

Abstract
Background
This aim of this study was to investigate the expression of BMP-7 in atrial fibrillation and illuminate the role of BMP-7 and TGF-β/Smads signaling in myocardial fibrosis.
Material/Methods
Fibrosis of myocardial fibroblasts was induced by TGF-β1 and the optimal condition was determined by the MTT assay. Cells with TGF-β1 treatment were sub-divided into 4 groups: TGF-β1 group, TGF-β1 + Smad3 siRNA group, TGF-β1 + BMP-7 group, and TGF-β1 + BMP-7 + Smad1/5 siRNA group. Cells were then analyzed by detecting the expression of epithelial cadherin (E-cadherin), collagen I, alpha smooth muscle cell actin (α-SMA), and activated Smads using Western blot. Mice were injected daily with Ach-CaCl2 with or without the addition of BMP-7 and Smad1/5 siRNA over a period of 4 weeks. Cardiac functions were tested by echocardiogram assay and fibrosis was diagnosed by histopathological examination. Finally, molecule biomarkers were detected using standard procedures.
Results
TGF-β1 treatment significantly down-regulated E-cadherin expression and up-regulated expressions of Collagen I, α-SMA, and pSmad3 (P<0.05). The effects of TGF-β1 treatment can be significantly suppressed by Smad3 siRNA (P<0.05). Cells in the BMP-7 group exhibited significantly higher expression levels of E-cadherin and pSmad1/5 together with lower expression levels of pSmad3, collagen I, and α-SMA (P<0.05). Moreover, Smad1/5 siRNA can substantially repress the effects of BMP-7 (P<0.05) and results from the mice model coincided with those in myocardial fibroblasts.
Conclusions
BMP-7 can regulate TGF-β1/Smad3 by targeting Smad1/5 to antagonize fibrosis in myocardial fibroblasts resulting from atrial fibrillation.
MeSH Keywords: Atrial Fibrillation, Bone Morphogenetic Protein Receptors, Endomyocardial Fibrosis, Latent TGF-beta Binding Proteins, Smad3 Protein
Background
Atrial fibrillation (AF) is a type of arrhythmia that affects millions of people around the world and has increasing prevalence and high mortality [1]. Although several risk factors for AF (e.g., sex, age, and hypertension status) have been identified, the underlying etiology requires further research [2]. Both aging and the presence of hypertension change the structure of extracellular matrix (ECM) and alter myocardial substrate, which results in an enhanced AF [3]. Fibrosis is an important substrate of ECM remodeling in AF because it subsequently changes connexin expression and then influences impulse propagation by interrupting the process of intermyocyte coupling [4]. Myofibroblasts and other inflammatory cells are thought to be the main source of the functional and structural changes in cardiac tissues during cardiac fibrosis [4].
The TGF-β super-family, which includes the transforming growth factor TGF-β1-3, Bone Morphogenetic Proteins (BMPs), and other actins, is one of the largest groups in which functions and structures of proteins are integrated [5]. Many researchers have shown that TGF-β1 induces epithelial mesenchymal transition (EMT), which is an important process in cardiac fibrosis [6,7]. TGF-β1 acts as an essential mediator and is able to induce EMT and affect ECM expression in many organs and tissues [5,8]. TGF-β1 binds with the receptors and then induces a multitude of responses, including the up-regulation of mesenchymal-related proteins such as collagen I and α-SMA, along with the down-regulation of intercellular adhesion proteins such as E-cadherin [9]. In addition, it has been shown that the effects of TGF-β1 on renal diseases are mediated by canonical transcription factors Smads, especially Smad3 [10]. Downstream Smads of TGF-β1, including Smad2 and Smad3, have been analyzed using rat models of myocardial pressure overload, but the mechanisms by which the TGF-β1/Smads pathway in cardiac fibrosis causes AF remain largely undefined [11,12].
Bone morphogenetic proteins, which belong to the TGF-β super-family, are multifunctional regulators of cell growth, differentiation, and morphogenesis [13]. Ghosh et al. revealed that BMP-2 down-regulated biological effects such as DNA synthesis in kidney cells induced by the epidermal growth factor [14]. BMP-7 also plays an important role in the EMP process [15]. It has been reported that BMP-7 can reverse fibrosis in many diseases affecting organs such as the liver and kidneys [8,15,16]. BMP-7 and TGF-β1 have homologous ligand structures, binding molecules and downstream signaling regulators, which control a balanced EMT [15], and BMP-7 might coordinate with cardiac interstitial fibrosis, but little evidence of this has been produced.
TGF-β1 and BMP-7 transfer their signals from cell surface to nucleus through binding with transmembrane kinase receptors (ALK receptors), and their downstream intracellular signaling pathways are mediated by Smad proteins [13]. TGF-β1 action is transduced by Smad2 and Smad3, BMP-7 signaling is regulated by Smad1/5/8, and both pathways are mediated by Smad-4 [17–19]. Vargha et al. reported that BMP-7 had a reversed EMT process presented as mesenchymal-to-epithelial phenotype in peritoneal mesothelial cells [20]. Another study has demonstrated that BMP-7 can antagonize TGF-β-induced EMT of mesothelial cells and reversed peritoneal fibrosis in rats exposed to peritoneal dialysis fluid [21]. However, the ability of BMP-7/Smads to prevent TGF-β1-mediated EMT and reverse cardiac fibrosis results from AF has not been assessed.
In the present study we investigated the molecular mechanism of BMP-7/Smad5 in TGF-β-induced fibrosis, which was conducted in cardiac fibroblasts, and then we evaluated the effect of BMP-7 as a treatment of cardiac fibrosis using a rat model.
Material and Methods
Cell culture
Myocardial fibroblasts were separated from SD mice as previously described [22]. Cells were characterized by the immunohistochemistry of vimentin and smooth muscle actin (SMA). Isolated cells were cultured in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal calf serum (FCS) at 37°C incubator with 5% CO2. Myocardial fibroblasts were cultured with TGF-β to induce fibrosis. Cells were treated with TGF-β for 24 h at different concentration levels (10−8, 10−7, 10−6, and 10−5 mol/L) to determine the optimal concentration level of TGF-β. As suggested by the dimethylthiahiazo (MTT) assay described below, 10−6 mol/L was considered as the optimal concentration. As a result, cells were treated with TGF-β for different times (6 h, 12 h, 24 h, and 48 h) using the optimal concentration level of 10−6 mol/L.
Immunohistochemistry and MTT assay
Immunohistochemical analysis of myocardial fibroblasts was established using the EnVision two-step method as previously described [23]. Rabbit polyclonal anti-vimentin and anti-SMA antibody (Zhongshan Biology Company, Beijing) were diluted 1:100 using the diluent. MTT assay was used to assess the proliferation status of myocardial fibroblasts in each group. A total of 3×103 cells were cultured in 96-well plates, incubated for 24 h, and then stained with 0.5 mg/ml MTT for 4 h. Samples were measured at 490 nm by an enzyme-linked immunosorbent assay (ELISA) reader (Synergy HT, BioTek Instruments, Winooski, VT) after supernatant was discarded and 200 μl of dimethylsulfoxide was added to dissolve precipitate.
Lentivirus transduction
A fragment containing Smad3 siRNA or Smad1/5 siRNA was cloned into the pCDH vector and then amplified by polymerase chain reaction (PCR). The pCDH plasmid and other essential plasmids were co-transfected into cells using the Lipofectamine LTX kit (Invitrogen, CA). Viral particles were collected after 48-htransfection.
Treatment of TGF-β, BMP-7 and Lentivirus in cells
Myocardial fibroblasts were divided into 5 groups: the control group (normal cells); the TGF-β group (cells treated with 10−6 mol/L TGF-b for 48 h); the Smad3 siRNA group (cells treated with 10−6 mol/L TGF-β for 48 h and then transfected with Smad3 siRNA); the BMP-7 group (cells treated with 10−6 mol/L TGF-β for 48 h and then cultured with 0.5 μg/mL recombinant human BMP-7 (rhBMP-7, Prospec, Israel); and the Smad1/5 siRNA group (cells treated with both TGF-β and rhBMP-7 and then transfected with Smad1/5 siRNA).
Animal model
A total of 60 male SD mice weighing 200 to 250 g (purchased from the Laboratory Animal Center of Chinese Medical University) were randomly allocated to the control group (n=10) and model group (n=50). For intentionally establishing the atrial fibrillation model, 1 ml/kg Ach-CaCl2 was injected daily into tail veins of mice in the model group and these mice were raised with regular feeding. Mice in the model group were inspected using electrocardiogram after 4 weeks and we randomly selected 40 mice in which atrial fibrillation models were successfully constructed.
We allocated the 50 selected mice (10 in the control group and 40 in the model group) in the experiment to 5 treatment groups: the control group (n=10, abdominal cavities of mice in the control group were injected with 1 ml saline every 2 days); the model group (n=10, abdominal cavities of mice in the model group were injected with 1 ml saline every 2 days); the low-dose BMP group (n=10, abdominal cavity of mice in the model group was injected with 80 μg/kg rhBMP-7 every 2 days); the high-dose BMP group (n=10, abdominal cavity of mice in the model group was injected with 300 μg/kg rhBMP-7 every 2 days); and the Smad1/5 siRNA group (n=10, left atrial of mice in the model group was injected with Smad1/5 siRNA transfected myocardial fibroblasts and then their abdominal cavities were injected with 300 μg/kg rhBMP-7 every 2 days). All mice were inspected on day 28 after treatments were established.
Western blot
Cells and tissues were harvested and lysed using the radioimmunoprecipitation assay (RIPA) buffer. Total protein was separated and calculated by the Bradford method. Then, total protein was denatured in boiling water and transferred onto polyvinylidene fluoride (PVDF) membranes after sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Membranes were blocked in Tris-buffered saline with tween (TBST) with 5% skim milk for 1 h and treated with primary antibodies (against E-cadherin, collagen I, α-SMA, Smad3, pSmad3, Smad1/5, pSmad1/5, 1:800 dilution, Zhongshan Biology Company, Beijing) at 4°C overnight. After membranes were washed, they were incubated with secondary antibodies (horseradish peroxidase-conjugated goat anti-goat, 1:2000 dilution, Zhongshan Biology Company, Beijing). Samples were processed with enhanced chemiluminescence and quantified by Lab Works 4.5 software in which β-actin was set as an endogenous control.
Cardiac function tests
Tests of cardiac function were carried out using the echocardiogram assay (ATL-HDI5000) in which ejection fraction (EF), fractional shortening (FS), left ventricular end-systolic dimension (LVESD), and left ventricular end-diastolic dimension (LVEDD) were detected.
Histopathological examination (HE)
Tissues from the left atria of mice were harvested and treated with 10% formalin. Then, 5-μm-thick paraffin sections were embedded and stained with HE assay. Samples were inspected under a microscope (Motic China Group Co. Ltd) and measured at 5 random sites.
Statistical analysis
All statistical analyses were performed with SPSS 18.0 software (Chicago, IL, USA). Data are presented as mean ± standard deviation (SD) for at least 3 experiments. The 2-tailed t test or one-way analysis of variance (ANOVA) was used to assess between-group comparisons, and P<0.05 provided evidence of statistical significance.
Results
Cell culture and TGF-β-treated cells
Cells were spindle-shaped or polygonal under an optical microscope (×400). Vimentin was stained as brown particles and they were evenly distributed in 95% of cell cytoplasm. Immunohistochemistry of SMA showed negative results (Figure 1).
Figure 1.

Immunohistochemistry results of isolated myocardial fibroblasts, ×400. (A) Vimentin was stained as brown particles in cell cytoplasm. (B) Negative result of immunohistochemistry for SMA.
Results from MTT assay in TGF-β–treated cells indicated that TGF-β can up-regulate the cell activity of myocardial fibroblasts in a dose- and time-dependent manner (Tables 1, 2, Figure 2). In this experiment, treatment of 10−6 mol/L TGF-β for 48 h resulted in a remarkable loss of the cobblestone-like growing appearance along with elongated fibroblast-like morphological features. A fibrosis model of cardiac fibroblasts was established in this control.
Table 1.
Influence of different density of TGF-β1 for cell activity.
| Control | 10−8 mol/L | 10−7 mol/L | 10−6 mol/L | 10−5 mol/L | |
|---|---|---|---|---|---|
| OD value | 0.116±0.007 | 0.133±0.007 | 0.148±0.007* | 0.175±0.005*#& | 0.190±0.007*#& |
P<0.05 versus control group;
P<0.05 versus 10−8 mol/L group;
P<0.05 versus 10−7 mol/L group.
Table 2.
Influence of different time of TGF-β1 for cell activity.
P<0.05 versus 0 h group;
P<0.05 versus 6 h group;
P<0.05 versus 12 h group.
Figure 2.

Cell activity was up-regulated by TGF-β in myocardial fibroblasts. (A) The effect of different concentrations of TGF-β on cell activity. * P<0.05 versus control group; # P<0.05 versus 10−8 mol/L group; & P<0.05 versus 10−7 mol/L group. (B) The effect of different treatment times of TGF-β on cell activity. * P<0.05 versus 0 h group; # P<0.05 versus 6 h group; & P<0.05 versus 12 h group. Data are presented as mean ±SD for 3 independent experiments.
TGF-β induced the expression of fibrosis markers by mediating Smad3
Compared with the control group, the expression level of epithelial marker E-cadherin in the TGF-β group was significantly decreased when expression levels of pSmad3 and mesenchymal markers. including collagen I and α-SMA were up-regulated (all P<0.05, Table 3, Figure 3). Compared with the TGF-β group, the TGF-β + Smad3 siRNA group exhibited significantly higher expressions of E-cadherin as well as down-regulated Smad3, pSmad3, collagen I, and α-SMA expressions (all P<0.05, Figure 3), indicating that TGF-β was capable of inducing fibrosis, which was suppressed by silencing Smad3.
Table 3.
Influence of TGF-β1 and Smad3 inhibitors on fibrosis markers.
| Group | Control group | TGF-β1 group | TGF-β1+Smad3 siRNA group |
|---|---|---|---|
| E-cadherin | 1.01±0.04 | 0.34±0.02* | 0.74±0.12 |
| Collagen I | 1.01±0.07 | 2.24±0.17* | 1.32±0.09# |
| α-sma | 0.98±0.05 | 8.25±0.96* | 2.34±0.48*# |
| Smad3 | 1.03±0.05 | 1.07±0.07 | 0.75±0.04 |
| pSmad3 | 1.04±0.11 | 2.26±0.24* | 1.57±0.19# |
P<0.05 versus control group;
P<0.05 versus TGF-β1 group.
Figure 3.

Expression levels of fibrosis markers in myocardial fibroblasts treated with TGF-β1 and Smad3 siRNA. (A) Western blot analysis of fibrosis markers in cells of different groups normalized to β-actin. (B–F) Quantitative data for protein levels of fibrosis markers in cells with different controls. Data are presented as mean ±SD for 3 independent experiments. * P<0.05 versus control group, # P<0.05 versus TGF-β1 group.
BMP-7 antagonized fibrosis of myocardial fibroblasts by mediating Smad1/5
Compared with cells with TGF-β treatment, cells in the BMP-7 + TGF-β group had significantly higher expression levels of E-cadherin and pSmad1/5 and lower expression levels of pSmad 3, collagen I, and α-SMA (all P<0.05, Table 4, Figure 4), suggesting the protective effects of BMP-7 on fibrosis induced by TGF-β. After transfection of Smad1/5 siRNA, expression levels of pSmad3, Smad1/5, pSmad1/5, E-cadherin, collagen I, and α-SMA for cells in the Smad1/5 siRNA group were ranked between the TGF-β and BMP-7 group. We found significant differences in the expressions of E-cadherin, collagen I, α-SMA, and pSmad1/5 among these groups (all P<0.05, Figure 4), indicating that the effect of BMP-7 was inhibited by Smad1/5 siRNA.
Table 4.
Influence of BMP-7 and Smad1/5 inhibitors on fibrosis markers.
| Group | TGF-β1 group | TGF-β1+BMP-7 group | TGF-β1+BMP-7+ Smad1/5 siRNA group |
|---|---|---|---|
| E-cadherin | 1.01±0.05 | 2.12±0.14* | 1.47±0.12*# |
| Collagen I | 1.02±0.07 | 0.45±0.09* | 0.72±0.10*# |
| a-sma | 0.98±0.04 | 0.41±0.07* | 0.74±0.06*# |
| Smad3 | 1.01±0.07 | 0.97±0.06 | 1.08±0.06 |
| pSmad3 | 1.03±0.06 | 0.68±0.07* | 0.82±0.08* |
| Smad1/5 | 0.97±0.04 | 1.02±0.07 | 0.87±0.04 |
| pSmad1/5 | 1.02±0.04 | 2.17±0.14* | 1.47±0.09*# |
P<0.05 versus TGF-β1 group;
P<0.05 versus TGF-β1 + BMP-7 group.
Figure 4.

Expression levels of fibrosis markers in myocardial fibroblasts treated with TGF-β1, BMP-7, and Smad1/5 siRNA. (A) Western blot analysis of fibrosis markers in cells of different groups normalized to β-actin. (C–H) Quantitative data for protein levels of fibrosis markers in cells with different controls. Data are presented as means ±SD for 3 independent experiments. * P<0.05 versus TGF-β1 group, # P<0.05 versus TGF-β1 + BMP-7 group.
Results of cardiac function tests
As shown in Figure 5, mice in the model group, which were treated with Ach-CaCl2, exhibited obvious atrial ectopy. Therefore, the atrial fibrillation model was successfully established and 40 mice in the model group were randomly selected for experimental purposes.
Figure 5.
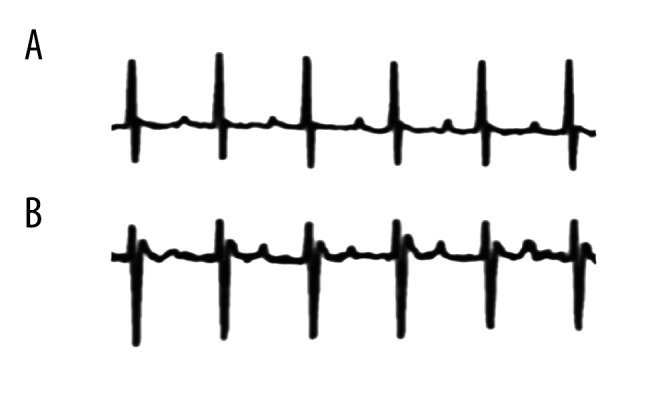
Representative telemetry ECG results showing spontaneous atrial ectopy in the model group (B), while no AF signals were observed in the control group (A).
After mice were treated with BMP-7 and Smad1/5 siRNA, all mice in the 5 groups were tested by several cardiac functions: EF, FS, LVESD, and LVEDD. There were significant differences in cardiac functions between any of the 2 groups in the control, model, low-dose BMP-7, or high-dose BMP-7 group (all P<0.05, Table 5, Figure 6). The control group exhibited the most robust cardiac functions, whereas the model group had the largest loss of cardiac functions. Cardiac functions in the low-dose BMP-7 group and high-dose BMP-7 group were ranked between the control and model group. Both the low-dose BMP-7 and high-dose BMP-7 groups exhibited higher EF and FS compared with the model group. Apart from that, the improvement in cardiac functions was more significant in the high-dose BMP-7 group in comparison to the low-dose BMP-7 group. For the Smad1/5 siRNA group, in which mice were treated with both high-dose BMP-7 and Smad1/5 siRNA, results were significantly lower compared with the control group, the low-dose BMP-7 group, and the high-dose BMP-7 group (all P<0.05, Figure 6). Results of the Smad1/5 siRNA group were similar to those in the model group and they were ranked between the model group and the low-dose BMP-7 group.
Table 5.
Results of echocardiographic measurement of atrial fibrillation model mice.
| Group | Control | Model | Low dose BMP-7 | High dose BMP-7 | High dose BMP-7 + Smad1/5 siRNA |
|---|---|---|---|---|---|
| EF/% | 63.45±3.21 | 27.3±4.83* | 37.94±3.14*# | 50.17±3.82*#& | 30.21±3.01*&@ |
| FS(%) | 36.3±2.4 | 13.6±3.22* | 20.14±3.31*# | 25.73±3.32*#& | 15.24±3.12*&@ |
| LVESD (mm) | 3.0±0.32 | 5.78±0.16 | 5.04±0.12 | 3.74±0.13 | 5.67±0.14 |
| LVEDD (mm) | 5.12±0.17 | 6.45±0.20 | 6.12±0.21 | 5.49±0.24 | 6.28±0.18 |
EF – ejection fraction; FS – fractional shortening; LVESD – left ventricular end-systolic dimension; LVEDD – left ventricular end-diastolic dimension.
P<0.05 versus control group;
P<0.05 versus model group;
P<0.05 versus model + medicine group;
P<0.05 versus transfection control group.
Figure 6.

Results of cardiac functions with echocardiographic measurement. Data are presented as mean ±SD for 3 independent experiments. EF – ejection fraction; FS – fractional shortening; LVESD – left ventricular end-systolic dimension; LVEDD – left ventricular end-diastolic dimension. * P<0.05 versus control group; # P<0.05 versus model group; & P<0.05 versus model + medicine group; @ P<0.05 versus transfection control group.
The above evidence suggests that Ach-CaCl2 treatment can interfere with cardiac functions of mice and that BMP-7 plays a protective role in cardiac functions. On the other hand, Smad1/5 siRNA inhibited the protective effect of BMP-7. These results were further assessed by histopathological examination of left atrium tissues in model mice. As shown in Figure 7, tissues in the control group exhibited neatly arranged and uniformly dyed muscle fibers, whereas muscle fibers of tissues in the model group were broken and disorganized in the white fibrosis area.
Figure 7.

HE staining of fibrous tissue in atria of rats. (A–E) Showed the results of rats in control group, model group, low-dose BMP-7 group, high-dose BMP-7 group, and high-dose BMP-7 + Smad1/5 siRNA group in order (×400).
The expression of fibrosis markers in cardiac tissues
We detected the expression level of fibrosis markers and related signaling molecules, including E-cadherin, Collagen I, α-SMA, Smad3/1/5, and pSmad3/1/5, after the experiment. For signaling molecules, the establishment of an atrial fibrillation model significantly up-regulated pSmad3 and down-regulated pSmad1/5. However, the above trend can be antagonized by both low- and high-dose BMP-7, while the effect of BMP-7 was significantly inhibited by Smad1/5 siRNA (all P<0.05, Table 6, Figure 8). Moreover, BMP-7 and Smad1/5 siRNA slightly affected the expression of Smad3 and Smad1/5. Both BMP-7 and Smad1/5 siRNA significantly influenced fibrosis markers, including E-cadherin, collagen I and α-SMA. Significant differences between each of the 2 groups in the 5 groups were observed (all P<0.05, Figure 8).
Table 6.
Expression level of fibrosis markers of model mice.
| Group | Control | Model | Low dose BMP-7 | High dose BMP-7 | High dose BMP-7 + Smad1/5 siRNA |
|---|---|---|---|---|---|
| E-cadherin | 1.01±0.04 | 0.21±0.02* | 0.46±0.03*# | 0.57±0.04*# | 0.40±0.03*# |
| Collagen I | 1.02±0.05 | 5.01±0.42* | 3.60±0.15*# | 3.26±0.15*#& | 4.77±0.34*#&@ |
| a-sma | 1.01±0.07 | 2.64±0.22* | 1.99±0.15*# | 1.71±0.17*#& | 2.23±0.22*#&@ |
| Smad3 | 1.00±0.06 | 1.23±0.07* | 0.94±0.05# | 0.87±0.05# | 1.12±0.07&@ |
| pSmad3 | 0.98±0.05 | 4.89±0.21* | 2.74±0.19*# | 2.36±0.17*#& | 3.97±0.20*#&@ |
| Smad1/5 | 1.01±0.06 | 0.94±0.04 | 1.07±0.06 | 1.17±0.08# | 1.04±0.05 |
| pSmad1/5 | 0.99±0.05 | 0.43±0.03* | 0.57±0.04* | 0.78±0.04*#& | 0.47±0.03*@ |
P<0.05 versus control group;
P<0.05 versus model group;
P<0.05 versus model + medicine group;
P<0.05 versus transfection control group.
Figure 8.

Expression levels of fibrosis markers in mice with different treatments. (A) Western blot analysis of fibrosis markers in cardiac tissues of different groups normalized to β-actin. (B–H) Quantitative data protein level of fibrosis markers in tissues with different control. Data are presented as mean ±SD for 3 independent experiments. * P<0.05 versus control group; # P<0.05 versus model group; & P<0.05 versus model + medicine group; @ P<0.05 versus transfection control group.
Tissues in the model group had lower E-cadherin and higher collagen I and α-SMA expressions, while BMP-7 treatments up-regulated E-cadherin and down-regulated collagen I and α-SMA expressions. Therefore, Smad1/5 can reverse the effects of BMP-7 on these expressions and in vivo results agreed with in vitro results.
Discussion
Our study investigated the effect of TGF-β and BMP-7 on myocardial fibrosis. We found that smad3 siRNA could suppress myocardial fibrosis induced by TGF-β and BMP-7 could also antagonize myocardial fibrosis induced by TGF-β, but the effect of BMP-7 was decreased when Smad1/5 siRNA was transfected. Our results from the atrial fibrillation model in mice show that BMP-7 elevated the cardiac function parameters in a dose-dependent manner and its protective effect in cardiac functions was inhibited when Smad1/5 siRNA was co-transfected.
A previous study has shown that TGF-β can promote procollagen formation and ECM synthesis, thus resulting in myocardial fibrosis [24]. Stimulation of TGF-β can activate fibroblasts, which were thereby transformed into myofibroblasts. Myofibroblasts, which are characterized by the acquired expression of α-SMA, are key effector cells in fibrotic states [25]. Important elements for evaluating myofibroblast differentiation include the expression of α-SMA and the secretion of extracellular matrix, such as collagen and fibronectin [26]. More importantly, the expression of α-SMA and collagen I are both coordinately regulated by TGF-β [27]. As suggested by our study, the expression of α-SMA and collagen I were both increased in myocardial fibroblasts when SD mice were treated with TGF-β. Furthermore, TGF-β exerts an important role in EMT, in which the Smad signaling pathway is involved [28]. Smad expression exhibits a distinct level in fibrosis tissues and Smad3 exerted its influence on regulating pathological fibrotic disorders (e.g., cystic fibrosis, scleroderma, and cirrhosis) induced by TGF-β [29]. Analogously, we demonstrated that Smad3 expression was increased in cardiac fibrosis when TGF-β treatments were introduced. As a result, both Smad3 siRNA and TGF-β were introduced in our experiment, which revealed that α-SMA and collagen I were decreased to a level between the control and TGF-β treatment group. Therefore, Smad3 siRNA can partially reverse the effect of TGF-β on α-SMA and collagen I.
It has been reported that BMP-7 was able to reverse EMT in proximal tubular cells [30], and there are several studies showing a negative relationship between BMP-7 and fibrosis of different organs. BMP-7 plays anti-inflammatory and anti-fibrogenic roles in the progression of chronic hepatitis as well as an anti-fibrotic role in the formation of hepatic fibrosis [31]. Furthermore, BMP-7 prevents fibrosis formation in the kidneys [8] and the expression of BMP-7 was largely decreased when idiopathic pulmonary fibrosis was triggered [32]. In our study, the addition of BMP-7 improved the cardiac function of mice in which the AF model was constructed, indicating that BMP-7 plays a protective role in myocardial fibrillation. Because it has also been reported that the activation of type I BMP receptor phosphorylated both Smad1 and Smad5 [33], we examined the expression of Smad1/5 and pSmad1/5. When BMP-7 was added to myocardial fibroblasts treated with TGF-β, pSmad1/5 was markedly increased. As suggested by the atrial fibrillation model, cardiac functions could be restored with the addition of Smad1/5 siRNA. All of the above results suggest that BMP-7 plays a protective role in myocardial fibrillation via its influence on the Smad1/5 pathway.
In addition to the studied BMP-7, TGF-β and Smads, there are many other cardiac biomarkers that should be emphasized, including interleukin-3 (IL-3), IL-4, IL-6, nuclear factor-κB (NF-κB) kinase (IKK) complex, and suppression of tumorigenicity 2 (ST2) [34–37]. Of note, ST2 mainly functions to protect myocardium viability, which suffers enormous pressure; ST2 is considered to be a reliable cardiac marker [29]. Downstream factors of TGF-β/Smad, such as dual-specificity phosphatase 9 (DUSP9), mitogen-activated protein kinase (MAPK), and T-box protein 3 (TBX3), also need to be studied because their modifications are the final results of regulated TGF-β/Smad expressions and they could be desirable treatment targets for myocardial fibrosis [38–40].
Conclusions
Although previous studies have shown that BMP-7 can antagonize fibrosis in different tissues and organs, it is unknown whether BMP-7 has similar effects on myocardial fibrosis. Our study shows that BMP-7 plays a positive role in myocardial fibrosis through the TGF-β/Smads signaling pathway and it is able to improve the shape, arrangement, and function of myocardial fibroblasts. Nevertheless, the specificity and efficacy of BMP-7 treatment for AF remain unknown and potential adverse effects of treatment using BMP-7 should be further investigated.
Footnotes
Source of support: Departmental sources
References
- 1.Lip GY, Brechin CM, Lane DA. The global burden of atrial fibrillation and stroke: A systematic review of the epidemiology of atrial fibrillation in regions outside North America and Europe. Chest. 2012;142:1489–98. doi: 10.1378/chest.11-2888. [DOI] [PubMed] [Google Scholar]
- 2.Fuster V, Ryden LE, Cannom DS, et al. ACC/AHA/ESC 2006 guidelines for the management of patients with atrial fibrillation: full text: A report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the European Society of Cardiology Committee for Practice Guidelines (Writing Committee to Revise the 2001 guidelines for the management of patients with atrial fibrillation) developed in collaboration with the European Heart Rhythm Association and the Heart Rhythm Society. Europace. 2006;8:651–745. doi: 10.1093/europace/eul097. [DOI] [PubMed] [Google Scholar]
- 3.Lau YF, Yiu KH, Siu CW, Tse HF. Hypertension and atrial fibrillation: Epidemiology, pathophysiology and therapeutic implications. J Hum Hypertens. 2012;26:563–69. doi: 10.1038/jhh.2011.105. [DOI] [PubMed] [Google Scholar]
- 4.Burstein B, Nattel S. Atrial fibrosis: Mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol. 2008;51:802–9. doi: 10.1016/j.jacc.2007.09.064. [DOI] [PubMed] [Google Scholar]
- 5.Borok Z. Role for alpha3 integrin in EMT and pulmonary fibrosis. J Clin Invest. 2009;119:7–10. doi: 10.1172/JCI38084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee KW, Everett THt, Rahmutula D, et al. Pirfenidone prevents the development of a vulnerable substrate for atrial fibrillation in a canine model of heart failure. Circulation. 2006;114:1703–12. doi: 10.1161/CIRCULATIONAHA.106.624320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cardin S, Libby E, Pelletier P, et al. Contrasting gene expression profiles in two canine models of atrial fibrillation. Circ Res. 2007;100:425–33. doi: 10.1161/01.RES.0000258428.09589.1a. [DOI] [PubMed] [Google Scholar]
- 8.Zeisberg M, Hanai J, Sugimoto H, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9:964–68. doi: 10.1038/nm888. [DOI] [PubMed] [Google Scholar]
- 9.Zhou Q, Yang M, Lan H, Yu X. miR-30a negatively regulates TGF-beta1-induced epithelial-mesenchymal transition and peritoneal fibrosis by targeting Snai1. Am J Pathol. 2013;183:808–19. doi: 10.1016/j.ajpath.2013.05.019. [DOI] [PubMed] [Google Scholar]
- 10.Meng XM, Chung AC, Lan HY. Role of the TGF-beta/BMP-7/Smad pathways in renal diseases. Clin Sci (Lond) 2013;124:243–54. doi: 10.1042/CS20120252. [DOI] [PubMed] [Google Scholar]
- 11.Bjornstad JL, Skrbic B, Marstein HS, et al. Inhibition of SMAD2 phosphorylation preserves cardiac function during pressure overload. Cardiovasc Res. 2012;93:100–10. doi: 10.1093/cvr/cvr294. [DOI] [PubMed] [Google Scholar]
- 12.Bujak M, Ren G, Kweon HJ, et al. Essential role of Smad3 in infarct healing and in the pathogenesis of cardiac remodeling. Circulation. 2007;116:2127–38. doi: 10.1161/CIRCULATIONAHA.107.704197. [DOI] [PubMed] [Google Scholar]
- 13.Yang YL, Liu YS, Chuang LY, et al. Bone morphogenetic protein-2 antagonizes renal interstitial fibrosis by promoting catabolism of type I transforming growth factor-beta receptors. Endocrinology. 2009;150:727–40. doi: 10.1210/en.2008-0090. [DOI] [PubMed] [Google Scholar]
- 14.Ghosh Choudhury G, Jin DC, Kim Y, et al. Bone morphogenetic protein-2 inhibits MAPK-dependent Elk-1 transactivation and DNA synthesis induced by EGF in mesangial cells. Biochem Biophys Res Commun. 1999;258:490–6. doi: 10.1006/bbrc.1999.0599. [DOI] [PubMed] [Google Scholar]
- 15.Kalluri R, Neilson EG. Epithelial-mesenchymal transition and its implications for fibrosis. J Clin Invest. 2003;112:1776–84. doi: 10.1172/JCI20530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang S, Chen Q, Simon TC, et al. Bone morphogenic protein-7 (BMP-7), a novel therapy for diabetic nephropathy. Kidney Int. 2003;63:2037–49. doi: 10.1046/j.1523-1755.2003.00035.x. [DOI] [PubMed] [Google Scholar]
- 17.Attisano L, Wrana JL. Signal transduction by the TGF-beta superfamily. Science. 2002;296:1646–47. doi: 10.1126/science.1071809. [DOI] [PubMed] [Google Scholar]
- 18.Wrana JL. Regulation of Smad activity. Cell. 2000;100:189–92. doi: 10.1016/s0092-8674(00)81556-1. [DOI] [PubMed] [Google Scholar]
- 19.Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–78. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 20.Vargha R, Endemann M, Kratochwill K, et al. Ex vivo reversal of in vivo transdifferentiation in mesothelial cells grown from peritoneal dialysate effluents. Nephrol Dial Transplant. 2006;21:2943–47. doi: 10.1093/ndt/gfl355. [DOI] [PubMed] [Google Scholar]
- 21.Loureiro J, Schilte M, Aguilera A, et al. BMP-7 blocks mesenchymal conversion of mesothelial cells and prevents peritoneal damage induced by dialysis fluid exposure. Nephrol Dial Transplant. 2010;25:1098–108. doi: 10.1093/ndt/gfp618. [DOI] [PubMed] [Google Scholar]
- 22.Melchior-Becker A, Dai G, Ding Z, et al. Deficiency of biglycan causes cardiac fibroblasts to differentiate into a myofibroblast phenotype. J Biol Chem. 2011;286:17365–75. doi: 10.1074/jbc.M110.192682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ou DL, Chen CL, Lin SB, et al. Chemokine receptor expression profiles in nasopharyngeal carcinoma and their association with metastasis and radiotherapy. J Pathol. 2006;210:363–73. doi: 10.1002/path.2053. [DOI] [PubMed] [Google Scholar]
- 24.Xiao H, Zhang YY. Understanding the role of transforming growth factor-beta signalling in the heart: overview of studies using genetic mouse models. Clin Exp Pharmacol Physiol. 2008;35:335–41. doi: 10.1111/j.1440-1681.2007.04876.x. [DOI] [PubMed] [Google Scholar]
- 25.Tomasek JJ, Gabbiani G, Hinz B, et al. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3:349–63. doi: 10.1038/nrm809. [DOI] [PubMed] [Google Scholar]
- 26.Dobaczewski M, de Haan JJ, Frangogiannis NG. The extracellular matrix modulates fibroblast phenotype and function in the infarcted myocardium. J Cardiovasc Transl Res. 2012;5:837–47. doi: 10.1007/s12265-012-9406-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Desmouliere A, Geinoz A, Gabbiani F, Gabbiani G. Transforming growth factor-beta 1 induces alpha-smooth muscle actin expression in granulation tissue myofibroblasts and in quiescent and growing cultured fibroblasts. J Cell Biol. 1993;122:103–11. doi: 10.1083/jcb.122.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xu J, Lamouille S, Derynck R. TGF-beta-induced epithelial to mesenchymal transition. Cell Res. 2009;19:156–72. doi: 10.1038/cr.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Flanders KC. Smad3 as a mediator of the fibrotic response. Int J Exp Pathol. 2004;85:47–64. doi: 10.1111/j.0959-9673.2004.00377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yang G, Zhu Z, Wang Y, et al. Bone morphogenetic protein 7 attenuates epithelial-mesenchymal transition induced by silica. Hum Exp Toxicol. 2016;35:69–77. doi: 10.1177/0960327115577550. [DOI] [PubMed] [Google Scholar]
- 31.Li S, Zeng XT, Ruan XL, Liu TZ, Wang XH. Association between XPD Lys751Gln polymorphism and bladder cancer susceptibility: An updated and cumulative meta-analysis based on 6,836 cases and 8,251 controls. Mol Biol Rep. 2014;41:3621–29. doi: 10.1007/s11033-014-3226-2. [DOI] [PubMed] [Google Scholar]
- 32.Gu P, Luo B, Yi X, et al. [The expressions and meanings of BMP-7 and TGF-beta in idiopathic pulmonary fibrosis and idiopathic nonspecific interstitial pneumonia]. Zhonghua Jie He He Hu Xi Za Zhi. 2014;37:664–70. [PubMed] [Google Scholar]
- 33.Hu W, Ye Y, Wang J, et al. Bone morphogenetic proteins induce rabbit bone marrow-derived mesenchyme stem cells to differentiate into osteoblasts via BMP signals pathway. Artif Cells Nanomed Biotechnol. 2013;41:249–54. doi: 10.3109/10731199.2012.731412. [DOI] [PubMed] [Google Scholar]
- 34.Schmitz J, Owyang A, Oldham E, et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity. 2005;23:479–90. doi: 10.1016/j.immuni.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 35.Kakkar R, Lee RT. The IL-33/ST2 pathway: Therapeutic target and novel biomarker. Nat Rev Drug Discov. 2008;7:827–40. doi: 10.1038/nrd2660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmieder A, Multhoff G, Radons J. Interleukin-33 acts as a pro-inflammatory cytokine and modulates its receptor gene expression in highly metastatic human pancreatic carcinoma cells. Cytokine. 2012;60:514–21. doi: 10.1016/j.cyto.2012.06.286. [DOI] [PubMed] [Google Scholar]
- 37.Meisel C, Bonhagen K, Lohning M, et al. Regulation and function of T1/ST2 expression on CD4+ T cells: induction of type 2 cytokine production by T1/ST2 cross-linking. J Immunol. 2001;166:3143–50. doi: 10.4049/jimmunol.166.5.3143. [DOI] [PubMed] [Google Scholar]
- 38.Li J, Ballim D, Rodriguez M, et al. The anti-proliferative function of the TGF-beta1 signaling pathway involves the repression of the oncogenic TBX2 by its homologue TBX3. J Biol Chem. 2014;289:35633–43. doi: 10.1074/jbc.M114.596411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Watanabe H, de Caestecker MP, Yamada Y. Transcriptional cross-talk between Smad, ERK1/2, and p38 mitogen-activated protein kinase pathways regulates transforming growth factor-beta-induced aggrecan gene expression in chondrogenic ATDC5 cells. J Biol Chem. 2001;276:14466–73. doi: 10.1074/jbc.M005724200. [DOI] [PubMed] [Google Scholar]
- 40.Morikawa M, Koinuma D, Mizutani A, et al. BMP sustains embryonic stem cell self-renewal through distinct functions of different Kruppel-like factors. Stem Cell Reports. 2016;6:64–73. doi: 10.1016/j.stemcr.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]