

Summary
Introduction
n‐3 Polyunsaturated fatty acids such as eicosapentaenoic acid (EPA), which are abundant in fish oil, have been shown to delay the onset of cardiovascular events. We previously established DahlS.Z‐Lepr fa/Lepr fa (DS/obese) rats, which are derived from a cross between Dahl salt‐sensitive and Zucker rats, as a model of metabolic syndrome. This study has now explored the influence of highly purified EPA on cardiac and adipose tissue pathophysiology in this animal model.
Materials and methods
DS/obese rats were administered EPA (300 or 1,000 mg kg−1 d−1, per os) or vehicle from age 9 to 13 weeks. Homozygous lean (DahlS.Z‐Lepr +/Lepr +, or DS/lean) littermates were studied as controls.
Results
Whereas EPA had no effect on body weight, food intake or systolic blood pressure in DS/obese rats, it attenuated cardiac fibrosis, diastolic dysfunction, oxidative stress and inflammation in these animals. In addition, EPA did not affect insulin resistance but reduced adipocyte hypertrophy and inflammation in visceral fat of DS/obese rats. Moreover, EPA increased circulating levels of adiponectin as well as attenuated both the down‐regulation of AMP‐activated protein kinase phosphorylation and the up‐regulation of phosphorylation of the p65 subunit of nuclear factor‐kB in the heart of DS/obese rats.
Conclusions
Treatment of DS/obese rats with EPA did not affect hypertension but reduced cardiac fibrosis and diastolic dysfunction, with the latter effects being accompanied by AMP‐activated protein kinase activation and inactivation of nuclear factor‐kB signalling in the heart, possibly as a result of an increase in adiponectin secretion. EPA may be suitable for the treatment of cardiac injury associated with metabolic syndrome.
Keywords: Adiponectin, eicosapentaenoic acid, inflammation, metabolic syndrome
Introduction
Given that metabolic syndrome (MetS) enhances the risk of atherosclerosis and can result in cardiovascular failure or other complications, the recent increase in the prevalence of this condition is a serious clinical problem 1. Dys‐regulated secretion of adipocytokines from adipocytes of visceral fat in obese individuals is implicated in the development of hypertension and complications associated with insulin resistance 2. Chronic inflammation has also been identified as a key link between obesity and insulin resistance or type 2 diabetes mellitus, with obesity‐related chronic inflammation being associated with up‐regulation of pro‐inflammatory cytokine production and inflammatory pathway activation in metabolic tissues 3.
Consumption of n‐3 polyunsaturated fatty acids (PUFAs) is thought to exert beneficial effects on arrhythmia, triglyceride levels, atherosclerotic plaque, endothelial dysfunction, platelet aggregation and inflammation 4, 5, 6. Eicosapentaenoic acid (EPA), a major n‐3 PUFA in fish oil, has been found to have various pharmacological effects. The Japan EPA Lipid Intervention Study, a large‐scale, prospective and randomized clinical trial, showed that the addition of EPA to statin treatment delays the onset of cardiovascular events in Japanese hypercholesterolaemic patients through cholesterol‐independent mechanisms, suggesting pleiotropic effects of EPA 7. EPA promotes the production of adiponectin, which protects against diabetes and atherosclerosis, as well as inhibits monocyte adhesion to endothelial cells 8, 9. Given that mammals are unable to synthesize PUFAs, the balance between n‐3 and n‐6 PUFAs in the body depends on the type of fatty acids ingested. A high n‐3 to n‐6 PUFA ratio is thought to protect against diseases related to inflammation 10. Dietary manipulation of PUFAs might thus provide both insight into the relevance of changes in fatty acid composition to adipose tissue inflammation and a basis for the development of new therapeutic strategies targeting MetS.
We recently established the DahlS.Z‐Lepr fa/Lepr fa (DS/obese) rat strain as the result of a cross between Dahl salt‐sensitive (DS) rats and Zucker rats, the latter of which harbour a missense mutation in the gene for the leptin receptor (Lepr). DS/obese rats maintained on normal laboratory chow develop a phenotype similar to that of human MetS, including hypertension, obesity, dyslipidaemia, glucose intolerance and insulin resistance 11. This study has here examined the effects of EPA on cardiac and adipose tissue pathophysiology as well as on glucose metabolism in these animals. It was expected that EPA would attenuate cardiac damage as well as improve hypertriglyceridaemia and insulin resistance in this model of MetS.
Materials and methods
Rats and protocols
Animal experiments were approved by the Animal Experiment Committee of Nagoya University Graduate School of Medicine (Daiko district, approval nos. 024‐40, 025‐015, 026‐014, 027‐006 and 028‐011). Eight‐week‐old male inbred DS/obese rats were obtained from Japan SLC Inc. (Hamamatsu, Japan) and were handled in accordance with the guidelines of Nagoya University Graduate School of Medicine as well as with the Guide for the Care and Use of Laboratory Animals (US National Institutes of Health publication no. 85‐23, revised 2011). The animals were fed a diet free of fish meal (fish meal‐free F1, 360 kcal/100 g, 4.4% of energy as fat; Funabashi Farm, Funabashi, Japan), with both the diet and tap water being freely available throughout the experimental period. The animals were randomly assigned to the low‐dose EPA administration group (300 mg kg−1 of body weight per day; EPA‐L group) or the high‐dose EPA administration group (1,000 mg kg−1 of body weight per day; EPA‐H group) or vehicle (MetS group) at an age of 9 weeks. Highly purified EPA (ethyl eicosapentate; Mochida Pharmaceutical Co. Ltd, Tokyo, Japan) was dissolved in 5% gum arabic solution and administered orally once daily via a gastric tube. The doses of EPA were determined on the basis of the results of previous studies 12, 13. Male homozygous lean littermates of DS/obese rats (DahlS.Z‐Lepr +/Lepr +, or DS/lean) were also treated with vehicle as controls (CONT group). Body weight and intake of both food and water were determined once a week. An oral glucose tolerance test (OGTT) and an insulin tolerance test (ITT) were carried out as previously described 14. At 13 weeks of age, rats were subjected to anaesthesia with intraperitoneal injection of ketamine (50 mg kg−1)–xylazine (10 mg kg−1) for echocardiographic and haemodynamic analyses. After subsequent intraperitoneal injection of an overdose of sodium pentobarbital (50 mg kg−1), the heart, liver, kidneys as well as visceral (retroperitoneal and epididymal) and subcutaneous (inguinal) fat were excised and weighed. Left ventricular (LV) and visceral adipose tissues were also separated for analysis.
Measurement of blood pressure, echocardiography and cardiac catheterization
Systolic blood pressure (SBP) and heart rate were measured once a week in conscious animals by tail‐cuff plethysmography (BP‐98A; Softron, Tokyo, Japan). Transthoracic echocardiography and cardiac catheterization were performed as previously described 15, 16, 17.
Histology and immunohistochemistry
The LV and visceral (retroperitoneal) fat tissue were fixed in ice‐cold 4% paraformaldehyde for 48 h, embedded in paraffin and processed for histology, as described 18, 19. For evaluation of macrophage infiltration into the LV myocardium and adipose tissue, paraffin‐embedded tissue sections were subjected to immunohistochemical staining for the monocyte–macrophage marker CD68, as described previously 14. All image analyses were performed with the use of nih scion image software (Scion, Frederick, MD, USA).
Biochemical analysis
Metabolic and hormonal parameters were measured as described previously 20, 21.
Assay of superoxide production
Reduced nicotinamide adenine dinucleotide phosphate (NADPH)‐dependent superoxide production by homogenates prepared from freshly frozen LV tissue was measured with the use of an assay based on lucigenin‐enhanced chemiluminescence as described previously 22, 23. Superoxide production in tissue sections was examined by staining with dihydroethidium (Sigma, St. Louis, MO, USA) as described 24, and the average of dihydroethidium fluorescence intensity values was calculated with the use of NIH Image (imagej) software 25.
Quantitative reverse transcription‐polymerase chain reaction analysis
Total RNA was extracted from LV and visceral (retroperitoneal) fat tissue and was subjected to reverse transcription and real‐time polymerase chain reaction analysis as described 21 with specific primers for cDNAs encoding atrial natriuretic peptide 15, brain natriuretic peptide 15, collagen type I or type III 26, connective tissue growth factor 22, transforming growth factor‐β1 15, monocyte chemoattractant protein‐1 (MCP‐1) 22, osteopontin 22, cyclooxygenase‐2 (COX‐2) 27 and the p22phox 28, gp91phox 28 and Rac1 29 subunits of NADPH oxidase. Reagents for detection of human glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) mRNA (Applied Biosystems, Foster City, CA, USA) were used to quantify rat GAPDH mRNA as an internal standard.
Immunoblot analysis
Total protein was isolated from LV tissue and quantitated as described previously 30. Equal amounts of protein were subjected to sodium dodecyl sulphate‐polyacrylamide gel electrophoresis, and the separated proteins were transferred to a polyvinylidene difluoride membrane, as described previously 31. The membrane was incubated overnight at 4 °C with a 1:1,000 dilution of rabbit polyclonal antibodies to total or Thr172‐phosphorylated forms of α‐AMP‐activated protein kinase (AMPK) (Cell Signaling Technology, Beverly, MA, USA) or to total or Ser536‐phosphorylated forms of nuclear factor‐kB (NF‐kB) (Cell Signaling Technology) and then with a 1:8,000 dilution of horseradish peroxidase conjugated goat antibodies to rabbit immunoglobulin G (Kirkegaard & Perry Laboratories, Gaithersburg, MD, USA), respectively. Detection and quantification of immune complexes were performed as described 31.
Statistical analysis
Data are shown as mean ± standard error of the mean values. Parameters were compared among groups of rats at 13 weeks of age with one‐way factorial analysis of variance (anova) followed by Fisher's multiple comparison test. Time courses for body weight, food intake and SBP as well as OGTT and ITT curves were compared among groups by two‐way repeated‐measures anova. Statistical significance was defined as a P value of <0.05.
Results
Physiological analyses and cardiac geometry and function
Body weight, food intake and SBP were significantly higher in the MetS group than in the CONT group from 9 to 13 weeks of age, but they did not differ between the MetS and either the EPA‐L or EPA‐H group (Figure 1A–C). At 13 weeks of age, the increases in the ratios of heart or LV weight to tibial length, indexes of cardiac and LV hypertrophy, respectively, in the MetS group were not altered by either dose of EPA (Table 1). The increases in the ratios of retroperitoneal or epididymal fat weight to tibial length in the MetS group were significantly attenuated in the EPA‐H group (Table 1).
Figure 1.
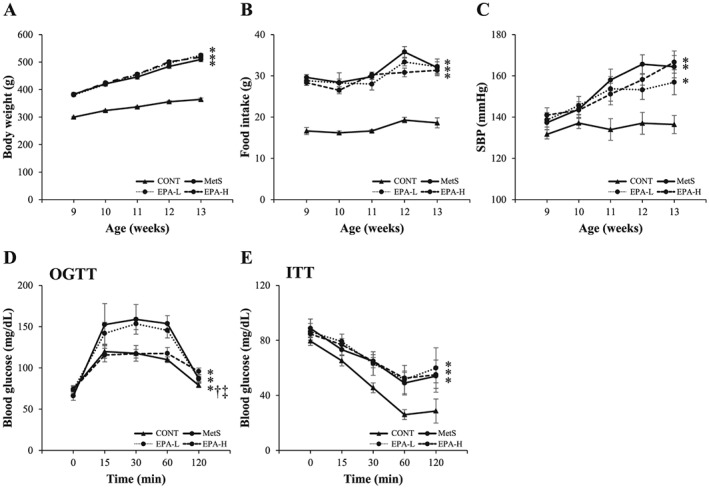
Age‐dependent changes in body weight (A), food intake (B) and systolic blood pressure (SBP) (C) as well as oral glucose tolerance test (OGTT) (D) and insulin tolerance test (ITT) (E) assays performed at 13 weeks of age for rats in the four experimental groups. Data are means ± standard error of the mean (n = 10, 10, 11 and 11 for control [CONT], metabolic syndrome [MetS], low‐dose eicosapentaenoic acid [EPA‐L] and high‐dose eicosapentaenoic acid [EPA‐H] groups, respectively). *P < 0.05 vs. CONT; †P < 0.05 vs. MetS; ‡P < 0.05 vs. EPA‐L.
Table 1.
Physiological parameters for 13‐week‐old rats in the four experimental groups
| Parameter | CONT | MetS | EPA‐L | EPA‐H |
|---|---|---|---|---|
| Body weight (g) | 364.3 ± 4.9 | 509.5 ± 8.1* | 518.1 ± 5.7* | 524.8 ± 6.1* |
| Tibial length (mm) | 37.6 ± 0.4 | 34.4 ± 0.2* | 34.3 ± 0.2* | 34.6 ± 0.2* |
| Food intake (g d−1) | 18.6 ± 1.2 | 32.1 ± 1.4* | 32.2 ± 1.9* | 31.3 ± 1.3* |
| SBP (mmHg) | 136.4 ± 4.4 | 164.4 ± 5.4* | 156.9 ± 6.1* | 166.7 ± 5.4* |
| Heart rate (beats/min) | 410.9 ± 2.6 | 356.8 ± 13.5* | 345.6 ± 12.8* | 352.4 ± 11.7* |
| Heart weight/tibial length (mg mm−1) | 29.9 ± 0.7 | 35.9 ± 8.9* | 37.2 ± 0.5* | 37.4 ± 1.1* |
| LV weight/tibial length (mg mm−1) | 22.4 ± 0.6 | 27.9 ± 0.7* | 28.4 ± 0.5* | 28.8 ± 0.8* |
| Liver weight/tibial length (mg mm−1) | 242.1 ± 6.7 | 526.7 ± 15.9* | 553.4 ± 13.6* | 570.3 ± 16.7* |
| Kidney weight/tibial length (mg mm−1) | 63.4 ± 2.6 | 87.7 ± 4.2* | 89.7 ± 5.2* | 98.4 ± 3.2* |
| Retroperitoneal fat weight/tibial length (mg mm−1) | 97.2 ± 5.1 | 482.1 ± 16.9* | 468.3 ± 9.9* | 442.4 ± 14.6*† |
| Epididymal fat weight/tibial length (mg mm−1) | 112.8 ± 3.1 | 375.8 ± 13.4* | 354.2 ± 10.9* | 332.9 ± 9.3*† |
| Inguinal fat weight/tibial length (mg mm−1) | 164.9 ± 9.8 | 1342.9 ± 43.7* | 1310.6 ± 34.8* | 1378.3 ± 23.2* |
| Fasting serum glucose (mg dL−1) | 117.8 ± 3.9 | 128.8 ± 5.6 | 117.8 ± 3.1 | 139.1 ± 15.9 |
| Fasting serum insulin (ng dL−1) | 0.81 ± 0.1 | 4.17 ± 1.2* | 3.79 ± 1.1* | 4.45 ± 1.5* |
| HOMA‐IR | 6.83 ± 0.76 | 60.4 ± 16.7* | 54.4 ± 5.91* | 76.9 ± 17.7* |
| HOMA‐β | 158.6 ± 21.2 | 867.6 ± 61.2* | 935.6 ± 196.0* | 1106.2 ± 167.5* |
| Serum adiponectin (µg mL−1) | 4.9 ± 0.2 | 7.2 ± 1.1* | 8.7 ± 1.1*† | 8.6 ± 0.5*† |
| Total cholesterol (mg dL−1) | 86.8 ± 3.4 | 278.6 ± 18.1* | 184.1 ± 6.4*† | 201.2 ± 23.7*† |
| LDL‐cholesterol (mg dL−1) | 20.6 ± 0.9 | 60.6 ± 10.2* | 28.4 ± 5.8† | 34.3 ± 7.1† |
| HDL‐cholesterol (mg dL−1) | 53.6 ± 2.1 | 147.6 ± 2.8* | 117.3 ± 4.9*† | 123.6 ± 4.1*† |
| Triglyceride (mg dL−1) | 43.8 ± 6.9 | 685.6 ± 136.1* | 491.5 ± 111.1* | 611.2 ± 55.5* |
Data are means ± SEM (n = 10, 10, 11 and 11 for CONT, MetS, EPA‐L and EPA‐H groups, respectively).
P < 0.05 vs. CONT.
P < 0.05 vs. MetS.
CONT, controls; EPA‐H, high‐dose eicosapentaenoic acid group; EPA‐L, low‐dose eicosapentaenoic acid group; HDL, high‐density lipoprotein; HOMA‐β, homeostasis model assessment of β‐cell function; HOMA‐IR, homeostasis model assessment of insulin resistance; LDL, low‐density lipoprotein; LV, left ventricular; MetS, metabolic syndrome; SBP, systolic blood pressure; SEM, standard error of the mean.
Echocardiography showed that the increases in the interventricular septum thickness, LV posterior wall thickness, LV mass, LV fractional shortening and LV ejection fraction in the MetS group were not affected in either EPA‐L or EPA‐H group (Table 2). The deceleration time, isovolumic relaxation time, time constant of isovolumic relaxation (tau) and the ratio of LV end‐diastolic pressure to LV end‐diastolic dimension, an index of LV diastolic stiffness, were all greater in the MetS group than in the CONT group. The early to late ventricular velocities were reduced in DS/obese rats than in DS/lean rats. All of these effects were alleviated in both the EPA‐L and EPA‐H groups (Table 2). These findings thus indicated that EPA did not affect LV hypertrophy but improved LV diastolic function in DS/obese rats.
Table 2.
Assessment of cardiac morphology and function for 13‐week‐old rats in the four experimental groups
| Parameter | CONT | MetS | EPA‐L | EPA‐H |
|---|---|---|---|---|
| IVST (mm) | 1.37 ± 0.01 | 1.63 ± 0.02* | 1.54 ± 0.02* | 1.58 ± 0.01* |
| LVPWT (mm) | 1.39 ± 0.02 | 1.57 ± 0.01* | 1.53 ± 0.02* | 1.57 ± 0.03* |
| LVDd (mm) | 8.37 ± 0.08 | 8.66 ± 0.07 | 8.42 ± 0.07 | 8.41 ± 0.16 |
| LVFS (%) | 39.59 ± 1.19 | 45.26 ± 0.73* | 43.78 ± 1.21* | 42.42 ± 0.88* |
| LVEF (%) | 67.06 ± 1.05 | 72.84 ± 0.94* | 72.81 ± 1.36* | 73.57 ± 0.94* |
| LV mass (mg) | 793.6 ± 21.8 | 1056.4 ± 17.5* | 984.5 ± 14.4* | 1051.5 ± 29.9* |
| E/A | 2.25 ± 0.07 | 1.91 ± 0.03* | 2.17 ± 0.06† | 2.18 ± 0.17† |
| DcT (ms) | 52.32 ± 0.34 | 62.21 ± 1.43* | 55.65 ± 0.91† | 55.31 ± 0.85† |
| IRT (ms) | 33.31 ± 0.65 | 48.41 ± 1.01* | 44.09 ± 0.52*, † | 43.91 ± 0.46*, † |
| Tau (ms) | 19.39 ± 0.51 | 42.49 ± 2.36* | 22.64 ± 2.45† | 25.66 ± 2.34† |
| LVEDP/LVDd (mmHg mm−1) | 0.23 ± 0.01 | 0.87 ± 0.11* | 0.76 ± 0.04*, † | 0.71 ± 0.05*, † |
Data are means ± SEM (n = 10, 10, 11 and 11 for CONT, MetS, EPA‐L and EPA‐H groups, respectively).
P < 0.05 vs. CONT.
P < 0.05 vs. MetS.
CONT, controls; DcT, deceleration time; E/A, ratio of peak flow velocity at the mitral level during rapid filling (E) to that during atrial contraction; EPA‐H, high‐dose eicosapentaenoic acid group; EPA‐L, low‐dose eicosapentaenoic acid group; IRT, isovolumic relaxation time; IVST, interventricular septum thickness; LVPWT; LV posterior wall thickness; LV, left ventricular; LVDd; LV end‐diastolic dimension; LVEDP, LV end‐diastolic pressure; LVEF, LV ejection fraction; LVFS; LV fractional shortening; MetS, metabolic syndrome; SEM, standard error of the mean; tau, time constant of isovolumic relaxation.
Glucose and lipid metabolism and hormonal parameters
The fasting concentration of glucose in serum did not differ among the four experimental groups (Table 1). The increases in the fasting serum insulin level as well as in homeostasis model assessment of insulin resistance and of β‐cell function indexes in the MetS group were not altered by EPA administration at either dose (Table 1). OGTT and ITT assays showed that the impaired glucose tolerance in the MetS group was attenuated in the EPA‐H group (Figure 1D) and that the insulin resistance in the MetS group was not ameliorated in either the EPA‐L or EPA‐H group (Figure 1E). The serum concentration of adiponectin was significantly increased in the MetS group that in the CONT group, and EPA further elevated this parameter at low and high doses (Table 1). Serum concentrations of total cholesterol, low‐density lipoprotein cholesterol, high‐density lipoprotein cholesterol and triglyceride were all increased in the MetS group, and, with the exception of triglyceride, these effects were reduced in both the EPA‐L and EPA‐H groups (Table 1).
Cardiomyocyte hypertrophy as well as cardiac fibrosis and gene expression
Microscopy showed that the increase in the cross‐sectional area of LV cardiomyocytes in the MetS group was not attenuated in the EPA‐L or EPA‐H group (Figure 2A,B). The significant up‐regulation of the expression of atrial natriuretic peptide and brain natriuretic peptide genes apparent in the heart of DS/obese rats was also insensitive to EPA treatment (Figure 2C,D). The Azan–Mallory staining showed that the increase in fibrosis in perivascular and interstitial regions of the LV myocardium in the MetS group was attenuated in both EPA‐L and EPA‐H groups (Figure 2E–H). The increases in the cardiac abundance of collagen types I and III, connective tissue growth factor and transforming growth factor‐β1 mRNAs in the MetS group were attenuated in both EPA‐L and EPA‐H groups (Figure 2I–L).
Figure 2.
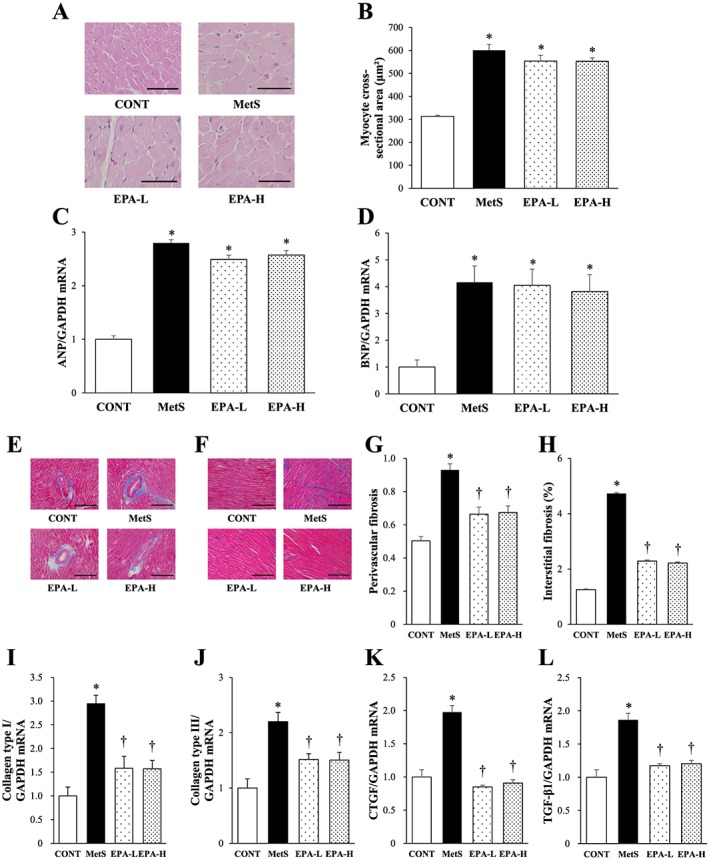
Size of cardiomyocytes, foetal‐type cardiac gene expression, cardiac fibrosis and fibrosis‐related gene expression in 13‐week‐old rats of the four experimental groups. (A) Transverse sections of the LV myocardium stained with haematoxylin–eosin. Scale bars, 100 µm. (B) Cross‐sectional area of cardiomyocytes as measured in sections similar to those in (A). (C,D) reverse transcription (RT) and real‐time polymerase chain reaction (PCR) analysis of atrial natriuretic peptide (ANP) (C) and brain natriuretic peptide (BNP) (D) mRNA abundance normalized by that of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) mRNA and expressed relative to the mean value for the control (CONT) group. (E,F) Azan–Mallory staining of perivascular (E) and interstitial (F) regions in the left ventricular (LV) myocardium to reveal collagen deposition. Scale bars, 100 µm. (G,H) Quantitation of the relative extents of perivascular (G) and interstitial (H) fibrosis in the LV myocardium in sections similar to those in (E) and (F). (I–L) RT and real‐time PCR analysis of collagen types I and III, connective tissue growth factor (CTGF) and transforming growth factor‐β1 (TGF‐β1) mRNAs, respectively. All quantitative data are means ± standard error of the mean (n = 10, 10, 11 and 11 for CONT, metabolic syndrome [MetS], low‐dose eicosapentaenoic acid [EPA‐L] and high‐dose eicosapentaenoic acid [EPA‐H] groups, respectively). *P < 0.05 vs. CONT; †P < 0.05 vs. MetS.
Cardiac oxidative stress and inflammation
The increases in superoxide generation in myocardial tissue sections shown by staining with dihydroethidium as well as in the activity of NADPH oxidase in LV homogenates in the MetS group were attenuated in both EPA‐L and EPA‐H groups (Figure 3A–C). EPA inhibited the up‐regulation of the expression of genes for the p22phox and gp91phox membrane components and for the Rac1 cytosolic component of NADPH oxidase apparent in the heart of the MetS group (Figure 3D–F).
Figure 3.
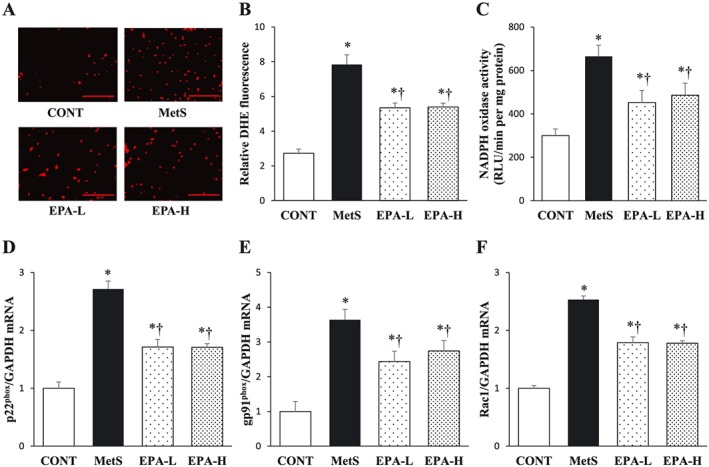
Cardiac oxidative stress in rats of the four experimental groups at 13 weeks of age. (A) Superoxide production as revealed by dihydroethidium staining in interstitial regions of the left ventricular (LV) myocardium. Scale bars, 100 µm. (B) Relative dihydroethidium (DHE) fluorescence intensity determined from sections similar to those in (A). (C) Nicotinamide adenine dinucleotide phosphate (NADPH)‐dependent superoxide production in LV homogenates. Results are expressed as relative light units (RLU) per minute per milligramme of protein. (D–F) Quantitative reverse transcription‐polymerase chain reaction (RT‐PCR) analysis of p22phox, gp91phox and Rac1 mRNAs, respectively. The amount of each mRNA was normalized by that of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) mRNA and then expressed relative to the mean value for the control (CONT) group. All quantitative data are means ± standard error of the mean (n = 10, 10, 11 and 11 for CONT, metabolic syndrome [MetS], low‐dose eicosapentaenoic acid [EPA‐L] and high‐dose eicosapentaenoic acid [EPA‐H] groups, respectively). *P < 0.05 vs. CONT; †P < 0.05 vs. MetS.
Immunohistochemical analysis of the LV myocardium for the monocyte–macrophage marker CD68 showed that the increase in the number of CD68‐positive cells in the MetS group was attenuated in both EPA‐L and EPA‐H groups (Figure 4A,B). EPA inhibited the up‐regulation of MCP‐1 and osteopontin mRNAs in the left ventricle of the MetS group (Figure 4C,D).
Figure 4.
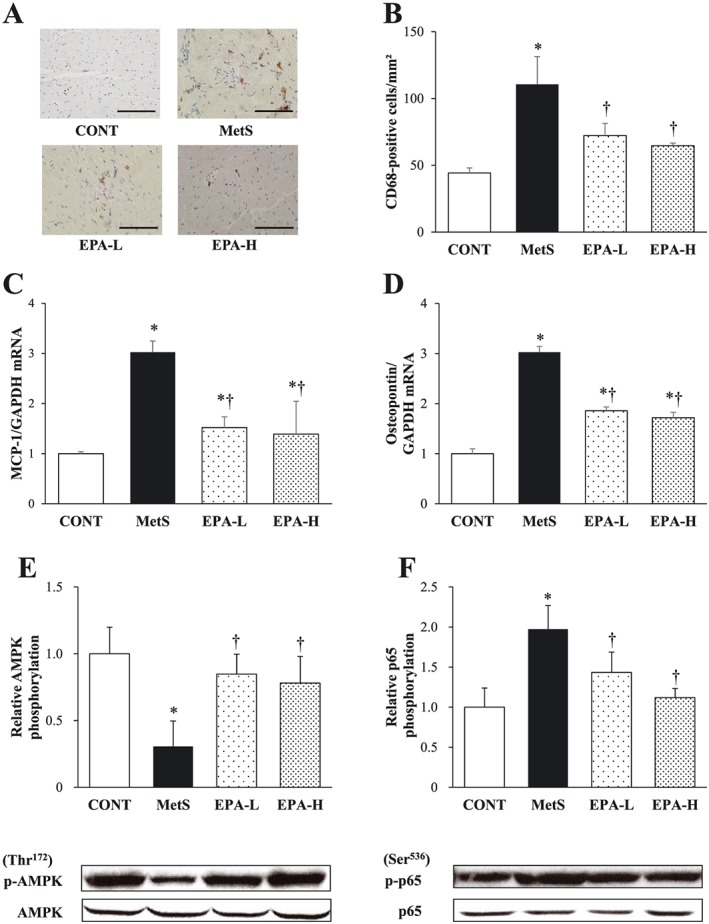
Cardiac inflammation as well as AMPK and NF‐kB activation status in the heart of 13‐week‐old rats in the four experimental groups. (A) Immunohistochemical staining for the monocyte–macrophage marker CD68. Scale bars, 100 µm. (B) Density of CD68‐positive cells in sections similar to those in (A). (C,D) RT and real‐time PCR analysis of MCP‐1 (C) and osteopontin (D) mRNA abundance in the left ventricle. (E,F) Immunoblot analysis of total and phosphorylated (p‐)forms of AMPK (E) and the p65 subunit of NF‐kB (F) in the left ventricle. Representative blots as well as the ratio of phosphorylated to total forms of AMPK or the p65 subunit of NF‐kB are shown. All quantitative data are means ± SEM (n = 10, 10, 11 and 11 for CONT, MetS, EPA‐L and EPA‐H groups, respectively). *P < 0.05 vs. CONT; †P < 0.05 vs. MetS. AMPK, AMP‐activated protein kinase; CONT, control; EPA‐H, high‐dose eicosapentaenoic acid; EPA‐L, low‐dose eicosapentaenoic acid; GAPDH, glyceraldehyde‐3‐phosphate dehydrogenase; MCP‐1, monocyte chemoattractant protein‐1; MetS, metabolic syndrome; NF‐kB, nuclear factor‐kB; PCR, polymerase chain reaction; RT, reverse transcription; SEM, standard error of the mean.
AMP‐activated protein kinase and nuclear factor‐kB activation
The amounts of phosphorylated (activated) forms of AMPK and the p65 subunit of NF‐kB in the left ventricle were decreased and increased, respectively, in the MetS group compared with the CONT group, and these effects were prevented by EPA treatment at either dose (Figure 4E,F).
Adipocyte hypertrophy as well as adipose inflammation and gene expression
The increase in the cross‐sectional area of retroperitoneal fat cells in the MetS group was attenuated in both EPA‐L and EPA‐H groups (Figure 5A,B). Immunohistochemical analysis of visceral fat for CD68 showed that EPA inhibited the increase in the number of macrophages apparent in the MetS group (Figure 5A,C). The number of areas of aggregated CD68‐positive cells surrounding adipocytes in a typical crown‐like pattern was also increased in the MetS group that in the other groups of rats (Figure 5A). EPA attenuated the up‐regulation of MCP‐1, osteopontin and COX‐2 mRNAs apparent in visceral adipose tissue of the MetS group (Figure 5D–F).
Figure 5.
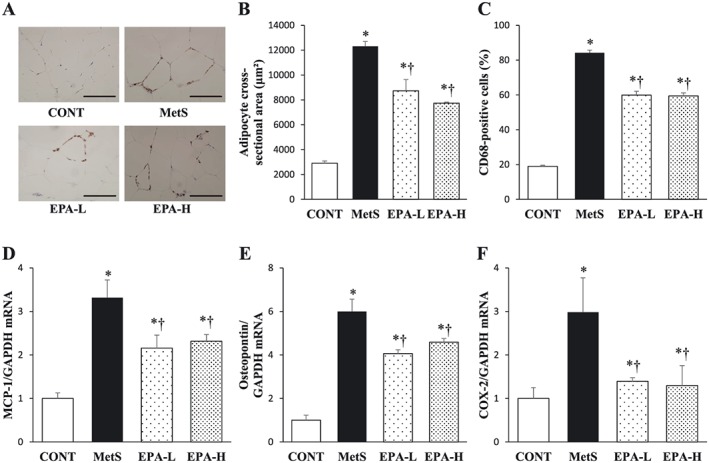
Retroperitoneal adipose tissue inflammation in rats of the four experimental groups at 13 weeks of age. (A) Immunohistochemical staining for the monocyte–macrophage marker CD68. Scale bars, 100 µm. (B) Cross‐sectional area of adipocytes determined from sections similar to those in (A). (C) The number of nuclei for CD68‐positive cells as a percentage of total nuclei as determined from sections similar to those in (A). (D–F) Quantitative reverse transcription‐polymerase chain reaction analysis of monocyte chemoattractant protein‐1 (MCP‐1), osteopontin and cyclooxygenase‐2 (COX‐2) mRNAs, respectively. The amount of each mRNA was normalized by that of glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) mRNA and then expressed relative to the mean value for the control (CONT) group. All quantitative data are means ± standard error of the mean (n = 10, 10, 11 and 11 for CONT, metabolic syndrome [MetS], low‐dose eicosapentaenoic acid [EPA‐L] and high‐dose eicosapentaenoic acid [EPA‐H] groups, respectively). *P < 0.05 vs. CONT; †P < 0.05 vs. MetS.
Discussion
The present study has here shown that highly purified EPA did not affect blood pressure or insulin resistance but attenuated adipocyte hypertrophy and inflammation in visceral fat as well as LV fibrosis, diastolic dysfunction, oxidative stress and inflammation in DS/obese rats. Furthermore, treatment of DS/obese rats with EPA reduced circulating levels of cholesterol but not of triglyceride, increased the circulating adiponectin concentration and induced activation of AMPK and inactivation of NF‐kB in the heart. The beneficial effects of EPA on the heart are likely due, at least in part, to reduced cardiac oxidative stress and inflammation associated with an adiponectin‐induced increase in AMPK activity and consequent down‐regulation of NF‐kB activity.
The influence of EPA on blood pressure and circulating triglyceride levels has been unclear. EPA suppressed the rise in blood pressure in a rat model of hyperinsulinaemia induced by dietary fructose intake 32, whereas n‐3 PUFAs (EPA and docosahexaenoic acid) from fish oil attenuated cardiac fibrosis and cardiac dysfunction without altering pressure gradients induced by aortic constriction in aortic‐banded mice 33, consistent with the present results. Also consistent with these data, insulin resistance was previously found to be accompanied by an elevation of plasma triglyceride levels 34. Unexpectedly, however, a significant decrease in the serum triglyceride concentration was not detected in DS/obese rats treated with EPA, in contrast to data from human clinical studies 35, 36. Although the reason for this discrepancy is uncertain, lipid metabolism may be altered in our rat model.
Glucose tolerance is impaired in association with insulin resistance in DS/obese rats. However, the high dose of EPA attenuated glucose intolerance but not insulin resistance, whereas the low dose had no effect on either of these parameters. Amelioration of glucose intolerance can be achieved through a reduction in insulin resistance or an increase in the insulin secretory capacity of pancreatic β‐cells. Insulin secretion comprises fasting basal secretion and secretion induced by elevated blood glucose levels because of food intake. The OGTT, ITT and homeostasis model assessment of insulin resistance data show that DS/obese rats are insulin resistant compared with DS/lean rats, with the former animals also manifesting an increased rate of basal insulin secretion. The glucose intolerance of DS/obese rats thus likely results from the inadequate insulin secretory response to glucose load 37. Type 2 diabetes is characterized by a delayed or diminished insulin secretory response to glucose rather than by altered basal insulin secretion 38. The results indicate that the high dose of EPA did not affect basal insulin secretion but enhanced the secretory response to glucose. It is thus possible that the high dose of EPA prevented the functional decline of β‐cells. Some previous studies have shown that EPA did not affect insulin resistance 39, 40, consistent with the present results, whereas one study found that EPA ameliorated this condition 41. The mechanisms by which EPA influences glucose metabolism warrant further study.
Obesity and MetS are associated with functional incompetence of adipose tissue accompanied by adipocyte enlargement and chronic inflammation 36. Macrophage accumulation in white fat plays a role in chronic inflammation through the production by these cells of free fatty acids and pro‐inflammatory molecules such as tumour necrosis factor‐α and MCP‐1 4, 39, 40. This study has now shown that the high dose of EPA decreased visceral (retroperitoneal and epididymal) but not subcutaneous fat mass in DS/obese rats. These results seem inconsistent with previous findings that EPA reduced both visceral and subcutaneous fat mass in mice with obesity induced by a high‐fat, high‐sucrose diet 42 and that it did not alter either visceral or subcutaneous fat mass in those with high‐fat diet‐induced obesity 42. The anti‐obesity effect of EPA may thus be dependent on the animal model studied. EPA also attenuated adipocyte hypertrophy as well as the accumulation of macrophages in visceral fat of DS/obese rats. These effects were accompanied by a decrease in gene expression for pro‐inflammatory proteins such as MCP‐1, osteopontin and COX‐2 in this tissue. It is thought that n‐3 PUFAs including EPA inhibit inflammation in adipose tissue by triggering apoptosis in enlarged adipocytes via activation of peroxisome proliferator‐activated receptor γ, by promoting the differentiation of preadipocytes, by suppressing the secretion of pro‐inflammatory cytokines and by up‐regulating the expression and secretion of anti‐inflammatory cytokines through an increase in the number of small adipocytes 20. Indeed, EPA at both low and high doses increased the serum adiponectin concentration in DS/obese rats, consistent with the previous finding that EPA increased the secretion of adiponectin in mice and obese humans 8, 43. In addition, EPA‐derived lipid mediators known as resolvins are also able to inhibit inflammation 44.
The AMPK serves as an important regulator of energy metabolism and functions as a survival factor during exposure to various metabolic stresses as well as during aging. AMPK signalling is also thought to inhibit inflammatory responses induced by activation of the NF‐kB signalling pathway 45. EPA attenuated both the down‐regulation of AMPK activity and the up‐regulation of NF‐kB activity apparent in the heart of DS/obese rats, with these findings being consistent with previous results showing that n‐3 PUFAs inhibit macrophage inflammation by inducing the deacetylation (inactivation) of NF‐kB through activation of an AMPK‐SIRT1 pathway 46. Adiponectin activates AMPK in the mouse heart, with this effect being thought to underlie the cardioprotective effect of adiponectin 47. EPA further increased the serum adiponectin concentration as well as attenuated the decrease in cardiac AMPK activity in DS/obese rats. Down‐regulation of AMPK activity and up‐regulation of NF‐kB signalling in the heart may thus play a role in cardiac remodelling and diastolic dysfunction in DS/obese rats, and the amelioration of heart damage by EPA in these animals may be mediated through activation of AMPK and inactivation of NF‐kB.
Increased oxidative stress is linked to obesity in experimental animals and humans and may play a role in the pathogenesis of MetS 14. Both macrophages and adipocytes are key sources of reactive oxygen species in adipose tissue in obesity. Infiltrated macrophages in such adipose tissue also produce increased amounts of tumour necrosis factor‐α 48. Treatment of DS/obese rats with EPA mitigated increases in both NADPH‐dependent superoxide generation and NADPH oxidase subunit gene expression in the heart, indicating that EPA attenuated cardiac oxidative stress in these animals. Given that AMPK inhibits reactive oxygen species production by NADPH oxidase 49, the present results support a model whereby activation of AMPK by EPA results in the inhibition of NF‐kB signalling and thereby down‐regulates the expression of NADPH oxidase subunit genes and alleviates oxidative stress in the heart of DS/obese rats.
In summary, this study has demonstrated that highly purified EPA ameliorated LV fibrosis, diastolic dysfunction, oxidative stress and inflammation, without lowering blood pressure, in DS/obese rats. EPA did not affect insulin resistance, but it diminished adipocyte hypertrophy and inflammation in visceral fat of these animals. The favourable cardiac effects of EPA are probably attributable, at least in part, to a reduction in cardiac oxidative stress and inflammation associated with increased AMPK activity and reduced NF‐kB activity. Elucidation of the molecular mechanisms of these actions of EPA will require further study.
Conflict of Interest Statement
No conflict of interest was declared.
Contributor Statements
S. I. acquired, analysed and interpreted data as well as drafted the manuscript. Y. S., K. Nagasawa and N. M. acquired and analysed data. Y. Y., A. U. and T. M. critically revised the manuscript for intellectual content. K. Nagata conceived and designed the research, interpreted data, handled funding and supervised the project.
Acknowledgements
We thank Riho Arai, Misato Kamiya, Yuuri Takeshita and Sae Ohura for technical assistance. This work was supported by unrestricted grants from Kyowa Hakko Kirin Co. Ltd (Tokyo, Japan), Mochida Pharmaceutical Co. Ltd (Tokyo, Japan), Mitsubishi Tanabe Pharma Corp. (Osaka, Japan), Takeda Pharmaceutical Co. Ltd (Osaka, Japan), Daiichi‐Sankyo Co. Ltd (Tokyo, Japan), MSD in Japan (Tokyo, Japan), Ajinomoto Pharmaceuticals Co. Ltd (Tokyo, Japan), Astellas Pharma Inc. (Tokyo, Japan) and Dr Kohzo Nagata (Nagoya University) as well as by Management Expenses Grants from the Japanese Government to Nagoya University.
Ito, S. , Sano, Y. , Nagasawa, K. , Matsuura, N. , Yamada, Y. , Uchinaka, A. , Murohara, T. , and Nagata, K. (2016) Highly purified eicosapentaenoic acid ameliorates cardiac injury and adipose tissue inflammation in a rat model of metabolic syndrome. Obesity Science & Practice, 2: 318–329. doi: 10.1002/osp4.50.
References
- 1. Romeo GR, Lee J, Shoelson SE. Metabolic syndrome, insulin resistance, and roles of inflammation – mechanisms and therapeutic targets. Arterioscler Thromb Vasc Biol 2012; 32: 1771–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Matsuzawa Y. Therapy insight: adipocytokines in metabolic syndrome and related cardiovascular disease. Nat Clin Pract Cardiovasc Med 2006; 3: 35–42. [DOI] [PubMed] [Google Scholar]
- 3. Osborn O, Olefsky JM. The cellular and signaling networks linking the immune system and metabolism in disease. Nat Med 2012; 18: 363–374. [DOI] [PubMed] [Google Scholar]
- 4. Hu FB, Bronner L, Willett WC, et al. Fish and omega‐3 fatty acid intake and risk of coronary heart disease in women. Jama 2002; 287: 1815–1821. [DOI] [PubMed] [Google Scholar]
- 5. Calder PC. n‐3 Fatty acids and cardiovascular disease: evidence explained and mechanisms explored. Clin Sci (Lond) 2004; 107: 1–11. [DOI] [PubMed] [Google Scholar]
- 6. Saravanan P, Davidson NC, Schmidt EB, Calder PC. Cardiovascular effects of marine omega‐3 fatty acids. Lancet 2010; 376: 540–550. [DOI] [PubMed] [Google Scholar]
- 7. Yokoyama M, Origasa H, Matsuzaki M, et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): a randomised open‐label, blinded endpoint analysis. Lancet 2007; 369: 1090–1098. [DOI] [PubMed] [Google Scholar]
- 8. Itoh M, Suganami T, Satoh N, et al. Increased adiponectin secretion by highly purified eicosapentaenoic acid in rodent models of obesity and human obese subjects. Arterioscler Thromb Vasc Biol 2007; 27: 1918–1925. [DOI] [PubMed] [Google Scholar]
- 9. Yamada H, Yoshida M, Nakano Y, et al. In vivo and in vitro inhibition of monocyte adhesion to endothelial cells and endothelial adhesion molecules by eicosapentaenoic acid. Arterioscler Thromb Vasc Biol 2008; 28: 2173–2179. [DOI] [PubMed] [Google Scholar]
- 10. Simopoulos AP. Evolutionary aspects of diet, the omega‐6/omega‐3 ratio and genetic variation: nutritional implications for chronic diseases. Biomed Pharmacother 2006; 60: 502–507. [DOI] [PubMed] [Google Scholar]
- 11. Hattori T, Murase T, Ohtake M, et al. Characterization of a new animal model of metabolic syndrome: the DahlS.Z‐Lepr fa/Lepr fa rat. Nutr Diabetes 2011; 1: e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kokura S, Nakagawa S, Hara T, et al. Enhancement of lipid peroxidation and of the antitumor effect of hyperthermia upon combination with oral eicosapentaenoic acid. Cancer Lett 2002; 185: 139–144. [DOI] [PubMed] [Google Scholar]
- 13. Hamazaki T, Urakaze M, Makuta M, et al. Intake of different eicosapentaenoic acid‐containing lipids and fatty acid pattern of plasma lipids in the rats. Lipids 1987; 22: 994–998. [DOI] [PubMed] [Google Scholar]
- 14. Takeshita Y, Watanabe S, Hattori T, et al. Blockade of glucocorticoid receptors with RU486 attenuates cardiac damage and adipose tissue inflammation in a rat model of metabolic syndrome. Hypertens Res 2015; 38: 741–750. [DOI] [PubMed] [Google Scholar]
- 15. Nagata K, Somura F, Obata K, et al. AT1 receptor blockade reduces cardiac calcineurin activity in hypertensive rats. Hypertension 2002; 40: 168–174. [DOI] [PubMed] [Google Scholar]
- 16. Matsuura N, Nagasawa K, Minagawa Y, et al. Restraint stress exacerbates cardiac and adipose tissue pathology via beta‐adrenergic signaling in rats with metabolic syndrome. Am J Physiol Heart Circ Physiol 2015; 308: H1275–1286. [DOI] [PubMed] [Google Scholar]
- 17. Hattori T, Murase T, Takatsu M, et al. Dietary salt restriction improves cardiac and adipose tissue pathology independently of obesity in a rat model of metabolic syndrome. J Am Heart Assoc 2014; 3: e001312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kato MF, Shibata R, Obata K, et al. Pioglitazone attenuates cardiac hypertrophy in rats with salt‐sensitive hypertension: role of activation of AMP‐activated protein kinase and inhibition of Akt. J Hypertens 2008; 26: 1669–1676. [DOI] [PubMed] [Google Scholar]
- 19. Miyachi M, Yazawa H, Furukawa M, et al. Exercise training alters left ventricular geometry and attenuates heart failure in Dahl salt‐sensitive hypertensive rats. Hypertension 2009; 53: 701–707. [DOI] [PubMed] [Google Scholar]
- 20. Matsuura N, Asano C, Nagasawa K, et al. Effects of pioglitazone on cardiac and adipose tissue pathology in rats with metabolic syndrome. Int J Cardiol 2015; 179: 360–369. [DOI] [PubMed] [Google Scholar]
- 21. Nagasawa K, Matsuura N, Takeshita Y, et al. Attenuation of cold stress‐induced exacerbation of cardiac and adipose tissue pathology and metabolic disorders in a rat model of metabolic syndrome by the glucocorticoid receptor antagonist RU486. Nutr Diabetes 2016; 6: e207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Nagata K, Obata K, Xu J, et al. Mineralocorticoid receptor antagonism attenuates cardiac hypertrophy and failure in low‐aldosterone hypertensive rats. Hypertension 2006; 47: 656–664. [DOI] [PubMed] [Google Scholar]
- 23. Takatsu M, Hattori T, Murase T, et al. Comparison of the effects of cilnidipine and amlodipine on cardiac remodeling and diastolic dysfunction in Dahl salt‐sensitive rats. J Hypertens 2012; 30: 1845–1855. [DOI] [PubMed] [Google Scholar]
- 24. Elmarakby AA, Loomis ED, Pollock JS, Pollock DM. NADPH oxidase inhibition attenuates oxidative stress but not hypertension produced by chronic ET‐1. Hypertension 2005; 45: 283–287. [DOI] [PubMed] [Google Scholar]
- 25. Miyata K, Rahman M, Shokoji T, et al. Aldosterone stimulates reactive oxygen species production through activation of NADPH oxidase in rat mesangial cells. J Am Soc Nephrol 2005; 16: 2906–2912. [DOI] [PubMed] [Google Scholar]
- 26. Sakata Y, Yamamoto K, Mano T, et al. Activation of matrix metalloproteinases precedes left ventricular remodeling in hypertensive heart failure rats: its inhibition as a primary effect of angiotensin‐converting enzyme inhibitor. Circulation 2004; 109: 2143–2149. [DOI] [PubMed] [Google Scholar]
- 27. Murase T, Hattori T, Ohtake M, et al. Effects of estrogen on cardiovascular injury in ovariectomized female DahlS.Z‐Lepr fa/Lepr fa rats as a new animal model of metabolic syndrome. Hypertension 2012; 59: 694–704. [DOI] [PubMed] [Google Scholar]
- 28. Yamada T, Nagata K, Cheng XW, et al. Long‐term administration of nifedipine attenuates cardiac remodeling and diastolic heart failure in hypertensive rats. Eur J Pharmacol 2009; 615: 163–170. [DOI] [PubMed] [Google Scholar]
- 29. Murase T, Hattori T, Ohtake M, et al. Cardiac remodeling and diastolic dysfunction in DahlS.Z‐Lepr fa/Lepr fa rats: a new animal model of metabolic syndrome. Hypertens Res 2012; 35: 186–193. [DOI] [PubMed] [Google Scholar]
- 30. Takatsu M, Nakashima C, Takahashi K, et al. Calorie restriction attenuates cardiac remodeling and diastolic dysfunction in a rat model of metabolic syndrome. Hypertension 2013; 62: 957–965. [DOI] [PubMed] [Google Scholar]
- 31. Xu J, Nagata K, Obata K, et al. Nicorandil promotes myocardial capillary and arteriolar growth in the failing heart of Dahl salt‐sensitive hypertensive rats. Hypertension 2005; 46: 719–724. [DOI] [PubMed] [Google Scholar]
- 32. Rousseau D, Helies‐Toussaint C, Moreau D, Raederstorff D, Grynberg A. Dietary n‐3 PUFAs affect the blood pressure rise and cardiac impairments in a hyperinsulinemia rat model in vivo . Am J Physiol Heart Circ Physiol 2003; 285: H1294–1302. [DOI] [PubMed] [Google Scholar]
- 33. Chen J, Shearer GC, Chen Q, et al. Omega‐3 fatty acids prevent pressure overload‐induced cardiac fibrosis through activation of cyclic GMP/protein kinase G signaling in cardiac fibroblasts. Circulation 2011; 123: 584–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hwang IS, Ho H, Hoffman BB, Reaven GM. Fructose‐induced insulin resistance and hypertension in rats. Hypertension 1987; 10: 512–516. [DOI] [PubMed] [Google Scholar]
- 35. Chen C, Yu X, Shao S. Effects of omega‐3 fatty acid supplementation on glucose control and lipid levels in type 2 diabetes: a meta‐analysis. PLoS One 2015; 10: e0139565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Reis CE, Landim KC, Nunes AC, Dullius J. Safety in the hypertriglyceridemia treatment with N‐3 polyunsaturated fatty acids on glucose metabolism in subjects with type 2 diabetes mellitus. Nutr Hosp 2014; 31: 570–576. [DOI] [PubMed] [Google Scholar]
- 37. Szkudelski T. Streptozotocin‐nicotinamide‐induced diabetes in the rat. Characteristics of the experimental model. Exp Biol Med (Maywood) 2012; 237: 481–490. [DOI] [PubMed] [Google Scholar]
- 38. Nugent DA, Smith DM, Jones HB. A review of islet of Langerhans degeneration in rodent models of type 2 diabetes. Toxicol Pathol 2008; 36: 529–551. [DOI] [PubMed] [Google Scholar]
- 39. Hartweg J, Perera R, Montori V, et al. Omega‐3 polyunsaturated fatty acids (PUFA) for type 2 diabetes mellitus. Cochrane Database Syst Rev 2008. Cd003205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Crochemore IC, Souza AF, de Souza AC, Rosado EL. Omega‐3 polyunsaturated fatty acid supplementation does not influence body composition, insulin resistance, and lipemia in women with type 2 diabetes and obesity. Nutr Clin Pract 2012; 27: 553–560. [DOI] [PubMed] [Google Scholar]
- 41. Kalupahana NS, Claycombe KJ, Moustaid‐Moussa N. (n‐3) Fatty acids alleviate adipose tissue inflammation and insulin resistance: mechanistic insights. Adv Nutr 2011; 2: 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sato A, Kawano H, Notsu T, et al. Antiobesity effect of eicosapentaenoic acid in high‐fat/high‐sucrose diet‐induced obesity: importance of hepatic lipogenesis. Diabetes 2010; 59: 2495–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Flachs P, Mohamed‐Ali V, Horakova O, et al. Polyunsaturated fatty acids of marine origin induce adiponectin in mice fed a high‐fat diet. Diabetologia 2006; 49: 394–397. [DOI] [PubMed] [Google Scholar]
- 44. Arita M, Yoshida M, Hong S, et al. Resolvin E1, an endogenous lipid mediator derived from omega‐3 eicosapentaenoic acid, protects against 2,4,6‐trinitrobenzene sulfonic acid‐induced colitis. Proc Natl Acad Sci U S A 2005; 102: 7671–7676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Salminen A, Hyttinen JM, Kaarniranta K. AMP‐activated protein kinase inhibits NF‐kappaB signaling and inflammation: impact on healthspan and lifespan. J Mol Med (Berl) 2011; 89: 667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Xue B, Yang Z, Wang X, Shi H. Omega‐3 polyunsaturated fatty acids antagonize macrophage inflammation via activation of AMPK/SIRT1 pathway. PLoS One 2012; 7: e45990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Dyck JR. The ischemic heart: starving to stimulate the adiponectin‐AMPK signaling axis. Circulation 2007; 116: 2779–2781. [DOI] [PubMed] [Google Scholar]
- 48. Hotamisligil GS, Arner P, Caro JF, Atkinson RL, Spiegelman BM. Increased adipose tissue expression of tumor necrosis factor‐alpha in human obesity and insulin resistance. J Clin Invest 1995; 95: 2409–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang S, Zhang M, Liang B, et al. AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26S proteasomes. Circ Res 2010; 106: 1117–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]