

ABSTRACT
The Paramyxoviridae comprise a large family of enveloped, negative-sense, single-stranded RNA viruses with significant economic and public health implications. For nearly all paramyxoviruses, infection is initiated by fusion of the viral and host cell plasma membranes in a pH-independent fashion. Fusion is orchestrated by the receptor binding protein hemagglutinin-neuraminidase (HN; also called H or G depending on the virus type) protein and a fusion (F) protein, the latter undergoing a major refolding process to merge the two membranes. Mechanistic details regarding the coupling of receptor binding to F activation are not fully understood. Here, we have identified the flexible loop region connecting the bulky enzymatically active head and the four-helix bundle stalk to be essential for fusion promotion. Proline substitution in this region of HN of parainfluenza virus 5 (PIV5) and Newcastle disease virus HN abolishes cell-cell fusion, whereas HN retains receptor binding and neuraminidase activity. By using reverse genetics, we engineered recombinant PIV5-EGFP viruses with mutations in the head-stalk linker region of HN. Mutations in this region abolished virus recovery and infectivity. In sum, our data suggest that the loop region acts as a “hinge” around which the bulky head of HN swings to-and-fro to facilitate timely HN-mediate F-triggering, a notion consistent with the stalk-mediated activation model of paramyxovirus fusion.
IMPORTANCE Paramyxovirus fusion with the host cell plasma membrane is essential for virus infection. Membrane fusion is orchestrated via interaction of the receptor binding protein (HN, H, or G) with the viral fusion glycoprotein (F). Two distinct models have been suggested to describe the mechanism of fusion: these include “the clamp” and the “provocateur” model of activation. By using biochemical and reverse genetics tools, we have obtained strong evidence in favor of the HN stalk-mediated activation of paramyxovirus fusion. Specifically, our data strongly support the notion that the short linker between the head and stalk plays a role in “conformational switching” of the head group to facilitate F-HN interaction and triggering.
INTRODUCTION
The Paramyxoviridae comprise a large family of enveloped, negative sense, single-stranded RNA viruses that are responsible for many important human and other animal diseases. Among these viruses are mumps virus, measles virus, respiratory syncytial virus, canine distemper virus, Newcastle disease virus (NDV), and parainfluenza viruses 1 to 5 (PIV1-5), as well as the Hendra and Nipah viruses (1–13). Paramyxoviruses cause significant threat to public health and safety and an economic loss to agribusiness. For entry into cells, all paramyxoviruses require fusion of their membrane with their target host cell plasma membrane (4, 14–21). A detailed understanding of the fusion mechanism is key to the development of therapeutic and vaccine strategies to prevent outbreaks.
Unlike enveloped viruses that require a single glycoprotein for fusion, such as influenza virus, human immunodeficiency virus, and Ebola virus (14, 22–30), nearly all paramyxoviruses require the concerted expression of the receptor binding protein (HN, H, or G) and the metastable fusion glycoprotein (F) to trigger membrane fusion (31–37). F and HN (H or G) physically interact to decrease the activation energy barrier required for membrane merger (4, 38). HN (H or G) binds to cell surface receptors which in turn activates the F protein to undergo irreversible refolding event which culminates in the mixing of virus and host membranes (12, 35–37, 39).
Recently, two models have been postulated to explain how F and HN/G/H work in concert to bring about membrane fusion. These include the “provocateur” or stalk-mediated F-activation model and the “clamp” or dissociation model (reviewed in references 12 and 13). The clamp model posits that HN/G/H remains associated with F in its metastable, prefusion form throughout its transport to the cell surface for incorporation into virions and that the proteins disengage after receptor binding to allow F activation. The formation of the F-HN/G/H complex is said to stabilize the prefusion F until receptor binding in order to prevent premature triggering of F. However, given that prefusion F can be expressed independently of HN/G/H and can also be triggered by heat as a surrogate for HN/H/G (40–45), the clamp model does not fully explain paramyxovirus fusion mechanism. Also, evidence suggests that the affinity of H/F glycoproteins interactions varies during intracellular transport (46), indicating a more complex mechanism governing fusion triggering. The provocateur mechanism alternatively suggests that HN/H/G actively associates with and triggers the metastable F protein only after receptor binding. Therefore, prior association of HN/H/G with F is not required for either stabilization or transport of prefusion F (34, 47).
In either of these models of paramyxovirus fusion triggering, the requirement for receptor engagement is indispensable for fusion triggering. The HN attachment protein from viruses such as NDV, mumps virus, PIV5, human parainfluenza viruses 1 through 4 (hPIV1-4), and Sendai virus use sialic acid as their receptor (48–52), whereas H proteins and G proteins use proteinaceous receptors (measles viruses uses multiple receptors, including CD150-Slam, PVRL4/nectin-4, and CD46 [53–57], and Nipah and Hendra viruses use Ephrin B2 or B3 [58–60]). The atomic structure of the receptor binding HN/G/H proteins have been solved for a number of different paramyxoviruses (61–68). The recent crystal structures of head and stalk domains reveal HN as tetrameric proteins with a receptor binding head and an extended ectodomain stalk that forms a four-helix bundle (4HB). The head and the stalk are linked by a flexible linker (Fig. 1). The crystal structure of NDV HN reveals a “four-heads-down” arrangement of the heads. Similarly, the PIV5 HN structures were shown to exhibit a “two-heads-down” and “two-heads-up” arrangement of the heads (61) or “four heads up” (50). The “two-heads-up/two-heads-down” arrangement is presumably an intermediate conformation. From these head arrangements, it has been hypothesized that in the prefusion state, the HN adopts a conformation that blocks F from interacting with the HN stalk. Receptor binding by the HN heads “swings” the head groups to the “up” position, allowing access to the stalk for F activation (13, 37, 39, 69). A derivative model proposed on the basis of cryotomography observations of hPIV3-infected cells suggests that HN exists in two organizations: (i) tetrameric arrangements of HN in the “heads down” position and without association with F and (ii) regions of complex surface density that contained HN in a “heads-up” state but without activating F, and that interaction with HN in this arrangement is not sufficient to activate F. It is proposed that only on receptor engagement by the HN globular head does HN activate F (70).
FIG 1.
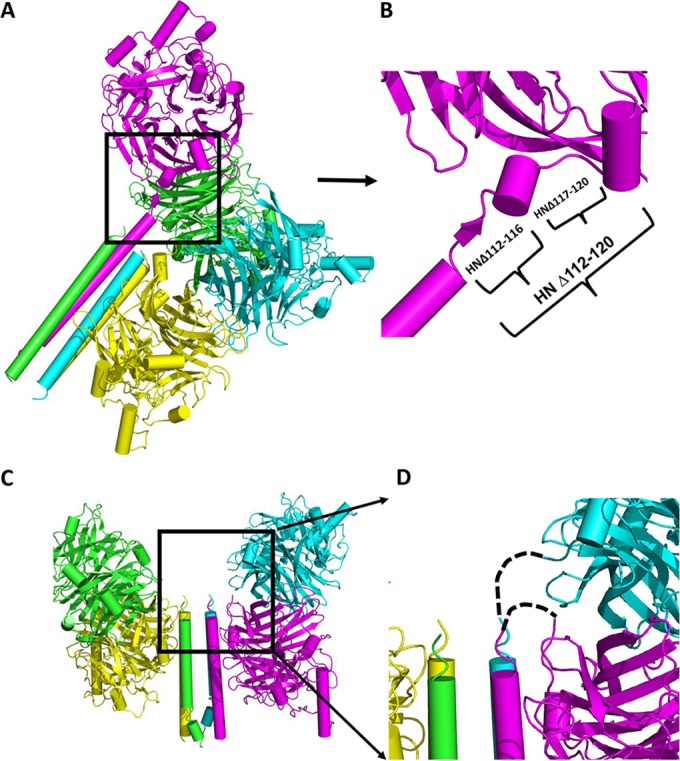
HN structure with head-stalk connecting loops. (A) Structure of PIV5 virus HN showing “two heads up” and “two heads down” with four helix bundle stalk conformation (PDB 4JF7) (61) shown in a 45° slanted representation. Each protomer is colored differently. (B) Expanded section of a single protomer in panel A showing connecting “flexible” helix and loops. The deletion mutations made are indicated by brackets. (C) Structure of NDV HN (PDB 3T1E) showing four heads down arrangement of the head and stalk. (D) Expanded section of panel C showing missing residues connecting heads to stalks indicated by dashed lines.
The factors which regulate this kind of “swinging motion” of the bulky head groups have not been investigated. Given the inherent flexibility of the connecting loop between the head and the stalk, we hypothesized that the residues which form this flexible loop are essential for the movement of the HN head in order to trigger F of paramyxoviruses.
We investigated the role of the flexible head-stalk linker region of HN protein of PIV5 and NDV. In addition to deleting the entire linker region, proline mutations were also introduced to rigidify the flexible regions due to its propensity to provide rigid separation between domains, as well as increased structural stiffness (71, 72). We observed that the introduction of proline residues inhibited HN-mediated activation of F for both PIV5 and NDV. Recombinant PIV5 virus harboring the same mutations in HN could not be rescued, presumably due to defects in virus-cell fusion. The results from this study strengthen the argument for the HN stalk-activation model of paramyxovirus fusion.
MATERIALS AND METHODS
Cells and antibodies.
HEK 293T, MDBK, CV-1, and Vero-ATCC cells were grown and maintained in Dulbecco modified eagle medium (DMEM) supplemented with 10% (vol/vol) fetal bovine serum (FBS) and 1% penicillin-streptomycin. BHK-21F cells were grown in DMEM containing 10% (vol/vol) FBS and 10% (vol/vol) tryptose phosphate broth. For BSR-T7/5 cells (BHK clone expressing T7 RNA polymerase), 500 μg/ml G418 was added during every third passage in DMEM–10% (vol/vol) FBS. Immunoprecipitation of PIV5 HN was carried out using a combination of monoclonal antibody 1b (MAb 1b) and polyclonal antibody R471 (PAb R471) as described previously (69). Immunoprecipitation of NDV HN and mutants were done using polyclonal antibody specific for the Australia-Victoria strain of NDV HN.
Cloning and mutagenesis.
PIV5 (W3A) F and Australia-Victoria NDV F were cloned into the pCAGGS vector for expression in mammalian cell lines (39). PCR fragments of PIV5 and NDV HN were first cloned into the pmEGFP-C1 vector using Sac1 and Kpn1 sites. Proline and deletion mutations were introduced into the pmEGFP-C1-HN construct using a QuikChange mutagenesis kit (Agilent Technologies, Santa Clara, CA) according to the manufacturer's instructions. The resultant mutants were subcloned into pCAGGS vector for mammalian cell expression. The nucleotide sequence of the entire open reading frame for each mutant was verified by sequencing at the Northwestern University Genomics Core sequencing facility.
Immunoprecipitation and SDS-PAGE.
To examine the expression of wild-type (Wt) and mutant HN proteins, 293T cells were transfected with pCAGGS-HN and mutant plasmids using Lipofectamine LTX with Plus reagent (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer's protocol. After 16 h posttransfection, the cells were starved for 30 min in methionine- and cysteine-free DMEM and radiolabeled with a 30-min pulse of a 50-μCi/well express protein labeling mix ([35S]Promix; Perkin-Elmer, Boston, MA), followed by a 1.5-h chase with DMEM–10% FBS. Proteins were immunoprecipitated using polyclonal and monoclonal antibodies specific for PIV5 and NDV HN proteins. Polypeptides were analyzed using reducing and nonreducing 10% SDS-PAGE gels.
Flow cytometry.
Cell surface expression of HN in 293T cells was assessed using flow cytometry. Briefly, overnight transfected cells were washed with phosphate-buffered saline (PBS) containing 0.02% NaN3, followed by incubation with a 1:100 dilution of PIV5 HN polyclonal (R471) or monoclonal (1b) antibody for PIV5 and, for NDV, AV-HN, polyclonal antibody at 4°C for 1 h. After a washing step with 5× PBS, the cells were incubated for 1 h with a 1:100 dilution of fluorescein isothiocyanate-conjugated goat anti-rabbit IgG or goat anti-mouse IgG (Jackson ImmunoResearch, West Grove, PA) as the secondary antibody for detection in a FACSCalibur flow cytometer (Becton Dickinson, Franklin Lakes, NJ). Raw data were processed using Cell ProQuest software to calculate the mean fluorescence intensity.
Hemadsorption assay.
To quantify the receptor binding activity of both Wt and mutant HNs, monolayer cultures of 293T cells in six-well plates were transfected with pCAGGS-HN or HN mutants (1 μg of DNA/well). After 18 h, the cells were washed gently with ice-cold PBS+ (i.e., PBS containing calcium and magnesium), followed by incubation for 2 h with 1% chicken red blood cells (RBCs) in PBS+ at 4°C to allow receptor binding but not fusion. The cells were washed with cold PBS+ 5× to remove unbound RBCs. Bound RBCs were lysed in 0.5 ml of ice-cold double-distilled H2O (ddH2O) and rocked for 2 h at 4°C. The lysate was clarified by centrifugation, and the absorbance of the supernatant was read at 410 nm on a Beckman Coulter DU 730 Life Sciences UV/Vis spectrophotometer (Beckman Coulter, Brea, CA).
Neuraminidase activity assay.
293T cells were transfected with Wt and mutant HN plasmids (1 μg/well) using Lipofectamine LTX reagent according to the manufacturer's protocol. The cells were detached from the plates using 530 μM EDTA (500 μl/well) in PBS. The cells were then centrifuged at 5,000 rpm for 1 min, resuspended in PBS+, and pelleted for 1 min at 5,000 rpm at 4°C to remove residual EDTA. Cell pellets were resuspended in 100 μl of 125 mM sodium acetate buffer (pH 4.75) containing 6.25 mM CaCl2. A 25-μl portion of 5 mM 4-methylumbelliferyl-N-acetyl-α-d-neuraminic acid (Sigma-Aldrich, St. Louis, MO) was added to the reaction. The reaction was allowed to proceed for 30 min at 37°C with occasional mixing. The, 75 μl of 20 mM sodium carbonate buffer (pH 10.4) was added to stop the reaction. The cells were centrifuged, and 180 μl of the supernatant was transferred to a 96-well plate. The fluorescence of the cleaved substrate was read at excitation and emission wavelengths of 356 and 450 nm, respectively, using a SpectraMax M5 plate reader (Molecular Devices, Sunnyvale, CA).
Luciferase reporter fusion assay.
To quantify the extent of fusion promotion by Wt HN and mutants, monolayer cultures of Vero cells (70 to 80% confluence) were transfected with 1 μg each of the pCAGGS-F, pCAGGS-HN, and pT7-luciferase (a plasmid that expresses firefly luciferase under T7 polymerase control). BSR-T7/5 cells, expressing T7 RNA polymerase, were removed with 50 mM EDTA, washed with PBS+, and resuspended in DMEM–10% FBS. Transfected Vero cells were overlaid with BSR-T7/5 cells, followed by incubation for 6 h at 37°C. Unbound BSR-T7/5 cells were gently washed with PBS+. Reporter Glo lysis buffer (Promega, Madison, WI) was used to lyse the cells. Subsequently, the cell lysates were pelleted by centrifugation, and 150 μl of the cleared lysates was added to a 96-well dish, along with 150 μl of the luciferase assay substrate (Promega). The luciferase activity of both Wt and mutants were quantified using a SpectraMax M5 plate reader (Molecular Devices). The luciferase activity was expressed as relative light units and normalized to that of Wt relative luciferase units.
Generation of recombinant viruses from cloned cDNA (reverse genetics).
rPIV5-EGFP is an infectious clone of PIV5 and a variant of the previously described pBH276 (73, 74). In this variant the intergenic regions were modified to contain the enhanced green fluorescent protein (EGFP) gene inserted between V/P and M genes. To construct recombinant virus with mutations in HN, an In-Fusion cloning kit (Clontech, Mountain View, CA) was used to insert PCR products of HN harboring the mutations in the head-stalk linker region into the rPIV5-EGFP genome. The cDNA construct was sequenced to ensure the presence of the desired HN mutation prior to transfection. To recover virus from the genomic cDNA, 1 μg of the genome plasmid and support plasmids (100 ng of pCAGGS-N, 20 ng of pCAGGS-P, and 0.5 μg of pCAGGS-L) were transfected into BRS-T7 cells using standard transfection protocols. On day 5 after transfection, the supernatants (P-1) were collected and centrifuged at 1,500 rpm to remove debris. BHK cells were infected with a 1:3 dilution of P-1 virus culture in DMEM–1% bovine serum albumin. After development of syncytia in BHK cells, the supernatant (P-2) was collected and further amplified in MDBK cells. The MDBK cell amplified virus (P-3) was used to determine the infectivity of recovered Wt and mutant virus. Syncytia from P-2 were stained using a Hema3 staining protocol (Fisher Scientific, Pittsburgh, PA) and photographed using an inverted phase-contrast microscope (Diaphot; Nikon, Melville, NY) connected to a digital camera (DCS 760; Kodak, Rochester, NY).
Confocal microscopy (virus entry) assay.
To assess the infectivity of the recovered viruses, 100% confluent CV-1 cells were infected with 1:3 dilution of P-3. After 6 h of incubation at 37°C, the virus was removed, and the cells were incubated overnight with DMEM–2% FBS media. At 18 h postinfection (p.i.), the cells were fixed in formaldehyde, permeabilized with 0.05% saponin solution, and stained with DAPI (4′,6′-diamidino-2-phenylindole). The cells were then imaged with a Zeiss LSM 800 with Airyscan microscope using a ×20 objective lens.
RESULTS
PIV5-HN mutagenesis and expression.
The atomic structure of PIV5 HN revealed a tetrameric head group attached to a 4HB stalk via a short linker. The apparent flexibility of the HN head-stalk linker results in a lack of electron density for this region for three of the four chains of PIV5 HN (Fig. 1A and B). For NDV there was no electron density for the presumed short linker between the stalk and the head groups (61, 62) (Fig. 1). The PIV5 HN linker for which there was interpretable electron density is made up of a short helix sandwiched by two short loops (Figure 1B). The flexibility in this region implies that it could serve as a “pivot” around which the bulky head can move away from the stalk to facilitate F-HN interaction. This hypothesis is consistent with the stalk-activated model of fusion. To test this notion, residues that span this region of HN were either deleted or substituted with proline, with the aim of restricting the movement of the protein backbone. Proline residues were introduced because of their restricted dihedral angles, which can lead to increased stability (72). To determine the biological activity of PIV5 Wt HN and HN mutants, protein expression was first assessed in 293T cells. As shown in Fig. 2A (upper panel), all of the mutants were expressed to levels similar to Wt HN when immunoprecipitated with a polyclonal antibody specific for PIV5 HN. However, immunoprecipitation with HN conformation-specific monoclonal antibody (MAb 1b) indicated that some of the mutants showed reduced amounts that could be recognized (Fig. 2A, lower panel, and Fig. 2B). This reduction in binding could be due to loss of specific epitopes or HN protein misfolding. To assess cell surface expression of the Wt and mutant HN proteins, flow cytometry was used to quantify the extent of cell surface expression. As shown in the histogram in Fig. 2B, proline-substituted residues at positions 118 to 120, as well as two of the three deletion mutants (HNΔ112-120 and HNΔ117-120), failed to be expressed at the cell surface. The defect in cell surface expression is consistent with reduction in specific monoclonal antibody binding, which is likely due, in part, to protein misfolding.
FIG 2.
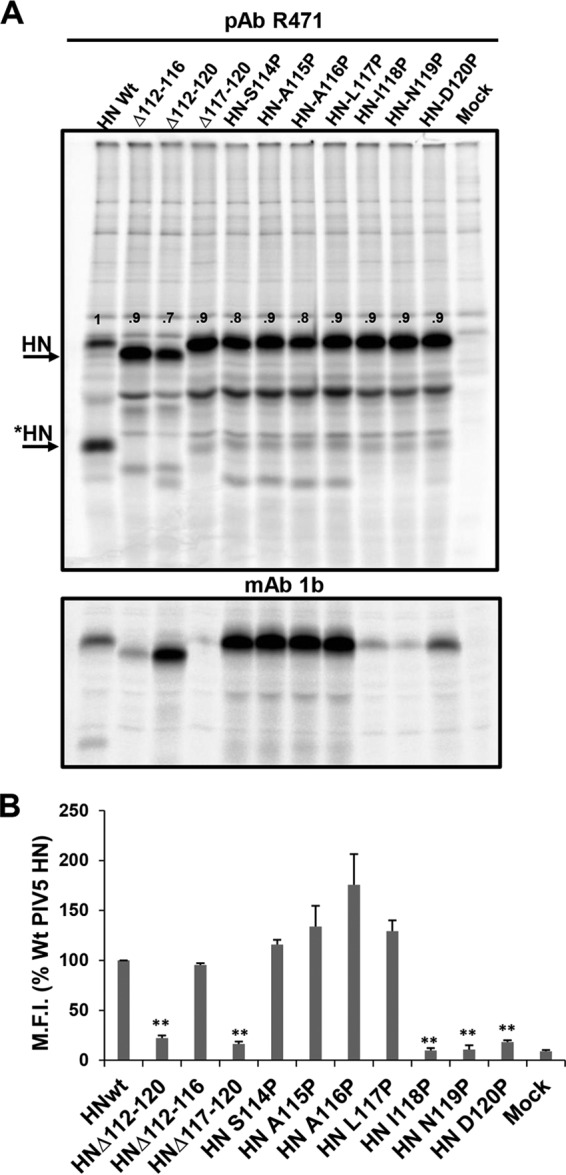
Expression of Wt and PIV5 HN mutants. Deletion mutants and point mutants of PIV5 HN domain were made in pCAGGS-HN vector. (A) Expression of HN mutants. 293T cells were transfected with plasmids expressing HN mutants and at 16 h p.i. transfection were 35S metabolically labeled for 2 h. The proteins were immunoprecipitated with PAb R471 (top) or MAb 1b (bottom). The proteins were analyzed by SDS-PAGE on a 10% gel. The band intensity was quantified using ImageJ and normalized by Wt HN. *HN indicates the previously characterized “clipped HN” (84). The band intensity values were normalized by total Wt HN, i.e., [HN + *HN]. (B) Cell surface expression of Wt and mutant HN proteins. The mean fluorescence intensity of each protein was normalized to that of the Wt. Error bars indicate the standard deviations of three independent values. P values were assessed using a Student t test (**, P < 0.01; otherwise, P > 0.05).
Substitution of proline into the flexible PIV5-HN head-stalk linker inhibits fusion.
HN is a multifunctional protein that is important for receptor binding, neuraminidase activity, and F activation. To evaluate whether mutations in the linker region influence HN functions, we assessed its ability to bind to sialic acid receptors using an RBC hemadsorption assay. 293T cells expressing Wt and mutant HNs were overlaid with 1% chicken RBCs and incubated at 4°C to allow binding without fusion. The amounts of bound RBCs were quantified and plotted (Fig. 3A). The neuraminidase activity shown in Fig. 3B mostly mirrored the receptor binding property of Wt and mutant HN (Fig. 3A). The exception was HNΔ112-116, where enhanced neuraminidase activity and retention of nearly 60% of receptor binding activity was observed. Proline substitution in this region, particularly, S114P and A115P, did not inhibit protein expression, receptor binding, and neuraminidase activity but inhibited fusion promotion (Fig. 3C). In sum, these data suggest that the flexible linker between the head and stalk is important for F activation.
FIG 3.

Biological activity of PIV HN with mutations in head-stalk connecting loop. (A) The receptor binding activity of Wt HN and various mutants was assessed using a hemadsorption assay. Bound RBCs were lysed and quantified by measuring the absorbance at 410 nm. Values are expressed as a percentage of the Wt HN. (B) Neuraminidase activity (NA) of Wt and mutants. Values are expressed as a percentage of the Wt HN activity. Error bars indicate the standard deviations of three experiments. (C) Fusion promotion expressed as a percentage of Wt HN relative luciferase units. P values were calculated using a Student t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001; otherwise, P > 0.05 or not determined).
Restriction in the degree of freedom of NDV-HN linker inhibits fusion promotion but not sialidase activity.
Unlike PIV5 HN in which the structure of the linker for one protomer has been determined, there is no structural description for the linker of any of the four NDV HN protomers (Fig. 1C and D). However, the sequences of NDV and PIV5 HNs are highly homologous. Residues spanning positions 116 to 123 of NDV HN were either deleted or individually substituted with proline. Protein expression was verified by 35S metabolic labeling in 293T cells transfected with respective plasmids. As shown in Fig. 4A, all but HN-A118P were expressed to levels comparable to those for Wt HN (Fig. 4A upper). All mutants were expressed as disulfide-linked dimers except those lacking Cys123, the residue that forms the disulfide-linked HN dimer (Fig. 4A, lower panel) (62). Similarly, all of the deletion and proline mutants except A118P exhibited cell surface expression to levels similar to those observed with the Wt (Fig. 4B). The lack of an HN species of normal electrophoretic mobility for HN mutant A118P may be due to a lack of proper folding and subsequent degradation. Although receptor binding of deletion and proline substituted mutants differed (Fig. 5A), there were comparable levels of neuraminidase activity (Fig. 5B). In addition to the deletion mutants, two proline mutants (positions 116 and 117) resulted in a significant reduction in fusion promotion (Fig. 5C) The HN mutants G116P and A117P exhibited no defects in receptor binding and neuraminidase activity. The lack of fusion promotion strongly indicated that these residues of the NDV HN may play an important role in fusion triggering. For example, NDV HN residues 116 to 123 may act as “hinge” which dictates receptor binding head movement consistent with the observations made above with PIV5 HN.
FIG 4.
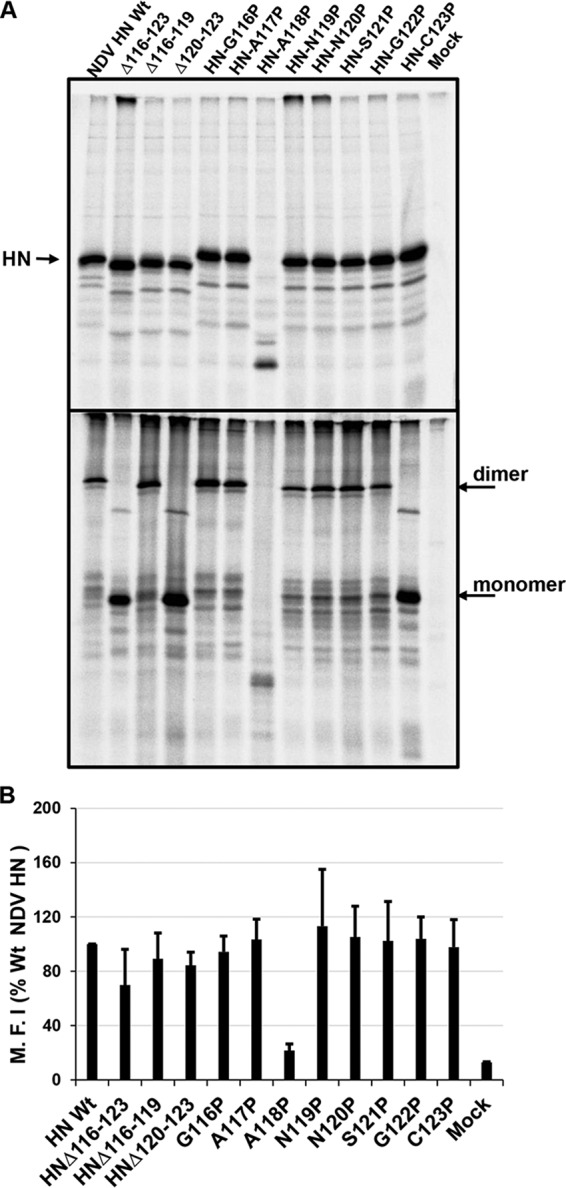
Expression of Wt and mutants NDV HN mutants. The expression of Wt and mutants of NDV HN loop (Fig. 1D) was assessed using the pCAGGS vector. Protein expression in 293T cells was evaluated as described in the legend to Fig. 2. Immunoprecipitation was performed using NDV-HN polyclonal antibody. The top (reduced) and bottom (nonreduced) panels show SDS-PAGE gels. (B) Cell surface expression of Wt and mutant NDV HN proteins. Expressed proteins were detected using NDV HN polyclonal antibodies. The mean fluorescence intensity (MFI) of each protein was normalized to that of the Wt. Error bars represent the standard deviations of three independent experiments. (Differences between the MFI values for NDV-HN Wt and mutants were not statistically significant.)
FIG 5.

Assessment of biological activity of NDV-HN mutants. (A) Receptor binding activity (hemadsorption) of Wt NDV HN and mutants. Bound RBCs were lysed in ddH2O, and the absorbance was read at a wavelength of 410 nm. Values are expressed as a percentage of the Wt HN. (B) Neuraminidase activity of Wt and mutants. Values are expressed as a percentage of the HN Wt activity. Error bars indicate the standard deviations of three independent experiments. (C) NDV HN and F-mediated cell-cell fusion. The extent of fusion promotion is expressed as percentage of Wt HN relative luciferase units. P values were calculated using Student t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001; otherwise, P > 0.05 or not determined).
Recovery of recombinant viruses with HN mutations.
The above-described experiments suggested that the flexible linker connecting the HN head to the stalk is essential for the HN conformational change necessary for F activation. To validate this observation in the context of virus entry and replication, recombinant plasmids containing the infectious virus cDNA and harboring the same HN mutations as outlined above were constructed. cDNA of the entire genome of PIV5 containing EGFP which has been found previously to replicate to the same extent as Wt PIV5 virus (74, 75) was used. The HN proline substitutions at the head-stalk linker of HN were introduced into the PIV5-EGFP genome. Upon transfection of the genome plasmid and helper genes (N, P, and L) into BSR-T7 cells, attempts were made to recover the viruses (P-1) and were subsequently amplified in BHK cells. After 3 days p.i., the growth of virus in BHK (P2) was observed as syncytia (Fig. 6A). Further amplification of recovered virus was done in MDBK cells. As shown in Fig. 6B, the deletion of residues 112 to 116 (HNΔ112-116) from the viral genome was found to block replication. Similarly, the presence of proline mutations in the HN head-stalk linker region (S114P and A115P) inhibited recovery of virus, whereas HN A116P was recovered and EGFP was expressed. This is consistent with the HN-A116P fusion promotion activity (Fig. 3). Taken together, the data from the above-described experiments strongly suggest that the flexible linker connecting the head and the stalk of paramyxovirus HN/G/H is essential for F triggering.
FIG 6.
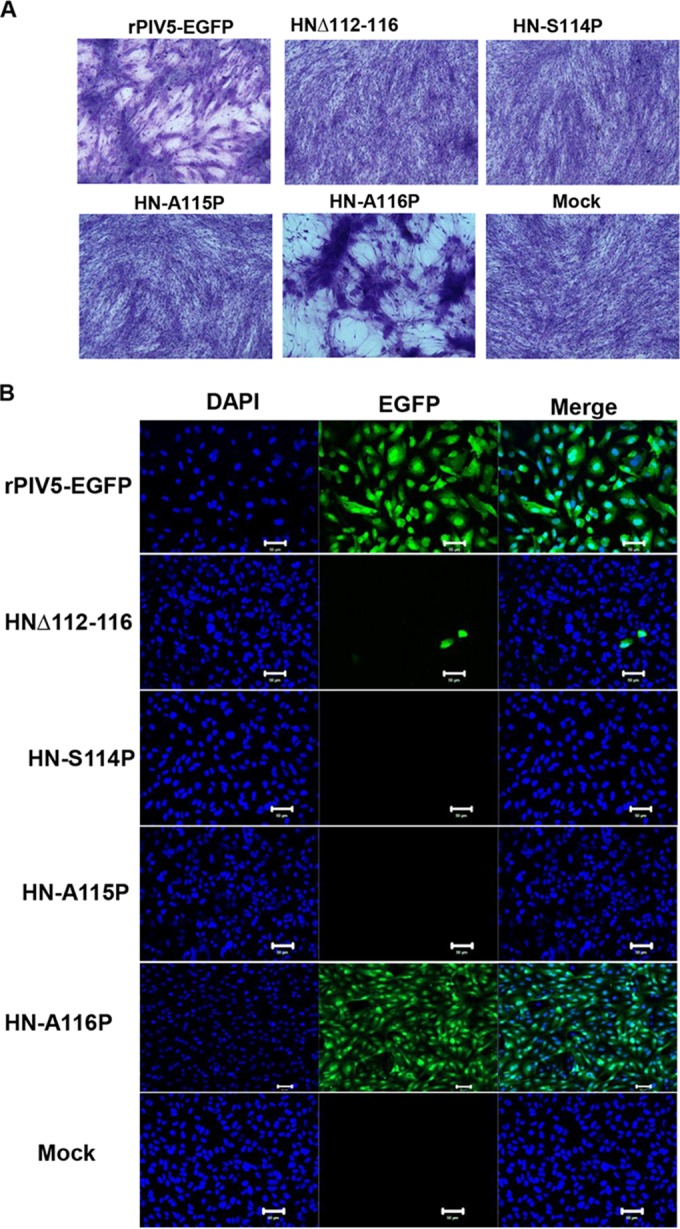
Recovery and entry of PIV5 virus and mutants: Attempts to recover Wt PIV5-EGFP and PIV5-EGFP containing HN mutations in the stalk-head connecting loop were made in BSR-T7 cells at 5 days posttransfection. Recovered Wt and mutant viruses were further amplified in BHK cells. (A) Representative images of virus-cell fusion (syncytia) in BHK cells infected with recovered virus. The cells were fixed after the removal of virus supernatant, stained, and photographed 72 h p.i. (B) Infectivity of PIV5-EGFP and mutant virus harboring HN mutations. Virus recovered from BHK cells was further amplified in MDBK cells prior to usage in the entry assay. CV-1 cells were infected with a 1:3 dilution of MDBK amplified virus. The cells were fixed, permeabilized with 0.05% saponin solution, and stained with DAPI at 18 h p.i. DAPI-stained CV-1 cells were imaged with an LSM 5 Zeiss confocal microscope (Thornwood, NY) or a Zeiss Axiovert 200M fluorescence microscope using a ×20 objective lens. Scale bar, 50 μm.
DISCUSSION
Virus-cell membrane fusion serves as a rate-determining step for entry into cells for many enveloped viruses, including paramyxoviruses. It has been known for many years that the fusion of most paramyxoviruses is orchestrated by two surface glycoproteins namely: HN/G/H and F (32, 76–80). For paramyxoviruses, it was hypothesized that the attachment proteins serve to lower the activation energy of F fusion activation (41). Recent studies have suggested that the “headless” stalk of the paramyxovirus receptor binding protein activates their cognate fusion proteins. This has been demonstrated for PIV5, NDV, mumps virus, measles virus, canine distemper virus, and henipavirus (39, 69, 81, 82). These data also show that the attachment function of the HN, G, or H can be decoupled from fusion promotion. However, in the context of virus replication and propagation, receptor binding is a crucial first step for merging virus and host membranes. The forces responsible for coupling receptor binding by the head to F activation by the stalk have not been elucidated. However, recent work by Navaratnarajah et al. (83) has indicated that the rigidity and length and of the measles H head-stalk linker segment influence H tetramerization and fusion promotion.
The biological activity of PIV5 glycoproteins have been studied by reconstituting cell-cell fusion assays (32, 47, 76). To test the functional significance of the linker that connects the HN head and stalk, cell-cell fusion assays involving PIV5 HN and NDV HN and mutants of both HNs were coexpressed with their cognate F proteins. We first deleted the entire PIV5 linker (HNΔ112-120), as well as made two shorter deletions (HNΔ112-116 and HNΔ117-120), to determine whether the length of the linker was important for fusion promotion (Fig. 1A). Deletion of the entire PIV5 HN linker reduced monoclonal antibody binding and cell surface expression. Similarly, the deletion of residues 117 to 120 severely inhibited surface expression, suggesting that these mutations drastically altered protein conformation.
Interestingly, deletion of residues 112 to 116 of PIV5 HN did not inhibit receptor binding and neuraminidase activity. Moreover, HN Δ112-116 had both Wt cell surface expression levels and Wt monoclonal antibody binding levels but lacked fusion promotion activity. We predict that this region (HN112-116) serves as a “hinge” around which the HN bulky head moves away from the stalk to promote fusion. Proline substitution for residues 114 and 115 (S114P and A115) inhibited F activation (Fig. 3). This is likely due to the restriction of the protein backbone, potentially limiting the degree of freedom of the linker that could interfere with a coil-helix or other required structural transition needed to activate F-mediated fusion. Our observation is consistent with the recent finding that interdomain restriction of movement of RSV F by proline substitution can increase protein stability (72). Taking the data together, we suggest that the amino acid residues, S114 and A115, act as a hinge around which the bulky heads “swing” the stalk to-and-fro to facilitate F-HN interactions and subsequent fusion.
We also made similar findings in experiments using NDV HN. Here, residues spanning positions 116 to 123 were either deleted or substituted with proline residues, amino acids that could restrict the free movement of the protein backbone (Fig. 4). Despite having similar expression and neuraminidase properties, the substitution of two proline residues into this region of NDV HN (residues P116 and P117) did not permit fusion promotion. This observation is consistent with that observed for PIV5 HN. Two mutants, Δ116-119 and S121P, showed enhanced receptor binding compared to Wt HN. This phenomenon may be a reflection of the indirect conformational changes or possibly an allosteric mechanism induced by the flexible linker that alters the sialidase binding pocket (or the second sialic acid binding site of NDV HN), resulting in increased receptor affinity. Therefore, this structurally undefined loop or linker in the NDV HN may serve to regulate the fusion-permissive conformation. The G116P and A117P HN mutants presumably inhibit fusion by limiting the movement of the HN head.
We used reverse genetics to generate PIV5 mutant viruses bearing mutations in the HN and expressing enhanced green fluorescent protein (EGFP). After the appearance of syncytia in BHK cells (Fig. 6A), further amplification in MDBK cells was carried out. Although Δ112-116, S114P, and A115P were not recovered, further passages in MDBK cells were performed to determine whether there could be a reversion of mutants generated. Most importantly, these data rule out inhibition due to mass action (kinetics). Although mutant viruses except A116P did not exhibit signs of virus recovery (syncytia), we assessed viral entry using fluorescence microscopy. PIV5-EGFP virus with S114P and A115P HN loop mutations could not infect cells due to a failure to recover virus. We hypothesize that the failure in virus recovery is due to defects in the ability of HN to regulate the conformational changes required for F activation. This conclusion is strongly supported by the fact that HN mutations in the head-stalk linker retained biological activity except membrane fusion promotion (Fig. 2 and 3).
Our present study fills an important gap in our mechanistic understanding of paramyxovirus fusion. Specifically, we have identified residues that regulate HN fusion promotion in the flexible linker connecting the head and the stalk of HN. We propose that these residues act as a “hinge” around which the receptor binding head move “up” to relieve the steric obstruction of the stalk to facilitate F interactions and F triggering. In conclusion, stabilization of the attachment glycoprotein of paramyxoviruses to prevent fusion may be useful from the perspective of vaccine design.
ACKNOWLEDGMENTS
We thank George P. Leser for assistance with microscopy.
This research was supported in part by National Institutes of Health Research Grants R01 AI 23173 (to R.A.L.) and R01 GM 61050 (to T.S.J.). R.A.L. is an Investigator of the Howard Hughes Medical Institute. E.A.-G. was supported by an UNCF-Merck Postdoctoral Science Research Fellowship.
REFERENCES
- 1.Choppin PW, Scheid A, Mountcastle WE. 1975. Proceedings: paramyxoviruses, membranes, and persistent infections. Neurology 25:494. [PubMed] [Google Scholar]
- 2.Peluso RW, Lamb RA, Choppin PW. 1978. Infection with paramyxoviruses stimulates synthesis of cellular polypeptides that are also stimulated in cells transformed by Rous sarcoma virus or deprived of glucose. Proc Natl Acad Sci U S A 75:6120–6124. doi: 10.1073/pnas.75.12.6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Galinski MS, Wechsler SL. 1991. The molecular biology of the paramyxovirus genus, p 41–82. In Kingsbury DW. (ed), The paramyxoviruses. Plenum Press, New York, NY. [Google Scholar]
- 4.Russell CJ, Jardetzky TS, Lamb RA. 2001. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J 20:4024–4034. doi: 10.1093/emboj/20.15.4024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iorio RM, Mahon PJ. 2008. Paramyxoviruses: different receptors—different mechanisms of fusion. Trends Microbiol 16:135–137. doi: 10.1016/j.tim.2008.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mountcastle WE, Compans RW, Choppin PW. 1971. Proteins and glycoproteins of paramyxoviruses: a comparison of simian virus 5, Newcastle disease virus, and Sendai virus. J Virol 7:47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Merz DC, Scheid A, Choppin PW. 1980. Importance of antibodies to the fusion glycoprotein of paramyxoviruses in the prevention of spread of infection. J Exp Med 151:275–288. doi: 10.1084/jem.151.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Choppin PW, Richardson CD, Merz DC, Hall WW, Scheid A. 1981. The functions and inhibition of the membrane glycoproteins of paramyxoviruses and myxoviruses and the role of the measles virus M protein in subacute sclerosing panencephalitis. J Infect Dis 143:352–363. doi: 10.1093/infdis/143.3.352. [DOI] [PubMed] [Google Scholar]
- 9.Lamb RA, Paterson RG. 1991. The nonstructural proteins of paramyxoviruses, p 181–214. In Kingsbury DW. (ed), The paramyxoviruses. Plenum Press, New York, NY. [Google Scholar]
- 10.Morrison T, Portner A. 1991. Structure, function, and intracellular processing of the glycoproteins of Paramyxoviridae, p 347–382. In Kingsbury DW. (ed), The paramyxoviruses. Plenum Press, New York, NY. [Google Scholar]
- 11.Blumberg BM, Chan J, Udem SA. 1991. Function of paramyxovirus 3′ and 5′ end sequences, p 235–247. In Kingsbury DW. (ed), The paramyxoviruses. Plenum Press, New York, NY. [Google Scholar]
- 12.Jardetzky TS, Lamb RA. 2014. Activation of paramyxovirus membrane fusion and virus entry. Curr Opin Virol 5:24–33. doi: 10.1016/j.coviro.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bose S, Jardetzky TS, Lamb RA. 2015. Timing is everything: Fine-tuned molecular machines orchestrate paramyxovirus entry. Virology 479-480:518–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huang RT, Wahn K, Klenk H-D, Rott R. 1980. Fusion between cell membrane and liposomes containing the glycoproteins of influenza virus. Arch Virol 65:123–133. doi: 10.1007/BF01317323. [DOI] [PubMed] [Google Scholar]
- 15.Hoekstra D, Klappe K, de Boer T, Wilschut J. 1985. Characterization of the fusogenic properties of Sendai virus: kinetics of fusion with erythrocyte membranes. Biochem 24:4739–4745. doi: 10.1021/bi00339a005. [DOI] [PubMed] [Google Scholar]
- 16.Paterson RG, Lamb RA. 1987. Ability of the hydrophobic fusion-related external domain of a paramyxovirus F protein to act as a membrane anchor. Cell 48:441–452. doi: 10.1016/0092-8674(87)90195-4. [DOI] [PubMed] [Google Scholar]
- 17.Di Simone C, Baldeschwieler JD. 1992. Membrane fusion of mumps virus with ghost erythrocytes and CV-1 cells. Virology 191:338–345. doi: 10.1016/0042-6822(92)90196-V. [DOI] [PubMed] [Google Scholar]
- 18.Bossart KN, Wang LF, Eaton BT, Broder CC. 2001. Functional expression and membrane fusion tropism of the envelope glycoproteins of Hendra virus. Virology 290:121–135. doi: 10.1006/viro.2001.1158. [DOI] [PubMed] [Google Scholar]
- 19.Muhlebach MD, Leonard VH, Cattaneo R. 2008. The measles virus fusion protein transmembrane region modulates availability of an active glycoprotein complex and fusion efficiency. J Virol 82:11437–11445. doi: 10.1128/JVI.00779-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zokarkar A, Connolly SA, Jardetzky TS, Lamb RA. 2012. Reversible inhibition of fusion activity of a paramyxovirus fusion (F) protein by an engineered disulfide bond in the membrane proximal external region. J Virol 86:12397–12401. doi: 10.1128/JVI.02006-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Brindley MA, Takeda M, Plattet P, Plemper RK. 2012. Triggering the measles virus membrane fusion machinery. Proc Nat Acad Sci U S A 109:E3018–E3027. doi: 10.1073/pnas.1210925109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.White J, Kartenbeck J, Helenius A. 1982. Membrane fusion activity of influenza virus. EMBO J 1:217–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stegmann T, Delfino JM, Richards FM, Helenius A. 1991. The HA2 subunit of influenza hemagglutinin inserts into the target membrane prior to fusion. J Biol Chem 266:18404–18410. [PubMed] [Google Scholar]
- 24.Melikyan GB, Jin H, Lamb RA, Cohen FS. 1997. The role of the cytoplasmic tail region of influenza virus hemagglutinin in formation and growth of fusion pores. Virology 235:118–128. doi: 10.1006/viro.1997.8686. [DOI] [PubMed] [Google Scholar]
- 25.Ivanovic T, Choi JL, Whelan SP, van Oijen AM, Harrison SC. 2013. Influenza-virus membrane fusion by cooperative fold-back of stochastically induced hemagglutinin intermediates. eLife 2:e00333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bieniasz PD, Fridell RA, Aramori I, Ferguson SSG, Caron MG, Cullen BR. 1997. HIV-1-induced cell fusion is mediated by multiple regions within both the viral envelope and the CCR-5 coreceptor. EMBO J 16:2599–2609. doi: 10.1093/emboj/16.10.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Doms RW, Moore JP. 2000. HIV-1 membrane fusion: targets of opportunity. J Cell Biol 151:F9–F14. doi: 10.1083/jcb.151.2.F9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Markosyan RM, Cohen FS, Melikyan GB. 2003. HIV-1 envelope proteins complete their folding into six-helix bundles immediately after fusion pore formation. Mol Biol Cell 14:926–938. doi: 10.1091/mbc.E02-09-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Weissenhorn W, Calder LJ, Wharton SA, Skehel JJ, Wiley DC. 1998. The central structural feature of the membrane fusion protein subunit from the Ebola virus glycoprotein is a long triple-stranded coiled coil. Proc Natl Acad Sci U S A 95:6032–6036. doi: 10.1073/pnas.95.11.6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ito H, Watanabe S, Sanchez A, Whitt MA, Kawaoka Y. 1999. Mutational analysis of the putative fusion domain of Ebola virus glycoprotein. J Virol 73:8907–8912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Paterson RG, Harris TJR, Lamb RA. 1984. Fusion protein of the paramyxovirus simian virus 5: nucleotide sequence of mRNA predicts a highly hydrophobic glycoprotein. Proc Natl Acad Sci U S A 81:6706–6710. doi: 10.1073/pnas.81.21.6706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Horvath CM, Paterson RG, Shaughnessy MA, Wood R, Lamb RA. 1992. Biological activity of paramyxovirus fusion proteins: factors influencing formation of syncytia. J Virol 66:4564–4569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lamb RA, Kolakofsky D. 1996. Paramyxoviridae: the viruses and their replication, p 1177–1204. In Fields BN, Knipe DM, Howley PM (ed), Fields virology, 3rd ed Lippincott-Raven Press, Philadelphia, PA. [Google Scholar]
- 34.Paterson RG, Johnson ML, Lamb RA. 1997. Paramyxovirus fusion (F) protein and hemagglutinin-neuraminidase (HN) protein interactions: intracellular retention of F and HN does not affect transport of the homotypic HN or F protein. Virology 237:1–9. doi: 10.1006/viro.1997.8759. [DOI] [PubMed] [Google Scholar]
- 35.Russell CJ, Kantor KL, Jardetzky TS, Lamb RA. 2003. A dual-functional paramyxovirus F protein regulatory switch segment: activation and membrane fusion. J Cell Biol 163:363–374. doi: 10.1083/jcb.200305130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lamb RA, Parks GD. 2013. Paramyxoviridae: the viruses and their replication, p 957–995. In Knipe DM, Howley PM (ed), Fields virology, 6th ed Wolters Kluwer/Lippincott, Philadelphia, PA. [Google Scholar]
- 37.Bose S, Heath CM, Shah PA, Jardetzky MATS, Lamb RA. 2013. Mutations in the paramyxovirus 5 fusion protein reveal domains important for fusion triggering and metastability. J Virol 87:13520–13531. doi: 10.1128/JVI.02123-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Russell CJ, Luque LE. 2006. The structural basis of paramyxovirus invasion. Trends Microbiol 14:243–246. doi: 10.1016/j.tim.2006.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bose S, Song AS, Jardetzky TS, Lamb RA. 2014. Fusion activation through attachment protein stalk domains indicates a conserved core mechanism of paramyxovirus entry into cells. J Virol 88:3925–3941. doi: 10.1128/JVI.03741-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poor TA, Song AS, Welch BD, Kors CA, Jardetzky TS, Lamb RA. 2015. On the stability of parainfluenza virus 5 F proteins. J Virol 89:3438–3441. doi: 10.1128/JVI.03221-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Paterson RG, Russell CJ, Lamb RA. 2000. Fusion protein of the paramyxovirus SV5: destabilizing and stabilizing mutants of fusion activation. Virology 270:17–30. doi: 10.1006/viro.2000.0267. [DOI] [PubMed] [Google Scholar]
- 42.Jurgens EM, Mathieu C, Palermo LM, Hardie D, Horvat B, Moscona A, Porotto M. 2015. Measles fusion machinery is dysregulated in neuropathogenic variants. mBio 6:e02528-14. doi: 10.1128/mBio.02528-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wong JJ, Paterson RG, Lamb RA, Jardetzky TS. 2016. Structure and stabilization of the Hendra virus F glycoprotein in its prefusion form. Proc Natl Acad Sci U S A 113:1056–1061. doi: 10.1073/pnas.1523303113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu K, Chan Y-P, Bradel-Tretheway B, Akyol-Ataman Z, Zhu Y, Dutta S, Yan L, Feng Y, Wang L-F, Skiniotis G, Lee B, Zhou ZH, Broder CC, Aguilar HC, Nikolov DB. 2015. Crystal structure of the prefusion Nipah virus fusion glycoprotein reveals a novel hexamer-of-trimers assembly. PLoS Pathog 11:e1005322. doi: 10.1371/journal.ppat.1005322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chan YP, Lu M, Dutta S, Yan L, Barr J, Flora M, Feng YR, Xu K, Nikolov DB, Wang LF, Skiniotis G, Broder CC. 2012. Biochemical, conformational, and immunogenic analysis of soluble trimeric forms of henipavirus fusion glycoproteins. J Virol 86:11457–11471. doi: 10.1128/JVI.01318-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brindley MA, Chaudhury S, Plemper RK. 2015. Measles virus glycoprotein complexes preassemble intracellularly and relax during transport to the cell surface in preparation for fusion. J Virol 89:1230–1241. doi: 10.1128/JVI.02754-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Connolly SA, Leser GP, Jardetzky TS, Lamb RA. 2009. Bimolecular complementation of paramyxovirus fusion and hemagglutinin-neuraminidase proteins enhances fusion: implications for the mechanism of fusion triggering. J Virol 83:10857–10868. doi: 10.1128/JVI.01191-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moscona A, Peluso RW. 1993. Relative affinity of the human parainfluenza virus type 3 hemagglutinin-neuraminidase for sialic acid correlates with virus-induced fusion activity. J Virol 67:6463–6468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Robach JG, Lamb RA. 2010. Analysis of parainfluenza virus-5 hemagglutinin-neuraminidase protein mutants that are blocked in internalization and degradation. Virology 406:189–201. doi: 10.1016/j.virol.2010.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuan P, Thompson T, Wurzburg BA, Paterson RG, Lamb RA, Jardetzky TS. 2005. Structural studies of the parainfluenza virus 5 hemagglutinin-neuraminidase tetramer in complex with its receptor, sialyllactose. Structure 13:1–13. doi: 10.1016/j.str.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 51.Tozawa H, Watanabe M, Ishida N. 1973. Structural components of Sendai virus. Serological and physiochemical characterization of hemagglutinin subunit associated with neuraminidase activity. Virology 55:242–253. [DOI] [PubMed] [Google Scholar]
- 52.Hoekstra D, Klappe K. 1986. Sendai virus-erythrocyte membrane interaction: quantitative and kinetic analysis of viral binding, dissociation, and fusion. J Virol 58:87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Erlenhoefer C, Wurzer WJ, Lèoffler S, Schneider-Schaulies S, ter Meulen V, Schneider-Schaulies J. 2001. CD150 (SLAM) is a receptor for measles virus but is not involved in viral contact-mediated proliferation inhibition. J Virol 75:4499–4505. doi: 10.1128/JVI.75.10.4499-4505.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yanagi Y, Ono N, Tatsuo H, Hashimoto K, Minagawa H. 2002. Measles virus receptor SLAM (CD150). Virology 299:155–161. doi: 10.1006/viro.2002.1471. [DOI] [PubMed] [Google Scholar]
- 55.Welstead GG, Hsu EC, Iorio C, Bolotin S, Richardson CD. 2004. Mechanism of CD150 (SLAM) down regulation from the host cell surface by measles virus hemagglutinin protein. J Virol 78:9666–9674. doi: 10.1128/JVI.78.18.9666-9674.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Naniche D, Varior-Krishnan G, Cervoni F, Wild TF, Rossi B, Rabourdidn-Combe C, Gerlier D. 1993. Human membrane cofactor protein (CD46) acts as a cellular receptor for measles virus. J Virol 67:6025–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Muhlebach MD, Mateo M, Sinn PL, Prufer S, Uhlig KM, Leonard VH, Navaratnarajah CK, Frenzke M, Wong XX, Sawatsky B, Ramachandran S, McCray PB Jr, Cichutek K, von Messling V, Lopez M, Cattaneo R. 2011. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature 480:530–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bonaparte MI, Dimitrov AS, Bossart KN, Crameri G, Mungall BA, Bishop KA, Choudhry V, Dimitrov DS, Wang LF, Eaton BT, Broder CC. 2005. Ephrin-B2 ligand is a functional receptor for Hendra virus and Nipah virus. Proc Natl Acad Sci U S A 102:10652–10657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Xu K, Rajashankar KR, Chan YP, Himanen JP, Broder CC, Nikolov DB. 2008. Host cell recognition by the henipaviruses: crystal structures of the Nipah G attachment glycoprotein and its complex with ephrin-B3. Proc Natl Acad Sci U S A 105:9953–9958. doi: 10.1073/pnas.0804797105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Negrete OA, Levroney EL, Aguilar HC, Bertolotti-Ciarlet A, Nazarian R, Tajyar S, Lee B. 2005. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 436:401–405. [DOI] [PubMed] [Google Scholar]
- 61.Welch BD, Yuan P, Bose S, Kors CA, Lamb RA, Jardetzky TS. 2013. Structure of the parainfluenza virus 5 (PIV5) hemagglutinin-neuraminidase (HN) ectodomain. PLoS Pathog 9:e1003534. doi: 10.1371/journal.ppat.1003534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yuan P, Swanson KA, Leser GP, Paterson RG, Lamb RA, Jardetzky TS. 2011. Structure of the Newcastle disease virus hemagglutinin-neuraminidase (HN) ectodomain reveals a four-helix bundle stalk. Proc Nat Acad Sci U S A 108:14920–14925. doi: 10.1073/pnas.1111691108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Colf LA, Juo ZS, Garcia KC. 2007. Structure of the measles virus hemagglutinin. Nat Struct Mol Biol 14:1227–1228. doi: 10.1038/nsmb1342. [DOI] [PubMed] [Google Scholar]
- 64.Santiago C, Celma ML, Stehle T, Casasnovas JM. 2010. Structure of the measles virus hemagglutinin bound to the CD46 receptor. Nat Struct Mol Biol 17:124–129. doi: 10.1038/nsmb.1726. [DOI] [PubMed] [Google Scholar]
- 65.Lawrence MC, Borg NA, Streltsov VA, Pilling PA, Epa VC, Varghese JN, McKimm-Breschkin JL, Colman PM. 2004. Structure of the haemagglutinin-neuraminidase from human parainfluenza virus type III. J Mol Biol 335:1343–1357. doi: 10.1016/j.jmb.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 66.Bowden TA, Crispin M, Harvey DJ, Aricescu AR, Grimes JM, Jones EY, Stuart DI. 2008. Crystal structure and carbohydrate analysis of Nipah virus attachment glycoprotein: a template for antiviral and vaccine design. J Virol 82:11628–11636. doi: 10.1128/JVI.01344-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bowden TA, Aricescu AR, Gilbert RJ, Grimes JM, Jones EY, Stuart DI. 2008. Structural basis of Nipah and Hendra virus attachment to their cell-surface receptor ephrin-B2. Nat Struct Biol 15:567–572. doi: 10.1038/nsmb.1435. [DOI] [PubMed] [Google Scholar]
- 68.Taylor G. 1996. Sialidases: structures, biological significance and therapeutic potential. Curr Opin Struct Biol 6:830–837. doi: 10.1016/S0959-440X(96)80014-5. [DOI] [PubMed] [Google Scholar]
- 69.Bose S, Zokarkar A, Welch BD, Leser GP, Jardetzky TS, Lamb RA. 2012. Fusion activation by a headless parainfluenza virus 5 hemagglutinin-neuraminidase stalk suggests a modular mechanism for triggering. Proc Nat Acad Sci U S A 109:E2625–E2634. doi: 10.1073/pnas.1213813109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gui L, Jurgens EM, Ebner JL, Porotto M, Moscona A, Lee KK. 2015. Electron tomography imaging of surface glycoproteins on human parainfluenza virus 3: association of receptor binding and fusion proteins before receptor engagement. mBio 6:e02393-14. doi: 10.1128/mBio.02393-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Papaleo E, Saladino G, Lambrughi M, Lindorff-Larsen K, Gervasio FL, Nussinov R. 2016. The role of protein loops and linkers in conformational dynamics and allostery. Chem Rev 116:6391–6423. doi: 10.1021/acs.chemrev.5b00623. [DOI] [PubMed] [Google Scholar]
- 72.Krarup A, Truan D, Furmanova-Hollenstein P, Bogaert L, Bouchier P, Bisschop IJ, Widjojoatmodjo MN, Zahn R, Schuitemaker H, McLellan JS, Langedijk JP. 2015. A highly stable prefusion RSV F vaccine derived from structural analysis of the fusion mechanism. Nat Commun 6:8143. doi: 10.1038/ncomms9143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.He B, Paterson RG, Ward CD, Lamb RA. 1997. Recovery of infectious SV5 from cloned DNA and expression of a foreign gene. Virology 237:249–260. doi: 10.1006/viro.1997.8801. [DOI] [PubMed] [Google Scholar]
- 74.He B, Leser GP, Paterson RG, Lamb RA. 1998. The paramyxovirus SV5 small hydrophobic (SH) protein is not essential for virus growth in tissue culture cells. Virology 250:30–40. doi: 10.1006/viro.1998.9354. [DOI] [PubMed] [Google Scholar]
- 75.Zhang L, Collins PL, Lamb RA, Pickles RJ. 2011. Comparison of differing cytopathic effects in human airway epithelium of parainfluenza virus 5 (W3A), parainfluenza virus type 3, and respiratory syncytial virus. Virology 421:67–77. doi: 10.1016/j.virol.2011.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bagai S, Puri A, Blumenthal R, Sarkar DP. 1993. Hemagglutinin-neuraminidase enhances F protein-mediated membrane fusion of reconstituted Sendai virus envelopes with cells. J Virol 67:3312–3318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Bagai S, Lamb RA. 1997. A glycine to alanine substitution in the paramyxovirus SV5 fusion peptide increases the initial rate of fusion. Virology 238:283–290. doi: 10.1006/viro.1997.8858. [DOI] [PubMed] [Google Scholar]
- 78.Paterson RG, Hiebert SW, Lamb RA. 1985. Expression at the cell surface of biologically active fusion and hemagglutinin-neuraminidase proteins of the paramyxovirus simian virus 5 from cloned cDNA. Proc Natl Acad Sci U S A 82:7520–7524. doi: 10.1073/pnas.82.22.7520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Paterson RG, Hiebert SW, Lamb RA. 1985. Primary structure of the fusion protein and the hemagglutinin-neuraminidase protein of the paramyxovirus SV5, p 303–308. In Modern approaches to vaccines. Cold Spring Harbor Publications, Cold Spring Harbor, NY. [Google Scholar]
- 80.Hu X, Ray R, Compans RW. 1992. Functional interactions between the fusion protein and hemagglutinin-neuraminidase of human parainfluenza viruses. J Virol 66:1528–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Brindley MA, Suter R, Schestak I, Kiss G, Wright ER, Plemper RK. 2013. A stabilized headless measles virus attachment protein stalk efficiently triggers membrane fusion. J Virol 87:11693–11703. doi: 10.1128/JVI.01945-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu Q, Stone JA, Bradel-Tretheway B, Dabundo J, Benavides Montano JA, Santos-Montanez J, Biering SB, Nicola AV, Iorio RM, Lu X, Aguilar HC. 2013. Unraveling a three-step spatiotemporal mechanism of triggering of receptor-induced Nipah virus fusion and cell entry. PLoS Pathog 9:e1003770. doi: 10.1371/journal.ppat.1003770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Navaratnarajah CK, Rosemarie Q, Cattaneo R. 2016. A Structurally unresolved head segment of defined length favors proper measles virus hemagglutinin tetramerization and efficient membrane fusion triggering. J Virol 90:68–75. doi: 10.1128/JVI.02253-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leser GP, Ector KJ, Lamb RA. 1996. The paramyxovirus simian virus 5 hemagglutinin-neuraminidase glycoprotein, but not the fusion glycoprotein, is internalized via coated pits and enters the endocytic pathway. Mol Biol Cell 7:155–172. doi: 10.1091/mbc.7.1.155. [DOI] [PMC free article] [PubMed] [Google Scholar]