

ABSTRACT
The study of phage-host relationships is essential to understanding the dynamic of microbial systems. Here, we analyze genome-wide interactions of Bacillus subtilis and its lytic phage ϕ29 during the early stage of infection. Simultaneous high-resolution analysis of virus and host transcriptomes by deep RNA sequencing allowed us to identify differentially expressed bacterial genes. Phage ϕ29 induces significant transcriptional changes in about 0.9% (38/4,242) and 1.8% (76/4,242) of the host protein-coding genes after 8 and 16 min of infection, respectively. Gene ontology enrichment analysis clustered upregulated genes into several functional categories, such as nucleic acid metabolism (including DNA replication) and protein metabolism (including translation). Surprisingly, most of the transcriptional repressed genes were involved in the utilization of specific carbon sources such as ribose and inositol, and many contained promoter binding-sites for the catabolite control protein A (CcpA). Another interesting finding is the presence of previously uncharacterized antisense transcripts complementary to the well-known phage ϕ29 messenger RNAs that adds an additional layer to the viral transcriptome complexity.
IMPORTANCE The specific virus-host interactions that allow phages to redirect cellular machineries and energy resources to support the viral progeny production are poorly understood. This study provides, for the first time, an insight into the genome-wide transcriptional response of the Gram-positive model Bacillus subtilis to phage ϕ29 infection.
INTRODUCTION
Due to their small dimension and limited size of genomes, bacteriophages have optimized the exploitation of host resources to increase the production of the viral progeny. A comprehensive understanding of these host-virus interactions requires the analysis of associated transcriptional changes in both organisms. Thus, we used the recently developed RNA sequencing (RNA-Seq) technology to monitor to a high level of accuracy and depth the genome-wide effect of the bacteriophage ϕ29 on Bacillus subtilis transcription. The transcriptome profiles were analyzed at two early infection time points (8 and 16 min postinfection) so that the identification of the bacterial genes corresponding to these stages could allow the identification of potential phage targets.
Phage ϕ29 is a well-characterized lytic virus that belongs to the Podoviridae family. Over the years, it has been the subject of many extensive studies that have contributed to the understanding of several molecular mechanisms of biological processes, such as transcription regulation, viral DNA packaging, viral morphogenesis, and DNA replication (1). Phage ϕ29 genome consists of a linear double-stranded DNA (dsDNA) molecule of 19,285 bp, which encodes 28 open reading frames (ORFs) transcribed from four early and one late promoters. The viral genes are expressed in a temporal sequence to ensure that DNA replication, and the production and assembly of viral components occur in an orderly fashion. Thus, bacterial cells infected with phage ϕ29 at a multiplicity of 5 initiates early viral expression immediately after infection when the host RNA polymerase begins to transcribe genes involved in DNA replication and transcription regulation. Most of these early transcripts reach maximum levels at about 15 min (2), coinciding with the activation of the late promoter, which is responsible to express genes coding for phage structural, morphogenetic, and lytic proteins (3). At the terminal stage of the phage reproduction cycle, the assembly of the components of the viral particle, which is composed of a prolate head, a neck formed by a connector (required for head assembly), and a lower collar from which the appendages necessary for phage adsorption to the cell wall are attached, and a tail knob, occurs. Finally, the viral production of holin and endolysin proteins promotes the lysis of the host cell, releasing the virus progeny.
Here, we assess the global effect of the phage on the B. subtilis transcription, a Gram-positive and endospore-forming bacteria that normally inhabits the soil or decaying plant material. Since the current knowledge of phage-host interactions is based largely on a small number of Escherichia coli phages (4), our study provides new insight into the viral effects in a different but also well-known bacterium that is a model organism for studies of Gram-positive bacteria. To our knowledge, this is the first time that RNA-Seq methodology was used in B. subtilis-infected cells.
MATERIALS AND METHODS
Sample collection.
Cultures of wild-type B. subtilis strain 168 were grown in Luria-Bertani (LB) medium containing 5 mM MgSO4 at 37°C until reaching an optical density at 600 nm (OD600) of 0.45. The cells were then infected with phage ϕ29 at a multiplicity of 5. Two independent biological replicates were performed for each condition. Samples for RNA extraction were taken from infected and noninfected cultures at time points of 0, 8, 16, 24, 32, 40, 48, 56, and 64 min postinfection. The cells were harvested by centrifugation and immediately frozen at −70°C. In addition, an aliquot of the cultures was taken at the indicated times to monitor the optical density, as well as to determine the number of PFU in infected cultures.
RNA extraction.
Total RNA was extracted using the MasterPure RNA purification kit supplied by Epicentre (MCR85102) according to the manufacturer's recommendations. Samples were treated with RNase-free DNase I (Roche) and then concentrated using the GeneJET RNA cleanup and concentration micro kit (K0841) provided by Thermo Scientific. The quality and quantity of total RNA were determined by NanoDrop ND-1000 UV spectroscopy (Thermo Scientific), and the RNA integrity was checked using a 2100 Bioanalyzer (Agilent Technologies). To determine the levels of genomic DNA contamination after the treatment, we performed quantitative PCR (qPCR) experiments with primers that amplify host or viral genome. In all of the samples the genomic DNA percentage was less than 0.02%.
RNA-Seq libraries preparation and sequencing.
Total RNA was enriched for mRNA by removing rRNA from the samples using a RiboZero magnetic kit (Bacteria; MRZMB126) supplied by Illumina. Approximately 2.5 μg of total RNA and 9 μl of Ribozero rRNA removal solution were used per reaction. The manufacturer's instructions were followed, except for an additional cleanup step with AmpureXP Beads (A63881) provided by Agencourt at an RNA/bead ratio of 1:1. Strand-specific RNA-Seq libraries were prepared using the NEBNext Ultra Directional RNA Library Prep kit for Illumina (E7420L) supplied by New England BioLabs, following the manufacturer's instructions with some modifications. Briefly, ribosome-depleted RNA samples were fragmented and then used for first-strand cDNA synthesis with random primers and the ProtoScript II reverse transcriptase but in the absence of actinomycin D. Then, we performed second-strand cDNA synthesis, end repair, 3′-end adenylation, and adaptor ligation. The adaptors allowed further PCR amplification with NEBnext Multiplex Oligos for Illumina (E7335) provided by New England BioLabs. These adaptors can also be used for multiplexing in the sequencing run since they contained short sequences referred to as indices. The number of PCR cycles was adjusted to 15, and the final amplified libraries were quality checked and quantified on a BioAnalyzer 2100. Finally, an equimolecular pool of libraries was titrated by quantitative PCR using the Kapa-SYBR FAST qPCR kit for LightCycler 480 (KK4610) from Kapa Biosystems and a reference standard for quantification. Each library was sequenced using TruSeq SBS kit v3-HS, in paired-end mode with the read length of 2×76 bp for the mRNA-Seq experiments, using the HiSeq2000 instrument (Illumina) according to the manufacturer's protocol. Images from the instrument were processed using the manufacturer's software to generate FASTQ sequence files.
Bioinformatics analysis of RNA-Seq data.
The analyzed data set was constituted by eight phage ϕ29-infected B. subtilis 168 samples corresponding to four different experimental conditions, with a total of 149,751,662 single-end reads 76 nucleotides in length. A preliminary analysis of the quality of the reads was performed using FastQC, a Java tool with graphic interface (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/). The reference genome, proteins, and annotation files of B. subtilis 168 and phage ϕ29 have been downloaded from NCBI ftp site (ftp://ftp.ncbi.nlm.nih.gov/genomes/Bacteria/Bacillus_subtilis_168_uid57675/ and ftp://ftp.ncbi.nlm.nih.gov/genomes/Viruses/Bacillus_phage_phi29_uid30615/). The reads were aligned using Bowtie2 aligner, which supports lax alignment parameters (5). Since the reads showed a good behavior in the alignment process, it was not necessary to trim or filter them due to poor quality reasons. To obtain a quantification of the differential gene expression pattern, we used Cuffdiff, a tool that calculates gene and transcript expression levels (in RPKM [reads per kilobase of transcript per million mapped reads]) under more than one condition and tests them for significant differences (6). Bacterial genes showing significant transcription changes (P ≤ 0.05) after phage ϕ29 infection were analyzed and classified using BsubCyc (http://bsubcyc.org/) (7), a model-organism database for the B. subtilis strain 168 that imports the GO terms (8) from the UniProtKB-GOA gene association files.
Accession number(s).
The sequencing data were deposited in the European Nucleotide Archive under accession number PRJEB13724 (http://www.ebi.ac.uk/ena/data/view/PRJEB13724).
RESULTS AND DISCUSSION
Analysis of virus-induced changes in the host cell.
Phage ϕ29 did not affect the growth rate of the B. subtilis culture during the first 16 min of infection since the turbidity (OD600) of ϕ29-infected and noninfected cultures rise with almost equal rates (Fig. 1A). Subsequently, the increase number of new viral infective particles (Fig. 1B) induced the lysis of infected cells, causing a progressive decline in the optical density. In fact, a small number of infective particles was already detected after 24 min of infection, reaching the maximum at approximately 48 min, when the infective cycle was completed. Previous studies showed that there was a correlation between the production of infective particles and the amount of ϕ29 DNA accumulated in infected cells as a consequence of replication. Comparison of the total nucleic acid content in cells infected or not with phage ϕ29 revealed a peak at 32 min and then a pronounced decrease (Fig. 1C), probably caused by the encapsulation of phage DNA in viral particles.
FIG 1.
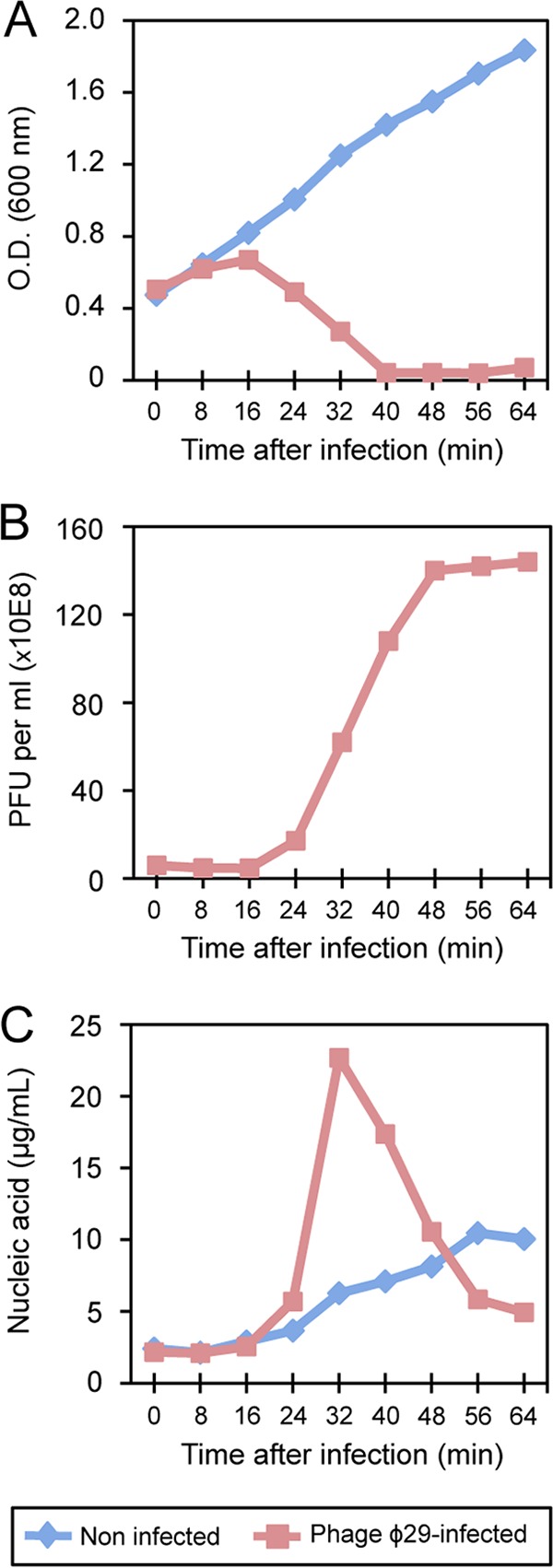
Analysis of host growth, phage virions, and nucleic acid content throughout the infection cycle of phage ϕ29 in B. subtilis. (A) Phage-mediated lysis of bacterial cultures. Lysis was monitored by measuring the OD600 values for samples taken at the indicated times after infection. (B) The ϕ29 virions were counted as PFU per ml of culture. One hundred-fold-diluted cultures were grown in LB medium containing magnesium at 37°C and then infected at an OD600 of 0.45 with ϕ29 at a multiplicity of 5. (C) The amount of nucleic acid detected at the indicated times postinfection was quantified by using a 2100 Bioanalyzer (Agilent Technologies), and values are expressed as micrograms of nucleic acid per ml of culture. The graphics are representative of four independent experiments.
Taking all of this into account, we decided to examine the transcriptional changes caused by phage ϕ29 in B. subtilis after 8 and 16 min of infection, since in these early times there is not a significant lysis of the cells that could interfere with the results. RNA-Seq technology was used to simultaneously analyze both host and viral transcriptomes. To distinguish the virus-induced changes from the B. subtilis growth-dependent changes, we compared the transcriptional levels of infected cells to those of noninfected samples at the same times. After performing an alignment of all the reads against B. subtilis and phage ϕ29 genomes using the Bowtie2 aligner (5), we observed that the percentage of transcripts from the ϕ29 genome at 8 min postinfection was between 1.9 and 3.7% of the total, whereas after 16 min of infection the percentage of viral RNA detected increased to 16.2 to 16.6% (Fig. 2). Genome-wide expression differences between infected and uninfected cells were visualized using box-and-whisker plots, showing that ϕ29 only induces relatively moderate changes in host gene expression at early times of infection (Fig. 3). Moreover, genes involved in cell growth and division were not affected during this period of infection (Tables 1 and 2), which is consistent with the profile of the growth curve shown in Fig. 1A. It is possible that most of the changes caused by ϕ29 take place later, during the production and assembly of the virus components and the transcription of lytic proteins. Although there is not a global transcriptional reprogramming of the host cell during the early stages of infection, a number of bacterial protein-coding genes showed significantly expression differences (Tables 1 and 2). In particular, about 2% of the approximately 4,242 protein-coding genes that comprise B. subtilis genome (9) were differentially expressed (P ≤ 0.05; fold change > 2) during the early stages of infection. Genes involved in nucleic acid metabolism, carbohydrate metabolism, and transport stand out among them (Fig. 4).
FIG 2.
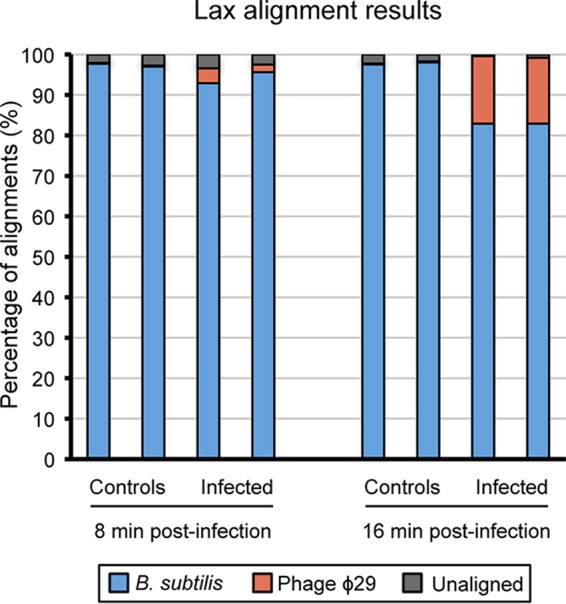
Alignment of RNA reads sets against B. subtilis or phage ϕ29 genome using the bioinformatics tool Bowtie2. The data for two individual experiments are shown.
FIG 3.
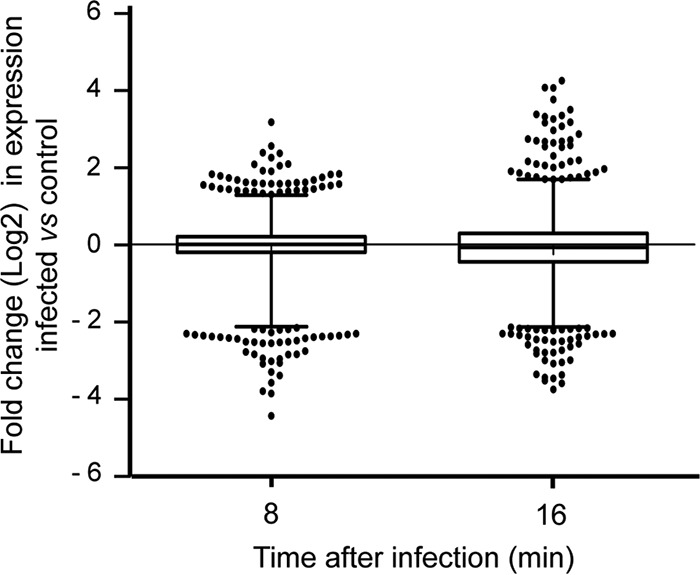
Box-and-whisker plots showing the relative fold change (log2) in the expression of B. subtilis genes after 8 or 16 min of ϕ29 infection compared to uninfected controls. The analysis considered 4,421 genes of B. subtilis. Each box represents the differential expression values from the lower to the upper quartile (corresponding to the 25 to 75% of the data set). The middle horizontal line represents the median. The whiskers extend from the boxes to 1 and 99% of the data set. Dots indicate outliers. Each data point represents the mean of two independent experiments.
TABLE 1.
GO biological process categories of B. subtilis protein-coding genes overexpressed in ϕ29 infection
| Function and gene | Annotation | Fold change (P) |
|
|---|---|---|---|
| 8 min | 16 min | ||
| Nucleic acid metabolism | |||
| helD | DNA 3′-5′ helicase IV | 9.9 (<0.01) | |
| ymaB | Putative enzyme involved in deoxyribonucleotide synthesis | 2.6 (0.04) | 9.0 (<0.01) |
| nrdE | Ribonucleoside-diphosphate reductase (major subunit) | 3.3 (0.01) | 7.8 (<0.01) |
| nrdF | Ribonucleoside-diphosphate reductase (minor subunit) | 2.8 (0.03) | 6.4 (<0.01) |
| ssbB | Single-strand DNA-binding protein | 2.8 (0.05) | |
| ywhA | Putative transcriptional regulator (MarR family) | 2.7 (0.02) | |
| rph | RNase PH | 2.6 (0.04) | |
| abrB | Transcriptional regulator for transition state genes | 2.5 (0.04) | |
| ycnC | Putative transcriptional regulator (TetR/AcrR family) | 2.7 (0.04) | 2.4 (0.04) |
| rpoC | RNA polymerase (β′ subunit) | 2.1 (0.05) | |
| Protein metabolism | |||
| yoaZ | Putative factor of the oxidative stress response | 4.3 (<0.01) | |
| gatC | Aspartyl/glutamyl-tRNA(Asn/Gln) amidotransferase (subunit C) | 3.4 (0.01) | |
| gatA | Aspartyl/glutamyl-tRNA(Asn/Gln) amidotransferase (subunit A) | 2.9 (0.01) | |
| gatB | Aspartyl/glutamyl-tRNA(Asn/Gln) amidotransferase (subunit B) | 2.7 (0.02) | |
| clpP | ATP-dependent Clp protease proteolytic subunit | 2.3 (0.05) | |
| Lipid metabolism | |||
| estA | Secreted alkaliphilic lipase | 3.4 (0.01) | |
| yqjD | Putative acyl-CoA carboxylase | 2.5 (0.04) | |
| fni | Isopentenyl diphosphate isomerase | 2.4 (0.04) | |
| Oxidation-reduction process | |||
| yetG | Heme-degrading monooxygenase | 3.1 (0.02) | |
| yfhC | Putative oxidoreductase (nitroreductase family) | 2.8 (0.02) | |
| trxA | Thioredoxin | 2.7 (0.03) | |
| yugK | Putative NADH-dependent butanol dehydrogenase | 2.6 (0.03) | |
| Transport | |||
| yodF | Putative Na+/metabolite permease | 2.8 (0.02) | |
| yvdB | Putative anion transporter | 2.5 (0.04) | |
| yfnC | Putative efflux transporter | 2.5 (0.03) | |
| Other | |||
| dhbF | Dimodular nonribosomal peptide synthase | 2.8 (0.02) | |
| proI | Pyrroline-5-carboxylate reductase | 2.7 (0.02) | |
| ywbC | Glyoxalase I | 2.6 (0.04) | |
| hemD | Uroporphyrinogen III cosynthase | 2.5 (0.04) | |
| yvgJ | Lipoteichoic acid primase | 2.5 (0.03) | |
| adeC | Adenine deaminase | 2.4 (0.03) | |
TABLE 2.
GO biological process categories of B. subtilis protein-coding genes downregulated in ϕ29 infection
| Function and genea | Annotation | Fold change (P) |
|
|---|---|---|---|
| 8 min | 16 min | ||
| Carbohydrate metabolism | |||
| rbsK* | Ribokinase | −5.0 (0.01) | −8.5 (<0.01) |
| manA | Mannose-6 phosphate isomerase; cupin family | −5.4 (<0.01) | −6.6 (<0.01) |
| amyE* | Alpha-amylase | −4.0 (0.01) | −4.9 (<0.01) |
| iolB* | 5-Deoxy-d-glucuronate isomerase | −3.1 (0.04) | −4.5 (<0.01) |
| uxaC* | Galacturonate isomerase | −4.5 (<0.01) | |
| iolC* | 2-Deoxy-5-keto-d-gluconic acid kinase | −3.4 (0.02) | −4.3 (<0.01) |
| treA* | Trehalose-6-phosphate hydrolase | −3.8 (0.01) | −4.3 (<0.01) |
| iolG* | Inositol dehydrogenase | −4.1 (<0.01) | |
| iolE* | 2-Keto-myo-inositol dehydratase | −4.0 (<0.01) | |
| iolD* | 3D-(3,5/4)-trihydroxycyclohexane-1,2-dione hydrolase | −3.4 (0.01) | |
| sacA* | Sucrase-6-phosphate hydrolase | −3.4 (0.03) | |
| xylB* | Xylulose kinase | −3.0 (0.02) | |
| licS (bglS) | Endo-β-1,3-1,4 glucanase | −2.9 (0.03) | |
| iolA (mmsA)* | (Methyl)malonate-semialdehyde dehydrogenase | −2.7 (0.03) | |
| licH | 6-Phospho-β-glucosidase | −2.7 (0.03) | |
| iolJ* | 2-Deoxy-5-keto-d-gluconic acid 6-phosphate aldolase | −2.7 (0.04) | |
| pel | Pectate lyase | −2.6 (0.02) | |
| gntK | Gluconate kinase | −3.8 (0.01) | |
| glpF* | Glycerol permease | −2.6 (0.04) | |
| Transport | |||
| rbsB* | Ribose ABC transporter (ribose-binding lipoprotein) | −5.1 (<0.01) | −11.5 (<0.01) |
| rbsA* | Ribose ABC transporter (ATP-binding protein) | −6.7 (<0.01) | −10.8 (<0.01) |
| rbsC* | Ribose ABC transporter (permease) | −5.7 (<0.01) | −10.3 (<0.01) |
| sunT | Sublancin 168 lantibiotic transporter | −5.5 (0.02) | |
| treP* | PTS system trehalose-specific EIIBC component | −4.9 (<0.01) | −5.2 (<0.01) |
| manP* | PTS system mannose-specific EIIBCA component | −4.6 (<0.01) | −4.5 (<0.01) |
| xynP* | Putative H+-xyloside symporter | −3.9 (0.01) | |
| iolF* | Inositol transport protein | −3.9 (<0.01) | |
| dctP* | C4-dicarboxylate transport protein | −9.8 (<0.01) | −3.8 (<0.01) |
| msmX* | Multiple sugar-binding transporter ATP-binding protein | −3.4 (0.01) | −3.5 (0.01) |
| nupC | Pyrimidine-nucleoside Na+(H+) cotransporter | −3.8 (0.01) | −3.3 (0.01) |
| mdxE | Maltose/maltodextrin-binding lipoprotein | −2.8 (0.04) | |
| maeN | Na+/malate symporter | −2.4 (0.03) | |
| licC | Lichenan permease IIC component | −2.7 (0.04) | −2.4 (0.04) |
| malP** | PTS system maltose-specific EIICB component | −3.2 (0.02) | |
| csbX** | Putative permease | −2.7 (0.04) | |
| cycB | Cyclodextrin-binding lipoprotein | −2.7 (0.04) | |
| Nucleic acid metabolism | |||
| rbsR* | Transcriptional regulator (LacI family) | −7.1 (<0.01) | −7.9 (<0.01) |
| manR* | Transcription activator | −3.0 (0.02) | −3.5 (<0.01) |
| yopS | Putative transcriptional regulator; phage SPβ | −3.0 (0.03) | |
| acoR* | Transcriptional regulator | −5.3 (<0.01) | −2.9 (0.02) |
| treR* | Transcriptional regulator (GntR family) | −2.3 (0.05) | |
| ykoM* | Putative transcriptional regulator (MarR family) | −5.2 (0.01) | |
| msmR** | Transcriptional regulator (LacI family) | −3.4 (0.02) | |
| deoC | Deoxyribose-phosphate aldolase | −3.4 (0.02) | |
| Oxidation-reduction process | |||
| yneN | Thioredoxin-like protein | −2.5 (0.04) | |
| yrbE | Putative oxidoreductase | −5.4 (<0.01) | −2.5 (0.04) |
| cccA* | Cytochrome c550 | −5.1 (0.02) | |
| yuxG** | Uncharacterized oxidoreductase | −2.6 (0.04) | |
| Other | |||
| ywsB* | Conserved hypothetical protein | −5.6 (<0.01) | −13.4 (<0.01) |
| ysbA** | Antiholin factor | −5.7 (<0.01) | −6.0 (<0.01) |
| ysbB** | Antiholin factor | −4.1 (<0.01) | −4.5 (<0.01) |
| cstA* | Carbon starvation-induced membrane protein | −2.7 (0.04) | −3.4 (0.01) |
| rocG* | Glutamate dehydrogenase | −2.9 (0.02) | |
| pdp | Pyrimidine-nucleoside phosphorylase | −3.3 (0.02) | −2.7 (0.03) |
| odhB** | 2-Oxoglutarate dehydrogenase complex (E2 subunit) | −2.5 (0.03) | |
*, The promoter contains high-affinity cre boxes; **, the promoter contains low-affinity cre boxes.
FIG 4.
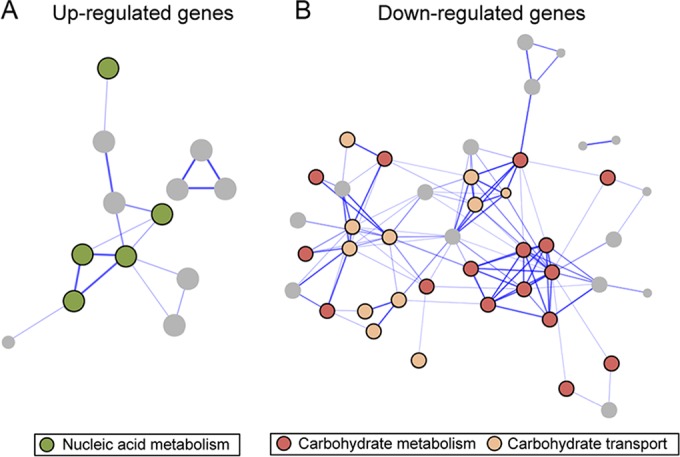
Interaction networks of the B. subtilis proteins encoded by genes that are upregulated (A) or downregulated (B) after 8 and 16 min of ϕ29 infection obtained from STRING database (v9.1) using default settings (confidence > 0.4). The proteins are represented by nodes, which are colored according to the GO biological process classification (nucleic acid metabolism in green and carbohydrate metabolism and carbohydrate transport in red and beige, respectively). Genes belonging to other GO categories are colored gray. Disconnected nodes are not shown. Lines represent the predicted functional associations between the proteins.
Upregulated genes after phage ϕ29 infection.
Only four genes were significantly upregulated by 8 min postinfection: ymaB, nrdE, and nrdF, which are members of the nrd operon, and ycnC (Table 1). YcnC is a putative transcriptional regulator of the TetR/AcrR family. The nrdE and nrdF genes encode the only ribonucleotide reductases known in B. subtilis. These proteins are essential for the biosynthesis of deoxyribonucleotides from the corresponding ribonucleotides (10). YmaB is a putative enzyme also involved in deoxyribonucleotide synthesis. The operon was induced by thymidine starvation and its expression is under the control of cell cycle and was directly or indirectly affected by the SOS regulator RecA (11). The DNA synthesis is a critical step of ϕ29 infection, and thus the upregulation of these genes will potentially increase the deoxyribonucleotide levels required for viral DNA replication. The larger amount of transcripts detected for trxA, a gene encoding a thioredoxin, could be related to nrdE and nrdF upregulation, since some studies suggest that TrxA is the only electron donor required for ribonucleotide reductase reduction in B. subtilis. However, trxA induction was detected only after 16 min of infection, perhaps because the increased levels of nrdE and nrdF transcripts at 8 min are not high enough to require an additional amount of TrxA that could promote its transcription. Moreover, thioredoxin is an essential protein involved in defense mechanisms against oxidative stress, although it is also induced by multiple stresses including heat, salt, or ethanol treatments (12).
Interestingly, the most upregulated gene is helD whose transcripts levels increase almost 10-fold after 16 min of ϕ29 infection. HelD is a helicase implicated in DNA repair and homologous recombination (13). In addition, the interaction of this protein with the RNA polymerase has a stimulatory effect on transcription cycling and elongation (14). There are plenty of phages that encode DNA helicases in order to drive the unwinding of its DNA helix. For example, protein 41, a helicase of bacteriophage T4, greatly stimulates the rate of strand displacement DNA synthesis at a viral replication fork (15). Other phages take advantage of host helicases, as in the case of bacteriophage P2, a temperate phage that infects Escherichia coli. During the lytic cycle, the P2 genome is replicated by a modified rolling circle mechanism that requires several host proteins such as DNA polymerase III, primase, and the replicative helicase DnaB. Since the helicases do not bind DNA in a sequence-specific manner, they have to be recruited by other proteins to the origin (16). Thus, P2 encodes a B protein that interacts with DnaB, allowing an efficient loading of this helicase on the viral genome and an optimal lagging-strand DNA synthesis (17). The genome of phage ϕ29 does not encode any helicase, but it is possible that host helicase HelD might be necessary for efficient ϕ29 DNA replication in vivo, in spite of the fact that the ϕ29 DNA polymerase has an intrinsic helicase-like activity (18).
After 16 min of infection, there is an induction of ssbB, which encodes a single-stranded DNA (ssDNA) binding protein responsible to protect incoming ssDNA from cellular nucleases, remove DNA secondary structures, and modulate RecA nucleation onto ssDNA (19). Phage ϕ29 encodes a ssDNA binding protein called p5, which is required for viral DNA replication in vivo (20). However, in vitro assays have shown that other SSB proteins, including E. coli SSB, bacteriophage T4 gp32, adenovirus DNA-binding protein, and the human replication factor A, can functionally substitute protein p5 (21). It is possible that the stimulation of viral DNA replication by p5 is carried out by a nonspecific mechanism, and thus it is possible that SsbB from B. subtilis could contribute to some extent to this process.
RNase PH plays a crucial role in the exonucleolytic degradation of CCA-containing tRNA precursors in B. subtilis (22), a process necessary to a proper tRNA maturation. In addition, this enzyme also plays a role in the secondary pathway of 23S rRNA 3′-end maturation (23), suggesting that RNase PH could have an impact in translation. The upregulation of rph might reflect a phage requirement to increase translational rates.
AbrB is a regulator of the transcription of genes expressed during the transition state between vegetative growth and the stationary phase. It is also a global modulator of catabolite repression. However, the level of abrB upregulation detected (Table 1) does not seem to be sufficient to activate the transcription of genes subjected to catabolite repression that are under its control, such as rbsA, gntK, or glpF (24) (Table 2). Interestingly, the expression of abrB is inhibited by phosphorylated Spo0A, the key regulator for sporulation activation, which is directly responsible for suppression of phage ϕ29 development (25).
The heterotrimeric complex formed by gatA, gatB, and gatC is involved in the formation of correctly charged Asn-tRNAAsn or Gln-tRNAGln through the transamidation of misacylated Asp-tRNAAsn or Glu-tRNAGln. In many bacteria, asparagine can be directly attached to the tRNA using an asparaginyl-tRNA synthetase. However, B. subtilis can also use a two-step indirect pathway to form Asn-tRNAAsn. This process uses a nondiscriminating aspartyl-tRNA synthetase to ligate Asp to tRNAAsn, and then the GatCAB complex transamidates Asp to Asn on the tRNA, forming Asn-tRNAAsn (26). In addition, the GatCAB complex is able to transamidate Glu-tRNAGln to Gln-tRNAGln (27). This upregulation could reveal a ϕ29 need to increase the amount of these aminoacyl-tRNAs to synthesize its own proteins. To check this possibility, we compared the amino acid content of the proteins from phage ϕ29 and B. subtilis strain 168 (Fig. 5). The analysis showed that glutamine content is quite similar between phage and bacteria but that, in contrast, ϕ29 proteins have an asparagine percentage higher than their host, and thus it might require a larger amount of Asn-tRNA to use in the translation process.
FIG 5.
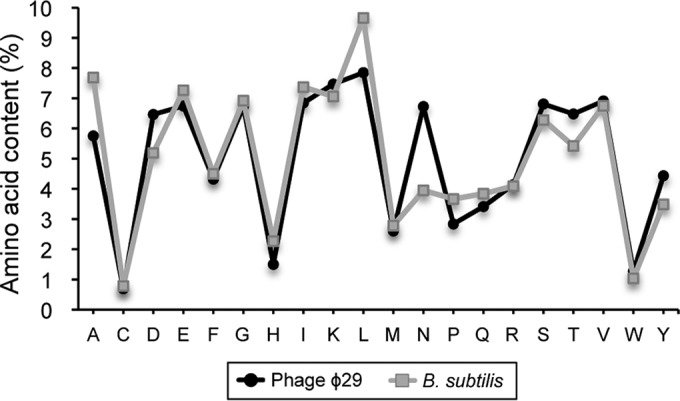
Average amino acid compositions of proteins encoded by the phage ϕ29 genome or by the B. subtilis genome. An in-house script written in Python language was used to compute the amino acid composition of the proteins of both organisms from the FASTA files obtained from the National Center for Biotechnology Information ftp site. The graphics were created using the R programming language and software environment.
ClpP is a protease involved in the degradation of misfolded proteins that is essential for stress tolerance in B. subtilis (28). It has been previously reported that the amount of unfolded polypeptides within the cell increases during the infection of phages such as PRD1 (29). On the other hand, several bacterial energy-dependent proteases have been shown to degrade regulatory proteins of bacteriophages. For example, E. coli ClpP degrades protein O of the λ phage, which has a main role in the replication of viral DNA (30). Similarly, it is possible that ClpP degrades some ϕ29 proteins required for viral development.
Three genes involved in lipid metabolism (estA, fni, and yqjD) are also upregulated after 16 min of ϕ29 infection. B. subtilis secretes estA, a lipase that catalyzes the hydrolysis of triglyceride. Fni is an enzyme implicated in the biosynthesis of isoprenoids, which serve as structural components of membranes and mediators of cellular redox chemistry (31). YqjD is a putative component of acetyl coenzyme A (acetyl-CoA) carboxylase complex involved in the synthesis of malonyl-CoA from acetyl-CoA, which is a central intermediate of the fatty acid biosynthesis pathway.
The dhbF gene encodes a dimodular nonribosomal peptide synthase involved in the synthesis of bacillibactin, a siderophore secreted by many Bacillus species (32). It is possible that the infected cells have higher iron requirements that could explain the high transcripts levels detected for dhbF and also for yetG (Table 1). YetG (also called HmoA) is a heme-degrading monooxygenase that is able to degrade the heme group, releasing the iron (33).
YvgJ is a lipoteichoic acid synthase-like protein. Lipoteichoic acids (LTAs) are cell envelope components widely distributed in Gram-positive bacteria. LTAs prevent phage sk11G adsorption to Lactococcus lactis subsp. cremoris SK110 by masking the actual receptor site (34).
YwbC (also called GlxA) is a glyoxalase I necessary for methylglyoxal detoxification. Methylglyoxal, a highly reactive dicarbonyl compound, is a toxic by-product of glycolysis. It is synthesized by methylglyoxal synthase under conditions of excessive carbon flux or phosphate limitation, which lead to an imbalance in the metabolism between the carbon rate acquisition and the glycolysis (35). The toxicity of methylglyoxal is due to its ability to interact with the nucleophilic centers of macromolecules. Thus, it has been shown that this compound can modify guanine bases in the genome, leading to DNA damage and increased mutational rates (36). In addition, methylglyoxal can also react with arginine, lysine, and cysteine residues in proteins, causing its inactivation and ultimately cell death. Its effect on viral development was studied using bacteriophage T4, which lost its infective capacity on E. coli after methylglyoxal treatment (37). Despite the fact that methylglyoxal has not been quantified in ϕ29 infected cells, it is possible that phage ϕ29 increases its cellular concentration; the upregulation of ywbC could be an attempt to counteract this damage that can potentially affect both bacteria and phage.
Downregulated genes after phage ϕ29 infection.
Transcriptional repression seems to occur earlier than inductions in phage ϕ29 infected cells, suggesting that the downregulation of specific bacterial genes is necessary to promote a rapid change in host metabolism for an optimal viral development. At 8 and 16 min after ϕ29 infection, the most downregulated pathways were related to the utilization of specific carbon sources of bacteria, including ribose, inositol, sucrose, lichenan, starch, trehalose, or galacturonate (Table 2). In fact, 39 of the 55 genes that are downregulated after viral infection are members of operons repressed by the catabolite control protein A (CcpA) (Table 2), a DNA-binding protein of the LacI/GalR family of transcriptional regulators (38, 39). Moreover, most of these CcpA-regulated genes (82%) contain high-affinity binding sequences for CcpA called cre (catabolite responsive elements). Since approximately 95 genes of B. subtilis are transcriptionally controlled by high-affinity cre boxes, there is a significant enrichment in these sequences in the promoters of genes downregulated after phage ϕ29 infection (P < 0.0001 [two-sided Fisher exact test]).
Like many other bacteria, B. subtilis is able to use a variety of carbohydrates as sources of carbon and energy. The expression of the genes required for its utilization depends on the presence of the specific substrate (induction) in the environment and the absence of preferred carbon sources, such as glucose, that can be well metabolized (catabolite repression). CcpA is responsible for the carbon catabolite control in B. subtilis and other Gram-positive bacteria. This protein is constitutively synthesized (40), and it does not change its expression either after 8 or 16 min of ϕ29 infection. The nutritional and physiological states of the bacterial host are important factors to determine the consequences of a phage infection. For example, the development of bacteriophage T4 (measured as the total number of PFU per infected cell) is strongly influenced by the carbon source present in the medium where its host E. coli grows (41). It has been also reported that λ phage reduces the transcription of bacterial pckA, a gluconeogenic gene involved in the utilization of alternative carbon sources such as succinate (42). It is possible that the effect of phage ϕ29 infection in the repression of genes involved in the utilization of specific carbon sources could maintain B. subtilis in a physiological state necessary for optimal viral development.
Particularly, there is a dramatic reduction in the levels of RNA from genes of the ribose operon (Table 2). d-Ribose is one of the metabolites that bacteria can actively transport into the cell to use as carbon and energy source. The high-affinity transport for this monosaccharide in B. subtilis is encoded by the ribose transport operon that consists in genes encoding RbsA (an ATP-binding transport protein), RbsB (a ribose-binding protein), RbsC (a permease), RbsD (a membrane transport protein), RbsK (a ribokinase that phosphorylates the sugar in the presence of ATP into d-ribose 5-phosphate as the first reaction in the metabolism of exogenous ribose), and RbsR (a transcription regulator that represses the operon expression) (43). The transcription of this operon is negatively controlled by the catabolite repressor protein CcpA (44). An additional ORF of unknown function called ywsB was detected in this operon downstream from rbsB (45). All genes except rbsD were downregulated at both 8 and 16 min postinfection.
Phage ϕ29 infection also affects the expression of genes involved in the catabolism of myo-inositol, a sugar alcohol abundant in soil that can be metabolized by several microorganisms. The structure of the iol operon for myo-inositol catabolism in B. subtilis consists of 10 genes (iolA to iolJ). Once myo-inositol is incorporated into the cell, the inositol dehydrogenase encoded by iolG catalyzes its oxidation to 2-keto-myo-inositol, which is the first reaction of the catabolic pathway that results in the conversion of myo-inositol to an equimolar mixture of dihydroxyacetone phosphate, acetyl-CoA, and CO2 (46, 47). Glucose repression of the operon is exerted through catabolite repression mediated by CcpA and also by IolR. Genes of iol operon were repressed after ϕ29 infection with the exception of iolH (a putative sugar-phosphate epimerase/isomerase) and iolI (a 2-keto-myo-inositol isomerase).
B. subtilis produces and secretes α-amylase (AmyE) to degrade extracellular starch, an abundant carbon source in nature. The enzyme hydrolyzes internal α-1,4-glycosidic linkages in starch and related molecules to yield products such as glucose or maltose (48). The gene sacA encodes an endocellular sucrose-6-phosphate hydrolase referred to as sucrase involved in sucrose degradation into α-d-glucose and β-d-fructose (49). UxaC is a protein required for the utilization of galacturonate as a carbon source (50). Polymethylgalacturonate (pectin) is a component of plant cell walls frequently found in the soil that can be converted to galacturonate, contributing to B. subtilis growing in its natural environment.
The three genes of the trehalose operon (treA, treP, and treR) were also downregulated following ϕ29 infection (Table 2). Trehalose is transported into the cell by a phosphotransferase system mediated by TreP, resulting in the phosphorylated trehalose-6-phosphate form (51), which is then hydrolyzed by trehalose-6-phosphate hydrolase (TreA), yielding glucose and glucose-6-phosphate (52). TreR is the specific repressor of the operon.
XylB is involved in the degradation of xylose although B. subtilis strain 168 is unable to effectively utilize xylose as sole carbon source. LicC is involved in the uptake of lichenan, a β-1,3-1,4-glucan, which is further degraded by LicH, a probable 6-phospho-β-glucosidase (53). In addition, licS encodes a β-1,3-1,4-endoglucanase necessary for lichenan utilization.
Two genes of the mannose operon are downregulated after ϕ29 infection: manA, a gene encoding a mannose-6-phosphate isomerase, and manP, which encodes a phosphotransferase system mannose-specific IIBCA transporter. The transcriptional activator ManR, whose gene also showed decreased levels of transcription, regulates this operon (54). Intriguingly, two subunits of the mannose transporter called IICMan and IIDMan are necessary for infection of E. coli cells by λ bacteriophage since they facilitate the penetration of viral DNA across the inner membrane (55).
Another repressed gene is pdp, which encodes a pyrimidine nucleoside phosphorylase that catalyzes the reversible phosphorolysis of the pyrimidine nucleosides uridine, thymidine, and 2′-deoxyuridine to the corresponding pyrimidine base and ribose-1-phosphate (56). In the same operon as pdp are included deoC and nupC. DeoC is a deoxyribose-phosphate aldolase involved in deoxyribonucleotide catabolic process. Its repression could be a phage ϕ29 attempt to diminish the cellular degradation of deoxyribonucleotides, which are necessary for replicating the viral genome.
Most of the downregulated genes that are involved in transport are sugar transporters except for nupC, which encodes a protein involved in transport of uridine (56). This reduction in the transcription levels could be related to the deleterious effect that the presence of uracil residues may have in ϕ29 genome integrity. Although phage ϕ29 DNA does not contain uracil residues, misincorporation during the replication process or spontaneous cytosine deamination may occur. If uracil residues appear in ssDNA regions of replicative intermediates, the action of host uracil DNA-glycosylase (UDG) will introduce a nick into the phosphodiester backbone producing the loss of the terminal region. To avoid this process, phage ϕ29 encodes a UDG inhibitor called p56 (57, 58).
Although the c-type cytochromes play an important role in electron transport systems, the cytochrome c550 encoded by the cccA gene is not essential for growth of B. subtilis (59). Instead, cytochrome c550 is related to the initiation of sporulation since a strain with this gene deleted showed delayed sporulation (60). Curiously, the lytic cycle of phage ϕ29 is suppressed when cells are infected during the early stages of sporulation and the viral genome becomes trapped into the spore (61).
The genes ysbA and ysbB encode putative antiholins predicted to finely direct the system by inhibiting holins and therefore regulating the accurate timing of cell lysis. At the end of its lytic cycle, dsDNA bacteriophages induce cell lysis through a holin-endolysin system to release new viral particles (62). Holins are small proteins, which form pores in the cytoplasmic membrane allowing endolysins to degrade cell wall peptidoglycan. The timing of lysis is precisely controlled to maximize the reproductive potential of the bacteriophage population (63). Some bacteria also include holin-antiholin-like genes in their genomes that resemble their phage counterparts. The induction of cell death in a part of the population in response to certain environmental stresses may ensure the survival of the remaining cells, and this may be considered a behavior similar to that in multicellular organisms. The downregulation of ysbA and ysbB might impede the cell to counteract cell lysis promoted by phage ϕ29.
It should be noted that some of the genes included in Table 2 such as dctP, acoR, and yrbE showed a decrease in downregulation at 16 min compared to that observed at 8 min postinfection. Probably, phage ϕ29 is directly responsible for causing this effect, because in our analysis infected cultures were compared to uninfected ones at the same times, but we cannot rule out the possibility that some of the bacterial transcriptional changes were not associated with infection. RNA-Seq technology monitors gene expression in both virus and host to a high level of accuracy and depth; however, the large number of data points generated by this technology could also introduce some false results.
Host differentially expressed genes response in certain environmental conditions.
We analyzed whether bacterial genes that are differentially expressed after ϕ29 infection also respond to certain environmental and nutritional conditions studied by Nicolas et al. (64). The comparison reveals that there is no significant overlap between upregulated genes during viral infection and those induced or repressed under several stress conditions such as heat, ethanol, salt, mitomycin, or oxidative stresses. The same occurs when we compared genes that respond to the presence of certain nutrients in the medium such as the specific carbon source that are used for B. subtilis growth (fructose, glycerol, and gluconate). Thus, it seems that the transcriptional changes detected for B. subtilis genes, which are shown in Tables 1 and 2, correspond to a virus-specific effect. However, ϕ29 induced genes showed a significant overlap (P < 0.01 [two-sided Fisher exact test]) compared to those repressed by cold stress when cells suffer a temperature downshift from 37 to 18°C. Since the B. subtilis cold shock response leads to a growth lag as a consequence of the inhibition of RNA, DNA, and protein synthesis (65), it could be expected that bacterial genes induced by ϕ29, which requires the host machinery to promote its multiplication, were repressed under such conditions. On the other hand, there is an interesting correlation (P < 0.005 [two-sided Fisher exact test]) between downregulated genes after ϕ29 infection and genes induced when glycerol or gluconate are the carbon sources used by the bacteria, suggesting a phage repression of the metabolism of a broad range of alternative carbon sources.
Transcriptional profile of the phage ϕ29 genome.
Phage ϕ29 DNA transcription is divided into early and late operons depending on the time when they are expressed during the infective cycle (Fig. 6A) (1, 66). The late operon is localized at the central region of the genome and is transcribed from the late promoter A3. Genes clustered into this operon (genes 7 to 16) encode phage structural, morphogenetic, and lytic proteins. Two early expressed operons located at both ends of the genome are divergently transcribed with respect to the late operon. The one on the left, which includes genes coding for essential DNA replication proteins and transcription regulators, is under the control of the tandemly organized promoters A2b and A2c. The right-side early operon is transcribed from the C2 promoter, and it encodes proteins required for the internalization of the phage DNA during the genome injection step and for the DNA replication process. This operon also contains the promoter C1, which is responsible of the transcription of ORFs 16.6 and 16.5. An additional promoter called A1 is located at the left early region and drives the expression of the pRNA, an RNA required for packaging of viral DNA. Phage ϕ29 DNA transcription requires the vegetative B. subtilis RNA polymerase containing the σA subunit to recognize the early promoters and, with the aid of the ϕ29 transcriptional regulator p4, the late A3 promoter that does not contain a typical −35 box (3).
FIG 6.

Genetic map and transcriptional profile of the phage ϕ29 genome. (A) Genetic and transcriptional map of the ϕ29 genome. Vertical bars indicate the locations of promoters A1, A2c, A2b, A3, B1, B2, C1, and C2. Arrows show the direction of transcription, and the transcriptional terminators TA1 and TD1 are indicated by red hairpin structures. Genes are indicated by numbers, and boxes are colored depending on the time when the samples were transcribed (salmon color refers to early transcripts, and light green to late transcripts). A black box indicates the region spanning the early A2b and A2c promoters and the late A3 promoter. (B) Transcriptional profile of the ϕ29 genome after 8 min of infection. Mapping of the reads from the strand-specific RNA-Seq analysis shows the expression of the early operons and the low levels of antisense transcription for the same regions. The depth is the number of reads detected for a particular genomic region. (C) Transcriptional profile of the ϕ29 genome after 16 min of infection. All of the genome was transcribed at this time, although most of the transcribed regions include early genes 6, 5.5, and 5, as well as late genes 7, 8, and 8.5. The amount of antisense transcripts (dark green) increased compared to panel B.
The transcript levels detected for each position of the phage ϕ29 genome at 8 and 16 min postinfection are shown in Fig. 6B and C, respectively, and the corresponding expression levels for each viral gene are shown at Table 3. The results indicate that the most abundant transcripts after 8 min of ϕ29 infection are derived from early operons. However, only a minor fraction of the transcripts initiated at the A2b and A2c promoters terminate at the left end of the genome. This is probably due to the presence of a Rho-independent transcriptional terminator called TA1, which is located within gene 4 (67). As a consequence, a larger amount of RNA from gene 6 (encoding a dsDNA-binding protein required for transcription regulation and DNA replication), gene 5.5 (a hypothetical gene), and gene 5 (encoding a ssDNA-binding protein necessary for viral DNA replication in vivo) was produced. This is consistent with previous data showing that proteins p6 and p5 were synthesized in far larger quantities in infected cells than were proteins p4 (a transcriptional regulator), p3 (the terminal protein needed for DNA replication), p2 (the DNA polymerase), and p1 (a DNA replication protein) (68, 69). Other genes also included in this early operon encode protein p56 (a uracil-DNA glycosylase inhibitor) and the hypothetical proteins p0.6 and p0.4. At 8 min postinfection, we also observed transcripts from the right early operon. In this case, the transcription levels are quite similar for all genes in the operon (Fig. 6B), which encodes p17 (a DNA replication protein), p16.7 (a protein involved in the distribution of in vivo phage DNA replication), and the hypothetical proteins p16.9, p16.8, p16.6, and p16.5. After 16 min of phage ϕ29 infection, there was a drastic decrease in the amount of RNA detected for these genes, whereas a moderate reduction in the levels of transcripts of genes 6, 5.5, and 5 and the rest of genes of the left early operon were observed (Table 3). This is because the activation of the late A3 promoter, which occurs at approximately 15 min after infection, requires the binding of the ϕ29 early protein p4 to specific DNA sequences in the intergenic region comprising promoters A2c, A2b, and A3. This binding also represses both early A2c and A2b promoters, leading to a decreased transcription levels of genes located at the early left operon (1). Moreover, transcripts expressed from the A1 promoter and the late A3 promoter were also identified at 16 min postinfection (Fig. 6C; Table 3). The higher transcriptional levels of the ϕ29 late genes detected at 16 min of infection compared to that at 8 min postinfection (Table 3) could cause the increment in the number of bacterial genes that showed a significant expression change at the later time (Tables 1 and 2). This transcriptional timing coincides with previous studies showing that the A3 promoter became active about 10 to 15 min after infection, whereas transcription from the early A2b and A2c promoters was already evident at 5 min postinfection (2). As previously described, there were smaller amounts of transcripts from the early C2 promoter than from the A2b and A2c promoters. Curiously, the expression profile of the phage ϕ29 genome at 16 min postinfection reveals a marked end of transcription between genes 8.5 and 9, suggesting that a transcriptional terminator that has not been previously identified is located at this region. As a result, a high level of transcripts of genes 7, 8, and 8.5 were produced compared to the rest of the genes of the late operon.
TABLE 3.
Expression levels of phage ϕ29 genes at 8 and 16 min postinfection
| Gene | Function of gene product | Value of expression (103)a |
|
|---|---|---|---|
| 8 min | 16 min | ||
| 0.2 | DNA packaging | 0.0 | 7.7 |
| 0.4 | Hypothetical protein | 235.4 | 67.8 |
| 0.6 | Hypothetical protein | 118.4 | 23.6 |
| 0.8 | Uracil-DNA glycosylase inhibitor | 104.1 | 16.5 |
| 1 | DNA replication | 194.9 | 34.1 |
| 2 | DNA polymerase | 175.5 | 31.7 |
| 3 | Terminal protein | 207.5 | 41.5 |
| 4 | Transcriptional regulator | 168.4 | 39.6 |
| 5 | Single stranded DNA-binding protein | 1,162.4 | 336.0 |
| 5.5 | Hypothetical protein | 658.8 | 147.9 |
| 6 | Double stranded DNA-binding protein | 1,074.0 | 288.1 |
| 7 | Head morphogenesis protein | 55.1 | 232.5 |
| 8 | Major head protein | 44.4 | 141.0 |
| 8.5 | Head fiber protein | 46.8 | 175.7 |
| 9 | Tail protein | 22.9 | 91.2 |
| 10 | Connector (upper collar protein) | 17.3 | 64.0 |
| 11 | Lower collar protein | 13.5 | 51.8 |
| 12 | Pre-neck appendage protein | 13.3 | 52.8 |
| 13 | Tail-associated protein | 8.6 | 26.0 |
| 14 | Holin | 6.2 | 13.8 |
| 15 | Peptidoglycan hydrolase | 8.3 | 22.5 |
| 16 | DNA packaging ATPase | 8.8 | 20.7 |
| 16.5 | Hypothetical protein | 20.5 | 4.3 |
| 16.6 | Hypothetical protein | 445.1 | 11.6 |
| 16.7 | Distribution of in vivo phage DNA replication | 648.5 | 15.5 |
| 16.8 | Hypothetical protein | 552.5 | 10.5 |
| 16.9 | Hypothetical protein | 772.6 | 20.4 |
| 17 | DNA replication | 529.8 | 11.3 |
Expression values are shown as RPKM (reads per kilobase of transcript per million mapped reads).
Interestingly, our studies also revealed the presence of antisense transcripts complementary to the well-known ϕ29 messenger RNAs. The antisense transcription was evident after 8 min of ϕ29 infection for the DNA strand opposite of the coding strand that serves as the template for expression of genes 5, 5.5, and 6 and those included in the right early operon (Fig. 6B); however, 8 min later both strands of the viral genome were transcribed, adding an additional layer to the transcriptome complexity. The promoters responsible for antisense transcription have not yet been identified since they may be produced by spurious expression events from promoterlike sequences that take advantage of the degenerate nature of bacterial transcription factor binding sites. Only two promoters, named B2 and B1, were previously known to give rise to antisense transcripts complementary to certain late genes. Since these transcripts did not contain ORFs of significant length with a reasonable ribosomal binding site, it has been proposed that they may have a role in transcriptional regulatory mechanisms. Nevertheless, promoters B2 and B1 were weakly expressed compared to other phage ϕ29 promoters with a maximum transcriptional rate at 30 min postinfection (2). Thus, is possible that other uncharacterized promoters were producing the antisense late transcripts shown in Fig. 6C. We observed distinct expression patterns for sense and antisense transcripts, which may suggest that they are independently regulated. Although further analysis will be needed to determine the physiological roles of the antisense transcripts and the underlying mechanisms, they might be potentially involved in global gene regulation of the ϕ29 genome. Antisense control of gene expression has been demonstrated in a wide variety of organisms, including some bacteriophages such as P22, P4, and T4. Thus, in the case of phage T4, the expression of gene 32 is regulated by an antisense transcript transcribed from the same DNA as the coding region but in the opposite orientation (70).
ACKNOWLEDGMENTS
We thank Ramón Peiró-Pastor of the Genomics and Massive Sequencing Service (CBMSO, Madrid, Spain) for processed RNA-Seq data. We are grateful to Rosa Ana Torremocha and Susana Ovalle of the Genomics Unit of the Parque Científico de Madrid for preparation of RNA-Seq libraries and Fernando Carrasco and Begoña Aguado (CBMSO) for helpful advice on RNA-Seq technology. We also thank the Centro Nacional de Análisis Genómico (Barcelona, Spain) for sequencing the samples.
Funding Statement
This study was also supported by an institutional grant from Fundación Ramón Areces to the Centro de Biología Molecular Severo Ochoa. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Meijer WJ, Horcajadas JA, Salas M. 2001. ϕ29 family of phages. Microbiol Mol Biol Rev 65:261–287. doi: 10.1128/MMBR.65.2.261-287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Monsalve M, Mencía M, Rojo F, Salas M. 1995. Transcription regulation in Bacillus subtilis phage ϕ29: expression of the viral promoters throughout the infection cycle. Virology 207:23–31. doi: 10.1006/viro.1995.1048. [DOI] [PubMed] [Google Scholar]
- 3.Sogo JM, Inciarte MR, Corral J, Viñuela E, Salas M. 1979. RNA polymerase binding sites and transcription map of the DNA of Bacillus subtilis phage ϕ29. J Mol Biol 127:411–436. doi: 10.1016/0022-2836(79)90230-4. [DOI] [PubMed] [Google Scholar]
- 4.Haüser R, Blasche S, Dokland T, Haggard-Ljungquist E, von Brunn A, Salas M, Casjens S, Molineux I, Uetz P. 2012. Bacteriophage protein-protein interactions. Adv Virus Res 83:219–298. doi: 10.1016/B978-0-12-394438-2.00006-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie2. Nat Methods 9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28:511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Caspi R, Altman T, Billington R, Dreher K, Foerster H, Fulcher CA, Holland TA, Keseler IM, Kothari A, Kubo A, Krummenacker M, Latendresse M, Mueller LA, Ong Q, Paley S, Subhraveti P, Weaver DS, Weerasinghe D, Zhang P, Karp PD. 2014. The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of Pathway/Genome Databases. Nucleic Acids Res 42:D459–D471. doi: 10.1093/nar/gkt1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. 2000. Gene ontology: tool for the unification of biology. Nat Genet 25:25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Barbe V, Cruveiller S, Kunst F, Lenoble P, Meurice G, Sekowska A, Vallenet D, Wang T, Moszer I, Medigue C, Danchin A. 2009. From a consortium sequence to a unified sequence: the Bacillus subtilis 168 reference genome a decade later. Microbiology 155:1758–1775. doi: 10.1099/mic.0.027839-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Härtig E, Hartmann A, Schatzle M, Albertini AM, Jahn D. 2006. The Bacillus subtilis nrdEF genes, encoding a class Ib ribonucleotide reductase, are essential for aerobic and anaerobic growth. Appl Environ Microbiol 72:5260–5265. doi: 10.1128/AEM.00599-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Scotti C, Valbuzzi A, Perego M, Galizzi A, Albertini AM. 1996. The Bacillus subtilis genes for ribonucleotide reductase are similar to the genes for the second class I NrdE/NrdF enzymes of Enterobacteriaceae. Microbiology 142(Part 11):2995–3004. doi: 10.1099/13500872-142-11-2995. [DOI] [PubMed] [Google Scholar]
- 12.Scharf C, Riethdorf S, Ernst H, Engelmann S, Volker U, Hecker M. 1998. Thioredoxin is an essential protein induced by multiple stresses in Bacillus subtilis. J Bacteriol 180:1869–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Carrasco B, Fernandez S, Petit MA, Alonso JC. 2001. Genetic recombination in Bacillus subtilis 168: effect of ΔhelD on DNA repair and homologous recombination. J Bacteriol 183:5772–5777. doi: 10.1128/JB.183.19.5772-5777.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wiedermannova J, Sudzinova P, Koval T, Rabatinova A, Sanderova H, Ramaniuk O, Rittich S, Dohnalek J, Fu Z, Halada P, Lewis P, Krasny L. 2014. Characterization of HelD, an interacting partner of RNA polymerase from Bacillus subtilis. Nucleic Acids Res 42:5151–5163. doi: 10.1093/nar/gku113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Venkatesan M, Silver LL, Nossal NG. 1982. Bacteriophage T4 gene 41 protein, required for the synthesis of RNA primers, is also a DNA helicase. J Biol Chem 257:12426–12434. [PubMed] [Google Scholar]
- 16.Odegrip R, Schoen S, Haggard-Ljungquist E, Park K, Chattoraj DK. 2000. The interaction of bacteriophage P2 B protein with Escherichia coli DnaB helicase. J Virol 74:4057–4063. doi: 10.1128/JVI.74.9.4057-4063.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Funnell BE, Inman RB. 1983. Bacteriophage P2 DNA replication: characterization of the requirement of the gene B protein in vivo. J Mol Biol 167:311–334. doi: 10.1016/S0022-2836(83)80338-6. [DOI] [PubMed] [Google Scholar]
- 18.Blanco L, Bernad A, Lázaro JM, Martín G, Garmendia C, Salas M. 1989. Highly efficient DNA synthesis by the phage ϕ29 DNA polymerase: symmetrical mode of DNA replication. J Biol Chem 264:8935–8940. [PubMed] [Google Scholar]
- 19.Yadav T, Carrasco B, Myers AR, George NP, Keck JL, Alonso JC. 2012. Genetic recombination in Bacillus subtilis: a division of labor between two single-strand DNA-binding proteins. Nucleic Acids Res 40:5546–5559. doi: 10.1093/nar/gks173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mellado RP, Peñalva MMA, Inciarte MR, Salas M. 1980. The protein covalently linked to the 5′ termini of the DNA of Bacillus subtilis phage ϕ29 is involved in the initiation of DNA replication. Virology 104:84–96. doi: 10.1016/0042-6822(80)90367-0. [DOI] [PubMed] [Google Scholar]
- 21.Gutiérrez C, Martín G, Sogo JM, Salas M. 1991. Mechanism of stimulation of DNA replication by bacteriophage ϕ29 single-stranded DNA-binding protein p5. J Biol Chem 266:2104–2011. [PubMed] [Google Scholar]
- 22.Wen T, Oussenko IA, Pellegrini O, Bechhofer DH, Condon C. 2005. Ribonuclease PH plays a major role in the exonucleolytic maturation of CCA-containing tRNA precursors in Bacillus subtilis. Nucleic Acids Res 33:3636–3643. doi: 10.1093/nar/gki675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Redko Y, Condon CC. 2010. Maturation of 23S rRNA in Bacillus subtilis in the absence of Mini-III. J Bacteriol 192:356–359. doi: 10.1128/JB.01096-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kobir A, Poncet S, Bidnenko V, Delumeau O, Jers C, Zouhir S, Grenha R, Nessler S, Noirot P, Mijakovic I. 2014. Phosphorylation of Bacillus subtilis gene regulator AbrB modulates its DNA-binding properties. Mol Microbiol 92:1129–1141. doi: 10.1111/mmi.12617. [DOI] [PubMed] [Google Scholar]
- 25.Meijer WJ, Castilla-Llorente V, Villar L, Murray H, Errington J, Salas M. 2005. Molecular basis for the exploitation of spore formation as survival mechanism by virulent phage ϕ29. EMBO J 24:3647–3657. doi: 10.1038/sj.emboj.7600826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nair N, Raff H, Islam MT, Feen M, Garofalo DM, Sheppard K. 2016. The Bacillus subtilis and Bacillus halodurans aspartyl-tRNA synthetases retain recognition of tRNA(Asn). J Mol Biol 428:618–630. doi: 10.1016/j.jmb.2016.01.014. [DOI] [PubMed] [Google Scholar]
- 27.Curnow AW, Hong K, Yuan R, Kim S, Martins O, Winkler W, Henkin TM, Soll D. 1997. Glu-tRNAGln amidotransferase: a novel heterotrimeric enzyme required for correct decoding of glutamine codons during translation. Proc Natl Acad Sci U S A 94:11819–11826. doi: 10.1073/pnas.94.22.11819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Krüger E, Witt E, Ohlmeier S, Hanschke R, Hecker M. 2000. The Clp proteases of Bacillus subtilis are directly involved in degradation of misfolded proteins. J Bacteriol 182:3259–3265. doi: 10.1128/JB.182.11.3259-3265.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Poranen MM, Ravantti JJ, Grahn AM, Gupta R, Auvinen P, Bamford DH. 2006. Global changes in cellular gene expression during bacteriophage PRD1 infection. J Virol 80:8081–8088. doi: 10.1128/JVI.00065-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bejarano I, Klemes Y, Schoulaker-Schwarz R, Engelberg-Kulka H. 1993. Energy-dependent degradation of lambda O protein in Escherichia coli. J Bacteriol 175:7720–7723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hahn FM, Hurlburt AP, Poulter CD. 1999. Escherichia coli open reading frame 696 is idi, a nonessential gene encoding isopentenyl diphosphate isomerase. J Bacteriol 181:4499–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.May JJ, Wendrich TM, Marahiel MA. 2001. The dhb operon of Bacillus subtilis encodes the biosynthetic template for the catecholic siderophore 2,3-dihydroxybenzoate-glycine-threonine trimeric ester bacillibactin. J Biol Chem 276:7209–7217. doi: 10.1074/jbc.M009140200. [DOI] [PubMed] [Google Scholar]
- 33.Gaballa A, Helmann JD. 2011. Bacillus subtilis Fur represses one of two paralogous haem-degrading monooxygenases. Microbiology 157:3221–3231. doi: 10.1099/mic.0.053579-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sijtsma L, Wouters JT, Hellingwerf KJ. 1990. Isolation and characterization of lipoteichoic acid, a cell envelope component involved in preventing phage adsorption, from Lactococcus lactis subsp. cremoris SK110 J Bacteriol 172:7126–7130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chandrangsu P, Dusi R, Hamilton CJ, Helmann JD. 2014. Methylglyoxal resistance in Bacillus subtilis: contributions of bacillithiol-dependent and independent pathways. Mol Microbiol 91:706–715. doi: 10.1111/mmi.12489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Papoulis A, al-Abed Y, Bucala R. 1995. Identification of N2-(1-carboxyethyl)guanine (CEG) as a guanine advanced glycosylation end product. Biochemistry 34:648–655. doi: 10.1021/bi00002a032. [DOI] [PubMed] [Google Scholar]
- 37.Krymkiewicz N, Dieguez E, Rekarte UD, Zwaig N. 1971. Properties and mode of action of a bactericidal compound (=methylglyoxal) produced by a mutant of Escherichia coli. J Bacteriol 108:1338–1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim JH, Yang YK, Chambliss GH. 2005. Evidence that Bacillus catabolite control protein CcpA interacts with RNA polymerase to inhibit transcription. Mol Microbiol 56:155–162. doi: 10.1111/j.1365-2958.2005.04496.x. [DOI] [PubMed] [Google Scholar]
- 39.Marciniak BC, Pabijaniak M, de Jong A, Duhring R, Seidel G, Hillen W, Kuipers OP. 2012. High- and low-affinity cre boxes for CcpA binding in Bacillus subtilis revealed by genome-wide analysis. BMC Genomics 13:401. doi: 10.1186/1471-2164-13-401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miwa Y, Saikawa M, Fujita Y. 1994. Possible function and some properties of the CcpA protein of Bacillus subtilis. Microbiology 140(Part 10):2567–2575. doi: 10.1099/00221287-140-10-2567. [DOI] [PubMed] [Google Scholar]
- 41.Hadas H, Einav M, Fishov I, Zaritsky A. 1997. Bacteriophage T4 development depends on the physiology of its host Escherichia coli. Microbiology 143(Part 1):179–185. doi: 10.1099/00221287-143-1-179. [DOI] [PubMed] [Google Scholar]
- 42.Chen Y, Golding I, Sawai S, Guo L, Cox EC. 2005. Population fitness and the regulation of Escherichia coli genes by bacterial viruses. PLoS Biol 3:e229. doi: 10.1371/journal.pbio.0030229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Woodson K, Devine KM. 1994. Analysis of a ribose transport operon from Bacillus subtilis. Microbiology 140(Part 8):1829–1838. doi: 10.1099/13500872-140-8-1829. [DOI] [PubMed] [Google Scholar]
- 44.Strauch MA. 1995. AbrB modulates expression and catabolite repression of a Bacillus subtilis ribose transport operon. J Bacteriol 177:6727–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kunst F, Ogasawara N, Moszer I, Albertini AM, Alloni G, Azevedo V, Bertero MG, Bessieres P, Bolotin A, Borchert S, Borriss R, Boursier L, Brans A, Braun M, Brignell SC, Bron S, Brouillet S, Bruschi CV, Caldwell B, Capuano V, Carter NM, Choi SK, Cordani JJ, Connerton IF, Cummings NJ, Daniel RA, Denziot F, Devine KM, Dusterhoft A, Ehrlich SD, Emmerson PT, Entian KD, Errington J, Fabret C, Ferrari E, Foulger D, Fritz C, Fujita M, Fujita Y, Fuma S, Galizzi A, Galleron N, Ghim SY, Glaser P, Goffeau A, Golightly EJ, Grandi G, Guiseppi G, Guy BJ, Haga K, Haiech J, Harwood CR, Henaut A, Hilbert H, Holsappel S, Hosono S, Hullo MF, Itaya M, Jones L, Joris B, Karamata D, Kasahara Y, Klaerr-Blanchard M, Klein C, Kobayashi Y, Koetter P, Koningstein G, Krogh S, Kumano M, Kurita K, Lapidus A, Lardinois S, Lauber J, Lazarevic V, Lee SM, Levine A, Liu H, Masuda S, Mauel C, Medigue C, Medina N, Mellado RP, Mizuno M, Moestl D, Nakai S, Noback M, Noone D, O'Reilly M, Ogawa K, Ogiwara A, Oudega B, Park SH, Parro V, Pohl TM, Portelle D, Porwollik S, Prescott AM, Presecan E, Pujic P, Purnelle B, Rapoport G, Rey M, Reynolds S, Rieger M, Rivolta C, Rocha E, Roche B, Rose M, Sadaie Y, Sato T, Scanlan E, Schleich S, Schroeter R, Scoffone F, Sekiguchi J, Sekowska A, Seror SJ, Serror P, Shin BS, Soldo B, Sorokin A, Tacconi E, Takagi T, Takahashi H, Takemaru K, Takeuchi M, Tamakoshi A, Tanaka T, Terpstra P, Togoni A, Tosato V, Uchiyama S, Vandebol M, Vannier F, Vassarotti A, Viari A, Wambutt R, Wedler H, Weitzenegger T, Winters P, Wipat A, Yamamoto H, Yamane K, Yasumoto K, Yata K, Yoshida K, Yoshikawa HF, Zumstein E, Yoshikawa H, Danchin A. 1997. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature 390:249–256. doi: 10.1038/36786. [DOI] [PubMed] [Google Scholar]
- 46.Yoshida KI, Aoyama D, Ishio I, Shibayama T, Fujita Y. 1997. Organization and transcription of the myo-inositol operon, iol, of Bacillus subtilis. J Bacteriol 179:4591–4598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yoshida K, Yamaguchi M, Morinaga T, Kinehara M, Ikeuchi M, Ashida H, Fujita Y. 2008. myo-Inositol catabolism in Bacillus subtilis. J Biol Chem 283:10415–10424. doi: 10.1074/jbc.M708043200. [DOI] [PubMed] [Google Scholar]
- 48.Yang M, Galizzi A, Henner D. 1983. Nucleotide sequence of the amylase gene from Bacillus subtilis. Nucleic Acids Res 11:237–249. doi: 10.1093/nar/11.2.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fouet A, Klier A, Rapoport G. 1986. Nucleotide sequence of the sucrase gene of Bacillus subtilis. Gene 45:221–225. doi: 10.1016/0378-1119(86)90258-1. [DOI] [PubMed] [Google Scholar]
- 50.Mekjian KR, Bryan EM, Beall BW, Moran CP Jr. 1999. Regulation of hexuronate utilization in Bacillus subtilis. J Bacteriol 181:426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Schöck F, Dahl MK. 1996. Analysis of DNA flanking the treA gene of Bacillus subtilis reveals genes encoding a putative specific enzyme IITre and a potential regulator of the trehalose operon. Gene 175:59–63. doi: 10.1016/0378-1119(96)00120-5. [DOI] [PubMed] [Google Scholar]
- 52.Helfert C, Gotsche S, Dahl MK. 1995. Cleavage of trehalose-phosphate in Bacillus subtilis is catalysed by a phospho-alpha-(1-1)-glucosidase encoded by the treA gene. Mol Microbiol 16:111–120. doi: 10.1111/j.1365-2958.1995.tb02396.x. [DOI] [PubMed] [Google Scholar]
- 53.Tobisch S, Glaser P, Kruger S, Hecker M. 1997. Identification and characterization of a new beta-glucoside utilization system in Bacillus subtilis. J Bacteriol 179:496–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sun T, Altenbuchner J. 2010. Characterization of a mannose utilization system in Bacillus subtilis. J Bacteriol 192:2128–2139. doi: 10.1128/JB.01673-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Esquinas-Rychen M, Erni B. 2001. Facilitation of bacteriophage lambda DNA injection by inner membrane proteins of the bacterial phosphoenol-pyruvate: carbohydrate phosphotransferase system (PTS). J Mol Microbiol Biotechnol 3:361–370. [PubMed] [Google Scholar]
- 56.Saxild HH, Andersen LN, Hammer K. 1996. dra-nupC-pdp operon of Bacillus subtilis: nucleotide sequence, induction by deoxyribonucleosides, and transcriptional regulation by the deoR-encoded DeoR repressor protein. J Bacteriol 178:424–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Serrano-Heras G, Bravo A, Salas M. 2008. Phage ϕ29 protein p56 prevents viral DNA replication impairment caused by uracil excision activity of uracil-DNA glycosylase. Proc Natl Acad Sci U S A 105:19044–19049. doi: 10.1073/pnas.0808797105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baños-Sanz JI, Mojardín L, Sanz-Aparicio J, Lázaro JM, Villar L, Serrano-Heras G, González B, Salas M. 2013. Crystal structure and functional insights into uracil-DNA glycosylase inhibition by phage ϕ29 DNA mimic protein p56. Nucleic Acids Res 41:6761–6773. doi: 10.1093/nar/gkt395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.von Wachenfeldt C, Hederstedt L. 1990. Bacillus subtilis 13-kilodalton cytochrome c-550 encoded by cccA consists of a membrane-anchor and a heme domain. J Biol Chem 265:13939–13948. [PubMed] [Google Scholar]
- 60.Shin I, Ryu HB, Yim HS, Kang SO. 2005. Cytochrome c550 is related to initiation of sporulation in Bacillus subtilis. J Microbiol 43:244–250. [PubMed] [Google Scholar]
- 61.Castilla-Llorente V, Salas M, Meijer WJ. 2009. Different responses to Spo0A-mediated suppression of the related Bacillus subtilis phages Nf and ϕ29. Environ Microbiol 11:1137–1149. doi: 10.1111/j.1462-2920.2008.01845.x. [DOI] [PubMed] [Google Scholar]
- 62.Allocati N, Masulli M, Di Ilio C, Laurenzi V. 2015. Die for the community: an overview of programmed cell death in bacteria. Cell Death Dis 6:e1609. doi: 10.1038/cddis.2014.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wang IN, Smith DL, Young R. 2000. Holins: the protein clocks of bacteriophage infections. Annu Rev Microbiol 54:799–825. doi: 10.1146/annurev.micro.54.1.799. [DOI] [PubMed] [Google Scholar]
- 64.Nicolas P, Mader U, Dervyn E, Rochat T, Leduc A, Pigeonneau N, Bidnenko E, Marchadier E, Hoebeke M, Aymerich S, Becher D, Bisicchia P, Botella E, Delumeau O, Doherty G, Denham EL, Fogg MJ, Fromion V, Goelzer A, Hansen A, Hartig E, Harwood CR, Homuth G, Jarmer H, Jules M, Klipp E, Le Chat L, Lecointe F, Lewis P, Liebermeister W, March A, Mars RA, Nannapaneni P, Noone D, Pohl S, Rinn B, Rugheimer F, Sappa PK, Samson F, Schaffer M, Schwikowski B, Steil L, Stulke J, Wiegert T, Devine KM, Wilkinson AJ, van Dijl JM, Hecker M, Volker U, Bessieres P, Noirot P. 2012. Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 335:1103–1106. doi: 10.1126/science.1206848. [DOI] [PubMed] [Google Scholar]
- 65.Konopásek I, Strzalka K, Svobodová J. 2000. Cold shock in Bacillus subtilis: different effects of benzyl alcohol and ethanol on the membrane organisation and cell adaptation. Biochim Biophys Acta 1464:18–26. doi: 10.1016/S0005-2736(99)00240-0. [DOI] [PubMed] [Google Scholar]
- 66.Rojo F, Mencía M, Monsalve M, Salas M. 1998. Transcription activation and repression by interaction of a regulator with the alpha subunit of RNA polymerase: the model of phage ϕ29 protein p4. Prog Nucleic Acid Res Mol Biol 60:29–46. doi: 10.1016/S0079-6603(08)60888-0. [DOI] [PubMed] [Google Scholar]
- 67.Barthelemy I, Salas M, Mellado RP. 1987. In vivo transcription of bacteriophage ϕ29 DNA: transcription termination. J Virol 61:1751–1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Abril AM, Salas M, Andreu JM, Hermoso JM, Rivas G. 1997. Phage ϕ29 protein p6 is in a monomer-dimer equilibrium that shifts to higher association states at the millimolar concentrations found in vivo. Biochemistry 36:11901–11908. doi: 10.1021/bi970994e. [DOI] [PubMed] [Google Scholar]
- 69.Gascón I, Lázaro JM, Salas M. 2000. Differential functional behavior of viral ϕ29, Nf and GA-1 SSB proteins. Nucleic Acids Res 28:2034–2042. doi: 10.1093/nar/28.10.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Belin D, Mudd EA, Prentki P, Yi-Yi Y, Krisch HM. 1987. Sense and antisense transcription of bacteriophage T4 gene 32. Processing and stability of the mRNAs. J Mol Biol 194:231–243. [DOI] [PubMed] [Google Scholar]