

SUMMARY
Paneth cells are a highly specialized population of intestinal epithelial cells located in the crypt adjacent to Lgr5+ stem cells, from which they differentiate through a process that requires downregulation of the Notch pathway. Their ability to store and release antimicrobial peptides protects the host from intestinal pathogens and controls intestinal inflammation. Here we show that PKCλ/ι is required for Paneth cell differentiation at the level of Atoh1 and Gfi1, through the control of EZH2 stability by direct phosphorylation. The selective inactivation of PKCλ/ι in epithelial cells results in the loss of mature Paneth cells, increased apoptosis and inflammation, and enhanced tumorigenesis. Importantly, PKCλ/ι expression in human Paneth cells decreases with progression of Crohn’s disease. Kaplan-Meier survival analysis of CRC patients revealed that low PRKCI levels correlated with significantly worse patient survival. Therefore, PKCλ/ι is a negative regulator of intestinal inflammation and cancer through its role in Paneth cell homeostasis.
Graphical Abstract
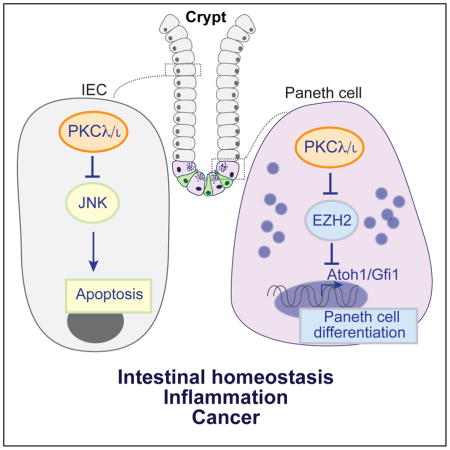
INTRODUCTION
The control of intestinal homeostasis relies on a perfectly orchestrated balance of interactions among the different cell types of the intestinal epithelium, the microbiota, and the immune system (Medema and Vermeulen, 2011). The gut epithelium undergoes continuous self-renewal from intestinal stem cells (ISCs) located in the proliferative crypt (van der Flier and Clevers, 2009). Paneth cells are critical to the control of the ISC niche and the intestinal barrier (Adolph et al., 2013; Clevers and Bevins, 2013). This is a highly specialized population of intestinal epithelial cells (IECs) located adjacent to Lgr5+ ISC, which differentiate from ISCs through upon downregulation of the Notch pathway (Fre et al., 2005; van Es et al., 2005). Paneth cells have large granules containing lysozyme and other peptides, such as defensins/cryptidins, that serve to protect the host from intestinal pathogens (Clevers and Bevins, 2013), and plays a critical role in the control of intestinal inflammation (Adolph et al., 2013). Alterations in the normal function of IECs, especially Paneth cells, contribute to pathologies like inflammatory bowel diseases (IBD), including Crohn’s disease (CD) and ulcerative colitis (UC) (Kaser et al., 2010). Importantly, patients with IBD are at an increased risk for colorectal cancer (CRC) (Jess et al., 2006). Patients with CD often exhibit a reduced number of healthy Paneth cells and decreased expression of defensins in areas of acute inflammation (Wehkamp et al., 2008). Therefore, understanding the signaling cascades that regulate Paneth cell differentiation and function, and their role in the control of intestinal homeostasis and pathology, is critical for the design of new therapies for these diseases.
Here we have addressed this biological problem in the context of the role and mechanisms of action of protein kinase C (PKC) λ/ι. This kinase, along with PKCζ, constitutes the atypical PKC family (Moscat et al., 2009). Both have been implicated in several oncogenic and inflammatory pathways in vitro, but their role in intestinal homeostasis and pathology has only recently begun to be investigated in physiologically relevant mouse models (Calcagno et al., 2010; Llado et al., 2015; Ma et al., 2013; Moscat et al., 2009). In this regard, our laboratory previously reported a specific tumor-suppressor role of PKCζ in intestinal carcinogenesis via the inhibition of metabolic stress reprogramming (Ma et al., 2013), as well as the β-catenin and Yap pathways in ISCs (Llado et al., 2015). In contrast, PKCλ/ι has been proposed by others to be a tumor promoter (Justilien et al., 2014). However, the recently reported analysis of PKC mutations identified in human cancers strongly suggested that most of these mutations led to loss of function, and none would result in a gain-of-function phenotype (Antal et al., 2015). These studies are in good agreement with our data demonstrating that PKCζ is a tumor suppressor in several types of neoplasia, including intestinal cancer (Galvez et al., 2009; Kim et al., 2013; Ma et al., 2013); but they are at odds with the pro-tumorigenic role of PKCλ/ι proposed by others (Murray et al., 2004).
However, when considering cancer etiology and progression, cross-talk between the tumor cells and the surrounding microenvironment must be taken into account. In this regard, the etiology of intestinal cancer is complex because disruption of the epithelial barrier, even under conditions in which inflammation is not the driver of the carcinogenic process, or as consequence of its erosion during IBD, inevitably results in the hyperproduction of inflammatory cytokines that enhance tumor progression (Grivennikov et al., 2012; Karin and Clevers, 2016). Therefore, at least in intestinal carcinogenesis, and most likely in all tumors, the existence of activating or inactivating mutations in a given gene should be put in the context of other factors driving the inflammatory and immunological landscape of the tumor. In this paper we have established the role of PKCλ/ι in the intestinal epithelium, and especially Paneth cells, in inflammation and cancer.
RESULTS
Paneth cell defects in PKCλ/ι-deficient intestinal epithelial cells
PKCλ/ι is widely expressed in the small intestine and colon, as determined by immunoblot analysis of IECs (Figure 1A), which suggested a role for PKCλ/ι in regulating homeostasis in the gastrointestinal tract. Although all IECs of the small intestine and colon expressed PKCλ/ι, its levels were much higher in Paneth cells at the crypt base (Figures 1B and S1A). This was confirmed by double immunofluorescence (IF) analysis of intestinal crypts in which we found an almost complete colocalization of PKCλ/ι with lysozyme (Lyz), a well-established marker of Paneth cells (Figures 1C and S1B). To investigate the potential role of PKCλ/ι in intestinal homeostasis, we crossed Prkcifl/fl mice with Villin-cre mice to generate a mouse line lacking PKCλ/ι specifically in IECs (PrkciIEC-KO mice). PrkciIEC-KO mice were born at the expected Mendelian ratios and developed normally. Epithelial PKCλ/ι deficiency had no effect on the overall structures of the ileum or the colon, and it is unlikely that this was due to compensation because expression of other PKCs was not altered in PrkciIEC-KO crypts (Figure S1C). However, we detected enlargement of the crypts and a severe reduction in the number of Paneth cells, as identified by the absence of granule-containing cells in H&E-stained sections, and a strong reduction in Lyz staining in the mutant mice (Figures 1D–1F). This was also accompanied by the presence of mislocalized Lyz+ cells in the upper crypt and villi of PrkciIEC-KO intestines (Figures 1E and 1G). Staining of markers of other differentiated cell populations did not show alterations in PrkciIEC-KO mice (Figure S1D). Electron microscopy (EM) analysis also showed loss of mature Paneth cells in PrkciIEC-KO crypts, and revealed alterations in the morphology of the remaining Paneth cells, which displayed reduced numbers of granules (Figures 1H and 1I). Further analysis by Lyz staining showed that in contrast to normal Paneth cells in which Lyz is packaged efficiently in the granules, in PrkciIEC-KO mice there was an increase in Paneth cells with reduced numbers of Lyz-containing granules and with diffuse Lyz staining (Figures 1J and 1K). Consistently, PrkciIEC-KO crypts showed decreased expression of Paneth cell-related gene transcripts (Figure 1L). Furthermore, genome-wide transcriptomic analysis identified downregulation of defensin transcripts in the PKCλ/ι-deficient crypts (Figures 1M). These results suggest that PKCλ/ι deficiency in IECs caused a disruption of Paneth cell homeostasis.
Figure 1. Severe alteration of Paneth cells in PKCλ/ι-deficient intestinal epithelia.

(A) Immunoblot analysis of PKCλ/ι in IECs from different intestinal regions of Prkcifl/fl and PrkciIEC-KO mice (n = 3): duodenum (D), jejunum (J), ileum (I) and colon (C). (B) IHC for PKCλ/ι of small intestine and colon sections (n = 6). Scale bars=50 μm (C) Double IF for PKCλ/ι (green) and Lysozyme (Lyz; red) of small intestine (n = 3). Scale bar=50 μm. (D) H&E staining of small intestine sections from Prkcifl/fl and PrkciIEC-KO mice (n = 6). Red arrows point to granulecontaining Paneth cells. Scale bars=50 μm. (E) IHC for Lyz in small intestine from Prkcifl/fl and PrkciIEC-KO mice (n = 6). Red arrow shows mislocalized Paneth cells. Scale bars=50 μm. (F) Quantification of Paneth (Lyz+) cells per crypt in Prkcifl/fl and PrkciIEC-KO mice (n = 6). (G) Quantification of mislocalized Paneth cells per crypt-villus unit in Prkcifl/fl and PrkciIEC-KO mice (n = 6). (H) EM images showing reduced number of healthy granules in Paneth cells in the ileum crypts of PrkciIEC-KO mice as compared to those in Prkcifl/fl mice (n = 3). Scale bar=10 μm. (I) Quantification of the number of granules per Paneth cell counted in EM images. (J) IF for Lyz (red) in Prkcifl/fl and PrkciIEC-KO ileum crypts. Scale bar=10 μm. (K) Quantification of the percent of Paneth cell phenotypes (granule number = or > 6, granule number < 6, or diffuse granule staining) counted in confocal microscopy images (n = 30). (L) qRT-PCR analysis of mRNA levels of Paneth cell-related genes in crypts of Prkcifl/fl and PrkciIEC-KO mice (n = 9). (M) Heat map of genome-wide transcriptomic analysis of crypts from PrkciIEC-KO mice as compared to Prkcifl/fl controls (n = 3). Results are shown as mean ±SEM. *p<0.05, **p<0.01, ***p<0.005, ****p<0.001. See also Figure S1.
PKCλ/ι deficiency increases intestinal epithelial cell death
Histological analysis of PrkciIEC-KO intestines showed a large number of dying cells with pyknotic nuclei at the crypt and increased numbers of apoptotic cells positive for cleaved caspase-3 and TUNEL, as compared to WT controls, in which apoptotic cells were mostly detected only at the villus tip (Figures 2A–2D and S2A and S2B). EM sections also revealed increased numbers of dead cells, including necrotic cells, in PrkciIEC-KO intestines, accompanied by infiltration of macrophages that engulfed dead cells (Figures 2E and 2F). Double IF of Lyz and TUNEL showed that most of the few remaining Paneth cells in the crypts of PrkciIEC-KO mice were TUNEL positive (Figures 2G and S2C), suggesting that immature PKCλ/ι-deficient Paneth cells also underwent cell death under basal conditions. Crypt apoptosis was also detected in the colon of PrkciIEC-KO mice (Figure S2D). NextBio analysis of the upregulated genes in the PrkciIEC-KO crypts showed a strong correlation with the GO category “positive regulation of programmed cell death” (Figure 2H). These results demonstrate that PKCλ/ι loss not only results in defective Paneth cells but also sensitizes crypt cells to apoptosis. Therefore, we next focused our attention on the molecular mechanisms whereby PKCλ/ι regulates crypt cell survival. Interestingly, gene set enrichment analysis (GSEA) identified “AP1_pathway” as a significantly upregulated category in PrkciIEC-KO crypts (Figure 2I). JNK activation is a major component of the AP1-driven cell death cascades (Davis, 2000). Interestingly, the staining of p-JNK and p-c-Jun was dramatically increased in the crypts of PrkciIEC-KO mice as compared to WT (Figure 2J). Immunoblot analysis of crypts confirmed increased JNK and caspase-3 activity associated with PKCλ/ι deficiency (Figure 2K). Since JNK is activated by TNF we hypothesized that PKCλ/ι deletion would sensitize IECs to TNF, which would further increase JNK activation and cell death, thus creating an amplifying loop resulting in increased inflammation. In keeping with this concept, when crypt organoid cultures from PrkciIEC-KO mice were treated with TNF or IFNγ, two cytokines highly abundant in the intestinal inflammatory milieu, the result was enhanced JNK activation and concomitant elevation of cleaved-caspase 3 (Figure 2L). To test whether this was a cell-autonomous effect, we used SW480 as a model system of human intestinal epithelial cells. Basal JNK activation was highly upregulated in SW480 cells in which PKCλ/ι was knockdown by shRNA, which was further enhanced by TNF (Figure 2M). To establish the role of JNK activation in apoptosis triggered by PKCλ/ι deficiency, PKCλ/ι-knocked down cells were treated with a cell-permeable potent and selective dual ATP and substrate mimetic JNK inhibitor, termed 126F4. Pre-incubation of PKCλ/ι-deficient cells with 126F4 rescued the increased apoptosis (Figure 2N and S2E). 126F4, like the previously reported DJNKI-1 (Barr et al., 2002; Bonny et al., 2001), attenuates JNK activation by interacting with the JIP binding site that is essential for both substrate recognition and activation of JNK by upstream kinases (Barr et al., 2002; Bonny et al., 2001; Stebbins et al., 2008). Furthermore, ex vivo cultures of PrkciIEC-KO organoids formed in lower numbers, with smaller size and lesser complexity than WT organoids, and displayed impaired viability (Figures 2O and S2F–S2H), which was rescued by incubation with 126F4 (Figure 2O), but with no effect on Lyz expression (Figure 2P). Collectively, these results demonstrate that the loss of PKCλ/ι in the intestinal epithelium results in impaired Paneth cell homeostasis associated with increased JNK activity and apoptosis of IECs.
Figure 2. PKCλ/ι deficiency increases intestinal epithelial cell death through JNK activation.

(A) IHC for cleaved-caspase 3 (C-Casp3) in Prkcifl/fl and PrkciIEC-KO small intestinal crypts. (n = 3). Scale bar=50 μm. (B) Quantification of the number of C-Casp3-positive cells per crypt. (C) IF for TUNEL (red) of small intestinal sections (n = 3). White dashed line marks the crypt base. Scale bar=50 μm. (D) Quantification of the number of TUNEL-positive cells per crypt. (E) EM images showing a number of dead cells (black circles and top, right) in the ileum crypts of PrkciIEC-KO mice (n = 3). Infiltrating macrophage engulfed the dead cells (right, bottom). Scale bars=10 μm. (F) Quantification of the number of dead cells per crypt. (G) Double IF for TUNEL (red) and Lysozyme (Lyz; green) in Prkcifl/fl and PrkciIEC-KO small intestine. White dashed line marks the crypt base. Scale bar=50 μm. (H) NextBio analysis of genes differentially expressed in PrkciIEC-KO intestinal crypts and Prkcifl/fl controls. Gene signatures corresponding to GO terms “Positive regulation of programmed cell death”. (I) GSEA enrichment in “PID AP1 Pathway” in crypts from Prkcifl/fl and PrkciIEC-KO mice using C2 MSigDB database. (J) IHC for p-JNK or p-c- Jun of small intestine sections from Prkcifl/fl and PrkciIEC-KO mice. (n = 3). Scale bar=50 μm. (K) Immunoblot using antibodies against indicated crypt proteins in Prkcifl/fl and PrkciIEC-KO mice. (L) Immunoblot of small intestinal organoids from Prkcifl/fl and PrkciIEC-KO mice using indicated antibodies. Organoids from each genotype were treated with either TNFα or IFNγfor 8 hr before collection. (M) SW480 shNT and shPKCλ/ι cells were stimulated with TNFα and analyzed by immunoblot at indicated time points. (N) SW480 shNT and shPKCλ/ι cells were preincubated with DMSO or JNK inhibitor (126F4), stimulated with TNFα and cycloheximide (CHX) for 4 hr, and analyzed by immunoblot. (O and P) Representative pictures and quantification of small intestinal organoids from Prkcifl/fl and PrkciIEC-KO mice cultured with or without 126F4 for 3 days (O). qRT-PCR analysis of mRNA level of Lyz in organoids (n = 3) (P). Scale Bar=100 μm. Results are shown as mean ±SEM. *p<0.05, **p<0.01. See also Figure S2.
Loss of PKCλ/ι in the intestinal epithelium results in increased inflammation
Paneth cells provide a first line of defense in the epithelial barrier by secreting antimicrobial peptides, which are important for protection against intestinal inflammation (Adolph et al., 2013). This, together with enhanced IEC apoptosis due to increased JNK activity results in intestinal inflammation. Indeed, NextBio analysis of genes downregulated in PrkciIEC-KO crypts revealed significant correlations with the GO terms “defense response” and “MHC class II antigen presentation” (Figure 3A). We also found spontaneous intestinal inflammation with bowel mucosa thickening, and increased immune cell infiltration in the small intestines of PrkciIEC-KO mice, accompanied by increased expression of inflammatory cytokines (Figures 3B–3D). Histological analysis of colonic sections of PrkciIEC-KO mice showed overall mild inflammation with focal areas of immune cell infiltration and microabscess and increased cytokine expression (Figures S3A–S3C). To investigate whether PKCλ/ι deficiency sensitizes mice to experimental inflammation in the large intestine, we subjected PrkciIEC-KO and control mice to dextran sodium sulphate (DSS). PrkciIEC-KO mice lost significantly more weight than Prkcifl/fl controls, concomitantly displaying colonic shortening with epithelial destruction and extensive intestinal ulceration (Figures 3E–3G). Reduced Ki67 staining was found in PrkciIEC-KO colonic sections, suggesting a defect in the regenerative response (Figure 3G). These results indicate that the loss of PKCλ/ι in the intestinal epithelium renders mice highly susceptible to spontaneous ileitis as well as to spontaneous and experimentally-induced colitis.
Figure 3. Loss of PKCλ/ι in the intestinal epithelium results in increased inflammation.

(A) NextBio analysis of genes differentially expressed in PrkciIEC-KO intestinal crypts and Prkcifl/fl controls. Venn diagrams show the number of common and unique genes in both sets. Gene signatures corresponding to GO terms “defense response” and “MHC protein complex”. (B) H&E staining of Prkcifl/fl and PrkciIEC-KO small intestines (n = 6). Red arrows point to immune cell infiltrates. (C) IF staining for CD45, Gr-1 or F4/80 in Prkcifl/fl and PrkciIEC-KO small intestinal sections (n = 3). (D) qRT-PCR analysis of mRNA levels of inflammatory cytokines in ileum crypts of Prkcifl/fl and PrkciIEC-KO mice (n = 6). (E-G) Percentage of change in body weight (E), representative pictures and quantification of colon length (F), and H&E and Ki67 staining (G) of Prkcifl/fl and PrkciIEC-KO colon sections (n = 4) subjected to a DSS-induced colitis model. (H) Phylum-level analysis of microbiota by 16SrDNA pyrosequencing of fecal pellets at basal conditions and after DSS treatment in Prkcifl/fl and PrkciIEC-KO mice (n = 8). Scale Bars=50 μm. Results are shown as mean ±SEM. *p<0.05, **p<0.01. See also Figure S3.
Because deregulation of intestinal homeostasis and susceptibility to inflammation are often associated with alterations in the commensal bacterial population, we investigated the extent to which bacterial communities were altered in PrkciIEC-KO mice. Analysis of PrkciIEC-KO mice revealed alterations in the microbiota composition, with increased abundance of Proteobacteria and reduction in Firmicutes (Figures 3H and S3D). Importantly, this observed dysbiosis in PrkciIEC-KO mice is consistent with that reported in human IBD patients (Frank et al., 2007). Differences in microbiota distributions were more pronounced after DSS treatment, with PrkciIEC-KO mice showing alterations in also Deferribacteres and Bacterioidetes (Figures 3H and S3D). These data suggest that alterations in Paneth cell and increased IEC apoptosis upon PKCλ/ι deficiency promotes intestinal inflammation through the loss of the mucosal barrier and by modification of the microbiota composition accompanied with bacterial translocation.
PKCλ/ι deficiency promotes tumorigenesis via increased inflammation and dysbiosis
Because inflammation is a well-established promoter of intestinal tumorigenesis, we tested whether the loss of PKCλ/ι in the intestinal epithelium could contribute to intestinal cancer. Of note, previously published data suggested that PKCλ/ι loss in the intestinal epithelium resulted in impaired tumorigenesis in ApcMin/+ mice (Murray et al., 2009). This is counterintuitive with the tumor promoter role of inflammation and the pro-inflammatory phenotype of PrkciIEC-KO mice. To address this question, we crossed PrkciIEC-KO mice with Apcfl/fl mice to generate Apcfl/+;PrkciIEC-KO mice. Interestingly, Apcfl/+;PrkciIEC-KO mice developed more and larger tumors than Apcfl/+ control mice both in the small intestine and colon (Figures 4A and 4B). Histological analysis revealed that tumors from Apcfl/+;PrkciIEC-KO mice were often accompanied by ulceration, with a concomitant increase in proliferation and cell death (Figures 4A–4D). Notably, PKCλ/ι-deficient tumors also showed increased expression of inflammatory cytokines (Figure 4E), indicating that the increased tumorigenesis could be related to the pro-inflammatory environment created by the lack of PKCλ/ι. To further test this hypothesis, we used a colitis-associated CRC model that combines the carcinogen azoxymethane (AOM) and DSS. AOM-DSS-treated PrkciIEC-KO mice developed more and larger tumors than identically treated Prkcifl/fl mice, which also displayed higher rates of proliferation and cell death (Figures S4A–S4D). Collectively, these results are consistent with a model whereby the loss of PKCλ/ι in the intestinal epithelium impairs Paneth cell function, increased IEC apoptosis, resulting in barrier disruption, commensal infiltration and severe inflammation, which promotes intestinal tumorigenesis. To rigorously test this hypothesis, we administered a cocktail of broad-spectrum antibiotics to mice of both genotypes and determined the effect on tumor development. Interestingly, antibiotic treatment impaired the increased tumorigenesis phenotype of Apcfl/+;PrkciIEC-KO mice, which showed a reduction in the number and size of tumors, as compared to untreated mice (Figures 4F and 4G). This effect was accompanied by a decrease in Tnf expression (Figure 4H). Similar results were obtained when the colons of these mice were analyzed (Figures S4F–S4H). These results established that antibiotic treatment diminished the pro-tumorigenic inflammatory conditions of PKCλ/ι-deficient intestines, supporting a role for PKCλ/ι as a non-cell-autonomous tumor suppressor in inflammation-induced intestinal tumorigenesis. Importantly, the reduction in inflammation and cell death promoted by antibiotic treatment did not restore Paneth cells to normal levels in Apcfl/+;PrkciIEC-KO mice (Figures 4I, 4J and S4E). This demonstrated that PKCλ/ι-deficiency in the intestinal epithelium results in defects in Paneth cell homeostasis independent of cell death and the presence of inflammation in the crypt microenvironment. This is in agreement with data in Figures 2O and 2P demonstrating that JNK inhibition in organoids rescued cell death, but not Paneth cell differentiation defects.
Figure 4. Increased inflammation and dysbiosis induced by PKCλ/ι deficiency promotes tumorigenesis.

(A) Macroscopic images and H&E staining of tumors (red dashed circles) in small intestine or colon from Apcfl/+ and Apcfl/+;PrkciIEC-KO mice (n = 10). (B) Quantification of the total tumor numbers and stratification of tumor numbers according to size (n = 10). (C) Double IF for TUNEL (red) and Ki67 (green) in small intestine tumors of Apcfl/+ and Apcfl/+;PrkciIEC-KO mice (n = 3). (D) Quantification of the number of TUNEL- or Ki67-positive cells per field. (E) qRT-PCR analysis of inflammatory cytokine mRNA levels in small intestinal tumors from Apcfl/+ and Apcfl/+;PrkciIEC-KO mice (n = 6). (F) H&E staining of small intestine from Apcfl/+ and Apcfl/+;PrkciIECKO mice with or without antibiotic treatment (n = 5). Black arrows point to tumors. (G) Quantification of the number of all tumors (left) or large tumors (right; size >2 mm) in Apcfl/+ and Apcfl/+;PrkciIEC-KO mice with or without antibiotic (Abx) treatment (n = 5–7). (H) qRT-PCR analysis of Tnf mRNA levels in the tumors of Apcfl/+ and Apcfl/+;PrkciIEC-KO small intestine (n = 6). (I) Double IF for Lysozyme (Lyz; green) and TUNEL (red) in non-tumor lesions of Apcfl/+ and Apcfl/+;PrkciIEC-KO small intestine (n = 3). (J) Quantification of the number of Lyz-positive Paneth cells and TUNEL-positive cells per crypt. Scale Bars=50 μm. Results are shown as mean ±SEM. *p<0.05, **p<0.01, ***p<0.005, ****p<0.001. See also Figure S4.
PKCλ/ι-deficiency impairs Paneth cell differentiation through Atoh1
Because PKCλ/ι’s effects on Paneth cell differentiation and intestinal homeostasis were not a consequence of increased cell death, we next asked whether Paneth cell defects in PrkciIEC-KO mice were actually due to impaired differentiation or to defective maintenance. To address this question, we characterized Paneth cell alterations in PrkciIEC-KO mice by double staining for Lyz and Alcian blue (AB), a marker of goblet cells. While in Prkcifl/fl intestines Lyz expression is localized in the crypt base and does not merge with AB, in PrkciIEC-KO intestines a significant number of Lyz+ cells were found in the villus and upper crypt co-stained for Lyz and AB (Figures 5A and 5B). These data suggest that PKCλ/ι deficiency leads to a failure of intestinal secretory progenitor cells to properly differentiate into Paneth cells, and to default to an intermediate goblet cell-like phenotype. In keeping with this hypothesis, the expression levels of Atoh1 and Gfi1 were lower in the crypts from PrkciIEC-KO mice (Figure 5C). Atoh1 and Gfi1 are critical transcription factors for the differentiation of Paneth cells (Shroyer et al., 2005; Yang et al., 2001). To test whether PKCλ/ι might also regulate Paneth cell maintenance or survival after differentiation, we crossed Prkcifl/fl mice with a Defa6-cre mouse line (Adolph et al., 2013) to create PrkciPC-KO, in which PKCλ/ι is selectively deleted in terminally differentiated Paneth cells. We confirmed the exclusive expression of this Cre line in Paneth cells by crossing with the Rosa-LacZ reporter (Figure S5A). Deletion of PKCλ/ι in PrkciPC-KO Paneth cells was also verified by double staining of PKCλ/ι and Lyz (Figure S5B). Importantly, no defects were found in Paneth cell numbers or Lyz content in PrkciPC-KO mice (Figures S5C–S5E). Of note, these mice did not show spontaneous enteritis or increased sensitivity to colitis (Figures S5F–S5H). These results strongly suggest that PKCλ/ι is important for proper Paneth cell differentiation but not for Paneth cell maintenance/survival in terminally differentiated cells.
Figure 5. PKCλ/ι-deficiency impairs Paneth cell differentiation through Atoh1.

(A) Alcian blue (AB) and Lysozyme (Lyz) staining of small intestine sections from Prkcifl/fl and PrkciIEC-KO mice (n = 5). (B) Quantification of AB and Lyz staining per area. (C) qRT-PCR analysis of Atoh1 and Gfi1 mRNA levels in small intestinal crypts from Prkcifl/fl and PrkciIEC-KO mice (n= 4). (D) Diagram of IEC differentiation. (E-G) qRT-PCR analysis of Paneth cell markers (E), other intestinal epithelial differentiation marker mRNA levels (F) or secretory cell transcriptional factors (G) in crypt organoids, either untreated (Ctrl) or treated with DAPT for 3 days (n = 4). Scale Bars=50 μm. Results are shown as mean ±SEM. *p<0.05, **p<0.01, ***p<0.005, ****p<0.001. See also Figure S5.
A critical event in Paneth cell differentiation is Notch inactivation in precursor cells (Fre et al., 2005; van Es et al., 2005; Yin et al., 2014) (Figure 5D). We next determined whether inactivation of Notch signaling could rescue the defect in Paneth cell differentiation caused by PKCλ/ι deficiency. For this, we incubated organoids of both genotypes with DAPT, a selective and widely used γ-secretase inhibitor of the Notch pathway (Dovey et al., 2001; Yin et al., 2014). Interestingly, incubation with DAPT led to a robust induction of Lyz in Prkcifl/fl but not in PrkciIEC-KO organoids (Figure 5E). The same results were obtained when the levels of Defa6, another marker of Paneth cell differentiation, or those of goblet (Muc2) or enteroendocrine (Chga) cells were determined (Figures 5E and 5F). In contrast, Alpi, which is a marker of enterocytes was not affected by DAPT treatment of PKCλ/ι-deficient organoids (Figure 5F). Notably, the addition of DAPT did not activate the expression of either Atoh1 or Gfi1 in PKCλ/ι-deficient organoids, although it induced a robust response in Prkcifl/fl organoids (Figure 5G). Collectively, these results demonstrate that PKCλ/ι is required for Paneth cell differentiation, most probably through the control of the transcriptional regulation of Atoh1 and Gfi1 upon Notch inactivation.
Epigenetic control of Atoh1 expression by PKCλ/ι
GSEA of the transcriptomic experiment described above provided an important clue in understanding the precise mechanisms whereby PKCλ/ι regulates Atoh1 expression. Interestingly, a category that was also significantly upregulated in PKCλ/ι-deficient crypts was “SUZ12_TARGETS” (Figure 6A). SUZ12 is a critical component of the polycomb repressive complex 2 (PRC2), which is well known for its ability to repress cell differentiation and regulate self-renewal in embryonic stem cells via histone methylation to produce H3K27me3 (Margueron and Reinberg, 2011). We hypothesized that epigenetic modifications controlled by PKCλ/ι could regulate Atoh1 expression. Importantly, ENCODE interrogation of the regulatory region of both Atoh1 and Gfi1 genes revealed the potential for recruitment of SUZ12 and EZH2, the enzymatic subunit of the PRC2 complex, along with the repression marker H3K27me3 (Figure 6B). Conceivably, PKCλ/ι could influence PRC2 activity to control Atoh1 expression. To test this possibility, we performed ChIP analysis in crypts from Prkcifl/fl and PrkciIEC-KO mice and found that both Atoh1 and Gfi1 regulatory regions were highly occupied by EZH2, which correlated with enrichment of the repression marker H3K27me3 (Figure 6C). Immunoblot analysis demonstrated increased levels of EZH2 in PKCλ/ι-deficient crypts (Figure 6D). Similar results were obtained when the levels of EZH2 were determined in 293 cells in which PKCλ/ι had been KO by CRISPR/Cas9 gene editing (Figure 6E). Notably, double IF analysis of intestinal samples from both mouse genotypes showed a strong accumulation of H3K27me3 and EZH2 signals in the crypts of PrkciIEC-KO mice (Figure 6F).
Figure 6. PKCλ/ι regulates Atoh1 by controlling EZH2 phosphorylation and stability.

(A) GSEA of SUZ12 targets in crypts from Prkcifl/fl and PrkciIEC-KO mice using C2 MSigDB database. (B) ENCODE schematic of epigenetic marks in ATOH1 gene. (C) Primer design for ChIP-qPCR analysis of Atoh1 and Gfi1 occupancy of EZH2 and H3K27me3. ChIP assay was performed with anti-EZH2 or anti-H3K27me3 antibodies. Co-immunoprecipitated DNA was examined using qPCR with primers specific for the Atoh1 or Gfi1 regulatory region (n = 3). (D, E) Immunoblot analysis of EZH2 in crypt proteins from Prkcifl/fl and PrkciIEC-KO mice (D) or 293 PKCλ/ι-KO cells generated by the CRISPR/CAS9 system (E). (F) Double IF for Lysozyme (Lyz; red) and H3K27me3 (upper panel) or EZH2 (lower panel) (green). White dashed line marks the crypt base. (n = 3) (G) In vitro phosphorylation of HA-tagged EZH2 immunoprecipitates by baculovirus-expressed recombinant PKCλ/ι with ATPγS followed by PNBM alkylation and immunoblotting for the indicated proteins. (H) CRISPR/CAS9-mediated 293 PKCλ/ι-KO or control cells were incubated with cycloheximide (CHX) and protein stability was determined by immunoblotting at indicated time points. EZH2 protein levels were normalized to actin (n = 5). (I) Small intestinal organoids from WT and PrkciIEC-KO mice were cultured and treated with or without GSK126 for 5 days and analyzed by IF for Lyz (n = 4). Representative pictures and quantification are shown. Scale Bars=25 μm. Results are shown as mean ±SEM. **p<0.01. See also Figure S6.
In vitro kinase assay using purified HA-tagged EZH2 and recombinant PKCλ/ι demonstrated that PKCλ/ι robustly phosphorylated EZH2 (Figure 6G). To map the PKCλ/ι phosphorylation sites in EZH2, we used titanium dioxide (TiO2)-based phosphopetide enrichment on enzymatic digests of in vitro phosphorylation reactions followed by high-performance liquid chromatography tandem mass spectrometry (MS/MS) analysis. Using this approach, we identified T487 as a major PKCλ/ι phosphorylation site and several sites of low abundance such as S690 and T345 (Figure S6). T487 and T345 are reported as sites important for protein degradation (Wu and Zhang, 2011). This suggests that PKCλ/ι might control EZH2 protein stability. Consistently, EZH2 was significantly more stable in cycloheximide-treated PKCλ/ι-deficient cells (Figure 6H). These data strongly suggest that loss of EZH2 phosphorylation upon PKCλ/ι-deficiency increases EZH2 stability and H3K27me3 levels, which represses Atoh1 and Gfi1 expression and Paneth cell differentiation. In support of this model, treatment with GSK126, an inhibitor of EZH2 activity, resulted in upregulation of the Paneth cell population in PrkciIEC-KO organoids (Figure 6I). Collectively, these results demonstrate that PKCλ/ι is required for Paneth cell differentiation through the regulation of PRC2 activity, and that this event is necessary to allow Atoh1 and Gfi1 transcription.
PKCλ/ι is expressed in human intestinal Paneth cells and its expression decreases with progression of Crohn’s disease
Paneth cell dysfunction and ileal inflammation have been linked to the etiology of IBD (Cadwell et al., 2008). Therefore, to investigate the relevance of PKCλ/ι to Paneth cell function in human patients, we used immunohistochemistry (IHC) for PKCλ/ι to analyze 40 ileum tissue sections derived from patients with CD. Expression of PKCλ/ι was first determined in normal mucosa from uninvolved proximal margins containing little or no inflammation. Importantly, PKCλ/ι staining strongly localized with that of Lyz, as demonstrated by double IF (Figure S7A). This indicated that PKCλ/ι expression, similar to our findings in mice, was enriched in human Paneth cells. To study the expression of PKCλ/ι upon disease progression, samples were subdivided by a pathologist, blinded to sample origins, into different categories (mild, moderate and severe) according to the severity of inflammation in the intestinal mucosa. Notably, there was a gradual reduction in PKCλ/ι with CD progression, with minimal expression in the most severe group (Figures 7A and 7B). Furthermore, active inflammatory lesions in the intestinal samples of CD patients showed that low PKCλ/ι/Lyz+ cells were not restricted to the crypt base and displayed a disorganized and diffuse pattern of Lyz expression not confined to the granule, as opposed to that in normal mucosa (Figure 7C, upper panel, and S7A). In addition, a large number of TUNEL+ cells were observed in the crypts of active lesions correlating with low PKCλ/ι expression (Figure 7C, lower panel). Interestingly, double Lyz and AB staining of moderate-severe CD patients demonstrated a large number of mislocalized immature Paneth cells that were positive for both Lyz and AB (Figure 7D). These findings suggest a defect in Paneth cells in CD samples similar to that observed in PrkciIEC-KO mice. Likewise, CD crypts with low PKCλ/ι levels showed enhanced expression of H3K27me3, similar to the phenotype of PKCλ/ι-deficient mice (Figure 7E).
Figure 7. PKCλ/ι levels are downregulated in clinical samples from inflammatory bowel disease patients.

(A) IHC for PKCλ/ι in ileum sections from patients with CD. Subgroups were assigned according to severity of the disease. (n = 10 for each subgroup). Scale Bars=50 μm. (B) Quantification of PKCλ/ι-positive cells per area or per crypt of tissue sections in (A). (C) Double IF for Lysozyme (upper panel; Lyz, red) or TUNEL (lower panel; red) and PKCλ/ι (green) White arrowheads point to TUNEL-positive cells. White dashed line marks the crypt base (n = 3). Scale Bars=25 μm. (D) Double staining for Alcian blue (AB) and Lyz in ileum sections from patients with CD (n = 3). Red arrowheads point the colocalization of AB and Lyz. Scale Bars=50 μm. (E) Double IF for PKCλ/ι (red) and H3K27me3 (green) in ileum sections of patients with CD (n = 3). White dashed line marks the crypt base. Scale Bars=25 μm. (F) PRKCI mRNA levels in IBD patient samples. Data were collected from public data sets of gene expression. (G) Pearson correlation analysis of gene expression between ATOH1 and PRKCI in three different IBD datasets. (H) GSEA of the correlation profile for PRKCI in an IBD clinical cohort (GSE59071) using the “Hallmarks” compilation from Molecular Signature Database (MSigDb, Broad Institute). (I) Enrichment plot corresponding to “INFLAMMATORY_RESPONSE” geneset of GSEA in (H). (J) Kaplan-Meier plot generated by median split of colorectal cancer patients from “Smith Colorectal” dataset according to their PRKCI expression (Precog, Stanford). Results are shown as mean ±SEM. **p<0.01, ***p<0.005, ****p<0.001. See also Figure S7.
To expand on these observations, we analyzed PRKCI mRNA expression in public gene datasets of CD and UC. Consistent with the results of the IHC of CD samples, PKCλ/ι was significantly downregulated in CD and UC samples, as compared to healthy controls, in all seven datasets analyzed (Figures 7F and S7B). Furthermore, there was a significant positive correlation between PRKCI and ATOH1 mRNA levels in three different datasets of IBD samples (Figure 7G), in agreement with a critical role for PKCλ/ι in Paneth cell differentiation through the control of Atoh1 expression. We next performed GSEA of correlation profiles in the IBD clinical dataset (GSE59071) that included the larger number of samples from both CD and UC patients in which PRKCI mRNA was downregulated. Analyses with the H compilation of MsigDB (Broad Institute) revealed that PRKCI levels inversely correlated with gene signatures related to inflammation (Figures 7H and 7I), consistent with a role for PKCλ/ι in controlling intestinal homeostasis.
Because PKCλ/ι-deficient mice had an inflammatory and protumorigenic phenotype, we next investigated the role of PKCλ/ι in human CRC in the context of inflammation. For this, we carried out a molecular concept map analysis of CRC microarray data in the Oncomine database (Rhodes et al., 2007) using signatures of PRKCI-correlated or PRKCI-anticorrelated genes derived from the IBD clinical cohort (GSE59071). A heatmap visualization of significant comparisons showed a strong overlap of PRKCI-correlated genes with those under-expressed in colon and colorectal tumors versus normal samples, in advanced clinical stage versus earlier stages and in patients with poor versus good prognoses (Figure S7C). Accordingly, PRKCI-anticorrelated genes showed significant overlap with those found to be over-expressed in these same categories (Figure S7C). Furthermore, Kaplan-Meier survival analysis of CRC patients stratified according to PRKCI expression revealed that low PRKCI levels correlated with significantly worse patient survival (Figure 7J). All these results support an unanticipated role of PRKCI as a non-cell autonomous tumor suppressor in the context of colon cancer associated with inflammation.
DISCUSSION
Alterations in intestinal epithelial homeostasis contribute to dysbiosis and intestinal inflammation, and are clearly involved in intestinal cancer initiation and progression (Medema and Vermeulen, 2011; Peterson and Artis, 2014). Paneth cells are critical for creating an anti-microbial and nurturing environment that aids in the survival and function of crypt cells and maintenance of the intestinal barrier (Clevers and Bevins, 2013; Sato et al., 2011). Here we show that PKCλ/ι is expressed in the whole intestinal epithelium and prominently in Paneth cells, and demonstrate that it plays a dual role by promoting Paneth cell differentiation and the survival of crypt cells. Thus, PKCλ/ι deficiency, by impairing Paneth cell differentiation, triggers an inflammatory response that is further amplified by increased apoptosis in the whole intestinal epithelium both in the small intestine and colon, and that is driven by the microbiota. Importantly, we also showed that human CD patients displayed reduced PKCλ/ι levels that correlated with impaired Paneth cell development, IEC death, and intestinal inflammation.
Germane to these observations is the fact that PKCλ/ι is required, in a cell-autonomous manner, for the optimal transcriptional activation of the two master regulators of Paneth cell differentiation, Atoh1 and Gf1, through their epigenetic control upon Notch inactivation. This explains why the phenotype of PrkciIEC-KO shared some characteristics with Atoh1 and Gfi1 KO mice (Shroyer et al., 2007; Shroyer et al., 2005; Yang et al., 2001). In this regard Atoh1 KO mice display a complete loss of all secretory lineages whereas Gfi1 KO mice only have a partial reduction in the number of Paneth and goblet cells, with a concomitant increase in enteroendocrine cells (Shroyer et al., 2007; Shroyer et al., 2005; Yang et al., 2001). In contrast PrkciIEC-KO mice, although dramatically affecting the levels of mature Paneth cells, did not show their complete ablation, consistent with the fact that PKCλ/ι inactivation resulted in the reduction but not complete loss of Atoh1 and Gfi1 expression. Furthermore, proof that Paneth cell defects are not secondary to the increased sensitivity of IECs to apoptosis or to inflammation, is that inhibition of JNK activity in organoid cultures rescued the increased apoptosis associated to PKCλ/ι ablation but did not rescue the defect in Paneth cell differentiation. Likewise, antibiotic treatment did not restore normal Paneth cell homeostasis in vivo, although completely abolished intestinal inflammation, which is in keeping with the notion that PKCλ/ι is a cell-autonomous epigenetic regulator of Paneth cell differentiation. Interestingly, treatment of PKCλ/ι-deficient organoids with a pharmacological PRC2 inhibitor fully normalized Paneth cell levels. Therefore, we propose here that epigenetic modification drugs, such as EZH2 inhibitors, could be a valid therapeutic strategy for IBD and intestinal cancer.
In this regard, our results also have important implications beyond the biology of intestinal inflammation, and resolve an important conundrum in the PKC field by definitively establishing a non-cell autonomous tumor suppressor role for PKCλ/ι in intestinal carcinogenesis. Intriguingly, previous data suggested that loss of PKCλ/ι in the intestinal epithelium prevented tumorigenesis in the ApcMin/+ mouse model (Murray et al., 2009), which correlated with more inflammation in the DSS model (Calcagno et al., 2010). These were paradoxical results if it is taken into consideration that inflammation is a tumor promoter state, and a risk factor for intestinal cancer in human patients (Karin and Clevers, 2016). Our results reported here resolve this puzzle by showing that the loss of PKCλ/ι in the epithelium results in more, not less, tumorigenesis. Of note, this effect is detected both under basal and DSS-induced conditions and, importantly, is abrogated in mice in which the microbiota has been obliterated by antibiotic treatment. We suggest that caution should be taken when considering the pharmacological inhibition of PKCλ/ι as an anti-cancer strategy since it will result in enhanced risk of CRC.
EXPERIMENTAL PROCEDURES
A detailed description of the Experimental Procedures utilized in this work can be found in the Supplemental Experimental Procedures. Primers used are described in Table S1.
Mice
All mouse strains were generated in a C57BL/6 background. All mice were born and maintained under pathogen-free conditions. Animal handling and experimental procedures conformed to institutional guidelines (SBP Medical Discovery Institute Institutional Animal Care and Use Committee).
CD Patient Samples
Surgically resected specimens were obtained from CD patients who had been admitted to Hyogo College of Medicine in Japan. Written informed consent was obtained from patients with the protocol approved by the Ethics Committee of Hyogo College of Medicine. De-identified samples were sent to SBP Medical Discovery Institute and used for histological analyses. The study was approved by the Ethics Committee of SBP Medical Discovery Institute.
Statistical Analyses
All the statistical tests are justified for every figure. Data are presented as the mean ± SEM. Significant differences between groups were determined using a Student’s t test (two-tailed unpaired) when the data met the normal distribution tested by D’Agostino test. If the data did not meet this test, a Mann-Whitney test was used. The significance level for statistical testing was set at p < 0.05. All experiments were performed at least two or three times.
Supplementary Material
Acknowledgments
Research was supported by grants from NIH (R01DK108743, R01CA172025 to J.M.; R01CA192642 to M.T.D.-M.; 5P30CA030199 to M.T.D-M. and J.M.; and DK088199, DK44319, and Harvard Digestive Diseases Center DK0034854 to R.S.B; P01CA081534, R01CA168517 to MP). Y.N. was supported by the JSPS Postdoctoral Fellowship for Research Abroad and by the Crohn’s & Colitis Foundation of America. MP holds the Daniel Hays Chair in Cancer Research at the School of Medicine at UCR. We thank Diantha LaVine for the artwork, Maryellen Daston for editing the manuscript, and Wei Liu, the personnel of the Proteomics, Histology, Cell Imaging, Bioinformatics, Genomics, Animal Facility, and Viral Vectors Shared Resources at SBP Medical Discovery Institute, and Malcolm R. Wood in the Core Microscopy Facility at the Scripps Research Institute for technical assistance. We also thank Prof. Hiroshi Seno for critical reading of the manuscript.
Footnotes
AUTHOR CONTRIBUTIONS
M.T.D.-M. and J.M. devised and coordinated the project. Y.N., M.R.-C., N.N., V.L., L.E., A.C., and S.K.D. performed the experiments. M.L., H.I., M.P., and R.S.B. provided materials that made the study possible. Y.N., M.R.-C., V.L., L.E., A.C., S.P., M.T.D.-M., and J.M. analyzed the data. Y.N., M.T.D.-M., and J.M. designed the experiments and wrote the manuscript with help from S.P., M.P., and R.S.B.
ACCESSION NUMBERS
The Gene Expression Omnibus accession number for the microarray data reported in this paper is GSE84118.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adolph TE, Tomczak MF, Niederreiter L, Ko HJ, Bock J, Martinez-Naves E, Glickman JN, Tschurtschenthaler M, Hartwig J, Hosomi S, et al. Paneth cells as a site of origin for intestinal inflammation. Nature. 2013;503:272–276. doi: 10.1038/nature12599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antal CE, Hudson AM, Kang E, Zanca C, Wirth C, Stephenson NL, Trotter EW, Gallegos LL, Miller CJ, Furnari FB, et al. Cancer-associated protein kinase C mutations reveal kinase’s role as tumor suppressor. Cell. 2015;160:489–502. doi: 10.1016/j.cell.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr RK, Kendrick TS, Bogoyevitch MA. Identification of the critical features of a small peptide inhibitor of JNK activity. J Biol Chem. 2002;277:10987–10997. doi: 10.1074/jbc.M107565200. [DOI] [PubMed] [Google Scholar]
- Bonny C, Oberson A, Negri S, Sauser C, Schorderet DF. Cell-permeable peptide inhibitors of JNK: novel blockers of beta-cell death. Diabetes. 2001;50:77–82. doi: 10.2337/diabetes.50.1.77. [DOI] [PubMed] [Google Scholar]
- Cadwell K, Liu JY, Brown SL, Miyoshi H, Loh J, Lennerz JK, Kishi C, Kc W, Carrero JA, Hunt S, et al. A key role for autophagy and the autophagy gene Atg16l1 in mouse and human intestinal Paneth cells. Nature. 2008;456:259–263. doi: 10.1038/nature07416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcagno SR, Li S, Shahid MW, Wallace MB, Leitges M, Fields AP, Murray NR. Protein kinase C iota in the intestinal epithelium protects against dextran sodium sulfateinduced colitis. Inflamm Bowel Dis. 2010 doi: 10.1002/ibd.21547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers HC, Bevins CL. Paneth cells: maestros of the small intestinal crypts. Annu Rev Physiol. 2013;75:289–311. doi: 10.1146/annurev-physiol-030212-183744. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Dovey HF, John V, Anderson JP, Chen LZ, de Saint Andrieu P, Fang LY, Freedman SB, Folmer B, Goldbach E, Holsztynska EJ, et al. Functional gamma-secretase inhibitors reduce beta-amyloid peptide levels in brain. J Neurochem. 2001;76:173–181. doi: 10.1046/j.1471-4159.2001.00012.x. [DOI] [PubMed] [Google Scholar]
- Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature. 2005;435:964–968. doi: 10.1038/nature03589. [DOI] [PubMed] [Google Scholar]
- Galvez AS, Duran A, Linares JF, Pathrose P, Castilla EA, Abu-Baker S, Leitges M, Diaz-Meco MT, Moscat J. Protein kinase Czeta represses the interleukin-6 promoter and impairs tumorigenesis in vivo. Mol Cell Biol. 2009;29:104–115. doi: 10.1128/MCB.01294-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 2012;491:254–258. doi: 10.1038/nature11465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jess T, Loftus EV, Jr, Velayos FS, Harmsen WS, Zinsmeister AR, Smyrk TC, Schleck CD, Tremaine WJ, Melton LJ, 3rd, Munkholm P, et al. Risk of intestinal cancer in inflammatory bowel disease: a population-based study from olmsted county, Minnesota. Gastroenterology. 2006;130:1039–1046. doi: 10.1053/j.gastro.2005.12.037. [DOI] [PubMed] [Google Scholar]
- Justilien V, Walsh MP, Ali SA, Thompson EA, Murray NR, Fields AP. The PRKCI and SOX2 oncogenes are coamplified and cooperate to activate Hedgehog signaling in lung squamous cell carcinoma. Cancer Cell. 2014;25:139–151. doi: 10.1016/j.ccr.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karin M, Clevers H. Reparative inflammation takes charge of tissue regeneration. Nature. 2016;529:307–315. doi: 10.1038/nature17039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaser A, Zeissig S, Blumberg RS. Inflammatory bowel disease. Annu Rev Immunol. 2010;28:573–621. doi: 10.1146/annurev-immunol-030409-101225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Valencia T, Abu-Baker S, Linares J, Lee SJ, Yajima T, Chen J, Eroshkin A, Castilla EA, Brill LM, et al. c-Myc phosphorylation by PKCzeta represses prostate tumorigenesis. Proc Natl Acad Sci U S A. 2013;110:6418–6423. doi: 10.1073/pnas.1221799110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llado V, Nakanishi Y, Duran A, Reina-Campos M, Shelton PM, Linares JF, Yajima T, Campos A, Aza-Blanc P, Leitges M, et al. Repression of Intestinal Stem Cell Function and Tumorigenesis through Direct Phosphorylation of beta-Catenin and Yap by PKCzeta. Cell reports. 2015 doi: 10.1016/j.celrep.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Tao Y, Duran A, Llado V, Galvez A, Barger JF, Castilla EA, Chen J, Yajima T, Porollo A, et al. Control of Nutrient Stress-Induced Metabolic Reprogramming by PKCzeta in Tumorigenesis. Cell. 2013;152:599–611. doi: 10.1016/j.cell.2012.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011;469:343–349. doi: 10.1038/nature09784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema JP, Vermeulen L. Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature. 2011;474:318–326. doi: 10.1038/nature10212. [DOI] [PubMed] [Google Scholar]
- Moscat J, Diaz-Meco MT, Wooten MW. Of the atypical PKCs, Par-4 and p62: recent understandings of the biology and pathology of a PB1-dominated complex. Cell Death Differ. 2009;16:1426–1437. doi: 10.1038/cdd.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray NR, Jamieson L, Yu W, Zhang J, Gokmen-Polar Y, Sier D, Anastasiadis P, Gatalica Z, Thompson EA, Fields AP. Protein kinase Ciota is required for Ras transformation and colon carcinogenesis in vivo. The Journal of cell biology. 2004;164:797–802. doi: 10.1083/jcb.200311011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray NR, Weems J, Braun U, Leitges M, Fields AP. Protein kinase C betaII and PKCiota/lambda: collaborating partners in colon cancer promotion and progression. Cancer Res. 2009;69:656–662. doi: 10.1158/0008-5472.CAN-08-3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LW, Artis D. Intestinal epithelial cells: regulators of barrier function and immune homeostasis. Nat Rev Immunol. 2014;14:141–153. doi: 10.1038/nri3608. [DOI] [PubMed] [Google Scholar]
- Rhodes DR, Kalyana-Sundaram S, Mahavisno V, Varambally R, Yu J, Briggs BB, Barrette TR, Anstet MJ, Kincead-Beal C, Kulkarni P, et al. Oncomine 3.0: genes, pathways, and networks in a collection of 18,000 cancer gene expression profiles. Neoplasia. 2007;9:166–180. doi: 10.1593/neo.07112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato T, van Es JH, Snippert HJ, Stange DE, Vries RG, van den Born M, Barker N, Shroyer NF, van de Wetering M, Clevers H. Paneth cells constitute the niche for Lgr5 stem cells in intestinal crypts. Nature. 2011;469:415–418. doi: 10.1038/nature09637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroyer NF, Helmrath MA, Wang VY, Antalffy B, Henning SJ, Zoghbi HY. Intestine-specific ablation of mouse atonal homolog 1 (Math1) reveals a role in cellular homeostasis. Gastroenterology. 2007;132:2478–2488. doi: 10.1053/j.gastro.2007.03.047. [DOI] [PubMed] [Google Scholar]
- Shroyer NF, Wallis D, Venken KJ, Bellen HJ, Zoghbi HY. Gfi1 functions downstream of Math1 to control intestinal secretory cell subtype allocation and differentiation. Genes & development. 2005;19:2412–2417. doi: 10.1101/gad.1353905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stebbins JL, De SK, Machleidt T, Becattini B, Vazquez J, Kuntzen C, Chen LH, Cellitti JF, Riel-Mehan M, Emdadi A, et al. Identification of a new JNK inhibitor targeting the JNK-JIP interaction site. Proc Natl Acad Sci U S A. 2008;105:16809–16813. doi: 10.1073/pnas.0805677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Flier LG, Clevers H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 2009;71:241–260. doi: 10.1146/annurev.physiol.010908.163145. [DOI] [PubMed] [Google Scholar]
- van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ, Radtke F, et al. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature. 2005;435:959–963. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- Wehkamp J, Koslowski M, Wang G, Stange EF. Barrier dysfunction due to distinct defensin deficiencies in small intestinal and colonic Crohn’s disease. Mucosal Immunol. 2008;1(Suppl 1):S67–74. doi: 10.1038/mi.2008.48. [DOI] [PubMed] [Google Scholar]
- Wu SC, Zhang Y. Cyclin-dependent kinase 1 (CDK1)-mediated phosphorylation of enhancer of zeste 2 (Ezh2) regulates its stability. J Biol Chem. 2011;286:28511–28519. doi: 10.1074/jbc.M111.240515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Bermingham NA, Finegold MJ, Zoghbi HY. Requirement of Math1 for secretory cell lineage commitment in the mouse intestine. Science. 2001;294:2155–2158. doi: 10.1126/science.1065718. [DOI] [PubMed] [Google Scholar]
- Yin X, Farin HF, van Es JH, Clevers H, Langer R, Karp JM. Nicheindependent high-purity cultures of Lgr5+ intestinal stem cells and their progeny. Nat Methods. 2014;11:106–112. doi: 10.1038/nmeth.2737. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.