

ABSTRACT
Thousands of endogenous retroviruses (ERV), viral fossils of ancient germ line infections, reside within the human genome. Evidence of ERV activity has been observed widely in both health and disease. While this is most often cited as a bystander effect of cell culture or disease states, it is unclear which signals control ERV transcription. Bioinformatic analysis suggests that the viral promoter of endogenous retrovirus K (ERVK) is responsive to inflammatory transcription factors. Here we show that one reason for ERVK upregulation in amyotrophic lateral sclerosis (ALS) is the presence of functional interferon-stimulated response elements (ISREs) in the viral promoter. Transcription factor overexpression assays revealed independent and synergistic upregulation of ERVK by interferon regulatory factor 1 (IRF1) and NF-κB isoforms. Tumor necrosis factor alpha (TNF-α) and LIGHT cytokine treatments of human astrocytes and neurons enhanced ERVK transcription and protein levels through IRF1 and NF-κB binding to the ISREs. We further show that in ALS brain tissue, neuronal ERVK reactivation is associated with the nuclear translocation of IRF1 and NF-κB isoforms p50 and p65. ERVK overexpression can cause motor neuron pathology in murine models. Our results implicate neuroinflammation as a key trigger of ERVK provirus reactivation in ALS. These molecular mechanisms may also extend to the pathobiology of other ERVK-associated inflammatory diseases, such as cancers, HIV infection, rheumatoid arthritis, and schizophrenia.
IMPORTANCE It has been well established that inflammatory signaling pathways in ALS converge at NF-κB to promote neuronal damage. Our findings suggest that inflammation-driven IRF1 and NF-κB activity promotes ERVK reactivation in neurons of the motor cortex in ALS. Thus, quenching ERVK activity through antiretroviral or immunomodulatory regimens may hinder virus-mediated neuropathology and improve the symptoms of ALS or other ERVK-associated diseases.
INTRODUCTION
Thousands of endogenous retroviruses (ERV), viral fossils of ancient germ line infections, reside within the human genome. Evidence of ERV activity has been observed widely in both health and disease. While this is most often cited as a bystander effect of cell culture or disease states, it is unclear which signals control ERV transcription and whether their expression is relevant to cellular processes. We previously proposed that the viral promoter of endogenous retrovirus K (ERVK) is responsive to inflammatory transcription factors (1) due to the presence of two conserved interferon-stimulated response elements (ISREs). Thus, select proinflammatory transcription factors may drive ERVK expression.
The transcription of integrated retroviral sequences within the human genome is regulated by viral promoters, called long terminal repeats (LTRs), flanking either side of the core viral genome. These LTRs contain transcriptional regulatory elements that are responsive to both viral and cellular transcription factors (TFs). Interferon regulatory factors (IRFs) and nuclear factor kappa B (NF-κB) have been shown to be crucial in the transcription of human immunodeficiency virus (HIV) proviruses (2, 3), thus promoting HIV replication in the context of inflammation (4). The human genome is already populated with numerous ERVK viral promoters containing IRF and NF-κB binding sites (1); however, their propensity to drive ERVK transcription and expression remains unclear.
In support of this paradigm for ERVK reactivation, proinflammatory cytokines have previously been shown to trigger viral expression in several ERVK-associated inflammatory diseases. For instance, tumor necrosis factor alpha (TNF-α) treatment has been demonstrated to enhance ERVK (HML-2) gag transcription in synoviocytes obtained from patients with rheumatoid arthritis (5). TNF-α and gamma interferon (IFN-γ) are able to enhance the expression of another human endogenous retrovirus, ERVW, in peripheral blood mononuclear cells (PBMCs) obtained from patients with multiple sclerosis (6). TNF-α has been shown to trigger ERVW syncytin protein expression by enhancing the binding of NF-κB subunit p65 to the ERVW promoter in a human astrocytic cell line (7). Nonetheless, how proinflammatory cytokines trigger ERVK expression, particularly in the cells of the central nervous system (CNS), remains to be studied.
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disease with the hallmark pathology of motor neuron damage within the cerebral cortex and spinal cord. While the concert of pathological pathways underlying the inception and progression of motor neuron death in ALS is unclear, mutations in several key genes have been implicated as risk factors for ALS (reviewed in reference 8). Emerging evidence for retroviral pathology in ALS stems from measurements of reverse transcriptase (RT) activity at levels similar to those found in individuals infected with human immunodeficiency virus (HIV) (9, 10). Indeed, ERVK RT expression is strongly enhanced in ALS, notably in prefrontal and motor cortex neurons (11). Moreover, overexpression of the ERVK envelope protein can promote motor neuron death in a murine model of ALS (12).
There is growing recognition that TNF-α and LIGHT play critical yet divergent roles in ALS neuropathology. Significantly higher levels of proinflammatory cytokines have been reported for the cerebrospinal fluid (CSF) and sera of ALS patients than for those of healthy controls (13–15). The pathogenic role of TNF-α in ALS is well documented and reviewed elsewhere (16, 17). In contrast, few studies have explored the contribution of LIGHT (homologous to lymphotoxin, exhibits inducible expression, and competes with HSV glycoprotein D for herpesvirus entry mediator, a receptor expressed by T lymphocytes) to ALS (18–20). Both cytokines are neurotoxic and have been associated with enhanced neuronal death. TNF-α is a potent activator of the canonical NF-κB signaling pathway, culminating in the activation of the p65 and p50 isoforms of this proinflammatory transcription factor (21). TNF-α-induced NF-κB has been shown to cause motor neuron death in vitro (22). In addition, IFN-γ has been demonstrated to synergize with TNF-α to induce NF-κB and enhance motor neuron death (23, 24). In line with this finding, anti-IFN-γ therapy is protective and delays motor neuron damage in ALS mouse models (20). Recently, elevated LIGHT signaling was shown to selectively contribute to motor neuron death in ALS spinal cords (18, 19). IFN-γ secreted by astrocytes is a key player in this process, as it leads to enhanced LIGHT production in spinal motor neurons (18, 19). Similar to TNF-α, LIGHT is also a potent activator of the canonical and noncanonical NF-κB pathways, leading to the activation of the alternative p52 NF-κB isoform (21). Additionally, TNF-α and IFN-γ are known to synergistically activate interferon regulatory factor 1 (IRF1) expression (24), but the role of IRF1 activation in ALS pathology remains unexplored. Overall, the sum of these augmented cytokine signaling pathways likely results in excessive activation of NF-κB and IRF1 in the brain.
Although exacerbated TNF-α, LIGHT, and IFN-γ signaling pathways in the CNS converge at NF-κB- and/or IRF1-dependent neuronal damage (25), the exact mechanism by which these proinflammatory transcription factors promote neuronal death is unclear. ERVK reactivation in neurons triggered by NF-κB/IRF1 may serve as the link between exacerbated proinflammatory cytokine signaling and neuronal damage in ALS.
MATERIALS AND METHODS
Ethics.
All research involving human autopsy tissue was approved by the University of Winnipeg Human Research Ethics Board under protocol HE-#791. Anonymized autopsy ALS (n = 5) and neuronormal control (n = 5) tissue specimens were obtained from the NIH NeuroBioBank.
Diagnosis and demographics for patient samples.
Pathological examination was used to confirm the clinical diagnosis of ALS. The postmortem interval (PMI) was <24 h for all patients. Table 1 indicates the patient diagnosis, age, gender, and PMI (in hours) for the individual samples used in this study. The brain regions analyzed were the prefrontal cortex (Brodmann area 9 [BA9]) and the motor cortex (Brodmann area 6 [BA6]).
TABLE 1.
Characteristics of cortical brain tissue specimensa
| Case no. | Diagnosis | Cause of death | Patient age (yr) | Patient genderb | Postmortem interval (h) |
|---|---|---|---|---|---|
| 2776 | ALS | ALS (progressive bulbar palsy) | 76 | F | 8.6 |
| 5215 | ALS | ALS | 59 | M | 12.5 |
| 5187 | ALS | ALS, hypertension, chronic urinary tract infection | 69 | M | 13.2 |
| 5212 | ALS | ALS | 50 | M | 21 |
| 5216 | ALS | ALS | 53 | M | 16.8 |
| 4660 | Neuronormal | Pancreatic cancer, diabetes, hypertension | 73 | F | 18.5 |
| 3371 | Neuronormal | Lung cancer | 52 | M | 16 |
| 4514 | Neuronormal | Lung cancer, chronic obstructive pulmonary disease | 66 | M | 17.3 |
| 3565 | Neuronormal | Cardiomyopathy | 76 | M | 11 |
| 3221 | Neuronormal | Chronic obstructive pulmonary disease | 90 | M | 17.8 |
Brodmann area 6 motor cortex and Brodmann area 9 prefrontal cortex samples were collected in all cases.
F, female; M, male.
Immunohistochemistry of autopsy tissue.
To determine the extent of ERVK RT, IRF1, and NF-κB expression patterns in the CNS of ALS patients, immunohistochemistry was performed to detect the levels and localization of these target proteins in autopsy human cortical brain tissue, as previously described (11). Primary antibodies used were mouse anti-human ERVK RT (1:750) (H00002087-A01; AbNova), rabbit anti-human IRF1 (1:100) (SC497; Santa Cruz), rabbit anti-human NF-κB p65 (1:100) (ab7970; Abcam), and rabbit anti-human NF-κB p50 (1:100) (ab7971; Abcam). Primary antibodies were detected using 1:250 AF488-conjugated goat anti-mouse (A11017; Molecular Probes) or AF594-conjugated goat anti-rabbit (11072; Molecular Probes) secondary antibodies. Neurons were identified using fluorescent Nissl stain (1:100) (Molecular Probes). Tissues were also counterstained with 1:50,000 DAPI (4′,6-diamidino-2-phenylindole). Free-floating tissues were mounted onto slides and stained in a 0.1% solution of Sudan Black B. Slides were rinsed and coverslips mounted using ProLong Gold antifade reagent (Molecular Probes). Controls were prepared by immunostaining without the primary antibodies. All samples were batch stained, and we used case-control matched tissues. Immunostained tissues were imaged with an Olympus FV1200 laser scanning confocal microscope fitted with the Olympus Fluoview, version 4.0B, software suite. This software was used to outline cellular as well as nuclear boundaries of ERVK+ neurons in neuronormal and ALS specimens. The mean intensities of total and nuclear ERVK RT, IRF1, NF-κB p50, and NF-κB p65 staining were recorded for these cells. These parameters were graphed to determine the correlation between the levels of total ERVK RT and nuclear translocation of each transcription factor in neuronormal versus ALS tissues. GraphPad Prism was used to compare the patient groups through the Mann-Whitney U test. Evaluation of correlation was performed using Pearson's rank correlation coefficient. To further determine the use of each of the target proteins as biomarkers in order to correctly discriminate between neuronormal controls and ALS patients, receiver operating characteristic (ROC) curves and the area under the curve (AUC) were calculated using GraphPad Prism.
Cell culture.
The SVGA cell line was previously derived from immortalized human fetal astrocytes (26) and was maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (HyClone). 293T cells were also maintained in complete DMEM. ReNcell CX cells (Millipore) are immortalized human neural progenitor cells (HNPCs) (27), and they were maintained in a proprietary ReNcell neural stem cell medium (Millipore) supplemented with 20 ng/ml human epidermal growth factor (EGF; PeproTech) and 20 ng/ml human basic fibroblast growth factor (bFGF; PeproTech). All cell lines were maintained in a 37°C incubator containing 5% CO2. SVGA cells were seeded into six-well plates and onto glass coverslips at densities of 300,000 cells/ml and 30,000 cells/ml, respectively, for 24 h. 293T cells were seeded into six-well plates at a density of 300,000 cells/ml. To differentiate HNPCs into neurons, ReNcell CX cells were seeded in laminin (20 μg/ml; Millipore)-coated six-well plates at a density of 50,000 cells/ml for 24 h. Adhered cells were rinsed with 1× phosphate-buffered saline (PBS) and allowed to differentiate in the presence of ReNcell medium lacking growth factors for 10 days. For imaging experiments, ReNcell CX cells were cultured and differentiated into neurons in Alvetex scaffolds. Alvetex membranes (Reinnervate) were treated with 70% ethanol for 1 min, rinsed with 1× PBS, and coated with 20 μg/ml laminin for 6 h. ReNcell CX cells were seeded onto each scaffold at a density of 5 × 105 cells/well for 1 h at 37°C and 5% CO2 per the manufacturer's instructions. The wells were then flooded with 2 ml of ReNcell medium supplemented with EGF and FGF. At 24 h postseeding, the cell culture medium was replaced with medium lacking growth factors. Cells were allowed to differentiate for 10 days, with partial medium changes performed every 3 days.
Transient transfection of cells with NF-κB and IRF constructs.
NF-κB p65-pCMVBL, NF-κB p50-pCMVBL, IRF1-pCMVBL, IRF2-pCMVBL, IRF3-pCMV2, IRF3 (5D)-pCMV2, IRF5-pCMV2, IRF5 (4D)-pCMV2, IRF7-pCMV2, IRF7 (6D)-pCMV2, IRF8-pCMV2, pCMVBL empty vector, and pCMV2 empty vector were generously provided by Rongtuan Lin (McGill University) (28). To determine whether IRF1 and NF-κB isoforms synergize to induce ERVK transcription, SVGA cells (or 293T cells) were transfected with 1-μg aliquots of these plasmids individually or in combinations by using 6 μl of Turbofect reagent per the manufacturer's instructions (Thermo Scientific). Cells were transfected in serum-free culture medium for 4 h, followed by addition of complete medium. Cells were harvested at 48 h posttransfection. Untransfected cells and those transfected with the empty vector were used as negative controls.
Q-PCR.
Total RNA was extracted and purified from cells by use of an Aurum total RNA minikit (Bio-Rad). The RNA concentration was measured with a NanoDrop spectrophotometer. The acceptable RNA purity was an A260/A280 value of 1.95 to 2.05. An iScript reverse transcription kit (Bio-Rad) was used to synthesize cDNA from the extracted RNA. A CFX Connect real-time system (Bio-Rad) was utilized to perform quantitative PCR (Q-PCR) in order to measure alterations in ERVK pol transcripts by using the SYBR green detection method. The primers used were as follows: ERVK pol F, 5′-TGATCCCMAAAGAYTGGCCTT-3′; and ERVK pol R, 5′-TTAAGCATTCCCTGAGGYAACA-3′. 18S rRNA was used as the endogenous control (Ambion kit). The data were analyzed using the ΔΔCT (Livak) method and were normalized to the appropriate negative control. All data were graphed as means ± standard errors of measurement. GraphPad Prism was used to carry out statistical analyses, including column statistics, one-way analysis of variance (ANOVA), and uncorrected Fisher's least significant difference (LSD) test.
Western blotting.
SVGA cells and ReNcell CX cell-derived neurons were treated with 0-, 0.1-, 0.5-, 1-, 5-, and 10-ng/ml doses of human TNF-α (PeproTech) or human LIGHT (PeproTech). At 24 h posttreatment, cells were harvested and lysed on ice with 50 μl of in-house lysis buffer (0.05 M Tris [pH 7.4], 0.15 M NaCl, 0.002 M EDTA, 10% glycerol, and 1% NP-40 in ultrapure water) to extract proteins. The lysis buffer was supplemented with 1× HALT protease and phosphatase inhibitor cocktail (Thermo Scientific). A bicinchoninic acid (BCA) assay (Thermo Scientific) was used to determine the protein content of each sample per the manufacturer's instructions. Cell lysates were prepared for SDS-PAGE and heated at 95°C for 10 min. Proteins (15 μg per lane) were separated by SDS-PAGE on a 10% polyacrylamide gel and then transferred to a nitrocellulose membrane. The membrane was blocked in 5% skim milk solution for 1 h and then probed with the desired primary antibody (1:1,000 dilution) overnight at 4°C, followed by incubation at room temperature for 3 h. Primary antibodies used were as follows: mouse anti-human ERVK2 RT (H00002087-A01; Abnova), rabbit anti-human IRF1 (SC497; Santa Cruz), rabbit anti-human NF-κB p65 (ab7970; Abcam), rabbit anti-human NF-κB p50 (ab32360; Abcam), rabbit anti-human NF-κB p52 (4882S; Cell Signaling), and mouse anti-human β-actin (MA5-15739; Thermo Pierce) (loading control). The membrane was then probed with horseradish peroxidase-conjugated goat anti-mouse or -rabbit IgG secondary antibody (1:5,000 dilution) (170-6516 and 170-6515; Bio-Rad) for 2 h at room temperature. The membrane was developed with 2 ml of Luminata Crescendo Western HRP substrate (Millipore) and imaged using a Bio-Rad ChemiDoc XRS+ or Protein Simple FluorChem M chemiluminescence imager. Image Lab software was used to determine the molecular weight and relative density (normalized to that of β-actin) of each band. The identity of each band was based on Gag-Pro-Pol processing, as previously described (29).
ChIP.
SVGA cells were seeded in 10-cm dishes at an approximate density of 3 × 106 cells/dish for 24 h at 37°C and 5% CO2. Laminin-coated dishes were used to seed ReNcell CX cells at a density of 3 × 105 cells/dish for 24 h at 37°C and 5% CO2. The culture medium on adhered ReNcell CX cells was then replaced with medium lacking EGF and bFGF, and cells were allowed to differentiate into neurons for 10 days. SVGA cells and neurons were treated with 10 ng/ml human TNF-α (PeproTech) or human LIGHT (PeproTech) for 8 h, fixed with 4% paraformaldehyde, and harvested. Untreated cells were used as the negative control. Chromatin immunoprecipitation (ChIP) was performed using a Pierce magnetic ChIP kit (Thermo Scientific) per the manufacturer's instructions. IRF1- and NF-κB-bound DNA segments were isolated using 3 μg of rabbit anti-human IRF1 (SC497; Santa Cruz), rabbit anti-human NF-κB p65 (ab7970; Abcam), rabbit anti-human NF-κB p50 (ab32360; Abcam), or rabbit anti-human NF-κB p52 (4882S; Cell Signaling) antibodies. Immunoprecipitation with an IgG antibody was used as the negative control. Q-PCR was performed on the immunoprecipitated DNA, with SYBR green detection, to amplify the ISREs in the ERVK 5′ LTR. Primers for the first ISRE (nucleotides [nt] 380 to 392) were 5′-TCACCACTCCCTAATCTCAAGT-3′ (forward) and 5′-TCAGCACAGACCCTTTACGG-3′ (reverse), and those for the second ISRE (nt 563 to 575) were 5′-CTGAGATAGGAGAAAAACCGCCT-3′ (forward) and 5′-GGAGAGGGTCAGCAGACAAA-3′ (reverse). Data were analyzed using the ΔΔCT method and were normalized to the input and IgG controls for each condition. All data were graphed as means ± standard errors of measurement. Statistical analyses were performed in GraphPad Prism, using two-way ANOVA and Tukey's multiple-comparison test.
Fluorescence microscopy.
SVGA cells and ReNcell CX cell-derived neurons in Alvetex scaffolds were treated with 10 ng/ml human TNF-α (PeproTech) or human LIGHT (PeproTech). Untreated cells were used as the negative control. At 24 h posttreatment, cells were fixed with methanol for 40 s and rinsed with 1× PBS. Cells were permeabilized with 250 μl of PBS-T (PBS with 0.25% Triton X-100) and blocked with 250 μl of 3% bovine serum albumin (BSA) in TBS-T (Tris-buffered saline with 0.25% Triton X-100) for 30 min. Immunocytochemistry was performed using 1:750 mouse anti-human ERVK RT (H00002087A01; Abnova) primary antibody for 3 h and 1:1,000 AF488-conjugated goat anti-mouse (A11017; Molecular Probes) secondary antibody for 2 h. Nuclei were counterstained with 1:50,000 DAPI. ReNcell CX cells were also stained with fluorescent Nissl to detect neurons (Molecular Probes). Coverslips or Alvetex membranes were mounted onto slides by use of ProLong Gold antifade reagent (Molecular Probes) and dried overnight. Controls were prepared by immunostaining without the primary antibodies. Confocal microscopy was performed using an Olympus FV1200 laser scanning confocal microscope. Olympus Fluoview, version 4.0B, software was used to outline cellular boundaries for assessment of the mean intensities of total cellular ERVK RT and DAPI staining, and the ERVK RT/DAPI staining ratio is reported for each representative micrograph.
RESULTS
TNF-α and LIGHT enhance ERVK polyprotein and RT expression in a cell-type-dependent manner.
Augmented levels of the proinflammatory cytokines TNF-α and LIGHT play a crucial role in ALS neuropathology (18–20, 30). Considering that ERVK reactivation coincides with proinflammatory signatures in ALS, we sought to determine whether these cytokines can enhance ERVK expression in human astrocytes and neurons. Treatment of the human astrocytic SVGA cell line and human neurons derived from the ReNcell CX cell line with TNF-α or LIGHT dose-dependently enhanced ERVK polyprotein and RT levels, albeit in a cell-type-dependent manner. LIGHT increased ERVK protein levels most prominently in astrocytes, whereas TNF-α was best able to induce ERVK expression in neurons (Fig. 1A and 2A). We also observed enhanced ERVK polyprotein processing in these cytokine-stimulated cells, which culminated in the production of a catalytic RT subunit containing an RNase H domain (RT-RH) and a structural RT subunit without an RNase H domain (Fig. 1A and 2A). These observations are in line with our previous finding of IFN-γ-mediated ERVK polyprotein cleavage to produce a heterodimeric mature and active ERVK RT (29). Interestingly, TNF-α-treated neurons exhibited marked cleavage of the ERVK polyprotein to generate the RT-RH catalytic subunit (Fig. 2A), suggesting that neuronal ERVK RT activity detected in ALS may optimally occur in the context of chronic TNF-α exposure.
FIG 1.
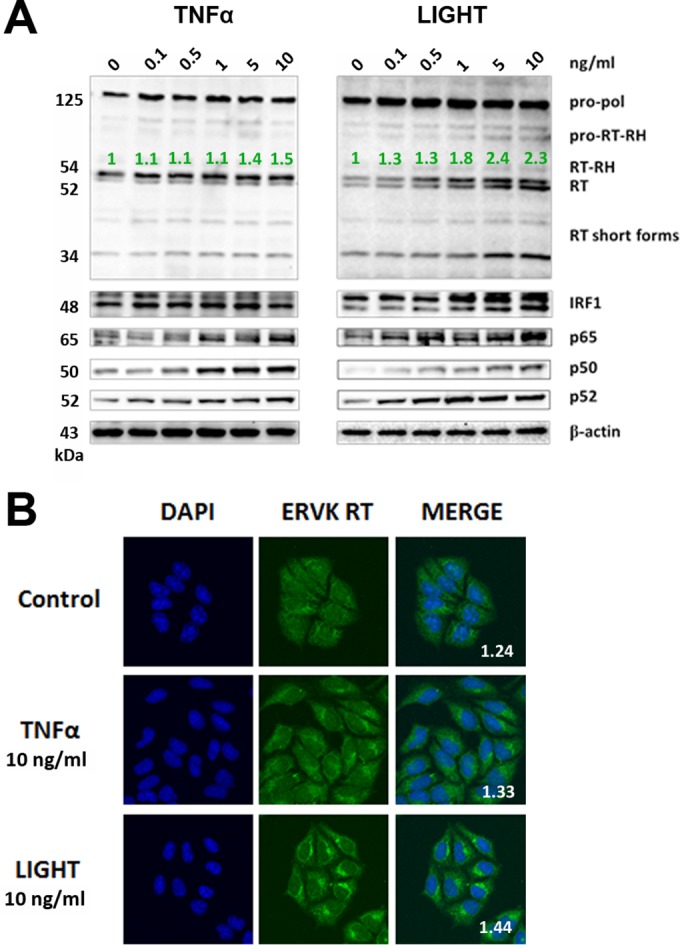
LIGHT, but not TNF-α, markedly enhances ERVK polyprotein and RT expression in astrocytes. SVGA cells were treated with various doses of TNF-α or LIGHT for 24 h. Western blotting and confocal microscopy were used to detect alterations in ERVK RT, IRF1, and NF-κB p65, p50, and p52 protein levels. (A) Western blot (cropped) showing that LIGHT strongly induced ERVK polyprotein (125 kDa) expression and cleavage to produce the small, 52/54-kDa RT without RNase H and the larger, 60-kDa RT with RNase H, concomitantly with upregulation of IRF1 and NF-κB protein levels. In comparison, TNF-α slightly enhanced ERVK polyprotein and RT subunit expression. β-Actin was used as the loading control (n = 3). Quantification of the 54-kDa band is depicted in green. (B) Representative confocal micrographs depicting LIGHT-mediated induction of ERVK RT expression. ERVK RT aggregates were deposited proximal to the nucleus and formed a perinuclear ring (n = 3). Quantification of the ERVK RT/DAPI staining ratio is presented in each merged micrograph.
FIG 2.
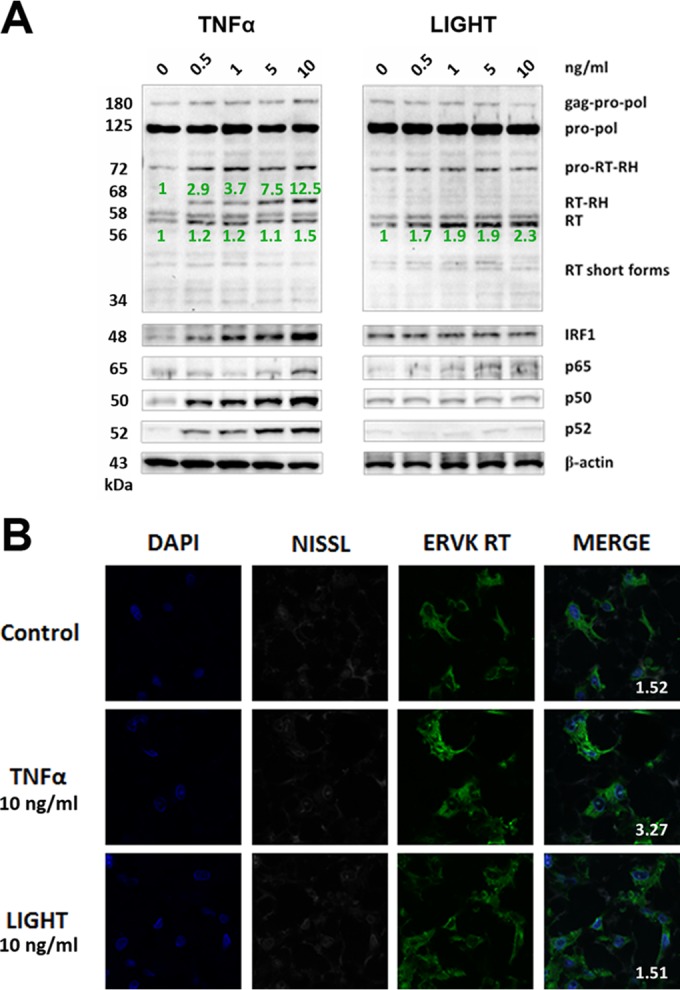
TNF-α, but not LIGHT, markedly enhances ERVK polyprotein and RT expression in neurons. ReNcell CX cell-derived neurons were treated with various doses of TNF-α or LIGHT for 24 h. Western blotting and confocal microscopy were used to detect alterations in ERVK RT, IRF1, and NF-κB p65, p50, and p52 protein levels. (A) Western blot (cropped) showing that TNF-α strongly induced ERVK polyprotein (180 and 125 kDa) expression and cleavage to produce the small, 56/58-kDa RT without RNase H and the larger, 68-kDa RT with RNase H, concomitantly with upregulation of IRF1 and NF-κB protein levels. In comparison, LIGHT enhanced the expression of the RT subunit without RNase H but not RT with the RNase H domain, suggesting that optimal RT activity may occur during exposure of neurons to TNF-α. β-Actin was used as the loading control (n = 3). Quantification of the 56-kDa and 68-kDa bands is depicted in green. (B) Representative confocal micrographs depicting TNF-α-mediated induction of ERVK RT expression in neurons. The fluorescent Nissl stain was used to identify neurons (n = 3). Quantification of the ERVK RT/DAPI staining ratio is presented in each merged micrograph.
Confocal microscopy revealed that under optimal stimulating conditions, LIGHT-treated astrocytes and TNF-α-treated neurons exhibited marked ERVK RT protein accumulation (Fig. 1B and 2B). In astrocytes, the ERVK polyprotein/RT formed a perinuclear ring and a large aggregate proximal to the nucleus (Fig. 1B). Nuclear ERVK RT expression also increased and exhibited a speckled pattern (Fig. 1B). We previously observed similar ERVK RT staining patterns in IFN-γ-treated cells (29), suggesting that ERVK polyprotein/RT aggregation is a common cellular feature occurring in the context of CNS inflammation. Moreover, the neurotoxic ERVK envelope protein was also upregulated in cytokine-stimulated cell cultures (data not shown). In addition, enhanced ERVK protein expression occurred concomitantly with the upregulation of the IRF1 and NF-κB p65, p50, and/or p52 transcription factors in astrocytes and neurons (Fig. 1A and 2A). This finding suggests that IRF1 and NF-κB isoforms likely play a crucial role in ERVK reactivation in astrocytes and neurons, and thus we sought to explore the mechanism behind this process.
IRF1 and NF-κB synergize to markedly enhance ERVK transcription.
Previously, in silico analysis revealed that two ISREs are a conserved feature of ERVK promoters called 5′ LTRs (Fig. 3A) (1). ISREs are known to bind interferon regulatory factors, such as IRF1 (31). The ERVK 5′ LTR also harbors numerous conserved putative NF-κB binding sites, including sites that partially overlap and are adjacent to IRF1 binding sites (Fig. 3A). The binding of IRF1 and NF-κB to their cognate sites is required for optimal transcriptional activation from the HIV-1 5′ LTR (32). Similarly, these proinflammatory transcription factors may be crucial for enhancing ERVK transcription in neuroinflammatory diseases, such as ALS.
FIG 3.

NF-κB and IRF1 interact with the ISREs in the ERVK 5′ LTR and synergize to enhance ERVK gene transcription. (A) In silico-predicted IRF1 and NF-κB binding sites, including two ISREs in the ERVK 5′ LTR consensus sequence. The data are from reference 1. (B) IRF1 and NF-κB p65 significantly enhance ERVK pol transcription in 293T cells. 293T cells were transfected with 1 μg of pCMVBL or pCMV2 empty vector (negative controls) or with plasmids encoding wild-type or phosphomimetic forms (5D, 4D, and 6D) of IRF and NF-κB for 48 h. The modulation of ERVK pol RNA levels was measured by using SYBR green detection and Q-PCR, and data were normalized to the negative control (**, P < 0.01; ****, P < 0.0001; n = 3). 18S rRNA was used as the endogenous control. Only IRF1 and NF-κB p65 significantly induced ERVK pol transcription. (C) IRF1 and NF-κB p65 and p50 synergize to significantly enhance ERVK pol transcription in astrocytes. SVGA cells were transfected with 1 μg of empty vector (negative control) or with plasmids encoding IRF1 and NF-κB isoforms, individually and in combinations, for 48 h. The modulation of ERVK pol RNA levels was measured by using SYBR green detection and Q-PCR, and data were normalized to the negative control (*, P < 0.05; ****, P < 0.0001; n = 3). 18S rRNA was used as the endogenous control. Although IRF1 or NF-κB p65 alone was sufficient to significantly induce ERVK pol transcription, IRF1 and NF-κB p65 and p50 synergized to further enhance ERVK pol RNA levels (up to 68-fold). (D) TNF-α and LIGHT markedly enhanced the binding of IRF1 and NF-κB p65 and p50 to both ISREs in the ERVK 5′ LTR, in a cell-type-dependent manner. Chromatin was extracted from SVGA cells and ReNcell CX cell-derived neurons treated with TNF-α (10 ng/ml) or LIGHT (10 ng/ml) for 8 h. Chromatin immunoprecipitation (ChIP) was performed with anti-human IRF1 or NF-κB p65, p50, or p52 antibody. Q-PCR was used to amplify immunoprecipitated ISRE sequences within the ERVK 5′ LTR, and products were detected using SYBR green detection. The fold enrichment of transcription factors at each ISRE was first normalized to the input control and then to the IgG negative control. All transcription factors were bound to the ISREs at basal levels. However, the binding of NF-κB p65 and p50 was significantly enhanced by LIGHT treatment, but not TNF-α treatment, in astrocytes (top panels) (n = 3; *, P < 0.05; ****, P < 0.0001). In contrast, the binding of NF-κB p65 and p50 was significantly enhanced by TNF-α treatment, but not LIGHT treatment, in neurons (bottom panels) (n = 2; *, P < 0.05; ***, P < 0.001).
To screen for which forms of NF-κB and IRFs modulate ERVK expression, 293T cells were transfected with a series of constructs overexpressing wild-type and/or phosphomimetic variants of p50, p65, IRF1, IRF2, IRF3, IRF5, IRF7, and IRF8 (Fig. 3B). In these cells, only NF-κB p65 and IRF1 significantly induced ERVK pol expression; thus, subsequent experiments focused on these select transcription factors.
To determine whether IRF1 and NF-κB cooperatively enhance ERVK transcription, we transiently transfected astrocytes with plasmids expressing constitutively active NF-κB (isoforms p65 and p50) and IRF1, individually or in combinations. The overexpression of IRF1 or NF-κB p65 alone was sufficient to significantly enhance ERVK pol transcription in astrocytes (Fig. 3C). We did not observe a perceivable effect of overexpression of NF-κB p50 alone on ERVK pol RNA levels. However, coexpression of IRF1 and NF-κB p65 and p50 in astrocytes produced a marked (68-fold) increase in ERVK pol RNA levels. These findings support the notion that IRF1 and the NF-κB p65/p50 heterodimer synergize to drive optimal transcriptional reactivation of ERVK in astrocytes. Enhanced expression of the Sp1 and Sp3 transcription factors, which have previously been shown to bind the ERVK promoter (33), can also regulate ERVK expression (data not shown), but not nearly to the extent seen with proinflammatory TFs.
TNF-α and LIGHT enhance the binding of IRF1 and NF-κB to ISREs in the ERVK 5′ LTR.
Proinflammatory cytokines, such as TNF-α and LIGHT, are generally potent activators of NF-κB and also lead to IRF1 activity (21, 24). Therefore, we sought to determine whether TNF-α- and LIGHT-mediated induction of ERVK expression is facilitated by enhanced interactions of NF-κB and IRF1 with their cognate binding sites on the ERVK 5′ LTR. For the first time, we showcase the biological importance of ISREs in the ERVK 5′ LTR, as they can interact with IRF1 and with NF-κB isoforms. Astrocytes and neurons exhibited basal IRF1 and NF-κB binding to both ISREs (Fig. 3D), which alludes to the basal ERVK expression observed in these cells. However, under optimal stimulating conditions, LIGHT-treated astrocytes (Fig. 3D) and TNF-α-treated neurons (Fig. 3D) exhibited markedly enhanced NF-κB p65 and p50 binding to each ISRE in the ERVK promoter. We did not observe any perceivable change in the binding of the NF-κB p52 isoform to the ISREs. This suggests that the canonical NF-κB p65/p50 complex or p50 homodimers predominantly bind the ERVK promoter and partake in proviral transcriptional reactivation. Furthermore, ChIP data did not support a role for the noncanonical p50/p52 NF-κB complex in ERVK transcription. Although the binding of IRF1 to the ISREs increased considerably with cytokine stimulation (9- and 7-fold in SVGA cells and neurons, respectively), the differences did not reach statistical significance. Overall, these findings support the notion that TNF-α- or LIGHT-induced IRF1 and NF-κB p65 and p50 binding to the ERVK promoter reactivates this endogenous retrovirus in the context of inflammation.
Interestingly, cytokine-mediated IRF1 and NF-κB binding to the ERVK promoter occurred in a cell-type-dependent manner. LIGHT, but not TNF-α, significantly enhanced NF-κB p65 and p50 binding to the ISREs in astrocytes (Fig. 3D). In contrast, TNF-α, but not LIGHT, significantly increased NF-κB p65 and p50 protein levels, as well as their interaction with the ISREs in the ERVK promoter, in neurons (Fig. 3D). Consistently, these results were associated with increased ERVK polyprotein/RT expression in LIGHT-treated astrocytes and TNF-α-treated neurons.
The ERVK pol gene was used as the negative control for ChIP Q-PCR (data not shown); however, transcription factor binding to the ERVK pol region was detected. This can be explained by extensive binding of NF-κB and IRF1 to regions other than promoters throughout the human genome (31). Cytokine treatment did not notably drive NF-κB and IRF1 binding to the ERVK pol gene, which confirms that transcription factor enrichment at the ERVK promoter region is not a random event under conditions of inflammation.
IRF1 and NF-κB expression is markedly increased in ERVK+ cortical neurons from ALS patients.
We previously demonstrated that ERVK RT expression is specifically increased in the cortical neurons of patients with ALS compared to those of neuronormal controls (11). However, the signals that lead to neuronal ERVK RT accumulation have remained unidentified. Augmented levels of TNF-α and LIGHT in the CNS are hallmarks of ALS (14, 16–20). We are the first to demonstrate that these proinflammatory cytokines lead to ERVK expression in human astrocytic and neuronal cell lines. In order to validate our in vitro findings, we sought to determine whether cortical brain tissue from patients with ALS exhibits increased NF-κB and IRF1 nuclear localization in ERVK+ neurons compared to that from neuronormal controls.
Here we highlight that ERVK RT expression predominantly accumulated in large pyramidal neurons in the third and fifth cortical layers of prefrontal and motor cortex tissue and was associated with a loss of cortical tissue organization in ALS (Fig. 4). Weak basal ERVK expression was observed in the neuronormal cortex, but a striking enhancement and expanded distribution of ERVK expression occurred in ALS cortical tissues. Interindividual differences in ERVK expression levels were maintained on comparing prefrontal and motor cortex samples, suggesting that either genetic background (polymorphisms in proteins of key cellular pathways) or disease severity (degree of inflammation) accounts for differential ERVK expression. Immunohistochemistry revealed that in comparison to those from neuronormal controls, motor cortex neurons from patients with ALS exhibited clear nuclear translocation of the proinflammatory transcription factors IRF1 and NF-κB p50 and, to a lesser degree, p65 (Fig. 5). Quantification of total neuronal ERVK RT and nuclear TF staining in cortical neurons revealed that the expression of each of these proteins was significantly upregulated in ALS tissues compared to neuronormal control tissues (P < 0.0001) (Fig. 6A). In addition, nuclear translocation of IRF1, NF-κB p50, and NF-κB p65 was strongly correlated with enhanced total ERVK RT levels in cortical neurons (Fig. 6B). Overall, our findings strongly support the premise that ERVK reactivation in the motor cortex in patients with ALS stems from enhanced interactions of cytokine-induced IRF1 and NF-κB transcription factors with the ERVK promoter.
FIG 4.

Representative images of ERVK reverse transcriptase (RT) in cortical brain tissue. Representative images show ERVK RT expression (green) in prefrontal cortex autopsy tissues (Brodmann area 9; NIH NeuroBioBank) from an individual with cancer (neuronormal; individual 3371) and a patient with ALS (individual 5215). Five total tissues were examined for each clinical group. (Left) Mosaic tiling (magnification, ×10) reveals enhanced ERVK RT expression in deep cortical tissue (cortical layer V) and upper cortical tissue (cortical layer III) in an ALS specimen. (Right) Nissl-stained cells reveal ERVK RT staining mainly in large pyramidal neurons. Nuclear DAPI staining is shown in blue. Bars, 10 μm for middle and right panels.
FIG 5.

The expression of ERVK RT is markedly enhanced in cortical neurons from patients with ALS and associates with increased levels and enhanced nuclear translocation of IRF1 and NF-κB. Representative confocal micrographs show ERVK RT, IRF1, and NF-κB p50 and p65 protein detection in Brodmann area 6 motor cortex tissues from an ALS patient (individual 5215) and a neuronormal control (individual 3371). ERVK RT expression was increased in the perinuclear region and in the axons of large pyramidal neurons in the ALS motor cortex. This occurred concomitantly with increased IRF1 (A), NF-κB p50 (B), and NF-κB p65 (C) nuclear translocation in cortical neurons. Five total tissues were examined for each clinical group.
FIG 6.

Augmented ERVK RT levels in ALS cortical neurons correlate with enhanced nuclear localization of proinflammatory transcription factors. (A) The mean optical intensity (relative intensity) was used as a metric to quantify the level of total ERVK RT, as well as the level of nuclear IRF1, NF-κB p50, or NF-κB p65, in cortical neurons from neuronormal individuals (n = 86 cells [10 to 29 cells per case]) and patients with ALS (n = 105 cells [10 to 27 cells per case]). Clinical groups (n = 5 each) were compared using the nonparametric Mann-Whitney U test. The expression of total ERVK RT and nuclear translocation of each transcription factor increased significantly in ALS cortical neurons compared to the controls (***, P < 0.0001). (B) Pearson's correlation between the mean relative intensities of total ERVK RT and nuclear IRF1, NF-κB p50, or NF-κB p65 measured in neuronormal (NN) (red) and ALS (blue) cortical neurons. ERVK RT expression exhibited a moderate (p65 [0.6863]) to strong (IRF1 [0.8574] and p50 [0.8405]) correlation with the extent of nuclear localization for each transcription factor. (C) ROC curve analyses depicting the accuracy (area under the curve [AUC]) of augmented ERVK RT and proinflammatory transcription factor levels in discriminating between cortical neurons in controls and those in ALS tissues. IRF1 and NF-κB p50 levels were excellent predictors of ALS. In contrast, total ERVK RT and nuclear NF-κB p65 levels were less accurate at distinguishing between the normal and diseased states.
We further utilized receiver operating characteristic (ROC) curve analyses to evaluate the ability of augmented ERVK RT and nuclear proinflammatory transcription factor levels to discriminate between neuronormal and ALS tissues (Fig. 6C). Based on the AUC values, total cellular ERVK RT expression in ALS motor cortex tissues was determined to be a biomarker of ALS (AUC, 0.8961). Enhanced nuclear localization of IRF1 and NF-κB p50 was also able to accurately distinguish between normal and ALS tissues (AUC of 0.9634 and 0.9834, respectively). In comparison, nuclear levels of NF-κB p65 were able to discriminate between the normal and diseased states, but to a lesser extent (AUC, 0.9137). Nonetheless, these findings support the notion that inflammation-induced ERVK is a novel biomarker of ALS, as previously proposed (11).
DISCUSSION
Several lines of evidence suggest that augmented levels of the cytokines TNF-α and LIGHT drive enhanced activity of proinflammatory transcription factors in neurological diseases (18, 19, 30). These signaling pathways may be important triggers of ERVK transcription in the CNS. Here we show that TNF-α and LIGHT are potent inducers of ERVK polyprotein and RT expression in neurons and astrocytes, respectively. To delineate the mechanism behind cytokine-induced ERVK reactivation, we utilized ChIP and confirmed that the ISREs in the ERVK promoter are transcription factor docking sites and that enhanced binding of IRF1 and NF-κB p65/p50 to these elements synergistically augments ERVK gene expression in response to proinflammatory stimuli (Fig. 7). The cooperative binding of IRF1 and NF-κB to their cognate sites is a conserved feature of many IRF1- and NF-κB-responsive gene promoters. For instance, synergy between IRF1 and NF-κB is required to induce transcription from the human inducible nitric oxide synthase, interleukin-15, major histocompatibility complex class I, vascular cell adhesion molecule I, and IFN-β promoters (22, 34–37). Accordingly, overlapping or adjacent IRF1 and NF-κB binding sites have been described for these promoters, similar to those observed for the ERVK promoter (1). IRF1 and NF-κB also synergistically activate transcription from the HIV-1 promoter, although IRF1 binding sites are not found adjacent to or overlapping NF-κB sites (32). In line with these findings, we have added the ERVK LTRs to the growing list of IRF1- and NF-κB-responsive enhancer elements. Future studies are required to perform mutational analysis of the various forms of ERVK promoters within the human genome to confirm the functionality of ISRE and κB sites in modulating ERVK transcription.
FIG 7.

IRF1 and NF-κB drive ERVK reactivation via ISREs in the LTR. This figure summarizes how the proinflammatory cytokines TNF-α and LIGHT promote the cellular expression of ERVK. Following engagement of IRF1 and NF-κB isoforms p50 and p65 with the ERVK LTR, viral RNA and protein production is induced. Additional κB sites in the ERVK LTR may host noncanonical p50 homodimers, thus facilitating recruitment of additional transcriptional activators, such as Sp1/Sp3. Accumulation of ERVK reverse transcriptase in human neurons forms a cytoplasmic aggresome as well as nuclear foci.
It is interesting that TNF-α and LIGHT enhance ERVK expression in a cell-type-dependent manner. TNF-α increased ERVK protein levels most prominently in neurons, whereas LIGHT was best able to induce ERVK in astrocytes. This effect can be explained by differential enrichment of NF-κB at the ERVK promoter during TNF-α or LIGHT stimulation of astrocytes and neurons. TNF-α, but not LIGHT, significantly increased the interaction of NF-κB p65 and p50 with the ISREs on the ERVK promoter in neurons. In contrast, LIGHT, but not TNF-α, significantly enhanced NF-κB p65 and p50 binding to the ISREs in astrocytes. The cell type specificity of TNF-α and LIGHT may also be explained by differential expression of their cognate cell surface receptors, as well as downstream signaling molecules, in astrocytes and neurons. TNF-α is known to be biologically active in both transmembrane and soluble forms (17, 38). Soluble TNF-α signals mainly through TNF receptor 1 (TNFR1) (17), which is found at a lower level in astrocytes than in neurons (The Human Protein Atlas [http://www.proteinatlas.org/ENSG00000067182-TNFRSF1A/tissue]). Overproduction of soluble TNF-α has been shown to cause neurodegeneration in the CNS (38). Transmembrane TNF-α, on the other hand, signals mainly through TNFR2, which is found primarily in microglial cells (17, 19). Since we utilized soluble TNF-α in our cell line models, it is not surprising that neurons, not astrocytes, were more responsive to this cytokine. Adaptor molecules that associate with TNF receptors, known as TRAFs, exert a second layer of control over cell-specific TNF-α and LIGHT signaling. TRAF3 is basally expressed in neurons but not in glial cells (The Human Protein Atlas [http://www.proteinatlas.org/ENSG00000131323-TRAF3/tissue]), and it has been shown to be much more inducible in neurons than in astrocytes (39). TRAF3 is a negative regulator of LIGHT signaling, as it inhibits the function of the LTβ receptor, which results in NF-κB inactivity (39). In contrast, TRAF3 has no effect on TNF-α-induced NF-κB signaling (39). Neuronal expression of TRAF3 may have inhibited LIGHT-induced NF-κB signaling, leading to a lack of any perceivable effect on ERVK expression in our neuronal models.
The transcription factors of the NF-κB class function as heterodimers or homodimers comprised of various combinations of the subunits p65, RelB, c-Rel, p50, and p52 (40). The most common NF-κB species found in human neurons are the canonical p65/p50 complex and the noncanonical p50/p50 complex (41). Different NF-κB dimers recognize slightly different binding sequences with high affinity (42). For instance, p50 homodimers bind the consensus decamer GGGGATYCCC, where Y is a pyrimidine base, while p65/p50 heterodimers have a high affinity for NF-κB sites with AT-rich centers (42). The majority of the NF-κB binding sites in the ERVK 5′ LTR are GC rich (60 to 80% GC content) (1), and thus the ERVK promoter is likely most responsive to p50 homodimers. Indeed, ChIP experiments revealed the most dramatic enrichment in the binding of NF-κB p50 at the ISREs in the ERVK promoter in both astrocytes and neurons. Nevertheless, p50 may be present in homodimeric complexes or in heterodimeric complexes with p65 at the ERVK promoter. Notably, NF-κB p50 homodimers are known to repress HIV-1 transcription, leading to retroviral latency (43). Thus, the type of p50 complex that forms at ERVK LTRs may either promote or repress the production of viral transcripts. In addition, the ERVK promoter harbors binding sites for coactivators, such as Sp1 and TFII-I, in the vicinity of NF-κB sites (1), suggesting that multiple transcription factors partake in the complexity of LTR-driven ERVK reactivation. Our results point to a synergistic effect of the IRF1-p50-p65 transcription factors in driving ERVK expression.
Compared to that in neuronormal controls, ERVK RT expression was markedly enhanced in large pyramidal neurons in BA9 prefrontal and BA6 motor cortex ALS tissues. Pyramidal neurons in the motor cortex normally exhibit constitutive basal NF-κB activity, which is required to maintain neuronal plasticity (44). In the presence of already active NF-κB, proinflammatory stimuli, including TNF-α and LIGHT, which culminate in IRF1 activation, may be sufficient to drive ERVK reactivation in pyramidal neurons. This is in contrast to IRF1 activation in cortical neurons during vesicular stomatitis virus (VSV) infection, which serves as a protective mechanism limiting viral replication and spread within the CNS (45). This highlights how select virus groups, including retroviruses (32), usurp inflammatory signaling as a means of self-promotion, whereas other types of infection are limited through the action of these signaling cascades.
Confocal microscopy revealed a unique pattern of ERVK RT expression in cells, consisting of punctate structures that accumulated in the perinuclear region along with the formation of a large aggregate proximal to the nucleus. This type of staining is typically seen for specialized inclusion bodies called aggresomes, which are compartments that sequester unwanted proteins and facilitate their clearance by autophagy, thereby dissipating the cytotoxic effects of protein aggregates (46, 47). Likewise, formation of ERVK RT aggresomes may be a cellular response to protect against toxic ERVK protein accumulation. Unfortunately, the appearance of aggresomes can impair vital cellular functions, e.g., inactivating the proteasomal pathway responsible for clearing protein aggregates (47). Interestingly, protein clearance pathways, such as the proteasome system and autophagy, are dysregulated in ERVK-associated neurological diseases, including ALS (48). Recently, we demonstrated that the proteasome system and autophagy are crucial for homeostatic clearance of ERVK proteins (49). In the absence of functional protein degradation pathways, inflammation-induced ERVK aggresomes may persist and propagate chronic neuronal damage.
In concert, the genetic background and transient insults can lead to chronic inflammation in the CNS (reviewed in references 50 and 51). It has been well established that exacerbated TNF-α and LIGHT signaling pathways in ALS converge at NF-κB to promote neuronal damage (25). Our findings suggest that inflammation-driven IRF1 and NF-κB activity promotes ERVK reactivation in neurons of the motor cortex in ALS (Fig. 7). A causative link between viral infection and the pathology of ALS is a highly debated topic. Our study suggests that chronic proinflammatory signals may perpetuate ERVK-mediated tissue damage through the action of select viral proteins (12). This may also prove to be a more general phenomenon occurring during lifetime inflammatory events. Thus, quenching ERVK activity through antiretroviral or immunomodulatory regimens may hinder virus-mediated neuropathology and improve the symptoms of ALS or other ERVK-associated chronic diseases (52–55).
ACKNOWLEDGMENTS
We have no conflicts of interest.
M.M. carried out the molecular biology experiments, acquired data, and performed statistical analyses. J.F.-P. performed immunohistochemistry and confocal microscopy assays. R.N.D. conceived the study, carried out ROC curve analyses, and prepared the figures. R.L. participated in the design of the study and provided plasmids. R.N.D. and M.M. drafted the manuscript. All authors read and approved the final manuscript.
This study was funded by the ALS Association (grant FTM479), the Manitoba Medical Services Foundation (grant 8-2013-12), the Manitoba Health Research Council (grants 270 and 1094), and the Canada Foundation for Innovation (grant 31503). M.M. was funded by a Canadian Institutes of Health Research Frederick Banting and Charles Best Canada graduate scholarship and a Manitoba graduate scholarship.
REFERENCES
- 1.Manghera M, Douville RN. 2013. Endogenous retrovirus-K promoter: a landing strip for inflammatory transcription factors? Retrovirology 10:16. doi: 10.1186/1742-4690-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Battistini A, Marsili G, Sgarbanti M, Ensoli B, Hiscott J. 2002. IRF regulation of HIV-1 long terminal repeat activity. J Interferon Cytokine Res 22:27–37. doi: 10.1089/107999002753452638. [DOI] [PubMed] [Google Scholar]
- 3.Sgarbanti M, Borsetti A, Moscufo N, Bellocchi MC, Ridolfi B, Nappi F, Marsili G, Marziali G, Coccia EM, Ensoli B, Battistini A. 2002. Modulation of human immunodeficiency virus 1 replication by interferon regulatory factors. J Exp Med 195:1359–1370. doi: 10.1084/jem.20010753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klatt NR, Chomont N, Douek DC, Deeks SG. 2013. Immune activation and HIV persistence: implications for curative approaches to HIV infection. Immunol Rev 254:326–342. doi: 10.1111/imr.12065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Freimanis G, Hooley P, Ejtehadi HD, Ali HA, Veitch A, Rylance PB, Alawi A, Axford J, Nevill A, Murray PG, Nelson PN. 2010. A role for human endogenous retrovirus-K (HML-2) in rheumatoid arthritis: investigating mechanisms of pathogenesis. Clin Exp Immunol 160:340–347. doi: 10.1111/j.1365-2249.2010.04110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Serra C, Mameli G, Arru G, Sotgiu S, Rosati G, Dolei A. 2003. In vitro modulation of the multiple sclerosis (MS)-associated retrovirus by cytokines: implications for MS pathogenesis. J Neurovirol 9:637–643. [DOI] [PubMed] [Google Scholar]
- 7.Mameli G, Astone V, Arru G, Marconi S, Lovato L, Serra C, Sotgiu S, Bonetti B, Dolei A. 2007. Brains and peripheral blood mononuclear cells of multiple sclerosis (MS) patients hyperexpress MS-associated retrovirus/HERV-W endogenous retrovirus, but not human herpesvirus 6. J Gen Virol 88:264–274. doi: 10.1099/vir.0.81890-0. [DOI] [PubMed] [Google Scholar]
- 8.Renton AE, Chio A, Traynor BJ. 2014. State of play in amyotrophic lateral sclerosis genetics. Nat Neurosci 17:17–23. doi: 10.1038/nn.3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McCormick AL, Brown RH Jr, Cudkowicz ME, Al-Chalabi A, Garson JA. 2008. Quantification of reverse transcriptase in ALS and elimination of a novel retroviral candidate. Neurology 70:278–283. doi: 10.1212/01.wnl.0000297552.13219.b4. [DOI] [PubMed] [Google Scholar]
- 10.MacGowan DJ, Scelsa SN, Imperato TE, Liu KN, Baron P, Polsky B. 2007. A controlled study of reverse transcriptase in serum and CSF of HIV-negative patients with ALS. Neurology 68:1944–1946. doi: 10.1212/01.wnl.0000263188.77797.99. [DOI] [PubMed] [Google Scholar]
- 11.Douville R, Liu J, Rothstein J, Nath A. 2011. Identification of active loci of a human endogenous retrovirus in neurons of patients with amyotrophic lateral sclerosis. Ann Neurol 69:141–151. doi: 10.1002/ana.22149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li W, Lee MH, Henderson L, Tyagi R, Bachani M, Steiner J, Campanac E, Hoffman DA, von Geldern G, Johnson K, Maric D, Morris HD, Lentz M, Pak K, Mammen A, Ostrow L, Rothstein J, Nath A. 2015. Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med 7:307ra153. doi: 10.1126/scitranslmed.aac8201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Poloni M, Facchetti D, Mai R, Micheli A, Agnoletti L, Francolini G, Mora G, Camana C, Mazzini L, Bachetti T. 2000. Circulating levels of tumour necrosis factor-alpha and its soluble receptors are increased in the blood of patients with amyotrophic lateral sclerosis. Neurosci Lett 287:211–214. doi: 10.1016/S0304-3940(00)01177-0. [DOI] [PubMed] [Google Scholar]
- 14.Tateishi T, Yamasaki R, Tanaka M, Matsushita T, Kikuchi H, Isobe N, Ohyagi Y, Kira J. 2010. CSF chemokine alterations related to the clinical course of amyotrophic lateral sclerosis. J Neuroimmunol 222:76–81. doi: 10.1016/j.jneuroim.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Babu GN, Kumar A, Chandra R, Puri SK, Kalita J, Misra UK. 2008. Elevated inflammatory markers in a group of amyotrophic lateral sclerosis patients from northern India. Neurochem Res 33:1145–1149. doi: 10.1007/s11064-007-9564-x. [DOI] [PubMed] [Google Scholar]
- 16.Olmos G, Llado J. 2014. Tumor necrosis factor alpha: a link between neuroinflammation and excitotoxicity. Mediators Inflamm 2014:861231. doi: 10.1155/2014/861231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McCoy MK, Tansey MG. 2008. TNF signaling inhibition in the CNS: implications for normal brain function and neurodegenerative disease. J Neuroinflammation 5:45. doi: 10.1186/1742-2094-5-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Aebischer J, Moumen A, Sazdovitch V, Seilhean D, Meininger V, Raoul C. 2012. Elevated levels of IFNgamma and LIGHT in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. Eur J Neurol 19:752–759, e45–e56. doi: 10.1111/j.1468-1331.2011.03623.x. [DOI] [PubMed] [Google Scholar]
- 19.Aebischer J, Cassina P, Otsmane B, Moumen A, Seilhean D, Meininger V, Barbeito L, Pettmann B, Raoul C. 2011. IFNgamma triggers a LIGHT-dependent selective death of motoneurons contributing to the non-cell-autonomous effects of mutant SOD1. Cell Death Differ 18:754–768. doi: 10.1038/cdd.2010.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Otsmane B, Moumen A, Aebischer J, Coque E, Sar C, Sunyach C, Salsac C, Valmier J, Salinas S, Bowerman M, Raoul C. 2014. Somatic and axonal LIGHT signaling elicit degenerative and regenerative responses in motoneurons, respectively. EMBO Rep 15:540–547. doi: 10.1002/embr.201337948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ware CF. 2005. Network communications: lymphotoxins, LIGHT, and TNF. Annu Rev Immunol 23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- 22.Neish AS, Read MA, Thanos D, Pine R, Maniatis T, Collins T. 1995. Endothelial interferon regulatory factor 1 cooperates with NF-kappa B as a transcriptional activator of vascular cell adhesion molecule 1. Mol Cell Biol 15:2558–2569. doi: 10.1128/MCB.15.5.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mir M, Asensio VJ, Tolosa L, Gou-Fabregas M, Soler RM, Llado J, Olmos G. 2009. Tumor necrosis factor alpha and interferon gamma cooperatively induce oxidative stress and motoneuron death in rat spinal cord embryonic explants. Neuroscience 162:959–971. doi: 10.1016/j.neuroscience.2009.05.049. [DOI] [PubMed] [Google Scholar]
- 24.Paludan SR. 2000. Synergistic action of pro-inflammatory agents: cellular and molecular aspects. J Leukoc Biol 67:18–25. [DOI] [PubMed] [Google Scholar]
- 25.Akizuki M, Yamashita H, Uemura K, Maruyama H, Kawakami H, Ito H, Takahashi R. 2013. Optineurin suppression causes neuronal cell death via NF-kappaB pathway. J Neurochem 126:699–704. doi: 10.1111/jnc.12326. [DOI] [PubMed] [Google Scholar]
- 26.Major EO, Miller AE, Mourrain P, Traub RG, de Widt E, Sever J. 1985. Establishment of a line of human fetal glial cells that supports JC virus multiplication. Proc Natl Acad Sci U S A 82:1257–1261. doi: 10.1073/pnas.82.4.1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Donato R, Miljan EA, Hines SJ, Aouabdi S, Pollock K, Patel S, Edwards FA, Sinden JD. 2007. Differential development of neuronal physiological responsiveness in two human neural stem cell lines. BMC Neurosci 8:36. doi: 10.1186/1471-2202-8-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sharma S, tenOever BR, Grandvaux N, Zhou GP, Lin R, Hiscott J. 2003. Triggering the interferon antiviral response through an IKK-related pathway. Science 300:1148–1151. doi: 10.1126/science.1081315. [DOI] [PubMed] [Google Scholar]
- 29.Manghera M, Ferguson J, Douville R. 2015. ERVK polyprotein processing and reverse transcriptase expression in human cell line models of neurological disease. Viruses 7:320–332. doi: 10.3390/v7010320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tolosa L, Caraballo-Miralles V, Olmos G, Llado J. 2011. TNF-alpha potentiates glutamate-induced spinal cord motoneuron death via NF-kappaB. Mol Cell Neurosci 46:176–186. doi: 10.1016/j.mcn.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 31.Rettino A, Clarke NM. 2013. Genome-wide identification of IRF1 binding sites reveals extensive occupancy at cell death associated genes. J Carcinog Mutagen 2013(Spec Iss Apoptosis):S6-009. doi: 10.4172/2157-2518.S6-009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sgarbanti M, Remoli AL, Marsili G, Ridolfi B, Borsetti A, Perrotti E, Orsatti R, Ilari R, Sernicola L, Stellacci E, Ensoli B, Battistini A. 2008. IRF-1 is required for full NF-kappaB transcriptional activity at the human immunodeficiency virus type 1 long terminal repeat enhancer. J Virol 82:3632–3641. doi: 10.1128/JVI.00599-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fuchs NV, Kraft M, Tondera C, Hanschmann KM, Lower J, Lower R. 2011. Expression of the human endogenous retrovirus (HERV) group HML-2/HERV-K does not depend on canonical promoter elements but is regulated by transcription factors Sp1 and Sp3. J Virol 85:3436–3448. doi: 10.1128/JVI.02539-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saura M, Zaragoza C, Bao C, McMillan A, Lowenstein CJ. 1999. Interaction of interferon regulatory factor-1 and nuclear factor kappaB during activation of inducible nitric oxide synthase transcription. J Mol Biol 289:459–471. doi: 10.1006/jmbi.1999.2752. [DOI] [PubMed] [Google Scholar]
- 35.Azimi N, Shiramizu KM, Tagaya Y, Mariner J, Waldmann TA. 2000. Viral activation of interleukin-15 (IL-15): characterization of a virus-inducible element in the IL-15 promoter region. J Virol 74:7338–7348. doi: 10.1128/JVI.74.16.7338-7348.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drew PD, Franzoso G, Becker KG, Bours V, Carlson LM, Siebenlist U, Ozato K. 1995. NF kappa B and interferon regulatory factor 1 physically interact and synergistically induce major histocompatibility class I gene expression. J Interferon Cytokine Res 15:1037–1045. [DOI] [PubMed] [Google Scholar]
- 37.Merika M, Williams AJ, Chen G, Collins T, Thanos D. 1998. Recruitment of CBP/p300 by the IFN beta enhanceosome is required for synergistic activation of transcription. Mol Cell 1:277–287. doi: 10.1016/S1097-2765(00)80028-3. [DOI] [PubMed] [Google Scholar]
- 38.Akassoglou K, Probert L, Kontogeorgos G, Kollias G. 1997. Astrocyte-specific but not neuron-specific transmembrane TNF triggers inflammation and degeneration in the central nervous system of transgenic mice. J Immunol 158:438–445. [PubMed] [Google Scholar]
- 39.Bista P, Zeng W, Ryan S, Bailly V, Browning JL, Lukashev ME. 2010. TRAF3 controls activation of the canonical and alternative NFkappaB by the lymphotoxin beta receptor. J Biol Chem 285:12971–12978. doi: 10.1074/jbc.M109.076091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hayden MS, Ghosh S. 2012. NF-kappaB, the first quarter-century: remarkable progress and outstanding questions. Genes Dev 26:203–234. doi: 10.1101/gad.183434.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou Z, Peng X, Insolera R, Fink DJ, Mata M. 2009. Interleukin-10 provides direct trophic support to neurons. J Neurochem 110:1617–1627. doi: 10.1111/j.1471-4159.2009.06263.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kunsch C, Ruben SM, Rosen CA. 1992. Selection of optimal kappa B/Rel DNA-binding motifs: interaction of both subunits of NF-kappa B with DNA is required for transcriptional activation. Mol Cell Biol 12:4412–4421. doi: 10.1128/MCB.12.10.4412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Williams SA, Chen LF, Kwon H, Ruiz-Jarabo CM, Verdin E, Greene WC. 2006. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J 25:139–149. doi: 10.1038/sj.emboj.7600900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kaltschmidt B, Kaltschmidt C. 2009. NF-kappaB in the nervous system. Cold Spring Harb Perspect Biol 1:a001271. doi: 10.1101/cshperspect.a001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nair S, Michaelsen-Preusse K, Finsterbusch K, Stegemann-Koniszewski S, Bruder D, Grashoff M, Korte M, Koster M, Kalinke U, Hauser H, Kroger A. 2014. Interferon regulatory factor-1 protects from fatal neurotropic infection with vesicular stomatitis virus by specific inhibition of viral replication in neurons. PLoS Pathog 10:e1003999. doi: 10.1371/journal.ppat.1003999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Boyault C, Zhang Y, Fritah S, Caron C, Gilquin B, Kwon SH, Garrido C, Yao TP, Vourc'h C, Matthias P, Khochbin S. 2007. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev 21:2172–2181. doi: 10.1101/gad.436407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Olzmann JA, Li L, Chin LS. 2008. Aggresome formation and neurodegenerative diseases: therapeutic implications. Curr Med Chem 15:47–60. doi: 10.2174/092986708783330692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Janssens J, Van Broeckhoven C. 2013. Pathological mechanisms underlying TDP-43 driven neurodegeneration in FTLD-ALS spectrum disorders. Hum Mol Genet 22:R77–R87. doi: 10.1093/hmg/ddt349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Manghera M, Ferguson-Parry J, Douville RN. 2016. TDP-43 regulates endogenous retrovirus-K viral protein accumulation. Neurobiol Dis 94:226–236. doi: 10.1016/j.nbd.2016.06.017. [DOI] [PubMed] [Google Scholar]
- 50.Lucas SM, Rothwell NJ, Gibson RM. 2006. The role of inflammation in CNS injury and disease. Br J Pharmacol 147(Suppl 1):S232–S240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dahm T, Rudolph H, Schwerk C, Schroten H, Tenenbaum T. 2016. Neuroinvasion and inflammation in viral central nervous system infections. Mediators Inflamm 2016:8562805. doi: 10.1155/2016/8562805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Majde JA. 2010. Neuroinflammation resulting from covert brain invasion by common viruses—a potential role in local and global neurodegeneration. Med Hypotheses 75:204–213. doi: 10.1016/j.mehy.2010.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Scarisbrick IA, Rodriguez M. 2003. Hit-hit and hit-run: viruses in the playing field of multiple sclerosis. Curr Neurol Neurosci Rep 3:265–271. doi: 10.1007/s11910-003-0087-9. [DOI] [PubMed] [Google Scholar]
- 54.Christensen T. 2010. HERVs in neuropathogenesis. J Neuroimmune Pharmacol 5:326–335. doi: 10.1007/s11481-010-9214-y. [DOI] [PubMed] [Google Scholar]
- 55.Robberecht W, Jubelt B. 2005. Reverse transcriptase takes ALS back to viruses. Neurology 64:410–411. doi: 10.1212/01.WNL.0000150927.53939.98. [DOI] [PubMed] [Google Scholar]