

ABSTRACT
Cell culture systems reproducing virus replication can serve as unique models for the discovery of novel bioactive molecules. Here, using a hepatitis C virus (HCV) cell culture system, we identified neoechinulin B (NeoB), a fungus-derived compound, as an inhibitor of the liver X receptor (LXR). NeoB was initially identified by chemical screening as a compound that impeded the production of infectious HCV. Genome-wide transcriptome analysis and reporter assays revealed that NeoB specifically inhibits LXR-mediated transcription. NeoB was also shown to interact directly with LXRs. Analysis of structural analogs suggested that the molecular interaction of NeoB with LXR correlated with the capacity to inactivate LXR-mediated transcription and to modulate lipid metabolism in hepatocytes. Our data strongly suggested that NeoB is a novel LXR antagonist. Analysis using NeoB as a bioprobe revealed that LXRs support HCV replication: LXR inactivation resulted in dispersion of double-membrane vesicles, putative viral replication sites. Indeed, cells treated with NeoB showed decreased replicative permissiveness for poliovirus, which also replicates in double-membrane vesicles, but not for dengue virus, which replicates via a distinct membrane compartment. Together, our data suggest that LXR-mediated transcription regulates the formation of virus-associated membrane compartments. Significantly, inhibition of LXRs by NeoB enhanced the activity of all known classes of anti-HCV agents, and NeoB showed especially strong synergy when combined with interferon or an HCV NS5A inhibitor. Thus, our chemical genetics analysis demonstrates the utility of the HCV cell culture system for identifying novel bioactive molecules and characterizing the virus-host interaction machinery.
IMPORTANCE Hepatitis C virus (HCV) is highly dependent on host factors for efficient replication. In the present study, we used an HCV cell culture system to screen an uncharacterized chemical library. Our results identified neoechinulin B (NeoB) as a novel inhibitor of the liver X receptor (LXR). NeoB inhibited the induction of LXR-regulated genes and altered lipid metabolism. Intriguingly, our results indicated that LXRs are critical to the process of HCV replication: LXR inactivation by NeoB disrupted double-membrane vesicles, putative sites of viral replication. Moreover, NeoB augmented the antiviral activity of all known classes of currently approved anti-HCV agents without increasing cytotoxicity. Thus, our strategy directly links the identification of novel bioactive compounds to basic virology and the development of new antiviral agents.
INTRODUCTION
Natural products possess a wide range of structural and functional diversity, with many of them exhibiting drug-like properties (1–4). Thus, natural products have been a rich source of new drugs for treating many diseases, while also serving as probes for characterizing molecules and pathways critical for biological processes. Among compounds approved by the U.S. FDA from 1981 to 2010, approximately 34% of the total, and 47% of the anti-infective small molecules, are compounds derived from natural products or their analogs (3). Isolation and identification of bioactive compounds are among the most fundamental steps of drug development, necessitating the screening of compounds via cell-based, in vitro, and/or in silico assays. Models that permit the identification of both bioactivity and modes of action are limited in number and therefore especially need to be developed. In the present study, we employed a viral replication cell culture system to screen a natural product library for novel bioactivities. This cell culture-based screen provided several advantageous features, as we note here. First, virus replication, which depends on a wide variety of cellular processes, is an especially sensitive indicator of bioactivity (5). Second, the use of different virus cell culture systems permits the determination of the step(s) in the viral life cycle that is targeted by novel bioactivities (6). Third, the targets of bioactive compounds can be readily identified using the information of a panel of cellular factors known to be involved in viral replication (5, 7). In the present study, we used the hepatitis C virus cell culture (HCVcc) system to identify the bioactivity and target molecule of a fungus-derived natural product known as neoechinulin B (NeoB).
Chronic HCV infection affects approximately 170 million people worldwide. HCV infection is a major cause of liver cirrhosis and hepatocellular carcinoma and constitutes a significant public health problem. In addition to the anti-HCV treatment using pegylated alpha interferon (IFN-α) combination with ribavirin, newly approved direct-acting antivirals (DAAs) that directly target HCV-derived proteins, including NS3 protease, NS5A, and NS5B polymerase, significantly improve clinical outcomes of HCV-infected patients (8, 9). However, the problems of these DAAs include the huge cost and thus the low availability of drugs, especially in disadvantaged countries. Another approach to antiviral drug development is to target cellular factors that are essential for HCV propagation. This line of trials has yielded promising developments of cyclophilin inhibitors and microRNA-122 inhibitors, which are classified as so-called host-targeting antivirals (HTAs) (8, 9). So far, we have characterized the anti-HCV activity of cyclophilin inhibitors and the role of cyclophilin in HCV replication (10, 11). Despite this progress in the development of novel antivirals, new medications, notably the DAAs, are very expensive, making these drugs difficult to use in disadvantaged countries with a significant HCV-infected population. To eradicate HCV infection worldwide, new low-cost anti-HCV drugs are greatly needed. So far, searches have been made for drug development using natural products as lead compounds (2, 12–16). However, most of these studies have not been successful in clarifying the mode of action of the studied compounds, especially with regard to target molecules.
In this study, we prepared an in-house natural product library consisting of compounds isolated from fungal strains and screened this library in cell-based functional assays using the HCV cell culture system (2, 17). We identified NeoB as a fungus-derived small molecule with the strongest potency to inhibit the production of infectious HCV. Further analysis demonstrated that NeoB is a novel inhibitor of liver X receptors (LXRs). Using NeoB as a probe, LXRs were revealed to be critical for HCV replication in host cells: LXR inactivation reduced the formation of double-membrane vesicles (DMVs), where the HCV replication complex is proposed to form. Thus, the linking of the HCV cell culture system to a chemical genetic analysis permits the identification of a new bioactive molecule which is a valuable tool for cell biological research and may serve as a lead for the development of novel antiviral drugs.
MATERIALS AND METHODS
Cell culture.
Huh-7, Huh7.5.1, Huh7-25, LucNeo#2, HuSE2, and 293T cells, as well as primary human hepatocytes, were cultured as described previously (2, 18–20). Huh7.5.1 and LucNeo#2 cells were kindly provided by Francis Chisari at The Scripps Research Institute and Kunitada Shimotohno at National Center for Global Health and Medicine. Huh7-25 cells, an Huh-7 cell clone deficient for CD81 expression, as well as a human hepatoma Huh-7 and a human embryonic kidney 293T cell line, were described previously (2). HuSE2 cells are immortalized human primary hepatocytes that can support HCV infection (18). Primary human hepatocytes were isolated from PXB mice (PhoenixBio). For detecting SREBP1 protein, Huh-7 cells were precultured for 24 h in medium supplemented with 10% lipoprotein-deficient serum (Sigma-Aldrich) and 10 μM compactin (Wako) and then incubated for 48 h with compounds and compactin (21).
Natural product library.
Natural products were extracted from culture broths of fungal strains isolated from seaweeds, mosses, and other plants as described previously (2, 17). We prepared an in-house natural product library consisting of approximately 200 isolated compounds.
NeoB is a secondary metabolite of Aspergillus amstelodami (22). We used a preparation of NeoB isolated from the fungus in all the experiments described in this study.
HCV cell culture assay.
Infectious HCV was recovered from the culture supernatant of Huh-7 cells transfected with HCV RNA, as described previously (2). Huh7.5.1 cells infected with HCV at a multiplicity of infection (MOI) of 0.15 for 4 h were left untreated or treated with compounds for 72 h. The infectivity of HCV and the amount of HCV core protein in the culture supernatant were quantified by infectious focus formation assay and chemiluminescent enzyme immunoassay (Lumipulse II HCV core assay), respectively (2). HuSE2 cells were infected with HCV at an MOI of 1 for 4 h.
Reagents.
Dimethyl sulfoxide (DMSO), cyclosporine, acriflavine, lamivudine, troglitazone, TO-901317, all-trans-retinoic acid (ATRA), 4-hydroxytamoxifen, GW6471, and GSK0660 were purchased from Sigma. Wy14643 was purchased from Cayman Chemical. 17β-Estradiol and FH535 were purchased from Merck. Ro41-5253 and GW501516 were from Enzo Life Sciences. Bafilomycin A1 and 5CPPSS-50 were from Wako. IFN-α was obtained from Schering-Plough. Recombinant full-length LXRβ protein was purchased from OriGene. Telaprevir, sofosbuvir, and daclatasvir were purchased from Selleckchem.
Compound screening.
Huh7.5.1 cells were infected with HCV at a multiplicity of infection (MOI) of 0.15 for 4 h. After free virus was washed out, cells were treated with compounds at 10 μM in growth medium for 72 h. Infectivity of HCV in the resultant culture supernatant and cell viability were quantified by infectious focus formation assay and MTT [3-(4,5-dimethylthiazol-2-yl)2 2,5-diphenyl tetrazolium bromide] assay, respectively (2). Normalized infectivity was calculated as HCV infectivity divided by cell viability. Compounds that decreased the normalized infectivity to less than 40% were selected as primary hits and were further evaluated for data reproducibility and dose dependency (2). Compounds that decreased cell viability to less than 50% of that observed for nontreated cells were considered cytotoxic and eliminated from evaluation.
Immunoblot analysis.
Immunoblot analysis was performed as described previously (10). Mouse anti-HCV core protein (2H9), rabbit anti-NS5A, rabbit anti-ISG56 (Abcam), rabbit anti-MxA (Santa Cruz Biotechnology), anti-LXR (Abcam), rabbit anti-SCD-1 (Cell Signaling Technology), mouse anti-SREBP1 (BD Pharmingen), and mouse anti-β-actin (Sigma) antibodies were used as primary antibodies. The intensities of the bands were quantified with ImageQuant TL (GE Healthcare).
Immunofluorescence analysis.
Indirect immunofluorescence analysis was performed essentially as described previously using anti-HCV core protein antibody (2H9) as the primary antibody (2).
MTT assay.
The cell viability was quantified by using a Cell Proliferation Kit II XTT (Roche Diagnostics) as described previously (23).
HCVpp assay.
An HCV pseudoparticle (HCVpp) assay, which evaluates HCV envelope-dependent entry activity, was performed as described previously (2, 24). HCVpps were prepared with expression plasmids for HCV JFH-1 E1E2, murine leukemia virus Gag-Pol, and luciferase protein, which were kindly provided by Francois-Loic Cosset at L'Université de Lyon. Compounds were evaluated by treating cells for 2 h prior to infection and for 4 h during HCV infection.
HCV replicon assay.
An HCV replicon assay, which reproduces HCV translation and replication, was performed as described previously using an HCV JFH-1 subgenomic replicon (SGR) RNA (SGR-JFH1/Luc) or cells carrying an HCV NN (genotype-1b) subgenomic replicon RNA (LucNeo#2 cells) (2, 19). An RNA polymerase I (Pol I)-based HCV expression system (pHH-JFH1) was used as a plasmid-driven replicon (25). For evaluation of HCV translation, an HCV JFH-1 subgenomic replicon encoding a GND substitution at its polymerase active GDD motif in the NS5B region (SGR-JFH1/Luc GND) (2, 26) was used.
Transcriptome analysis.
Huh-7.5.1 cells were treated with compounds (0.2% DMSO and 20 μM NeoB) for 24 h, and total RNA was recovered. Total RNA (500 ng) was amplified using a total RNA amplification kit (Ambion, Carlsbad, CA). cRNA was hybridized to an Illumina Human HT-12, version 4, array (Illumina, San Diego, CA). Scanning of the chip was performed using Illumina BeadScan and BeadStudio software packages, and data were generated using BeadStudio (version 1.6). Three biological replicates were analyzed for the microarray experiments. Bioconductor packages limma and lumi combined with Illumina's Human HT-12, version 4.0, annotation set were used for differential expression analysis (27, 28). Expression levels were background corrected and quantile normalized using the neqc function of the limma package. Probes annotated as being low quality by HT-12, version 4.0, were removed. Nonresponsive probes with interquantile ranges less than or equal to 0.15 across all samples were removed as well, leaving 21,737 probes in the end. Thresholds for differential expression analysis were set at a log2 fold change of 1 and a minimum false discovery rate of 0.05. Gene ontology (GO) analysis was performed using the mean log P value (29).
Luciferase reporter assay.
Huh7-25 cells were transfected with a reporter plasmid carrying an interferon-sensitive response element (ISRE) and the binding elements for peroxisome proliferator-activated receptor (PPAR), liver X receptor (LXR), retinoic acid receptor (RAR), or estrogen receptor (ER) upstream of the firefly luciferase and a reporter carrying the herpes simplex virus thymidine kinase promoter upstream of the Renilla luciferase, with or without an expression plasmid for a nuclear hormone receptor (either PPARα, PPARβ/δ, PPARγ, RARα, LXRα, or LXRβ) along with the heterodimeric partner retinoid X receptor alpha (RXRα) or ERα (30–32). The cells were then either left untreated or treated with compounds in the presence or absence of their agonist for 48 h. The cells were lysed, and the luciferase activity was measured (23).
For quantification of Gaussia luciferase (Gluc) activity, Huh7-25 cells were transfected with the reporter vector carrying a stearoyl coenzyme A (CoA) desaturase-1 (SCD-1) gene promoter region upstream of the Gluc gene (GeneCopoeia), with or without the LXR expression plasmid, and then either left untreated or treated with NeoB for 72 h. Gluc activity was measured using a Secrete-Pair dual luminescence assay kit (GeneCopoeia) according to the manufacturer's protocol.
RT-PCR analysis.
Reverse transcription-PCR (RT-PCR) analysis was performed as described previously, using RNA prepared from Huh-7 cells treated for 48 h with NeoB or 5CPPSS-50 in the presence or absence of an LXR ligand, TO-901317 (10). The primers used in this study were 5′-GATCAAAGAGGAGCCAGTGC-3′ and 5′-TAGATGGTGGCTGCTGAGTG-3′ for SREBP-1c transcripts, 5′-CCTGCTGTACTTGGGGATCGGGAACG-3′ and 5′-CCAGCGCGGCAAACAGCACAAAG-3′ for ABCG1 transcripts, 5′-GCGGAGCCATGGATTGCAC-3′ and 5′-CTCTTCCTTGATACCAGGCCC-3′ for SCD-1 transcripts, and 5′-CCATGGAGAAGGCTGGGG-3′ and 5′-CAAAGTTGTCATGGATGACC-3′ for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) transcripts. The intensities of the bands were quantified with ImageJ software.
siRNA.
Small interfering RNAs (siRNAs) used in this study were the following: 5′-GGAUGCUAAUGAAACUGGU-3′ and 5′-GAACAGAUCCGGAAGAAGA-3′ (si-LXRα); 5′-AGCUAACAGCGGCUCAAGA-3′ and 5′-AGAUCGUGGACUUCGCUAA-3′ (si-LXRβ) (Applied Biosystems). For siRNA screening of putative LXR downstream genes, we used a mixture of four different siRNAs against each gene (siGENOME SMARTpool; Dharmacon). siRNAs used were those against SCD-1 (si-SCD-1; siGENOME SMARTpool [Dharmacon]) and randomized control siRNAs (siGENOME Nontargeting siRNA Pool 1; Dharmacon). Cells were transfected with siRNAs using Lipofectamine RNAiMAX (Life Technologies) or DharmaFECT 4 transfection reagent (Dharmacon) according to the manufacturers' protocols.
Surface plasmon resonance.
The kinetics of NeoA and NeoB binding to LXRα and LXRβ were analyzed using a Biacore 3000 instrument (GE Healthcare). LXRα and LXRβ were immobilized onto the surface of a CM5 sensor chip using an amine coupling kit (GE Healthcare). Different concentrations of NeoA and NeoB diluted with HBS-P (10 mM HEPES, 0.15 M NaCl, 0.005% surfactant P20) containing 5% DMSO were injected for 120 s at a flow rate of 20 μl/min at 25°C. The bulk effect of DMSO was subtracted using reference flow cells. Kinetic parameters were determined by analyzing the data using BIAevaluation, version 4.1, software (GE Healthcare).
Oil red O staining.
Huh7-25 and Huh7.5.1 cells, either uninfected or infected with HCV, and primary human hepatocytes were either left untreated or treated with 20 μM NeoB for 72 h in medium with or without 25 μM chloroquine to facilitate lipid accumulation. Cellular lipids were then evaluated with a steatosis assay kit (Cayman Chemical) according to the manufacturer's protocol.
Electron microscopic analysis.
Electron microscopic analysis was performed essentially as described previously (33). In brief, the cells were prefixed with 2.5% glutaraldehyde, 2% paraformaldehyde, and 0.1 M phosphate buffer (pH 7.4) for 2 h at room temperature, postfixed with 1% osmium tetroxide, and embedded in Epon. Ultrathin sections were stained with uranyl acetate and lead citrate and then examined with a transmission electron microscope (HT7700; Hitachi, Ltd., Japan). For the quantification data, we counted the cells that carried clear double- or multimembrane vesicles (DMVs or MMVs) over 200 observed cells and calculated the percentage of DMV- or MMV-positive cells.
DENV replication assay.
Huh-7 cells infected with dengue virus type 1 (DENV-1) at an MOI of 0.125 for 1 h were treated with compounds for 72 h. DENV RNA in the culture supernatant was quantified by real-time RT-PCR using a TaqMan Fast Virus 1-Step Master Mix (Applied Biosystems) with 5′-GAACATGGRACAAYTGCAACYAT-3′ and 5′-CCGTAGTCDGTCAGCTGTATTTCA-3′ as primers and 5′-ACACCTCAAGCTCC-3′ as a probe (34).
Poliovirus replication assay.
Poliovirus replication was assessed as described previously using a poliovirus replicon RNA carrying the luciferase gene (35).
Hepatitis B virus (HBV) replication assay.
HepG2.2.15 cells (23) were treated with compounds for 6 days in medium that was changed every 3 days. Compounds were supplemented in the fresh medium when the medium was changed. Intracellular DNA was recovered with a QIAamp DNA minikit (Qiagen), and HBV DNA was quantified by real-time PCR as described previously (23).
Synergy analysis.
To examine whether the effects of the drug combinations were synergistic, additive, or antagonistic, MacSynergy (kindly provided by Mark Prichard), a mathematical model based on Bliss independence theory, was conducted as described previously (2). The theoretical additive effects were compared to the actual experimental effects at various concentrations of the two compounds and were plotted as a three-dimensional differential surface. On the plot, additive effects show the zero plane at the z axis. Synergistic and antagonistic effects show peaks above and below this plane, respectively. The 95% confidence interval of the experimental results is shown.
Statistics.
Statistical significance was determined using Student's t tests. P values of less than 0.05 were considered statistically significant.
Accession number(s).
Microarray data were deposited in the Gene Expression Omnibus (GEO) database under accession number GSE63026.
RESULTS
Screening for natural products that suppress the production of infectious HCV.
We prepared an in-house natural product library consisting of approximately 200 isolated compounds derived from fungal secondary metabolites, as described in Materials and Methods (2, 17). We employed the infectious HCV cell culture (HCVcc) system, which reproduces the whole HCV life cycle (36–38), to identify bioactive natural organic compounds. Huh7.5.1 cells persistently infected with HCV were treated with compounds for 72 h, and the infectivity of HCV produced from these cells was quantified (2). In the chemical screening, compounds that reduced the infectivity of the HCV produced to less than 40% were regarded as primary hits, which were then validated by evaluating their reproducibility, dose dependency, and cytotoxicity. Neoechinulin B [(3Z)-3-[[2-(2-methylbut-3-en-2-yl)-1H-indol-3-yl]methylidene]-6-methylidenepiperazine-2,5-dione] (NeoB) (22) (Fig. 1A) was among the most potent compounds in reducing viral infectivity and was the focus of the following analyses.
FIG 1.
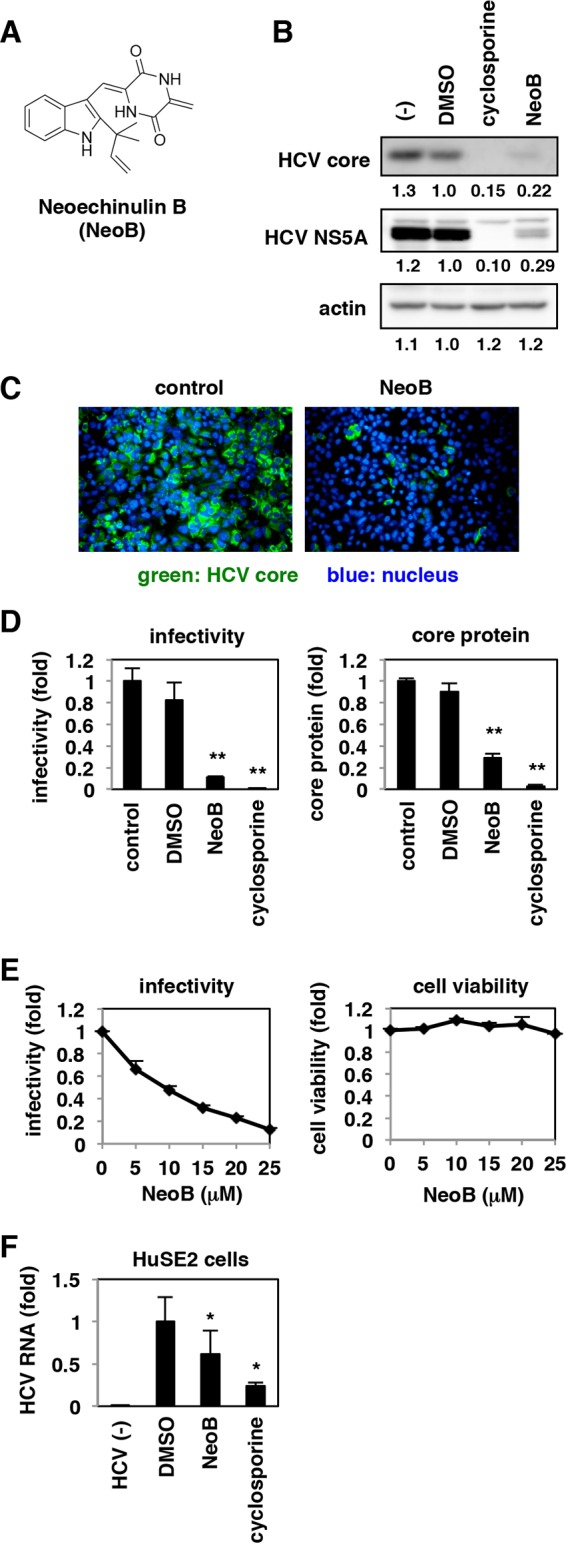
Neoechinulin B (Neo B) reduced the production of hepatitis C virus (HCV) from infected cells. (A) Chemical structure of NeoB. (B and C) Huh7.5.1 cells were infected with HCV at an MOI of 0.15 for 4 h and then incubated for 72 h with or without 0.5% DMSO, 20 μM NeoB, or 5 μg/ml cyclosporine. The resulting culture supernatants were inoculated onto naive Huh7.5.1 cells to detect intracellular HCV core protein, NS5A, and actin protein as an internal control, as indicated, at 48 h postinoculation by immunoblotting and immunofluorescence. The values below the panels indicate the relative band intensities (see Materials and Methods) for these proteins by setting those treated with DMSO as 1.0. (D) HCV infectivity and HCV core protein in the culture supernatant, prepared as described for panel B, were quantified by an infectious focus formation assay and chemiluminescent enzyme immunoassay, respectively, as described in Materials and Methods. (E) HCV infectivity was determined as described for panel D at various concentrations of NeoB (0 to 25 μM). Cell viability was quantified by MTT assay. (F) HuSE2 cells infected with HCV or uninfected were treated for 72 h with or without 0.5% DMSO, 10 μM NeoB, or 5 μg/ml cyclosporine. HCV RNA in the cells was quantified by real-time RT-PCR analysis. The data indicate the means of three independent experiments. Statistical analyses were performed by t test. *, P < 0.05; **, P < 0.01.
NeoB reduces the production of HCV.
To characterize the anti-HCV activity of compounds, Huh7.5.1 cells were infected with HCV at a multiplicity of infection (MOI) of 0.15 for 4 h and treated with the compounds for 72 h. Infectivity of HCV in the resulting culture supernatant was evaluated by reinoculating the supernatant into naive Huh7.5.1 cells and detecting HCV core and NS5A proteins by immunoblotting (Fig. 1B) and immunofluorescence (Fig. 1C). The infectivity of HCV produced from NeoB-treated Huh7.5.1 cells was significantly reduced, similar to results in cells treated with cyclosporine, a known HCV replication inhibitor (Fig. 1B and C). In quantitative analysis, HCV infectivity and core protein level were reduced to approximately 11% and 29% of the control levels, respectively, by NeoB treatment (Fig. 1D). This effect was dose dependent without significant cytotoxicity (Fig. 1E). The anti-HCV activity of NeoB was also observed in immortalized primary human hepatocytes, HuSE2 cells (Fig. 1F). Thus, NeoB decreased the production of infectious HCV particles.
NeoB reduces the levels of HCV RNA replication.
We next investigated which step in the HCV life cycle was inhibited by NeoB. The life cycle of HCV can be divided into the following steps: (i) early steps, attachment and entry; (ii) middle steps, translation and RNA replication; and (iii) late steps, assembly and release (5) (Fig. 2A). We first evaluated the effects of NeoB on the early steps using an HCV pseudoparticle (HCVpp) system that models the HCV envelope-mediated attachment and entry (see Materials and Methods) (24). As shown in Fig. 2B, NeoB had no significant effect on the luciferase activity driven by HCVpp infection, in contrast to bafilomycin A1, a known HCV entry inhibitor (39) (Fig. 2B, left). The middle steps of the HCV life cycle, including translation and RNA replication, were evaluated using an HCV subgenomic replicon (see Materials and Methods) (40). The replicon carrying a mutation in the NS5B polymerase active motif (replicon GND) that allows translation but not RNA replication (40) was used to evaluate HCV translation, without replication (Fig. 2A). NeoB was shown to reduce the activity of the replication-competent replicon (Fig. 2B, middle) but not that of the replication-deficient replicon GND (Fig. 2B, right), suggesting that NeoB inhibits the RNA replication step of the HCV life cycle. IFN-α is known to suppress HCV replication through activation of IFN-sensitive response element (ISRE)-mediated transcription and the resultant induction of downstream IFN-stimulated genes (ISGs) (41–44). As shown in Fig. 2C, NeoB did not enhance the level of ISRE-mediated transcription in either the presence or absence of IFN-α (Fig. 2C, left) and had no significant effect on protein expression levels of ISGs, ISG56 and MxA (Fig. 2C, right). Thus, NeoB inhibits RNA replication in a manner independent of the IFN pathway.
FIG 2.
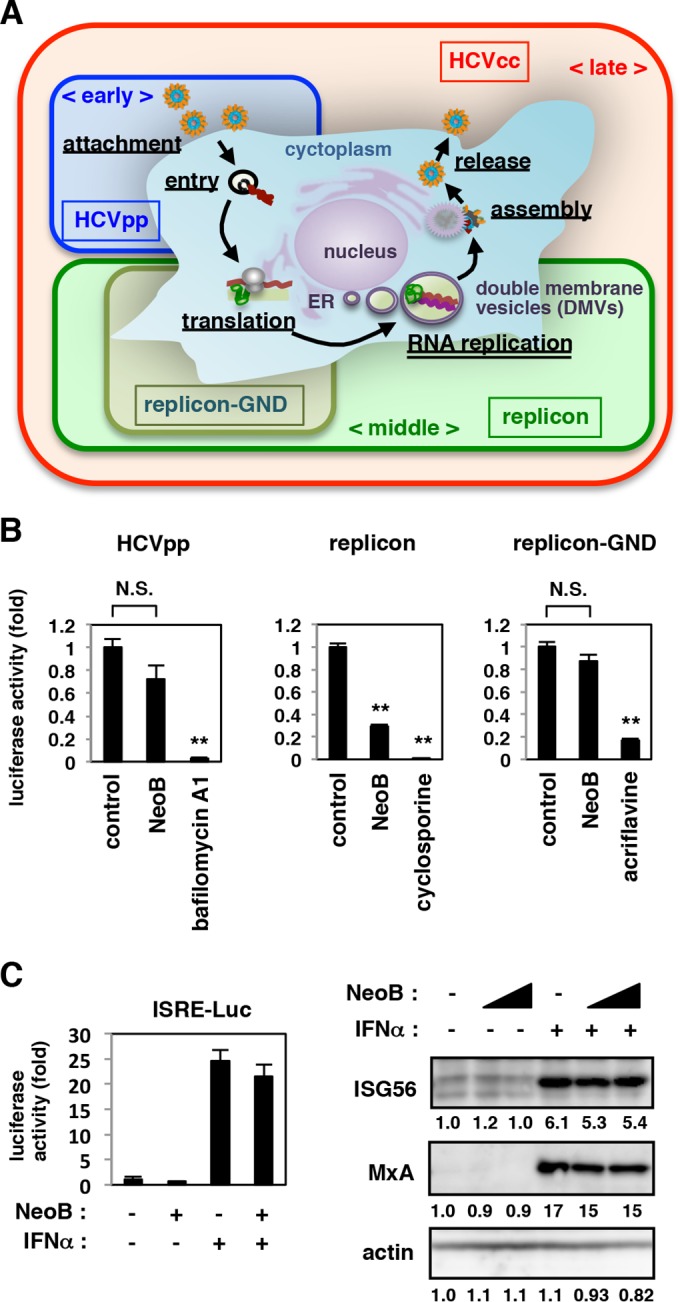
NeoB suppressed HCV RNA replication. (A) Schematic representation for the HCV life cycle and the assay systems used to evaluate each step of the life cycle. HCV pseudoparticle (HCVpp) assay permits evaluation of effects on the early step of the life cycle including attachment and entry. HCV replicon reproduces the middle step, consisting of translation and RNA replication. An HCV replicon carrying a GND substitution within the polymerase active motif of NS5B (replicon-GND) reproduces translation in the absence of RNA replication. The HCVcc assay permits evaluation of effects on the whole HCV life cycle including the late steps, assembly and release. Double-membrane vesicles (DMVs) are proposed to be sites for the formation of viral replication complexes. (B) For an HCVpp assay, Huh7.5.1 cells were left untreated or pretreated with 20 μM NeoB or 2 nM bafilomycin A1 (a known inhibitor of viral entry) for 2 h and then infected with HCVpp for 4 h in the presence or absence of compounds (left). After free virus and compounds were washed out, cells were incubated for an additional 72 h and then lysed for measurement of luciferase activity driven by HCVpp infection. Replicon assays evaluating translation/replication (middle) and translation (right) were also performed. Huh-7 cells were transfected with HCV JFH-1 subgenomic replicon RNA or the RNA encoding a GND substitution in the NS5B polymerase active motif GDD, and then cells were cultured in the presence or absence of 20 μM NeoB or 5 μg/ml cyclosporine (a known inhibitor of replication) for 48 h or in the presence or absence of 5 μM acriflavine (a known inhibitor of translation) for 4 h. Luciferase activity was quantified, and the relative values are indicated. (C) Effect of NeoB on the IFN signaling pathway. Huh7.5.1 cells transfected with a reporter plasmid carrying IFN-sensitive responsive elements (ISRE) upstream of the luciferase gene were treated with or without 20 μM NeoB and/or 100 IU/ml IFN-α for 24 h, and the luciferase activity was measured (left). HCV-infected Huh7.5.1 cells were treated with or without 20 and 30 μM NeoB and/or 100 IU/ml IFN-α to detect IFN-α downstream genes, ISG56 and MxA, and actin as an internal control by immunoblotting (right). Band intensities are indicated as described in the legend of Fig. 1B. **, P < 0.01, N.S., not significant.
NeoB inhibits the host transcription mediated by LXRs.
HCV RNA replication is regulated by numerous cellular factors and environments (6, 7, 45). To identify the target molecules of NeoB, we performed a transcriptome analysis that compared cellular gene expression between NeoB-treated and untreated Huh-7.5.1 cells. In NeoB-treated cells, 36 and 25 genes out of 17,509 genes showed more than 2-fold upregulation and downregulation, respectively (Table 1). Gene ontology (GO) enrichment analysis based on a mean log P value indicates that the GO terms such as drug metabolism, lipid metabolism, and hormone metabolism are significantly overrepresented among genes downregulated by NeoB (Table 2). Many of the genes involved in these metabolic processes are regulated by transcription factors, including nuclear hormone receptors (46, 47). Moreover, the HCV life cycle is known to be modulated by multiple nuclear hormone receptors such as peroxisome proliferator-activated receptor (PPAR), estrogen receptor (ER), and farnesoid X receptor (32, 48–50), leading us to question whether NeoB affected the function of nuclear hormone receptors that, in turn, modulate HCV replication.
TABLE 1.
Differentially expressed genes between NeoB-treated and DMSO-treated Huh-7.5.1 cellsa
| Gene group and genomic location | Gene symbol | Ensembl gene identification no.b | log2 fold change | Adjusted P value |
|---|---|---|---|---|
| Genes upregulated in NeoB-treated cells | ||||
| chr2:220074601:220074650:− | ABCB6 | ENSG00000198925 | 1.27 | 8.63E−06 |
| chr14:23080980:23081029:+ | ABHD4 | ENSG00000100439 | 1.28 | 7.40E−05 |
| chr3:64501536:64501585:− | ADAMTS9 | ENSG00000163638 | 1.04 | 1.23E−03 |
| chr7:16832449:16832498:− | AGR2 | ENSG00000106541 | 1.08 | 4.50E−03 |
| chr7:134223746:134223769:+, chr7:134225799:134225824:+ | AKR1B10 | ENSG00000198074 | 1.69 | 5.02E−06 |
| chr10:5032031:5032080:− | AKR1C2 | ENSG00000151632 | 2.01 | 8.10E−07 |
| chr10:5242219:5242268:+ | AKR1C4 | ENSG00000187134 | 1.67 | 2.52E−06 |
| chr9:75784996:75785045:+ | ANXA1 | ENSG00000135046 | 1.26 | 1.41E−04 |
| chr11:27148940:27148989:+ | BBOX1 | ENSG00000129151 | 1.20 | 1.43E−04 |
| chr3:9908433:9908482:− | CIDEC | ENSG00000187288 | 1.82 | 8.78E−06 |
| chr11:111779543:111779592:− | CRYAB | ENSG00000109846 | 1.11 | 1.68E−03 |
| chr15:75012151:75012199:− | CYP1A1 | ENSG00000140465 | 3.87 | 2.87E−07 |
| chr2:38295101:38295150:− | CYP1B1 | ENSG00000138061 | 2.05 | 3.59E−05 |
| chr11:30886573:30886622:− | DCDC5 | ENSG00000170959 | 1.29 | 1.11E−03 |
| chr19:40354005:40354054:− | FCGBP | ENSG00000090920 | 1.63 | 1.22E−05 |
| chr10:95066558:95066607:− | MYOF | ENSG00000138119 | 1.43 | 5.92E−06 |
| chr10:95072831:95072880:− | MYOF | ENSG00000138119 | 1.31 | 5.78E−04 |
| chr15:59912059:59912108:+ | GCNT3 | ENSG00000140297 | 1.65 | 4.14E−06 |
| chr19:18499687:18499736:+ | GDF15 | ENSG00000130513 | 1.98 | 4.52E−07 |
| chr7:23314321:23314370:+ | GPNMB | ENSG00000136235 | 1.02 | 1.19E−03 |
| chr22:35789915:35789964:+ | HMOX1 | ENSG00000100292 | 1.43 | 3.90E−06 |
| chr9:110247501:110247550:− | KLF4 | ENSG00000136826 | 1.46 | 9.36E−05 |
| chr15:43823563:43823612:+ | MAP1A | ENSG00000166963 | 1.14 | 7.15E−03 |
| chr1:151040632:151040681:+ | MLLT11 | ENSG00000213190 | 1.01 | 1.49E−04 |
| chr10:95066645:95066694:− | MYOF | ENSG00000138119 | 1.39 | 7.40E−05 |
| chr16:69743411:69743460:− | NQO1 | ENSG00000181019 | 1.03 | 1.41E−04 |
| chr16:83999581:83999630:+ | OSGIN1 | ENSG00000140961 | 1.76 | 5.92E−06 |
| chr6:37142789:37142838:+ | PIM1 | ENSG00000137193 | 1.07 | 2.61E−04 |
| chr2:238483068:238483117:− | RAB17 | ENSG00000124839 | 1.38 | 8.78E−06 |
| chr4:25749247:25749296:− | SEL1L3 | ENSG00000091490 | 1.03 | 3.99E−05 |
| chr3:53925890:53925939:− | SELK | ENSG00000113811 | 1.90 | 1.07E−05 |
| chr11:2943785:2943786:+, chr11:2946240:2946287:+ | SLC22A18 | ENSG00000110628 | 1.34 | 1.65E−05 |
| chr11:62648437:62648486:+ | SLC3A2 | ENSG00000168003 | 1.10 | 7.99E−04 |
| chr4:139100417:139100466:− | SLC7A11 | ENSG00000151012 | 1.02 | 8.90E−04 |
| chr4:88903786:88903835:+ | SPP1 | ENSG00000118785 | 1.71 | 5.15E−06 |
| chr4:88902806:88902855:+ | SPP1 | ENSG00000118785 | 1.97 | 5.89E−05 |
| chr20:627643:627692:− | SRXN1 | ENSG00000234516 | 1.19 | 1.36E−04 |
| chr2:234669616:234669665:+ | UGT1A1 | ENSG00000241635 | 2.30 | 4.52E−07 |
| chr2:234600466:234600466:+, chr2:234602445:234602493:+ | UGT1A1 | ENSG00000167165 | 1.06 | 1.57E−04 |
| chrX:48549686:48549735:+ | WAS | ENSG00000015285 | 1.27 | 2.79E−05 |
| Genes that are downregulated in NeoB-treated cells | ||||
| chr4:100197724:100197773:− | ADH1A | ENSG00000187758 | −1.51 | 2.91E−06 |
| chr17:7952205:7952254:+ | ALOX15B | ENSG00000179593 | −1.12 | 9.36E−05 |
| chr7:127639957:127640006:+ | NAG8 | NA | −1.22 | 1.32E−02 |
| chr4:74736642:74736691:+ | CXCL1 | ENSG00000163739 | −1.16 | 3.81E−03 |
| chr4:76942608:76942657:− | CXCL10 | ENSG00000169245 | −1.53 | 2.37E−05 |
| chr10:94836994:94837043:+ | CYP26A1 | ENSG00000095596 | −1.51 | 6.65E−05 |
| chr7:99303110:99303159:− | CYP3A7 | ENSG00000160870 | −1.01 | 7.38E−04 |
| chr10:54076976:54077025:+ | DKK1 | ENSG00000107984 | −1.02 | 3.64E−02 |
| chr8:13072083:13072130:− | DLC1 | ENSG00000164741 | −1.19 | 2.66E−02 |
| chr6:46188715:46188764:− | RCAN2 | ENSG00000172348 | −1.42 | 5.78E−04 |
| chr4:155505393:155505442:− | FGA | ENSG00000171560 | −1.12 | 6.06E−05 |
| chr12:109715925:109715974:− | FOXN4 | ENSG00000139445 | −1.25 | 1.53E−05 |
| chr13:111546687:111546736:+ | NA | NA | −1.16 | 1.47E−02 |
| chr9:127422749:127422798:+ | AB074162 | NA | −1.09 | 2.91E−03 |
| chr4:47440113:47440162:+ | AK094850 | NA | −1.16 | 1.21E−03 |
| chr7:45952203:45952252:− | IGFBP3 | ENSG00000146674 | −1.03 | 1.36E−04 |
| chr5:135527232:135527281:− | LOC389332 | NA | −1.02 | 6.29E−04 |
| chr16:19422294:19422343:+ | TMC5 | ENSG00000103534 | −1.18 | 6.79E−04 |
| chr1:206280923:206280971:− | BF034544 | ENSG00000196533 | −1.50 | 1.07E−05 |
| chr9:34665320:34665369:− | LOC730098 | ENSG00000187186 | −1.02 | 7.50E−03 |
| chr9:103947413:103947462:+ | LPPR1 | ENSG00000148123 | −1.11 | 2.30E−04 |
| chr22:24126201:24126250:+ | MMP11 | ENSG00000099953 | −1.00 | 8.90E−04 |
| chr2:16086639:16086688:+ | MYCN | ENSG00000134323 | −1.11 | 2.26E−05 |
| chr2:16086313:16086362:+ | MYCN | ENSG00000134323 | −1.05 | 4.99E−04 |
| chr7:98258792:98258841:+ | NPTX2 | ENSG00000106236 | −1.24 | 8.63E−06 |
| chr16:67696578:67696627:+ | PARD6A | ENSG00000102981 | −1.03 | 1.57E−04 |
| chr12:54975763:54975812:− | PPP1R1A | ENSG00000135447 | −1.17 | 1.21E−03 |
| chrX:105277317:105277366:− | SERPINA7 | ENSG00000123561 | −1.12 | 1.87E−03 |
| chr6:134491070:134491119:− | SGK1 | ENSG00000118515 | −1.01 | 2.15E−04 |
| chr20:10279945:10279994:+ | SNAP25 | ENSG00000132639 | −1.48 | 1.39E−04 |
| chr20:10287988:10288037:+ | SNAP25 | ENSG00000132639 | −1.71 | 5.15E−06 |
Based on Illumina HT-12 v4.0 annotation (accessed March 2014; 27). Each row corresponds to an Illumina bead array probe. Multiple probes can be associated with a given gene; not all probes have associated gene annotations. NA, not available.
Ensembl annotations are from Illumina (now outdated).
TABLE 2.
Gene ontology enrichment analysis using mean log P value for biological process terms
| Total no. of genes in set | No. of tested genes in seta | Gene set statisticb | Gene set P valuec | Gene set description |
|---|---|---|---|---|
| 156 | 100 | 1.903920114 | 1.46E−28 | Xenobiotic metabolic process |
| 137 | 99 | 1.620075009 | 2.68E−12 | Response to nutrient |
| 41 | 24 | 2.680057789 | 1.21E−11 | Secondary metabolic process |
| 14 | 10 | 3.896239669 | 1.23E−10 | Glycoside metabolic process |
| 91 | 55 | 1.879901718 | 1.95E−10 | Terpenoid metabolic process |
| 8 | 6 | 5.132219556 | 3.30E−10 | Aminoglycoside antibiotic metabolic process |
| 8 | 6 | 5.132219556 | 3.30E−10 | Daunorubicin metabolic process |
| 8 | 6 | 5.132219556 | 3.30E−10 | Doxorubicin metabolic process |
| 80 | 48 | 1.962377737 | 4.01E−10 | Diterpenoid metabolic process |
| 110 | 69 | 1.72724906 | 4.74E−10 | Isoprenoid metabolic process |
| 60 | 42 | 2.044646234 | 6.84E−10 | Tetrapyrrole metabolic process |
| 9 | 7 | 4.598087516 | 7.69E−10 | Polyketide metabolic process |
| 12 | 7 | 4.582972329 | 8.76E−10 | Flavonoid metabolic process |
| 39 | 28 | 2.353678461 | 1.09E−09 | Porphyrin-containing compound metabolic process |
| 101 | 68 | 1.701032533 | 2.89E−09 | Unsaturated fatty acid metabolic process |
| 88 | 60 | 1.758042245 | 4.39E−09 | Icosanoid metabolic process |
| 88 | 60 | 1.758042245 | 4.39E−09 | Fatty acid derivative metabolic process |
| 76 | 46 | 1.912375696 | 8.92E−09 | Retinoid metabolic process |
| 94 | 67 | 1.668312943 | 2.07E−08 | Cellular hormone metabolic process |
| 10 | 9 | 3.494980169 | 1.01E−07 | Xenobiotic catabolic process |
| 35 | 22 | 2.346522543 | 1.86E−07 | Drug metabolic process |
| 21 | 14 | 2.747511772 | 3.61E−07 | Retinoic acid metabolic process |
| 23 | 17 | 2.498626389 | 7.22E−07 | Quinone metabolic process |
| 9 | 6 | 3.99034837 | 9.37E−07 | Omega-hydroxylase P450 pathway |
| 6 | 5 | 4.310759589 | 1.04E−06 | Porphyrin-containing compound catabolic process |
| 6 | 5 | 4.310759589 | 1.04E−06 | Tetrapyrrole catabolic process |
| 6 | 5 | 4.310759589 | 1.04E−06 | Heme catabolic process |
| 6 | 5 | 4.310759589 | 1.04E−06 | Pigment catabolic process |
| 67 | 46 | 1.756731978 | 1.40E−06 | Regulation of smooth muscle cell proliferation |
Tested genes are those in the gene set for which a gene statistic has been submitted.
Mean −log10 value of the tested genes in the gene set.
P value corresponding to the gene set statistic.
We performed reporter assays to examine the effect of NeoB on the transcriptional activity of specific nuclear hormone receptors, including PPARs, retinoic acid receptor (RAR), ER, and LXR, in uninfected Huh7-25 cells (Fig. 3A). NeoB did not have a significant effect on the transcriptional activity of PPARα, PPARβ/δ, PPARγ, RAR, or ER in either the presence or absence of receptor-specific agonists, in contrast to the transcriptional repression by known antagonists as positive controls (GW6471, GSK0660, FH535, Ro41-5253, and 4-hydroxytamoxifen) (Fig. 3A). Among these, NeoB decreased the reporter activity mediated by LXRα and LXRβ (Fig. 3A, panels f and g) without affecting the level of the LXR protein itself (Fig. 3A, panel h). Consistent with this observation, transcripts for LXR downstream genes, including ATP-binding cassette subfamily G member 1 (ABCG1) (Fig. 3B), sterol regulatory element-binding protein 1c (SREBP1c) (Fig. 3B; see also Fig. 5B), and stearoyl-CoA desaturase-1 (SCD-1) (see Fig. 7D), clearly decreased with NeoB treatment. The inhibition of LXRα and LXRβ upon NeoB treatment was also observed in HCV-infected Huh-7 cells and HuSE2 cells (Fig. 3C and D). The half-maximal inhibitory concentrations (IC50s) for inhibition of LXRα and LXRβ were 5.5 and 7.6 μM, respectively, in HuSE2 cells (Fig. 3D) although these values varied among cell types. These data suggest that NeoB specifically inhibits the transcriptional activity of LXRs.
FIG 3.

NeoB inhibited liver X receptor (LXR)-mediated transcription. (A) Transcription reporter assay. Huh7-25 cells were transfected with a reporter plasmid carrying binding elements for PPAR, RAR, ER, or LXR upstream of the firefly luciferase, a Renilla luciferase reporter construct, for internal control and the following expression plasmids for nuclear hormone receptors: PPARα/RXRα (a), PPARβ/δ/RXRα (b), PPARγ/RXRα (c), RARα/RXRα (d), ERα (e), LXRα/RXRα (f), and LXRβ/RXRα (g). The cells were treated with or without NeoB at 20 μM (2.2× 50% effective concentration; 0.76× 90% effective concentration of anti-HCV activity) or the indicated inhibitor of the respective nuclear receptor, as indicated (20 μM GW6471, 30 μM GSK0660, 10 μM FH535, 10 μM Ro41-5253, 20 μM 4-hydroxytamoxifen, or 30 μM 5CPPSS-50), together with or without their agonist (25 μM Wy14643, 10 μM GW501516, 30 μM troglitazone, 1 μM ATRA, 1 μM 17β-estradiol, or 30 μM TO-901317, as indicated). Following treatment, cells were lysed and assayed for luciferase activities. LXR and actin proteins in the cells treated with or without 20 μM NeoB (h) were detected by immunoblotting. (B) In Huh-7 cells treated with or without 20 μM NeoB or 30 μM 5CPPSS-50 in the presence or absence of 10 μM TO-901317, an LXR agonist, mRNAs for LXR downstream genes, sterol regulatory element binding protein 1c (SREBP1c) and ATP-binding cassette subfamily G member 1 (ABCG1), and GAPDH as an internal control were detected by RT-PCR. (C and D) HCV-infected Huh-7 cells or HuSE2 cells were transfected with a reporter plasmid carrying the LXR binding elements and the expression plasmid for LXRα or LXRβ and RXRα and then treated with or without 0.5% DMSO and 20 μM NeoB (C) or 2.5, 5, 10, and 20 μM NeoB (D). Cell lysates were assessed for luciferase activity as described in panel A. *, P < 0.05; **, P < 0.01; NS, not significant.
FIG 5.

NeoB reduced lipid accumulation in hepatocyte-derived cell lines. (A) Oil red O staining was performed as described in Materials and Methods with Huh7.5.1 and Huh7-25 cells treated with 0.2% DMSO (control; a and e), 20 μM NeoB (b and f), 20 μM NeoA (c and g), or 30 μM 5CPPSS-50 (d and h) for 72 h. (B) mRNAs for SREBP1c and GAPDH were detected in Huh-7.5.1 cells treated with or without the indicated compounds in the presence of 5 μM TO-901317 for 24 h. (C) Uncleaved and cleaved forms of SREBP1 and actin proteins were detected by immunoblotting using Huh-7 cells treated with or without NeoB (5, 10, and 20 μM) as described in Materials and Methods. The arrow indicates the cleaved SREBP1 protein. The values below the panels indicate the band intensities of cleaved SREBP1 and actin proteins. (D and E) Oil red O staining was performed on HCV-infected Huh7 cells or primary human hepatocytes treated with 0.2% DMSO (control; i, i-2, and l), 20 μM NeoB (j, j-2, and m), or 20 μM NeoA (k, k-2, and n) for 72 h. Panels i-2, j-2, and k-2 are higher-magnification images of the boxed areas shown in panels i, j, and k, respectively.
FIG 7.

NeoB reduced the formation of double-membrane vesicles. (A) HCV-infected (c to h) or uninfected (a and b) Huh7.5.1 cells treated with DMSO or NeoB were observed by electron microscopy. Images in panels b, d, and g show the boxed areas from panels a, c, and f, respectively, at higher magnification. Images in panels e and h show distinct fields from the respective samples. N, nucleus; LD, lipid droplets; DMV, double-membrane vesicle; MMV, multimembrane vesicle; SV, small vesicle. (B) The number of cells containing DMVs or MMVs was counted over the observed 200 cells, and the percentages for DMV- or MMV-positive cells are indicated. (C) In a dengue virus replication assay, Huh-7 cells infected with dengue virus type 1 were treated with or without NeoB or IFN-α as a positive control for 72 h, and viral RNA in the culture supernatant was quantified by real-time RT-PCR. For a poliovirus replication assay, Huh-7 cells pretreated with or without NeoB or guanidine as a positive control were transfected with a poliovirus replicon RNA and treated with or without compounds for 9 h to measure luciferase activity. Hepatitis B virus replication was evaluated in HepG2.2.15 cells treated with or without NeoB or lamivudine as a positive control for 6 days by quantifying viral DNA in culture supernatant by real-time PCR. *, P < 0.05; **, P < 0.01; NS, not significant.
Molecular interaction of NeoB with LXRs is correlated with the inhibition of LXR and the modulation of lipid metabolism.
A derivative analysis of NeoB revealed that a series of analogs, including neoechinulin A (NeoA), fumitremorgin C (FTMC), 3-indoleethanol (3-IET), and N-acetyltryptamine (N-ATTP), did not inhibit LXR activity (Fig. 4A and B). NeoA is the closest derivative among these, with a double bond in the diketopiperazine structure replaced by a single bond (Fig. 4A). Intriguingly, we found by surface plasmon resonance analysis that NeoB interacted with recombinant LXRα and LXRβ, with KD (equilibrium dissociation constant) values of 2.9 ± 1.4 and 12.5 ± 1.1 μM, respectively (Fig. 4C). In contrast, NeoA did not show any in vitro affinity to either LXRα or LXRβ (Fig. 4C), in agreement with the lack of an NeoA effect on the transcriptional activity of these receptors (Fig. 4B).
FIG 4.

Characterization of LXR antagonistic activity for NeoB analogs. (A) Chemical structures of NeoB analogs. (B) Effect of NeoB and its analogs on LXR-mediated transcriptional activity. Huh7-25 cells transfected with a reporter plasmid carrying the binding elements of LXR, an expression plasmid encoding LXRβ, and that encoding RXRα were treated with or without 20 μM NeoB or the indicated derivatives or 30 μM 5CPPSS-50 in the presence of TO-901317 as described in the legend of Fig. 3A. Relative luciferase activities are shown. (C) In vitro binding assay by surface plasmon resonance. The binding kinetics of NeoB or NeoA to recombinant LXRα and LXRβ were analyzed as described in Materials and Methods, using recombinant LXRα or LXRβ protein immobilized on a sensor chip and NeoA or NeoB as the analyte in surface plasmon resonance analysis. RU, resonance units; **, P < 0.01; NS, not significant.
LXRs are implicated in a wide variety of physiological events, including lipid metabolism, cholesterol metabolism, glucose homeostasis, inflammation, and neurological homeostasis (51–53). LXRs induce the expression of SREBP1c, a master regulator of lipid and cholesterol metabolism (51, 54), and mediate lipid accumulation in hepatocytes, as can be observed by oil red O staining (55, 56). Employing this methodology, we treated uninfected Huh7.5.1 and Huh7-25 cells with compounds and then stained them with oil red O to observe intracellular lipid accumulation. As shown in Fig. 5A, lipid staining was markedly reduced in both Huh7.5.1 and Huh7-25 hepatocyte-derived cell lines that were treated with 5CPPSS-50, a known pharmacological inhibitor of LXR (Fig. 5A, panels d and h). Treatment with NeoB similarly reduced lipid accumulation in these cells, but NeoA did not (Fig. 5A, panels b, c, f, and g). These phenotypes were accompanied by, and correlated with, the decreased accumulation of SREBP1c mRNA (Fig. 5B) and of the cleaved form of SREBP1 protein (Fig. 5C, arrow) (57). Similar observations were obtained in HCV-infected Huh-7 cells and primary human hepatocytes (Fig. 5D and E). We therefore concluded that NeoB is a novel LXR inhibitor that impairs LXR-mediated transcription, thereby modulating LXR-dependent cellular physiology.
LXR's role in supporting HCV replication.
We then examined whether the repression of LXR was related to the observed reduction in HCV replication by NeoB treatment. To date, the significance of LXRs in the regulation of the HCV life cycle has not been fully understood: one paper showed that LXR depletion resulted in a slight reduction in HCV RNA levels in the replicon system although no further analysis was presented (48); other studies have reported that TO-901317 and GW3965, known as LXR antagonists, modulate HCV entry (58, 59). We showed that NeoB decreased HCV replicon activity but that NeoB derivatives lacking LXR inhibitory activity did not affect HCV replication (Fig. 4B and 6A). LXR was inactivated by another antagonist, 5CPPSS-50, which also supported reduced levels of HCV replication without cytotoxicity (Fig. 6B). Similarly, knockdown of endogenous LXR in HCV-infected cells yielded decreased levels of HCV proteins such as HCV core and NS5A, consistent with decreased replication activity (Fig. 6C). Moreover, in cells overexpressing LXR and RXR, HCV replication levels were increased in a dose-dependent manner (Fig. 6D). The above results clearly suggest that LXR transcriptional activity supports efficient HCV replication and that dysregulation of LXR activity by antagonists results in decreased HCV replication in host cells.
FIG 6.
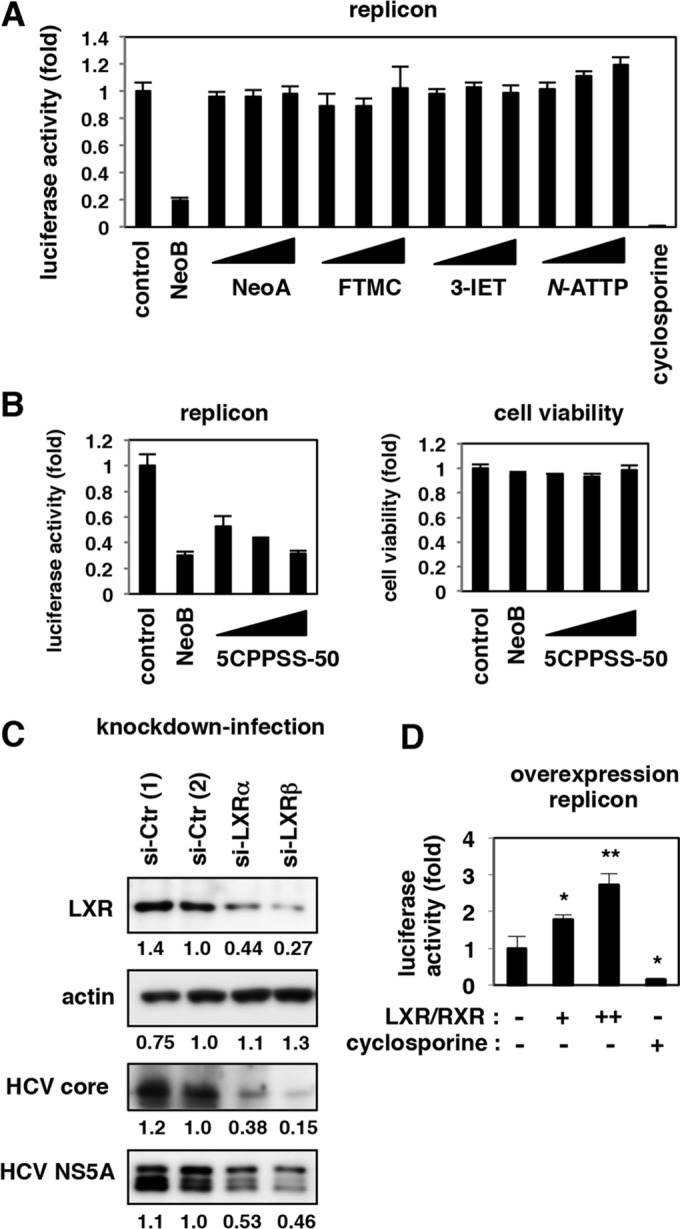
LXRs supported efficient HCV replication. (A and B) Huh7.5.1 cells transfected with the HCV replicon RNA were treated with or without 0.4% DMSO, 20 μM NeoB, and its derivatives (A) and with various concentrations of an LXR antagonist, 5CPPSS-50 (20, 30, and 40 μM). Luciferase activities were quantified as described in the legend of Fig. 2B. Cell viability was also measured as described in the legend of Fig. 1E. (C) HCV-infected Huh7.5.1 cells were transfected with randomized siRNAs [si-Ctr (1) and si-Ctr (2)] or siRNAs against LXRα (si-LXRα) and LXRβ (si-LXRβ) and subjected to immunoblotting to detect LXR, actin, HCV core, and HCV NS5A proteins in the cells. (D) Huh7.5.1 cells were transfected with an RNA Pol I-driven HCV replicon plasmid (see Materials and Methods) together with or without expression plasmids for both LXRβ and RXRα. The cells transfected with LXRβ/RXRα were also treated with 5 μM TO-901317 to activate LXR signaling. As a positive control, cyclosporine was added at 24 h after transfection. At 5 days posttransfection, the replication activity was determined by reporter assay. *, P < 0.05; **, P < 0.01.
LXR inactivation disrupts double-membrane vesicles, putative sites of viral replication.
We further examined the mechanisms whereby LXR mediates efficient HCV replication. As previously reported, electron microscopic analysis of HCV-infected cells showed remarkable accumulations of membrane compartments, known as double-membrane vesicles (DMVs) or multimembrane vesicles (MMVs), which are putative sites of the viral replication complex (60–64), while these membrane compartments were never seen in uninfected cells (Fig. 7A, compare panels a and b to panels c to e). However, upon inactivation of LXRs of HCV-infected cells by NeoB, DMVs and MMVs were severely dispersed, and small vesicles (SVs) were remarkably increased (Fig. 7A, compare panels c to e to panels f to h). The frequency of DMV/MMV-positive cells relative to the total number of cells was quantified to be significantly reduced by NeoB treatment (Fig. 7B). A similar observation was obtained in cells transfected with a mutant HCV RNA [JFH1(GND)] that produces HCV proteins but is deficient for viral replication (data not shown), suggesting that the NeoB effect was not the result of a decrease in HCV RNA replication but, rather, a direct effect on the machinery for producing and/or maintaining these membrane structures. In support of this, we observed that LXR inhibition also attenuated the efficient replication of DMV-associated viruses but not DMV-unrelated viruses. Specifically, poliovirus is known to replicate in the DMVs, while another plus-stranded RNA virus, dengue virus, replicates via a different type of membrane structure, the invaginated vesicles (64). Notably, NeoB reduced the replication level of poliovirus but not that of dengue virus (Fig. 7C, left and center panels). Replication of hepatitis B virus, a hepatotropic DNA virus that replicates independently of intracellular membrane compartments, was not affected by NeoB treatment (Fig. 7C, right). These data are in agreement with the observation that the antiviral effect of NeoB is specific for viruses that replicate in association with DMVs.
The observed effects of NeoB treatment were accompanied by the downregulation of stearoyl CoA desaturase-1 (SCD-1), a key enzyme in the synthesis of monounsaturated fatty acids from saturated fatty acids, thereby altering membrane fluidity (63, 65). Among 22 genes that were reported as downstream genes of LXR (51–53), SCD-1 was suggested to contribute to efficient HCV replication: knocking down endogenous SCD-1 levels reduced the replication of HCV in both replicon (Fig. 8A) and HCV-infected cells (Fig. 8B) without apparent cytotoxic effects (Fig. 8A and B, lower graphs). Actually, SCD-1 was transcriptionally regulated by LXRs (Fig. 8C and D). Overproduction of SCD-1 significantly rescued the reduction in HCV replication in LXR-depleted cells although it did not fully restore the replication level (Fig. 8E), suggesting that SCD-1 was at least one of the targets through which LXR supports HCV replication. Indeed, SCD-1 has been proposed to be important for the formation of DMVs (63, 65), but it has not been known which stimulus and signal regulate DMV formation. Considered together, our results support the possibility that LXR regulates the formation of DMVs at least in part through induction of SCD-1, thereby promoting the efficient replication of HCV.
FIG 8.

An LXR downstream gene, SCD-1, mediated efficient HCV replication. (A) LucNeo#2 cells treated with siRNAs against the indicated genes were tested for HCV replication activity as described in Materials and Methods. Cell viability was also measured by MTT assay. (B) HCV-infected Huh7.5.1 cells transfected with or without randomized siRNAs (si-Ctr) or an siRNA against SCD-1 (si-SCD-1) were subjected to detection for SCD-1, actin, HCV core, and NS5A proteins by immunoblotting. Cell viability was also measured as described for panel A. (C) Huh7.5.1 cells transfected with a reporter plasmid carrying the SCD-1 promoter together with or without expression plasmids for both LXRβ and RXRα were treated with or without NeoB to measure the luciferase activity. (D) Huh7.5.1 cells treated with or without NeoB in the presence or absence of TO-901317 for 24 h were detected for SCD-1 and GAPDH mRNAs. (E) LucNeo#2 cells transfected with or without siRNA against LXRβ and the expression plasmid encoding SCD-1 were assayed to detect luciferase activity at 72 h posttransfection. *, P < 0.05.
Synergistic antiviral profiles of NeoB in combination with known anti-HCV agents.
To explore the significance of NeoB-mediated LXR inhibition as an antiviral strategy, we investigated the effect of NeoB on the replication of different HCV genotypes, 1b (NN) and 2a (JFH-1) (19, 37). As shown in Fig. 9A, NeoB decreased the replication activity of both of these genotypes (Fig. 9A).
FIG 9.

NeoB had a multigenotypic anti-HCV effect and exhibited a synergistic anti-HCV activity when combined with known anti-HCV agents. (A) An HCV replicon assay was performed using replicons of genotypes 1b (NN) and 2a (JFH-1). (B to E) Cotreatment of NeoB with IFN-α, telaprevir (an HCV protease inhibitor), sofosbuvir (a polymerase inhibitor), or daclatasvir (an NS5A inhibitor). HCV-infected Huh7.5.1 cells were treated with the indicated concentrations and combinations of compounds for 72 h. HCV infectivity in the culture supernatant and cell viability were determined as described in the legend of Fig. 1E. The results were analyzed for a synergy plot as described in Materials and Methods. The three-dimensional plots show the difference between theoretical additive effects and the actual experimental effects provided by combination treatments. The zero plane across the z axis indicates the theoretical additive effects. Positive and negative values in the z axis indicate synergy and antagonism, respectively. *, P < 0.05; **, P < 0.01.
We then evaluated the anti-HCV activity of all the major classes of clinically available anti-HCV agents when combined with NeoB in HCV-infected cells: interferon (Fig. 9B), a protease inhibitor (telaprevir) (Fig. 9C), a polymerase inhibitor (sofosbuvir) (Fig. 9D), and an NS5A inhibitor (daclatasvir) (Fig. 9E). Supplementation with NeoB led to a greater reduction in HCV infectivity without a significant increase in cytotoxicity at any given concentration, compared with results achieved by these anti-HCV agents alone (Fig. 9B to E). Notably, synergy/antagonism analysis with the Bliss independence model showed that IFN-α and an NS5A inhibitor especially exerted strong synergistic anti-HCV effects in combination with NeoB at most of the tested doses (in this analysis, a peak above the zero plane in the z axis indicates a synergistic antiviral effect by cotreatment of two drugs, while a valley under the zero and zero plane itself designate antagonistic and additive antiviral effects, respectively) (Fig. 9B and E). Thus, NeoB is highly effective in treating multiple HCV genotypes and in multidrug treatment combinations with currently approved anti-HCV agents.
DISCUSSION
Identification of relevant bioactivities of natural products is a fundamental and critical step in linking natural source compounds to biological applications that can lead to development of new drugs. HCV is highly dependent on cellular factors for its efficient propagation, and many compounds that target cellular factors therefore inhibit the HCV life cycle (5, 7, 8). We here took advantage of this fact by using an HCV cell culture system to screen a natural product library for novel antiviral activities. This screen identified NeoB as an anti-HCV compound and demonstrated that this effect is mediated by inhibition of LXR. This study additionally revealed that LXRs regulate the formation of membrane structures that are associated with HCV replication. LXRs are known to be involved in a wide variety of physiological processes, including lipid metabolism, cholesterol metabolism, glucose homeostasis, inflammation, and neurological homeostasis, making it a potential drug target for diseases, including atherosclerosis, diabetes, hepatic steatosis, and Alzheimer's disease (51–53). The compound identified here, NeoB, was originally isolated from Aspergillus amstelodami (22). Although NeoA, a NeoB derivative (Fig. 4A), has been reported to show multiple physiological activities, including antioxidant and cytoprotective activities in neuronal cells and anti-inflammatory effects in macrophages (66, 67), there are no reports on the bioactivity of NeoB so far. In the present work, we demonstrated that NeoB, but not NeoA, interacted with LXRs and inhibited their transcriptional activities. While the use of LXR ligands for drug development has been an area of interest for over a decade (51–53), it will be interesting to observe in the future whether NeoB or related compounds affect LXR-mediated cellular functions other than those implicated in HCV replication. For instance, the present work demonstrates that NeoB modulates lipid accumulation in hepatocytes, independently of HCV infection (Fig. 5).
Our analysis using NeoB as a probe has clarified the significance of LXR in HCV replication (Fig. 10). Inhibition of LXR correlated with anti-HCV activity, suggesting that an LXR downstream gene(s) is involved in HCV replication. One such target gene is SCD-1 (Fig. 8) although other genes may also contribute to the modulation of HCV replication. Recent reports proposed that unsaturated fatty acids produced by SCD-1 are important for the formation and/or maintenance of the membrane structures used by the HCV replication complex (63, 65). However, the factor(s) that triggers or regulates the formation of the HCV replication site has remained poorly understood. Our results suggest that LXR inactivation and the resulting attenuation of LXR downstream gene induction disrupted the formation of the membrane compartments that are associated with HCV replication.
FIG 10.
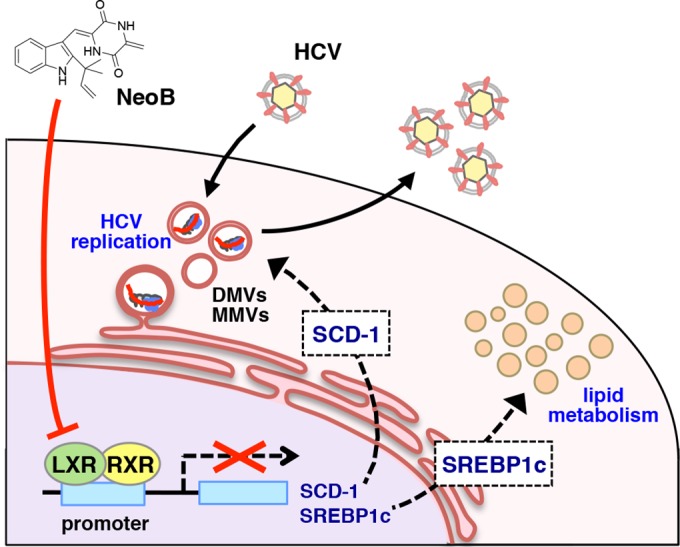
Schematic representation of the role of LXRs in mediating HCV replication and of an antagonizing effect of NeoB. The LXR/RXR heterodimer induces its downstream genes, including SCD-1 and SREBP1c. This transactivation creates a host cell environment that allows efficient HCV replication, including the formation or maintenance of double-membrane (DMV) or multimembrane (MMV) vesicles. SCD-1 is one of the LXR downstream genes that support efficient HCV replication. LXR also critically regulates lipid metabolism, a process that likely involves SREBP1c as a key LXR downstream gene. NeoB inhibits LXR-mediated transcription and thereby impairs the induction of these downstream genes. Thus, NeoB inhibition of LXR transcriptional activity modulates lipid accumulation while also reducing the host permissiveness to HCV replication in the absence of apparent cytotoxicity.
The primary strategy for developing anti-HCV drugs has been the targeting of virus-derived factors such as NS3 protease, NS5A, and NS5B polymerase (8, 9). However, since the huge cost of the resulting anti-HCV agents constitutes a significant social problem, the identification of new molecular targets for the development of additional antiviral agents is an appealing approach. To date, natural products reported to inhibit HCV replication/production include silymarin/silibinin, epigallocatechin gallate, ladanein, naringenin, and quercetin (12–16). However, while these compounds likely act upon cellular factors, their actual modes of action, structure-activity relationships, and especially their target molecules remain poorly defined. The identification of LXRs as a cellular target of NeoB raises the possibility that LXRs can serve as potential targets for the development of antiviral agents, not only against HCV but also against other DMV-associated viruses. Moreover, the fact that the LXR inhibitor identified here displays enhanced antiviral effects, including synergy, in combination with other antiviral drugs is a significant finding for drug development. In conclusion, viral cell culture systems are powerful tools for identifying unknown bioactivities of natural products and for linking such compounds to the development of new classes of antiviral agents.
ACKNOWLEDGMENTS
Huh7.5.1 and LucNeo#2 cells were kindly provided by Francis Chisari at The Scripps Research Institute and Kunitada Shimotohno at National Center for Global Health and Medicine, respectively. The expression plasmids for preparing HCVpp were generous gifts from Francois-Loic Cosset at L'Université de Lyon.
This study was supported by grants-in-aid from the following: the Ministry of Health, Labor, and Welfare, Japan; the Ministry of Education, Culture, Sports, Science, and Technology, Japan; Japan Society for the Promotion of Science (KAKENHI 26460565 and 26102747); Core Research for Evolutional Science and Technology, Japan Science and Technology Agency; a research grant from the ONO Medical Research Foundation; and Research Program on Hepatitis from the Japan Agency for Medical Research and Development.
Funding Statement
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
REFERENCES
- 1.Cragg GM, Newman DJ. 2013. Natural products: a continuing source of novel drug leads. Biochim Biophys Acta 1830:3670–3695. doi: 10.1016/j.bbagen.2013.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakajima S, Watashi K, Kamisuki S, Tsukuda S, Takemoto K, Matsuda M, Suzuki R, Aizaki H, Sugawara F, Wakita T. 2013. Specific inhibition of hepatitis C virus entry into host hepatocytes by fungi-derived sulochrin and its derivatives. Biochem Biophys Res Commun 440:515–520. doi: 10.1016/j.bbrc.2013.09.100. [DOI] [PubMed] [Google Scholar]
- 3.Newman DJ, Cragg GM. 2012. Natural products as sources of new drugs over the 30 years from 1981–2010. J Nat Prod 75:311–335. doi: 10.1021/np200906s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Osada H. 2010. Introduction of new tools for chemical biology research on microbial metabolites. Biosci Biotechnol Biochem 74:1135–1140. doi: 10.1271/bbb.100061. [DOI] [PubMed] [Google Scholar]
- 5.Scheel TK, Rice CM. 2013. Understanding the hepatitis C virus life cycle paves the way for highly effective therapies. Nat Med 19:837–849. doi: 10.1038/nm.3248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lohmann V, Bartenschlager R. 2014. On the history of hepatitis C virus cell culture systems. J Med Chem 57:1627–1642. doi: 10.1021/jm401401n. [DOI] [PubMed] [Google Scholar]
- 7.Khattab MA. 2009. Targeting host factors: a novel rationale for the management of hepatitis C virus. World J Gastroenterol 15:3472–3479. doi: 10.3748/wjg.15.3472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bartenschlager R, Lohmann V, Penin F. 2013. The molecular and structural basis of advanced antiviral therapy for hepatitis C virus infection. Nat Rev Microbiol 11:482–496. doi: 10.1038/nrmicro3046. [DOI] [PubMed] [Google Scholar]
- 9.Liang TJ, Ghany MG. 2013. Current and future therapies for hepatitis C virus infection. N Engl J Med 368:1907–1917. doi: 10.1056/NEJMra1213651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Watashi K, Hijikata M, Hosaka M, Yamaji M, Shimotohno K. 2003. Cyclosporin A suppresses replication of hepatitis C virus genome in cultured hepatocytes. Hepatology 38:1282–1288. doi: 10.1053/jhep.2003.50449. [DOI] [PubMed] [Google Scholar]
- 11.Watashi K, Ishii N, Hijikata M, Inoue D, Murata T, Miyanari Y, Shimotohno K. 2005. Cyclophilin B is a functional regulator of hepatitis C virus RNA polymerase. Mol Cell 19:111–122. doi: 10.1016/j.molcel.2005.05.014. [DOI] [PubMed] [Google Scholar]
- 12.Ciesek S, von Hahn T, Colpitts CC, Schang LM, Friesland M, Steinmann J, Manns MP, Ott M, Wedemeyer H, Meuleman P, Pietschmann T, Steinmann E. 2011. The green tea polyphenol, epigallocatechin-3-gallate, inhibits hepatitis C virus entry. Hepatology 54:1947–1955. doi: 10.1002/hep.24610. [DOI] [PubMed] [Google Scholar]
- 13.Ferenci P, Scherzer TM, Kerschner H, Rutter K, Beinhardt S, Hofer H, Schoniger-Hekele M, Holzmann H, Steindl-Munda P. 2008. Silibinin is a potent antiviral agent in patients with chronic hepatitis C not responding to pegylated interferon/ribavirin therapy. Gastroenterology 135:1561–1567. doi: 10.1053/j.gastro.2008.07.072. [DOI] [PubMed] [Google Scholar]
- 14.Gonzalez O, Fontanes V, Raychaudhuri S, Loo R, Loo J, Arumugaswami V, Sun R, Dasgupta A, French SW. 2009. The heat shock protein inhibitor Quercetin attenuates hepatitis C virus production. Hepatology 50:1756–1764. doi: 10.1002/hep.23232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haid S, Novodomska A, Gentzsch J, Grethe C, Geuenich S, Bankwitz D, Chhatwal P, Jannack B, Hennebelle T, Bailleul F, Keppler OT, Poenisch M, Bartenschlager R, Hernandez C, Lemasson M, Rosenberg AR, Wong-Staal F, Davioud-Charvet E, Pietschmann T. 2012. A plant-derived flavonoid inhibits entry of all HCV genotypes into human hepatocytes. Gastroenterology 143:213–222 e215. doi: 10.1053/j.gastro.2012.03.036. [DOI] [PubMed] [Google Scholar]
- 16.Nahmias Y, Goldwasser J, Casali M, van Poll D, Wakita T, Chung RT, Yarmush ML. 2008. Apolipoprotein B-dependent hepatitis C virus secretion is inhibited by the grapefruit flavonoid naringenin. Hepatology 47:1437–1445. doi: 10.1002/hep.22197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Myobatake Y, Takeuchi T, Kuramochi K, Kuriyama I, Ishido T, Hirano K, Sugawara F, Yoshida H, Mizushina Y. 2012. Pinophilins A and B, inhibitors of mammalian A-, B-, and Y-family DNA polymerases and human cancer cell proliferation. J Nat Prod 75:135–141. doi: 10.1021/np200523b. [DOI] [PubMed] [Google Scholar]
- 18.Aly HH, Watashi K, Hijikata M, Kaneko H, Takada Y, Egawa H, Uemoto S, Shimotohno K. 2007. Serum-derived hepatitis C virus infectivity in interferon regulatory factor-7-suppressed human primary hepatocytes. J Hepatol 46:26–36. doi: 10.1016/j.jhep.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 19.Goto K, Watashi K, Murata T, Hishiki T, Hijikata M, Shimotohno K. 2006. Evaluation of the anti-hepatitis C virus effects of cyclophilin inhibitors, cyclosporin A, and NIM811. Biochem Biophys Res Commun 343:879–884. doi: 10.1016/j.bbrc.2006.03.059. [DOI] [PubMed] [Google Scholar]
- 20.Ishida Y, Yamasaki C, Yanagi A, Yoshizane Y, Fujikawa K, Watashi K, Abe H, Wakita T, Hayes CN, Chayama K, Tateno C. 2015. Novel robust in vitro hepatitis B virus infection model using fresh human hepatocytes isolated from humanized mice. Am J Pathol 185:1275–1285. doi: 10.1016/j.ajpath.2015.01.028. [DOI] [PubMed] [Google Scholar]
- 21.Shao W, Machamer CE, Espenshade PJ. 2016. Fatostatin blocks ER exit of SCAP but inhibits cell growth in a SCAP-independent manner. J Lipid Res 57:1564–1573. doi: 10.1194/jlr.M069583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marchelli R, Dossena A, Pochini A, Dradi E. 1977. The structures of five new didehydropeptides related to neoechinulin, isolated from Aspergillus amstelodami. J Chem Soc Perkin 1 7:713–717. [PubMed] [Google Scholar]
- 23.Watashi K, Liang G, Iwamoto M, Marusawa H, Uchida N, Daito T, Kitamura K, Muramatsu M, Ohashi H, Kiyohara T, Suzuki R, Li J, Tong S, Tanaka Y, Murata K, Aizaki H, Wakita T. 2013. Interleukin-1 and tumor necrosis factor-alpha trigger restriction of hepatitis B virus infection via a cytidine deaminase activation-induced cytidine deaminase (AID). J Biol Chem 288:31715–31727. doi: 10.1074/jbc.M113.501122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bartosch B, Dubuisson J, Cosset FL. 2003. Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J Exp Med 197:633–642. doi: 10.1084/jem.20021756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Masaki T, Suzuki R, Saeed M, Mori K, Matsuda M, Aizaki H, Ishii K, Maki N, Miyamura T, Matsuura Y, Wakita T, Suzuki T. 2010. Production of infectious hepatitis C virus by using RNA polymerase I-mediated transcription. J Virol 84:5824–5835. doi: 10.1128/JVI.02397-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kato T, Date T, Miyamoto M, Sugiyama M, Tanaka Y, Orito E, Ohno T, Sugihara K, Hasegawa I, Fujiwara K, Ito K, Ozasa A, Mizokami M, Wakita T. 2005. Detection of anti-hepatitis C virus effects of interferon and ribavirin by a sensitive replicon system. J Clin Microbiol 43:5679–5684. doi: 10.1128/JCM.43.11.5679-5684.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Du P, Kibbe WA, Lin SM. 2008. lumi: a pipeline for processing Illumina microarray. Bioinformatics 24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 28.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, Hornik K, Hothorn T, Huber W, Iacus S, Irizarry R, Leisch F, Li C, Maechler M, Rossini AJ, Sawitzki G, Smith C, Smyth G, Tierney L, Yang JY, Zhang J. 2004. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol 5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Raghavan N, Amaratunga D, Cabrera J, Nie A, Qin J, McMillian M. 2006. On methods for gene function scoring as a means of facilitating the interpretation of microarray results. J Comput Biol 13:798–809. doi: 10.1089/cmb.2006.13.798. [DOI] [PubMed] [Google Scholar]
- 30.Tsukuda S, Watashi K, Iwamoto M, Suzuki R, Aizaki H, Okada M, Sugiyama M, Kojima S, Tanaka Y, Mizokami M, Li J, Tong S, Wakita T. 2015. Dysregulation of retinoic acid receptor diminishes hepatocyte permissiveness to hepatitis B virus infection through modulation of sodium taurocholate cotransporting polypeptide (NTCP) expression. J Biol Chem 290:5673–5684. doi: 10.1074/jbc.M114.602540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Watashi K, Hijikata M, Tagawa A, Doi T, Marusawa H, Shimotohno K. 2003. Modulation of retinoid signaling by a cytoplasmic viral protein via sequestration of Sp110b, a potent transcriptional corepressor of retinoic acid receptor, from the nucleus. Mol Cell Biol 23:7498–7509. doi: 10.1128/MCB.23.21.7498-7509.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watashi K, Inoue D, Hijikata M, Goto K, Aly HH, Shimotohno K. 2007. Anti-hepatitis C virus activity of tamoxifen reveals the functional association of estrogen receptor with viral RNA polymerase NS5B. J Biol Chem 282:32765–32772. doi: 10.1074/jbc.M704418200. [DOI] [PubMed] [Google Scholar]
- 33.Yamada S, Fukuchi S, Hashimoto K, Fukui Y, Tsuda M, Kataoka M, Katano H, Inoue N. 2014. Guinea pig cytomegalovirus GP129/131/133, homologues of human cytomegalovirus UL128/130/131A, are necessary for infection of monocytes and macrophages. J Gen Virol 95:1376–1382. doi: 10.1099/vir.0.064527-0. [DOI] [PubMed] [Google Scholar]
- 34.Moi ML, Lim CK, Kotaki A, Takasaki T, Kurane I. 2011. Detection of higher levels of dengue viremia using FcγR-expressing BHK-21 cells than FcγR-negative cells in secondary infection but not in primary infection. J Infect Dis 203:1405–1414. doi: 10.1093/infdis/jir053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Arita M, Wakita T, Shimizu H. 2012. Valosin-containing protein (VCP/p97) is required for poliovirus replication and is involved in cellular protein secretion pathway in poliovirus infection. J Virol 86:5541–5553. doi: 10.1128/JVI.00114-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lindenbach BD, Evans MJ, Syder AJ, Wolk B, Tellinghuisen TL, Liu CC, Maruyama T, Hynes RO, Burton DR, McKeating JA, Rice CM. 2005. Complete replication of hepatitis C virus in cell culture. Science 309:623–626. doi: 10.1126/science.1114016. [DOI] [PubMed] [Google Scholar]
- 37.Wakita T, Pietschmann T, Kato T, Date T, Miyamoto M, Zhao Z, Murthy K, Habermann A, Krausslich HG, Mizokami M, Bartenschlager R, Liang TJ. 2005. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791–796. doi: 10.1038/nm1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhong J, Gastaminza P, Cheng G, Kapadia S, Kato T, Burton DR, Wieland SF, Uprichard SL, Wakita T, Chisari FV. 2005. Robust hepatitis C virus infection in vitro. Proc Natl Acad Sci U S A 102:9294–9299. doi: 10.1073/pnas.0503596102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tscherne DM, Jones CT, Evans MJ, Lindenbach BD, McKeating JA, Rice CM. 2006. Time- and temperature-dependent activation of hepatitis C virus for low-pH-triggered entry. J Virol 80:1734–1741. doi: 10.1128/JVI.80.4.1734-1741.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lohmann V, Korner F, Koch J, Herian U, Theilmann L, Bartenschlager R. 1999. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110–113. doi: 10.1126/science.285.5424.110. [DOI] [PubMed] [Google Scholar]
- 41.Daito T, Watashi K, Sluder A, Ohashi H, Nakajima S, Borroto-Esoda K, Fujita T, Wakita T. 2014. Cyclophilin inhibitors reduce phosphorylation of RNA-dependent protein kinase to restore expression of IFN-stimulated genes in HCV-infected cells. Gastroenterology 147:463–472. doi: 10.1053/j.gastro.2014.04.035. [DOI] [PubMed] [Google Scholar]
- 42.Levy DE, Kessler DS, Pine R, Reich N, Darnell JE Jr. 1988. Interferon-induced nuclear factors that bind a shared promoter element correlate with positive and negative transcriptional control. Genes Dev 2:383–393. doi: 10.1101/gad.2.4.383. [DOI] [PubMed] [Google Scholar]
- 43.Saito T, Gale M Jr. 2008. Regulation of innate immunity against hepatitis C virus infection. Hepatol Res 38:115–122. [DOI] [PubMed] [Google Scholar]
- 44.Sun J, Desai MM, Soong L, Ou JH. 2011. IFN-alpha/beta and autophagy: tug-of-war between HCV and the host. Autophagy 7:1394–1396. doi: 10.4161/auto.7.11.17514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bishe B, Syed G, Siddiqui A. 2012. Phosphoinositides in the hepatitis C virus life cycle. Viruses 4:2340–2358. doi: 10.3390/v4102340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guengerich FP. 2001. Common and uncommon cytochrome P450 reactions related to metabolism and chemical toxicity. Chem Res Toxicol 14:611–650. doi: 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- 47.Wang YM, Ong SS, Chai SC, Chen T. 2012. Role of CAR and PXR in xenobiotic sensing and metabolism. Expert Opin Drug Metab Toxicol 8:803–817. doi: 10.1517/17425255.2012.685237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Garcia-Mediavilla MV, Pisonero-Vaquero S, Lima-Cabello E, Benedicto I, Majano PL, Jorquera F, Gonzalez-Gallego J, Sanchez-Campos S. 2012. Liver X receptor alpha-mediated regulation of lipogenesis by core and NS5A proteins contributes to HCV-induced liver steatosis and HCV replication. Lab Invest 92:1191–1202. doi: 10.1038/labinvest.2012.88. [DOI] [PubMed] [Google Scholar]
- 49.Rakic B, Sagan SM, Noestheden M, Belanger S, Nan X, Evans CL, Xie XS, Pezacki JP. 2006. Peroxisome proliferator-activated receptor alpha antagonism inhibits hepatitis C virus replication. Chem Biol 13:23–30. doi: 10.1016/j.chembiol.2005.10.006. [DOI] [PubMed] [Google Scholar]
- 50.Scholtes C, Diaz O, Icard V, Kaul A, Bartenschlager R, Lotteau V, Andre P. 2008. Enhancement of genotype 1 hepatitis C virus replication by bile acids through FXR. J Hepatol 48:192–199. doi: 10.1016/j.jhep.2007.09.015. [DOI] [PubMed] [Google Scholar]
- 51.Calkin AC, Tontonoz P. 2012. Transcriptional integration of metabolism by the nuclear sterol-activated receptors LXR and FXR. Nat Rev Mol Cell Biol 13:213–224. doi: 10.1038/nrm3312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Steffensen KR, Gustafsson JA. 2004. Putative metabolic effects of the liver X receptor (LXR). Diabetes 53(Suppl 1):S36–S42. doi: 10.2337/diabetes.53.2007.S36. [DOI] [PubMed] [Google Scholar]
- 53.Tontonoz P, Mangelsdorf DJ. 2003. Liver X receptor signaling pathways in cardiovascular disease. Mol Endocrinol 17:985–993. doi: 10.1210/me.2003-0061. [DOI] [PubMed] [Google Scholar]
- 54.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. 2000. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRα and LXRβ. Genes Dev 14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fang DL, Wan Y, Shen W, Cao J, Sun ZX, Yu HH, Zhang Q, Cheng WH, Chen J, Ning B. 2013. Endoplasmic reticulum stress leads to lipid accumulation through upregulation of SREBP-1c in normal hepatic and hepatoma cells. Mol Cell Biochem 381:127–137. doi: 10.1007/s11010-013-1694-7. [DOI] [PubMed] [Google Scholar]
- 56.Kim KH, Lee GY, Kim JI, Ham M, Won Lee J, Kim JB. 2010. Inhibitory effect of LXR activation on cell proliferation and cell cycle progression through lipogenic activity. J Lipid Res 51:3425–3433. doi: 10.1194/jlr.M007989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang X, Sato R, Brown MS, Hua X, Goldstein JL. 1994. SREBP-1, a membrane-bound transcription factor released by sterol-regulated proteolysis. Cell 77:53–62. doi: 10.1016/0092-8674(94)90234-8. [DOI] [PubMed] [Google Scholar]
- 58.Bocchetta S, Maillard P, Yamamoto M, Gondeau C, Douam F, Lebreton S, Lagaye S, Pol S, Helle F, Plengpanich W, Guerin M, Bourgine M, Michel ML, Lavillette D, Roingeard P, le Goff W, Budkowska A. 2014. Up-regulation of the ATP-binding cassette transporter A1 inhibits hepatitis C virus infection. PLoS One 9:e92140. doi: 10.1371/journal.pone.0092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zeng J, Wu Y, Liao Q, Li L, Chen X, Chen X. 2012. Liver X receptors agonists impede hepatitis C virus infection in an Idol-dependent manner. Antiviral Res 95:245–256. doi: 10.1016/j.antiviral.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 60.Chang K, Wang T, Luo G. 2009. Proteomics study of the hepatitis C virus replication complex. Methods Mol Biol 510:185–193. doi: 10.1007/978-1-59745-394-3_14. [DOI] [PubMed] [Google Scholar]
- 61.Chao TC, Su WC, Huang JY, Chen YC, Jeng KS, Wang HD, Lai MM. 2012. Proline-serine-threonine phosphatase-interacting protein 2 (PSTPIP2), a host membrane-deforming protein, is critical for membranous web formation in hepatitis C virus replication. J Virol 86:1739–1749. doi: 10.1128/JVI.06001-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gouttenoire J, Penin F, Moradpour D. 2010. Hepatitis C virus nonstructural protein 4B: a journey into unexplored territory. Rev Med Virol 20:117–129. doi: 10.1002/rmv.640. [DOI] [PubMed] [Google Scholar]
- 63.Lyn RK, Singaravelu R, Kargman S, O'Hara S, Chan H, Oballa R, Huang Z, Jones DM, Ridsdale A, Russell RS, Partridge AW, Pezacki JP. 2014. Stearoyl-CoA desaturase inhibition blocks formation of hepatitis C virus-induced specialized membranes. Sci Rep 4:4549. doi: 10.1038/srep04549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Romero-Brey I, Merz A, Chiramel A, Lee JY, Chlanda P, Haselman U, Santarella-Mellwig R, Habermann A, Hoppe S, Kallis S, Walther P, Antony C, Krijnse-Locker J, Bartenschlager R. 2012. Three-dimensional architecture and biogenesis of membrane structures associated with hepatitis C virus replication. PLoS Pathog 8:e1003056. doi: 10.1371/journal.ppat.1003056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nguyen LN, Lim YS, Pham LV, Shin HY, Kim YS, Hwang SB. 2014. Stearoyl coenzyme A desaturase 1 is associated with hepatitis C virus replication complex and regulates viral replication. J Virol 88:12311–12325. doi: 10.1128/JVI.01678-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Akashi S, Shirai K, Okada T, Konishi K, Takeuchi T, Kuramochi K, Takahashi M, Nakagawa T, Ogura Y, Fujieda S, Shibata Y, Sugawara F, Kobayashi S, Watanabe N, Arai T. 2012. Neoechinulin A imparts resistance to acute nitrosative stress in PC12 cells: a potential link of an elevated cellular reserve capacity for pyridine nucleotide redox turnover with cytoprotection. Biol Pharm Bull 35:1105–1117. doi: 10.1248/bpb.b12-00055. [DOI] [PubMed] [Google Scholar]
- 67.Kim KS, Cui X, Lee DS, Sohn JH, Yim JH, Kim YC, Oh H. 2013. Anti-inflammatory effect of neoechinulin a from the marine fungus Eurotium sp. SF-5989 through the suppression of NF-κB and p38 MAPK pathways in lipopolysaccharide-stimulated RAW264.7 macrophages. Molecules 18:13245–13259. doi: 10.3390/molecules181113245. [DOI] [PMC free article] [PubMed] [Google Scholar]