

Abstract
Objective
Vilazodone is a selective serotonin reuptake inhibitor and 5-HT1A receptor partial agonist approved for the treatment of major depressive disorder (MDD). This report summarizes the safety and tolerability of vilazodone 40 mg/day during short- and long-term treatment of adult MDD.
Methods
Pooled data from two 8-week, double-blind studies of vilazodone (n = 436) vs placebo (n = 433) and data from one 52-week, open-label study (n = 616, vilazodone only) were analyzed. Patients aged 18-70 with DSM-IV-TR-defined MDD received vilazodone or placebo (8-week studies only) once daily, with food, titrated to 40 mg/day over 2 weeks. Safety and tolerability assessments included adverse events (AEs), laboratory tests, vital signs, electrocardiograms, and weight.
Results
The most common AEs in all studies were diarrhea, nausea, and headache. Vilazodone-associated AEs in the two 8-week studies, defined as an incidence rate of ≥5% in the vilazodone group and at least twice that for placebo, were diarrhea (28.0% vs 9.2%), nausea (23.4% vs 5.1%), and insomnia (6.0% vs 2.1%), with the majority reported as mild to moderate and <5% of those patients requiring concomitant (directed) treatment for these conditions. Discontinuation rates due to AEs were 7.1% (vilazodone) and 3.2% (placebo) in the 8-week studies and 20.7% in the 52-week study. Vilazodone had no clinically significant effects on vital signs, laboratory tests, or electrocardiograms.
Conclusion
Vilazodone 40 mg/day was well tolerated during short- and long-term MDD treatment in these trials. Safety profiles associated with 8- and 52-week exposure were consistent.
Keywords: treatment emergent adverse event, adverse event, sexual side effects, weight gain, antidepressant tolerability, antidepressant-induced suicidality
Introduction
Features of the safety and tolerability profile of antidepressants often influence prescribing paradigms for major depressive disorder (MDD).1 The American Psychiatric Association practice guideline for the treatment of MDD indicates initial antidepressant selection should be based largely on the individual’s presenting symptoms and anticipated adverse events (AEs).2 The AE profile is also cited as a reason for noncompliance and premature discontinuation of treatment.3,4
Although practice guidance suggests that extended treatment is of benefit,2 as many as 35% of patients discontinue antidepressant therapy within the first few months,5 with weight gain, sexual dysfunction, and sleep disorders most often reported as the primary reason.3,4 Strategies for determining the most appropriate treatment in relation to management of AEs are often unsuccessful but include changes in the daily drug schedule (fixed vs flexible schedule), symptom-directed therapies, antidepressant switches, drug holidays, and dose reduction.6 Consequently, there is an unmet need for alternative antidepressants that offer reliable efficacy with an improved tolerability profile overall and for individual patients.
The AE profile of an individual antidepressant generally can be predicted by its receptor-binding profile.7 For example, selective serotonin reuptake inhibitor (SSRI) therapy is associated with nausea, which is thought to be caused by stimulation of the 5-HT3 receptors. Similarly, the sexual dysfunction associated with long-term SSRI therapy is thought to be attributable to chronic, unopposed stimulation of the postsynaptic 5-HT2 receptors.8
Vilazodone is a new molecule that is an SSRI and 5-HT1A receptor partial agonist.9,10 Vilazodone has a 30-fold greater potency for serotonin reuptake inhibition than the SSRI fluoxetine with selectivity for the inhibition of norepinephrine or dopamine reuptake inhibition comparable to that of fluoxetine.11 The unique, dual mechanism of vilazodone has been shown, in nonclinical studies, to enhance serotonin levels. Additionally, it has been suggested that the high selectivity of vilazodone for the 5-HT1A receptor may result in improved tolerability.10
Vilazodone has been approved by the United States (US) Food and Drug Administration (FDA) for the treatment of MDD. The efficacy of treatment with vilazodone 40 mg once daily (qd) with food was established in two 8-week placebo-controlled studies, which showed significant improvements in depressive symptoms compared with placebo as measured by the Montgomery-Asberg Depression Rating Scale (MADRS).12,13 The efficacy of vilazodone was further supported by significant changes in MADRS-based therapeutic response rates and significant improvement on multiple other measures relating to depression and global clinical improvement compared with placebo.12,13 Results from efficacy measures from an open-label, 40 mg qd 52-week study supported findings of the 8-week placebo-controlled studies.12-14
This work summarizes findings from these three phase 3 studies of vilazodone and characterizes the safety and tolerability profile of vilazodone during both short- and long-term treatment of MDD in adults.
Methods
Details of study design and conduct for the two phase 3, 8-week placebo-controlled studies (NCT00285376 and NCT00683592) and the 52-week safety study (NCT00644358) have been reported previously.12-14 All studies were conducted in accordance with the Declaration of Helsinki and with Good Clinical Practice guidelines. Protocols were approved by each center’s institutional review board in accordance with the US Code of Federal Regulations. All patients provided informed consent before any study procedures and were compensated for their time. The 52-week study was not a continuation of the 8-week studies.
Patients
In brief, all three phase 3 studies included men and women 18 to 65 or 70 years old with a diagnosis of MDD according to the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Text Revision (DSM-IV-TR) criteria,15 with a 17-item Hamilton Rating Scale for Depression (HAMD-D17) score ≥22 (8-week studies) or ≥18 (52-week study) at screening and baseline. For the two 8-week studies, patients had to have a current major depressive episode (single or recurrent) with a duration of ≥4 weeks and <2 years. Patients with a history of Axis 1 post-traumatic stress disorder (PTSD), eating disorder, or obsessive compulsive disorder (OCD) within the 6 months prior to screening were excluded as were patients with any history of schizophrenia, schizoaffective disorder, or bipolar disorder. Patients with generalized anxiety disorder, social phobia, or simple phobia were permitted to enroll. Additionally, patients exhibiting MDD with postpartum onset, psychotic features, or seasonal pattern; or those meeting the criteria for DSM-IV-TR substance abuse (alcohol or drugs) within 3 months of screening; or judged to pose a serious suicidal risk were also excluded. Other exclusionary conditions included psychotherapy within the preceding 12 weeks, failure to respond to an adequate trial of 2 antidepressants of different drug classes during the same episode, or concurrent use of drugs, including psychotropics or migraine medications, with a serotonergic mechanism of action. Patients with significant comorbid conditions that might interfere with trial participation were excluded at the investigators’ discretion. Exclusion criteria in the 52-week study were similar to those in the 8-week studies, except that patients were required to have a HAM-D score ≥18, were not excluded because of PTSD, eating disorder, OCD or a DSM-IV-TR MDD specifier, and they may not have met DSM-IV-TR criteria for substance abuse or dependence within 1 year of the baseline visit.
Study Design
Each study consisted of 3 periods: washout, screening, and a treatment period, which was either 8 weeks, double-blind, placebo-controlled or 52 weeks, open-label. The washout period in all studies was 4 weeks for monoamine oxidase inhibitors and fluoxetine, 12 weeks for depot neuroleptics, and 2 weeks for all other antidepressants. After the washout and screening periods, eligible patients underwent baseline assessments and, in the 8-week studies, were randomly assigned (1:1) to receive vilazodone or placebo orally once daily. All patients in the 52-week study received vilazodone. In all studies, patients were treated with vilazodone according to a fixed-titration schedule of 10 mg qd for 7 days, followed by 20 mg qd for the next 7 days, and then at the targeted dose of 40 mg qd, and were instructed to take study drug in the morning with food.
Assessments
Safety measures for all studies included reported AEs, clinical laboratory evaluations (chemistry, hematology, and urinalysis), electrocardiograms (ECG), and vital signs (systolic blood pressure [SBP]; diastolic blood pressure [DBP], heart rate, respiration, and weight). AEs were collected via patient self-reporting as well as general investigator solicitation. AEs starting on or after the date of initial study drug administration, preexisting events that worsened in severity after the start of study drug, and AEs that started within 30 days of the discontinuation of study medication were considered treatment emergent (TEAE) for these integrated evaluations. AE coding was based on the Medical Dictionary for Regulatory Activities (MedDRA; version 11.1).
Additional safety assessments relevant to antidepressant use were conducted and included (1) suicidality as evaluated via AE reporting and comprehensive text string search (TSS) results;16 (2) the development of serotonin syndrome as identified through evaluations of TEAEs associated with the clinical features defined by Hunter Serotonin Toxicity Criteria17; (3) the development of mania or hypomania as identified via review of TEAEs associated with substance-induced mood disorder with manic features; and (4) changes in sexual function as assessed by either the Arizona Sexual Experience Scale (ASEX)18 or the Change in Sexual Functioning Questionnaire (CSFQ).19
Analyses
For these analyses, the safety population was defined as all randomized or enrolled patients who received study drug and had at least 1 post-baseline safety assessment. Data for the two 8-week trials were pooled, and treatment group evaluations were based on treatment as received. Informational P values from statistical tests from the pooled 8-week study data are also provided.
TEAEs, serious adverse events (SAEs), and TEAEs leading to study discontinuation were tabulated by MedDRA System Organ Class (SOC) and Preferred Term for each treatment group. Stratifications by sex, age (<55 vs ≥55), and race (Caucasian vs not Caucasian) were evaluated to identify any potential demographic differences in AE profiles. In the pooled 8-week database, TEAEs attributed to vilazodone were defined as events that occurred at an incidence rate of 5% or more in the 40-mg/day vilazodone treatment group and at least twice the rate for placebo.
Safety analyses included summarization of quantitative and qualitative changes from baseline to end of treatment (EOT). EOT was defined as each patient’s last study visit. This corresponded to week 8 or 52 for patients completing the short- or long-term studies, respectively. The last visit at or prior to study termination was used for those patient who discontinued prematurely.
For the analyses of clinical laboratory evaluations, vital signs, and ECGs, the percentage of patients with potentially clinically significant (PCS) high or low values while on study were summarized according to predefined criteria. Treatment group comparisons of the incidence of TEAEs and PCS occurrences for vital signs and weight were evaluated using the Fisher exact test.
For continuous safety measures in the pooled 8-week studies, treatment effects were estimated by the difference in least-squares mean (LSM) changes from baseline to EOT. LSM changes were estimated from an analysis of covariance (ANCOVA) model containing terms for treatment group and study, with the baseline measurement included as a covariate.
The incidences of suicidal ideation and behavior were derived by mapping comprehensive text string search (TSS) results16 to the Columbia Classification Algorithm for Suicide Assessment (C-CASA).20 AE verbatim terms, MedDRA Preferred Terms, and all comment fields within the study databases were included in the text string search. Odds ratios (ORs) for suicidal behavior and ideation were estimated using a conditional logistic regression model containing terms for treatment group and study for the pooled placebo-controlled data.
Results
Baseline Demographics and Primary Disease Characteristics
Demographic and baseline disease characteristics are summarized in Table 1. Mean age at baseline was 41 years for the 8-week studies and 43 years for the 52-week study. Women were more common among the study population (59.5% in the 8-week studies and 67.9% in the 52-week study), and the majority of patients were Caucasian (81.2% in the 8-week studies and 80.0% in the 52-week study). Within the 8-week studies, differences in demographics and disease characteristics between the placebo and vilazodone groups were not considered clinically relevant. Mean baseline MADRS scores were 31.4 in the 8-week studies and 29.9 in the 52-week study, indicating depression of moderate severity on average. The duration of the current episode was more than 6 months for approximately 50% of patients in both treatment groups in the 8-week studies and 64% of patients in the 52-week study.
Table 1. Baselinedemographics Anddisease Characteristics (Safety Populations).
| POOLED 8-WK STUDIES | 52-WK STUDY | ||
|---|---|---|---|
| CHARACTERISTIC | VILAZODONE n = 436 | PLACEBO n = 433 | VILAZODONE N = 599 |
| Sex, n (%) | |||
| Men | 170 (39.0) | 182 (42.0) | 192 (32.1) |
| Women | 266 (61.0) | 251 (58.0) | 407 (67.9) |
| Race, n (%) | |||
| Caucasian | 360 (82.6) | 346 (79.9) | 479 (80.0) |
| Black or African American | 54 (12.4) | 66 (15.2) | 101 (16.9) |
| Asian | 10 (2.3) | 10 (2.3) | 10 (1.7) |
| Other/Unknown | 12 (2.8) | 11 (2.5) | 9 (1.5) |
| Age, mean (SD), y | 40.6 (12.2) | 41.3 (12.6) | 42.8 (12.5) |
| 18–39, n (%) | 196 (45.0) | 197 (45.5) | 234 (39.1) |
| 40–54, n (%) | 180 (41.3) | 157 (36.3) | 252 (42.1) |
| ≥55, n (%) | 60 (13.8) | 79 (18.2) | 113 (18.9) |
| Mean BMI, mean (SD), kg/m2 | 30.2 (7.9) | 30.1 (6.8) | 31.6 (8.0) |
| Underweight (BMI <18.5), n (%) | 5 (1.1) | 3 (0.7) | 3 (0.5) |
| Normal (BMI 18.5–<25.0), n (%) | 128 (29.4) | 105 (24.2) | 119 (19.9) |
| Overweight (BMI 25.0–<30.0), n (%) | 111 (25.5) | 138 (31.9) | 163 (27.2) |
| Obese (BMI ≥ 30.0), n (%) | 192 (44.0) | 182 (42.0) | 311 (51.9) |
| Unknown | 0 | 5 (1.2) | 3 (0.5) |
| Duration of current episode, months, n (%) | |||
| <1 | 1 (0.2) | 0 | 0 |
| 1–<6 | 214 (49.1) | 226 (52.2) | 215 (35.9) |
| 6–12 | 128 (29.4) | 137 (31.6) | 172 (28.7) |
| >12 | 93 (21.3) | 70 (16.2) | 212 (35.4) |
| Baseline MADRS and HAMD-D17 total scores, mean (SD) | |||
| MADRS | 31.4 (3.7) | 31.4 (3.8) | 29.9 (4.4) |
| HAMD-D17 | 24.9 (2.4) | 25.2 (2.5) | 23.1 (3.3) |
BMI, body mass index; HAMD-D17, 17-item Hamilton Rating Scale for Depression;
MADRS, Montgomery-Asberg Depression Rating Scale; SD, standard deviation; wk, week.
Patient Disposition and Vilazodone Exposure
In the 8-week studies, 436 patients were exposed to vilazodone for a total of 59.5 patient-years. During the 52-week study, 599 patients were exposed to vilazodone for a total of 348.2 patient-years, with 314 patients (52.4%) exposed for a minimum of 6 months and 118 patients (19.7%) exposed for a minimum of 12 months.
Figure 1 shows the patient disposition for the 8-week studies and the 52-week study. For the 8-week studies, the percentages of patients discontinued prematurely (20.9% vilazodone and 19.4% placebo) were similar between groups. In the 52-week study, the percentage of patients who completed the study was 42.4%.
Figure 1.
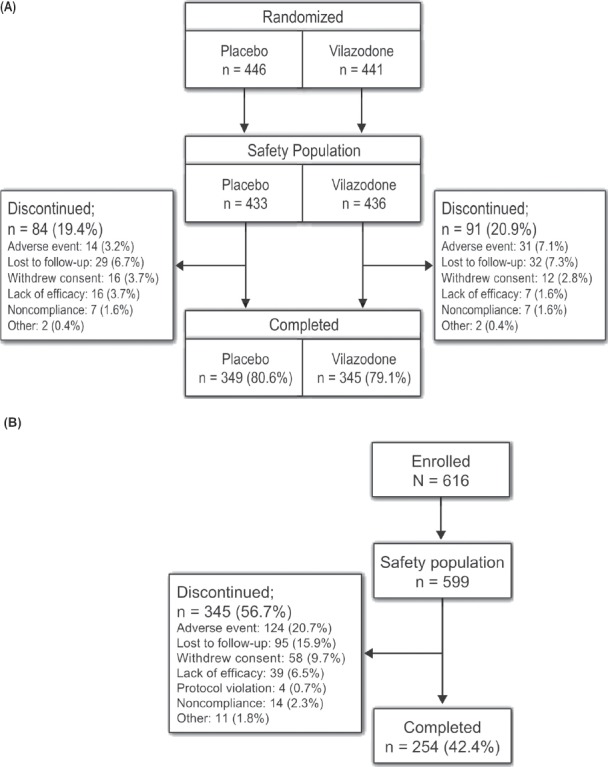
Patient Disposition Through (A) The Pooled 8-Week Studies and (B) The 52-Week Study
Patients who received vilazodone discontinued early from the 8-week studies primarily because they were lost to follow-up (7.3%), had an AE (7.1%), or withdrew consent (2.8%). Patients in the placebo group discontinued primarily because they were lost to follow-up (6.7%), withdrew consent (3.7%), had a lack of efficacy (3.7%) or had an AE (3.2%). The most frequent reasons for early discontinuation from the 52-week study were AEs (20.7%), lost to follow-up (15.9%), withdrawal of consent (9.7%), and lack of efficacy (6.5%).
Treatment-Emergent Adverse Events
The incidence of patients reporting TEAEs in the 8-week studies was significantly greater (P < 0.001) in the vilazodone group (358 of 436 patients, 82.1%) than in the placebo group (280 of 433 patients, 64.7%). Table 2 shows all TEAEs that occurred in ≥5.0% of patients. In the placebo group, 91.4% of patients reporting TEAEs reported a maximum severity of mild to moderate compared with 90.8% of patients in the vilazodone group, with 55.4% of all TEAEs reported as mild in the placebo group compared with 60.4% in the vilazodone group. The 3 most common TEAEs overall in the vilazodone group were diarrhea (28.0% vs 9.2% in the placebo group), nausea (23.4% vs 5.1% in the placebo group), and headache (13.3% vs 12.0% in the placebo group). Diarrhea and nausea occurred in ≥5% of vilazodone patients and at least at twice the rate of placebo, as did insomnia (6.0% vs 2.1% in the placebo group).
Table 2. Treatment-Emergent Adverse Events Occurring In ≥5.0% Of Patients In Any Treatment Group In Phase 3 Studies By Preferred Term.
| POOLED 8-WK STUDIES | ||||||
|---|---|---|---|---|---|---|
| PREFERRED TERMa | VILAZODONE n = 436 | PLACEBO n = 433 | ||||
| Patients with ≥1 TEAE, n (%) | 358 (82.1) | 280 (64.7) | ||||
| Diarrhea | 122 (28.0) | 40 (9.2) | ||||
| Nausea | 102 (23.4) | 22 (5.1) | ||||
| Headache | 58 (13.3) | 52 (12.0) | ||||
| Dizziness | 37 (8.5) | 20 (4.6) | ||||
| Dry mouth | 35 (8.0) | 22 (5.1) | ||||
| Insomnia | 26 (6.0) | 9 (2.1) | ||||
| Nasopharyngitis | 23 (5.3) | 20 (4.6) | ||||
| Upper respiratory tract infection | 19 (4.4) | 33 (7.6) | ||||
| 52-WK STUDY—VILAZODONE, N = 599 | ||||||
| Patients with ≥1 TEAE, n (%) | 562 (93.8) | |||||
| PREFERRED TERM,a n (%) | ||||||
| Diarrhea | 215 (35.9) | Dizziness | 64 (10.7) | Nasopharyngitis | 45 (7.5) | |
| Nausea | 189 (31.6) | Abnormal dreams | 62 (10.4) | Vomiting | 44 (7.3) | |
| Headache | 122 (20.4) | Somnolence | 59 (9.8) | Anxiety | 36 (6.0) | |
| Upper respiratory tract infection | 82 (13.7) | Weight increase | 57 (9.5) | Urinary tract infection | 35 (5.8) | |
| Insomnia | 76 (12.7) | Increased appetite | 54 (9.0) | Back pain | 33 (5.5) | |
| Dry mouth | 66 (11.0) | Fatigue | 49 (8.2) | |||
MedDRA, Medical Dictionary for Regulatory Activities; TEAE, treatment-emergent adverse event; wk, week.
MedDRA, version 11.1.
In the 52-week study, 562 of 599 (93.8%) patients experienced one or more TEAE. The most frequent TEAEs were diarrhea (35.9%), nausea (31.6%), and headache (20.4%). TEAEs occurring in the 52-week study were mostly mild (55.6%) with 84.2% of patients reporting a TEAE reporting a maximum severity of mild to moderate.
Initial Onset and Duration of the Most Common Adverse Events
The time to initial onset of either diarrhea or nausea was significantly shorter on average for the vilazodone group compared with the placebo group (P < 0.001 for both TEAEs). Most cases of diarrhea and nausea in the vilazodone group had onset during the titration period (i.e., within the first 2 weeks). The median onset of the first occurrence of diarrhea for vilazodone and placebo was 2 days and 8 days, with a median duration of 8 days and 4 days, respectively. Only 3.7% of vilazodone patients experiencing diarrhea required treatment with concomitant medications and none of these episodes were reported as severe. Four patients discontinued due to diarrhea (0.9%). Median onset of the first occurrence of nausea was 2 days for vilazodone and 6.5 days for placebo (P < 0.001) with a median duration, respectively, of 5 days and 4.5 days. Few patients (n = 5; 1.1%) who experienced nausea required treatment for the AE and few discontinued as a result (n = 2; 0.5%).
In the 52-week study, median onset of the first occurrence and median duration were 4 days and 7 days for diarrhea and 5 days and 7 days for nausea. As in the 8-week studies, <5% of patients (n = 29) received concomitant drug therapy for diarrhea; 2 of these patients experienced diarrhea reported as severe. Similarly, 3.7% of patients took concomitant medications to treat nausea and 8 of these patients reported nausea as severe.
For the evaluations of onset and duration of insomnia, TEAEs coding to the MedDRA Preferred Terms of “insomnia,” “initial insomnia,” “middle insomnia,” “early morning waking,” and “hyposomnia” were grouped together. The median onset of the first occurrence of insomnia for the vilazodone and placebo groups was 11 days and 9 days, respectively, and the median duration was 10 days and 14 days, respectively, in the 8-week studies. In the 52-week study, median onset of the first insomnia occurrence and median insomnia duration were 19 days and 25 days, respectively. No cases of hyposomnia occurred in any of the 3 studies.
Serious Adverse Events
In the 8-week studies, 9 SAEs were reported for 7 of 433 patients (1.6%) receiving placebo and 11 SAEs were observed for 9 of 436 patients (2.1%) receiving vilazodone. The only SAEs that occurred in more than 1 patient were suicidal ideation (0 with vilazodone; 3 with placebo) and depression (1 with vilazodone; 2 with placebo). Depression was the only SAE deemed possibly related and all others were judged unlikely or not related to study medication.
In the 52-week study, 23 of 599 patients (3.8%) experienced 33 SAEs while taking vilazodone. Infections and infestations was the most common system organ class (5 patients, 0.8%), including pneumonia in 2 patients (0.3%). All other SAEs occurred in single patients. Serotonin syndrome and panic attack were deemed probably and chest discomfort was deemed possibly related to study medication. All other SAEs were judged unlikely or not related to study medication. There was no pattern to SAEs with regard to MedDRA Preferred Terms or SOCs.
Treatment-Emergent Adverse Events Leading to Discontinuation
TEAEs led to discontinuation for 31 of 436 patients (7.1%) receiving vilazodone and 14 of 433 patients (3.2%) receiving placebo in the 8-week studies and no specific TEAE led to discontinuation in ≥1% of patients receiving vilazodone. TEAEs leading to discontinuation in more than 1 patient in the vilazodone group were diarrhea and palpitations (4 patients each) and nausea, depression, and fatigue (2 patients each). In the placebo group, suicidal ideation and headache (3 patients each) were reported in >1 patient.
During the 52-week study, 124 of 599 patients (20.7%) experienced a TEAE that led to discontinuation. Specific TEAEs leading to discontinuation in ≥1% of patients were nausea (8 patients, 1.3%), diarrhea (7 patients, 1.2%), and anxiety (6 patients, 1.0%).
Clinical Laboratory Evaluations
In the 8-week studies, few patients (n ≤ 6 for any parameter) in either group had PCS abnormalities for any of the evaluated clinical chemistry, hematology, or urinalysis parameters. PCS elevations in liver function tests were observed in 4 patients in the vilazodone group: aspartate aminotransferase and gamma glutamyl transpeptidase, 1 patient (0.3%) each (>3 times the upper limit of normal [ULN] range); bilirubin, 2 patients (0.5%) (>1.5 × ULN), compared with 3 patients in the placebo group: gamma glutamyl transpeptidase, 1 patient (0.3%); bilirubin, 2 patients (0.5%).
Similarly, few patients treated with vilazodone (n ≤ 9 for any parameter) had PCS abnormalities for any of the evaluated laboratory parameters in the 52-week study (including liver function abnormalities). No patients in the 8-week or 52-week studies met the criteria for Hy’s law (>3 × ULN for aspartate aminotransferase and >2 × ULN for total bilirubin, a prognostic indicator of drug-induced liver injury).21
Vital Signs and ECG
Changes from baseline to EOT in vital signs are tabulated by treatment group for the 8- and 52-week studies in Table 3. The incidences of PCS high or low vital signs and ECG values while on study are shown in Table 4 along with the PCS criteria that were utilized. LSM changes from baseline to EOT in PR interval (PR), QRS interval (QRS), and QTc (Fridericia) interval corrected for heart rate (QTc) for vilazodone compared with placebo are shown in Table 5.
Table 3. Mean Change From Baseline To End Of Treatment In Vital Signs In The Pooled 8-Week Or 52-Week Studies.
| POOLED 8-WK STUDIES | 52-WK STUDY N = 599 | |||||||
|---|---|---|---|---|---|---|---|---|
| VILAZODONE n = 436 | PLACEBO n = 433 | |||||||
| PARAMETER | BASELINE, MEAN (SD) | LSM (SE), CHANGE AT WK 8/EOT | BASELINE, MEAN (SD) | LSM (SE), CHANGE AT WK 8/EOT | P VALUEa | BASELINE, MEAN (SD) | CHANGE AT WK 52/EOT, MEAN (SD) | |
| SBP, mm Hg | 120.6 (12.8) | −0.2 (0.5) | 119.5 (11.6) | −0.6 (0.5) | 0.629 | 121.6 (13.6) | 0.4 (12.0) | |
| DBP, mm Hg | 75.9 (9.2) | 0.5 (0.3) | 76.4 (8.7) | −0.5 (0.3) | 0.046 | 78.4 (8.5) | 0.3 (8.3) | |
| Pulse, bpm | 72.9 (9.8) | 0.5 (0.4) | 73.0 (9.6) | 1.0 (0.4) | 0.362 | 73.3 (9.7) | 1.6 (11.0) | |
| Weight, kg | 85.8 (23.6) | 0.2 (0.1) | 86.5 (20.9) | 0.2 (0.1) | 0.886 | 89.6 (23.8) | 1.0 (4.7) | |
ANCOVA, analysis of covariance; bpm, beats per minute; DBP, diastolic blood pressure; EOT, end of treatment; LSM, least squares mean; SBP, systolic blood pressure; SD, standard deviation; SE, standard error; wk, week.
P value for treatment group difference based on difference in LSM change from baseline from an ANCOVA model with terms for treatment group and study with baseline value included as a covariate.
Table 4. Patients With Potentially Clinically Significant Values For Vital Signs And Ecg Parameters In The 8-Week And 52-Week Studies.
| FREQUENCY, n/n AT RISK (%) | |||||||
|---|---|---|---|---|---|---|---|
| POOLED 8-WK STUDIES | 52-WK STUDY | ||||||
| PARAMETER | PCS LEVEL | DEFINITIONa | VILAZODONE n = 436 | PLACEBO n = 433 | P VALUEb | N = 599 | |
| SBP, mm Hg | High | ≥180 and ↑ ≥20 | 2/431 (0.5) | 0/432 (0) | 0.249 | 4/596 (0.7) | |
| Low | ≤80 and ↓ ≥20 | 0/431 (0) | 0/432 (0) | >0.999 | 0/597 (0) | ||
| DBP, mm Hg | High | ≥110 and ↑ ≥10 | 0/430 (0) | 1/431 (0.2) | >0.999 | 2/597 (0.3) | |
| Low | ≤50 and ↓ ≥15 | 0/430 (0) | 0/432 (0) | >0.999 | 5/596 (0.8) | ||
| Pulse, bpm | High | ≥120 and ↑ ≥15 | 2/432 (0.5) | 0/432 (0) | 0.499 | 1/597 (0.2) | |
| Low | ≤50 and ↓ ≥15 | 0/431 (0) | 2/432 (0.5) | 0.499 | 1/595 (0.2) | ||
| Weight | High | >7% ↑ | 4/432 (0.9) | 5/430 (1.2) | 0.752 | 92/597 (15.4) | |
| Low | >7% ↓ | 6/432 (1.4) | 6/430 (1.4) | >0.999 | 36/597 (6.0) | ||
| Heart rate, bpm | High | ≥120 and ↑ ≥15 | 0/390 (0) | 0/388 (0) | >0.999 | 0/541 (0) | |
| Low | ≤50 and ↓ ≥15 | 1/375 (0.3) | 1/374 (0.3) | >0.999 | 2/516 (0.4) | ||
| PR interval, ms | High | ≥250 | 0/388 (0) | 0/384 (0) | >0.999 | 0/540 (0) | |
| QRS interval, ms | High | ≥150 | 0/388 (0) | 0/386 (0) | >0.999 | 1/541 (0.2) | |
| QTcF, ms | High | ≥500 | 0/388 (0) | 0/386 (0) | >0.999 | 0/541 (0) | |
bpm, beats per minute; ms, millisecond; QTcF, QT interval corrected (Fridericia); wk, week.
Increases and decreases are with respect to baseline values.
P values for treatment group difference based on the Fisher exact test for the pooled 8-week studies.
Table 5. Mean Change From Baseline To Eot In Ecg Parameters In The Pooled 8-Week Or 52-Week Studies.
| POOLED 8-WK STUDIES | ||||||||
|---|---|---|---|---|---|---|---|---|
| VILAZODONE n = 436 | PLACEBO n = 433 | 52-WK STUDY N = 599 | ||||||
| PARAMETER | BASELINE, MEAN (SD) | LSM (SE), CHANGE AT WK 8/EOT | BASELINE, MEAN (SD) | LSM (SE), CHANGE AT WK 8/EOT | P VALUEa | BASELINE, MEAN (SD) | CHANGE AT WK 52/EOT, MEAN (SD) | |
| Heart rate, bpm | 67.3 (11.1) | 1.9 (0.5) | 66.9 (10.0) | 3.0 (0.4) | 0.088 | 66.7 (10.5) | 3.6 (10.2) | |
| PR interval, ms | 155.2 (20.5) | −0.9 (0.6) | 157.0 (20.5) | −0.2 (0.6) | 0.428 | 158.4 (22.0) | −1.7 (13.3) | |
| QRS interval, ms | 88.6 (9.7) | −1.3 (0.4) | 87.7 (9.6) | −0.1 (0.4) | 0.022 | 87.5 (10.2) | −0.9 (6.9) | |
| QTcB, ms | 405.6 (24.6) | −0.6 (0.9) | 403.1 (24.4) | 1.7 (0.9) | 0.072 | 401.6 (24.6) | 2.1 (22.0) | |
| QTcF, ms | 398.9 (21.7) | −2.4 (0.8) | 396.7 (21.7) | −1.3 (0.7) | 0.309 | 395.6 (21.4) | −1.3 (18.2) | |
| QT interval, ms | 386.2 (30.2) | −5.7 (1.1) | 384.3 (28.0) | −6.8 (1.1) | 0.481 | 383.9 (28.0) | −8.0 (24.0) | |
ANCOVA, analysis of covariance; bpm, beats per minute; EOT, end of treatment; LSM, least-squares mean; ms, millisecond; QTcB, QT interval corrected (Bazett); QTcF, QT interval corrected (Fridericia); SD, standard deviation; SE, standard error; wk, week.
P value for treatment group difference based on difference in LSM change from baseline from an ANCOVA model with terms for treatment group and study with baseline value included as a covariate.
Mean body weight change from baseline to EOT was 0.2 kg in both treatment groups in the 8-week studies (Table 3). PCS weight increases >7% were observed in 4 vilazodone patients (0.9%) and 5 placebo patients (1.2%), and weight PCS decreases >7% were seen in 6 patients (1.4%) for both groups (Table 4). In the 52-week study, the mean change in body weight at EOT was 1.0 kg for all patients and, for patients remaining in the study at week 52, was 1.7 kg. PCS weight increases were observed in 92 patients (15.4%), and PCS weight reductions were observed in 36 patients (6.0%). Assessed at 6 months, mean (SD) weight change for patients with normal baseline weight (body mass index [BMI] of 18.5 to <25.0 kg/m2; n = 95) was 1.3 (3.5) kg; for overweight (BMI of 25.0 to <30.0 kg/m2; n = 136) and obese (BMI ≥30.0 kg/m2; n = 258) patients, mean (SD) weight increases were 1.6 (3.7) kg and 1.0 (5.9) kg, respectively. The reported incidences of TEAEs of weight increase and weight decrease were 9.5% (57 patients) and 1.7% (10 patients), respectively.14
Adverse Events of Special Interest
In the 8-week studies, 1 patient (0.2%) each in the vilazodone and placebo groups met the C-CASA criteria for suicidal behavior based on TSS. C-CASA-defined suicidal ideation was identified for 1 patient (0.2%) in the vilazodone group and 3 patients (0.7%) in the placebo group based on TSS. There was no increased likelihood in either suicidal ideation (OR = 0.3, P = 0.337) or behavior (OR = 1.0, P = 0.997) associated with vilazodone treatment. In the 52-week study, suicidal ideation and suicidal behavior were identified for 7 patients (1.2%) and 2 patients (0.3%), respectively. No completed suicides occurred.
No patients in the 8-week studies experienced serotonin syndrome according to Hunter’s criteria. One patient in the 52-week study experienced severe serotonin syndrome due to an overdose with study drug associated with non-adherence to study drug scheduling. No patients in the 8-week studies were identified as potentially having mania or manic symptoms associated with vilazodone. Two patients (0.3%) in the 52-week study experienced mania or manic symptoms associated with vilazodone. Both patients withdrew from the study.
In the first 8-week study,12 LSM change from baseline to EOT in ASEX total score in the vilazodone group vs the placebo group was 0.8 versus –1.0 (P = 0.011) for men and –1.2 versus 0.1 (P = 0.054) for women (decrease indicates improved score). In the second 8-week study, LSM change from baseline to EOT in CSFQ score in the vilazodone group vs the placebo group was 0.5 versus 1.7 (P = 0.211) for men and 1.9 versus 2.2 (P = 0.682) for women (increase indicates improved score). In the 52-week study, mean change in CSFQ total score during vilazodone treatment was 1.5 in men and 2.7 in women.
During the 8-week studies, 8.0% of patients in the vilazodone group and 0.9% in the placebo group reported at least one TEAE related to sexual function, with libido decrease the most frequently reported: 3.7% of the vilazodone group (males, 4.7%; females, 3.0%) and 0.2% of the placebo group (males, 0%; females, 0.4%). In the 52-week study, 15.1% of patients reported at least one TEAE related to sexual function. As in the 8-week studies, libido decreased was the most commonly reported: 4.3% of the total population (males, 5.7%; females, 3.7%).
Safety Evaluations of Demographic Subgroups
In Phase 3 studies, there were no clinically meaningful differences in subgroups defined by age, sex, and race from the patterns observed in the total population for TEAEs, SAEs, or TEAEs leading to discontinuation. Diarrhea was more frequent in older patients (≥55 years of age) and nausea was more frequent in younger patients. Dizziness was also reported more frequently in younger patients. Vomiting and somnolence were observed at a higher percentage in non-Caucasians compared with Caucasians. Female patients reported gastrointestinal TEAEs more often than male patients (nausea was reported more often for females and diarrhea more often for males). Females also reported nervous system TEAEs (especially headache) and infections and infestations (particularly nasopharyngitis) more often than males.
With regard to SAEs and TEAEs leading to discontinuation, SAEs occurred more often in patients ≥55 years of age during 8 weeks of vilazodone treatment and more often in males during 52 weeks of vilazodone treatment. TEAEs leading to discontinuation in the 8-week studies were greater for patients who were ≥55 years of age or Caucasian. In the 52-week study, TEAEs leading to discontinuation occurred more often for patients who were ≥55 years of age, female, or Caucasian.
Discussion
This analysis of safety and tolerability data from the phase 3 studies found that in adult patients with MDD, vilazodone 40 mg/day is generally well tolerated during up to 52 weeks of treatment. In the 8-week studies, the most common TEAEs associated with vilazodone occurring at least at twice the rate as with placebo were diarrhea, nausea, and insomnia, which are commonly reported with serotonergic medications. TEAEs were typically mild to moderate and most patients did not require directed treatment with concomitant medications. Most diarrhea and/or nausea first occurred during the initial 2 weeks of titration and were not a cause for study discontinuation. Similarly, the percentage of subjects who experienced an SAE while taking vilazodone was low, and there was no apparent pattern to SAEs based on SOC or preferred term.
No clinically concerning effect of vilazodone on laboratory test results or vital signs was observed. Changes from baseline in SBP, DBP, and pulse were similar between the vilazodone and placebo groups in the 8-week studies and incidences of PCS high or low values during both short- and long-term treatment were low in the vilazodone groups with all incidence rates ≤0.5% in the 8-week studies and ≤0.8% in the 52-week study. Change in weight with vilazodone in the 8-week studies was the same as that with placebo (0.2 kg). Mean weight change in the 52-week study was 1.0 kg for the safety population and 1.7 kg for the subset of patients completing the study, which compares favorably with average gains of 6.8 to 10.8 kg reported after 6 to 12 months of therapy with other SSRIs.22 However, the 15.4% of patients who experienced a >7% increase in body weight during up to 1 year of exposure suggests that a subset of patients may be prone to weight gain. Further study is needed to determine whether this weight gain is a direct result of vilazodone treatment or an indirect result of clinical improvement.
Vilazodone did not demonstrate any clinically significant effects on ECG parameters or any signal of proarrhythmic potential. This finding is consistent with those from a phase 1, thorough QT study designed to more rigorously evaluate the effects of supertherapeutic doses of vilazodone on the ECG compared with placebo and moxifloxacin.22
Evaluations of drug class effects were specifically conducted for suicidality, serotonin toxicity, and activation of mania/hypomania. The findings for these topics were not of clinical concern and were consistent with those for other antidepressants.
Understanding the impact of vilazodone on sexual function based on interpretation of currently available data is challenging. Results from both AE reporting and scale data suggest a differential effect by sex associated with vilazodone treatment. Men were more likely to experience AEs associated with sexual functioning and less overall improvement based on scale results when compared with women. Further studies with appropriate active comparators are needed to determine whether the impact of vilazodone on sexual function is benign or at least less severe than that of other antidepressants known to negatively impact sexual function.
It should be noted that the safety profiles for the 8-week and 52-week studies were similar. No new safety findings or concerns emerged during the 52-week study compared with the 8-week studies, and no longer-term safety concerns were identified.
This work summarizes phase 3 information from both short- and longer-term vilazodone use, all at the indicated dose of 40 mg qd. Although a considerable number of patients (n = 2177; 551.7 subject years of exposure) were studied, with any safety assessment the ability to detect or to find signals for rare AEs is limited. Only a small percentage of patients were ≥65 years of age; therefore, findings may not accurately reflect the safety and tolerability profile of vilazodone in the elderly. Nevertheless, the findings presented here support that vilazodone is generally well tolerated during both short-term and long-term treatment of MDD in adults.
Acknowledgments
The authors acknowledge the writing and editorial assistance provided by Norma Padilla, PhD, an employee of ApotheCom (supported by funding from Dogwood Pharmaceuticals, a subsidiary of Forest Laboratories, Inc.), in the development and submission of this manuscript.
Footnotes
Author Disclosures
M. Liebowitz—Equity: Pherin Pharmaceuticals and Liebowitz Social Anxiety Scale; Consultancy: AstraZeneca, Pfizer, Takeda, Pherin, Otsuka, and Eisai; Speaker: Wyeth, Pfizer, and Pherin Pharmaceuticals; Licensing software or LSAS: GlaxoSmithKline, Pfizer, Avera, Tikvah, Endo, Eli Lilly, Indevus, and Servier; Clinical Trial Contracts: Pfizer, GlaxoSmithKline, AstraZeneca, Forest, Tikvah, Avera, Eli Lilly, Novartis, Sepracor, Horizon, Johnson & Johnson, Allergan, PGX Health, Abbott, Jazz, MAP, Takeda, Wyeth, Cephalon, Indevus, Endo, Ortho-McNeil, Gruenthal, Purdue Pharma, and Lundbeck; Investigator-Initiated Trial Support: Pfizer.
H.A. Croft—Research Grants: Croft Research Center from Boehringer-Ingelheim, Bristol-Myers-Squibb, Cephalon, Forest, GlaxoSmithKline, Eli Lilly, Labopharm, Merck, Organon, Otsuka, Pfizer, sanofi-aventis, and Takeda; Speaking Honoraria: AstraZeneca, Bristol-Myers Squibb, Forest, GlaxoSmithKline, Eli Lilly, Angelini-Labopharm, Pfizer, Sanofi-Aventis, and Wyeth; Consultancy: Forest, GlaxoSmithKline, Clin Data, Forest, Eli Lilly, and Pfizer; Advisory Board: GlaxoSmithKline, Clin Data, Forest, Eli Lilly, and Pfizer.
D.K. Kajdasz, H. Whalen, S. Gallipoli, M. Athanasiou, and C.R. Reed are all former employees of Dogwood Pharmaceuticals, a subsidiary of Forest Laboratories, Inc.
References
- 1.Zimmerman M, Posternak M, Friedman M et al. Which factors influence psychiatrists’ selection of antidepressants? Am JP sychiatry. 2004;161(7):1285–1289. doi: 10.1176/appi.ajp.161.7.1285. [DOI] [PubMed] [Google Scholar]
- 2.Gelenberg AJ, Freeman MP, Markowitz JC . Practice Guideline for the Treatment of Patients With Major Depressive Disorder. 3rd. Washington, DC: American Psychiatric Association; 2010. [Google Scholar]
- 3.Gardner DM, MacKinnon N, Langille DB, Andreou P. A comparison of factors used by physicians and patients in the selection of antidepressant agents. Psychiatr Serv. 2007;58(1):34–40. doi: 10.1176/ps.2007.58.1.34. [DOI] [PubMed] [Google Scholar]
- 4.Ashton AK, Jamerson BD, Weinstein WL, Wagoner C. Antidepressant-related adverse effects impacting treatment compliance: results of a patient survey. Curr Ther Res. 2005;66(2):96–106. doi: 10.1016/j.curtheres.2005.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bull SA, Hunkeler EM, Lee JY et al. Discontinuing or switching selective serotonin-reuptake inhibitors. Ann Pharmacother. 2002;36(4):578–584. doi: 10.1345/aph.1A254. [DOI] [PubMed] [Google Scholar]
- 6.Khan A, Schwartz K, Redding N, Kolts RL, Brown WA. Psychiatric diagnosis and clinical trial completion rates: analysis of the FDA SBA reports. Neuropsychopharmacology. 2007;32(11):2422–2430. doi: 10.1038/sj.npp.1301361. [DOI] [PubMed] [Google Scholar]
- 7.Cassano P, Fava M. Tolerability issues during long-term treatment with antidepressants. Ann Clin Psychiatry. 2004;16(1):15–25. doi: 10.1080/10401230490281618. [DOI] [PubMed] [Google Scholar]
- 8.Ferguson JM. SSRI antidepressant medications: adverse effects and tolerability. Prim Care Companion J Clin Psychiatry. 2001;3(1):22–27. doi: 10.4088/pcc.v03n0105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dawson LA, Watson JM. Vilazodone: a 5-HT1A receptor agonist/serotonin transporter inhibitor for the treatment of affective disorders. CNS Neurosci Ther. 2009;15(2):107–117. doi: 10.1111/j.1755-5949.2008.00067.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Khan A. Vilazodone, a novel dual-acting serotonergic antidepressant for managing major depression. Expert Opin Invest Drugs. 2009;18(11):1753–1764. doi: 10.1517/13543780903286396. [DOI] [PubMed] [Google Scholar]
- 11.Kehne JH, Bartoszyk GD, Greiner HE . Poster presented at: 65th Annual Meeting of the Society of Biological Psychiatry. New Orleans, LA: May 20-22, 2010. In vitro characterization of vilazodone as a dual-acting serotonin reuptake inhibitor and 5-HT1A receptor partial agonist. [Google Scholar]
- 12.Rickels K, Athanasiou M, Robinson DS, Gibertini M, Whalen H, Reed CR. Evidence for efficacy and tolerability of vilazodone in the treatment of major depressive disorder: a randomized, double-blind, placebo-controlled trial. J Clin Psychiatry. 2009;70(3):326–333. doi: 10.4088/jcp.08m04637. [DOI] [PubMed] [Google Scholar]
- 13.Khan A, Cutler AJ, Kajdasz DK et al. A randomized, double-blind, placebo-controlled, 8-week study of vilazodone, a serotonergic agent for the treatment of major depressive disorder. J Clin Psychiatry. 2011;72(4):441–447. doi: 10.4088/JCP.10m06596. [DOI] [PubMed] [Google Scholar]
- 14.Robinson DS, Kajdasz DK, Gallipoli S, Whalen H, Wamil A, Reed CR. A one-year, open-label study assessing the safety and tolerability of vilazodone in patients with major depressive disorder. J Clin Psychiatry. 2011;31(5):643–646. doi: 10.1097/JCP.0b013e31822c6741. [DOI] [PubMed] [Google Scholar]
- 15.Diagnostic and Statistical Manual of Mental Disorders. Washington, DC: American Psychiatric Association; 2000. American Psychiatric Association. Fourth Edition, Text Revision. [Google Scholar]
- 16. Stone M, Laughren T, Jones ML et al. Risk of suicidality in clinical trials of antidepressants in adults: analysis of proprietary data submitted to US Food and Drug Administration. BMJ. 2009;339:b2880. doi: 10.1136/bmj.b2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dunkley EJ, Isbister GK, Sibbritt D, Dawson AH, Whyte IM. The Hunter Serotonin Toxicity Criteria: simple and accurate diagnostic decision rules for serotonin toxicity. QJM. 2003;96(9):635–642. doi: 10.1093/qjmed/hcg109. [DOI] [PubMed] [Google Scholar]
- 18.McGahuey CA, Gelenberg AJ, Laukes CA et al. The Arizona Sexual Experience Scale (ASEX): reliability and validity. J Sex Marital Ther. 2000;26(1):25–40. doi: 10.1080/009262300278623. [DOI] [PubMed] [Google Scholar]
- 19.Clayton A, Kornstein S, Prakash A, Mallinckrodt C, Wohlreich M. Changes in sexual functioning associated with duloxetine, escitalopram, and placebo in the treatment of patients with major depressive disorder. J Sex Med. 2007;4(4 Pt 1):917–929. doi: 10.1111/j.1743-6109.2007.00520.x. [DOI] [PubMed] [Google Scholar]
- 20.Posner K, Oquendo MA, Gould M, Stanley B, Davies M. Columbia Classification Algorithm of Suicide Assessment (C-CASA): classification of suicidal events in the FDA’s pediatric suicidal risk analysis of antidepressants. Am J Psychiatry. 2007;164(7):1035–1043. doi: 10.1176/appi.ajp.164.7.1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.US Department of Health and Human Services Guidance for industry. Drug-induced liver injury: premarketing clinical evaluation. US Department of Health and Human Services Web site. www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM174090.pdf Accessed October 7, 2010.
- 22.Morganroth J, Thorn MD, Gallipoli S . Poster presented at: 45th Annual Meeting of the American Society of Health-System Pharmacists Midyear Clinical Meeting and Exhibition. Anaheim, CA: Dec 5-9, 2010. An evaluation of the effect of vilazodone on cardiac safety. [Google Scholar]