Protein Labeling
Ammonium sulfate is commonly used in protein purification and has little to no effect on protein function, such as enzymatic activity.27 Therefore, we used ammonium sulfate as a protein-compatible salt to further explore its effect on protein labeling. Two protein G B1 domain (pGB1) variants in which the π-clamp was incorporated at the N- or C-terminus were used as model proteins to evaluate the effect of ammonium sulfate on the reaction rate. The N-terminal π-clamp pGB1 (protein 3a) and the C-terminal π-clamp pGB1 (protein 3b) were reacted with perfluoroaryl probe 2 with and without 2 M ammonium sulfate, and the yields at different time points were characterized by liquid chromatography–mass spectrometry (LC–MS) analysis of the crude reaction mixtures (Figure 2 and Figures S16–S17). The rates of the labeling reactions were significantly enhanced in the presence of ammonium sulfate, as evidenced by a 90% product formation for the labeling of protein 3a over 120 min, while only 18% yield was achieved without salt (Figure 2a). The yields for the labeling of protein 3b with and without 2 M ammonium sulfate were 83% and 10% respectively after 240 min (Figure 2b). The rate constants of reactions with ammonium sulfate were 13-fold and 19-fold greater when compared to those of reactions without ammonium sulfate for the labeling of 3a and 3b, indicating that adding ammonium sulfate may be a general approach to enhance protein labeling efficiency via π-clamp mediated conjugation. Circular dichroism analysis of protein 3a and 3b showed that ammonium sulfate had little effect on the secondary structure of the two proteins (Figure S18), suggesting that the rate enhancement was not due to significant changes in protein structure.
Figure 2.

Ammonium sulfate accelerated π-clamp mediated protein labeling. Shown are the structures (left), the yield-time curves for the protein labeling reaction (middle), and the deconvoluted mass spectra from LC−MS analysis of the crude reaction mixture (right) for pGB1 variants 3a (a) and 3b (b) reacting with perfluoroaryl probe 2 with and without 2 M ammonium sulfate. Reaction conditions: 0.1 mM protein 3a or 3b, 1 mM probe 2, 200 mM phosphate, 20 mM TCEP, with or without 2 M ammonium sulfate, 37 °C.
Regioselectivity of Cysteine Arylation in the Presence of Ammonium Sulfate Is Maintained
A key feature of the π-clamp is that the chemistry is regioselective at cysteine: only the cysteine residue in the π-clamp motif is arylated, while other cysteines within the polypeptide chain or external thiols in solution are unchanged. We investigated if the regioselective properties of this reaction in the presence of ammonium sulfate was maintained. We found the reactivity between a double glycine mutant (Gly-Cys-Pro-Gly, peptide 1N) and probe 2 did not change significantly with 2 M ammonium sulfate (Figure S19). This finding indicates the observed rate enhancement with ammonium sulfate is not a general effect for all cysteine arylation reactions. Furthermore, reacting both the π-clamp peptide 1A and the double glycine mutant 1N (10 equiv relative to 1A) with probe 2 in the same solution with 2 M ammonium sulfate produced the π-clamp product quantitatively, while the double glycine mutant remained unchanged (Figure 3a and Figure S20).
Figure 3.

Ammonium sulfate did not perturb regioselectivity of the π-clamp mediated cysteine arylation. (a) Selective labeling of the π-clamp peptide 1A in the presence of 10 equiv of double glycine mutant 1N with ammonium sulfate. Reaction conditions: 0.05 mM 1A, 0.5 mM 1N, 0.1 mM probe 2, 200 mM phosphate, 20 mM TCEP, 2 M ammonium sulfate, 37 °C. (b) Regioselective labeling of the π-clamp cysteine in the presence of a competing N-terminal cysteine in the same protein molecule with ammonium sulfate. Reaction conditions: (1) 32 μM 4a, 1 mM probe 2, 200 mM phosphate, 20 mM TCEP, 2 M ammonium sulfate, 37 °C, 210 min. (2) 0.1 mg/mL TEV protease, 50 mM Tris, 0.1 mM EDTA, 10 mM TCEP, pH 8, room temperature, 16 h. The deconvoluted mass spectra for the whole protein peak of 4a–4c and mass spectrum of 4d obtained from LC–MS analysis of the crude reactions were shown.
The regioselectivity of the reaction with ammonium sulfate was also confirmed in the selective labeling of a protein containing both a π-clamp and a competing N-terminal cysteine. We prepared an engineered anthrax toxin lethal factor protein 1–263 (LFN) with a π-clamp moiety at the C-terminus and a cysteine at the N-terminus (4a, see Figure S21 for the protein preparation). A TEV protease cleavage site was positioned between the C-terminal π-clamp and the rest of the protein for unambiguous verification of the regioselectivity. Protein 4a was almost quantitatively converted to the monolabeled protein 4b after reacting with probe 2 in the presence of 2 M ammonium sulfate (Figure 3b). Upon cleavage by TEV protease and analysis by LC–MS, protein 4c with a free N-terminal cysteine was generated along with the arylated C-terminal π-clamp, confirming selective chemistry occurred in the presence of ammonium sulfate.
Ammonium Sulfate Accelerated Site-Specific Antibody Modification
Antibody-drug conjugates (ADCs)28,29 are targeted therapies that combine the selective delivery capacity of antibodies with the high cytotoxicity of drug payloads. Most procedures to make ADCs yield heterogeneous products containing a mixture of species with different positions of drug conjugation and varied drug-to-antibody ratios.28 Each species could show distinct pharmacokinetics and pharmacodynamics and different efficacies and safety profiles,30 making it difficult to systematically study ADCs and achieve consistent batch-to-batch synthesis. Several strategies have been developed to create site-specific antibody-drug conjugates,31−33 such as enzymatic conjugation,34 unnatural amino acid incorporation,35 drug conjugation through glycoengineering36 and engineered thio-antibody (THIOMAB).37 We previously applied the π-clamp-mediated conjugation to produce site-specific ADCs by selectively conjugating drug molecules to only the π-clamp cysteine residue.26 We envisioned that adding ammonium sulfate would increase the rate of antibody conjugation while not perturbing antibody function (Figure 4a).
Figure 4.

Ammonium sulfate accelerated site-specific modification of antibody. (a) Scheme for the site-specific biotin/drug conjugation to π-clamp trastuzumab 5. Val-Cit-PABC is the valine-citrulline p-aminobenzyl carbamate linker. (b) The deconvoluted mass spectra for antibody before conjugation (left), after conjugation in the presence of ammonium sulfate (middle), and after conjugation without ammonium sulfate (right). Reaction conditions: 40 μM π-clamp trastuzumab 5, 500 μM probe 6, 100 mM phosphate, 10 mM TCEP, ± 1.25 M (NH4)2SO4, 37 °C, 3 h. (c) Biotinylated trastuzumab (5-biotin) binds to recombinant HER2 in Octet BioLayer Interferometry assay (KD = 122 ± 1.5 pM). 5-biotin was immobilized on the streptavidin biosensors and sampled with serially diluted concentrations of recombinant HER2; see Figure S24 for fitting and data analysis. (d) The deconvoluted mass spectra for 5-MMAF and 5-MMAE synthesis. Reaction conditions for 5-MMAF: 40 μM π-clamp trastuzumab 5, 500 μM probe 6B, 100 mM phosphate, 10 mM TCEP, ± 1.25 M (NH4)2SO4, 37 °C, 210 min. Reaction condition for 5-MMAE: 40 μM π-clamp trastuzumab 5, 500 μM probe 6C, 100 mM phosphate, 10 mM TCEP, ± 1.25 M (NH4)2SO4, 37 °C, 16 h. (e) 5-MMAF and 5-MMAE prepared with ammonium sulfate were functional, selectively killing HER2 positive BT474 cells, while they were significantly less toxic for HER2 negative HEK 293T cells. Experiments were performed in triplicate. Error bars indicate the standard deviation from the average of three experiments.
We first determined the optimal concentration of ammonium sulfate since antibody tended to salt out under high ammonium sulfate concentration. We investigated reactions between the π-clamp trastuzumab (protein 5) and a biotin-perfluoroaryl probe 6A under different concentrations of ammonium sulfate, and the optimum reaction rate was achieved with 1.25 M ammonium sulfate (Figure S22). The π-clamp trastuzumab (protein 5, 40 μM) was completely converted to the heavy chain monolabeled product (5-biotin) within 3 h in the presence of 1.25 M ammonium sulfate, while only 33% of product was formed without ammonium sulfate (Figure 4b and Figure S23). Although we observed some precipitation of the antibody with 1.25 M ammonium sulfate, the modified product (5-biotin) was redissolved in buffer solution and showed full binding affinity to recombinant HER2 protein in an Octet BioLayer Interferometry assay (KD = 122 ± 2 pM, consistent with the binding affinity of native trastuzumab,26Figure 4c and Figure S24).
Ammonium sulfate significantly enhanced the labeling yields for the conjugation reaction between π-clamp trastuzumab (protein 5) and perfluoroaryl linked monomethyl auristatin F (6B) or perfluoroaryl linked monomethyl auristatin E (6C, see Figure S25 for the probe synthesis). Almost complete conversion was achieved for both ADCs (5-MMAF and 5-MMAE) with the addition of 1.25 M ammonium sulfate, while the yields were significantly lower for reactions without ammonium sulfate (Figure 4d, Figure S23 and Figure S26, < 20% and <1% for 5-MMAF and 5-MMAE respectively). Native trastuzumab (without π-clamp) led to no biotin or drug conjugation under the same conditions (Figure S27), showing the specificity of the conjugation. The two ADCs prepared with ammonium sulfate killed HER2-positive BT474 cells (EC50 = 0.16 nM and 0.47 nM for 5-MMAF and 5-MMAE respectively) and showed little effect on HER2-negative HEK 293T cells (Figure 4e). These experiments further confirmed the regioselectivity and protein-compatibility of our ammonium sulfate-promoted antibody labeling method.
Mechanism of the Salt Effect
Computational and structure–reactivity studies were carried out to understand the salt effect on π-clamp-mediated conjugation. We first tried to understand the key factors that affected the reaction rate in a quantitative manner and then investigated how salts may change those factors. On the basis of our hypothesis that interactions between the side chains and the perfluoroaryl probe are critical for the unique reactivity of the π-clamp, we designed a series of peptide mutants and measured the rates of their reactions with perfluoroaryl probe 2 (Figure 5a, Figures S28–40 and Table S3). In these peptides, the two phenylalanines in the π-clamp sequence (Phe-Cys-Pro-Phe) were mutated to unnatural amino acids containing extended aromatic side chains (peptides 1B-1E), polar substituted aromatic side chains (peptides 1F-1I), and aliphatic side chains (peptides 1J-1M). In order to quantify the interactions between the amino acid side chains and the perfluoroaryl probe, we proposed a simplified computational binding model (Figure 5b) consisting of only the perfluoroaryl group and the amino acid side chain. The binding geometry for each side chain-perfluoroaryl pair was optimized to obtain the lowest energy structure (Figure S41), and then high level density function theory (DFT) calculations were performed in an implicit solvent model to obtain the free binding energy between side chains of peptides 1A to 1M and perfluoroaryl moiety (see Computational Studies section in Methods for details). We found a linear relationship between the experimental rate constant (logarithmic scale) and the free energy of binding (Figure 5c and Table S4). This linear free energy relationship (LFER)38 supported the importance of the side chain-perfluoroaryl interaction.
Figure 5.

Mutation and computational studies on salt effect and π-clamp mediated arylation. (a) Rate constants for reactions between π-clamp mutants and perfluoroaryl probe 2. Reaction conditions: 200 mM phosphate, 20 mM TCEP, pH 8.0, 37 °C. (See Figures S28–S40 for detailed conditions of each reaction.) (b) Schematic of the binding model used for calculating the binding energy without an ion. (c) Linear free energy relationship (LFER) between the experimental reaction rate constant (k) and computational binding energy relative to baseline (ΔΔG, defined as ΔG – ΔG0). log10(k/k0) was found linearly correlated with ΔΔG. To serve as baseline, the term k0 refers to the rate constant of arylation reaction between peptide 1A and probe 2 without additional salt, and ΔG0 refers to the calculated binding energy in the binding model for 1A without an ion. (d) Schematic of the binding model used for calculating the binding energy with an ion. (e) Linear free energy relationship (LFER) between the experimental reaction rate constant (k) and computational binding energy with an ion incorporated relative to baseline (ΔΔG, defined as ΔG – ΔG0). log10(k/k0) was found linearly correlated with ΔΔG. The term k0 refers to the rate constant of the arylation reaction between 1A and 2 with NaCl, and ΔG0 refers to the calculated binding energy with Na+ incorporated in the binding model for 1A.
Because of the observed linear free energy relationship, we hypothesized that the salt effect may be a result of ions directly participating and affecting the binding events. The ions were added to the binding model for peptide 1A (Figure 5d), and the binding energies were calculated upon lowest energy structures (Figure S42 and Table S5). We assumed that, out of the cation and anion in a particular salt, the rate-changing effect would be determined mostly by the ion with stronger effect on binding energy. So citrate, sulfate, sodium cation and guanidinium were used to represent the salts used in this study since they showed a stronger effect on the binding energy than their counterions. We found a linear free energy relationship between the experimental rate constant and the free energy of binding in the presence of salt for all three salt concentrations studied (Figure 5e). These data provided evidence that ions could directly affect binding between amino acid side chains and the perfluoroaryl probe to tune the rate of the conjugation reaction. We are working on exploring the mechanism of how ions affect the binding interaction.
Salt Promoted Regioselective Cysteine Alkylation
Salts can promote other chemical reactions besides perfluoroarylation. The presence of unique hydrophobic environment in the π-clamp allowed us to expand the salt-promoted arylation (SNAr) chemistry to salt-promoted alkylation (SN2) reactions. We hypothesized that a hydrophobic alkylating probe might interact with the phenyl rings in the π-clamp to enhance the selectivity of cysteine alkylation reaction, and this reaction may be promoted with salt. To evaluate the hypothesis, the alkylation reagent 7 was synthesized with a structure reminiscent of probe 2 (Figure S43). Under the same conditions, π-clamp peptide 1A and the double glycine mutant 1N were reacted with probe 7. Dithiothreitol (DTT) was used as reducing reagent instead of TCEP because TCEP reacted with probe 7. We found 48% yield for π-clamp peptide 1A toward the production of alkylated product, while only 10% yield was observed for the double glycine mutant 1N after 15 min (Figure 6 and Figure S44), preliminarily showing the selective alkylation of π-clamp cysteine over the control cysteine.
Figure 6.
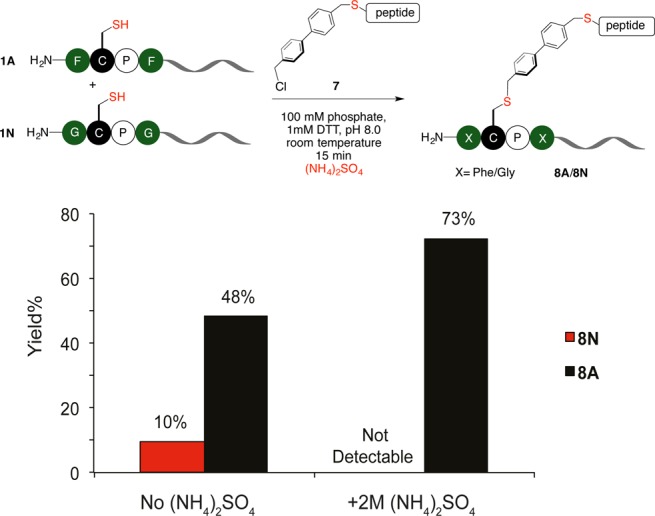
Salt effect enabled site-selective cysteine alkylation. The competing alkylation reaction between the π-clamp peptide 1A and the double glycine mutant 1N using a hydrophobic alkylating probe 7. Reaction conditions: 0.1 mM 1A, 0.1 mM 1N, 1 mM DTT, 0.5 mM probe 7, 100 mM phosphate, 0.25% DMSO, pH 8.0, with or without 2 M ammonium sulfate, room temperature, 15 min.
We next evaluated whether or not the salt effect can be applied to the selective cysteine alkylation reaction. 2 M ammonium sulfate was added to the alkylation reactions with probe 7. We found ammonium sulfate promoted the reaction between probe 7 and the π-clamp peptide 1A to give a 73% yield in 15 min. Surprisingly ammonium sulfate reduced the yield for the double glycine mutant 1N to almost zero (Figure 6 and Figure S44). The addition of ammonium sulfate significantly improved the regioselectivity of alkylation reaction on π-clamp using probe 7. In contrast to the hydrophobic probe 7, the widely used alkylation reagent bromoacetamide is not regioselective. Reacting the nonhydrophobic bromoacetamide with peptide 1A and 1N gave similar yields for the two peptides, and ammonium sulfate did not significantly change the yield (Figure S45). Collectively, regioselective alkylation of π-clamp cysteine over the control cysteine was achieved with the assistance of ammonium sulfate.
Discussion
The use of salt solutions to accelerate a reaction in small molecule chemistry for a Diels–Alder reaction22−24 was discovered several decades ago. Yet, the applications in bioconjugation involving large biomolecules have not been explored. Here we described the discovery of salts to remarkably change the reaction rate of the π-clamp mediated site-selective conjugation in aqueous solution. As the first case of applying Hofmeister series to bioconjugation, we varied the reaction rate constant of site-specific π-clamp arylation by 4 orders of magnitude without changing the peptide tag, the probe, or the temperature, or adding a catalyst. The salt’s influence depends on its identity and concentration. Compared to accelerating reaction rates by increasing the temperature or concentration, the advantage of the salt effect is significant as well as practical for the modification of biomolecules, considering heating and concentrating are generally not compatible with delicate or easy-to-aggregate proteins.
To demonstrate the utility of salt effect, we promoted the labeling of model proteins and trastuzumab without affecting antibody function using the protein-compatible salt ammonium sulfate. The selectivity of the π-clamp mediated arylation was not impaired by ammonium sulfate, enabling accelerated and site-specific modification of proteins that contain multiple cysteine residues. A trade-off between the enhanced hydrophobic interaction and the reduced solubility may need to be considered when using ammonium sulfate to accelerate protein labeling, especially for large proteins such as antibodies. The best salt concentration for protein labeling is related to the protein of interest. For example, the optimal ammonium sulfate concentration for π-clamp C225 antibody labeling is 1 M (Figure S46), which is different from 1.25 M for π-clamp trastuzumab. The concentration of salt should be varied according to the properties of the substrate protein. However, complete conversion could be achieved even when the protein partially salted out, as exemplified by 5-biotin, 5-MMAF, and 5-MMAE synthesis.
The salt effect is specific to reactions between a π-clamp and hydrophobic electrophile. The arylation reaction rate between the cyclohexylalanine mutant of π-clamp (peptide 1J) and probe 2 was also tunable with salts. Similar to the case in π-clamp peptide 1A, different salts influenced the reaction rates, and the trend followed the Hofmeister series (Figures S47–S52). However, it will abolish the salt effect by changing either one of the reaction partners to a nonhydrophobic molecule, which is supported by the observation that the reaction yields were not significantly changed when ammonium sulfate was added to the reactions involving the double glycine mutant (nonhydrophobic peptide sequence) or bromoacetamide (nonhydrophobic probe). The reactions between maleimide and cysteine were not promoted by ammonium sulfate (Table S6 and Figures S53–S55). To demonstrate salt effect is not limited to arylation, we applied the salt effect to promote two fundamentally different chemical reactions: π-clamp mediated cysteine arylation (SNAr reaction) and alkylation (SN2 reaction). Cysteine alkylation is widely used in bioconjugation, but selective alkylation is rarely reported.39 As a proof of concept, we developed a π-clamp mediated selective alkylation reaction that was promoted by ammonium sulfate. On the basis of the versatility and biocompatibility of salts, we envision that the salt effects could offer a tunable chemical feature for the rapidly expanding bioconjugation toolbox.
Methods
Synthesis of the Arylation and Alkylation Probes
Probe 2, 6A, and 6B were prepared as previously described.26,40 The synthesis of probe 6c was summarized in Figure S25. Probe 7 was synthesized as described below. To 7-Cys (19 μmol, 16.7 mg) dissolved in 1 mL of DMF in a plastic Eppendorf tube was added 4,4′-bis(chloromethyl)-1,1′-biphenyl (380 μmol, 95.4 mg) and diisopropylethylamine (190 μmol, 32 μL). The tube was vortexed and sonicated to ensure complete reagent mixing. The reaction mixture was left at room temperature for 60 min. 1 μL of reaction mixture was quenched by addition of 20 μL of 50% water/50% acetonitrile/0.5% TFA and was then analyzed by LC–MS. Resulting reaction mixtures were quenched by addition of 20 mL of 95% water/5% acetonitrile/0.1% TFA, and excess 4,4′-bis(chloromethyl)-1,1′-biphenyl was precipitated as a white solid. The resulting sample was centrifuged at 4000 rpm for 5 min. The supernatant was filtered through 0.22 μm nylon syringe filter and purified by RP-HPLC. Scheme for the reaction and LC–MS analysis for the product is shown in Figure S43.
Reaction Yield Determination
Yields for peptide substrates were determined by integrating total ion current (TIC) spectra. First, using the Agilent MassHunter software package, the peak area for all relevant peptidic species on the chromatogram were integrated. In cases where no side product was generated in the experiments, the conversion of the limiting reagent equals the yield of the product. Conversion was calculated by integrating the total ion current (TIC) of the same limiting peptide species within the dynamic linear range of the LC–MS instrument. Then the yield was calculated as following: % yield = % conversion = 1 – St/S0 where St is the peak area of the limiting reagent at time t, and S0 is the peak area of the limiting reagent at time 0. For protein substrate, the yield was calculated from the relative peak intensity of starting material and products in the deconvoluted mass spectrum as follows: % yield = Idesired product/Iall relevant species where Idesired product is the peak intensity of desired protein product, and Iall relevant species is the sum of the peak intensities of all relevant species (starting material and products) in the deconvoluted mass spectra.
Kinetics Study
The pH of salt stock solution was adjusted to 8.0 before use. To measure the second order rate constants, reaction mixture was prepared on ice and divided into several 10-μL aliquots. All aliquots were immediately put into a 37 °C water bath unless otherwise noted. For reactions that take more than 1 h to monitor, all aliquots were heated in a PCR machine set at 37 °C to prevent solvent evaporation. Reactions were quenched by addition of 100 μL of 50% water/50% acetonitrile/0.5% TFA at different time points and then subjected to LC–MS analysis. The initial concentration of probe and substrate were known. The second-order rate constants were determined by fitting the following kinetics eq 1:
| 1 |
Error of reaction rate constant was obtained from the linear fitting of the kinetics curves for measuring the reaction rate.
Antibody Expression and Labeling
The π-clamp trastuzumab 5 and π-clamp C225 were expressed as previously described.26 The antibody was labeled in 100 mM phosphate, 10 mM TCEP at pH 8.0 to keep the interchain disulfide reduced during the reaction. All the reactions were heated at in a PCR machine to maintain the temperature and prevent solvent evaporation. The reaction mixture was diluted and then buffer exchanged with 0.1 M Tris buffer, pH 8.0 using 10K Amicon Ultra centrifugal filters to remove the probe and ammonium sulfate. The concentrated antibody can be directly used for cell assays or binding assays. For LC−MS analysis of trastuzumab and its conjugates, the antibody was first treated with PNgase F (5000 u/mL) in 0.1 M Tris buffer (pH 8.0) at 45 °C for 1 h to remove N-glycans and then incubated with 20 mM TCEP to reduce the interchain disulfide.
Octet BioLayer Interferometry Binding Assay
In vitro binding assays were performed using the Fortebio Octet RED96 Bio-Layer Interferometry system at 30 °C. Streptavidin biosensors were dipped into 200 μL of 20 nM 5-biotin in PBS with 0.02% Tween and 0.1% BSA for the loading. The biosensors loaded with antibody were sampled with recombinant HER2 (R&D Systems) at various HER2 concentrations in PBS with 0.02% Tween and 0.1% BSA to obtain the association curve. Buffer only served as the reference. After association, the biosensors were dipped into buffer to obtain the dissociation curve. The association and dissociation curves were fitted with Fortebio Biosystems (global fitting algorithm) to obtain the KD.
Cell Viability Assay
Cells were seeded in a 96-well white opaque plate at a density of 4 × 103/well (HEK 293T) or 10 × 103/well (BT474). Cells were allowed to attach for 24 h at 37 °C and 5% CO2 in humidified atmosphere. Cells were then treated with serial dilutions of drug or ADCs for 96 h (BT474) or 72 h (HEK 293T). The viability of cells was measured using CellTiter Glo reagents following the manufacturer’s protocol and was normalized to the viability of cells without any treatment. The data were plotted using OriginLab software, and the half-maximal effective concentration (EC50) values were obtained by fitting the viability curves with a sigmoidal Boltzmann fit.
Computational Studies
Calculations without an ion (mutations studies, Figure 5c) were initialized from the six possible stacked geometries: perfluoroaryl moiety face-on with the side chain face, perfluoroaryl moiety face-on with the side chain edge, side chain face-on with perfluoroaryl moiety edge, perfluoroaryl moiety edge-on with side chain edge, side chain face-on with reactive fluorine in perfluoroaryl moiety, and side chain edge-on with reactive fluorine in perfluoroaryl moiety. Calculations with an ion (salt effect studies, Figure 5e) started with one initial structure: a sandwich of the ion between the phenylalanine side chain and perfluoroaryl moiety. The lowest energy structure was chosen after optimization. Geometries were not constrained through the calculation, and final binding structures were checked to ensure they conformed to two conditions. First, side chains should not fill volume that is occupied by the peptide part of the probe 2 that linked to the perfluoroaryl, which was not present in the simulation. Second, the methyl group on the side chain representing the attachment point to the peptide backbone should not significantly interact with the ligand due to steric restriction. For calculations involving an ion, the thiol in the perfluoroaryl moiety was replaced with a fluorine atom to prevent spurious interactions that would not be present in the full system. Because this change is far from, and electronically unconjugated to, the reaction center, we expect this modification to have little impact on the binding energy.
Density functional calculations were performed using the QCHEM 4.3 software package.41 For calculation without an ion, an initial coarse geometry optimization was performed with the B3LYP functional42 using Grimme’s D1 corrections43 in a 6-311+g* basis set.44,45 Then, a final optimization was performed using the VV1046 (rpw86/PBE) functional47 in a 6-311+g* basis with a B term of 5.9, a C term of 0.0093, and an SG1 nonlocal grid.48 The XC grid used was according to a Lebedev (75, 302) scheme.49 For calculation with an ion, the geometry optimization on initial structure was first performed in an STO-3g basis set using the B3LYP functional with Grimme’s dispersion corrections and then optimized in a 6-311+g* basis set again using the B3LYP functional with Grimme’s dispersion corrections. An implicit solvent model was used, namely a conducting polarizable continuum model with a dielectric of 78.4.50 Vibrational analysis was performed to account for zero-point energy and vibrational free energy within the harmonic approximation.
Acknowledgments
This work was supported by an MIT start-up fund, the National Institutes of Health (NIH; R01GM110535), and the Sontag Foundation Distinguished Scientist Award (to B.L.P.). C.Z. is a recipient of the George Büchi Research Fellowship, the Koch Graduate Fellowship in Cancer Research of MIT, and the Bristol-Myers Squibb Graduate Fellowship in Synthetic Organic Chemistry. The theory work was funded by a grant from the NSF (CHE-1464804). M.W. was supported by the NSF Graduate Research Fellowship Program. J.J.S. would like to thank the Royal Commission for the Exhibition of 1851 for a research fellowship. The Biophysical Instrumentation Facility for the Study of Complex Macromolecular Systems (NSF-0070319) and the Octet Bio-Layer Interferometry System (NIH S10 OD016326) are gratefully acknowledged. The authors acknowledge Alexander Vinogradov (MIT) for technical assistance and fruitful discussions and R .J. Collier (Harvard) for contributing laboratory equipment used in this study.
Glossary
Abbreviations
- TCEP
tris(2-carboxyethyl)phosphine
- DTT
dithiothreitol
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.6b00180.
Supplementary experimental procedures; reaction rate constant analysis, calculated lowest energy binding structures, and LC–MS characterization (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tobias D. J.; Hemminger J. C. Getting Specific about Specific Ion Effects. Science 2008, 319, 1197–1198. 10.1126/science.1152799. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Cremer P. S. Chemistry of Hofmeister Anions and Osmolytes. Annu. Rev. Phys. Chem. 2010, 61, 63–83. 10.1146/annurev.physchem.59.032607.093635. [DOI] [PubMed] [Google Scholar]
- Ru M. T.; Hirokane S. Y.; Lo A. S.; Dordick J. S.; Reimer J. a.; Clark D. S. On the Salt-Induced Activation of Lyophilized Enzymes in Organic Solvents: Effect of Salt Kosmotropicity on Enzyme Activity. J. Am. Chem. Soc. 2000, 122, 1565–1571. 10.1021/ja9935198. [DOI] [Google Scholar]
- Yang Z.; Liu X.-J.; Chen C.; Halling P. J. Hofmeister Effects on Activity and Stability of Alkaline Phosphatase. Biochim. Biophys. Acta, Proteins Proteomics 2010, 1804, 821–828. 10.1016/j.bbapap.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Pinna M. C.; Salis A.; Monduzzi M.; Ninham B. W. Hofmeister Series: The Hydrolytic Activity of Aspergillus Niger Lipase Depends on Specific Anion Effects. J. Phys. Chem. B 2005, 109, 5406–5408. 10.1021/jp050574w. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Cremer P. S. The Inverse and Direct Hofmeister Series for Lysozyme. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 15249–15253. 10.1073/pnas.0907616106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis R. A.; Ulrich J.; Montaser A.; Prausnitz J. M.; Blanch H. W. Protein-Protein Interactions in Concentrated Electrolyte Solutions: Hofmeister-Series Effects. Biotechnol. Bioeng. 2002, 79, 367–380. 10.1002/bit.10342. [DOI] [PubMed] [Google Scholar]
- Lloyd G. O.; Steed J. W. Anion-Tuning of Supramolecular Gel Properties. Nat. Chem. 2009, 1, 437–442. 10.1038/nchem.283. [DOI] [PubMed] [Google Scholar]
- Collins K. D. Ions from the Hofmeister Series and Osmolytes: Effects on Proteins in Solution and in the Crystallization Process. Methods 2004, 34, 300–311. 10.1016/j.ymeth.2004.03.021. [DOI] [PubMed] [Google Scholar]
- Lo Nostro P.; Ninham B.; Milani S.; Fratoni L.; Baglioni P. Specific Anion Effects on the Optical Rotation of Glucose and Serine. Biopolymers 2006, 81, 136–148. 10.1002/bip.20389. [DOI] [PubMed] [Google Scholar]
- Hofmeister F. Zur Lehre von Der Wirkung Der Salze. Naunyn-Schmiedeberg's Arch. Pharmacol. 1888, 24, 247–260. 10.1007/BF01918191. [DOI] [Google Scholar]
- Duong-Ly K. C.; Gabelli S. B. Salting out of Proteins Using Ammonium Sulfate Precipitation. Methods Enzymol. 2014, 541, 85–94. 10.1016/B978-0-12-420119-4.00007-0. [DOI] [PubMed] [Google Scholar]
- Zangi R.; Hagen M.; Berne B. J. Effect of Ions on the Hydrophobic Interaction between Two Plates. J. Am. Chem. Soc. 2007, 129, 4678–4686. 10.1021/ja068305m. [DOI] [PubMed] [Google Scholar]
- Zangi R.; Berne B. J. Aggregation and Dispersion of Small Hydrophobic Particles in Aqueous Electrolyte Solutions. J. Phys. Chem. B 2006, 110, 22736–22741. 10.1021/jp064475+. [DOI] [PubMed] [Google Scholar]
- Dill K. A. Dominant Forces in Protein Folding. Biochemistry 1990, 29, 7133–7155. 10.1021/bi00483a001. [DOI] [PubMed] [Google Scholar]
- Meyer E. E.; Rosenberg K. J.; Israelachvili J. Recent Progress in Understanding Hydrophobic Interactions. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15739–15746. 10.1073/pnas.0606422103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovchuk P.; Protozanova E.; Frank-Kamenetskii M. D. Base-Stacking and Base-Pairing Contributions into Thermal Stability of the DNA Double Helix. Nucleic Acids Res. 2006, 34, 564–574. 10.1093/nar/gkj454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai C. J.; Lin S. L.; Wolfson H. J.; Nussinov R. Studies of Protein-Protein Interfaces: A Statistical Analysis of the Hydrophobic Effect. Protein Sci. 1997, 6, 53–64. 10.1002/pro.5560060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones S.; Thornton J. M. Principles of Protein-Protein Interactions. Proc. Natl. Acad. Sci. U. S. A. 1996, 93, 13–20. 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W.; Hecht M. H. Generic Hydrophobic Residues Are Sufficient to Promote Aggregation of the Alzheimer’s Abeta42 Peptide. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 15824–15829. 10.1073/pnas.0605629103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschberg J. H. K. K.; Brunsveld L.; Ramzi A.; Vekemans J. A. J. M.; Sijbesma R. P.; Meijer E. W. Helical Self-Assembled Polymers from Cooperative Stacking of Hydrogen-Bonded Pairs. Nature 2000, 407, 167–170. 10.1038/35025027. [DOI] [PubMed] [Google Scholar]
- Kumar A. Salt Effects on Diels–Alder Reaction Kinetics. Chem. Rev. 2001, 101, 1–19. 10.1021/cr990410+. [DOI] [PubMed] [Google Scholar]
- Rizzo C. Salt Effects on a Hydrophobically Accelerated Diels-Alder Reaction Follow the Hofmeister Series. J. Org. Chem. 1992, 57, 6382–6384. 10.1021/jo00050a004. [DOI] [Google Scholar]
- Rideout D. C.; Breslow R. Hydrophobic Acceleration of Diels-Alder Reactions. J. Am. Chem. Soc. 1980, 102, 7816–7817. 10.1021/ja00546a048. [DOI] [Google Scholar]
- Kool E. T.; Breslow R. Dichotomous Salt Effects in the Hydrophobic Acceleration of the Benzoin Condensation. J. Am. Chem. Soc. 1988, 110, 1596–1597. 10.1021/ja00213a036. [DOI] [Google Scholar]
- Zhang C.; Welborn M.; Zhu T.; Yang N. J.; Santos M. S.; Van Voorhis T.; Pentelute B. L. π-Clamp-Mediated Cysteine Conjugation. Nat. Chem. 2016, 8, 120–128. 10.1038/nchem.2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao L.; van Rantwijk F.; Sheldon R. a. Cross-Linked Enzyme Aggregates: A Simple and Effective Method for the Immobilization of Penicillin Acylase. Org. Lett. 2000, 2, 1361–1364. 10.1021/ol005593x. [DOI] [PubMed] [Google Scholar]
- Sievers E. L.; Senter P. D. Antibody-Drug Conjugates in Cancer Therapy. Annu. Rev. Med. 2013, 64, 15–29. 10.1146/annurev-med-050311-201823. [DOI] [PubMed] [Google Scholar]
- Webb S. Pharma Interest Surges in Antibody Drug Conjugates. Nat. Biotechnol. 2011, 29, 297–298. 10.1038/nbt0411-297. [DOI] [PubMed] [Google Scholar]
- Hamblett K. J.; Senter P. D.; Chace D. F.; Hamblett K. J.; Senter P. D.; Chace D. F.; Sun M. M. C.; Lenox J.; Cerveny C. G.; Kissler K. M.; et al. Effects of Drug Loading on the Antitumor Activity of a Monoclonal Antibody Drug Conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. 10.1158/1078-0432.CCR-04-0789. [DOI] [PubMed] [Google Scholar]
- Chudasama V.; Maruani A.; Caddick S. Recent Advances in the Construction of Antibody-Drug Conjugates. Nat. Chem. 2016, 8, 114–119. 10.1038/nchem.2415. [DOI] [PubMed] [Google Scholar]
- Akkapeddi P.; Azizi S.-A.; Freedy A. M.; Cal P. M. S. D.; Gois P. M. P.; Bernardes G. J. L. Construction of Homogeneous Antibody–drug Conjugates Using Site-Selective Protein Chemistry. Chem. Sci. 2016, 7, 2954–2963. 10.1039/C6SC00170J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal P.; Bertozzi C. R. Site-Specific Antibody–Drug Conjugates: The Nexus of Bioorthogonal Chemistry, Protein Engineering, and Drug Development. Bioconjugate Chem. 2015, 26, 176–192. 10.1021/bc5004982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunbul M.; Yin J. Site Specific Protein Labeling by Enzymatic Posttranslational Modification. Org. Biomol. Chem. 2009, 7, 3361–3371. 10.1039/b908687k. [DOI] [PubMed] [Google Scholar]
- Tian F.; Lu Y.; Manibusan A.; Sellers A.; Tran H.; Sun Y.; Phuong T.; Barnett R.; Hehli B.; Song F.; et al. A General Approach to Site-Specific Antibody Drug Conjugates. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 1766–1771. 10.1073/pnas.1321237111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q.; Stefano J. E.; Manning C.; Kyazike J.; Chen B.; Gianolio D. a.; Park A.; Busch M.; Bird J.; Zheng X.; et al. Site-Specific Antibody-Drug Conjugation through Glycoengineering. Bioconjugate Chem. 2014, 25, 510–520. 10.1021/bc400505q. [DOI] [PubMed] [Google Scholar]
- Junutula J. R.; Raab H.; Clark S.; Bhakta S.; Leipold D. D.; Weir S.; Chen Y.; Simpson M.; Tsai S. P.; Dennis M. S.; et al. Site-Specific Conjugation of a Cytotoxic Drug to an Antibody Improves the Therapeutic Index. Nat. Biotechnol. 2008, 26, 925–932. 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- Hammett P. Linear Free Energy Relationships in Rate and Equilibrium Phenomena. Trans. Faraday Soc. 1938, 34, 156–165. 10.1039/tf9383400156. [DOI] [Google Scholar]
- Kawakami T.; Ogawa K.; Goshima N.; Natsume T. DIVERSE System: De Novo Creation of Peptide Tags for Non-Enzymatic Covalent Labeling by in Vitro Evolution for Protein Imaging Inside Living Cells. Chem. Biol. 2015, 22, 1671–1679. 10.1016/j.chembiol.2015.10.016. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Spokoyny A. M.; Zou Y.; Simon M. D.; Pentelute B. L. Enzymatic “click” ligation: Selective Cysteine Modification in Polypeptides Enabled by Promiscuous Glutathione S-Transferase. Angew. Chem., Int. Ed. 2013, 52, 14001–14005. 10.1002/anie.201306430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao Y.; Gan Z.; Epifanovsky E.; Gilbert A. T. B.; Wormit M.; Kussmann J.; Lange A. W.; Behn A.; Deng J.; Feng X.; et al. Advances in Molecular Quantum Chemistry Contained in the Q-Chem 4 Program Package. Mol. Phys. 2015, 113, 184–215. 10.1080/00268976.2014.952696. [DOI] [Google Scholar]
- Becke A. D. Density-Functional thermochemistry.III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648. 10.1063/1.464913. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Clark T.; Chandrasekhar J.; Spitznagel G. W.; Schleyer P. V. R. Efficient Diffuse Function-Augmented Basis Sets for Anion Calculations. III. The 3-21+G Basis Set for First-Row Elements, Li-F. J. Comput. Chem. 1983, 4, 294–301. 10.1002/jcc.540040303. [DOI] [Google Scholar]
- Krishnan R.; Binkley J. S.; Seeger R.; Pople J. A. Self-Consistent Molecular Orbital Methods. XX. A Basis Set for Correlated Wave Functions. J. Chem. Phys. 1980, 72, 650–654. 10.1063/1.438955. [DOI] [Google Scholar]
- Vydrov O. A.; Van Voorhis T. Nonlocal van Der Waals Density Functional: The Simpler the Better. J. Chem. Phys. 2010, 133, 244103. 10.1063/1.3521275. [DOI] [PubMed] [Google Scholar]
- Perdew J. P.; Burke K.; Ernzerhof M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- Gill P. M. W.; Johnson B. G.; Pople J. A. A Standard Grid for Density Functional Calculations. Chem. Phys. Lett. 1993, 209, 506–512. 10.1016/0009-2614(93)80125-9. [DOI] [Google Scholar]
- Lebedev V. I. Spherical Quadrature Formulas Exact to Orders 25–29. Sib. Math. J. 1977, 18, 99–107. 10.1007/BF00966954. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.