

ABSTRACT
The emergence of transmissible HIV-1 strains with resistance to antiretroviral drugs highlights a continual need for new therapies. Here we describe a novel acylguanidine-containing compound, 1-(2-(azepan-1-yl)nicotinoyl)guanidine (or SM111), that inhibits in vitro replication of HIV-1, including strains resistant to licensed protease, reverse transcriptase, and integrase inhibitors, without major cellular toxicity. At inhibitory concentrations, intracellular p24Gag production was unaffected, but virion release (measured as extracellular p24Gag) was reduced and virion infectivity was substantially impaired, suggesting that SM111 acts at a late stage of viral replication. SM111-mediated inhibition of HIV-1 was partially overcome by a Vpu I17R mutation alone or a Vpu W22* truncation in combination with Env N136Y. These mutations enhanced virion infectivity and Env expression on the surface of infected cells in the absence and presence of SM111 but also impaired Vpu's ability to downregulate CD4 and BST2/tetherin. Taken together, our results support acylguanidines as a class of HIV-1 inhibitors with a distinct mechanism of action compared to that of licensed antiretrovirals. Further research on SM111 and similar compounds may help to elucidate knowledge gaps related to Vpu's role in promoting viral egress and infectivity.
IMPORTANCE New inhibitors of HIV-1 replication may be useful as therapeutics to counteract drug resistance and as reagents to perform more detailed studies of viral pathogenesis. SM111 is a small molecule that blocks the replication of wild-type and drug-resistant HIV-1 strains by impairing viral release and substantially reducing virion infectivity, most likely through its ability to prevent Env expression at the infected cell surface. Partial resistance to SM111 is mediated by mutations in Vpu and/or Env, suggesting that the compound affects host/viral protein interactions that are important during viral egress. Further characterization of SM111 and similar compounds may allow more detailed pharmacological studies of HIV-1 egress and provide opportunities to develop new treatments for HIV-1.
INTRODUCTION
The development and scale-up of effective combination antiretroviral therapies have significantly reduced human immunodeficiency virus type 1 (HIV-1)-related morbidity and mortality globally (1); however, selection and transmission of drug-resistant HIV-1 strains remain a concern (2, 3). Thus, new antivirals that overcome drug resistance or act via novel mechanisms are needed. Acylguanidines are a broad class of antiviral compounds that target ion channels and other host and viral factors (4–8). Their anti-HIV-1 activity is exemplified by 5-(N,N-hexamethylene)amiloride (HMA; Fig. 1A) (5, 6), as well as N-(5-(1-methyl-1H-pyrazol-4-yl)naphthalene-2-carbonyl)guanidine (BIT-225; Fig. 1B) (7). HMA and BIT-225 inhibit HIV-1 replication in monocyte-derived macrophages at low micromolar concentrations (6, 7). Moreover, treatment of infected cells with these compounds results in reduced budding and release of viral particles without affecting intracellular viral protein production, suggesting that blockade occurs at a late stage of viral replication (5, 7).
FIG 1.
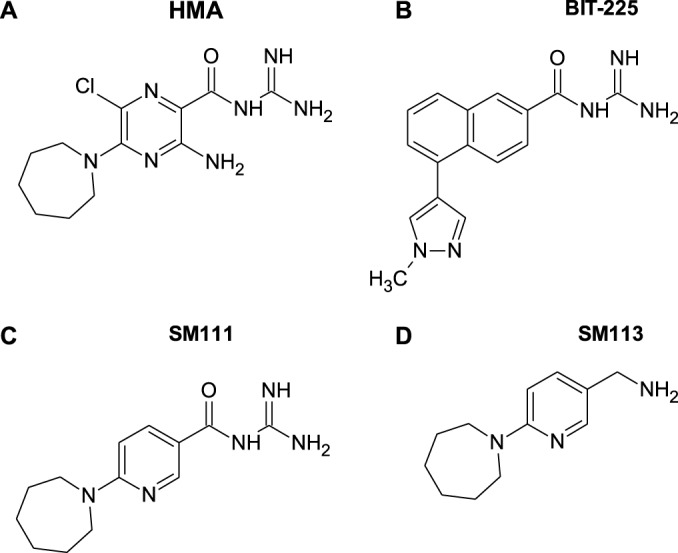
Chemical structures of acylguanidine compounds, namely, 5-(N,N-hexamethylene)amiloride (HMA) (A), N-(5-(1-methyl-1H-pyrazol-4-yl)naphthalene-2-carbonyl)guanidine (BIT-225) (B), 1-(2-(azepan-1-yl)nicotinoyl)guanidine (SM111) (C), and C-(6-azepan-1-yl-pyridin-3-yl)-methylamine (SM113) (D).
HMA and BIT-225 are both reported to target the HIV-1 accessory protein Vpu, an ∼81-amino-acid transmembrane protein that is found in HIV-1 and some simian immunodeficiency virus (SIV) lineages and enhances viral replication through multiple mechanisms (9–11). Most notably, Vpu augments virion release by downregulating CD4 (12–15), whose presence on the cell surface would otherwise promote elimination of infected cells through antibody-dependent cellular cytotoxicity (16–18), as well as the host restriction factor tetherin (also known as BST2 or CD317), whose presence would otherwise capture mature virions at the cell surface (19–22). Vpu is also reported to form cation-selective ion channels (11, 23–30); however, this property is controversial (31, 32), and its role in HIV-1 replication is unknown (11). Since acylguanidines can also act on host ion channels, host transporters, and viral polymerases (4, 8), additional antiviral mechanisms are possible. Unfortunately, the high in vitro cytotoxicity of most acylguanidines with antiviral activity has precluded more detailed molecular and cellular studies (4–6). For example, while BIT-225 has shown promise in early clinical trials (33), it displays lower activity in T cell lines, where inhibitory concentrations frequently cause cell death (7, 34, 35).
To further assess this class of HIV-1 inhibitors, we examined a novel acylguanidine-containing compound, 1-(2-(azepan-1-yl)nicotinoyl)guanidine (or SM111), which is structurally similar to HMA (Fig. 1C). We previously identified SM111 as a potent inhibitor of the influenza A/M2 ion channel that also blocks in vitro replication of influenza A virus without obvious cytotoxicity (36). Here, we describe SM111's ability to inhibit in vitro HIV-1 replication, including that of drug-resistant strains, with substantially less toxicity than that of HMA or BIT-225 in both a T cell line and primary cells. Growth of HIV-1 in the presence of SM111 resulted in the rapid selection of viruses harboring I17R or W22* mutations in Vpu's transmembrane domain, the latter mutation in combination with an N136Y mutation in Env. Our data indicate that SM111 inhibits HIV-1 particle release and impairs virion infectivity, supporting acylguanidines as antiretrovirals with a novel mechanism of action.
MATERIALS AND METHODS
Reagents, cell lines, and compounds.
The following reagents were obtained from the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: human interleukin-2 (IL-2) (from Maurice Gately, Hoffmann-La Roche Inc.) (37), pNL4-3 (from Malcolm Martin) (38), p210-13 ΔVpu plasmid (from Ronald Desrosiers) (39), pHEF-VSVG from Lung-Ji Chang (40), TZM-bl cells (from John C. Kappes and Xiaoyun Wu) (41), and human anti-Env antibody 2G12 (from Hermann Katinger) (42).
CEM-GXR25 (here termed CEM-GXR) is a CD4+ T cell line containing an HIV-1 long terminal repeat (LTR)-driven green fluorescent protein (GFP) reporter construct that allows the identification of infected cells by flow cytometry (43). These cells were maintained in R10+ medium (RPMI 1640 with HEPES and l-glutamine [Lonza], 10% fetal calf serum [FCS], 100 U of penicillin/ml, and 100 μg of streptomycin/ml [Sigma]). Peripheral blood mononuclear cells (PBMC) from healthy HIV-negative subjects were purchased commercially (StemCell Technologies) and maintained in R10+ medium supplemented with 100 U/ml human IL-2. HEK-293T cells were purchased commercially (Lenti-X 293T; Clontech Laboratories) and maintained in D10+ medium (Dulbecco's modified Eagle medium with 4.5 g/liter glucose and l-glutamine [Lonza] supplemented with 10% FCS, 100 U penicillin/ml, and 100 μg streptomycin/ml [Sigma]).
SM111, SM113, and BIT-225 were synthesized as reported previously (35, 36). For each compound, a 100 mM stock solution was prepared in dimethyl sulfoxide (DMSO), aliquoted, and stored at −20°C until use. Azidothymidine, efavirenz, indinavir, and HMA were obtained from Sigma. Raltegravir was kindly provided by Peter Cheung (BC Centre for Excellence in HIV/AIDS, Vancouver, Canada).
Virus construction.
HIV-1ΔVpu was generated by digesting p210-13 ΔVpu with EcoRI and NheI, and the resulting DNA fragment was ligated into wild-type pNL4.3 plasmid digested with the same enzymes. Vpu and Env mutations were introduced into the same EcoRI/NheI region of pNL4.3 by site-directed mutagenesis using standard overlap PCR extension methods. All strains and mutations were validated by DNA sequencing. Viral stocks were generated by transfection of HEK-293T cells using Lipofectamine 3000 (Life Technologies). Viruses pseudotyped with the vesicular stomatitis virus G protein (VSV-G) were produced by cotransfection of HEK-293T cells with pNL4.3 and pHEF-VSVG. At 48 h posttransfection, cell-free culture supernatants were collected and stored at −80°C until use. Viral titers were determined using CEM-GXR cells as described previously (43).
Recombinant HIV-1NL4.3 strains encoding major patient-derived drug resistance mutations to protease inhibitors (PI-RS), nucleoside reverse transcriptase inhibitors (NRTI-RS), nonnucleoside reverse transcriptase inhibitors (NNRTI-RS), and integrase inhibitors (INI-RS) were constructed from patient-derived HIV-1 pol plasma RNA sequences using a homologous recombination procedure as described previously (44). Cultures were monitored daily by flow cytometry (Guava easyCyte 8HT; Millipore). Once GFP expression (denoting HIV-1 infection) was detected in >15% of reporter cells, cell-free culture supernatants were harvested and frozen at −80°C. The HIV-1 pol gene for all viral stocks was confirmed by DNA sequencing. Written informed consent was provided by participants enrolled at the Massachusetts General Hospital (Boston, USA) or the BC Centre for Excellence in HIV/AIDS (Vancouver, Canada) by following protocols approved by the Research Ethics Board.
Cell viability.
The cytotoxicity of SM111 and control compounds in the absence of HIV-1 infection was evaluated in CEM-GXR cells and PBMC. Briefly, 1.5 × 105 CEM-GXR cells or PBMC that had been activated with 5 μg/ml of phytohemagglutinin (PHA) 3 days earlier were cultured in the presence of 0.1% DMSO containing compounds at various concentrations for 6 days. Half of the culture was removed for analysis of cell viability every 2 days; the remainder was replenished with fresh culture medium with or without compounds to maintain the desired concentrations. Cell viability was measured every 2 days using the Guava ViaCount assay (Millipore), which employs two dyes of different fluorescent wavelengths and membrane permeabilities to discriminate viable from nonviable populations. At day 6, an ATP luminescent viability assay (ViaLight Plus; Lonza) was also employed in accordance with the manufacturer's instructions. The cytotoxic effects of SM111 in the presence of HIV-1 were investigated on the final day of replication assays in GXR-CEM cells and PBMC by use of the same methods.
Viral replication.
CEM-GXR cells were infected with HIV-1 stocks at a multiplicity of infection (MOI) of 0.003 in triplicate unless otherwise stated. Cells were washed at 16 h postinfection and resuspended in R20+ medium (RPMI 1640 with HEPES and l-glutamine, 20% FCS, 100 U of penicillin/ml, and 100 μg of streptomycin/ml) containing SM111 or control compounds. Viral spread was monitored daily by measuring the percent cellular GFP expression by flow cytometry. Viral replication capacity (vRC) was calculated as described previously (45). Briefly, for each virus, the natural-log slope of the percentage of GFP-positive (GFP+) cells was calculated during the exponential phase of viral spread (days 3 to 6). This value was then divided by the mean rate of spread of wild-type or no-drug-treated HIV-1 to generate a normalized measure of the replication capacity. A vRC of 1.0 indicates a rate of viral spread that was equal to that of the control, while vRC values of <1.0 indicate inhibition of viral spread. vRCs are calculated from the average replication rates of at least three independent experiments.
The ability of SM111 to inhibit HIV-1 replication was also assessed in primary PBMC from healthy donors. Cells were activated with 5 μg/ml PHA and infected 3 days later with viral stocks at an MOI of 0.003. At 6 h postinfection, cells were washed and resuspended in R10+ medium supplemented with 100 U/ml of IL-2 in the presence of 0.1% DMSO plus SM111 or a control compound for an additional 6 days. The accumulation of p24Gag was quantified in culture supernatants at days 3 and 6 using an enzyme-linked immunosorbent assay (ELISA; Xpress Bio), and the increase in p24Gag values was used as the measure of viral replication.
Outgrowth and sequencing of SM111-resistant HIV-1 strains.
To isolate SM111-resistant HIV-1 strains, 15 cultures of CEM-GXR cells were infected with HIV-1NL4.3 at an MOI of 0.003, followed by washing 16 h later to remove the viral inoculum. Fourteen cultures were resuspended in R20+ medium containing 100 μM SM111, and one culture was maintained as a no-drug control. Viral spread was monitored daily by measuring the percentage of infected (GFP+) cells using flow cytometry. Supernatant was collected from two SM111-treated cultures that exhibited viral outgrowth above 15%. HIV-1 RNA was extracted from both cultures using PureLink Total RNA kits (Invitrogen), and the entire HIV-1 protein coding region was amplified by nested reverse transcription (RT)-PCR using gene-specific primers (sequences available on request). The resulting amplicons were directly sequenced using a 3130xl automated DNA sequencer (Applied Biosystems, Inc.). Chromatograms were analyzed using Sequencher v5.0 (Genecodes).
Vpu-mediated CD4 and tetherin downregulation.
For transfection assays, a CD4+ CEM T cell line was electroporated with pSELECT-GFPzeo plasmids (InvivoGen) encoding HIV-1NL4.3, Vpu mutants, or empty vector (ΔVpu) control. At 24 h postelectroporation, cells were stained with allophycocyanin (APC)-conjugated CD4 (BD Biosciences) and phycoerythrin (PE)-conjugated tetherin antibodies (anti-CD317; BioLegend). The cell surface expression of these proteins was measured using flow cytometry, and the mean fluorescence intensity (MFI) of CD4 and tetherin staining was determined.
For HIV-1 infection assays, CEM-GXR cells were infected with VSV-G-pseudotyped HIV-1NL4.3 or Vpu mutant viruses at an MOI of 0.3. Cells were washed 16 h postinfection and resuspended in culture medium for an additional 48 h, after which CD4 or tetherin expression was measured by flow cytometry as described above. To assess receptor downregulation, the MFI of CD4 or tetherin among GFP+ (HIV-1-infected) cells was divided by the corresponding MFI for GFP− (uninfected) cells.
To assess the effects of SM111 on cell surface CD4 and tetherin expression in the absence of HIV-1 infection, CEM-GXR cells were cultured in R10+ medium with or without SM111 at various concentrations. Cells were collected after 48 h posttreatment, and cell surface CD4 and tetherin were measured using flow cytometry as described above.
Viral egress, infectivity, and envelope staining.
CEM-GXR cells were infected with VSV-G-pseudotyped HIV-1NL4.3 or Vpu mutant viruses at an MOI of 0.3. Cells were washed 16 h postinfection and resuspended in medium with or without SM111 at defined concentrations for 48 h. Cell-free supernatants were quantified for HIV-1 p24Gag protein using ELISA (Xpress Bio), and aliquots were frozen for subsequent experiments to assess virion infectivity (see below). Env expression on the surface of infected CEM-GXR cells was measured using human anti-Env antibody 2G12 (42), followed by a goat anti-human antibody conjugated to Alexa Fluor-647 (Invitrogen). Virion infectivity was determined by exposing 104 TZM-bl cells to culture supernatant corresponding to 0.5 ng p24Gag for 48 h, followed by chemiluminescence detection as described previously (41).
Intracellular and extracellular p24Gag production.
HEK-293T cells were transfected with wild-type pNL4.3 using Lipofectamine 3000. Six hours posttransfection, lipid complexes were removed, and fresh D10+ medium with or without SM111 in 0.1% DMSO was added. Cells were collected 48 h later, stained for intracellular HIV-1 p24Gag using the PE-conjugated KC57 antibody (Beckman Coulter), and analyzed using flow cytometry. Supernatant p24Gag was quantified using ELISA as described above.
RESULTS
SM111 is an acylguanidine-containing compound with limited cytotoxicity.
Two acylguanidine-containing compounds, HMA (Fig. 1A) and BIT-225 (Fig. 1B), are reported to inhibit HIV-1 replication; however, both display substantial in vitro toxicity, particularly to T cells (5, 6, 34, 35). We therefore sought to design an acylguanidine-based derivative with a reduced cytotoxic component. We observed that the heterocyclic amino-pyrazine core of HMA resembles the heterocyclic core of methotrexate, which is also cytotoxic (46), and speculated that the high nitrogen density in these cores might be a cause. Based on this, we synthesized SM111, which resembles HMA except for the removal of one ring nitrogen and the exocyclic amino group, along with the chloro atom (Fig. 1C) (36). As the acylguanidine moiety is common to HMA, BIT-225, and other reported antiviral compounds (4), and is thus presumably required for anti-HIV-1 activity, we also assessed C-(6-azepan-1-yl-pyridin-3-yl)-methylamine (SM113; Fig. 1D) for use as a control analogue in which SM111's acylguanidine was replaced with a primary amine (36).
To assess the cytotoxicities of these compounds, we first compared the effects of SM111 and control SM113 to those of HMA and BIT-225 on CEM-GXR cells, a GFP reporter CD4+ T cell line that is permissive to HIV-1 (Fig. 2) (43). Substantial changes in cell viability were observed only after incubation with more than 100 μM SM111 for 6 days, as measured by flow cytometry based on differential labeling with cell-permeable dyes (50% cytotoxic concentration [CC50] for SM111 = 162 ± 5 μM) (Fig. 2A) or bioluminescent detection of cellular ATP (CC50 for SM111 = 128 ± 28 μM) (Fig. 2B). In contrast, the control compound SM113 was tolerated at concentrations up to 200 μM (Fig. 2A and B). Furthermore, CEM-GXR cells treated with SM111 for 6 days following infection with HIV-1 strain NL4.3 (HIV-1NL4.3) at a multiplicity of infection (MOI) of 0.003 tolerated SM111 at similar concentrations (CC50 = 161 ± 22 μM) (Fig. 2C), suggesting that viral infection did not influence the toxicity of SM111 in these cells. In contrast, substantial reductions in cell viability and cellular ATP were detected at concentrations above 18 μM for HMA and at all tested concentrations of BIT-225 (Fig. 2A and B), and cell death was visible by microscopy (data not shown), indicating that these compounds were more toxic to CEM-GXR cells than SM111.
FIG 2.

SM111 has minimal toxicity in comparison to that of reported acylguanidine compounds. (A to C) Effects of SM111 (blue), SM113 (green), HMA (purple), and BIT-225 (red) on viability of uninfected CEM-GXR cells (A) and cellular ATP in uninfected (B) and infected (C) CEM-GXR cells after 6 days. (D to F) Effects of compounds on viability of uninfected PBMC (D) and cellular ATP in uninfected (E) and infected (F) PBMC after 6 days. For panels A, B, D, and E, data are representative of three independent experiments performed in triplicate. For panels C and F, histograms and error bars represent means ± standard deviations (SD) calculated from three independent experiments (C) and using cells from three different donors (F), performed in triplicate.
In peripheral blood mononuclear cells (PBMC), we observed reduced cell viability after 6 days at concentrations above 100 μM SM111 by use of the ViaCount assay (CC50 = 112 ± 13 μM) (Fig. 2D) and above 50 μM SM111 by use of the ATP-based assay (CC50 = 96 ± 21 μM) (Fig. 2E). The control compound SM113 was tolerated at concentrations up to 200 μM. In PBMC infected with HIV-1NL4.3 for 6 days, substantial reductions in cellular ATP relative to that of no-drug controls were observed at SM111 concentrations above 100 μM (CC50 = 120 ± 30 μM) (Fig. 2F), suggesting that viral infection did not affect the toxicity of SM111 in PBMC. In contrast, both HMA and BIT-225 were highly toxic to PBMC after 6 days at all concentrations tested (Fig. 2D and E).
Taken together, these results demonstrate that SM111 has an improved cytotoxicity profile compared to those of HMA and BIT-225 in both a T cell line and PBMC. Based on these observations, we performed subsequent cell culture experiments with maximal doses of 100 μM for SM111 and control compound SM113 in CEM-GXR cells and 56 μM in PBMC.
SM111 inhibits in vitro HIV-1 replication.
We next assessed the impact of SM111 on viral replication capacity (vRC) using a multicycle GFP reporter T cell assay (43). CEM-GXR cells were infected with HIV-1NL4.3 at an MOI of 0.003 and then washed to remove viral inoculum 16 h later. Following this, viral spread in the absence or presence of SM111 was assessed by monitoring the proportion of GFP-expressing (HIV-1-infected) cells by flow cytometry. After 6 days, we observed that viral replication was substantially inhibited in the presence of 44 μM or higher concentrations of SM111 (Fig. 3A). To combine data across experiments, the slopes of viral spread were log transformed and normalized to wild-type HIV-1NL4.3 in the absence of SM111 to generate a quantitative measure of vRC as described previously (43). This revealed that inhibition of viral replication by SM111 was dose dependent, with a calculated EC50 of 55 ± 2 μM in this assay (Fig. 3B). In contrast, 100 μM control compound SM113 did not affect viral replication (Fig. 3A), indicating that the antiviral activity of SM111 required the acylguanidine moiety. Notably, no antiviral activity was observed when this T cell assay was used with HMA or BIT-225 at concentrations that did not also induce substantial cytotoxicity (data not shown).
FIG 3.

SM111 inhibits in vitro replication of HIV-1NL4.3. (A) CEM-GXR cells were infected with HIV-1NL4.3 for 24 h (MOI = 0.003), washed, and cultured in the absence or presence of SM111 or SM113 at the indicated concentrations. Cellular GFP, a marker of viral infection, was monitored on days 2 to 6 by flow cytometry. (B) Viral replicative capacity (vRC) of HIV-1NL4.3 in CEM-GXR cells in the presence of indicated SM111 concentrations, normalized to viral spread in the absence of compound. (C) Replication of HIV-1NL4.3 in CEM-GXR cells infected at MOIs of 0.03, 0.01, and 0.003 in the absence or presence of 100 μM SM111. (D) Schematic of experimental plan for results shown in panel E, indicating the times of addition and removal of 100 μM SM111 in CEM-GXR cells relative to infection with HIV-1NL4.3 (MOI = 0.003) at time zero. (E) vRCs of HIV-1NL4.3 in the absence or presence of 100 μM SM111, annotated (i to v) as shown in panel D. (F) PBMC were infected with HIV-1NL4.3 for 24 h (MOI = 0.003), washed, and cultured in the absence or presence of 50 μM SM111 or SM113. Supernatant levels of p24Gag were monitored by ELISA on days 3 and 6 postinfection. For panels A, C, and F, data are representative of three independent experiments performed in triplicate (from three different donors in panel F). For panels B and E, histograms and error bars represent means ± SD calculated from three independent experiments performed in triplicate.
To further investigate the effect of SM111 on HIV-1NL4.3, we next tested its ability to inhibit HIV replication in cells infected with virus at higher titers. We observed that 100 μM SM111 also blocked viral spread in cells infected at MOIs of 0.01 and 0.03 (Fig. 3C). We next performed time-of-addition studies in which CEM-GXR cells were continuously treated with 100 μM SM111 beginning either 24 h prior to infection, simultaneously with infection, or 24 h postinfection (Fig. 3D and E, conditions ii, iv, and v). No viral spread was observed under any of these conditions. However, complete HIV-1 inhibition required continuous exposure to SM111, as removal of SM111 at the time of infection reduced vRC by an average of only 33% ± 12% relative to untreated infected cells (Fig. 3D and E, condition i). Similarly, addition of SM111 at infection and removal of it 24 h later reduced vRC by an average of only 27% ± 12% relative to untreated infected cells (Fig. 3D and E, condition iii). These data support SM111 as a reversible inhibitor of viral replication.
SM111 also inhibited HIV-1 replication in PBMC based on production of extracellular p24Gag (Fig. 3F). In contrast, no antiviral activity was observed in control PBMC treated with SM113. While we note that these measurements in PBMC are not directly comparable to those for CEM-GXR cells due to the different assay readouts used (extracellular p24Gag production and Tat-induced GFP expression occur at different steps of viral replication), these results nevertheless indicate that SM111 can inhibit HIV-1NL4.3 replication substantially in both CEM-GXR cells and PBMC at concentrations that do not induce observable cytotoxicity.
SM111 inhibits HIV-1 strains resistant to licensed antiretroviral drugs.
To investigate SM111's mechanism of action, we next tested its ability to inhibit recombinant HIV-1NL4.3 strains carrying patient-derived sequences that harbor major resistance mutations to indinavir (protease inhibitor [PI]), azidothymidine (nucleoside reverse transcriptase inhibitor [NRTI]), efavirenz (nonnucleoside reverse transcriptase inhibitor [NNRTI]), or raltegravir (integrase inhibitor [INI]), designated, respectively, HIV-1PI-RS, HIV-1NRTI-RS, HIV-1NNRTI-RS, and HIV-1INI-RS (Fig. 4A) (35). As expected, wild-type HIV-1NL4.3 failed to replicate in the presence of 100 nM of each antiretroviral (ARV) (Fig. 4B), while each resistant HIV-1 strain replicated in CEM-GXR cells in the presence of its respective drug (Fig. 4C to F). Of note, SM111 inhibited the replication of all ARV-resistant strains by >95%, a level comparable to that of wild-type HIV-1NL4.3. These data indicate that HIV-1's mechanism of resistance to SM111 is distinct from those of existing ARVs, suggesting a unique mode of action for SM111.
FIG 4.

SM111 inhibits in vitro replication of antiretroviral-resistant HIV-1 strains in CEM-GXR cells. (A) Properties of drug-resistant viruses, including primary mutations and representative antiretroviral drugs. (B) Replication of HIV-1NL4.3 in the absence or presence of 100 nM antiretroviral drugs or 100 μM SM111. (C) Replication of HIV-1PI-RS in the absence or presence of 100 nM indinavir or 100 μM SM111. (D) Replication of HIV-1NRTI-RS in the absence or presence of 100 nM azidothymidine or 100 μM SM111. (E) Replication of HIV-1NNRTI-RS in the absence or presence 100 nM efavirenz or 100 μM SM111. (F) Replication of HIV-1INI-RS in the absence or presence 100 nM raltegravir or 100 μM SM111. For panels B to F, error bars represent means ± SD. Data are representative of two independent experiments performed in triplicate.
SM111 selects for mutations in Vpu and Env.
To identify viral factors that might modulate SM111 activity, we propagated HIV-1NL4.3 in the presence of SM111 to select resistance mutations. Of 14 CEM-GXR cultures infected with HIV-1NL4.3 in the presence of 100 μM SM111, two (denoted strain C and strain H) exhibited viral breakthrough by 9 days postinfection (Fig. 5), compared to outgrowth of control HIV-1NL4.3 at day 4 in the absence of SM111. Bulk (direct) Sanger sequencing of the culture supernatant indicated that strain C featured both a premature stop codon in place of tryptophan 22 (W22*) in Vpu's transmembrane domain and a highly dominant N136Y variant substitution in Env gp120 that is predicted to abolish a common N-linked glycosylation site (47). Neither wild-type Vpu nor wild-type Env sequences were detected. Strain H encoded a nonconservative substitution of isoleucine to arginine at Vpu codon 17 (I17R), which is also located in the transmembrane domain. No additional mutations were observed in either strain.
FIG 5.

SM111 selects for mutations in Vpu and Env. Culture of HIV-1NL4.3 in CEM-GXR cells in the presence of 100 μM SM111 is shown. Strains A to N represent 14 independent infected cultures. Viral outgrowth was observed in 2 of 14 (14.3%) cultures (strains C and H) by day 9. Mutations identified in each strain by full-genome viral sequencing are annotated.
Validation of SM111 resistance mutations.
To verify that the selected mutations conferred resistance to SM111, we reengineered them into wild-type HIV-1NL4.3 singly and in observed combinations, yielding strains HIV-1VpuW22*, HIV-1EnvN136Y, HIV-1VpuW22*/EnvN136Y, and HIV-1VpuI17R. Because we postulated that the Vpu W22* substitution would resemble a Vpu deletion mutation, we also generated a strain lacking the Vpu coding region (HIV-1ΔVpu) as an additional control (35, 39).
We began by assessing vRC of all mutant viruses in the absence of SM111 using CEM-GXR reporter cells. Notably, the spread of HIV-1VpuW22*/EnvN136Y, HIV-1VpuI17R, and HIV-1ΔVpu was enhanced by ∼20% compared to that of wild-type HIV-1NL4.3 (Fig. 6A to C). Similar enhancement of viral spread was also observed for the HIV-1VpuW22* single mutant (Fig. 6D). These results indicate that Vpu is dispensable for HIV-1 replication in this reporter cell assay and are consistent with studies demonstrating that deletion of Vpu can enhance viral cell-to-cell spread in immortalized CD4+ T cell lines (48). In contrast, replication of the HIV-1EnvN136Y single mutant was reduced 45% ± 11% relative to that of HIV-1NL4.3 (Fig. 6E), suggesting that loss of this N-linked glycosylation site in the absence of the concomitant Vpu mutation affected viral fitness in this cell line.
FIG 6.

Validation of SM111 resistance mutations in CEM-GXR cells and PBMC. (A to F) Replication of HIV-1VpuW22*/EnvN136Y (A), HIV-1VpuI17R (B), HIV-1ΔVpu (C), HIV-1VpuW22* (D), and HIV-1EnvN136Y (E) in CEM-GXR cells in comparison to HIV-1NL4.3 in the absence and presence of 100 μM SM111. (F) Percent inhibition of each virus strain at day 6 postinfection at various SM111 concentrations. (G to I) Replication of HIV-1VpuW22*/EnvN136Y (G), HIV-1VpuI17R (H), and HIV-1ΔVpu (I) in PBMC in comparison to HIV-1NL4.3 in the absence and presence of 56 μM SM111. Data are representative of three independent experiments performed in triplicate in CEM-GXR cells (A to F) or using PBMC from three different donors in triplicate (G to I). Data for HIV-1NL4.3 are shown repetitively to facilitate comparisons with replication of mutant strains. Error bars represent means ± SD.
As expected based on results shown in Fig. 3, replication of wild-type HIV-1NL4.3 was inhibited by >95% in the presence of 100 μM SM111; however, mutant viruses and HIV-1ΔVpu displayed differential levels of resistance to this compound in CEM-GXR cells (Fig. 6A to F). While replication of HIV-1VpuW22*/EnvN136Y and HIV-1VpuI17R was inhibited minimally (<20%) by SM111 compared to the same viruses in the absence of drug, 100 μM SM111 reduced replication of HIV-1ΔVpu by 72% ± 6% (Fig. 6C), HIV-1VpuW22* by 52% ± 19% (Fig. 6D), and HIV-1EnvN136Y single mutant by 64% ± 14% (Fig. 6E). Together these data suggest that the combination of Vpu W22* and Env N136Y mutations leads to improved resistance. These resistance profiles were consistent over multiple concentrations of SM111, with HIV-1VpuI17R and HIV-1VpuW22*/EnvN136Y exhibiting the highest resistance to SM111, followed by HIV-1VpuW22*, HIV-1ΔVpu, and HIV-1EnvN136Y (Fig. 6F).
We next assessed replication of HIV-1VpuW22*/EnvN136Y, HIV-1VpuI17R, andHIV-1ΔVpu strains in primary PBMC (Fig. 6G to I). In contrast to the enhanced viral spread observed in CEM-GXR cells, HIV-1VpuI17R, HIV-1VpuW22*/EnvN136Y, and HIV-1ΔVpu replicated less well than HIV-1NL4.3 in PBMC in the absence of SM111: extracellular p24Gag levels for these strains were 12 to 67% lower than that of wild-type HIV-1NL4.3 6 days postinfection. This result is consistent with Vpu's role in enhancing viral spread by counteracting tetherin in primary cells (19). Notably, in the presence of 56 μM SM111, wild-type HIV-1NL4.3 was inhibited 35% ± 3%, while HIV-1VpuW22*/EnvN136Y, HIV-1VpuI17R, and HIV-1ΔVpu strains all displayed levels of replication that were comparable (>90%) to their respective no-drug controls.
Taken together, these data confirm that the Vpu I17R mutation as well as the Vpu W22* mutation in combination with Env N136Y conferred resistance to SM111 in both CEM-GXR cells and PBMC. Notably, both mutant viruses appeared to be less sensitive to SM111 than either HIV-1VpuW22* or HIV-1ΔVpu in CEM-GXR cells, suggesting that their mechanisms of resistance are distinct from simple deletion of Vpu. Moreover, mutant viruses replicated substantially less well than wild-type HIV-1NL4.3 in PBMC, suggesting that the barrier to SM111 resistance may be higher in primary cells.
Vpu W22* and VPU I17R impair CD4 and tetherin downregulation.
Vpu's transmembrane domain is critical for antagonism of CD4 and tetherin (30, 49–52). To assess whether SM111-selected mutations in Vpu altered Vpu's ability to modulate these cellular proteins, we transfected an immortalized CD4+ T cell line (CEM) with dual expression plasmids encoding GFP and either wild-type or mutant Vpu sequences and then measured endogenous CD4 and tetherin cell surface expression 24 h later using flow cytometry (Fig. 7A). Downregulation of CD4 and tetherin was observed for wild-type Vpu; however, mutant sequences containing W22* or I17R were unable to downregulate either protein and appeared indistinguishable from those of empty vector (GFP-only) control.
FIG 7.

SM111-induced Vpu mutations abrogate CD4 and tetherin downregulation functions. (A) Representative flow cytometry data showing cell surface expression of CD4 (top) or tetherin (bottom) in CEM T cells transfected with a plasmid encoding GFP and wild-type (WT) or mutant Vpu sequences. Numbers in each plot indicate MFIs of CD4 or tetherin staining. Data are representative of three independent experiments. (B and C) CEM-GXR cells were infected with VSV-G-pseudotyped HIV-1NL4.3 or Vpu/Env mutant viruses as indicated, and cell surface expression of CD4 (B) or tetherin (C) was measured after 48 h by flow cytometry. Data are normalized to CD4 or tetherin expression on uninfected cells (dashed lines). (D) Uninfected CEM-GXR cells were treated with SM111, and cell surface expression of CD4, tetherin, or CXCR4 was measured after 2 days. Data are normalized to CD4, tetherin, or CXCR4 expression on untreated cells (dashed lines). For panels B to D, the means ± SD of results from three independent experiments are shown. *, P < 0.05 (two-tailed Student's t test).
To confirm these results, we infected CEM-GXR cells with HIV-1NL4.3, HIV-1VpuW22*/EnvN136Y, HIV-1VpuI17R, or HIV-1ΔVpu and measured CD4 and tetherin cell surface expression 48 h later by flow cytometry (Fig. 7B and C). As expected, infection with HIV-1NL4.3 resulted in lower CD4 expression (48% ± 6%, compared to the MFI of uninfected cells); however, the HIV-1ΔVpu strain also maintained the ability to downregulate CD4, presumably due to the dominant effects of intact Nef and Env proteins that also display this activity (53). Thus, while modest differences in CD4 downregulation were seen among the viruses tested, we could not assess the impact of Vpu mutations on this phenotype by this assay. On the other hand, while infection with HIV-1NL4.3 was associated with reduced expression of tetherin (43% ± 2%, compared to MFI of uninfected controls), HIV-1ΔVpu failed to display this activity (104% ± 5%). Of note, tetherin levels were elevated on cells infected with HIV-1VpuW22*/EnvN136Y (151% ± 8.2% compared to uninfected cells). This level was significantly higher than that of cells infected with HIV-1ΔVpu (P < 0.05), suggesting that the combination of Vpu W22* and Env N136Y mutations may stimulate greater intrinsic cellular responses (54). Together, these results indicate that SM111-selected mutations abrogated Vpu's ability to downregulate CD4 and to counteract tetherin, demonstrating that their SM111-resistant phenotypes come at a cost to these canonical Vpu activities.
Interestingly, treatment of uninfected CEM-GXR cells with SM111 led to a partial yet dose-dependent reduction of CD4 and tetherin cell surface expression (e.g., 87% ± 3% and 52% ± 6% residual staining of CD4 and tetherin, respectively, with 100 μM SM111, relative to MFI of untreated cells) (Fig. 7D), while cell surface expression of the HIV-1NL4.3 coreceptor CXCR4 was unaffected by up to 100 μM SM111. It is thus conceivable that SM111-mediated downregulation of HIV-1's entry receptor CD4 could contribute in part to its antiviral effect, though the modest reduction in cell surface expression of this molecule (only 13% at 100 μM SM111) is unlikely to fully account for the >95% viral inhibition observed at this concentration. The SM111-mediated reduction in tetherin expression is also unlikely to explain its antiviral effect, since downregulation of this host restriction factor would be expected to enhance, rather than inhibit, viral replication. However, since SM111-mediated tetherin downregulation alleviates pressure to maintain this key Vpu function, this observation might explain in part the selection of SM111-resistant strains lacking functional Vpu.
SM111 inhibits virion release.
The observation that SM111-mediated HIV-1 inhibition could be overcome by mutations in Vpu and/or Env is consistent with reports that another acylguanidine compound, BIT-225, targets HIV-1 egress (7). To further assess this, we measured the effects of SM111 on virion production by quantifying extracellular p24Gag levels in the supernatants of CEM-GXR cells infected with HIV-1NL4.3 or mutant viruses (Fig. 8A). As expected based on their inability to downregulate tetherin, extracellular p24Gag levels in the absence of SM111 were substantially lower for HIV-1VpuI17R (63 ± 13 ng/ml) and HIV-1ΔVpu (42 ± 10 ng/ml) than for wild-type HIV-1NL4.3 (162 ± 31 ng/ml). Of note, while p24Gag levels for the single mutant HIV-1VpuW22* were similar to those for HIV-1ΔVpu (data not shown), the double mutant HIV-1VpuW22*/EnvN136Y displayed an intermediate phenotype (141 ± 18 ng/ml), indicating that abolition of the N-linked glycosylation at Env codon 136 partially rescues the defect in virion release mediated by the loss of Vpu's tetherin downregulation function. The addition of SM111 resulted in a dose-dependent reduction in supernatant p24Gag levels for all viruses tested. These declines were most dramatic for HIV-1NL4.3 (50% reduction in virion release with addition of 100 μM SM111), though supernatant p24Gag levels nevertheless remained substantially higher for HIV-1NL4.3 than for mutant viruses at all SM111 concentrations tested. Together, these results suggest that SM111-mediated impairment of HIV-1 replication is due, at least in part, to impairment of virion release.
FIG 8.

SM111 inhibits particle release, virion infectivity, and Env surface expression. (A) Viral p24Gag levels were measured by ELISA in supernatants from CEM-GXR cells infected with VSV-G-pseudotyped HIV-1 strains following treatment with the indicated concentrations of SM111 at 48 h. (B) The infectivities of culture supernatants of strains shown in panel A were measured by incubating TZM-bl cells with 0.5 ng p24Gag. Results are shown as luminescence in arbitrary light units. (C) Env surface expression on CEM-GXR cells was detected by flow cytometry at 48 h postinfection in the absence or presence of SM111 at the indicated concentrations. MFI of infected cells minus background staining (based on uninfected cells) is shown. Data for all panels are representative of three independent experiments performed in triplicate.
SM111 resistance mutations enhance virion infectivity.
To assess SM111's impact on virion infectivity, we infected TZM-bl reporter cells with a defined viral inoculum (0.5 ng p24Gag harvested from the CEM-GXR cell supernatants [Fig. 8A]) and measured cellular luminescence (in arbitrary light units) 48 h later. We observed that treatment of CEM-GXR HIV-1NL4.3 producer cells with increasing concentrations of SM111 resulted in a dose-dependent reduction in infectivity; specifically, 100 μM SM111 inhibited HIV-1NL4.3 infectivity by ∼99% compared to that of no-drug controls (Fig. 8B). Importantly, in the absence of SM111, HIV-1VpuW22*/EnvN136Y, HIV-1VpuI17R, and HIV-1ΔVpu viruses exhibited >3-fold-higher infectivity than HIV-1NL4.3 (Fig. 8B). Moreover, in contrast with HIV-1NL4.3 and consistent with their resistant phenotypes in viral replication assays, infectivity of HIV-1VpuW22*/EnvN136Y, HIV-1VpuI17R, and to a lesser extent HIV-1ΔVpu was maintained at higher SM111 concentrations. While some SM111 would have been carried over in the inoculum, these concentrations (less than 10 μM in all cases) would have been insufficient to inhibit viral replication or to reduce viral infection using this single-cycle entry assay (Fig. 3A and data not shown). These results indicate that HIV-1VpuW22*/EnvN136Y and HIV-1VpuI17R, and to a lesser extent HIV-1ΔVpu, displayed enhanced virion infectivity in both the absence and presence of SM111, suggesting that this contributed to their relative resistance to the compound.
SM111 reduces cell surface expression of Env.
Vpu deletion is reported to enhance Env expression at the cell surface (55–59), which could account for the increase in virion infectivity observed for mutant strains. To investigate this, CEM-GXR reporter cells were infected with VSV-G-pseudotyped HIV-1NL4.3, HIV-1VpuW22*/EnvN136Y, HIV-1VpuI17R, or HIV-1ΔVpu at an MOI of 0.3 in the absence or presence of SM111. Robust HIV-1 infection (measured in terms of GFP+ cells) was detected in all cultures at all SM111 concentrations tested (data not shown). Forty-eight hours following infection, cell surface Env expression was measured in GFP+ (HIV-infected) versus GFP− (HIV-uninfected) cells by flow cytometry (Fig. 8C). In the absence of SM111, Env expression was elevated on the surface of cells infected with mutant strains HIV-1VpuW22*/EnvN136Y, HIV-1VpuI17R, and HIV-1ΔVpu compared to those infected with HIV-1NL4.3. Upon treatment with increasing concentrations of SM111, Env expression was reduced in a dose-dependent manner for all HIV-1 strains tested. However, while HIV-1NL4.3-infected cultures exhibited envelope staining equivalent to background (uninfected cells) at SM111 concentrations of ≥44 μM, Env expression on cells infected with HIV-1VpuW22*/EnvN136Y or HIV-1VpuI17R, and to a lesser extent HIV-1ΔVpu, was maintained well above background levels at all SM111 concentrations tested. Finally, while Env protein was also detected at low levels on GFP-negative bystander cells, consistent with Env shedding, this was not enhanced in the presence of SM111 (data not shown), suggesting that SM111 did not promote Env release. Together, these results indicate that SM111 blocked wild-type HIV-1 Env expression at the surface of infected CEM-GXR cells. Moreover, cells infected with HIV-1 encoding Vpu I17R or Vpu W22* plus Env N136Y mutations (or complete Vpu deletion, to a lesser degree) retained Env expression, which likely contributed to their ability to produce infectious progeny virions in the presence of this acylguanidine compound.
SM111 does not affect intracellular Gag protein expression.
While our results suggest that SM111 acts, at least in part, by impairing viral release and reducing virion infectivity, these experiments do not rule out effects of SM111 on viral protein synthesis. To investigate this directly, we transfected HEK-293T cells with wild-type pNL4.3 plasmid in the absence or presence of SM111. At 48 h posttransfection, de novo intracellular p24Gag expression was examined using flow cytometry and extracellular (supernatant) p24Gag production was quantified using ELISA. While the MFI of intracellular p24Gag was unaffected by SM111 (Fig. 9A and B), we observed that extracellular p24Gag was inhibited by SM111 in a dose-dependent manner (Fig. 9B). While some toxicity, as measured by cellular ATP levels, was observed in HEK-293T cells at higher SM111 concentrations (Fig. 9C), this was unlikely to account for SM111-mediated reductions in extracellular p24Gag. Taken together, these results suggest that SM111 did not substantially impair viral protein synthesis.
FIG 9.
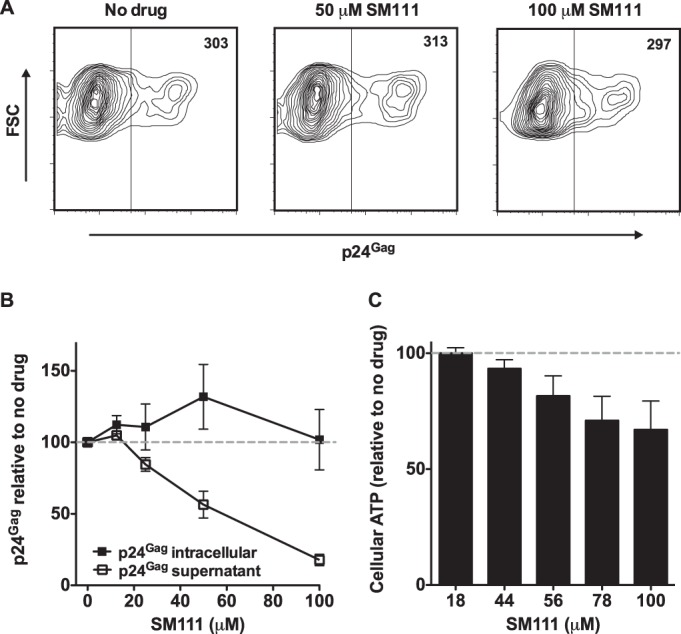
SM111 does not inhibit intracellular p24Gag expression. HEK-293T cells were transfected with wild-type pNL4.3 plasmid, followed by detection of intracellular p24Gag by flow cytometry and extracellular p24Gag by ELISA. (A) Representative flow cytometry data showing intracellular p24Gag expression in the absence or presence of 50 μM or 100 μM SM111. Mean fluorescence intensity (MFI) of p24Gag staining is indicated. (B) Dose-dependent effects of SM111 on intracellular and extracellular p24Gag. (C) Cellular ATP levels for untransfected cells treated with various concentrations of SM111. For panels B and C, means ± SD of results from three independent experiments are shown.
DISCUSSION
We have characterized a novel acylguanidine-based compound, 1-(2-(azepan-1-yl)nicotinoyl)guanidine (SM111), that inhibits replication of both wild-type and drug-resistant HIV-1 strains and that is substantially less cytotoxic than the previously reported acylguanidine compounds HMA and BIT-225. We observed that SM111 selected for mutations in Vpu's transmembrane domain, alone or in combination with a mutation at Env codon 136 that is predicted to abolish a common N-linked glycosylation site adjacent to the V1-V2 loop (47). We observed that SM111 reduced extracellular, but not intracellular, p24Gag production and that it impaired the infectivity of cell-free virions. Further, SM111-resistant strains displayed higher relative virion infectivity and higher surface Env expression than wild-type HIV-1NL4.3 in both the absence and presence of SM111. Taken together, our results suggest that SM111 acts at a late stage of HIV-1 replication by reducing both viral egress and virion infectivity (potentially by reducing Env incorporation into the virion) and that SM111-mediated inhibition of HIV-1 can be overcome in large part via selection of mutations that restore both cell surface Env expression and viral infectivity.
It is notable that both SM111-resistant strains selected in culture (HIV-1VpuW22*/EnvN136Y and HIV-1VpuI17R) encoded mutations in the transmembrane domain of Vpu. Although loss of Vpu is a common cell culture adaptation (48, 58), and Env mutations have been previously observed to compensate for Vpu deletion mutations in vitro (58, 60), several lines of evidence suggest that the observed Vpu mutations specifically conferred SM111 resistance. First, both HIV-1VpuW22*/EnvN136Y and HIV-1VpuI17R demonstrated greater resistance to 100 μM SM111 than HIV-1ΔVpu in CEM-GXR cells (although their levels of resistance were broadly comparable in PBMC, where all three mutants replicated substantially less well than HIV-1NL4.3 in the absence of SM111). Second, both SM111-resistant strains displayed enhanced Env surface expression in comparison to HIV-1ΔVpu, which in turn corresponded with improved virion infectivity, and in the case of HIV-1VpuW22*/EnvN136Y, partially restored p24Gag release. Notably however, both W22* and I17R mutants abolished Vpu's CD4 and tetherin downregulation activities, indicating that SM111 resistance comes at a cost to these canonical Vpu activities. The selection of resistant mutants lacking these key activities was likely facilitated, at least in part, by SM111's ability to downregulate cell surface tetherin directly, which alleviated pressure to maintain this key Vpu function.
While our data support SM111 as a novel inhibitor of HIV-1 egress pathways that involve Vpu and Env, specific mechanisms remain to be determined. In particular, it will be critical to determine whether SM111 acts directly on these viral proteins or indirectly through interactions with cellular proteins. Vpu is proposed by some groups to possess an ion channel (or viroporin) function, and both HMA and BIT-225 are reported to block Vpu-dependent ion currents (4, 5, 7). If true, SM111 would be anticipated to inhibit this Vpu activity, given its structural similarity to HMA and our previous evidence that SM111 is a potent inhibitor of the influenza A/M2 viroporin (36). Interestingly, Vpu I17 is proposed to project into the center of a pentameric Vpu pore domain (61). Moreover, molecular docking studies predict that both HMA and BIT-225 interact within the deep transmembrane pore of a Vpu pentamer with the acylguanidine moiety interacting near S23 (62–65), the residue that was replaced by a premature stop codon in W22*. However, the Vpu transmembrane domain is also reported to interfere with the subunits of endogenous ion channels such as TASK-1 and promote their degradation (66), so it is also possible that SM111 and/or the mutations identified here might affect the ability of Vpu to interact with TASK-1 or other cellular transmembrane proteins. These hypotheses warrant further investigation.
Acylguanidines like SM111 might also modulate the expression of viral and host cell factors through indirect effects on protein trafficking. This possibility is supported by studies using the acylguanidine-containing compound amiloride, which functions as a potassium-sparing diuretic by inhibiting the sodium-proton exchanger of the secretory pathway as well as other ion transporters, enzymes, and receptors that underlie protein export (8). Indeed, we observed that SM111 reduced the surface expression of tetherin (and to a lesser extent CD4) in uninfected CEM-GXR cells, though CXCR4 expression was unaffected. The former observation suggests that tetherin is not critically involved in SM111's antiviral activity, which is consistent with reports that BIT-225's anti-HIV-1 properties are independent of tetherin expression in Sup-T1 cell lines (34).
Vpu and Env are bicistronically translated from the same mRNA (57), and Vpu mutations are often associated with increased Env expression on the infected cell surface (57–59). Consistent with this, strains encoding Vpu I17R or Vpu W22* alongside Env N136Y displayed higher envelope staining in both the absence and presence of SM111. Interestingly, mutations in Vpu and Env have been reported to overcome antiviral pressures in other contexts, including cellular host restriction factors. For example, Env G367E enhanced the ability of mutations at Vpu codon Q35 to overcome restriction by IFITM1 (60). Deletion of Vpu also conferred resistance to the GBP5 restriction factor in macrophages, which also associated with higher Env expression (59). While mutation of Vpu and/or Env may be beneficial to in vitro replication in some instances, it is less clear whether these adaptations will enhance in vivo replication.
In conclusion, we have characterized a novel acylguanidine compound (SM111) that inhibits HIV-1 replication through impairment of viral release and reduction of virion infectivity. The latter appears to be accomplished, at least in part, by blocking the expression of Env on the infected cell surface, which in turn might result in the release of poorly infectious progeny virions lacking the viral surface protein. In the CEM-GXR T cell line, resistance to SM111 occurred through mutations in Vpu and Env that increased Env surface expression and thus likely promoted virion infectivity. We note that SM111's efficacy on HIV-1NL4.3 (EC50s of ∼55 μM in CEM-GXR and ∼33 μM in PBMC) is insufficient for it to be used directly as a therapeutic. Moreover, although no obvious effects on cell viability were observed in our studies at the concentrations tested, SM111's low therapeutic index (CC50/EC50 = 2.3 to 3.6) prevents us from fully excluding the contribution of cytotoxicity to the antiviral activities observed. Optimized derivatives of SM111 are thus desired for future preclinical studies. Nevertheless, SM111 and similar compounds may allow more detailed pharmacological studies of Vpu's role in viral egress and provide opportunities to develop new therapies to treat HIV-1.
ACKNOWLEDGMENTS
We thank Raymond Andersen, Pouria H. Jalily, and Luping Yan (University of British Columbia) for chemistry technical assistance and Bruce Walker (Ragon Institute of MGH, MIT, and Harvard) and Richard Harrigan (British Columbia Centre for Excellence in HIV/AIDS) for providing HIV-1 sequences containing Pol resistance mutations.
P.M., I.T., S.C.M., A.S., K.C., N.N.K., B.B., J.R., A.F., Z.L.B., and M.A.B. designed and/or performed the experiments. P.M., I.T., D.F., Z.L.B., and M.A.B. analyzed the data and wrote the manuscript.
We declare that no competing interests exist.
Funding Statement
This study was supported by the following: the Canadian Institutes for Health Research (CIHR; operating grants MOP-93536 and HOP-115700 to Z.L.B. and M.A.B.); the Canadian HIV Cure Enterprise Team grant HIG-133050 from CIHR, in partnership with CANFAR and IAS (to Z.L.B. and M.A.B.); the Grand Challenges Canada Stars in Global Health (I.T.); the International Development Research Centre Small Grants Program (I.T., Z.L.B., and M.A.B.); the Canada-Sub Saharan Africa (CANSSA) HIV/AIDS Network, through funding provided by the Global Health Research Initiative, itself a collaborative partnership of IDRC, CIHR, and Global Affairs Canada (Z.L.B. and M.A.B.); CIHR postdoctoral fellowships (P.M. and J.R.) and a Michael Smith Foundation for Health Research (MSFHR) postdoctoral fellowship (P.M.); a CIHR undergraduate summer studentship award (N.N.K.); CIHR New Investigator and MSFHR Scholar Awards (Z.L.B.); a Canada Research Chair on Retroviral Entry (A.F.); and a Canada Research Chair in Viral Pathogenesis and Immunity (M.A.B.). Funding was also provided by a CIHR Industry-Partnered Collaborative Research operating grant (D.F.), which was cofunded by Cardiome Pharma Corp., Vancouver, Canada. The funding agencies and Cardiome Pharma Corp. played no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- 1.UNAIDS. 2013. UNAIDS report on the global AIDS epidemic. UNAIDS, Geneva, Switzerland. [Google Scholar]
- 2.Menendez-Arias L. 2013. Molecular basis of human immunodeficiency virus type 1 drug resistance: overview and recent developments. Antiviral Res 98:93–120. doi: 10.1016/j.antiviral.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Ammaranond P, Sanguansittianan S. 2012. Mechanism of HIV antiretroviral drugs progress toward drug resistance. Fund Clin Pharmacol 26:146–161. doi: 10.1111/j.1472-8206.2011.01009.x. [DOI] [PubMed] [Google Scholar]
- 4.Gazina EV, Petrou S. 2012. Viral targets of acylguanidines. Drug Discov Today 17:1039–1043. doi: 10.1016/j.drudis.2012.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ewart GD, Mills K, Cox GB, Gage PW. 2002. Amiloride derivatives block ion channel activity and enhancement of virus-like particle budding caused by HIV-1 protein Vpu. Eur Biophys J 31:26–35. doi: 10.1007/s002490100177. [DOI] [PubMed] [Google Scholar]
- 6.Ewart GD, Nasr N, Naif H, Cox GB, Cunningham AL, Gage PW. 2004. Potential new anti-human immunodeficiency virus type 1 compounds depress virus replication in cultured human macrophages. Antimicrob Agents Chemother 48:2325–2330. doi: 10.1128/AAC.48.6.2325-2330.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Khohury G, Ewart G, Luscombe C, Miller M, Wilkinson J. 2010. Antiviral efficacy of the novel compound BIT225 against HIV-1 release from human macrophages. Antimicrob Agents Chemother 54:835–845. doi: 10.1128/AAC.01308-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kleyman TR, Cragoe EJ. 1988. Amiloride and its analogs as tools in the study of ion transport. J Membr Biol 105:1–21. doi: 10.1007/BF01871102. [DOI] [PubMed] [Google Scholar]
- 9.Dubé M, Bego MG, Paquay C, Cohen ÉA. 2010. Modulation of HIV-1-host interaction: role of the Vpu accessory protein. Retrovirology 7:114. doi: 10.1186/1742-4690-7-114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matheson NJ, Sumner J, Wals K, Weekes MP, Vigan R, Weinelt J, Schindler M, Antrobus R, Costa AS, Frezza C, Clish DB, Neil SJ, Lehner PJ. 2015. Cell surface proteomic map of HIV infection reveals antagonism of amino acid metabolism by Vpu and Nef. Cell Host Microbe 18:409–423. doi: 10.1016/j.chom.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Strebel K. 2014. HIV-1 Vpu—an ion channel in search of a job. Biochim Biophys Acta 1838:1074–1081. doi: 10.1016/j.bbamem.2013.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Willey RL, Maldarelli F, Martin MA, Strebel K. 1992. Human immunodeficiency virus type 1 Vpu protein regulates the formation of intracellular gp160-CD4 complexes. J Virol 66:226–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Willey RL, Maldarelli F, Martin MA, Strebel K. 1992. Human immunodeficiency virus type 1 Vpu protein induces rapid degradation of CD4. J Virol 66:7193–7200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Geleziunas R, Bour S, Wainberg MA. 1994. Cell surface down-modulation of CD4 after infection by HIV-1. FASEB J 8:593–600. [DOI] [PubMed] [Google Scholar]
- 15.Margottin F, Bour SP, Durang H, Selig L, Benichou S, Richard V, Thomas D, Strebel K, Benarous R. 1998. A novel human WD protein, h-βTrCP, that interacts with HIV-1 Vpu connects CD4 to the ER degradation pathway through an F-box motif. Mol Cell 1:565–574. doi: 10.1016/S1097-2765(00)80056-8. [DOI] [PubMed] [Google Scholar]
- 16.Veillette M, Richard J, Pazgier M, Lewis GK, Parsons MS, Finzi A. 2016. Role of HIV-1 envelope glycoproteins conformation and accessory proteins on ADCC responses. Curr HIV Res 14:9–23. [DOI] [PubMed] [Google Scholar]
- 17.Veillette M, Désormeaux A, Medjahed H, Gharsallah NE, Coutu M, Baalwa J, Guan Y, Lewis G, Ferrari G, Hahn BH, Haynes BF, Robinson JE, Kaufmann DE, Bonsignori M, Sodroski J, Finzi A. 2014. Interaction with cellular CD4 exposes HIV-1 envelope epitopes targeted by antibody-dependent cell-mediated cytotoxicity. J Virol 88:2633–2644. doi: 10.1128/JVI.03230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pham TN, Lukhele S, Hajjar F, Routy JP, Cohen ÉA. 2014. HIV Nef and Vpu protect HIV-infected CD4+ T cells from antibody-mediated cell lysis through down-modulation of CD4 and BST2. Retrovirology 11:15. doi: 10.1186/1742-4690-11-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Neil SJD, Zang T, Bieniasz PD. 2008. Tetherin inhibits retrovirus release and is antagonized by HIV-1 Vpu. Nature 451:425–431. doi: 10.1038/nature06553. [DOI] [PubMed] [Google Scholar]
- 20.Van Damme N, Goff D, Katsura C, Jorgenson RL, Mitchell R, Johnson MC, Stephens EB, Guatelli J. 2008. The interferon-induced protein BST-2 restricts HIV-1 release and is downregulated from the cell surface by the viral Vpu protein. Cell Host Microbe 3:245–252. doi: 10.1016/j.chom.2008.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dubé M, Roy BB, Guiot-Guillain P, Binette J, Mercier J, Chiasson A, Cohen ÉA. 2010. Antagonism of tetherin restriction of HIV-1 release by Vpu involves binding and sequestration of the restriction factor in a perinuclear compartment. PLoS Pathog 6:e1000856. doi: 10.1371/journal.ppat.1000856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sauter D, Schindler M, Specht A, Landford WN, Münch J, Kim K-A, Votteler J, Schubert U, Bibollet-Ruche F, Keele BF, Takehisa J, Ogano Y, Ochsenbauer C, Kappes JC, Ayouba A, Peeters M, Learn GH, Shaw G, Sharp PM, Bieniasz P, Hahn BH, Hatziioannou T, Kirchhoff F. 2009. Tetherin-driven adaptation of Vpu and Nef function and the evolution of pandemic and nonpandemic HIV-1 strains. Cell Host Microbe 6:409–421. doi: 10.1016/j.chom.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fischer WB, Li LH, Mahato DR, Wang YT, Chen C. 2014. Viral channel proteins in intracellular protein-protein communication: Vpu of HIV-1, E5 of HPV16 and p7 of HCV. Biochim Biophys Acta 1838:1113–1121. doi: 10.1016/j.bbamem.2013.08.017. [DOI] [PubMed] [Google Scholar]
- 24.Ewart GD, Sutherland T, Gage PW, Cox GB. 1996. The Vpu protein of human immunodeficiency virus type 1 forms cation-selective ion channels. J Virol 70:7108–7115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schubert U, Ferrer-Montiel AV, Oblatt-Montal M, Henklein P, Strebel K, Montal M. 1996. Identification of an ion channel activity of the Vpu transmembrane domain and its involvement in the regulation of virus release from HIV-1-infected cells. FEBS Lett 398:12–18. doi: 10.1016/S0014-5793(96)01146-5. [DOI] [PubMed] [Google Scholar]
- 26.Marassi FM, Ma C, Gratkowski H, Straus SK, Strebel K, Oblatt-Montal M, Montal M, Opella SJ. 1999. Correlation of the structural and functional domains in the membrane protein Vpu from HIV-1. Proc Natl Acad Sci U S A 96:14336–14341. doi: 10.1073/pnas.96.25.14336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ma C, Marassi FM, Jones DH, Straus SK, Bour S, Strebel K, Schubert U, Oblatt-Montal M, Montal M, Opella SJ. 2002. Expression, purification, and activities of full-length and truncated versions of the integral membrane protein Vpu from HIV-1. Protein Sci 11:546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mehnert T, Rough A, Judge PJ, Lam YH, Fischer D, Watts A, Fischer WB. 2008. Biophysical characterization of Vpu from HIV-1 suggests a channel-pore dualism. Proteins 70:1488–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sauter D, Schwarz S, Wang K, Zhang R, Sun B, Schwarz W. 2014. Genistein as antiviral drug against HIV ion channel. Planta Med 80:682–687. doi: 10.1055/s-0034-1368583. [DOI] [PubMed] [Google Scholar]
- 30.Bolduan S, Votteler J, Lodermeyer V, Greiner T, Koppensteiner H, Schindler M, Thiel G, Schubert U. 2011. Ion channel activity of HIV-1 Vpu is dispensable for counteraction of CD317. Virology 416:75–85. doi: 10.1016/j.virol.2011.04.009. [DOI] [PubMed] [Google Scholar]
- 31.Lamb RA, Pinto LH. 1997. Do Vpu and Vpr of human immunodeficiency virus type 1 and NB of influenza B virus have ion channel activities in the viral life cycles? Virology 229:1–11. doi: 10.1006/viro.1997.8451. [DOI] [PubMed] [Google Scholar]
- 32.Coady MJ, Daniel NG, Tiganos E, Allain B, Friborg J, Lapointe JY, Cohen ÉA. 1998. Effects of Vpu expression on Xenopus oocyte membrane conductance. Virology 244:39–49. doi: 10.1006/viro.1998.9087. [DOI] [PubMed] [Google Scholar]
- 33.Wilkinson J, Ewart G, Luscombe C, McBride K, Ratanasuwan W, Miller M, Murphy RL. 2016. A phase 1b/2a study of the safety, pharmacokinetics and antiviral activity of BIT225 in patients with HIV-1 infection. J Antimicrob Chemother 71:731–738. doi: 10.1093/jac/dkv389. [DOI] [PubMed] [Google Scholar]
- 34.Kuhl BD, Cheng V, Donahue DA, Sloan RD, Liang C, Wilkinson J, Wainberg MA. 2011. The HIV-1 Vpu viroporin inhibitor BIT225 does not affect Vpu-mediated tetherin antagonism. PLoS One 6:e27660. doi: 10.1371/journal.pone.0027660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tietjen I, Ntie-Kang F, Mwimanzi P, Onguéné PA, Scull MA, Idowu TO, Ogundaini AO, Meva'a LM, Abegaz BM, Rice CM, Andrae-Marobela K, Brockman MA, Brumme ZL, Fedida D. 2015. Screening of the pan-African Natural Product Library identifies ixoratannin A-2 and boldine as novel HIV-1 inhibitors. PLoS One 10:e0121099. doi: 10.1371/journal.pone.0121099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jalily PH, Eldstrom J, Miller SC, Kwan DC, Tai SHS, Chou D, Niikura M, Tietjen I, Fedida D. 2016. Mechanisms of action of novel influenza A/M2 viroporin inhibitors derived from hexamethylene amiloride. Mol Pharmacol 90:80–95. doi: 10.1124/mol.115.102731. [DOI] [PubMed] [Google Scholar]
- 37.Lahm HW, Stein S. 1985. Characterization of recombinant human IL-2 with micromethods. J Chromatogr 326:357–361. doi: 10.1016/S0021-9673(01)87461-6. [DOI] [PubMed] [Google Scholar]
- 38.Adachi A, Gendelman HE, Koenig S, Folks T, Willey R, Rabson A, Martin MA. 1986. Production of acquired immunodeficiency syndrome-associated retrovirus in human and nonhuman cells transfected with an infectious molecular clone. J Virol 59:284–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gibbs JS, Regier DA, Desrosiers RC. 1994. Construction and in vitro properties of HIV-1 mutants with deletions in “nonessential” genes. AIDS Res Hum Retroviruses 10:343–350. doi: 10.1089/aid.1994.10.343. [DOI] [PubMed] [Google Scholar]
- 40.Chang L-J, Urlacher V, Iwakuma T, Cui Y, Zucali J. 1999. Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Gene Ther 6:715–728. doi: 10.1038/sj.gt.3300895. [DOI] [PubMed] [Google Scholar]
- 41.Wei X, Decker JM, Liu H, Zhang Z, Arani RB, Kilby M, Saag MS, Wu X, Shaw GM, Kappes JC. 2002. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob Agents Chemother 46:1896–1905. doi: 10.1128/AAC.46.6.1896-1905.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trkola A, Purtscher M, Muster T, Ballaun C, Buchacher A, Sullivan N, Srinivasin K, Sodroski J, Moore JP, Katinger H. 1996. Human monoclonal antibody 2G12 defines a distinctive neutralization epitope on the gp120 glycoprotein of human immunodeficiency virus type 1. J Virol 70:1100–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Brockman MA, Tanzi GO, Walker BD, Allen TM. 2006. Use of a novel GFP reporter cell line to examine replication capacity of CXCR4- and CCR5-tropic HIV-1 by flow cytometry. J Viral Methods 131:134–142. doi: 10.1016/j.jviromet.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 44.Brumme ZL, Li C, Miura T, Sela J, Rosato PC, Brumme CJ, Markle TJ, Martin E, Block BL, Trocha A, Kadie CM, Allen TM, Pereyra F, Heckerman D, Walker BD, Brockman MA. 2011. Reduced replication capacity of NL4.3 recombinant viruses encoding reverse transcriptase-integrase sequences from HIV-1 elite controllers. J Acquir Immune Defic Syndr 56:100–108. doi: 10.1097/QAI.0b013e3181fe9450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Brockman MA, Brumme ZL, Brumme CJ, Miura T, Sela J, Rosato PC, Kadie CM, Carlson JM, Markle TJ, Streeck H, Kelleher AD, Markowitz M, Jessen H, Rosenberg E, Altfeld M, Harrigan PR, Heckerman D, Walker BD, Allen TM. 2010. Early selection in Gag by protective HLA alleles contributes to reduced HIV-1 replication capacity that may be largely compensated for in chronic infection. J Virol 84:11937–11949. doi: 10.1128/JVI.01086-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang T, Weigt SS, Belperio JA, Lynch JP III. 2011. Immunosuppressive and cytotoxic therapy: pharmacology, toxicities, and monitoring. Semin Respir Crit Care Med 32:346–370. doi: 10.1055/s-0031-1279831. [DOI] [PubMed] [Google Scholar]
- 47.O'Rourke SM, Sutthent R, Phung P, Mesa KA, Frigon NL, To B, Horthongkham N, Limoli K, Wrin T, Berman PW. 2015. Glycans flanking the hypervariable connecting peptide between the A and B strands of the V1/V2 domain of HIV-1 gp120 confer resistance to antibodies that neutralize CRF01_AE viruses. PLoS One 10:e0119608. doi: 10.1371/journal.pone.0119608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gummuluru S, Kinsey CM, Emerman M. 2000. An in vitro rapid-turnover assay for human immunodeficiency virus type 1 replication selects for cell-to-cell spread of virus. J Virol 74:10882–10891. doi: 10.1128/JVI.74.23.10882-10891.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pickering S, Hue S, Kim EY, Reddy S, Wolinsky SM, Neil SJD. 2014. Preservation of tetherin and CD4 counter-activities in circulating Vpu alleles despite extensive sequence variation within HIV-1 infected individuals. PLoS Pathog 10:e1003895. doi: 10.1371/journal.ppat.1003895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vigan R, Neil SJD. 2010. Determinants of tetherin antagonism in the transmembrane domain of the human immunodeficiency virus type 1 Vpu protein. J Virol 84:12958–12970. doi: 10.1128/JVI.01699-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tiganos E, Friborg J, Allain B, Daniel NG, Yao XJ, Cohen ÉA. 1998. Structural and functional analysis of the membrane-spanning domain of the human immunodeficiency virus type 1 Vpu protein. Virology 251:96–107. doi: 10.1006/viro.1998.9368. [DOI] [PubMed] [Google Scholar]
- 52.Magadán JG, Bonifacino JS. 2012. Transmembrane domain determinants of CD4 downregulation by HIV-1 Vpu. J Virol 86:757–772. doi: 10.1128/JVI.05933-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chen BK, Gandhi RT, Baltimore D. 1996. CD4 down-modulation during infection of human T cells with human immunodeficiency virus type 1 involves independent activities of vpu, env, and nef. J Virol 70:6044–6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bego MG, Mercier J, Cohen ÉA. 2012. Virus-activated interferon regulatory factor 7 upregulates expression of the interferon-regulated BST2 gene independently of interferon signaling. J Virol 86:3513–3527. doi: 10.1128/JVI.06971-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Stephens EB, McCormick C, Pacyniak E, Griffin D, Pinson DM, Sun F, Nothnick W, Wong SW, Gunderson R, Berman NEJ, Singh DK. 2002. Deletion of the vpu sequences prior to the env in a simian-human immunodeficiency virus results in enhanced Env precursor synthesis but is less pathogenic for pig-tailed macaques. Virology 293:252–261. doi: 10.1006/viro.2001.1244. [DOI] [PubMed] [Google Scholar]
- 56.Arias JF, Heyer LN, von Bredow B, Weisgrau KL, Moldt B, Burton DR, Rakasz EG, Evans DT. 2014. Tetherin antagonism by vpu protects HIV-infected cells from antibody-dependent cell-mediated cytotoxicity. Proc Natl Acad Sci U S A 111:6425–6430. doi: 10.1073/pnas.1321507111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schwartz S, Felber BK, Fenyö EM, Pavlakis GN. 1990. Env and Vpu proteins of human immunodeficiency virus type 1 are produced from multiple bicistronic mRNAs. J Virol 64:5448–5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schubert U, Bour S, Willey RL, Strebel K. 1999. Regulation of virus release by the macrophage-tropic human immunodeficiency virus type 1 AD8 isolate is redundant and can be controlled by either Vpu or Env. J Virol 73:887–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Krapp C, Hotter D, Gawanbacht A, McLaren PJ, Kluge SF, Stürzel CM, Mack K, Reith E, Engelhart S, Ciuffi A, Hornung V, Sauter D, Telenti A, Kirchoff F. 2016. Guanylate binding protein (GBP) 5 is an interferon-inducible inhibitor of HIV-1 infectivity. Cell Host Microbe 19:504–514. doi: 10.1016/j.chom.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 60.Ding S, Pan Q, Liu SL, Liang C. 2014. HIV-1 mutates to evade IFITM1 restriction. Virology 454-455:11–24. doi: 10.1016/j.virol.2014.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lu JX, Sharpe S, Ghirlando R, Yau WM, Tycko R. 2010. Oligomerization state and supramolecular structure of the HIV-1 Vpu protein transmembrane segment in phospholipid bilayers. Protein Sci 19:1877–1896. doi: 10.1002/pro.474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Patargias G, Ewart G, Luscombe C, Fischer WB. 2010. Ligand-protein docking studies of potential HIV-1 drug compounds using the algorithm FlexX. Anal Bioanal Chem 396:2559–2563. doi: 10.1007/s00216-010-3498-x. [DOI] [PubMed] [Google Scholar]
- 63.Kim CG, Lemaitre V, Watts A, Fischer WB. 2006. Drug-protein interaction with Vpu from HIV-1: proposing binding sites for amiloride and one of its derivatives. Anal Bioanal Chem 386:2213–2217. doi: 10.1007/s00216-006-0832-4. [DOI] [PubMed] [Google Scholar]
- 64.Lemaitre V, Ali R, Kim CG, Watts A, Fischer WB. 2004. Interaction of amiloride and one of its derivatives with Vpu from HIV-1: a molecular dynamics simulation. FEBS Lett 563:75–81. doi: 10.1016/S0014-5793(04)00251-0. [DOI] [PubMed] [Google Scholar]
- 65.Rosenberg MR, Weaver LM, Casarotto MG. 2016. Probing interactions of Vpu from HIV-1 with amiloride-based compounds. Biochim Biophys Acta 1858:733–739. doi: 10.1016/j.bbamem.2015.12.028. [DOI] [PubMed] [Google Scholar]
- 66.Hsu K, Seharaseyon J, Dong P, Bour S, Marban E. 2004. Mutual functional destruction of HIV-1 vpu and host TASK-1 channel. Mol Cell 14:259–267. doi: 10.1016/S1097-2765(04)00183-2. [DOI] [PubMed] [Google Scholar]