

ABSTRACT
We sought to determine the possibility of an interrelationship between primary virus replication in the eye, the level of viral DNA in the trigeminal ganglia (TG) during latency, and the amount of virus reactivation following ocular herpes simplex virus type 1 (HSV-1) infection. Mice were infected with virulent (McKrae) or avirulent (KOS and RE) strains of HSV-1, and virus titers in the eyes and TG during primary infection, level of viral gB DNA in TG on day 28 postinfection (p.i.), and virus reactivation on day 28 p.i. as measured by explant reactivation were calculated. Our results suggest that the avirulent strains of HSV-1, even after corneal scarification, had lower virus titers in the eye, had less latency in the TG, and took a longer time to reactivate than virulent strains of HSV-1. The time to explant reactivation of avirulent strains of HSV-1 was similar to that of the virulent LAT(−) McKrae-derived mutant. The viral dose with the McKrae strain of HSV-1 affected the level of viral DNA and time to explant reactivation. Overall, our results suggest that there is no absolute correlation between primary virus titer in the eye and TG and the level of viral DNA in latent TG and time to reactivation.
IMPORTANCE Very little is known regarding the interrelationship between primary virus replication in the eye, the level of latency in TG, and the time to reactivate in the mouse model. This study was designed to answer these questions. Our results point to the absence of any correlation between the level of primary virus replication and the level of viral DNA during latency, and neither was an indicator of how rapidly the virus reactivated following explant TG-induced reactivation.
INTRODUCTION
Following ocular infection with herpes simplex virus type 1 (HSV-1), the virus travels in a retrograde direction toward neuronal cell bodies and establishes latency in trigeminal ganglia (TG) of infected mice (1–3). After establishment of latency in neurons, the latency-associated transcript (LAT) is the only viral product consistently detected in abundance in infected mouse, rabbit, and human TG (4–9). LAT is important for the high, wild-type (WT) rate of in vivo spontaneous (10) and induced (4) reactivation from latency. At various times throughout the life of the latently infected individual, the latent virus spontaneously reactivates and returns to the eye in an anterograde direction (1–3). The eye disease that is broadly referred to as herpes stromal keratitis (HSK) or corneal scarring (CS) occurs mainly as the result of virus reactivation rather than primary ocular infection in humans (11–15).
Neurovirulence of HSV-1 strains can influence the eye disease in ocularly infected mice (16, 17). Based on neurovirulence in animal studies, HSV-1 strains can be classified into two main categories: (i) avirulent HSV-1 strains, such as strains KOS and RE, which do not kill BALB/c mice or New Zealand White (NZW) rabbits following ocular infection and do not replicate efficiently in the eye without corneal scarification, and (ii) virulent HSV-1 strains, such as McKrae and its LAT(−)-derived virus, dLAT2903, that kill ∼80% and ∼50% of BALB/c mice and NZW rabbits, respectively, following ocular infection (18–21) and do not require corneal scarification for efficient ocular infection. Thus, in addition to the presence of LAT, the degree of HSV-1 virulence can also influence the level of recurrence.
Following ocular infection, latent HSV genomes express LAT in a portion of those neurons maintaining them, and virus can be recovered by cocultivation of ex-planted ganglia (22–24). In contrast to spontaneous reactivation in rabbits and humans, spontaneous reactivation in the murine model of ocular HSV-1 is rare (25). Previously, it was suggested that the HSV-1 genome is maintained in a quiescent state in sensory neurons during latency in mice due to the absence of detectable viral protein synthesis. However, studies from mice indicate that lytic transcripts and proteins are expressed at very low levels in latently infected ganglia (26–28). There is also human observational data based on tissue sampling, which demonstrated expression of HSV-1 genes during latency (29–32). Previously we have shown that this low level of HSV-1 gene expression in the presence of LAT is associated with T cell exhaustion in TG of latently infected mice (28).
We have previously demonstrated a positive correlation between virus replication in the eye and eye disease following infection of both susceptible strains of mouse, such as BALB/c, and more resistant strains of mouse, such as C57BL/6, with a virulent HSV-1 strain of McKrae (33–35). We also have reported that the presence of LAT correlated with a higher level of latency and T cell exhaustion in WT C57BL/6 mice infected with McKrae (28), while an absence of CD8+ T cells correlated with lower latency (36). However, very little is known with regard to the interrelationship of virus replication in the eye and TG during primary ocular infection, the level of viral DNA during latency, and the time to reactivation following explant TG reactivation. To address these issues, we examined latency reactivation in TG of WT mice versus CD8α−/− mice following infection with virulent LAT(+) versus LAT(−) viruses. We also determined whether the dose of virus following infection with LAT(+) versus LAT(−) viruses impacted latency reactivation in TG of latently infected mice. In addition, we determined if the combination of corneal scarification with the use of avirulent strains of HSV-1 can influence latency reactivation in infected mice. Finally, the contribution of primary virus replication to latency reactivation was determined in vitro and in vivo.
Here, we report that despite a significant decrease in the level of latency in CD8α−/− mice infected with LAT(+) or LAT(−) virus, the duration of explant reactivation for LAT(+)- or LAT(−)-infected mice that were initially infected with 2 × 105 PFU/eye were similar to that of their WT-infected counterpart. The infectious dose altered the differences in the level of latency reactivation between LAT(+) and LAT(−) viruses. The levels of latency in RE- and KOS-infected mice were different from each other; however, the duration to reactivation was similar between them. The levels of latency in RE- and KOS-infected mice were drastically lower than those of LAT(+) and LAT(−) McKrae viruses, and their reactivation was similar to that of the LAT(−) McKrae virus. Primary virus replication in the eye and TG of infected mice did not influence the level of latency reactivation in latent TG. Thus, these studies point to the absence of any relationship between the level of primary virus replication and the level of resulting viral DNA during latency, and neither was an indicator of how rapidly the virus reactivated following explant TG-induced reactivation.
MATERIALS AND METHODS
Virus and mice.
Plaque-purified virulent HSV-1 strains McKrae [WT, LAT(+)], avirulent KOS [WT, LAT(+)], dLAT2903 [virulent, LAT(−)] (10), and avirulent HSV-1 strain RE [WT, LAT(+)] were grown in rabbit skin (RS) cell monolayers in minimal essential medium (MEM) containing 5% fetal calf serum (FCS) as described previously (10, 37). HSV-1 strains KOS and RE were kindly provided by Richard Longnecker (Northwestern University, Chicago, IL) and Robert Hendricks (University of Pittsburg, Pittsburg, PA), respectively. In this study, mice were ocularly infected with HSV-1 strain McKrae, KOS, dLAT2903, or RE. Throughout the study, McKrae and dLAT2903 viruses are called LAT(+) and LAT(−), respectively.
Female WT C57BL/6 and C57BL/6-CD8α−/− mice were purchased from Jackson Laboratories. All animal procedures adhered to the Association for Research in Vision and Ophthalmology (ARVO) statement for the Use of Animals in Ophthalmic and Vision Research and according to institutional animal care and use guidelines.
Ocular infection.
Mice were infected ocularly with 2 × 105, 1 × 105, 2 × 104, 1 × 104, 2 × 103, or 1 × 103 PFU per eye of HSV-1 strain McKrae [LAT(+)] or dLAT2903 [LAT(−)] without corneal scarification. Some mice were ocularly infected with 2 × 105 PFU per eye of HSV-1 strain KOS or RE with and without corneal scarification. Each virus was suspended in 2 μl of tissue culture medium and administered as an eye drop.
Titration of virus in tears of infected mice.
Tear films were collected from both eyes of 10 mice per group on days 3 and 5 p.i. using a Dacron-tipped swab (19). Each swab was placed in 1 ml of tissue culture medium and squeezed, and the amount of virus was determined by a standard plaque assay on RS cells.
Titer of infectious virus in the trigeminal ganglia.
On day 3, 5, or 7 postinfection, mice were euthanized and the trigeminal ganglia (TG) were isolated and homogenized individually as described previously (11). The cell debris was removed by centrifugation at 3,000 rpm for 10 min using a Beckman TA10 rotor. The virus titer in the supernatant was then measured using a plaque assay on RS cells as we have described previously (16).
In vitro explant reactivation assay.
Mice were sacrificed at 28 days p.i., and individual TG were removed and cultured in tissue culture medium as we described previously (38). Aliquots of medium were removed from each culture daily for up to 15 days and plated on indicator cells (RS cells) to assay for the appearance of reactivated virus. As the media from the explanted TG cultures were plated daily, the time at which reactivated virus first appeared in the explanted TG cultures could be determined.
DNA extraction and PCR analysis for HSV-1 genomic DNA.
DNA was isolated from homogenized individual TG using the commercially available DNeasy blood & tissue kit (Qiagen, Stanford, CA) according to the manufacturer's instructions. PCR analyses were done using gB-specific primers (forward, 5′-AACGCGACGCACATCAAG-3′; reverse, 5′-CTGGTACGCGATCAGAAAGC-3′; probe, 5′-FAM-CAGCCGCAGTACTACC-3′) as we described previously (23, 24, 36, 39). The amplicon length for this primer set is 72 bp. The relative copy number of gB DNA was calculated using standard curves generated from the plasmid pAc-gB1 as we described previously (40). In all experiments glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used for normalization of transcripts.
Statistical analysis.
Student's t test and chi-squared tests were performed using the computer program Instat (GraphPad, San Diego, CA). Results were considered statistically significant when the P value was <0.05.
RESULTS
Reactivation correlates with the amount of latency in WT but not in CD8α−/− mice.
Previously we reported that latency in CD8α−/− mice following ocular infection with HSV-1 strain McKrae was significantly lower than that of WT C57BL/6 mice infected similarly (36, 41). However, the role of LAT in the establishment of latency reactivation in CD8α−/− mice is not known. To determine if CD8+ T cells also affect the level of latency in LAT(−)-infected mice, WT and CD8α−/− mice were ocularly infected with 2 × 105 PFU/eye of WT HSV-1 strain McKrae [LAT(+)] or dLAT2903 [LAT(−)] virus. Individual TG from surviving mice were isolated on day 28 p.i. Total DNA was extracted and TaqMan quantitative PCR (qPCR) was performed to quantitate viral gB DNA as described in Materials and Methods. Cellular GAPDH DNA was used as an internal control. Consistent with previous reports (28, 36, 41, 42), there was significantly more HSV-1 DNA in TG from WT mice latently infected with LAT(+) than LAT(−) virus (P < 0.0001) (Fig. 1, WT), characteristic of more latency with LAT(+) than LAT(−) virus in WT mice. In contrast to WT mice, in CD8α−/− mice LAT(+) and LAT(−) viruses had similar levels of latency, and this was similar to the amount of latency seen in WT mice infected with LAT(−) virus (P = 0.6) (Fig. 1, CD8α−/−). Thus, LAT(−)-infected WT mice and CD8α−/− mice infected with either LAT(+) or LAT(−) virus had significantly fewer latent viral genomes than did WT mice infected with LAT(+) virus (P < 0.0001) (Fig. 1).
FIG 1.
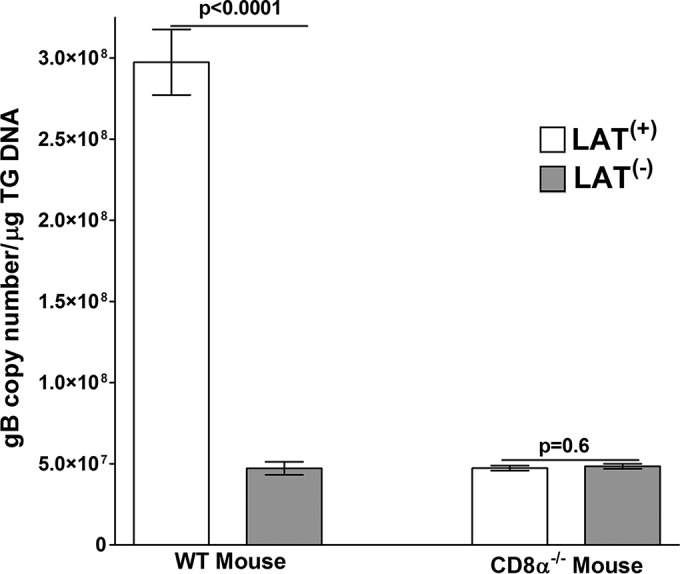
Effect of LAT on the level of latency in different strains of mice. WT C57BL/6 and C57BL/6-CD8α−/− mice were infected with LAT(+) or LAT(−) virus at 2 × 105 PFU per eye. Twenty-eight days p.i., TG from infected mice were isolated and quantitative PCR was performed on each individual mouse TG. In each experiment, an estimated relative copy number of the HSV-1 gB for viral DNA was calculated using standard curves generated from pGem-gB1. Briefly, DNA template was serially diluted 10-fold such that 5 μl contained from 103 to 1011 copies of gB, and then it was subjected to TaqMan PCR with the same set of primers. By comparing the normalized threshold cycle of each sample to the threshold cycle of the standard, the copy number for each reaction was determined. GAPDH expression was used to normalize the relative expression of viral (gB) DNA in the TG. Each bar for WT mice is based on 40 TG from 2 separate experiments, while each bar for CD8α−/− mice is based on 20 TG. Data are presented as means ± standard errors of the means (SEM).
We next tested whether lower latency in TG of the above-described mice correlated with lower explant reactivation from latency. WT and CD8α−/− mice were ocularly infected with 2 × 105 PFU/eye of LAT(+) or LAT(−) virus. Virus reactivation was analyzed by explanting individual TG from infected mice on day 28 p.i. as described in Materials and Methods. Consistent with previous studies, the time to reactivation in WT mice was significantly shorter with LAT(+) virus than LAT(−) virus (5.6 ± 0.2 days versus 6.9 ± 0.2 days; P = 0.0001) (Fig. 2, WT). Surprisingly, the time to reactivation was also significantly shorter with LAT(+) than LAT(−) virus in CD8α−/− mice (5.5 ± 0.4 days versus 7.9 ± 0.4 days; P < 0.0001) (Fig. 2, CD8α−/−), even though LAT(+) and LAT(−) viruses had similar levels of latency in these mice (Fig. 1). In addition, the time to reactivation with LAT(+) virus was similar in WT- and CD8α−/−-infected mice (P = 0.8) (Fig. 2). The time to reactivation of LAT(−) virus was also similar in WT and CD8α−/− mice (P = 0.3) (Fig. 2). Thus, in CD8α−/− mice the time to explant reactivation did not correlate with the amount of latency that was established.
FIG 2.
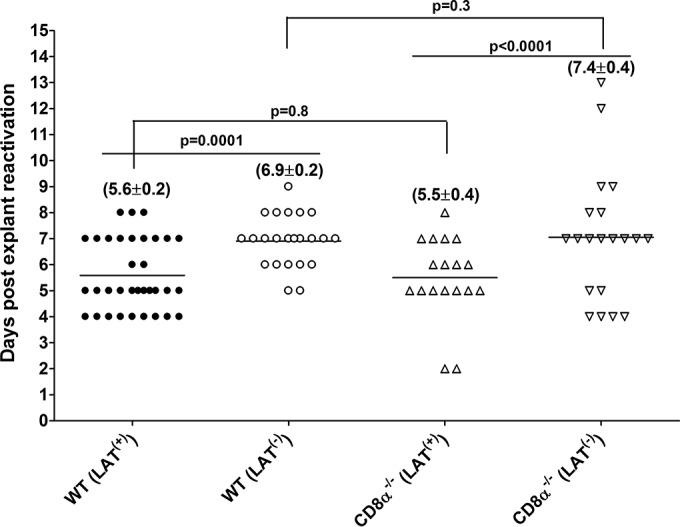
Effect of LAT on induced reactivation in different strains of mice. WT C57BL/6 and C57BL/6-CD8α−/− mice were infected with 2 × 105 PFU per eye of LAT(+) or LAT(−) virus as described for Fig. 1. On day 28 p.i., TG from infected mice were isolated and each individual TG was incubated in 1.5 ml of tissue culture medium at 37°C. A 100-μl aliquot was removed from each culture daily for 15 days and used to infect RS cell monolayers. The RS cells were monitored daily for the appearance of CPE to determine the time of first appearance of reactivated virus from each TG. The results are plotted as the number of TG that reactivated daily. Numbers indicate the average time (±SEM) that the TG from each group first showed CPE. Each point represents the means ± SEM from 34 TG for WT [LAT(+)], 23 TG for WT [LAT(−)], 18 for CD8α−/− [LAT(+)], and 18 for CD8α−/− [LAT(−)] mice.
In contrast to latent infection, analysis of the TG for infectious virus at days 3, 5, and 7 p.i. indicated that the average amount of HSV-1 was similar between LAT(+)- and LAT(−)-infected WT or CD8α−/− mice (P > 0.05 by Student t test) (Fig. 3). Virus was detectable at day 3 and the titers increased from day 3 to day 5, attaining levels similar to those of the control groups (P > 0.05 by Student t test) (Fig. 3). Between days 5 and 7 postinfection, the virus titers in the TGs of all groups exhibited a decline (P > 0.05) (Fig. 3). Thus, our results suggest that the absence of LAT or CD8+ T cells had no statistically significant effect on virus replication during the acute phase of infection in TG of infected mice (days 3, 5, and 7 p.i.). These results are similar to our published studies showing that the absence of LAT had no effect on primary virus replication in TG of mice and rabbits infected with LAT(+) and LAT(−) viruses (10, 43).
FIG 3.
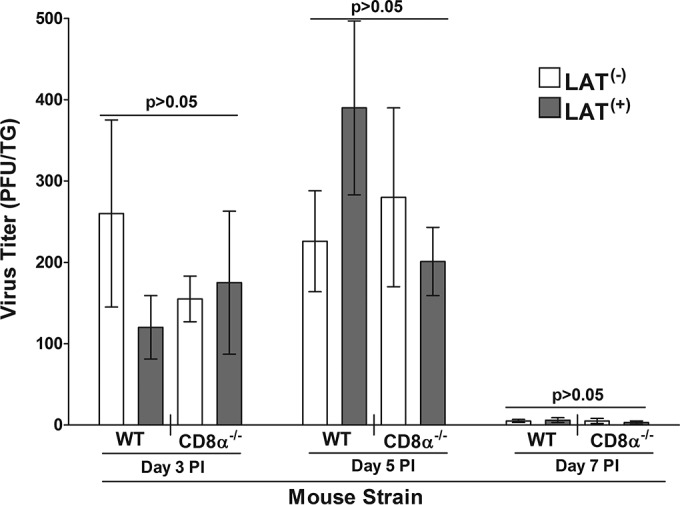
Virus titers in TG after ocular infection. WT C57BL/6 and CD8α−/− mice were infected ocularly with 2 × 105 PFU per eye of dLAT2903 [LAT(−)] or WT HSV-1 strain McKrae [LAT(+)] as described in Materials and Methods. Mice were euthanized on the indicated days. TG were removed and homogenized, and virus titers were determined as described in Materials and Methods. Each point represents the means ± SEM of titers from extracts of 10 TG.
Effect of initial virus dose on the level of viral latency and time of explant reactivation in infected mice.
To investigate the effect of the dose of infectious virus, we infected WT mice with 1 × 105, 2 × 104, 1 × 104, 2 × 103, or 1 × 103 PFU per of eye of LAT(+) or LAT(−) virus. Individual TG were isolated on day 28 p.i. Total DNA was isolated, and TaqMan qPCR was performed to quantitate viral gB DNA as described above. The data from Fig. 1 for WT mice infected with 2 × 105 PFU per eye is reproduced in Fig. 4A for comparison. The amount of latency in mice infected with 2 × 105 PFU of LAT(+) virus was significantly higher than that in mice infected with 1 × 105, 2 × 104, or 1 × 104 PFU per eye of LAT(+) virus (Fig. 4A and B, compare LAT(+) virus at 2 × 105 with LAT(+) virus at 1 × 105, 2 × 104, or 1 × 104) (P < 0.0001). In contrast, the level of latency in LAT(−)-infected mice was similar between the 2 × 105-PFU group and the 1 × 105- or 2 × 104-PFU group (Fig. 4A, compare LAT(−) virus at 2 × 105 with LAT(−) virus at 1 × 105 and 2 × 104) (P = 0.8), except for the 1 × 104-PFU group (Fig. 4B). However, the amount of latency in mice infected with 1 × 104 PFU of LAT(+) virus was significantly higher than that in mice infected with 1 × 104 PFU per eye of LAT(−) virus (Fig. 4B) (P = 0.0005). At 2 × 105, 1 × 105, 2 × 104, and 1 × 104 PFU per eye, the amount of latency with LAT(+) virus was significantly higher than that of the LAT(−) group (Fig. 4A and B). At the lowest dose of 2 × 103 or 1 × 103 PFU per eye, the amounts of latency with both the LAT(+) and LAT(−) viruses were reduced compared to those of the higher infectious doses (Fig. 4A and B). Surprisingly, however, in contrast to the 2 × 105-, 1 × 105-, 2 × 104-, and 1 × 104-PFU/eye groups, the level of viral latency was similar for LAT(+) and LAT(−) viruses in mice infected with 2 × 103 PFU/eye (Fig. 4B, 2 × 103) (P = 0.25) and with 1 × 103 PFU/eye (Fig. 4B, 1 × 103) (P = 0.10). These results suggest that at low dose, LAT infection does not significantly influence the amount of latency that is established.
FIG 4.

Effect of dose of virus on the level of latency in TG of latently infected mice. WT C57BL/6 mice were ocularly infected with 1 × 105, 2 × 104, 1 × 104, 2 × 103, or 1 × 103 PFU/eye of HSV-1 strain McKrae [LAT(+)] or dLAT2903 [LAT(−)] virus. On day 28 p.i., TG were harvested from the latently infected mice. Quantitative PCR was performed on each individual mouse TG. GAPDH expression was used to normalize the relative expression of viral (gB) DNA in the TG. Each point represents the means ± SEM from 20 TG. The Y-scale for 1 × 104, 2 × 103, and 1 × 103 PFU is different from the Y-scale for 2 × 105, 1 × 105, and 2 × 104 PFU. Data for 2 × 105 PFU are repeated from the data shown in Fig. 1. (A) Latency in mice infected with 2 × 105, 1 × 105, or 2 × 104 PFU/eye of LAT(+) or LAT(−) virus. (B) Latency in mice infected with 1 × 104, 2 × 103, or 1 × 103 PFU/eye of LAT(+) or LAT(−) virus.
Latency data shown in Fig. 4 were also analyzed in detail across groups and are shown in Table 1. There was a significant difference in mean latency values overall between LAT(+) and LAT(−) groups (P < 0.0001) (Fig. 4). There was also a significant difference in the dose-response curves between LAT(+) and LAT(−) groups (P < 0.0001) (Fig. 4). At dosages below 2 × 103, no difference was observed between LAT(+) and LAT(−) groups. At dosages at or above 1 × 104, there were significant differences between LAT(+) and LAT(−) groups (P < 0.0001 for each dose) (Fig. 4). Within both the LAT(+) and LAT(−) groups, there was a significant dose-response difference at dosages at or below 1 × 104 (P < 0.0001 for each contrast) (Table 1). Within the LAT(+) group, the dose-response curve plateaued at 2 × 104 with no difference observed between the 2 × 104 and 1 × 105 doses (P = 0.0745) or between the 1 × 105 and 2 × 105 doses (P = 0.1864) (Table 1, left). Within the LAT(−) group, the dose-response curve also plateaued at 2 × 104, with no difference observed between the 2 × 104 and 1 × 105 doses (P = 0.4245) or between the 1 × 105 and 2 × 105 doses (P = 0.1001) (Table 1, right). Thus, within both the LAT(+) and LAT(−) groups, the dose-response curve flattened at the dose level of 2 × 104. This suggests an optimal dose difference between LAT(+) and LAT(−) is somewhere in the range of 1 × 104 to 2 × 104.
TABLE 1.
P values for differences within LAT(+)- and LAT(−)-infected TG across dosesa
| Dose (PFU/eye) |
P value by strain and dose |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LAT(+) |
LAT(−) |
|||||||||
| 1 × 103 | 2 × 103 | 1 × 104 | 2 × 104 | 1 × 105 | 1 × 103 | 2 × 103 | 1 × 104 | 2 × 104 | 1 × 105 | |
| 2 × 103 | <0.0001 | <0.0001 | ||||||||
| 1 × 104 | <0.0001 | <0.0001 | <0.0001 | 0.8892 | ||||||
| 2 × 104 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | ||||
| 1 × 105 | <0.0001 | <0.0001 | <0.0001 | 0.0745 | <0.0001 | <0.0001 | <0.0001 | 0.4245 | ||
| 2 × 105 | <0.0001 | <0.0001 | <0.0001 | <0.0001 | 0.1864 | <0.0001 | <0.0001 | <0.0001 | 0.9999 | 0.1001 |
Data from Fig. 1 and 4 with regard to WT C57BL/6 mice were analyzed across groups with analysis of variance (ANOVA) followed by Tukey's test to correct for multiple comparisons. Data were transformed prior to analysis with the Box-Cox method in order to meet the assumptions necessary for parametric assessment. Residuals were inspected to confirm the overall fit and to confirm the absence of influential outliers. All analyses were performed at a two-sided alpha level of 0.05. SAS v9.3 software was used.
To determine if virus reactivation was affected at different virus doses, WT mice were ocularly infected as described above with 1 × 105, 2 × 104, 1 × 104, 2 × 103, or 1 × 103 PFU/eye of LAT(+) and LAT(−) viruses, and virus reactivation was analyzed as described for Fig. 2. The reactivation data and the number of TG showing cytopathic effects (CPE) are shown in Fig. 5 and Table 2. Consistent with an infectious dose of 2 × 105 PFU/eye (Fig. 2), time of reactivation in mice infected with 1 × 105 PFU/eye of LAT(+) virus was significantly shorter than that for LAT(−) virus (5.1 ± 0.3 days versus 6.4 ± 0.2 days; P = 0.002) (Fig. 5). The time to reactivate at an infectious dose of 2 × 104 PFU/eye tended to be faster with LAT(+) virus than LAT(−) virus (5.4 ± 0.4 days versus 6.6 ± 0.5 days) (Fig. 5). However, the difference did not reach statistical significance (P = 0.09). This may have been a result of less TG showing reactivation compared to the experiment using 2 × 105 or 1 × 105 PFU/eye (Table 2). Interestingly, while 100% of TG of mice infected with 2 × 105 PFU/eye of LAT(+) and LAT(−) viruses eventually reactivated, 90% and 75% of TG infected with 1 × 105 PFU/eye of LAT(+) and LAT(−) viruses, respectively, reactivated, while only 80% and 60% of TG infected with 2 × 104 PFU/eye of LAT(+) and LAT(−) viruses, respectively, did so (Table 2). The time to reactivate at an infectious dose of 1 × 104 PFU/eye was also faster with LAT(+) virus than LAT(−) virus (6.2 ± 0.3 days versus 7.1 ± 0.4 days) (Fig. 5), but the difference did not reach statistical significance (P = 0.09). Similar to the lack of significant differences between LAT(+) and LAT(−) virus at the infection dose of 2 × 104 PFU/eye, the lack of differences at 1 × 104 PFU/eye between the two viruses could be associated with less reactivation in which only 50% and 45% of TG infected with LAT(+) and LAT(−) viruses were reactivated, respectively (Table 2). Although the means of the time to reactivate among different doses for each virus were not statistically significant, the numbers of TG showing reactivation were not the same and declined as the dose of infection reduced (Fig. 5 and Table 2). Additionally, no viral reactivation was detected with either virus in any of the TG from mice infected with the lowest doses of 2 × 103 or 1 × 103 PFU/eye during the 15-day monitoring period (Fig. 5 and Table 2). This suggests that, at least in C57BL/6 mice infected with WT McKrae and LAT(−) McKrae viruses, there is a minimum threshold of latency that must be reached before viral reactivation can be readily detected by explant TG-induced reactivation. This threshold appears to require (in the absence of corneal scarification) an initial ocular infection of more than 2 × 103 PFU/eye and more than 4 × 104 copies of viral gene/μg of total TG DNA. Finally, the above-described results suggest that at an ocular infection dose of 2 × 103 or 1 × 103 PFU/eye and without corneal scarification in C57BL/6 mice infected with HSV-1 strain McKrae, the presence of LAT does not significantly affect the amount of latency that is established in TG.
FIG 5.
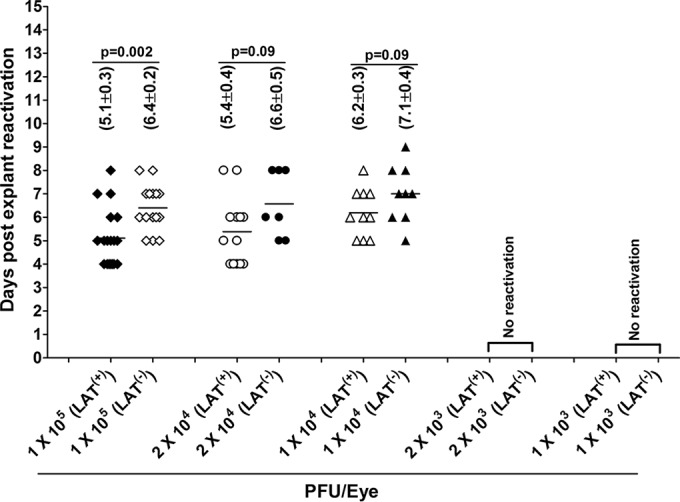
Effect of dose of HSV-1 on explant reactivation in TG of latently infected mice. WT C57BL/6 mice were ocularly infected with 1 × 105, 2 × 104, 1 × 104, 2 × 103, or 1 × 103 PFU/eye of HSV-1 strain McKrae [LAT(+)] or dLAT2903 [LAT(−)] virus as described for Fig. 4. On day 28 p.i., TG from infected mice were harvested and explant reactivation was performed as described for Fig. 2. The presence of infectious virus in each culture was monitored daily for 15 days and used to infect RS cell monolayers. The RS cells were monitored daily for the appearance of CPE to determine the time of first appearance of reactivated virus from each TG. The results are plotted as the number of TG that reactivated daily. Numbers indicate the average time (±SEM) that the TG from each group first showed CPE. Each point represents the means ± SEM from 20 TG for each virus from two separate experiments.
TABLE 2.
TG reactivation detected by explant cultures in mice infected with different doses of LAT(+) and LAT(−) virusesa
| Virus | No. (%) of TG showing virus/total TG by doseb (PFU/eye) |
|||||
|---|---|---|---|---|---|---|
| 2 × 105 | 1 × 105 | 2 × 104 | 1 × 104 | 2 × 103 | 1 × 103 | |
| McKrae [LAT(+)] | 34/34 (100) | 18/20 (90) | 13/16 (80) | 10/20 (50) | 0/16 (0) | 0/10 (0) |
| dLAT2903 [LAT(−)] | 23/23 (100) | 15/20 (75) | 7/12 (60) | 9/20 (45) | 0/12 (0) | 0/10 (0) |
| KOS [LAT(+)] | 10/20 (50) | ND | ND | ND | ND | ND |
| RE [LAT(+)] | 18/20 (90) | ND | ND | ND | ND | ND |
TG from latently infected mice were isolated on day 28 p.i., and the presence of infectious virus for each TG culture was monitored daily for up to 15 days postculture as described in Materials and Methods.
Numbers for each virus indicate the number of TG showing CPE per total number of tested TG. Numbers in parentheses indicate the percentage of TG that showed CPE. ND, not done.
Relationship of latency and reactivation following infection with avirulent strains of HSV-1.
Both McKrae [LAT(+)] and McKrae-derived dLAT2903 [LAT(−)] viruses are virulent in BALB/c mice and do not require corneal scarification for efficient replication in the eye of both susceptible strains of mice, such as BALB/c, and mice that are refractory to infection, such as C57BL/6, which was used in the above-described studies. In contrast, both KOS and RE strains of HSV-1 are avirulent in BALB/c mice and need corneal scarification for efficient in vivo replication. Thus, to determine the pattern of latency reactivation with these avirulent strains of HSV-1 in C57BL/6 mice, corneas were scarified and mice were ocularly infected with 2 × 105 PFU per eye of KOS or RE. Individual TG were isolated on day 28 p.i., DNA was isolated, and TaqMan qPCR was performed to quantitate viral gB DNA as described above. The amount of viral DNA during latency in RE-infected mice was 3-fold higher than that for KOS-infected mice (P < 0.0001) (Fig. 6A). However, despite corneal scarification and an infectious dose of 2 × 105 PFU/eye, the amount of latency in both KOS- and RE-infected mice was at least 104-fold lower than when mice were infected with dLAT2903 [LAT(−)] virus [compare Fig. 1, LAT(−), with 6A] (P < 0.0001). In addition, the amount of latency established with KOS and RE was even less than that seen with the McKrae infectious dose of 1 × 103 PFU/eye (compare Fig. 6A to 4B, right side).
FIG 6.
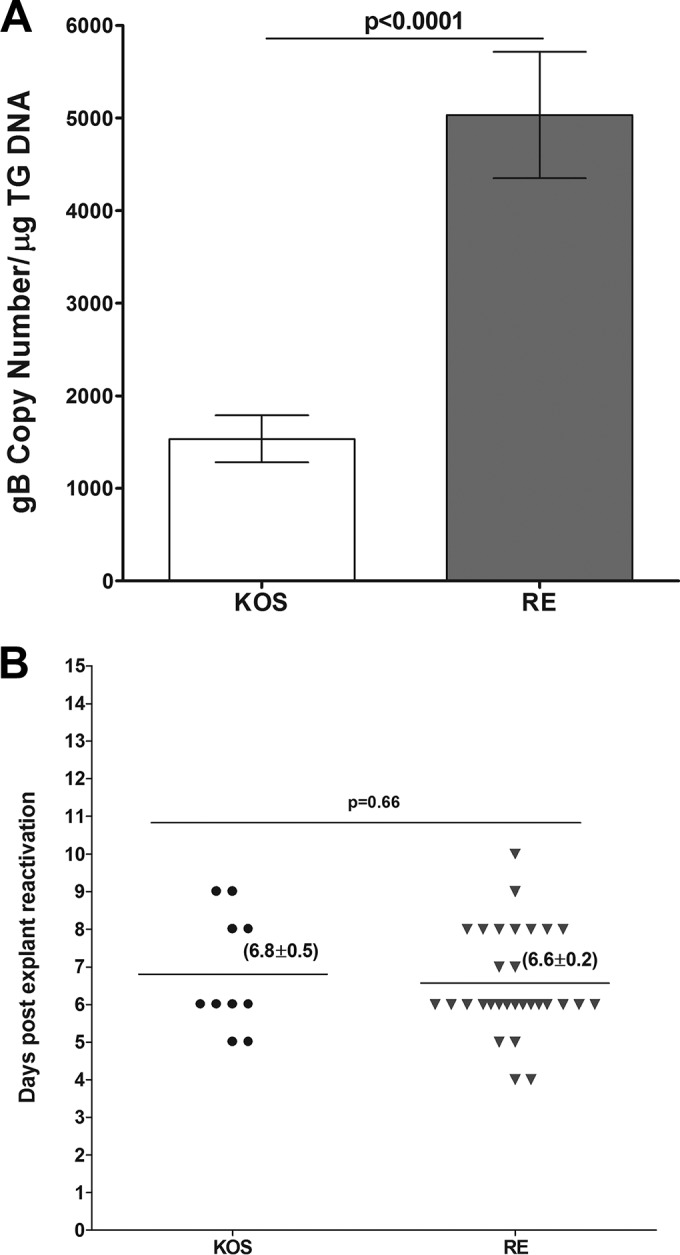
Level of latency and duration of explant reactivation following ocular infection of mice with avirulent strains of HSV-1. Cornea from wt C57BL/6 mice were scarified before ocular infection and then were infected ocularly with 2 × 105 PFU per eye of HSV-1 strains KOS and RE. On day 28 p.i., TG from infected mice were harvested for qPCR and explant reactivation. (A) gB DNA in latent TG. Quantitative PCR was performed on each mouse TG. GAPDH expression was used to normalize the relative expression of viral (gB) DNA in the TG. Each point represents the means ± SEM from 20 TG. (B) Explant reactivation in latent TG. Each individual TG from infected mice was incubated in 1.5 ml of tissue culture medium at 37°C, and the presence of infectious virus was monitored for 15 days as described for Fig. 2. For each virus, 20 TG from 10 mice were used. In KOS-infected mice, only 50% of latent TG were positive for the appearance of CPE, while in the RE strain 90% of TG showed CPE. Numbers indicate the average time (±SEM) that the TG from each group first showed CPE. (A) gB DNA; (B) explant reactivation.
We examined reactivation of KOS and RE in C57BL/6 mice using the same approach as that described above for LAT(+) and LAT(−) McKrae, except that for KOS and RE corneal scarification was performed again just prior to infection. Mice were ocularly infected with 2 × 105 PFU/eye of KOS or RE. Time of reactivation of KOS was similar to that of RE (6.8 ± 0.5 days versus 6.6 ± 0.2 days) (P = 0.66) (Fig. 6B). However, reactivation was detected in only 50% of TG from KOS-infected mice during the 15 days of the monitoring period, while reactivation was detected in 90% of TG from RE-infected mice (Table 2). These differences in the rate of reactivation between KOS and RE were statistically significant (Table 2) (P = 0.01). These results suggest a lack of correlation between the level of latency and reactivation of KOS compared to RE in C57BL/6 mice. In addition, reactivation was readily detected with both KOS and RE (Fig. 6B), even though the latency copy number was approximately 1.5 × 103 and 4 × 103, respectively, per μg of total TG DNA (Fig. 6B). In contrast, with McKrae, no reactivation was detected even when the latency copy number was ∼10-fold higher (3 × 104 to 4 × 104 per μg of total TG DNA) (Fig. 4B, right). This suggests that in C57BL/6 mice different strains of HSV-1 require different minimal levels of latency to be established for reactivation to be readily detected by explant TG-induced reactivation.
Relationship of virus replication in the eye with the level of latency and time of reactivation.
The above-described results using both virulent and avirulent strains of HSV-1 suggest that there is no correlation between the levels of latency in TG of latently infected mice and the time of explant reactivation. To assess the effects of primary infection on the level of latency reactivation, mice were infected ocularly with 2 × 105 PFU/eye of McKrae [LAT(+)], LAT2903 [LAT(−)], KOS, or RE. Corneal scarification was not performed in mice infected with LAT(+) and LAT(−) viruses, while for both KOS and RE mice were infected both with and without corneal scarification. Tear films were collected on days 3 and 5 p.i., and the amount of infectious virus was determined by plaque assay. As expected, virus titers with KOS and RE were significantly higher on both days when corneal scarification was employed (Fig. 7) (P < 0.05). The virus titer in the eyes of mice infected with KOS and RE strains on day 3 and day 5 p.i. were similar following corneal scarification (Fig. 7) (P > 0.05), and both were significantly lower than those for LAT(+)- and LAT(−)-infected mice on both days 3 and 5 p.i. (Fig. 7) (P < 0.05). Virus replication in the eyes of LAT(+)-infected mice was similar to that of LAT(−)-infected mice (Fig. 7) (P > 0.05). These results suggest that virus replication in the eye of virulent strains of HSV-1 is greater than that of avirulent HSV-1 strains even after corneal scarification.
FIG 7.
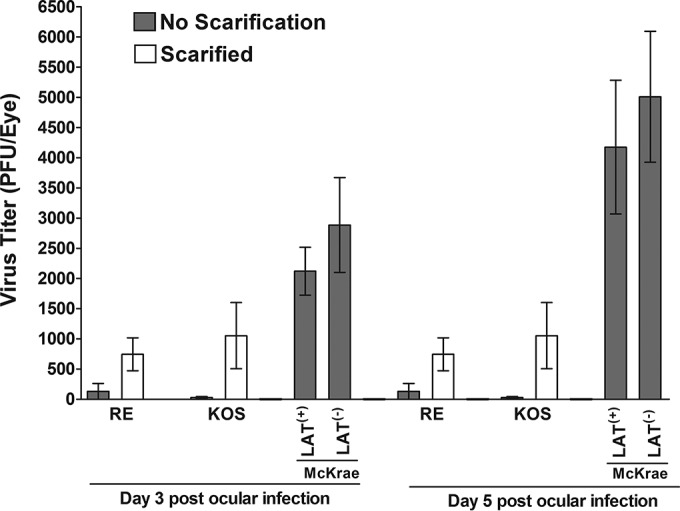
Virus titers in the eyes of infected mice. WT C57BL/6 mice were ocularly infected with 2 × 105 PFU per eye of HSV-1 strains McKrae [LAT(+)], dLAT2903 [LAT(−)], RE [LAT(+)], and KOS [LAT(+)] as described in Materials and Methods. The presence of infectious virus in the eyes of infected mice was monitored on days 3 and 5 p.i. by collecting tear films from 20 eyes for each virus. Each point represents the means ± SEM from 20 eyes.
DISCUSSION
Following primary herpes simplex virus type 1 (HSV-1) ocular infection, the virus replicates in the eye. The longer it takes for the immune system to clear the virus from the eye and the higher the ocular viral load, the more extensive and protracted the consequent ocular disease (19, 44–46). Following ocular infection, the virus establishes latency in ganglia of the infected animals and humans (4–9). During the life of the latently infected individual the virus can occasionally reactivate, travel back to the eye, and cause recurrent eye disease. Indeed, a major cause of CS is the scarring induced by HSV-1 following reactivation from latency (11, 47). Because of the preexisting immune response and the fact that CS is mostly due to a pathogenic immune response, CS is more likely to occur following recurrent rather than primary infection (9, 48–50).
We have previously shown a direct correlation between virus load in the eye and the duration of primary virus replication in the eye with the severity of CS in ocularly infected mice (19, 44, 51). We also have shown that severity of CS is associated with higher latency in TG of latently infected mice (52) and higher programmed death 1 (PD-1) expression, a marker of T cell exhaustion (28). Finally, we have shown that CD8α+ DCs contribute to the enhancement of latency and T cell exhaustion in the TG of infected mice (23, 24, 36, 38, 39, 53), while CD8+ T cells play no role in maintaining latency or blocking reactivation in TG of latently infected mice (41). Furthermore, the level of latency and T cell exhaustion increased in the presence of LAT (24, 28). However, very little is known regarding the interrelationship between primary virus replication in the eye and TG, the level of latency in TG, and the time to reactivation of the latent virus in the explant TG-induced reactivation mouse model. This study sought to determine if such a relationship exists.
In contrast to human and rabbit, spontaneous reactivation of latent HSV-1 in mice is limited (21, 54). Sawtell and Thompson performed a direct comparison of in vivo reactivation in mice following hyperthermic stress versus explant reactivation (55). The authors reported quantitative and qualitative differences between HSV-1 reactivation in vivo and in vitro (explant). However, more recently, it was reported that in a rabbit ocular model of infection, the spontaneous reactivation phenotype of LAT(+) virus was similar to that in the mouse TG explant-induced reactivation model, and that both were significantly higher than that of LAT(−) viruses (56). Based on a combination of in vivo and explant reactivation models, various groups have reported that LAT mutants that do not express detectable amounts of LAT have a reduced reactivation phenotype in both mice and rabbits (4, 10, 57, 58). The explant TG-induced reactivation phenotype in the mouse is not always predictive of the in vivo spontaneous reactivation phenotype in rabbits and induced reactivation in mice. In this study and in our previously published works, we have used the intact TG model of explant reactivation as previously reported (59), while in other studies TG were dissociated to look at explant virus reactivation (57). In the intact model of TG explant reactivation, virus is not observed until at least day 4 after explant culture, but when TG is dissociated virus reactivation is observed earlier after culture. Although explant TG-induced reactivation is not an ideal way to look at virus reactivation in vivo, we believe the intact TG model of reactivation more closely resembles the conditions in vivo than the dissociated TG model. In this study, we have shown that while there is no direct correlation between the primary virus titer in the eye and TG with the subsequent level of latency in the TG of infected mice, the level of viral DNA in latent TG is directly correlated with time of reactivation in a virus- and dose-dependent manner. The level of latency descended by approximately 15-, 7-, 4-, and 2-fold at 2 × 105, 1 × 105, 2 × 104, and 1 × 104 PFU/eye, respectively, between LAT(+)- and LAT(−)-infected TG, while no specific patterns were detected in mice infected with 2 × 103 or 1 × 103 of each virus. In addition, previously it was shown that the effect of LAT on the latency reactivation phenotype is mouse strain dependent (60). Similar to our study, it was previously reported that the explant reactivation kinetics in rabbit TG reflected the behavior of LAT in vivo in the rabbit eye model (61).
In the present study, we confirmed that explant reactivation occurred faster in McKrae-infected mice than in LAT(−)-infected mice. The time to reactivation in McKrae LAT(−)-infected mice was similar to that of LAT(+) KOS and RE strains of HSV-1. This is consistent with previous studies showing that RE, KOS, and dLAT2903 [LAT(−)] strains of HSV-1 had low spontaneous and induced reactivation in rabbits, while McKrae had high reactivation (10, 62, 63). The murine model of latency and reactivation is both mouse strain and virus strain specific (60, 64). In our studies with McKrae viruses in C57BL/6 mice, the percentage of explanted TG from which virus reactivated was dose dependent with both LAT(+) and LAT(−) viruses. At the highest dose used, all TG reactivated with both viruses by the end of the study period. At the intermediate dose, the percentage of TG with reactivations declined for both LAT(+) and LAT(−) viruses with a faster decline for LAT(−) virus. At the lowest doses no reactivation was detected with either virus. This was despite the fact that at these doses of McKrae the level of viral DNA (latency) was significantly higher than it was with strains KOS or RE, which both reactivated in the same strain of mice. Previously we showed that LAT(−) virus reactivated poorly in Swiss Webster mice compared to LAT(+) virus, while only minimal or no significant difference was seen between LAT(+) and LAT(−) viruses in BALB/c mice (60). In this study, we found that following our standard ocular infectious virus dose of 2 × 105 PFU/eye, the time to reactivation of the KOS and RE WT strains [i.e., LAT(+) virus] of HSV-1 in C57BL/6 mice was similar to that of McKrae LAT(−) virus.
LAT is the only viral gene abundantly transcribed during HSV-1 neuronal latency (4–6, 9). Previously in the murine model of HSV-1 latency it was shown that the presence of LAT increases the number of neurons in which latency is established, but LAT has not been demonstrated to be required for establishment of latency (10, 65). In this study, we have shown that the level of latency in the avirulent KOS and RE strains of HSV-1 is dramatically lower than that with the virulent McKrae strain of HSV-1, even in the absence of LAT and even when the infectious dose of McKrae was 1,000-fold less than that for KOS and RE. At higher infectious doses, the amount of latency with McKrae LAT(+) was higher than that with LAT(−) virus, while at a low infectious dose no differences in the amount of latency were detected between LAT(+) and LAT(−) McKrae. In summary, LAT expression does not significantly affect the levels of viral genomes in TG of latently infected C57BL/6 mice when the initial infection was performed at a low multiplicity of infection (MOI). In contrast, at an MOI of higher than 1 × 104, more neurons harbor latent virions and/or may have more copies of the virus; thus, LAT demonstrates its phenotype with regard to latency reactivation. Finally, our results suggest that in the absence of LAT, the dose-response curve between LAT(+) and LAT(−) viruses flattened at 2 × 104 PFU per eye, suggesting an optimal dose of infection between LAT(+) and LAT(−) viruses would be somewhere in the higher range of 2 × 104 PFU per eye.
Overall, our results suggest that in C57BL/6 mice, (i) the amount of latency established by the avirulent HSV-1 strains KOS and RE is lower than that with the virulent McKrae strain; (ii) the time to reactivation following TG explantation is similar with the two avirulent strains examined and the virulent LAT(−) strain, but they all are slower to reactivate than the virulent LAT(+) strain McKrae; (iii) the initial infectious dose affects the level of latency reactivation in C57BL/6 mice; and (iv) virus replication during primary ocular infection does not always correlate with the level of latency reactivation in C57BL/6 mice [McKrae LAT(+) and LAT(−) grow similarly in mouse eyes and TG yet establish latency and reactivate differently].
ACKNOWLEDGMENT
We thank Nigel Fraser for critical reading of the manuscript.
REFERENCES
- 1.Diefenbach RJ, Miranda-Saksena M, Douglas MW, Cunningham AL. 2008. Transport and egress of herpes simplex virus in neurons. Rev Med Virol 18:35–51. doi: 10.1002/rmv.560. [DOI] [PubMed] [Google Scholar]
- 2.Wisner TW, Sugimoto K, Howard PW, Kawaguchi Y, Johnson DC. 2011. Anterograde transport of herpes simplex virus capsids in neurons by both separate and married mechanisms. J Virol 85:5919–5928. doi: 10.1128/JVI.00116-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Taylor MP, Kobiler O, Enquist LW. 2012. Alphaherpesvirus axon-to-cell spread involves limited virion transmission. Proc Natl Acad Sci U S A 109:17046–17051. doi: 10.1073/pnas.1212926109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hill JM, Sedarati F, Javier RT, Wagner EK, Stevens JG. 1990. Herpes simplex virus latent phase transcription facilitates in vivo reactivation. Virology 174:117–125. doi: 10.1016/0042-6822(90)90060-5. [DOI] [PubMed] [Google Scholar]
- 5.Rock DL, Nesburn AB, Ghiasi H, Ong J, Lewis TL, Lokensgard JR, Wechsler SL. 1987. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J Virol 61:3820–3826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stevens JG. 1989. Human herpesviruses: a consideration of the latent state. Microbiol Rev 53:318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wechsler SL, Nesburn AB, Watson R, Slanina S, Ghiasi H. 1988. Fine mapping of the major latency-related RNA of herpes simplex virus type 1 in humans. J Gen Virol 69:3101–3106. doi: 10.1099/0022-1317-69-12-3101. [DOI] [PubMed] [Google Scholar]
- 8.Wechsler SL, Nesburn AB, Watson R, Slanina SM, Ghiasi H. 1988. Fine mapping of the latency-related gene of herpes simplex virus type 1: alternative splicing produces distinct latency-related RNAs containing open reading frames. J Virol 62:4051–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Roizman B, Whitley RJ. 2013. An inquiry into the molecular basis of HSV latency and reactivation. Annu Rev Microbiol 67:355–374. doi: 10.1146/annurev-micro-092412-155654. [DOI] [PubMed] [Google Scholar]
- 10.Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol 68:8045–8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barron BA, Gee L, Hauck WW, Kurinij N, Dawson CR, Jones DB, Wilhelmus KR, Kaufman HE, Sugar J, Hyndiuk RA, Laibson PR, Stulting RD, Asbell PA. 1994. Herpetic eye disease study. A controlled trial of oral acyclovir for herpes simplex stromal keratitis. Ophthalmology 101:1871–1882. [DOI] [PubMed] [Google Scholar]
- 12.Wilhelmus KR, Dawson CR, Barron BA, Bacchetti P, Gee L, Jones DB, Kaufman HE, Sugar J, Hyndiuk RA, Laibson PR, Stulting RD, Asbell PA. 1996. Risk factors for herpes simplex virus epithelial keratitis recurring during treatment of stromal keratitis or iridocyclitis. Herpetic Eye Disease Study Group. Br J Ophthalmol 80:969–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gordon YJ. 1990. Pathogenesis and latency of herpes simplex virus type 1 (HSV-1): an ophthalmologist's view of the eye as a model for the study of the virus-host relationship. Adv Exp Med Biol 278:205–209. doi: 10.1007/978-1-4684-5853-4_21. [DOI] [PubMed] [Google Scholar]
- 14.Kaufman HE, Azcuy AM, Varnell ED, Sloop GD, Thompson HW, Hill JM. 2005. HSV-1 DNA in tears and saliva of normal adults. Investig Ophthalmol Vis Sci 46:241–247. doi: 10.1167/iovs.04-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Steiner I. 1996. Human herpes viruses latent infection in the nervous system. Immunol Rev 152:157–173. doi: 10.1111/j.1600-065X.1996.tb00915.x. [DOI] [PubMed] [Google Scholar]
- 16.Koelle DM, Ghiasi H. 2005. Prospects for developing an effective vaccine against ocular herpes simplex virus infection. Curr Eye Res 30:929–942. doi: 10.1080/02713680500313153. [DOI] [PubMed] [Google Scholar]
- 17.Stroop WG, Schaefer DC. 1987. Severity of experimentally reactivated herpetic eye disease is related to the neurovirulence of the latent virus. Investig Ophthalmol Vis Sci 28:229–237. [PubMed] [Google Scholar]
- 18.Dix RD, McKendall RR, Baringer JR. 1983. Comparative neurovirulence of herpes simplex virus type 1 strains after peripheral or intracerebral inoculation of BALB/c mice. Infect Immun 40:103–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ghiasi H, Bahri S, Nesburn AB, Wechsler SL. 1995. Protection against herpes simplex virus-induced eye disease after vaccination with seven individually expressed herpes simplex virus 1 glycoproteins. Investig Ophthalmol Vis Sci 36:1352–1360. [PubMed] [Google Scholar]
- 20.Thompson RL, Cook ML, Devi-Rao GB, Wagner EK, Stevens JG. 1986. Functional and molecular analyses of the avirulent wild-type herpes simplex virus type 1 strain KOS. J Virol 58:203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berman EJ, Hill JM. 1985. Spontaneous ocular shedding of HSV-1 in latently infected rabbits. Investig Ophthalmol Vis Sci 26:587–590. [PubMed] [Google Scholar]
- 22.Allen SJ, Rhode-Kurnow A, Mott KR, Jiang X, Carpenter D, Rodriguez-Barbosa JI, Jones C, Wechsler SL, Ware CF, Ghiasi H. 2014. Regulatory interactions between herpesvirus entry mediator (TNFRSF14) and latency associated transcript (LAT) during HSV-1 latency. J Virol 88:1961–1971. doi: 10.1128/JVI.02467-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mott KR, Allen SJ, Zandian M, Akbari O, Hamrah P, Maazi H, Wechsler SL, Sharpe AH, Freeman GJ, Ghiasi H. 2014. Inclusion of CD80 in HSV targets the recombinant virus to PD-L1 on DCs and allows productive infection and robust immune responses. PLoS One 9:e87617. doi: 10.1371/journal.pone.0087617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mott KR, Allen SJ, Zandian M, Ghiasi H. 2014. Coregulatory interactions between CD8α DCs, LAT, and PD-1 contribute to higher HSV-1 latency. J Virol 88:6599–6610. doi: 10.1128/JVI.00590-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Webre JM, Hill JM, Nolan NM, Clement C, McFerrin HE, Bhattacharjee PS, Hsia V, Neumann DM, Foster TP, Lukiw WJ, Thompson HW. 2012. Rabbit and mouse models of HSV-1 latency, reactivation, and recurrent eye diseases. J Biomed Biotech 2012:612316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Feldman LT, Ellison AR, Voytek CC, Yang L, Krause P, Margolis TP. 2002. Spontaneous molecular reactivation of herpes simplex virus type 1 latency in mice. Proc Natl Acad Sci U S A 99:978–983. doi: 10.1073/pnas.022301899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kramer MF, Coen DM. 1995. Quantification of transcripts from the ICP4 and thymidine kinase genes in mouse ganglia latently infected with herpes simplex virus. J Virol 69:1389–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Allen SJ, Hamrah P, Gate DM, Mott KR, Mantopoulos D, Zheng L, Town T, Jones C, von Andrian UH, Freeman GJ, Sharpe AH, Benmohamed L, Ahmed R, Wechsler SL, Ghiasi H. 2011. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus type 1. J Virol 85:4184–4197. doi: 10.1128/JVI.02290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robert PY, Adenis JP, Denis F, Alain S, Ranger-Rogez S. 2003. Herpes simplex virus DNA in corneal transplants: prospective study of 38 recipients. J Med Virol 71:69–74. doi: 10.1002/jmv.10454. [DOI] [PubMed] [Google Scholar]
- 30.Remeijer L, Maertzdorf J, Doornenbal P, Verjans GM, Osterhaus AD. 2001. Herpes simplex virus 1 transmission through corneal transplantation. Lancet 357:442. doi: 10.1016/S0140-6736(00)04011-3. [DOI] [PubMed] [Google Scholar]
- 31.Derfuss T, Arbusow V, Strupp M, Brandt T, Theil D. 2009. The presence of lytic HSV-1 transcripts and clonally expanded T cells with a memory effector phenotype in human sensory ganglia. Ann N Y Acad Sci 1164:300–304. doi: 10.1111/j.1749-6632.2009.03871.x. [DOI] [PubMed] [Google Scholar]
- 32.Hufner K, Horn A, Derfuss T, Glon C, Sinicina I, Arbusow V, Strupp M, Brandt T, Theil D. 2009. Fewer latent herpes simplex virus type 1 and cytotoxic T cells occur in the ophthalmic division than in the maxillary and mandibular divisions of the human trigeminal ganglion and nerve. J Virol 83:3696–3703. doi: 10.1128/JVI.02464-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ghiasi H, Hofman FM, Cai S, Perng GC, Nesburn AB, Wechsler SL. 1999. Vaccination with different HSV-1 glycoproteins induces different patterns of ocular cytokine responses following HSV-1 challenge of vaccinated mice. Vaccine 17:2576–2582. doi: 10.1016/S0264-410X(99)00056-0. [DOI] [PubMed] [Google Scholar]
- 34.Ghiasi H, Pemg GC, Hofman FM, Cai S, Nesburn AB, Wechsler SL. 1999. Specific and nonspecific immune stimulation of MHC-II-deficient mice results in chronic HSV-1 infection of the trigeminal ganglia following ocular challenge. Virology 258:208–216. doi: 10.1006/viro.1999.9710. [DOI] [PubMed] [Google Scholar]
- 35.Ghiasi H, Perng G, Nesburn AB, Wechsler SL. 1999. Either a CD4(+) or CD8(+) T cell function is sufficient for clearance of infectious virus from trigeminal ganglia and establishment of herpes simplex virus type 1 latency in mice. Microb Pathog 27:387–394. doi: 10.1006/mpat.1999.0314. [DOI] [PubMed] [Google Scholar]
- 36.Mott KR, Allen SJ, Zandian M, Konda B, Sharifi BG, Jones C, Wechsler SL, Town T, Ghiasi H. 2014. CD8a dendritic cells drive establishment of HSV-1 latency. PLoS One 9:e93444. doi: 10.1371/journal.pone.0093444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Osorio Y, Ghiasi H. 2003. Comparison of adjuvant efficacy of herpes simplex virus type 1 recombinant viruses expressing TH1 and TH2 cytokine genes. J Virol 77:5774–5783. doi: 10.1128/JVI.77.10.5774-5783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mott KR, Ghiasi H. 2008. Role of dendritic cells in enhancement of herpes simplex virus type 1 latency and reactivation in vaccinated mice. Clin Vaccine Immunol 15:1859–1867. doi: 10.1128/CVI.00318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Matundan H, Mott KR, Ghiasi H. 2014. Role of CD8+ T cells and myeloid DCs in protection from ocular HSV-1 challenge in immunized mice. J Virol 88:8016–8027. doi: 10.1128/JVI.00913-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1992. Expression of herpes simplex virus type 1 glycoprotein B in insect cells. Initial analysis of its biochemical and immunological properties. Virus Res 22:25–39. [DOI] [PubMed] [Google Scholar]
- 41.Mott KR, Gate D, Matundan HH, Ghiasi YN, Town T, Ghiasi H. 2016. CD8+ T cells play a bystander role in mice latently infected with herpes simplex virus 1. J Virol 90:5059–5067. doi: 10.1128/JVI.00255-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Branco FJ, Fraser NW. 2005. Herpes simplex virus type 1 latency-associated transcript expression protects trigeminal ganglion neurons from apoptosis. J Virol 79:9019–9025. doi: 10.1128/JVI.79.14.9019-9025.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghiasi H, Osorio Y, Perng GC, Nesburn AB, Wechsler SL. 2002. Overexpression of interleukin-2 by a recombinant herpes simplex virus type 1 attenuates pathogenicity and enhances antiviral immunity. J Virol 76:9069–9078. doi: 10.1128/JVI.76.18.9069-9078.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ghiasi H, Kaiwar R, Nesburn AB, Slanina S, Wechsler SL. 1994. Expression of seven herpes simplex virus type 1 glycoproteins (gB, gC, gD, gE, gG, gH, and gI): comparative protection against lethal challenge in mice. J Virol 68:2118–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ghiasi H, Wechsler SL, Kaiwar R, Nesburn AB, Hofman FM. 1995. Local expression of tumor necrosis factor alpha and interleukin-2 correlates with protection against corneal scarring after ocular challenge of vaccinated mice with herpes simplex virus type 1. J Virol 69:334–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ghiasi H, Wechsler SL, Cai S, Nesburn AB, Hofman FM. 1998. The role of neutralizing antibody and T-helper subtypes in protection and pathogenesis of vaccinated mice following ocular HSV-1 challenge. Immunology 95:352–359. doi: 10.1046/j.1365-2567.1998.00602.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dawson CR. 1984. Ocular herpes simplex virus infections. Clin Dermatol 2:56–66. doi: 10.1016/0738-081X(84)90066-X. [DOI] [PubMed] [Google Scholar]
- 48.Streilein JW, Dana MR, Ksander BR. 1997. Immunity causing blindness: five different paths to herpes stromal keratitis. Immunol Today 18:443–449. doi: 10.1016/S0167-5699(97)01114-6. [DOI] [PubMed] [Google Scholar]
- 49.Liesegang TJ. 1999. Classification of herpes simplex virus keratitis and anterior uveitis. Cornea 18:127–143. doi: 10.1097/00003226-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 50.Liesegang TJ. 2001. Herpes simplex virus epidemiology and ocular importance. Cornea 20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 51.Ghiasi H, Slanina S, Nesburn AB, Wechsler SL. 1994. Characterization of baculovirus-expressed herpes simplex virus type 1 glycoprotein K. J Virol 68:2347–2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mott KR, Bresee CJ, Allen SJ, BenMohamed L, Wechsler SL, Ghiasi H. 2009. Level of herpes simplex virus type 1 latency correlates with severity of corneal scarring and exhaustion of CD8+ T cells in trigeminal ganglia of latently infected mice. J Virol 83:2246–2254. doi: 10.1128/JVI.02234-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mott KR, Underhill D, Wechsler SL, Ghiasi H. 2008. Lymphoid-related CD11c+ CD8a+ dendritic cells are involved in enhancing HSV-1 latency. J Virol 82:9870–9879. doi: 10.1128/JVI.00566-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kumar M, Hill JM, Clement C, Varnell ED, Thompson HW, Kaufman HE. 2009. A double-blind placebo-controlled study to evaluate valacyclovir alone and with aspirin for asymptomatic HSV-1 DNA shedding in human tears and saliva. Investig Ophthalmol Vis Sci 50:5601–5608. doi: 10.1167/iovs.09-3729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sawtell NM, Thompson RL. 2004. Comparison of herpes simplex virus reactivation in ganglia in vivo and in explants demonstrates quantitative and qualitative differences. J Virol 78:7784–7794. doi: 10.1128/JVI.78.14.7784-7794.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jin L, Perng GC, Carpenter D, Mott KR, Osorio N, Naito J, Brick DJ, Jones C, Wechsler SL. 2007. Reactivation phenotype in rabbits of a herpes simplex virus type 1 mutant containing an unrelated antiapoptosis gene in place of latency-associated transcript. J Neurovirol 13:78–84. doi: 10.1080/13550280601164333. [DOI] [PubMed] [Google Scholar]
- 57.Leib DA, Bogard CL, Kosz-Vnenchak M, Hicks KA, Coen DM, Knipe DM, Schaffer PA. 1989. A deletion mutant of the latency-associated transcript of herpes simplex virus type 1 reactivates from the latent state with reduced frequency. J Virol 63:2893–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sawtell NM, Thompson RL. 1992. Herpes simplex virus type 1 latency-associated transcription unit promotes anatomical site-dependent establishment and reactivation from latency. J Virol 66:2157–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tal-Singer R, Lasner TM, Podrzucki W, Skokotas A, Leary JJ, Berger SL, Fraser NW. 1997. Gene expression during reactivation of herpes simplex virus type 1 from latency in the peripheral nervous system is different from that during lytic infection of tissue cultures. J Virol 71:5268–5276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perng GC, Slanina SM, Ghiasi H, Nesburn AB, Wechsler SL. 2001. The effect of latency-associated transcript on the herpes simplex virus type 1 latency-reactivation phenotype is mouse strain-dependent. J Gen Virol 82:1117–1122. doi: 10.1099/0022-1317-82-5-1117. [DOI] [PubMed] [Google Scholar]
- 61.Trousdale MD, Steiner I, Spivack JG, Deshmane SL, Brown SM, MacLean AR, Subak-Sharpe JH, Fraser NW. 1991. In vivo and in vitro reactivation impairment of a herpes simplex virus type 1 latency-associated transcript variant in a rabbit eye model. J Virol 65:6989–6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hill JM, Rayfield MA, Haruta Y. 1987. Strain specificity of spontaneous and adrenergically induced HSV-1 ocular reactivation in latently infected rabbits. Curr Eye Res 6:91–97. doi: 10.3109/02713688709020074. [DOI] [PubMed] [Google Scholar]
- 63.Danaher RJ, Jacob RJ, Steiner MR, Allen WR, Hill JM, Miller CS. 2005. Histone deacetylase inhibitors induce reactivation of herpes simplex virus type 1 in a latency-associated transcript-independent manner in neuronal cells. J Neurovirol 11:306–317. doi: 10.1080/13550280590952817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kennedy DP, Clement C, Arceneaux RL, Bhattacharjee PS, Huq TS, Hill JM. 2011. Ocular herpes simplex virus type 1: is the cornea a reservoir for viral latency or a fast pit stop? Cornea 30:251–259. doi: 10.1097/ICO.0b013e3181ef241d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Thompson RL, Sawtell NM. 1997. The herpes simplex virus type 1 latency-associated transcript gene regulates the establishment of latency. J Virol 71:5432–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]