

ABSTRACT
While combined antiretroviral therapy (cART) can result in undetectable plasma viral loads, it does not eradicate HIV infection. Furthermore, HIV-infected individuals while on cART remain at an increased risk of developing serious comorbidities, such as cancer, neurological disease, and atherosclerosis, suggesting that during cART, tissue-based HIV may contribute to such pathologies. We obtained DNA and RNA env, nef, and pol sequences using single-genome sequencing from postmortem tissues of three HIV+ cART-treated (cART+) individuals with undetectable viral load and metastatic cancer at death and performed time-scaled Bayesian evolutionary analyses. We used a sensitive in situ hybridization technique to visualize HIV gag-pol mRNA transcripts in cerebellum and lymph node tissues from one patient. Tissue-associated virus evolved at similar rates in cART+ and cART-naive (cART−) patients. Phylogenetic trees were characterized by two distinct features: (i) branching patterns consistent with constant viral evolution and dispersal among tissues and (ii) very recently derived clades containing both DNA and RNA sequences from multiple tissues. Rapid expansion of virus near death corresponded to wide-spread metastasis. HIV RNA+ cells clustered in cerebellum tissue but were dispersed in lymph node tissue, mirroring the evolutionary patterns observed for that patient. Activated, infiltrating macrophages were associated with HIV RNA. Our data provide evidence that tissues serve as a sanctuary for wild-type HIV during cART and suggest the importance of macrophages as an alternative reservoir and mechanism of virus spread.
IMPORTANCE Combined antiretroviral therapy (cART) reduces plasma HIV to undetectable levels; however, removal of cART results in plasma HIV rebound, thus highlighting its inability to entirely rid the body of infection. Additionally, HIV-infected individuals on cART remain at high risk of serious diseases, which suggests a contribution from residual HIV. In this study, we isolated and sequenced HIV from postmortem tissues from three HIV+ cART+ individuals who died with metastatic cancer and had no detectable plasma viral load. Using high-resolution evolutionary analyses, we found that tissue-based HIV continues to replicate, evolve, and migrate among tissues during cART. Furthermore, cancer onset and metastasis coincided with increased HIV expansion, suggesting a linked mechanism. HIV-expressing cells were associated with tissue macrophages, a target of HIV infection. Our results suggest the importance of tissues, and macrophages in particular, as a target for novel anti-HIV therapies.
INTRODUCTION
Although current combined antiretroviral therapy (cART) regimens can extend the lives of individuals infected with human immunodeficiency virus (HIV) by years or decades, two major problems remain. First, HIV is never eradicated in treated patients and plasma viral loads (VL) will rebound if treatment is stopped. Subsets of T cells (1–7) and lymph (8, 9) are considered likely reservoirs of virus during cART. Virus obtained from circulating T cells during suppressive therapy showed a lack of evolution compared to pretherapy virus, suggesting maintenance through latency rather than replication (10, 11). On the other hand, virus from lymph nodes appears to continue to evolve throughout therapy (9) and is a heterogeneous population (8, 9). These contrasting patterns suggest multiple evolutionary strategies for maintenance during cART.
Second, significant HIV comorbidities, including cancer, lipid disorders, and neurological diseases, develop in cART-treated (cART+) patients at a higher rate than in the general population, despite fully suppressed VL and restored immunity. Of the major HIV-related comorbidities, cancer is the leading cause of death for HIV-infected (HIV+), cART-treated patients (12–14). However, the relationship between HIV infection, tumor growth, and metastasis is still largely unknown. In a previous study, we sequenced HIV DNA from tumor and nontumor postmortem tissues from two cART-naive (cART−) patients and found that virus in tumors was evolutionarily distinct from virus in nontumor tissues (15). We also recently quantified viral DNA in more than 65% of postmortem tissue samples obtained from a cohort of 20 HIV-infected patients who were virally suppressed at death, 15 of whom were diagnosed with cancer during treatment and/or at autopsy. HIV DNA was detected in 12 different anatomical locations, including tumor sites. Complete clinical details of the study are reported elsewhere (16).
From this unique cohort, we selected a subset of five cancer patients to characterize tissue-based virus. We extracted HIV DNA and RNA from tissues and used single-genome sequencing to obtain a 3,000-bp region of the HIV genome spanning env through nef. We investigated whether viral populations were dispersed among tissues, if the viral populations were evolving during cART, and whether viral dynamics were associated with development and spread of cancer.
MATERIALS AND METHODS
NNAB participants.
National Neurological AIDS Bank (NNAB) participants are recruited from the greater Los Angeles, CA, area. An institutional review board (IRB)-approved informed consent is obtained from each participant or his/her legal guardian. Eligibility criteria are as follows. Participants must be aged 18 years or older and agree to participate in study examinations and donation of blood, urine, and (optional) cerebrospinal fluid (CSF) during life and to donate their brain and other tissues for research in the event of their death. HIV-positive (HIV+) participants are selected because they have one or more of the following: a CD4+ cell count of <50 cells/mm3, systemic lymphoma or another widespread malignancy, Mycobacterium avium complex infection, wasting with loss of ≥30% of body weight, primary central nervous system (CNS) lymphoma, progressive multifocal leukoencephalopathy, congestive heart failure, chronic renal failure with potential or current need for dialysis, chronic obstructive pulmonary disease, end-stage liver disease, serum albumin of ≤3.2 g/dl, or any condition which, in the opinion of the study physician, is likely to lead to death within the period of study. In 2013, the criteria were amended to recruit a limited number of older (age > 60 years) HIV+ participants. There are no exclusions based on race, ethnicity, language, immigration status, socioeconomic status, gender, substance use, or sexual preference. Most participants are recruited premortem; however, the remains of very recently deceased individuals may be donated by legal heirs, in which case information is obtained from guardians and/or by authorized release of medical records. Extensive details concerning NNAB guidelines, questionnaires and medical assessments are available in reference 16.
ACSR.
The AIDS and Cancer Specimen Resource (ACSR) is a National Cancer Institute-funded program that supplies specimens from HIV+ participants with cancer to investigators interested in AIDS malignancy research. For this study, the ACSR independently verified the tissue diagnoses made by the NNAB and evaluated levels of HIV DNA. All of the cases described here reside at both the ACSR and the NNAB. The use of these materials is acknowledged to be a product of both NIH-funded programs. This study has University of California at San Francisco IRB approval (number 13-12020).
Patient cohort.
For the present study, we selected a subset of five participants from the NNAB studies with tissues available from the postmortem autopsy. These patients were chosen because they had substantial cancer pathology at death and an undetectable (<40 HIV copies/mm of fluid) VL at autopsy from the CSF and/or the blood (as measured from the cardiac aspirate). Extensive medical histories were available and are described in detail elsewhere (16). All tissues used in this study were histologically examined by ACSR pathologists and classified based on the pathology that was present in tissues extracted for the HIV DNA studies. For a subset of analyses, we also included viral sequences from two additional patients (designated KS1 and KS3), who died prior to the availability of cART from advanced AIDS, including Kaposi's sarcoma (KS). Similar to the participants in the current study, KS1 and KS3 had multiple postmortem tissues available through the ACSR. Fewer clinical details were available for the two KS patients, although we also had exact dates of death and self-reported dates of infection. Details on all patients are reported in Table 1.
TABLE 1.
Clinical details of NNAB/ART+ patient cohort
| NNAB identifier | Study no. | No. of yrs of known HIV infectiona | PMIc (h) | CSF VL (no. of copies/mm3) | Blood VL (no. of copies/mm3) | Last living blood VL (no. of copies/mm3) | Cancer diagnosis |
|---|---|---|---|---|---|---|---|
| 4149 | C01 | 20 | 20.5 | <40 | <40 | <40 | Non-Hodgkin's lymphoma |
| 5095 | C02 | 3.6 | 8 | <400 | <40 | <400 | Plasmacytoma/myeloma, meningoencephalitis |
| 4154 | C03 | 13 | 13 | <40 | <40 | <40 | Anal carcinoma, non-Hodgkin's lymphoma |
| 4143 | C04 | 4.6 | 4 | <40 | <400 | <40 | Kaposi's sarcoma, non-Hodgkin's lymphoma, kidney tumor |
| 1010 | C05 | 12.1 | 3.5 | <400 | <40 | <400 | Basal cell carcinoma, prostate cancer, anal carcinoma, hepatic metastasis |
| NAb | KS1 | 7.7 | NA | NA | NA | NA | Kaposi's sarcoma |
| NA | KS3 | 8.5 | NA | NA | NA | NA | Kaposi's sarcoma |
Based on date of diagnoses in clinical records.
NA, not applicable.
Postmortem interval.
RNA and DNA extraction.
Tissues were chosen for sequence analysis from the NNAB cohort participants if they were HIV DNA positive using digital droplet PCR (ddPCR; details for ddPCR experiments are presented in reference 16). An additional two tissues were included (lung from patient C02 and aorta from patient C05) that were ddPCR negative. KS1 tissues studied included skin tumor, lymph node tumor, lymph node, and kidney. KS3 tissues included skin tumor, small bowel tumor, liver, and stomach. All tissues from KS1 and KS3 were HIV+. Total RNA and genomic DNA were isolated simultaneously from each tissue section (30 to 50 ng) using the AllPrep DNA/RNA minikit (Qiagen; number 80204). Tissues were homogenized just prior to extraction in Buffer RLT Plus (lysis buffer) using a TissueRupter rotor-stator homogenizer (Qiagen; number 9001271) with a fresh sterile disposable probe (Qiagen; number 990890) for each sample. Manufacturer's guidelines were followed, with the exception of two 50-μl final elutions using RNase-free water during the RNA isolation. The 100-μl final volume of RNA generated was concentrated using an RNeasy MinElute Cleanup kit (Qiagen; number 74204). RNA and DNA extractions, cDNA synthesis, and first-round PCR setup were conducted in a restricted-access, amplicon-free room with separate air handling and laboratory equipment where no amplified PCR products or recombinant cloned plasmids were allowed, and work surfaces and equipment were thoroughly cleaned before and after use with Eliminase (Decon Labs, Inc.). cDNA was synthesized using the SuperScript III first-strand synthesis System (Invitrogen Life Technologies; number 18080-051) using the provided oligo(dT)20 primer according to the manufacturer's recommendations. RNA was incubated at 65°C for 5 min with deoxynucleoside triphosphates (0.5 mM) and 5 μM oligo(dT)20 and then cooled quickly to 4°C. First-strand cDNA synthesis was performed in a 40-μl reaction volume containing 1× reverse transcription buffer (10 mM Tris-HCl [pH 8.4], 25 mM KCl), 5 mM MgCl2, 10 mM dithiothreitol, 2 U/μl of RNase-OUT (RNase inhibitor), and 10 U/μl of SuperScript III reverse transcriptase (RT). The reaction was heated to 45°C for 90 min, followed by 85°C for 5 min. The reaction was cooled to 37°C, and 0.1 U/μl of Escherichia coli RNase H was added, followed by a 20-min incubation. cDNA was stored at −20°C.
Single-genome sequencing.
A modified single-genome sequencing protocol based on previously published methods was used to prevent PCR resampling (17). cDNA and genomic DNA were serially diluted until an average of 30% or less of the nested PCRs were positive. During the first-round PCR, diluted cDNA or genomic DNA was amplified in 20-μl reaction mixtures containing 1× Platinum Blue PCR SuperMix (Invitrogen Life Technologies) and 0.2 μM each primer: BEF1, 5′-TAATAGCAATAGTTGTGTGG-3′, and BNR1, 5′-AGCTCCCAGGCTCAGATCT-3′ (6111 to 6130 and 9558 to 9576 of HIV-1 HXB2, respectively), with the following cycling parameters: initial denaturation 94°C for 3 min and then 40 cycles of 94°C for 30 s, 56°C for 30 s, and 72°C for 4 min, followed by 72°C for 10 min. Second-round glycoprotein 120 (gp120) PCR consisted of 2 μl of the first-round PCR mixture added to a 20-μl second-round reaction mixture consisting of 1× Platinum Blue PCR SuperMix (Invitrogen Life Technologies) and 0.2 μM each primer: BEF2, 5′-CAATAGTTGTGTGGTCCATAG-3′, and BER2, 5′-CAACAGATGCTGTTGCGC-3′ (6117 to 6137 and 7905 to 7922 of HIV-1 HXB2, respectively), with the following cycling parameters: initial denaturation 94°C for 3 min and then 40 cycles of 94°C for 30 s, 56°C for 30 s, and 72°C for 3 min, followed by 72°C for 10 min. This second-round PCR generates a 1.8-kb product containing a complete envelope gp120 (env) sequence. Second-round env PCR products were visualized on 1% agarose gels stained with ethidium bromide, and reaction mixtures containing a single 1.8-kb product were considered positive and selected for sequencing with BEF2 and BER2. Subsequently, the first-round reactions that corresponded to positive second-round env PCRs were then used to amplify the nef gene sequence; the second-round nef PCR mixture consisted of 2 μl of the first-round PCR added to a 20-μl second-round reaction mixture consisting of 1× Platinum Blue PCR SuperMix (Invitrogen Life Technologies) and 0.2 μM each primer: BNF1, 5′-CTGGCTGTGGAAAGATACCT-3′, and BNR2, 5′-ATCTGAGGGCTCGCCACT-3′ (7965 to 7984 and 9488 to 9505 of HIV-1 HXB2, respectively), with the following cycling parameters: initial denaturation 94°C for 3 min and then 40 cycles of 94°C for 30 s, 58°C for 30 s, and 72°C for 2 min, followed by 72°C for 10 min. Second-round nef PCR products were visualized on 1% agarose gels stained with ethidium bromide, and reaction mixtures containing single 1.5-kb products were considered positive and selected for sequencing with BNF1 and BNR2. The primers were designed using Primer3 and observing regions of conservation in alignments of published HIV-1 subtype B sequences downloaded from the Los Alamos HIV Sequence Database (http://www.hiv.lanl.gov).
In order to examine tissue-based HIV for drug resistance mutations, a 1.3-kb fragment of genomic DNA encompassing HIV-1 protease and the first 300 codons corresponding to RT (pol) was sequenced from DNA isolated from each tissue using a limiting-dilution PCR modification of previously described methods (18, 19). During the first-round PCR, diluted genomic DNA was amplified in 20-μl reaction mixtures containing 1× Platinum Blue PCR SuperMix (Invitrogen Life Technologies) and 0.2 μM each primer: MAW26, 5′-TTGGAAATGTGGAAAGGAAGGAC-3′, and RT21, 5′-CTGTATTTCTGCTATTAAGTCTTTTGATGGG-3′ (2028 to 2050 and 3509 to 3539 of HIV-1 HXB2, respectively) with the following cycling parameters: initial denaturation 95°C for 3 min and then 35 cycles of 94°C for 15 s, 55°C for 30 s, and 72°C for 2 min, followed by 72°C for 10 min. The second-round PCR mixture consisted of 2 μl of the first-round PCR mixture added to a 20-μl second-round reaction mixture consisting of 1× Platinum Blue PCR SuperMix (Invitrogen Life Technologies) and 0.2 μM each primer: PRO1, 5′-CAGAGCCAACAGCCCCACCA-3′, and RT20, 5′-CTGCCAGTTCTAGCTCTGCTTC-3′ (2147 to 2166 and 3441 to 3462 of HIV-1 HXB2, respectively), with the following cycling parameters: initial denaturation 95°C for 3 min and then 35 cycles of 94°C for 15 s, 63°C for 30 s, and 72°C for 2 min, followed by 72°C for 10 min. Second-round gp120 PCR products were visualized on 1% agarose gels stained with ethidium bromide, and reactions yielding a single 1.3-kb product were considered positive and selected for sequencing with PRO1 and RT20.
Sequencing was performed on an Applied Biosystems 3730xl DNA analyzer (Life Technologies) at the University of Florida Interdisciplinary Center for Biotechnology Research (UF ICBR). All sequences were assembled with the Geneious R7 software package (Biomatters; http://www.geneious.com).
Sequence alignment.
Sequences were aligned using ClustalW (20) in MEGA5 (21), with further optimization performed by hand. The final env and nef alignments spanned from positions 6213 to 7823 and positions 8797 to 9411 relative to the HXB2 genome, respectively. Due to a large number of insertions and deletions that are typically problematic to align and may bias phylogenetic analysis, hypervariable regions in env (V1, V2, and V4 domains) were excluded. A preliminary maximum-likelihood (ML) phylogeny for each gene was estimated using sequences from all participants to ensure no cross-contamination of patient samples was present. Sequences were tested for the presence of hypermutations using the HYPERMUTE tool (http://www.lanl.gov); sequences with a P value of <0.01 were removed from the alignments.
RNA and DNA population structure.
Pairwise genetic distances (PGD) were computed using the Tamura-Nei 93 (TN93) model of nucleotide substitution using a publically available script (https://github.com/veg/tn93). We used the Fst statistic, which measures population subdivision, to determine whether the RNA and DNA populations were significantly different in patients C02 and C05. One thousand bootstrap replicates were used to assess significance of the Fst values. This analysis was implemented in HYPHY (22).
Phylogenetic analysis.
To determine relationships among sequences, individual maximum-likelihood phylogenies for each patient were estimated using PhyML (23) under a general time-reversible (GTR) nucleotide substitution model and gamma distributed rate variation among sites (+Γ). Statistical support was assessed with 200 bootstrap replicates.
Bayesian time-scaled analysis.
The Bayesian phylogenetic approach implemented in BEAST v.1.8.2 (24) was used to generate rooted, time-measured phylogenies for three ART-treated patients (C02, C04, and C05). To compare evolutionary patterns among cART-treated and cART-naive cancer patients, we also undertook Bayesian phylogenetic analyses on tissue virus from two additional ART-naive cancer patients (KS1 and KS3). A relaxed molecular clock model was used with a log- normal model of among-branch rate variation. For each patient a normally distributed prior distribution was placed on the root height parameter; the mean of this distribution was set to the reported date of infection (Table 1) with a standard deviation of 0.5. For Bayesian phylogenetic analyses we used the Hasegawa-Kishino-Yano (HKY) model of nucleotide substitution with gamma distributed among site rate variation (+Γ). Separate substitution models were specified for 1 plus 2 codon positions versus 3rd codon positions. The Bayesian skygrid coalescent prior was used (99 categories). Analyses were run for 100 million steps or until chain convergence, as assessed using Tracer (ESS values of >200). In some cases, the results from multiple analyses were combined. The maximum clade credibility (MCC) tree was reconstructed from the posterior distribution, minus a 10% burn-in. Each MCC tree was visualized and annotated in FigTree v.1.4.2 (programs available at http://tree.bio.ed.ac.uk/software/).
RNAscope and IHC.
We used an in situ hybridization technique (RNAscope) developed by Advanced Cell Diagnostics (ACD) to identify tissue-based cells expressing HIV gag-pol mRNA transcripts. This technology uses a “double Z” probe design, which greatly increases signal-to-noise ratio with visualization of single mRNA transcripts. RNAscope was performed using the 2.0 HD reagent kit-RED kit (ACD; catalog number 310036) according to the manufacturer's instructions. HIV mRNA was detected using an HIV gag-pol probe (ACD; catalog number 317691). The RNAscope assay was followed by standard immunohistochemistry (IHC) for macrophage markers using mouse monoclonal antibody (MAb) to CD163 (Novocastra) and CD68 (Dako) (both brown), CD68 and CD163 nuclei were counterstained with hematoxylin QS (Vector Laboratories; catalog number H-3404). Tissue sections were analyzed with a Leica DM6000 B microscope equipped with a Leica DFC 500 camera and LAS v4.3 software.
Accession number(s).
Sequences have been submitted to GenBank (accession numbers KU708874 to KU709831).
RESULTS
HIV RNA and DNA sequences were obtained from tissues for four of the five participants.
Total DNA and RNA was extracted from each ddPCR+ tissue for five participants and subjected to first-round single-genome amplification of the HIV env-nef domains, followed by second-round amplification of separate env and nef gene sequences. HIV pol DNA was amplified from a subset of these tissues to examine sequence data for drug resistance mutations. Sequences were generated from 54% of the ddPCR HIV+ tissues (n = 19) and at least one tissue for four of the five patients (Table 2).
TABLE 2.
Number of sequences obtained for each patient
| Patient | Site | No. of sequences |
||||
|---|---|---|---|---|---|---|
| RNA env | DNA env | RNA nef | DNA nef | DNA pol | ||
| C01 | Cerebellum | 0 | 0 | 0 | 0 | NAa |
| Frontal cortex | 0 | 0 | 0 | 0 | NA | |
| Basal ganglia | 0 | 0 | 0 | 0 | NA | |
| Meninges | 0 | 0 | 0 | 0 | NA | |
| Occipital lobe | 1 | 0 | 1 | 0 | NA | |
| Liver | 0 | 0 | 0 | 0 | NA | |
| Lymph node (tumor) | 0 | 0 | 0 | 0 | NA | |
| Lung | 0 | 3 | 0 | 3 | NA | |
| C02 | Cerebellum | 29 | 34 | 30 | 31 | 14 |
| Frontal cortex | 0 | 17 | 0 | 16 | 16 | |
| Temporal lobe | 0 | 6 | 0 | 6 | NA | |
| Meninges | 0 | 2 | 0 | 2 | NA | |
| Occipital lobe | 0 | 20 | 0 | 17 | 10 | |
| Lymph node (tumor) | 18 | 22 | 18 | 22 | 14 | |
| Spleen | 0 | 0 | 0 | 0 | NA | |
| Colon | 0 | 0 | 0 | 0 | NA | |
| Kidney | 0 | 0 | 0 | 0 | NA | |
| Lung | 0 | 16 | 0 | 19 | NA | |
| C03 | Kidney | 0 | 0 | 0 | 0 | NA |
| Liver | 0 | 0 | 0 | 0 | NA | |
| C04 | Cerebellum | 0 | 0 | 0 | 0 | NA |
| Basal ganglia | 0 | 0 | 0 | 0 | NA | |
| Aorta | 0 | 0 | 0 | 0 | NA | |
| Kidney | 0 | 0 | 0 | 2 | NA | |
| Colon | 0 | 9 | 0 | 9 | 1 | |
| Lung | 0 | 26 | 0 | 18 | 8 | |
| C05 | Frontal cortex | 0 | 0 | 0 | 0 | NA |
| Temporal lobe | 0 | 0 | 0 | 1 | NA | |
| Basal ganglia | 0 | 1 | 0 | 1 | NA | |
| Liver (tumor) | 0 | 0 | 0 | 0 | NA | |
| Lymph node | 22 | 21 | 15 | 8 | 9 | |
| Spleen | 17 | 25 | 15 | 10 | 9 | |
| Kidney | 0 | 3 | 0 | 8 | 7 | |
| Pancreas | 0 | 2 | 0 | 7 | NA | |
| Aorta | 0 | 0 | 0 | 0 | NA | |
NA, not applicable.
RNA and DNA sequences arose from same population.
For participants C02 and C05, we used the Fst statistic to test whether sequences obtained from RNA and DNA represented genetically distinct subpopulations. No significant differences were found in either participant for either env or nef (Fst = −0.012 and P = 0.894 for C02; Fst = 0.020 and P = 0.23 for C05). Therefore, DNA and RNA sequences from each tissue were grouped together for the remainder of analyses. We did not apply this test between tissues, as in some cases too few sequences were present for meaningful analysis.
env and nef from both cART+ and cART− patients showed similar levels of diversity.
For each patient, we then calculated PGD among all env and nef sequences (Fig. 1). For the env gene, patients C02, C04, and C05 exhibited lower mean PGD than the two cART-naive patients, although the interquartile range (IQR) for C02 and C05 overlapped those for KS1 and KS3, while the IQR for C04 was less than for both KS patients. However, for the nef gene, the mean PGD for C05 was similar to that of KS1 and KS3, and the IQR for all participants overlapped.
FIG 1.
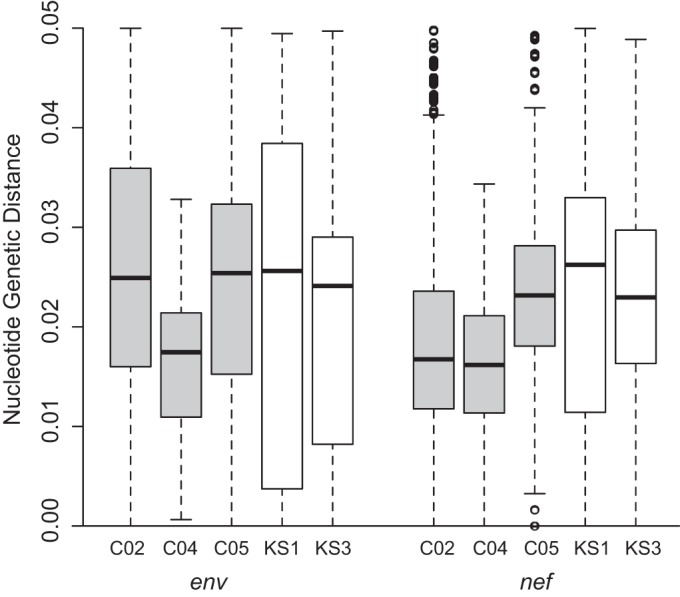
Pairwise genetic distance in env (left) and nef (right) for cART-treated patients (in gray) and cART-naive patients (in white). Shown are the mean (thick black bar), interquartile range (white or gray box), and range (dotted lines). Outliers are shown as open circles.
Anatomically dispersed viral populations evolve via different modes of evolution.
In the env gene phylogeny of patient C02 (Fig. 2, left), many brain-derived sequences cluster in three distinct clades (A to C), each of which has a very recent common ancestor, and most of the remaining sequences from brain, lymph node tumor, and lung have only a few or no closely related sequences and therefore descend from long terminal branches. The majority of DNA and RNA sequences in clades A to C are identical. This pattern is consistent with a sudden and recent expansion of a population of viruses that has been genetically distinct from the remainder. However, note that these clades are not exclusively comprised of virus from the brain, as three lymph node tumor sequences are among them, and both RNA and DNA sequences are present. A few lymph node tumor-derived sequences are present in clades A and B, with a potential migration event(s) between lymph node tumor and brain. The nef gene showed a similar pattern (Fig. 2, right). This patient was reportedly on cART throughout infection, although clinical records indicate that adherence may have been suboptimal. However, the patient was in a managed-care facility, where medication was administered by staff, before death. The patient's last VL measurement at death was undetectable and no virus was detected at autopsy in either plasma or CSF. His only cancer diagnosis occurred at autopsy, when plasmacytoma was diagnosed in multiple tissues, along with meningeal infiltrate into the brain. The lack of detection of this cancer while alive suggests a late onset and rapid spread. We did not apply a test for significance of population structure among tissues, as clearly there are multiple patterns of gene evolution within the tree, which would be masked by a simple statistic.
FIG 2.

Maximum-likelihood trees of the env (left) and nef (right) sequences for patient C02. Branches are drawn in substitutions/site according to the scale at the bottom. Circles are colored by the tissue of origin. Open circles, RNA; filled circles, DNA. Asterisks represent branches with support of >70.
In the env phylogeny of patient C04, there were two well-supported clades, one containing only lung sequences (E) and the other containing lung and colon sequences (F) (Fig. 3). Note that the scale of substitution for env is much different from that for C02 and reflects the relatively low diversity within this patient (Fig. 3, left). The nef gene showed more variation and longer branches, and the lung-exclusive clade is now within the rest of the tree and contains a kidney sequence that is identical to the lung sequence (Fig. 3, right). Additionally, clade G contained identical sequences from lung and colon, suggesting minimal barriers to gene flow among tissues. This patient, who was receiving fully suppressive cART since being diagnosed with HIV infection, also experienced treated yet unresolved lymphoma until death. No RNA sequences were obtained for this patient, suggesting that proviruses were not being expressed at the time of death (or the RNA recovered from the autopsy tissue was degraded).
FIG 3.

Maximum-likelihood trees of the env (left) and nef (right) sequences for patient C04. Branches are drawn in substitutions/site according to the scale at the bottom. Circles are colored by the tissue of origin. Open circles, RNA; filled circles, DNA. Asterisks represent branches with support of >70.
The patient C05 env phylogeny (Fig. 4, left) shows a pattern similar to that in patient C02, in which some clades (e.g., H) contain identical RNA and DNA sequences from, in this case, the spleen (H), or spleen and tumor lymph node (I), while other clades contain virus from multiple tissues that appears to be accumulating mutations (J). A clade with a very long branch (K) is in both trees and contains DNA sequences from kidney and pancreas, which suggests a potential compartmentalization and lack of replication, although in nef one spleen sequence is closely related and other kidney sequences are found scattered in the nef tree. The clades of identical sequences present in env did not have a nef counterpart, and thus, those clades disappear in the nef tree. This patient received a lymphoma diagnosis around 2 years before death. Late in life, this patient's cancer spread to the liver, leading to death.
FIG 4.

Maximum-likelihood trees of the env (left) and nef (right) sequences for patient C05. Branches are drawn in substitutions/site according to the scale at the bottom. Circles are colored by the tissue of origin. Open circles, RNA; filled circles, DNA. Asterisks represent branches with support of >70.
Evolutionary rates in env and nef are similar in ART-infected and ART-naive viral populations.
We then estimated rates of HIV sequence evolution for these three patients using a Bayesian analysis in which a strong prior was placed on the root of the tree, corresponding to the patients' reported date of diagnosis. We performed the same analysis using two additional patients, who died with KS prior to the era of cART. These patients also had known dates of death and HIV diagnosis. These results are shown in Fig. 5. For comparison, we included on the graph viral evolutionary rates obtained from an earlier study (8) that analyzed HIV gag gene evolution between pretherapy and during suppressive therapy. Finally, we included the within-host evolutionary rates reported in references 25 and 26.
FIG 5.

BEAST estimated evolutionary rates. Mean (circles) and 95% confidence interval (vertical bars) evolutionary rates were estimated for the env and nef regions from the cART-infected and cART-naive patients in this study (C02 to KS3). Also shown are the rates reported in reference 26 (env1); rates for progressors (env2) and nonprogressors (env3) from reference 25; and pretherapy and posttherapy rates from reference 8. The y axis on log scale. Gray boxes indicate estimates from cART-treated patients.
The evolutionary rates for both env and nef in patients KS1 and KS3 were similar. Estimated env gene rates for patients C02 and C05 were not significantly different from the env rates for KS1 and KS3. C04 did exhibit a significantly lower rate than the other patients; however, this rate was still >1 × 10−3. All three cART-treated patients had higher mean rates for nef than the cART-naive patients, although the distributions overlapped for all patients (Fig. 5). These rates are all similar to those reported in two previous studies (shown in the figure). However, these results are in stark contrast to those found by Josefsson et al. (8), who reported that the HIV evolutionary rate was reduced by orders of magnitude when HIV in T cells during suppressive cART was compared to plasma-derived virus sampled pre-cART. The rates estimated from our patients are similar to that estimated from one ART-naive patient using a similar approach (27).
In addition, in our study, we found that among-branch rate variation was extremely high in nearly all patients and genes (Table 3), consistent with the patients harboring viruses that evolve at different speeds. We note that while the viral population sampled at death may have a more recent temporal origin than the date of HIV diagnoses, the evolutionary rates would then be expected to increase, since the same amount of variation would need to have arisen in a shorter period. Although the date of HIV diagnosis may have been later than the true date of HIV infection, the precise clinical records of these patients indicates major health complications at the time of diagnosis, suggesting frequent health care and opportunities for testing.
TABLE 3.
Coefficient of variation estimated by BEAST
| Patient | Gene | Mean | 95% CIa |
|---|---|---|---|
| C02 | env | 0.77 | 0.60, 0.96 |
| C04 | env | 0.54 | 0.002, 0.94 |
| C05 | env | 0.88 | 0.55, 1.23 |
| KS1 | env | 0.86 | 0.62, 1.13 |
| KS3 | env | 0.85 | 0.51, 1.23 |
| C02 | nef | 0.91 | 0.55, 1.30 |
| C04 | nef | 0.97 | 0.043, 1.62 |
| C05 | nef | 0.96 | 0.22, 1.74 |
| KS1 | nef | 0.28 | 0.0006, 0.59 |
| KS3 | nef | 0.81 | 0.55, 1.13 |
CI, confidence interval.
Wild-type viruses evolve in tissues in cART-treated patients.
We sequenced the pol gene from a subset of the tissues for each of the three patients to determine whether the viral populations in tissues were wild-type virus, and therefore shielded from the effects of cART, or whether they were drug resistant, which would suggest some mechanism by which viruses replicated in tissues but not in blood T cells. Only one sequence (in participant C05) out of a total 88 pol sequences analyzed carried a drug resistance mutation (against nonnucleoside reverse transcriptase inhibitors [NNRTIs]). This patient was on nevirapine early in his treatment but was quickly switched to non-NNRTIs.
HIV RNA gag-pol transcripts are present in cellular clusters in cerebellum and singularly in lymph node tumor.
Images from the RNAscope analysis showed that HIV mRNA transcripts for gag-pol were present within cells and in extracellular space in both the lymph node tumor and cerebellum in patient C02, the same tissues in which both viral DNA and RNA sequences were also recovered (Fig. 6). In the lymph node tumor, HIV-expressing cells were typically localized distinct from each other (Fig. 6a and b). In contrast, HIV-expressing cells were found in “clusters” in the cerebellum and appeared to be expressing high levels of viral transcripts (Fig. 6c and d). In both tissues, HIV-expressing cells were surrounded by cells that costained for CD68/CD163, indicative of infiltrating activated macrophages. Furthermore, some of these macrophages appeared to be associated with HIV RNA in the brain (Fig. 6e and f). These results are consistent with the phylogenetic patterns, in which viral variants were dispersed among tissues and a rapid expansion of identical DNA and RNA sequences was evident in the cerebellum.
FIG 6.

RNAscope of cells expressing HIV gag-pol transcripts. Each punctum (pink dot) represents a single HIV gag-pol transcript in lymph node tumor (a and b) and cerebellum (c to f). Note that many puncta fuse together and appear as dense red staining of the entire cell. Cells were stained for markers of macrophages (CD68) and their activation/infiltration phenotype (CD163) (brown). Panel b is a ×630 magnification of the region outlined in panel a. Panels d and f are ×400 magnifications of the regions outlined in panels c and e, respectively. (Panels 6c and d are republished from reference 31 with permission of the publisher.)
DISCUSSION
A striking result from this study was the lack of a substantial reduction in HIV evolutionary rates in anatomical tissues collected from fully suppressed cART-treated patients compared to viral populations in cART-naive patients. Several studies that have sampled T cells or plasma after therapy interruption have found a genetically homogeneous HIV population that was similar, if not identical, to the pretherapy virus (10, 11). One study, which estimated the evolutionary rate of post-ART T cells compared with pre-cART plasma virus using a method similar to that described here, found that the evolutionary rate was reduced orders of magnitude in post-cART virus, suggesting little to no virus replication or evolution in the T-cell population (8). Distinct subsets of T cells are known to act as cellular reservoirs of HIV during cART (1–7), and the maintenance of nonevolving viral populations has been shown to occur through cellular expansion of memory cells with integrated but nonexpressing provirus (28, 29). Therefore, an alternative site of viral maintenance during cART, apart from memory T cells, must be present in order to explain the results presented here.
It is possible that lymphatic tissues, including lymph node and spleen, may be conducive to viral replication during cART. A recent study used the same analytical method as the one in this study to estimate evolutionary history and found evidence of ongoing replication of wild-type virus in lymph nodes and continual seeding of the blood from lymph node sanctuary sites where drugs are not fully suppressive (9). Another group reported evidence for HIV RNA+ cells in lymph nodes prior to therapy interruption and a diverse population of rebound virus, suggesting that the rebounding viral population originated from multiple sources, including lymph nodes (8). Lower concentrations of drugs are also found in lymphatic tissues (30). Our finding of ongoing replication of HIV RNA and DNA in lymph node and spleen is consistent with these observations. The lymph system may serve as an alternative route for HIV-infected cells to migrate (31), which would explain our observation of the dispersal of viral population among tissues (i.e., no compartmentalization), as well the lack of detectable virus in the blood. Furthermore, a recent study suggested that the meninges may be part of the lymphatic system (32), which could provide an explanation of late infection in the brain noted previously (33) and shown in this study in the recent and rapid expansion in the cerebellum. We note that our intention in this study was not to determine routes of migration among tissues or the extent of compartmentalization among tissues, as the numbers of sequences among sites were unequal. In addition, clearly there were several types of evolution occurring among these sequences, which could result from infection of different cell types with various dynamics.
For example, in addition to T cells, tissue-resident macrophages are long-lived cells capable of harboring virus (34, 35). Macrophages are less resistant to the cytopathic effects of the virus (36), contain lower intracellular concentrations of cART (37–39), and participate in efficient cell-to-cell transfer of virus among macrophages and from macrophages to T cells (40), thus undermining the effectiveness of cART (41). We did not test the identity of the cells from which virus was extracted; however, we found evidence of activated infiltrating macrophages in very close proximity to HIV-expressing cells in two tissues, and potential HIV expression by these macrophages in the brain. Infection of multiple cell types, i.e., T cells and macrophages, may provide explanations for the two distinct branching patterns found in all patients in this study, consistent with ongoing evolution in long-lived cells and clonal expansion.
All of the participants in this study, as well as the two cART-naive participants, were diagnosed with at least one type of cancer. In a previous study, we found that tumors harbored viral strains that were distinct from those in nontumor tissues and that macrophages were identified as productively infected cells in the tumors (15). Tumor-associated macrophages may contribute to ongoing HIV replication by orchestrating tumor progression, angiogenesis, tumor growth, and metastasis (42–45) and migration of cells (46). The presence of activated infiltrating macrophages surrounding HIV-expressing cells in the two tissues studied in this work is consistent with recent migration of these cells, and potentially of the HIV-infected cells as well. If these cells were derived from the tumor as a result of metastasis, this again would explain the ability of the virus to replicate despite cART, dispersal of virus among tissues, and evidence of rapid and recent expansion of a replicating viral population. Furthermore, cancers can induce type I IFN production, which may play a role in anti-tumor immunity (47), drive T cell dysfunction (48), and contribute to other aspects of HIV disease pathogenesis, for example, virus control (MX2 postentry inhibition [49], APOBEC3 restriction of replication [50], BST-2 virus release [51], and NK cell activation [52]) or immune activation or dysfunction (CD38 expression on CD8+ T cells [53], CD4+ T cell/Fas-dependent apoptosis [54], or decreased interleukin 7 [IL-7]/increased activation-induced T cell proliferation [55]). These beneficial or deleterious effects may or may not relate to the evolution and dispersion of virus populations among tissues during the onset of cancer.
There are, of course, limitations to this study. In particular, the individuals selected do not represent a random or representative sample of all HIV-infected persons, as the NNAB preferentially selects participants thought to have serious life-threatening illnesses. Furthermore, one selection criterion for inclusion was a short postmortem interval, which could exclude healthier participants who expire at some distance from the study site, while preferentially including participants who expire due to poor health in a medical facility. Although the medical information reported here is complete to the best of our ability, some clinical variables are unknown. The precise location of tissue sampling could clearly impact HIV recovery; for example, Lamers et al. (16) found high variability in the amount and detection of HIV in tissues could within an individual. We used a sequencing technique (single-genome sequencing) and multiple extractions and amplifications in order to ensure that we sampled the broadest variability possible, but deep-sequencing studies could provide even more resolution. Additional studies including more patients are clearly required to completely understand the trajectory of viral evolution during cART.
ACKNOWLEDGMENTS
This study depended on the generous voluntary efforts of human participants and their caregivers to donate time and tissues.
S.L.L., D.J.N., R.R., M.S., E.J.S., and M.S.M. were funded by National Institute of Mental Health grant NIH R01 MH100984. E.M., C.A.S., and M.S.M. were funded by National Cancer Institute grant UM1 CA181255. E.J.S. was funded by National Institute of Mental Health grant NIMH U24MH100929. S.L.L., D.J.N., and M.S. were funded by National Institute of Neurological Disorders and Stroke grant NS063897.
REFERENCES
- 1.Buzon MJ, Sun H, Li C, Shaw A, Seiss K, Ouyang Z, Martin-Gayo E, Leng J, Henrich TJ, Li JZ, Pereyra F, Zurakowski R, Walker BD, Rosenberg ES, Yu XG, Lichterfeld M. 2014. HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat Med 20:139–142. doi: 10.1038/nm.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, Schacker TW, Hill BJ, Douek DC, Routy JP, Haddad EK, Sékaly RP. 2009. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat Med 15:893–900. doi: 10.1038/nm.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chun T, Carruth L, Finzi D, Shen X, DiGiuseppe J, Taylor H, Hermankova M, Chadwick K, Margolick J, Quinn T, Kuo Y, Brookmeyer R, Zeiger M, Barditch-Crovo P, Siliciano R. 1997. Quantification of latent tissue reservoirs and total body viral load in HIV-1 infection. Nature 387:183–188. doi: 10.1038/387183a0. [DOI] [PubMed] [Google Scholar]
- 4.Finzi D, Blankson J, Siliciano JD, Margolick JB, Chadwick K, Pierson T, Smith K, Lisziewicz J, Lori F, Flexner C, Quinn TC, Chaisson RE, Rosenberg E, Walker B, Gange S, Gallant J, Siliciano RF. 1999. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat Med 5:512–517. doi: 10.1038/8394. [DOI] [PubMed] [Google Scholar]
- 5.Finzi D, Hermankova M, Pierson T, Carruth L, Buck C, Chaisson R, Quinn T, Chadwick K, Margolick J, Brookmeyer R, Gallant J, Markowitz M, Ho D, Richman D, Siliciano R. 1997. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278:1295–1300. doi: 10.1126/science.278.5341.1295. [DOI] [PubMed] [Google Scholar]
- 6.Gattinoni L, Lugli E, Ji Y, Pos Z, Paulos CM, Quigley MF, Almeida JR, Gostick E, Yu Z, Carpenito C, Wang E, Douek DC, Price DA, June CH, Marincola FM, Roederer M, Restifo NP. 2011. A human memory T cell subset with stem cell-like properties. Nat Med 17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Persaud D, Pierson T, Ruff C, Finzi D, Chadwick K, Margolick J, Ruff A, Hutton N, Ray S, Siliciano R. 2000. A stable latent reservoir for HIV-1 in resting CD4(+) T lymphocytes in infected children. J Clin Invest 105:995–1003. doi: 10.1172/JCI9006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Josefsson L, von Stockenstrom S, Faria NR, Sinclair E, Bacchetti P, Killian M, Epling L, Tan A, Ho T, Lemey P, Shao W, Hunt PW, Somsouk M, Wylie W, Douek DC, Loeb L, Custer J, Hoh R, Poole L, Deeks SG, Hecht F, Palmer S. 2013. The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc Natl Acad Sci U S A 110:E4987–E4996. doi: 10.1073/pnas.1308313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lorenzo-Redondo R, Fryer HR, Bedford T, Kim EY, Archer J, Kosakovsky Pond SL, Chung YS, Penugonda S, Chipman JG, Fletcher CV, Schacker TW, Malim MH, Rambaut A, Haase AT, McLean AR, Wolinsky SM. 2016. Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 530:51–56. doi: 10.1038/nature16933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson JA, Archin NM, Ince W, Parker D, Wiegand A, Coffin JM, Kuruc J, Eron J, Swanstrom R, Margolis DM. 2011. Clonal sequences recovered from plasma from patients with residual HIV-1 viremia and on intensified antiretroviral therapy are identical to replicating viral RNAs recovered from circulating resting CD4+ T cells. J Virol 85:5220–5223. doi: 10.1128/JVI.00284-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kearney MF, Spindler J, Shao W, Yu S, Anderson EM, O'Shea A, Rehm C, Poethke C, Kovacs N, Mellors JW, Coffin JM, Maldarelli F. 2014. Lack of detectable HIV-1 molecular evolution during suppressive antiretroviral therapy. PLoS Pathog 10:e1004010. doi: 10.1371/journal.ppat.1004010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bonnet F, Burty C, Lewden C, Costagliola D, May T, Bouteloup V, Rosenthal E, Jougla E, Cacoub P, Salmon D, Chêne G, Morlat P, Agence Nationale de Recherches sur le Sida et les Hépatites Virales EN19 Mortalité Study Group, Mortavic Study Group . 2009. Changes in cancer mortality among HIV-infected patients: the Mortalité 2005 Survey. Clin Infect Dis 48:633–639. doi: 10.1086/596766. [DOI] [PubMed] [Google Scholar]
- 13.Antiretroviral Therapy Cohort Collaboration. 2010. Causes of death in HIV-1-infected patients treated with antiretroviral therapy, 1996–2006: collaborative analysis of 13 HIV cohort studies. Clin Infect Dis 50:1387–1396. doi: 10.1086/652283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simard EP, Engels EA. 2010. Cancer as a cause of death among people with AIDS in the United States. Clin Infect Dis 51:957–962. doi: 10.1086/656416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Salemi M, Lamers SL, Huysentruyt LC, Galligan D, Gray RR, Morris A, McGrath MS. 2009. Distinct patterns of HIV-1 evolution within metastatic tissues in patients with non-Hodgkins lymphoma. PLoS One 4:e8153. doi: 10.1371/journal.pone.0008153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lamers SL, Rose R, Maidji E, Agsalda-Garcia M, Nolan DJ, Fogel GB, Salemi M, Garcia DL, Bracci P, Yong W, Commins D, Said J, Khanlou N, Hinkin CH, Sueiras MV, Mathisen G, Donovan S, Shirimizu B, Stoddart CA, Mcgrath MS, Singer EJ. 2016. HIV DNA is frequently present within pathologic tissues evaluated at autopsy from combined antiretroviral therapy-treated patients with undetectable viral loads. J Virol 90:8968–8983. doi: 10.1128/JVI.00674-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palmer S, Kearney M, Maldarelli F, Halvas EK, Bixby CJ, Bazmi H, Rock D, Falloon J, Davey RT, Dewar RL, Metcalf JA, Hammer S, Mellors JW, Coffin JM. 2005. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J Clin Microbiol 43:406–413. doi: 10.1128/JCM.43.1.406-413.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Palmer S, Shafer RW, Merigan TC. 1999. Highly drug-resistant HIV-1 clinical isolates are cross-resistant to many antiretroviral compounds in current clinical development. AIDS 13:661–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shafer RW, Warford A, Winters MA, Gonzales MJ. 2000. Reproducibility of human immunodeficiency virus type 1 (HIV-1) protease and reverse transcriptase sequencing of plasma samples from heavily treated HIV-1-infected individuals. J Virol Methods 86:143–153. doi: 10.1016/S0166-0934(00)00144-0. [DOI] [PubMed] [Google Scholar]
- 20.Thompson JD, Gibson TJ, Plewniak F, Jeanmougin F, Higgins DG. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882. doi: 10.1093/nar/25.24.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. 2011. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pond SL, Frost SD, Muse SV. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679. doi: 10.1093/bioinformatics/bti079. [DOI] [PubMed] [Google Scholar]
- 23.Guindon S, Delsuc F, Dufayard J, Gascuel O. 2009. Estimating maximum likelihood phylogenies with PhyML. Methods Mol Biol 537:113–137. doi: 10.1007/978-1-59745-251-9_6. [DOI] [PubMed] [Google Scholar]
- 24.Drummond AJ, Rambaut A. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214. doi: 10.1186/1471-2148-7-214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edo-Matas D, Lemey P, Tom JA, Serna-Bolea C, van den Blink AE, van 't Wout AB, Schuitemaker H, Suchard MA. 2011. Impact of CCR5delta32 host genetic background and disease progression on HIV-1 intrahost evolutionary processes: efficient hypothesis testing through hierarchical phylogenetic models. Mol Biol Evol 28:1605–1616. doi: 10.1093/molbev/msq326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lemey P, Rambaut A, Pybus O. 2006. HIV evolutionary dynamics within and among hosts. AIDS Rev 8:125–140. [PubMed] [Google Scholar]
- 27.Alizon S, von Wyl V, Stadler T, Kouyos RD, Yerly S, Hirschel B, J Böni Shah C, Klimkait T, Furrer H, Rauch A, Vernazza PL, Bernasconi E, Battegay M, Bürgisser P, Telenti A, Günthard HF, Bonhoeffer S, Swiss HIV Cohort Study . 2010. Phylogenetic approach reveals that virus genotype largely determines HIV set-point viral load. PLoS Pathog 6(9):e1001123. doi: 10.1371/journal.ppat.1001123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Maldarelli F, Wu X, Su L, Simonetti FR, Shao W, Hill S, Spindler J, Ferris AL, Mellors JW, Kearney MF, Coffin JM, Hughes SH. 2014. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science 345:179–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wagner TA, McLaughlin S, Garg K, Cheung CY, Larsen BB, Styrchak S, Huang HC, Edlefsen PT, Mullins JI, Frenkel LM. 2014. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science 345:570–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fletcher CV, Staskus K, Wietgrefe SW, Rothenberger M, Reilly C, Chipman JG, Beilman GJ, Khoruts A, Thorkelson A, Schmidt TE, Anderson J, Perkey K, Stevenson M, Perelson AS, Douek DC, Haase AT, Schacker TW. 2014. Persistent HIV-1 replication is associated with lower antiretroviral drug concentrations in lymphatic tissues. Proc Natl Acad Sci U S A 111:2307–2312. doi: 10.1073/pnas.1318249111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lamers SL, Rose R, Ndhlovu LC, Nolan DJ, Salemi M, Maidji E, Stoddart CA, McGrath MS. 16 November 2015. The meningeal lymphatic system: a route for HIV brain migration? J Neurovirol. doi: 10.1007/s13365-015-0399-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Louveau A, Smirnov I, Keyes TJ, Eccles JD, Rouhani SJ, Peske JD, Derecki NC, Castle D, Mandell JW, Lee KS, Harris TH, Kipnis J. 2015. Structural and functional features of central nervous system lymphatic vessels. Nature 523:337–341. doi: 10.1038/nature14432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lamers SL, Salemi M, Galligan DC, Morris A, Gray R, Fogel G, Zhao L, McGrath MS. 2010. Human immunodeficiency virus-1 evolutionary patterns associated with pathogenic processes in the brain. J Neurovirol 16:230–241. doi: 10.3109/13550281003735709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar A, Abbas W, Herbein G. 2014. HIV-1 latency in monocytes/macrophages. Viruses 6:1837–1860. doi: 10.3390/v6041837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sharova N, Swingler C, Sharkey M, Stevenson M. 2005. Macrophages archive HIV-1 virions for dissemination in trans. EMBO J 24:2481–2489. doi: 10.1038/sj.emboj.7600707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, Phipps T, Wahl LA, Lane HC, Fauci AS, Burke DS. 1988. Efficient isolation and propagation of human immunodeficiency virus on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med 167:1428–1441. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gavegnano C, Detorio MA, Bassit L, Hurwitz SJ, North TW, Schinazi RF. 2013. Cellular pharmacology and potency of HIV-1 nucleoside analogs in primary human macrophages. Antimicrob Agents Chemother 57:1262–1269. doi: 10.1128/AAC.02012-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gavegnano C, Schinazi RF. 2009. Antiretroviral therapy in macrophages: implication for HIV eradication. Antivir Chem Chemother 20:63–78. doi: 10.3851/IMP1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schinazi RF, Bassit L, Gavegnano C. 2010. HCV drug discovery aimed at viral eradication. J Viral Hepat 17:77–90. doi: 10.1111/j.1365-2893.2009.01246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Groot F, Welsch S, Sattentau QJ. 2008. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 111:4660–4663. doi: 10.1182/blood-2007-12-130070. [DOI] [PubMed] [Google Scholar]
- 41.Duncan CJ, Williams JP, Schiffner T, Gartner K, Ochsenbauer C, Kappes J, Russell RA, Frater J, Sattentau QJ. 2014. High-multiplicity HIV-1 infection and neutralizing antibody evasion mediated by the macrophage-T cell virological synapse. J Virol 88:2025–2034. doi: 10.1128/JVI.03245-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Alfano M, Graziano F, Genovese L, Poli G. 2013. Macrophage polarization at the crossroad between HIV-1 infection and cancer development. Arterioscler Thromb Vasc Biol 33:1145–1152. doi: 10.1161/ATVBAHA.112.300171. [DOI] [PubMed] [Google Scholar]
- 43.Condeelis J, Pollard JW. 2006. Macrophages: obligate partners for tumor cell migration, invasion, and metastasis. Cell 124:263–266. doi: 10.1016/j.cell.2006.01.007. [DOI] [PubMed] [Google Scholar]
- 44.Qian BZ, Pollard JW. 2010. Macrophage diversity enhances tumor progression and metastasis. Cell 141:39–51. doi: 10.1016/j.cell.2010.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sica A, Larghi P, Mancino A, Rubino L, Porta C, Totaro MG, Rimoldi M, Biswas SK, Allavena P, Mantovani A. 2008. Macrophage polarization in tumour progression. Semin Cancer Biol 18:349–355. doi: 10.1016/j.semcancer.2008.03.004. [DOI] [PubMed] [Google Scholar]
- 46.Noy R, Pollard JW. 2014. Tumor-associated macrophages: from mechanisms to therapy. Immunity 41:49–61. doi: 10.1016/j.immuni.2014.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. 2015. Type I interferons in anticancer immunity. Nat Rev Immunol 15:405–414. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 48.Cha L, Berry CM, Nolan D, Castley A, Fernandez S, French MA. 2014. Interferon-alpha, immune activation and immune dysfunction in treated HIV infection. Clin Transl Immunology 3:e10. doi: 10.1038/cti.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goujon C, Moncorgé O, Bauby H, Doyle T, Ward CC, Schaller T, Hué S, Barclay WS, Schulz R, Malim MH. 2013. Human MX2 is an interferon-induced post-entry inhibitor of HIV-1 infection. Nature 502:559–562. doi: 10.1038/nature12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Krisko JF, Martinez-Torres F, Foster JL, Garcia JV. 2013. HIV restriction by APOBEC3 in humanized mice. PLoS Pathog 9:e1003242. doi: 10.1371/journal.ppat.1003242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J, Liang C. 2010. BST-2 diminishes HIV-1 infectivity. J Virol 84:12336–12343. doi: 10.1128/JVI.01228-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Naranbhai V, Altfeld M, Karim SS, Ndung'u T, Karim QA, Carr WH. 2013. Changes in natural killer cell activation and function during primary HIV-1 Infection. PLoS One 8:e53251. doi: 10.1371/journal.pone.0053251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benito JM, López M, Lozano S, Martinez P, González-Lahoz J, Soriano V. 2004. CD38 expression on CD8 T lymphocytes as a marker of residual virus replication in chronically HIV-infected patients receiving antiretroviral therapy. AIDS Res Hum Retroviruses 20:227–233. doi: 10.1089/088922204773004950. [DOI] [PubMed] [Google Scholar]
- 54.Ahr B, Robert-Hebmann V, Devaux C, Biard-Piechaczyk M. 2004. Apoptosis of uninfected cells induced by HIV envelope glycoproteins. Retrovirology 1:12. doi: 10.1186/1742-4690-1-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tan JT, Dudl E, LeRoy E, Murray R, Sprent J, Weinberg KI, Surh CD. 2001. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci U S A 98:8732–8737. doi: 10.1073/pnas.161126098. [DOI] [PMC free article] [PubMed] [Google Scholar]