

Abstract
Several evidences showed that patients with gastrointestinal stromal tumors (GISTs) develop additional malignancies. However, thorough incidence of second tumors remains uncertain as the possibility of a common molecular pathogenesis.
A retrospective series of 128 patients with histologically proven GIST treated at our institution was evaluated. Molecular analysis of KIT and PDGFR-α genes was performed in all patients. Following the involvement of KRAS mutation in many tumors’ pathogenesis, analysis of KRAS was performed in patients with also second neoplasms.
Forty-six out of 128 GIST patients (35.9%) had a second neoplasm. Most second tumors (52%) raised from gastrointestinal tract and 19.6% from genitourinary tract. Benign neoplasms were also included (21.7%). Molecular analysis was available for 29/46 patients with a second tumor: wild-type GISTs (n. 5), exon 11 (n. 16), exon 13 (n. 1), exon 9 (n. 1) KIT mutations, exon 14 PDGFR-α mutation (n. 2) and exon 18 PDGFR-α mutation (n. 4). KIT exon 11 mutations were more frequent between patients who developed a second tumor (P = 0.0003). Mutational analysis of KRAS showed a wild-type sequence in all cases. In metachronous cases, the median time interval between GIST and second tumor was 21.5 months.
The high frequency of second tumors suggests that an unknown common molecular mechanism might play a role, but it is not likely that KRAS is involved in this common pathogenesis. The short interval between GIST diagnosis and the onset of second neoplasms asks for a careful follow-up, particularly in the first 3 years after diagnosis.
Keywords: GIST, KIT, KRAS, PDGFR, second malignancies
1. Introduction
Gastrointestinal stromal tumors (GISTs) are the most common mesenchymal tumors of the digestive tract (about 15/106 people/year)[1]; GISTs occur anywhere along the gastrointestinal tract, though they are most common in stomach (50–60%) and small intestine (30–35%) and less frequent in colon, rectum, and esophagus.[2] Common sites of metastases are liver and peritoneum and rarely lymph nodes, bone, and lung. The malignant potential of a GIST is mainly determined by its size, mitotic count, and the site of origin.[3] Disease-related symptoms depend on tumor size and localization (e.g., gastrointestinal bleeding, abdominal pain), but generally GISTs’ diagnosis occurs incidentally either during abdominal surgery, at autopsy or during other procedures for unrelated diseases.[4]
Over the last 2 decades, scientific interest about this tumor has greatly increased as well as the knowledge of its complex biology. Since the discovery of c-Kit gene (KIT) mutations in GISTs in 1998,[5] this neoplasm represent a paradigm of molecular target therapies for solid tumors on the basis of the successful treatment with imatinib, a tyrosine kinase inhibitor (TKI) able to inhibit the growth of cells expressing KIT-mutant isoforms.[6] Although most GISTs are sporadic and have no established risk factors, in some cases they are observed in the context of hereditary autosomical dominant syndromes due to germ-line KIT or platelet-derived growth factor receptor α (PDGFRA) mutations or other hereditary syndromes (Carney's triad, Carney–Stratakis syndrome, and Type 1 Neurofibromatosis).[7–9] The most frequent driver mutations observed in GISTs involve KIT and PDGFRA genes (85–90%); they lead to a constitutive activation of KIT and/or PDGFRA receptors which, in turn, upregulate 2 main signal pathways (RAS-RAF-MEK-ERK and PI3K-AKT-mTOR transducer protein kinases).[10] It is noteworthy that KIT and PDGFRA genes are both located on chromosome 4q11-q12 and might be evolved from a common ancestral gene through a mechanism of duplication.[11] In addition, other genes whose expression is relatively increased in GISTs compared to other soft tissue tumors have been identified in several recent studies. Particularly, 1 GIST-specific gene, encoding for the hypothetical protein FLJ10261, named “Discovered On GIST 1” (DOG1) was identified not only in typical GISTs but also in KIT-mutation-negative GISTs.[12]
Recent evidences have demonstrated the complex biology of GISTs and their heterogeneous nature, suggesting the need of different diagnostic and therapeutic approaches. In 2013, Ricci et al[13] proposed a schematic representation of GISTs based on immunohistochemical and molecular profile, defined “GISTOGRAM.” Starting from the well-established evidence of immunohistochemical expression of KIT (95%)[14] and DOG-1 (98%),[12] as well as KIT (77%) and PDGFRA mutations (6.5%),[15] the peculiarity of this work was to redefine the amount of GISTs included in the designation “wild type.” Recently, mutually exclusive mutations occurring in activating genes other than KIT and PDGFRA have been found: 1.3% of GISTs have a mutation of NF-1 gene [16] and 2% of GISTs harbor a mutation in the gene encoding for succinate dehydrogenase (SDH), mainly occurring in patients with Carney–Stratakis diad or Carney triad.[17]BRAFV600E and KRAS mutations were observed in 2% and 4% of GISTs, respectively, with concurrent KIT or PDGFRA mutations in a study conducted by Miranda et al[18] on 2 cohorts coming from Italy and Ticino. In conclusion, the rate of GISTs that are really wild type is below 10% of all GISTs, including those cases in which a driver mutation has not yet been identified.
Emerging data suggest that the association of GISTs and secondary neoplasms, either synchronous or not, is not infrequent as several cases have been described, mostly as case reports, but also in large case series and reviews.[19,20] The overall frequency of second tumors in different series varied from 4.5% to 43%, a value higher than expected in the general population.[19] The most frequent GIST-associated cancers are gastrointestinal carcinomas, followed by extra-intestinal tumors (lymphoma/leukemia, carcinomas of prostate, breast, kidney, lung, female genital tract, carcinoid tumors, soft tissue and bone sarcomas, malignant melanomas and seminomas). In spite of these data, it has not yet been established whether the coexistence of GIST with other tumors is stochastic or a result of related pathogenetic mechanisms. Several hypotheses have been proposed, including some cancerogenic agents which influence neighboring tissues (e.g., N-methyl-N-nitro-N-nitrosoguanidine—MNNG), Helicobacter pylori infection, and mutations of proto-oncogenes encoding tyrosine kinases such as c-MET.[21–24] However, available data are insufficient for supporting any hypothesis, and no clinical study systematically analyzed the molecular profile of GISTs associated with other malignancies.
The objectives of the present paper are to assess the prevalence and histotype of second tumors in our GIST series, to compare clinico-pathologic characteristics of patients affected by GIST and other neoplasms to those affected by GIST only and, finally, to evaluate gene mutations that could explain the coexistence of GISTs and second tumors.
2. Patients and methods
2.1. Patients, malignancies, and follow-up
A retrospective analysis was conducted on 128 patients with histologically proven diagnosis of GIST treated in our Center between July 2002 and June 2014. We reviewed clinical records in order to identify the cases with GISTs and associated tumors, benign neoplasms included. Secondary tumors were defined as synchronous if diagnosed during staging or surgery of GIST or not-synchronous if previously or subsequently diagnosed. GIST diagnosis was revised according to current diagnosis criteria.[25] Immunohistochemical staining was performed with CD117, CD34, desmin, and S100. Since 2010 also molecular analysis of KIT (exon 9, 11 and 13) and PDGFRA (exon 12, 14 and 18) genes was performed. Mutational analysis of KRAS (exons 2 and 3) was also performed in the same cases for which KIT and PDGFRA mutational status was available. The risk category was defined assessing the tumor size and mitotic count following Miettinen's criteria.[26] Associated malignancies were classified according to current World Health Organization (WHO) classification of malignant neoplasms. Neurofibromatosis type 1 (NF-1) and familiar GIST were also included in this study. Age, sex, tumor localization, morphological variant (epithelioid, spindle-cell, and mixed), malignant potential (risk classification), and selected immunohistochemical parameters were assessed. The median follow-up of patients was 48.7 months (range: 2–141 months).
The study has been conducted in accordance with the rules of the local Ethics Committee and the Declaration of Helsinki. All patients provided a written consent for use of their clinical data; a separate consent for molecular analyses was obtained.
2.2. Immunohistochemistry and PCR
The histological diagnosis of all GISTs has been confirmed at the Department of Pathology of Università Cattolica del Sacro Cuore. DNA was extracted from three 10 μm-slides from paraffin-embedded tissues using QIAamp DNA mini kit (Qiagen, Milan, Italy), following the manufacturer's protocol. KIT gene (9, 11, 13, and 17 exons), PDGFRA gene (12, 14, and 18 exons), and KRAS gene (2 and 3 exons) were amplified using the same primers and polymerase chain reaction (PCR) conditions previously described.[27,28] Briefly, DNA (100–200 ng) was amplified in a mixture containing 1 × PCR buffer (20 mM Tris, pH 8.3; 50 mMKCl; 1.5 mM MgCl2), deoxyribonucleotide triphosphates (200 mM each), primers (20 pM each), and 0.5 U GoTaq (Promega, Milan, Italy) in a final volume of 25 μL. After visualization onto agarose gel, stained with ethidium bromide and visualized under UV light, PCR products were treated with ExoSAP-IT (GE Healthcare, Milan, Italy) following the manufacturer's protocol, amplified with the BigDye Terminator cycle-sequencing kit (version 3.1; Applied Biosystems, Milan, Italy) using forward and reverse primers, and sequenced with an ABI PRISM 3100-Avant Genetic Analyzer (Applied Biosystems). Water was used as a negative control. The sensitivity of this method is 15% in our laboratory.
2.3. Statistical analyses
Exact Fisher test and Chi-squared test were used to establish the significance of the association between patients’ characteristics and the coexistence of GIST and second tumors and the relationship between GIST and associated tumors’ characteristics. The differences in median age at diagnosis between all patients affected by GIST and in patients with associated tumors as well as the difference in median size of GIST at diagnosis between these 2 groups were compared using Student's t test. All reported P values are 2-tailed and a level of 0.05 or less was considered statistically significant.
3. Results
We extracted clinical data of 128 patients with histologically proven diagnosis of GIST treated in our Center. In total, 69 out of 128 patients were women; median age at diagnosis was 65 years (range: 23–86 years). Median size of GIST at diagnosis was 50 mm (range: 3–240 mm). The median mitotic rate was 4 (range: 1–100). The most common primary sites were: stomach (59%), small intestine (29%), omentum (5%), and rectum (4%); anus, gastro-oesophageal junction, and vagina were involved in <1% of cases. Between small intestine GISTs, the most frequent locations were jejunum (46%), duodenum (35%), and ileum (22%). Two women affected by GISTs related to NF-1 syndrome (1.7%) and 3 cases of multiple GISTs (2.5%) were also found. Nine patients were metastatic at diagnosis (7.5%) and the most frequent sites of metastases were liver (78%) and peritoneum (56%); lymph node metastases were detected in 2 patients (22%), in association with other metastatic sites. Thirty-two GISTs (25%) belong to low or very low-risk category, 38 to the intermediate category (30%) and 58 GISTs expressed a high malignant potential (45%). Median follow-up time for all GIST patients was 48.7 months (range: 2–141 months). Characteristics of all patients affected by GIST are summarized in Table 1.
Table 1.
Characteristics of all patients affected by GIST.

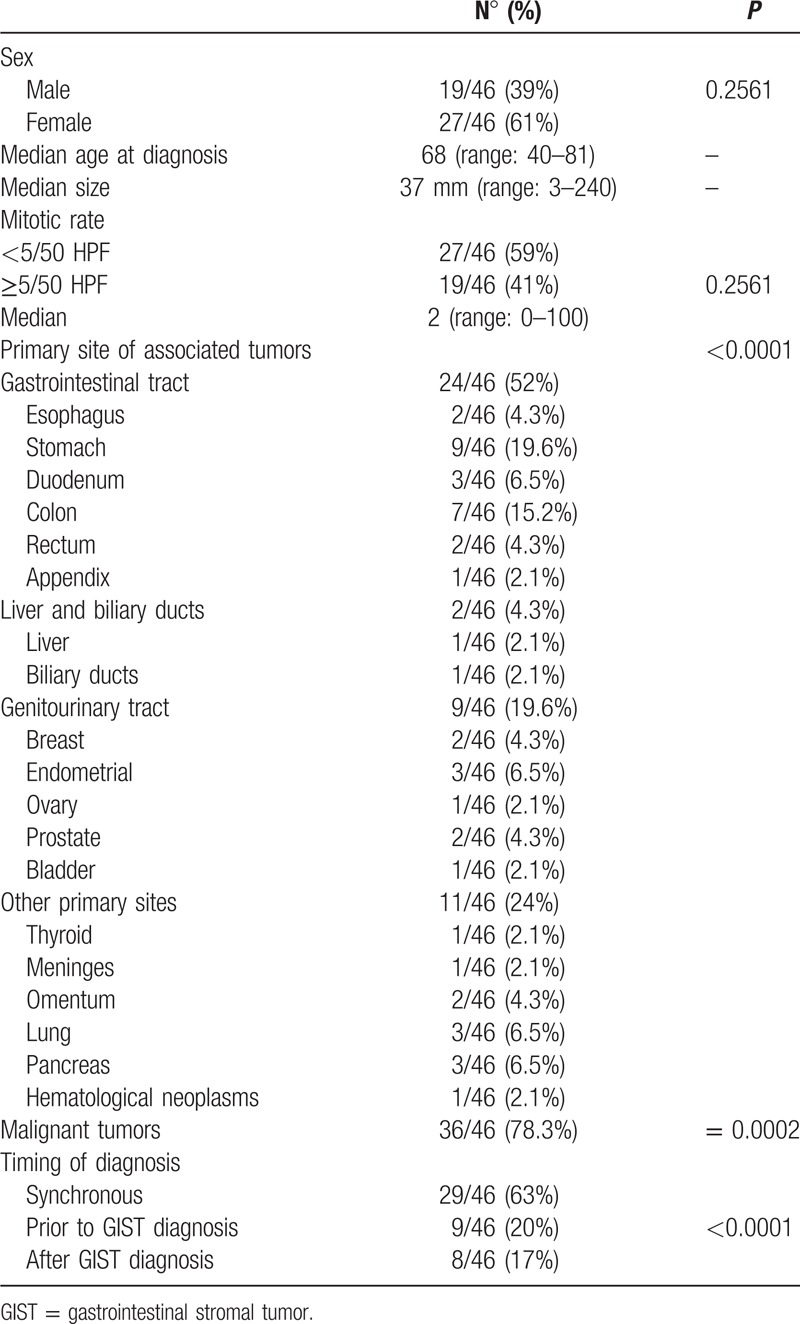
In 46 out of 128 patients affected by GIST (36%), another primary tumor was diagnosed; 37 were women (61%) but the sex was not related to the presence of second malignancies (P = 0.2561); the median age at diagnosis was 68 years (range 44–80), not statistically significant when patients with GIST and other primary malignancies were compared to patients with isolated GISTs (P = 0.06). The median size of GISTs in patients with second tumors was 50 mm (range: 3–240 cm), not significantly different from the isolated GIST group (P = 0.10). The median mitotic rate was 2 (range: 1–100) and also in this case no statistically significant difference was seen when the mitotic rate of patients affected by GIST and another primary tumor was compared to the mitotic rate of all 128 GISTs (P = 0.48); high mitotic rate (≥5/50 HPF) was not associated to second malignancies even when we compared GIST plus associated tumor group to all study population. Associated tumors most frequently raised from gastrointestinal tract (52%; P = 0.0001) and genitourinary tract (19.6%), whereas only 4.3% of associated tumors had an hepatobiliary origin. Thirty-six out of 46 cases were malignant tumors (P = 0.0002). The histotypes of associated malignant tumors were the following: gastric adenocarcinoma (n = 6), gastric diffuse large B cell lymphoma (n = 1), colorectal adenocarcinoma (n = 6), esophageal squamous carcinoma (n = 2), duodenal neuroendocrine tumor (n = 1), adenocarcinoma of the papilla of Vater (n = 2), hepatocellular carcinoma (n = 1), biliary duct carcinoma (n = 1), pancreatic adenocarcinoma (n = 1), pancreatic neuroendocrine tumor (n = 1), lung adenocarcinoma (n = 3), breast ductal carcinoma (n = 2), endometrial sarcoma (n = 1), endometrial adenocarcinoma (n = 1), prostatic adenocarcinoma (n = 2), urothelial carcinoma of the bladder (n = 1), thyroid papillary carcinoma (n = 1), peritoneal mesothelioma (n = 1), retroperitoneal liposarcoma (n = 1), Hodgkin's Lymphoma (n = 1). The benign neoplasms included in our series were: gastrointestinal multiple fibrous inflammatory polyps (n = 2), gastric tubular adenoma (n = 2), colic adenoma (n = 1), pancreatic intrapapillar mucinous neoplasm (n = 1), meningioma (n = 1), ovarian cystadenoma (n = 1), uterine leiomyoma (n = 1), mucinous cystadenoma of appendix (n = 1).
Diagnosis of associated malignancies was performed prior to GIST diagnosis in 20% of patients, after GIST diagnosis in 17% and it was synchronous in 63% of cases. In metachronous cases, the median time interval between GIST and second tumor diagnosis was 21.5 months. Sixteen out of 29 patients (55%) with synchronous diagnosis of GISTs and other primary tumors had symptoms related to GIST (abdominal pain, bleeding); thus, second malignancy was detected only at the time of GIST workup or resection. In the remaining 13 cases, GIST was an incidental finding during pre-operatory CT scan or abdominal surgery for the associated primary tumor, and in most cases, it was a micro-GISTs (<1 cm).
Characteristics of patients affected by GIST and other primary tumors are summarized in Table 2.
Table 2.
Characteristics of patients affected by GIST and other primary tumors.
A third tumor was diagnosed during the follow-up period in 4 out of 46 patients (8.7%). In 1 case, 7 years after the diagnosis of breast cancer, a gastric GIST was surgically resected and 19 months later also a gastric adenocarcinoma was found. A second patient developed a multifocal hepatocellular carcinoma 2 years after diagnosis of gastric GIST, synchronous to an esophageal adenocarcinoma. The third patient was affected either by synchronous gastric GIST, gastric leiomyoma, and duodenal NET. The last one developed synchronous prostatic adenocarcinoma and bladder cancer and 10 years later a jejuneal GIST.
Overall 13 out of 46 patients received adjuvant Imatinib. During the follow up period, none of these patients developed metastases from GIST, whereas 4 patients with a synchronous tumor died because of the other malignancy (colangiocarcinoma, HCC, pancreatic adenocarcinoma, gastric adenocarcinoma). No patient died because of GIST.
Twenty-nine out of 46 patients with GIST and associated malignancies were evaluated for mutational analysis of KIT (exons 9, 11, 13) and PDGFRA (exons 12, 14, 18). Five GISTs were wild type, whereas 14 patients had a mutation in the KIT gene (12 in exon 11, 1 in exon 9 and 1 in exon 13) and 4 patients had mutation in the PDGFRA gene, 2 in exon 14 and other 2 in exon 18 (D842V). In those patients harboring a mutation in exon 11 of KIT gene, 8 had heterozygous mutations, 2 patients had a deletion, and in 2 cases was found a complex mutation. Interestingly, KIT mutations seems to be related to the presence of other primary tumors (P = 0.0003) and specifically exon 11 mutations (P < 0.0001). Both cases of PDFGRA mutation in exon 14 were germline mutations and were associated with fibrous gastrointestinal polyps; in addition, in 1 case, it was associated with exon 11 deletion of KIT. Mutational analysis of KRAS (exons 2 and 3) was performed in all 29 cases evaluated for KIT and PDGFRA mutation and in all patients it resulted wild type (Table 3). No relationship between genetic mutations and risk categories of GISTs was found (P = 0.26).
Table 3.
GIST mutations and associated malignancies.

4. Discussion
Over the past decade, several evidences have demonstrated the heterogeneous genetic pattern of GIST; most GISTs are driven by a pathological activation of KIT or PDGFRA, however, other genetic changes—including gain of function BRAF mutations[29] and loss of succinate dehydrogenase (SDH) complex activity[30]—have been identified in the subset of wild-type GISTs. Recently, KRAS codon 12 and 13 mutations were reported in a small subset of KIT or PDGFRA mutant GISTs.[18] No specific genetic alterations have been identified in sporadic GISTs associated to secondary malignancies as opposed to syndromic GISTs (e.g., NF-1 and triad of Carney–Stratakis) in which peculiar gene mutations were found driving pathogenesis both of GISTs and other related tumors.[16,17]
Recent studies have shown a high frequency of second malignancies in GIST patients, ranging from 2.95 to 43% in different series.[21,31–34] This discrepancy could be explained by selection criteria used in each study, in particular, regarding the inclusion of benign neoplasms. In the present study, other tumors were associated to GIST in 46 out of 120 patients (38.3%); this is one of the largest monoinstitutional series on this topic and the high level of association might be explained by inclusion of benign neoplasms and long period of observation and follow-up. The most frequent neoplasms associated with concomitant GISTs are located in gastrointestinal tract with gastric and colorectal adenocarcinomas being leading types according to literature.[20] To our knowledge, no cases of GISTs coexisting with pancreatic IPMN and peritoneal mesothelioma have been previously reported. In our series, GISTs diagnosed synchronously to second malignancy were more frequently symptomatic than reported in literature (16/29; 55%); thus, the associated tumor was mostly diagnosed incidentally. On the other hand, 13 cases in this group (45%) were incidental findings during abdominal surgery for the second tumor and classified as microGISTs (<1 cm). This result differs from most of the studies available in which almost all cases of synchronous GISTs were microGISTs or asymptomatic GISTs detected incidentally during surgery. Three possible reasons could be proposed for this event: (1) very thorough abdominal cavity inspection was performed during surgery or on examination of resection specimen to detect other lesions, even of small size; (2) more attention was paid to GIST-related symptoms which could be overlooked during diagnostic investigations for other malignancies (3) some microGISTs may not have been addressed by the surgeon to oncologist attention if also second cancer belonged to early stage and did not require any additional treatment, so that cases are not included in the database.
The follow-up period ranged from 2 to 141 months with a median of 48.7 months. In 8 patients (17%), second tumor was detected after diagnosis of GIST; the median time interval between GIST and second tumor was 22.8 months. Second malignancies of this subgroup not only raised from gastrointestinal tract (2 gastric adenocarcinoma, 1 gastric diffuse large B cell lymphoma and 1 colic adenocarcinoma), but also involved lung (2 cases), breast and prostate (1 case). Two cases of gastric adenocarcinoma growth in the same organ of previous high risk GIST both within 18 months from GIST diagnosis. This result suggests a possible common etiologic agent acting on neighbor tissues and should lead us to perform gastroscopy more frequently than annually. Ultimately, the relative short interval between GIST diagnosis and the onset of second neoplasms suggests the need to perform strict follow-up particularly in the first year after diagnosis; screening exams for breast and prostate cancer should not be overlooked in these cases. Only 1 patient in cohort of GISTs associated with second malignancies was metastatic and no one died for GIST, whereas 4 patients died for related tumors (colangiocarcinoma, HCC, pancreatic adenocarcinoma, gastric adenocarcinoma), that negatively affected the prognosis.
The occurrence of both metachronous and concomitant GISTs with other neoplasms has raised the question of whether such an event is a stochastic one or expression of a causal relationship. Several hypotheses have been proposed, but the question is still matter of debate: a possible explanation could be the existence of a unique genetic mutation involving different cells and leading to 2 different neoplasms. In our study, KIT exon 11 mutations were more frequent between patients who developed a second tumor (P = 0.0003), but the sample is too small to have a clinical relevance; in addition, exon 11 mutations are the most frequent genetic alterations in GISTs, so a definitive conclusion cannot be drawn. Only fibrous gastrointestinal polyps seem closely related to germline mutations of PDGFRA exon 14, as reported in previous studies.[35]
KRAS and the other members of RAS family (HRAS, NRAS) encode for important proteins which release mitogenic growth signals into cytoplasm and nucleus. Gain-of-function mutations of RAS caused alterations of RAS-RAF-MAPK signaling pathway leading to increased cell proliferation, that plays an important role in the pathogenesis of a wide variety of human malignancies, including colon-rectum, pancreas, lung, breast cancer, and, rarely, sarcomas. In 2012, Miranda et al[18] reported 3 cases of concurrent KRAS mutations in KIT or PDGFRA mutant GIST in 2 cohorts of patients tested independently in Italy and Ticino.[18] None of the patients carrying concomitant mutations of KIT and KRAS gene was treated with imatinib; these patients underwent surgical eradication of the tumor and were classified as disease-free subjects at the last follow-up. For this reason, the authors explored the biologic consequences of the concomitant presence of KIT and KRAS mutations through in vitro experiments and found that KRAS mutations can cause resistance of KIT-mutant GISTs to imatinib; in fact, imatinib was able to switch off the mutated receptor KIT but not the downstream signaling triggered by RAS-RAF effectors. More recently, Lasota et al[36] performed KRAS mutational analysis in a larger cohort of GISTs (514 cases) without finding any case of KRAS mutations. However, none of the 2 above-cited studies evaluated GISTs coexisting with other malignancies. Considering the role of RAS-RAF-MAPK signaling pathway as effector of activation of KIT and PDGFRA tyrosine kinases and the implication of RAS mutations in cancerogenesis of several tumors, some KRAS gene aberration might be shared by GISTs and associated tumors. In our study, we did not find any mutation in KRAS among cases for which KIT and PDGFRA mutational status was available, suggesting that probably KRAS mutations do not play a key-role in cancerogenesis of GISTs and associated malignancies.
On the other hand, it has been suggested that patients affected by GIST with a second primary neoplasm—either synchronous or metachronous—had GISTs with increased mitotic rate (≥5 per 50 high-power fields) (P = 0.0006).[37] In our series, no difference in the median mitotic rate was seen between GIST associated to second neoplasms and all study population (P = 0.49) and high mitotic rate (≥5/50 HPF) seems not related too.
5. Conclusions
This is one of the largest series on second tumors and GISTs as monoinstitutional experience. As previously considered,[38] the high frequency of second tumors, synchronous or metachronous, in patients affected by GISTs suggests that GIST could be considered as a “sentinel tumor.” Thus, after diagnosis of GIST, the risk of a second concomitant neoplasm, especially in gastrointestinal tract, should be considered and a careful surgical and pathological inspection is suggested. Surveillance not only for GIST but also for second malignancies is an important component of the management of patients affected by GIST, particularly in the first years after diagnosis. In addition, esophagogastroduodenoscopy should be planned more frequently than annually and screening for breast and prostate cancer should also be considered. KIT, PDGFRA, and KRAS mutational status does not seem to play a key-role in a common pathogenesis of GIST and second malignancies, but no definitive conclusions can be drawn due to the lack of molecular analysis in second tumors of our sample. Thus, more studies are required to investigate the genetic mechanism of cancerogenesis and progression associating GIST and coexisting tumors.
Footnotes
Abbreviations: GIST = gastrointestinal stromal tumor, KIT = cKIT gene, KRAS = KRAS gene, PDGFRA = platelet-derived growth factor receptor α, SDH = succinate dehydrogenase.
The authors have no funding and conflicts of interest to disclose.
References
- 1.Duffaud F, Blay JY. Gastrointestinal stromal tumors: biology and treatment. Oncology 2003; 65:187–197. [DOI] [PubMed] [Google Scholar]
- 2.Miettinen M, Lasota J. Gastrointestinal stromal tumors (GISTs): definition, occurrence, pathology, differential diagnosis and molecular genetics. Pol J Pathol 2003; 54:3–24. [PubMed] [Google Scholar]
- 3.Joensuu H, Vehtari A, Riihimaki J, et al. Risk of recurrence of gastrointestinal stromal tumor after surgery: an analysis of pooled population-based cohorts. Lancet Oncol 2012; 13:265–274. [DOI] [PubMed] [Google Scholar]
- 4.Lau S, Tam KF. Imaging of gastrointestinal stromal tumor (GIST). Clin Radiol 2004; 59:487–498. [DOI] [PubMed] [Google Scholar]
- 5.Hirota S, Isozaki K, Moriyama Y, et al. Gain of function mutations of c-kit in human gastrointestinal stromal tumors. Science 1998; 297:577–580. [DOI] [PubMed] [Google Scholar]
- 6.Joensuu H, Roberts PJ, Sarlomo-Rikala M, et al. Effect of the tyrosine kinase inhibitor STI571 in a patient with a metastatic gastrointestinal stromal tumor. N Engl J Med 2001; 344:1052–1056. [DOI] [PubMed] [Google Scholar]
- 7.Gupta P, Tewari M, Shukla HS. Gastrointestinal stromal tumor. Surg Oncol 2008; 17:129–138. [DOI] [PubMed] [Google Scholar]
- 8.Fuller CE, Williams GT. Gastrointestinal stromal tumor. Surg Oncol 2008; 17:129–138. [DOI] [PubMed] [Google Scholar]
- 9.Carney JA. The triad of gastric epithelioid leiomyosarcoma, pulmonary chondroma, and functioning extra-adrenal paraganglioma: a five-year review. Medicine (Baltimore) 1983; 62:159–169. [DOI] [PubMed] [Google Scholar]
- 10.Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumors: origin and molecular oncology. Nat Rev Cancer 2011; 11:865–878. [DOI] [PubMed] [Google Scholar]
- 11.Roberts WM, Look AT, Roussel MF, et al. Tandem linkage of human CSF-1 receptor (c-fms) and PDGF receptor genes. Cell 1989; 55:655–661. [DOI] [PubMed] [Google Scholar]
- 12.West RB, Corless CL, Chen X, et al. The novel marker, DOG1, is expressed ubiquitously in gastrointestinal stromal tumors irrespective of KIT e PDGFRA mutational status. Am J Pathol 2004; 165:107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ricci R, Dei Tos AP, Rindi G. GISTogram: a graphic presentation of the growing GIST complexity. Virchows Arch 2013; 463:481–487. [DOI] [PubMed] [Google Scholar]
- 14.Sarlomo-Rikala M, Kovatich AJ, Barusevicius A, et al. CD117: a sensitive marker for gastrointestinal stromal tumors that is more specific than CD34. Mod Pathol 1998; 11:728–734. [PubMed] [Google Scholar]
- 15.Heinrich MC, Corless CL, Duensing A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science 2003; 299:708–710. [DOI] [PubMed] [Google Scholar]
- 16.Maertens O, Prenen H, Debiec-Rychter M, et al. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol gent 2006; 15:1015–1023. [DOI] [PubMed] [Google Scholar]
- 17.Pantaleo MA, Astolfi A, Indio V, et al. SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst 2011; 103:983–987. [DOI] [PubMed] [Google Scholar]
- 18.Miranda C, Nucifora M, Molinari F, et al. KRAS and BRAF mutations predict primary resistance to imatinib in gatrointestinal stromal tumors. Clin Cancer Res 2012; 26:1769–1776. [DOI] [PubMed] [Google Scholar]
- 19.Agaimy A, Wunsch PH, Miettinen M. Occurrence of other malignancies in patients with gastrointestinal stromal tumors. Semin Diagn Pathol 2006; 23:120–129. [DOI] [PubMed] [Google Scholar]
- 20.Vassos N, Agaimy A, Hohenberger W, et al. Coexistence of gastrointestinal stromal tumors (GIST) and malignant neoplasms of different origin: prognostic implications. Int J Surg 2014; 12:371–377. [DOI] [PubMed] [Google Scholar]
- 21.Scheiman J, Cutler A. Helicobacter pylori and gastric cancer. Am J Med 1999; 106:222–226. [DOI] [PubMed] [Google Scholar]
- 22.Moss SF, Malfertheiner P. Helicobacter and gastric malignancies. Helicobacter 2007; 12:23–30. [DOI] [PubMed] [Google Scholar]
- 23.Cohen A, Geller SA, Horowitz LS. Experimental models for gastric leiomyosarcoma. The effects of N-methyl-N-nitrsoguanidine in combination with stress, aspirin, or sodium taurocholate. Cancer 1984; 53:1088–1092. [DOI] [PubMed] [Google Scholar]
- 24.Au WY, Ho KM, Shek TW. Papillary renal cell carcinoma and gastrointestinal stromal tumor: a unique association. Ann Oncol 2004; 15:843–844. [DOI] [PubMed] [Google Scholar]
- 25.Miettinen M, Lasota J. Gastrointestinal stromal tumors: pathology and prognosis at different sites. Semin Diagn Pathol 2006; 23:669–676. [DOI] [PubMed] [Google Scholar]
- 26.Miettinen M, Lasota J. Review on morphology, molecular pathology, prognosis and differential diagnosis. Arch Pathol Lab Med 2006; 130:1466–1478. [DOI] [PubMed] [Google Scholar]
- 27.Ricci R, Martini M, Cenci T, et al. Case of rectal GI stromal tumor demonstrating that KIT and PDGFRA mutations are not always mutually exclusive. J Clin Oncol 2016; 34:e107–109. [DOI] [PubMed] [Google Scholar]
- 28.Basso M, Strippoli A, Orlandi A, et al. KRAS mutational status affects oxaliplatin-based chemotherapy independently from basal mRNA ERCC-1 expression in metastatic colorectal cancer patients. Br J Cancer 2013; 108:115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Agaimy A, Terracciano LM, Dirnhofer S, et al. V600E BRAF mutations are alternative early molecular events in a subset of KIT/PDGFRA wild-type gastrointestinal stromal tumours. J Clin Pathol 2009; 62:613–616. [DOI] [PubMed] [Google Scholar]
- 30.Pantaleo MA, Astolfi A, Indio V, et al. SDHA loss-of-function mutations in KIT-PDGFRA wild-type gastrointestinal stromal tumors identified by massively parallel sequencing. J Natl Cancer Inst 2011; 103:983–987. [DOI] [PubMed] [Google Scholar]
- 31.Miettinen M, Sobin LH, Lasota J. Gastrointestinal stromal tumors of the stomach: a clinopathological and molecular genetic study of 1765 cases with long term follow-up. Am J Surg Pathol 2005; 29:52–68. [DOI] [PubMed] [Google Scholar]
- 32.Kramer K, Wolf S, Mayer B, et al. Frequence, spectrum and prognostic impact of additional malignancies in patients with gastrointestinal stromal tumors. Neoplasia 2015; 1:134–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hechtman JF, DeMatteo R, Nafa K, et al. Additional primary malignancies in patients with gastrointestinal stromal tumor (GIST): a clinicopathologic study of 260 patients with molecular analysis and review of the literature. Ann Surg Oncol 2015; 22:2633–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ricci R, Martini M, Cenci T, et al. PDGFRA-mutant syndrome. Mod Pathol 2015; 28:954–964. [DOI] [PubMed] [Google Scholar]
- 35.Lasota J, Xi L, Coates T, et al. No KRAS mutations found in gastrointestinal stromal tumors (GISTs): molecular genetic study of 514 cases. Mod Pathol 2013; 26:1488–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hechtman JF, DeMatteo R, Nafa K, et al. Additional primary malignancies in patients with gastrointestinal stromal tumor (GIST): clinicopathologic study of 260 patients with molecular analysis and review of the literature. Ann Surg Oncol 2015; 22:2633–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.M.G. Rodriquenz, R. Ricci, M. Iezzi, M. Martini, M. Basso, V. Dadduzio, L.M. Larocca, C. Barone, A. Cassano. 1431P Gastrointestinal Stromal Tumors (Gist) and Second Malignancies: A Novel ‘Sentinel Tumor”? doi:10.1093/annonc/mdu354.20. [Google Scholar]