

Abstract
Antibodies derived from non-human sources must be modified for therapeutic use so as to mitigate undesirable immune responses. While complementarity-determining region (CDR) grafting-based humanization techniques have been successfully applied in many cases, it remains challenging to maintain the desired stability and antigen binding affinity upon grafting. We developed an alternative humanization approach called CoDAH (“Computationally-Driven Antibody Humanization”) in which computational protein design methods directly select sets of amino acids to incorporate from human germline sequences to increase humanness while maintaining structural stability. Retrospective studies show that CoDAH is able to identify variants deemed beneficial according to both humanness and structural stability criteria, even for targets lacking crystal structures. Prospective application to TZ47, a murine anti-human B7H6 antibody, demonstrates the approach. Four diverse humanized variants were designed, and all possible unique VH/VL combinations were produced as full-length IgG1 antibodies. Soluble and cell surface expressed antigen binding assays showed that 75% (6 of 8) of the computationally designed VH/VL variants were successfully expressed and competed with the murine TZ47 for binding to B7H6 antigen. Furthermore, 4 of the 6 bound with an estimated KD within an order of magnitude of the original TZ47 antibody. In contrast, a traditional CDR-grafted variant could not be expressed. These results suggest that the computational protein design approach described here can be used to efficiently generate functional humanized antibodies and provide humanized templates for further affinity maturation.
Keywords: antibody, computational protein design, human string content, humanization, pareto optimization, protein structure analysis
Abbreviations
- MHC
major histocompatibility complex
- HSC
Human String Content
- RMSD
root mean square deviation
- BLI
bio-layer interferometry
- IDT
Integrated DNA Technologies; PBS, phosphate-buffered saline
- PBS-T
PBS + 0.1% Tween-20
- PBS-F
PBS + 0.1% Bovine serum albumin
Introduction
The inherent ability of antibodies to be raised against almost any antigen at high affinity and specificity has contributed to their immense success as therapeutics.1 This success critically depends on the development of techniques to overcome the immunogenicity challenges that initially hampered the clinical application of non-human antibodies (e.g., murine origin).2 One set of techniques seeks to “humanize” an exogenous antibody, e.g., by complementarity determining region (CDR) grafting,3 resurfacing,4 specificity-determining residue (SDR) grafting,5 superhumanization,6 human string content optimization 7 and various library-based methods.8-11 Such techniques have been shown to generate antibodies with significantly reduced immunogenicity.12 Another set of techniques seeks to generate “fully human” antibodies, e.g., via display of human antibody libraries with phage 13 or yeast,14 immunization of humanized mice,15,16 or B cell cloning.17,18 The resulting antibodies also often have relatively low immunogenicity, though there are notable exceptions.19,20 While fully human antibody development is leading to an increasing number of approved and promising clinical candidates, the majority of FDA approved antibodies are humanized murine antibodies,21,22 and humanization is still widely performed due to its relative maturity and low cost.23
Antibody humanization pursues the complementary goals of increasing acceptability to the immune system while maintaining stability, specificity, and affinity. In general, humanization relies on the fact that the human immune system is relatively tolerant of human antibodies,24 and that antibodies of any given isotype have similar modular structure and sequence-structure-function relationships. For example, standard grafting-based humanization techniques leverage antibody structural modularity to incorporate key functional parts of the exogenous antibody on a human antibody framework, with the aim of maximizing the human portion of the antibody while maintaining antigen binding. Though this technique has been widely and effectively used, it remains a demanding task that can fail entirely or lead to a substantial decrease in binding affinity or stability that must be overcome by extensive mutagenesis.25,26 Notably, a good starting point is essential to improve the chance for success via mutagenesis.
While traditional grafting seeks to transfer function from the exogenous antibody to a human framework by grafting key antigen recognition residues, an alternative method described by Lazar et al.7 seeks to transfer humanness to the functional antibody by substituting selected amino acids from human germline antibodies. This approach is based on the recognition that T cell-dependent responses are a primary driver of the immunogenicity of biologics,27 and the core unit of detection in this pathway is a small, linear peptide epitope. Thus, to avoid immune recognition, it may be sufficient to ensure that the constituent peptides of the target variant are all sufficiently human-like. It follows that instead of being limited to a single human germline antibody, the humanization process can incorporate different amino acids from different human germline antibodies at different positions, each selected so that the peptides in the target variant correspond to aligned peptides in various human germlines, and, as a result, are likely to be acceptable as self peptides, despite originating in the context of a different protein.
None of these prior humanization techniques attempts to directly model and optimize the structural impact of substitutions as an integral part of their selection; the structural context is implicit in the choice of positions available for grafting, or assessed in a post-processing step by which point it may be too late to sufficiently guide the design process. While the lack of a crystal structure for a target may have limited the potential utility of structure-based methods, in recent years, modeling techniques (and the structural databases upon which they rely, according to their characteristic fold and overall high sequence identity) have improved sufficiently such that antibody structures can routinely be reliably modeled,28-31 including the hypervariable CDRs.32-34 Furthermore, structure-based computational protein design has already been widely and successfully used in other protein engineering contexts,35-37 and rich computational tools specialized for antibodies 38-40 have also enabled antibody engineering and humanization to be more accessible. Olimpieri et al.41 recently released a comprehensive webserver that provides helpful tools for antibody humanization. These advances now provide the opportunity to incorporate structural modeling and design in the humanization process, which in turn can enable efficient generation of functionally humanized candidates, as demonstrated here.
In order to optimize an antibody for humanness and structural quality simultaneously, we developed a novel structure-based computational protein design method called CoDAH (“Computationally-Driven Antibody Humanization”). CoDAH (Fig. 1) selects sets of mutations making the best trade-offs between the competing criteria of humanness, assessed via human string content score,7 and structural energy, assessed via the AMBER force field.42 We show in retrospective tests that the method generates diverse designs that achieve desired levels of humanness and elucidate the energetic and mutational consequences of humanization, and further that designs based on homology models are quite similar to those based on crystal structures. In prospective application to the humanization of TZ47, a murine anti-B7H6 antibody,44 we show that CoDAH yields a very high hit rate of functional humanized variants, while an initial attempt at humanization via CDR grafting produced a variant that failed to express. We thus conclude that the approach developed here is valuable to initiating the humanization process with a diverse set of stable, functional, human-like variants.
Figure 1.

Overview of the structure-based antibody humanization method. (left) Humanness is evaluated by the human string content (HSC) score,7 which calculates aligned nonamer matches against human antibody germline sequences. (right) Structural stability is evaluated by rotameric energy, comprised of position-specific terms for rotamers at individual positions (side-chain internal energy plus backbone interaction) and pairs of positions (side-chain interactions). (center) Sets of mutations are chosen so as to make optimal trade-offs between the competing humanness and energy scores. Fab structures are colored bluer for higher HSC.
Results
Human String Content characterizes humanness
To assess the humanness of a target antibody, we adopt the Human String Content (HSC) score,7 which computes for each peptide in the target sequence the number of residues identical to their counterparts in the most similar aligned peptide from a human germline antibody (Fig. 1). Here, we consider overlapping 9mers, representing the extent of the core peptide binding unit to major histocompatibility complex (MHC)-II molecules responsible for T cell-mediated recognition of a foreign protein.45 To confirm the utility of this score with the expansion of sequences since the publication of the initial study,7 we recapitulated the distributions of scores of 513 murine, 32 chimeric, 61 humanized, and 279 human antibody sequences (VH and Vκ) from the international ImMunoGeneTics information system® (IMGT; www.imgt.org) database,46,47 according to a database of 212 unique VH and 85 unique Vκ germline sequences from the current VBASE (www.vbase2.org) and IMGT databases.48-50 As shown in Fig. 2 and Table S1, human and humanized antibody sequences have significantly higher HSC scores than do murine and chimeric antibodies (p-value < 10−10 by 2-sided t-tests). In general, the light chain scores were higher, perhaps due to relatively less diversity among light chains than among heavy chains.
Figure 2.
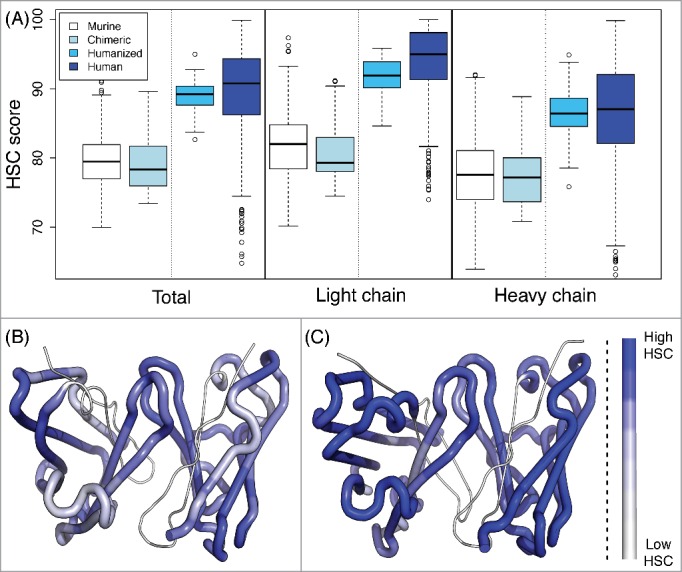
HSC scores of antibody sequences. (A) The HSC scores of human (279 sequences), humanized (61), chimeric (32), and murine (513) antibodies are distinct. In general, scores for light chains are higher than those for heavy chains. Plots illustrate HSC distributions with the upper and lower quartiles as a box, the median as a thick center line, 1.5 times the interquartile range as whiskers, and outliers as circles. (B and C) Visualization of HSC scores within a Fab, where the higher the HSC score is, the more blue. (B) Murine antibody (aL2, 1I3G) with median murine HSC score of 79.4. (C) Human antibody (scFv 9004G, 2YC1) with median human HSC score of 90.7.
We conclude that HSC is a valuable metric of humanness, and seek to select mutations that improve HSC while maintaining favorable structural energies. There are trade-offs between the 2 objectives, and our method identifies all Pareto optimal designs (Fig. 1), i.e., those with the best energy for a given HSC score (or vice versa). The optimization can also be iterated to find the second-best at each score, the third-best, and so on. We first assessed the ability of this method to effectively optimize variants of previously developed antibodies, and then applied it to humanize a murine antibody.
Designs for homology models are similar to those for crystal structures
A potential limitation of structure-based humanization is that a crystal structure of the target antibody may not be available. In order to demonstrate that the structure-based method is applicable in such cases, we applied CoDAH to generate designs for corresponding crystal structure and homology model pairs (Table 1). Models were built using the PIGS webserver based on template structures of moderate similarity (<80% sequence identity to the target) followed by side chain energy minimization; the range of root mean square deviation (RMSD) values between the model structures and corresponding crystal structures was 0.9–1.3 Å (Cα atoms), similar to previously reported values.28 The median HSC score of murine antibodies was ∼80, and we observed that about 15 mutations were sufficient to reach the median of about 89 for humanized antibodies. Accordingly, the mutational load was fixed to 15 and one Pareto optimal and 4 near-optimal sets of designs were generated for each target.
Table 1.
Retrospective test sets
| Proprietary or code name | INN (Target) | Species | HSC score of original clone | Starting structure |
|---|---|---|---|---|
| Model vs. crystal structure | ||||
| CAMPATH-1G | CD52 | Rat | 82.87 | 1BFO |
| GNC92H2 | GNC | Chimeric | 81.69 | 1I7Z |
| Remicade | Infliximab (TNF) | Chimeric | 79.91 | 4G3Y |
| Simulect | Basiliximab (IL2RA) | Chimeric | 75.8 | 3IU3 |
| cT84.66 | CEACAM5 | Chimeric | 74.85 | 1J05 |
| Lazar et al.7 | ||||
| AC10 | CD30 | Chimeric | 79.66 | — |
| Erbitux (225) | Cetuximab (EGFR) | Chimeric | 75.23 | 1YY8 |
| Re-humanization | ||||
| CAMPATH-1H | Alemtuzumab (CD52) | Humanized | 82.87 | — |
| Avastin | Bevacizumab (VEGF-A) | Humanized | 77.31 | — |
| Herceptin | Trastuzumab (ERBB-2) | Humanized | 74.72 | — |
Table 2 shows that there were many homology model-based designs that were identical or nearly identical to crystal structure-based designs, and that overall the 2 sets of designs used the same mutations at the same frequencies. The two cases where identical designs were not observed (1BFO and 3IU3) also exhibited the highest RMSD between model and crystal structures, while the most similar model/crystal structure pair (1J05) had the highest mutation frequency correlation and the largest fraction of identical designs. While this test artificially eliminated templates of high sequence identity to the target, it is expected that in practice, higher-quality models will be employed; indeed, recent reports on antibody structure modeling have shown near sub-angstrom accuracy in modeling framework regions.29,31 Thus homology model-based designs should be much like those based on crystal structures.
Table 2.
Humanization using homology models vs. crystal structures
| Proprietary or code name | Cα RMSD | Max HSC | Total designs | Identical designs | Up to 1 different mutation | Up to 2 different mutations | Spearman's ρ |
|---|---|---|---|---|---|---|---|
| CAMPATH-1G | 1.3 | 87.84 | 45 | 0 (0%) | 10 (22%) | 32 (71%) | 0.72 |
| GNC92H2 | 1.09 | 88.83 | 229 | 30 (13%) | 90 (39%) | 154 (67%) | 0.86 |
| Remicade | 1.05 | 87.83 | 184 | 2 (1%) | 32 (17%) | 100 (54%) | 0.63 |
| Simulect | 1.3 | 83.90 | 208 | 0 (0%) | 17 (8%) | 59 (28%) | 0.5 |
| cT84.66 | 0.91 | 82.57 | 115 | 29 (25%) | 84 (73%) | 106 (92%) | 0.87 |
HSC score and structural energy are competing criteria, driving different mutational choices
In order to evaluate the interplay between incorporating humanizing mutations and potentially destabilizing the antibody, we applied CoDAH to 5 additional antibodies (Table 1): 2 previously targeted by Lazar et al.7 and 3 previously humanized by other techniques. A crystal structure (1YY8) was available for Ab 225, but homology modeling was necessary for the others; the PIGS server was employed, but now used the best possible templates. Designs were optimized over a range of mutational loads from 10 up (“unrestricted”, indicating that the mutational load constraint in eq. 9 was omitted).
Fig. 3 depicts the Pareto optimal designs, among which there are clear trade-offs between the HSC and energy scores. (Fig. S1 depicts mutation frequencies over all the designs.) At a given mutational load (i.e., moving along one curve), in order to reach a higher HSC score, an energetic penalty is incurred. It is generally possible to make a substantial increase in HSC with a relatively minor energetic penalty, before hitting a “knee” beyond which humanizing mutations are much more energetically unfavorable. Increasing the mutational load allows selection of variants that are deemed better by the energy evaluation, thereby reaching a given HSC score with less energetic penalty. However, making more mutations does increase the risk of inconsistency between the energetic model and experimental reality. Thus, these trends should be considered primarily as guidelines to inform the selection of diverse designs making different trade-offs. It should be noted that the predecessor sequence may not always be energetically optimal for a given backbone structure according to the force field and the considered rotamers, perhaps due to the limitations of incomplete representation of discrete rotamers and rotamer energy calculations on the fixed backbone structure, and the lack of the antigen in the modeling.
Figure 3.

Pareto optimal variants, trading off HSC score and energy at a range of mutational loads from predecessor sequences. (A) anti-CD30 (AC10); (B) anti-EGFR (Ab 225); (C) trastuzumab; (D) bevacizumab; (E) alemtuzumab. All humanized designs generated have energies that are better than or only marginally higher than the predecessor sequences.
A rough comparison to the original HSC optimization method is possible for AC10 and Ab 225, though we note that the original HSC optimization method optimized and scored light and heavy chains separately and the germline sequence sets differ. For AC10, the maximally humanized variant (light HSC 86 and heavy HSC 91) employed 22 mutations (5 light chain and 17 heavy), in comparison to their final variant (88 and 83) at a total of 42 mutations (15 and 27). Similarly for Ab 225, CoDAH employed 40 mutations (18 and 22) to attain a maximally humanized variant at HSC scores of 94 and 86, as compared to 53 mutations (27 and 26) for the previous final variant with HSC scores of 95 and 84. Thus, in general, this method can identify comparably humanized antibodies with a lower mutational load, forming the theoretical basis for potentially improved performance over other methods. Because we optimize the HSC scores for the entire structure as a whole including the VH/VL interface region, it is expected that this new method gives structurally more stable variants than the sequence-based optimization employed by Lazar et al.7
The generation of the entire set of Pareto optimal designs enables elucidation of trends in mutations balancing HSC and energy, and ultimately selection of a diverse set of representative designs for experimental evaluation. To study this process in one of the targets, we further explored the 55 designs generated for Ab 225 with an unrestricted mutational load (an average of 36 mutations each). As Fig. 4A shows, the mutations common to all designs, as well as those that appear in designs of lower HSC scores, generally appear on the antibody surface. In contrast, in order to achieve high HSC scores, mutations of buried residues are required, and thus an energetic penalty results. This tendency was also observed in other test cases (Table S2). Humanized designs along the Pareto optimal curve generally share similar mutations at similar HSC scores, with some marked transitions to different mutational choices (Fig. 4B). Thus, clusters of variants with similar mutations are apparent. A small number of diverse representative variants from the clusters could be selected to experimentally evaluate the mutational patterns driving the HSC/energy trade-offs.
Figure 4.
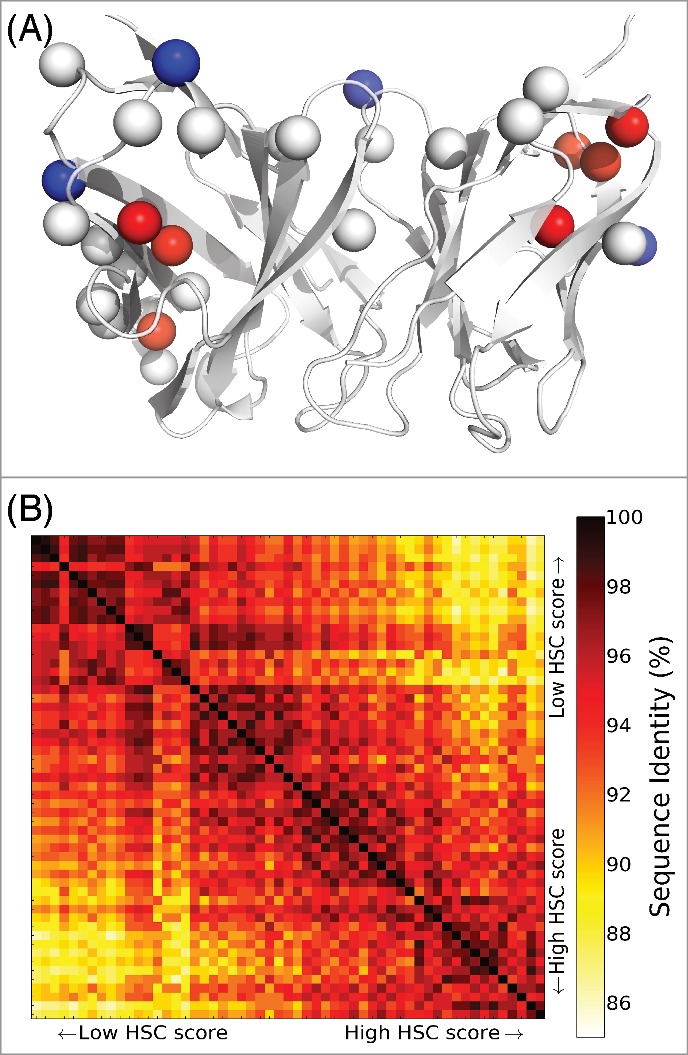
Sequence characteristics of humanized Ab 255 variants with unrestricted mutational load. (A) Mutation locations, with respect to a cartoon rendering of the Ab 255 model. There are 18 single mutations common to all 55 humanized designs (Cβ atoms as white spheres), 4 mutations unique to the 20 lowest HSC score designs (blue spheres), and 7 unique to the 20 highest HSC score designs (red spheres). (B) Pairwise sequence identities among the 55 humanized designs.
Computational protein design enables efficient TZ47 humanization
TZ47 is a murine antibody against the tumor antigen B7H6.44 We initially attempted to humanize the antibody using traditional CDR grafting. The CDR-grafted sequence was designed by selecting separate fully human antibody VH and VL regions with the highest sequence identity to the murine TZ47 VH and VL (Ms TZ47) using BLAST.51 CDRs (defined by IMGT numbering) were replaced with TZ47 CDRs to generate a CDR-grafted humanized TZ47 variant. The human VH used contained 63.6% overall identity to the murine TZ47 VH (67.0% excluding CDRs), and the human VL used contained 72.0% overall identity to the murine TZ47 VL (75.3% identity excluding CDRs). Canonical class structures were conserved between the grafts for CDR L1, L2, L3, and H1. The CDR H2 for both human VL and TZ47 VL could not be assigned into a single class. The CDR-grafted variant, however, could not be expressed as a full-length antibody. We then applied CoDAH as an alternative approach.
Computational design
Because a crystal structure of the antibody was not available, homology modeling was employed and humanized designs were built for the resulting model structure. CDR residues (defined by Kabat numbering) were fixed, and CoDAH was utilized to generate designs making a range of trade-offs between HSC and energy by substitution of framework residues. We observed that mutating 25 positions of the murine antibody was minimally sufficient to reach near the median humanized HSC score of ∼89, whereas the HSC score of the murine predecessor TZ47 was ∼76. Twelve variants with HSC scores higher than 88 were identified (Fig. 5A). In order to sub-select a diverse group, the designs were organized into a tree-based on sequence identity, and 4 representative variants were chosen, denoted 4, 9, 10, and 11 (Fig. 5B). While all heavy chains were distinct, 2 pairs of the computationally designed light chains were identical (4L = 9L and 10L = 11L). The two distinct light chains differ by only one amino acid, but they were kept separate. Each of the 8 distinct combinations of heavy chain and light chain pairings were experimentally evaluated for antigen binding.
Figure 5.

Humanized variants of TZ47. Initially 12 designs with 25 mutations were generated (A) and 4 diverse designs were selected according to a tree-based representation of sequence identity (B). Humanized variants are named according to the combination of (design # VH) / (design # VL). There are 2 unique light chains and 4 heavy chains; their possible combinations were also considered (A). Design 4 has 7 light-chain and 18 heavy-chain mutations, whereas Design 10 and 11 have 8 light and 17 heavy mutations. Thus the combinations 4H/10L and 9H/10L have 26 mutations in total (24 mutations for 11H/4L and 10H/4L) and are not on the Pareto optimal curve of designs with 25 mutations.
Of note, some structurally important residues in Fv region were conserved without specifically forcing them to be fixed. For example, the residue at H:71 (according to Kabat and Chothia numbering) of Ms TZ47 is alanine, which was conserved in all the designs, although we allowed all 10 amino acids found in human germline antibody sequences (A, I, V, K, P, T, M, L, R and E) for this position. The first few residues of CDR-H1 (H:26 to H:30) were also all conserved (GYTFT). As we did not allow mutations at the CDR residues, regions that are known to be in interactions with CDRs might be also captured by the rotameric-based energy.
Expression of humanized TZ47 variants
Each variable region sequence was synthesized and grafted into full-length human IgG1 heavy/light chain plasmids as appropriate, and all 8 unique combinations of heavy chains and light chains were used to transfect human embryonic kidney (HEK) cells. Six of the 8 antibody variants were successfully expressed (Fig. S2, Table 3). The two variants that failed to express (9H/4L and 9H/10L) utilized the 9H design, suggesting an expression issue specific to this heavy chain. As mentioned above, the CDR-grafted variant also failed to express. For the 6 expressed antibody variants, protein yields ranged from 20–40 mg/L.
Table 3.
Summary of expression and binding results for all humanized variants of the original murine TZ47 antibody
| Antibody | Expression | Soluble B7H6 Binding (BLI) | Cell Surface Expressed B7H6 Binding | Competes with Ms TZ47 |
|---|---|---|---|---|
| Ms TZ47 | + | + | + | NA |
| CDR Grafted | − | NA | NA | NA |
| 4H/4L | + | + | + | + |
| 4H/10L | + | + | + | + |
| 9H/4L | − | NA | NA | NA |
| 9H/10L | − | NA | NA | NA |
| 10H/4L | + | + | + | + |
| 10H/10L | + | + | + | + |
| 11H/4L | + | − | + | + |
| 11H/10L | + | − | + | + |
Binding activity of humanized TZ47 variants
The six expressed variants were further tested for binding to the soluble B7H6 antigen extracellular domain expressed as a murine Fc fusion protein and to cell-surface expressed B7H6. Bio-layer interferometry (BLI), a form of label-free kinetic analysis, was used to assess antibody recognition of the soluble B7H6-Fc antigen. In these tests, the original Ms TZ47 antibody was found to exhibit a KD of ∼290 nM (Fig. 6A), whereas no binding of VRC01, a human IgG1 antibody specific to HIV-1 envelope protein, was observed (Fig. 6B). In contrast, 4 of the 6 humanized variants demonstrated detectable binding signals to human B7H6 (Fig. 6C). Steady state kinetic analyses of the 4 variants estimated effective KDs ranging from high nM to low μM, placing them approximately within an order of magnitude of the original murine TZ47 KD (Figure 6). The two variants utilizing the 11HC design did not bind soluble B7H6 within the detectable range of BLI.
Figure 6.
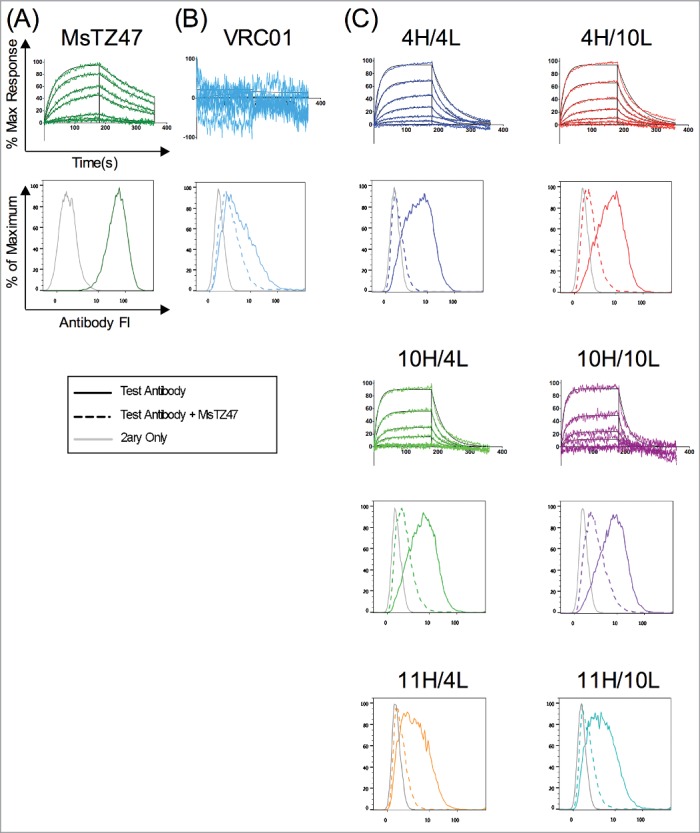
Validation of TZ47 variant binding activity and epitope specificity to soluble and cell-surface expressed B7H6. Shown are the normalized response curves for BLI testing at concentrations ranging from 26–1667 nM and histograms of fluorescence intensities of RMA-B7H6 cells stained with test antibody alone (solid color), test antibody +Ms TZ47 (dashed color), and secondary reagents only (solid gray) of the murine TZ47 (A), negative control VRC01 antibody (B), and humanized TZ47 variants (C).
To test antibody variant binding to cell-surface expressed B7H6, 2 cell lines were used: RMA-B7H6, which was stably transfected to express B7H6, and the parental RMA cell line, which was evaluated in order to verify that binding was specific to surface-expressed B7H6 rather than other surface proteins present. Initial staining experiments showed significantly higher staining of RMA-B7H6 cells relative to B7H6-negative RMA cells for all antibodies tested, including negative control antibody VRC01 (Table 3). It is possible that induction of B7H6 expression in these cells altered cellular expression of other cell-surface proteins, or expression of some level of misfolded B7H6 resulted in new non-specific targets for binding to occur besides faithful B7H6 epitopes. Thus, to further evaluate and confirm the fine-epitope specificity of the humanized variants, a competition experiment against the original murine TZ47 was performed.
Binding specificity of humanized TZ47 variants
To determine whether the humanized variants compete with the original murine TZ47 antibody for the same epitope on B7H6, RMA-B7H6 cells were pre-incubated with humanized antibody variants or VRC01 for one hour before the addition of a roughly equimolar concentration of murine TZ47. The histograms in Fig. 6C show significant shifts in antibody fluorescence intensity for all 6 humanized variants upon addition of the original Ms TZ47. In contrast, the histogram for VRC01 (Fig. 6B) remains relatively unchanged upon addition of TZ47, indicating that VRC01 failed to be effectively competed by TZ47. This data further suggests that VRC01 binds either to non-B7H6 proteins on the RMA-B7H6 cells, or to misfolded B7H6, as hypothesized above. Thus, these competition experiments indicate that all 6 expressible variants bind the B7H6 antigen at the same site as the original TZ47 antibody and qualitatively differ from the observed binding of RMA-B7H6 cells by the negative control VRC01 antibody. Good correlation between BLI and competition assay results were seen for all antibodies tested (Table 3), including the 11H-containing combinations. Although these variants did not bind soluble B7H6 antigen by BLI, they also stained RMA-B7H6 cells the most weakly compared to the other variants (which all bind with KDs near the limits of BLI detection).
Discussion
The process of antibody humanization has 2 potentially competitive goals: increasing humanness and retaining the desired function. Grafting methods increase humanness by converting to a human framework, but they can then require extensive mutagenesis to regain affinity and stability. Conversely, introducing humanizing mutations into the original framework risks destabilizing the protein, and post-analysis of the structural suitability of sets of humanizing mutations depends on having generated a good candidate pool. To directly address the trade-off between humanness and stability, we developed a novel structure-based antibody humanization algorithm that generates diverse variants simultaneously optimizing the 2 properties. Our method, CoDAH, leverages advances in antibody structural modeling (now often reaching sub-Ångström accuracy), and integrates established methods assessing the potential effects of mutations on protein stability and humanness, within a powerful combinatorial optimization framework that identifies beneficial combinations of mutations.
After demonstrating the effectiveness of CoDAH on retrospective cases, we applied it to humanizing TZ47, a murine antibody targeting B7H6. Humanization based on HSC score alone 7 has previously been shown to generate functional antibodies when applied to antibodies with low nanomolar KDs. In contrast, the antibody TZ47 binds its antigen with KD = 290 nM, leaving a narrow margin for error and demonstrating the use of CoDAH in humanizing even low-affinity antibodies.
We found that 6 of 8 computational designs were successfully expressed as full-length human IgG1 antibodies in HEK cells, while the CDR-grafted humanized TZ47 variant was undetectable in supernatants of transfected HEK cells. Notably, all expressible humanized variants were shown to bind the same B7H6 epitope as the original murine TZ47. Of the 4 computationally designed antibodies selected for testing, 2 (designs 4 and 10) successfully retained detectable binding to both soluble B7H6 antigen and cell-surface expressed antigen. In addition, 2 mismatched combinations of the humanized designs 4 and 10, 4H/10L and 10H/4L, also retained antigen binding. The permissibility of swapping the heavy and light chains for these 2 variants while maintaining antigen binding may be explained by their mutation positions. The full designs 4 and 10 differ by only 3 amino acids, which are all on the surface. On the other hand, there are differences in interface regions between 4H and 11H, which may affect structural orientations and thus potentially disturb antigen binding of their combinations 52 (Fig. S3).
One strategy to restore antigen binding to CDR-grafted humanized antibodies involves back-mutating Vernier Zone Residues, a set of 30 total framework residues in VH/VL that may contribute to the conformation of CDR loops.53 However, there is a risk of restoring murine immunogenicity along with antigen binding by retaining such residues.54 The humanization method proposed here solves this problem by simultaneously predicting key structural residues, analogous to the Vernier zone residues, and taking into account trade-offs between the role of such residues in both structure and immunogenicity. In the case of TZ47, computationally designed variants were found to retain all Vernier zone residues from the original murine TZ47, whereas the CDR-grafted variant had 5 Vernier zone mutations (not shown).
The intuition driving antibody humanization is that, due to self-tolerance, making a target more “human” is likely to reduce the risk of a detrimental immune response. Therefore a suitable measure of “humanness” is critical. While global sequence identity metrics have shown good overall discrimination of human vs. exogenous antibodies,55-57 they are less suitable for residue-level redesign. Furthermore, immunogenicity may not only be a matter of overall sequence identity to human antibodies, as humanized and even fully human antibodies may be immunogenic.58 Because T cell dependent responses against an exogenous protein are initiated by recognition of constituent epitopes in MHC proteins, peptide-level analysis provides the appropriate granularity. Instead of directly predicting and then seeking to mutagenically delete T cell epitopes, as is done for non-immunoglobulin proteins,59-63 the shared sequence-structure-function relationships of antibodies enables direct incorporation of germline amino acids so as to render constituent peptides human-like. While the subsequent demonstration of reduced clinical immunogenicity of a humanized target is beyond the scope of the present paper, we note that a recent report showed an HSC-optimized anti-CD30 antibody could be safely administered in a Phase 1 clinical study.64
The success of our computational method in generating beneficial designs for a range of retrospective targets, along with its high experimental hit rate in prospective humanization of TZ47, suggest that structure-based protein design may provide an efficient and effective technique for initiating antibody humanization. In particular, computational design may yield a number of different variants maintaining a high degree of stability and antigen binding affinity or avidity. The identification of such a diverse set of stable and active starting points may also increase the chance of success of subsequent rounds of affinity-improving mutagenesis, compared to starting from a non-binding CDR-grafted variant.
Materials and Methods
As illustrated in Fig. 1, our method unifies sequence-based evaluation of humanness with structure-based evaluation of energy (i.e., stability), integrating them within a protein redesign algorithm that optimizes variants making the best trade-offs between these 2 aspects. The components of the algorithm are now described, followed by details of the retrospective and prospective applications.
Evaluation of humanness
The humanness of an antibody is assessed in terms of its human string content score 7 (Fig. 1, left). Consider a single l-length peptide p starting at position i in an antibody chain (light or heavy). The results presented here, as well as by Lazar et al.,7 use l = 9, as this represents the typical length of the core peptide binding an MHC.65,66 The position-specific humanness score contributed by that peptide is its maximum identity to a corresponding peptide (i.e., also starting at position i) in an aligned human germline g from a set G of considered germlines.
| (1) |
The indicator function I{} is 1 if the predicate is true, i.e., the amino acid in the target antibody is the same as the corresponding one in the germline, and 0 otherwise. So the maximum score contributed by a peptide is 9, if all 9 of its amino acids are identical to corresponding amino acids in some germline sequence.
The HSC score of an entire antibody chain v is then the average over all its constituent peptides, scaled to a percentage (0–100).
| (2) |
Evaluation of structural energy
Antibody stability (Fig. 1, right) is assessed by rotamer-based structural energies, with position-specific single and pairwise terms based on the AMBER force field.42 A rotamer library 67 specifies a discrete set of possible side-chain conformations for both the original protein and possible substitutions, and an energy matrix is constructed by the OSPREY protein redesign software package.68,69 OSPREY eliminates rotamers necessitating steric clashes, as well as those that are provably not part of optimal or sufficiently near-optimal designs.70 Only the Fv region is considered in the energy calculations.
Identification of possible substitutions
The CDRs are not allowed to mutate during humanization. To identify CDRs, antibody sequences are annotated by the Kabat numbering scheme using the AbNum program.71 Light chain CDRs are defined as residues 24–34 (L1), 50–56 (L2) and 89–97 (L3), and heavy chain CDRs are composed of residues 31–35 (H1), 50–65 (H2) and 95–102 (H3).
At each position, any amino acid that appears in some human germline antibody sequence at the corresponding aligned position is considered as a possible substitution. It is also possible to filter the set based on those that are sufficiently frequent, appear in particular germlines, or based on other criteria as desired.
Structure-based antibody humanization algorithm
The humanization algorithm identifies the Pareto optimal protein designs,72 i.e., those making the best trade-offs between the 2 competing objectives of HSC score and rotamer-based energy. The algorithm follows a “sweep” approach analogous to that previously developed for enzyme deimmunization.43 Initially, the HSC score (Eq. 1) is calculated for the original target antibody sequence. The lowest-energy variant with a better HSC score than that of the original is then optimized, moving one step to the right on the curve in Fig. 1. The process is then repeated, finding the minimum-energy variant among those with better HSC scores than the first variant, and so forth along the curve. After a complete curve has been generated, the process can be repeated to produce a next-to-optimal curve, constraining the sequences to differ by at least one mutation from those already identified. Further increasingly suboptimal curves can likewise be generated.
In order to optimize the minimum-energy variant achieving at least a specified HSC score, the integer programming formulation of Parker et al.43 is adapted to use HSC. The integer program represents a variant as a set of rotamers, some from the original sequence and some as substitutions, encoded by a set of binary variables: si,r indicates whether or not the variant uses rotamer at position . In order to incorporate pairwise energy terms, the integer program also includes pair variables: pir,jt indicates whether or not the variant uses both rotamer r at i and t at j. Finally, in order to assess and thereby constraint HSC score, the “window” binary variable wi,p is defined to indicate whether or not amino acids spanning positions i to i + l − 1 (l = 9 in this case) correspond to linear peptide p (i.e., a sequence of 9 amino acids). Only peptides with HSC scores better than the original sequence are allowed.
The objective function for the integer program is to minimize the energy , computed as:
| (3) |
where is the energy of rotamer r at position i and the pairwise energy between rotamers r at i and t at j.
During the sweep, the next variant is constrained to improve the HSC score beyond the value H for the previous variant:
| (4) |
The variables are further constrained as follows, in order to ensure that the pair and window variables are consistent with the singleton variables, and that only one rotamer is taken at each position:
| (5) |
| (6) |
| (7) |
| (8) |
where is the amino acid type at position h in peptide p, and is that of rotamer .
Additionally the mutation load m can be constrained, specifying the number of rotamers that are not of the corresponding original amino acid type:
| (9) |
where is the amino acid at position i in the original target.
The algorithm is implemented in Python with the IBM ILOG CPLEX API. Source code is freely available for academic use, upon request.
Sequence databases
Human germline antibody sequences were retrieved from the VBASE 49,50 and the IMGT databases.48 Pseudogenes were excluded. In total, there are 212 unique VH, 80 Vλ, and 85 Vκ sequences.
Deposited antibodies of different classes (murine, chimeric, humanized, and human) were selected from the IMGT database.48 Since Vκ is most common among humanized antibodies, VL-containing antibodies were discarded from all classes.
Retrospective test sets
Retrospective assessment of the humanization performance of our method was performed on antibodies selected from 3 different sources (Table 1): 1) Five chimeric/nonhuman antibodies with available crystal structures were used for the comparison between designs on homology models and crystal structures; 2) AC10 and Ab 225 were previously targeted by Lazar et al.7 (detailed results for the other 2 targets in that paper, Ab 4D5 and 17-1A, were not reported, so they were not considered here.); and 3) Three additional marketed therapeutic antibodies of nonhuman origin, bevacizumab (Avastin®), alemtuzumab (Campath®, Lemtrada®) and trastuzumab (Herceptin®), were re-humanized here using only homology models of the original exogenous sequences.
Homology models were prepared using the PIGS antibody modeling server.28 For the model vs. crystal structure comparisons, model structures were built from template framework structures with sequence identity to the target antibody lower than 80% for each of the light and heavy chains. All CDRs were grafted and initial side chains were added by SCWRL 4.0.73 For other cases, the best possible template structures available in the modeling server were used.
CDR-H3 loops were remodeled using FREAD.32,74,75 For the construction of homology models to compare with crystal structures, loops from the corresponding crystal structures were discarded. When FREAD was unable to find any matches in the database, the prediction by PIGS was retained. The TINKER molecular dynamics package 76 was employed to minimize the model structure using the AMBER-f99sb 77 force field parameters with the generalized Born solvent model (GB/SA).78
Prospective Application
Structure modeling
The structure of the TZ47 Fv region was modeled by a hand-curated process rather than the server-based process employed in the retrospective tests, in order to more carefully control the model construction. Template structures of VH/VL were identified using BLAST.51 The best template identified for both light and heavy chains was PDB code 1D5I, which had light chain sequence identity of 90.87% (95.9% without CDRs) and heavy chain sequence identity of 85.83% (95.88%).
One hundred model structures were generated using MODELLER 79 and the best model was chosen by the DOPE potential.80 Non CDR-H3 loops were grafted on the above model. Structures for CDR sequences identical to those in the target were identified for L1 (1AXS), L2 (1BOG), L3 (1BOG) and H2 (1KN2) based on the canonical structures.33,81-83 For CDR-H1, the structure from 1A14, with nearly identical sequence, was grafted (TZ47: GYTFTGY, 1A14: GYTFTNY).
CDR-H3 was separately modeled using FREAD.32,74 The CDR-H3 structure in 1BAF was found to be the best match (CAIPGPMDYWG, 1BAF: CARGWPLAYWG). The sequence identity is 54.55% and the environment specific substitution score 32,74,84 is 51, well exceeding the cut-off value of >25 for accurate structure prediction.
The structure with grafted CDRs was energy-minimized against AMBER-f99sb 77 with GB/SA 78 using the TINKER package.76
Production of Humanized TZ47 Variant Antibodies
The CDR-grafted sequence was designed by selecting fully human antibody VH and VL regions with highest sequence identity to the murine TZ47 (Ms TZ47) VH and VL using NCBI BLAST. CDRs, as determined by VBASE2,49 of each homologous human VH & VL of interest were replaced with the Ms TZ47 CDRs to generate the CDR-grafted Humanized TZ47 variant.
VH and VL regions from computationally designed and CDR-grafted variants were ordered as IDT gBlocks with appropriate restriction sites for digestion and ligation into corresponding heavy and light chain plasmids for the human IgG1 antibody, VRC01: pCMVR-VRC01-HC and pCMVR-VRC01-LC. Full-length antibodies were produced and purified as previously described.85 Briefly, HEK-293F cells at a density of 106 cells/mL were co-transfected with various combinations of heavy chain and light chain plasmids at a concentration of 0.5 mg/L each and cultured for 5 days before Protein A purification of cell culture supernatants. Cultures were centrifuged at 3,000 × g for 15 min and supernatants passed over 0.3 mL Protein A resin columns. Columns were then washed with 3 mL phosphate-buffered saline (PBS) before elution with 100 mM Glycine, pH 3. Eluants were then buffer-exchanged back into PBS using Amicon-10K ultracentrifugation tubes.
Bio-Layer Interferometry (BLI) Testing for Binding to Soluble B7H6
A ForteBio Octet Red 96 machine was used for BLI studies. Soluble B7H6-Mouse Fc fusion antigen was biotinylated using ThermoScientific EZ-Link Sulfo-NHS-LC-Biotin at a targeted rate of 5 biotin / B7H6-Fc. Streptavidin-coated tips were allowed to equilibrate in PBS + 0.1% Tween-20 (PBS-T) for 100 s before activation in 0.1 mg/mL biotinylated B7H6-Fc for 200s, association in humanized TZ47 variant antibodies at 7 concentrations ranging from 0–1667 nM for 180s, dissociation in PBS-T for 180 s, and regeneration in 10 mM Glycine pH 3 for 10s. The murine TZ47 was tested at a concentration range of 0–1200 nM and the negative control VRC01 at 0–1667 nM. A second set of streptavidin tips was run using the same protocol without the activation step in biotinylated buffer to serve as a reference for non-specific binding. Data was processed by double-referencing against signal observed in wells containing PBS-T only and non-activated streptavidin tips. Steady state analysis provided estimates for KDs.
Cell Binding and Competition Assays
A Miltenyi Biotech MACSQuant Flow Cytometer was used for cell-binding assays. For the competition assay, 2.5E5 RMA-B7H6 cells/well were washed 3x with PBS + 0.1% BSA (PBS-F) before pre-incubation with 667 nM of either humanized antibody variants or VRC01 for 1 h. After preincubation, some wells were additionally treated with 500 nM murine TZ47 ‘spiked’ in for 20 min, while others were left in the humanized TZ47 variant preincubation for an extra 20 min. Cells were then washed 3x with PBS-F before secondary detection reagents were added to measure the gain or loss of signal for humanized variants and the competing murine TZ47. After a final wash with PBS-F, cells were resuspended in 200 μL PBS-F/well before reading on a Miltenyi MACSQuant flow cytometer.
Disclosure of Potential Conflicts of Interest
In addition to his role as a Dartmouth faculty member, Chris Bailey-Kellogg is co-founder and CTO of Stealth Biologics, LLC, a Delaware biotechnology company. He acknowledges that there is a potential financial conflict of interest related to his association with this company, and hereby affirms that the data presented in this paper is free of any bias. This work has been reviewed and approved as specified in his Dartmouth conflict of interest management plans. Charles Sentman is an inventor on a patent application for the use of the TZ47 antibody used in this study, and this has been licensed by Cardio3 BioSciences. This is managed in compliance with the policies of Dartmouth College. The remaining authors declare no conflict of interest.
Supplemental Material
Supplemental data for this article can be accessed on the publisher's website.
Funding
This work was supported in part by NIH grant R01-GM-098977 and a pilot grant associated with 5P30GM10345. We also gratefully acknowledge computational resources provided by NSF grant CNS-1205521.
References
- 1.Brooks M. Top 100 selling drugs of 2013. Medscape Medical News, 2014. Available at http://www.medscape.com/viewarticle/820011 [Google Scholar]
- 2.Carter PJ. Potent antibody therapeutics by design. Nat Rev Immunol 2006; 6:343-57; PMID:16622479; http://dx.doi.org/ 10.1038/nri1837 [DOI] [PubMed] [Google Scholar]
- 3.Jones PT, Dear PH, Foote J, Neuberger MS, Winter G. Replacing the complementarity-determining regions in a human antibody with those from a mouse. Nature 1986; 321:522–5; PMID:37138318302875 [DOI] [PubMed] [Google Scholar]
- 4.Roguska MA, Pedersen JT, Keddy CA, Henry AH, Searle SJ, Lambert JM, Goldmacher VS, Blättler WA, Rees AR, Guild BC. Humanization of murine monoclonal antibodies through variable domain resurfacing. Proc Natl Acad Sci 1994; 91:969-73; PMID:8302875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzales NR, Padlan EA, De Pascalis R, Schuck P, Schlom J, Kashmiri SV. SDR grafting of a murine antibody using multiple human germline templates to minimize its immunogenicity. Mol Immunol 2004; 41:863-72; PMID:15261458; http://dx.doi.org/ 10.1016/j.molimm.2004.03.041 [DOI] [PubMed] [Google Scholar]
- 6.Khee Hwang WY, Almagro JC, Buss TN, Tan P, Foote J. Use of human germline genes in a CDR homology-based approach to antibody humanization. Methods 2005; 36:35-42; PMID:15848073; http://dx.doi.org/ 10.1016/j.ymeth.2005.01.004 [DOI] [PubMed] [Google Scholar]
- 7.Lazar GA, Desjarlais JR, Jacinto J, Karki S, Hammond PW. A molecular immunology approach to antibody humanization and functional optimization. Mol Immunol 2007; 44:1986-98; PMID:17079018; http://dx.doi.org/ 10.1016/j.molimm.2006.09.029 [DOI] [PubMed] [Google Scholar]
- 8.Baca M, Presta LG, O'Connor SJ, Wells JA. Antibody humanization using monovalent phage display. J Biol Chem 1997; 272:10678-84; PMID:9099717; http://dx.doi.org/ 10.1074/jbc.272.16.10678 [DOI] [PubMed] [Google Scholar]
- 9.Dall'Acqua WF, Damschroder MM, Zhang J, Woods RM, Widjaja L, Yu J, Wu H. Antibody humanization by framework shuffling. Methods 2005; 36:43-60; PMID:15848074; http://dx.doi.org/ 10.1016/j.ymeth.2005.01.005 [DOI] [PubMed] [Google Scholar]
- 10.Dennis MS. CDR repair: A novel approach to antibody humanization In Shire SJ, Gombotz W, Bechtold-Peters K, Andya J (Eds.), Current Trends in Monoclonal Antibody Development and Manufacturing. New York: Springer-Verlag, 2010; 9-28. [Google Scholar]
- 11.Osbourn J, Groves M, Vaughan T. From rodent reagents to human therapeutics using antibody guided selection. Methods 2005; 36:61-8; PMID:15848075; http://dx.doi.org/ 10.1016/j.ymeth.2005.01.006 [DOI] [PubMed] [Google Scholar]
- 12.Hwang WYK, Foote J. Immunogenicity of engineered antibodies. Methods 2005; 36:3-10; PMID:15848070; http://dx.doi.org/ 10.1016/j.ymeth.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 13.McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nat 1990; 348:552-4; PMID:2247164; http://dx.doi.org/ 10.1038/348552a0 [DOI] [PubMed] [Google Scholar]
- 14.Feldhaus MJ, Siegel RW, Opresko LK, Coleman JR, Feldhaus JMW, Yeung YA, Cochran JR, Heinzelman P, Colby D, Swers J, et al.. Flow-cytometric isolation of human antibodies from a nonimmune Saccharomyces cerevisiae surface display library. Nat Biotechnol 2003; 21:163-70; PMID:12536217; http://dx.doi.org/ 10.1038/nbt785 [DOI] [PubMed] [Google Scholar]
- 15.Lee E-C, Liang Q, Ali H, Bayliss L, Beasley A, Bloomfield-Gerdes T, Bonoli L, Brown R, Campbell J, Carpenter A, et al.. Complete humanization of the mouse immunoglobulin loci enables efficient therapeutic antibody discovery. Nat Biotechnol 2014; 32:356-63; PMID:24633243; http://dx.doi.org/ 10.1038/nbt.2825 [DOI] [PubMed] [Google Scholar]
- 16.Lonberg N. In Chernajovsky Y, Nissim A (Eds.), Human monoclonal antibodies from transgenic mice Therapeutic Antibodies. Berlin: Springer-Verlag; 2008; 69-97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duvall M, Bradley N, Fiorini RN. A novel platform to produce human monoclonal antibodies: The next generation of therapeutic human monoclonal antibodies discovery. mAbs 2011; 3(2):203-8; PMID:21285537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li J, Sai T, Berger M, Chao Q, Davidson D, Deshmukh G, Drozdowski B, Ebel W, Harley S, Henry M, et al.. Human antibodies for immunotherapy development generated via a human B cell hybridoma technology. Proc Natl Acad Sci U S A 2006; 103:3557-62; PMID:16505368; http://dx.doi.org/ 10.1073/pnas.0511285103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Clark M. Antibody humanization: a case of the ‘Emperor's new clothes’? Immunology Today 2000; 21:397-402; PMID:10916143; http://dx.doi.org/ 10.1016/S0167-5699(00)01680-7 [DOI] [PubMed] [Google Scholar]
- 20.Hansel TT, Kropshofer H, Singer T, Mitchell JA, George AJ. The safety and side effects of monoclonal antibodies. Nat Rev Drug Dis 2010; 9:325-38; PMID:20305665; http://dx.doi.org/ 10.1038/nrd3003 [DOI] [PubMed] [Google Scholar]
- 21.Almagro JC, Fransson J. Humanization of antibodies. Front Biosci 2008; 13:1619-33; PMID:17981654 [DOI] [PubMed] [Google Scholar]
- 22.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Dis 2010; 9:767-74; PMID:20811384; http://dx.doi.org/ 10.1038/nrd3229 [DOI] [PubMed] [Google Scholar]
- 23.Lu Z-J, Deng S-J, Huang D-G, He Y, Lei M, Zhou L, Jin P. Frontier of therapeutic antibody discovery: the challenges and how to face them. World J Biol Chem 2012; 3:187; PMID:23275803; http://dx.doi.org/ 10.4331/wjbc.v3.i12.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bogen B, Ruffini P. Review: to what extent are T cells tolerant to immunoglobulin variable regions? Scandinavian J Immunol 2009; 70:526-30; PMID:19906193; http://dx.doi.org/ 10.1111/j.1365-3083.2009.02340.x [DOI] [PubMed] [Google Scholar]
- 25.Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol 2009; 157:220-33; PMID:19459844; http://dx.doi.org/ 10.1111/j.1476-5381.2009.00190.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pavlinkova G, Colcher D, Booth BJ, Goel A, Wittel UA, Batra SK. Effects of humanization and gene shuffling on immunogenicity and antigen binding of anti-tag-72 single-chain Fvs. Inter J Cancer 2001; 94:717-26; PMID:11745468; http://dx.doi.org/ 10.1002/ijc.1523 [DOI] [PubMed] [Google Scholar]
- 27.Jawa V, Cousens LP, Awwad M, Wakshull E, Kropshofer H, De Groot AS. T-cell dependent immunogenicity of protein therapeutics: preclinical assessment and mitigation. Clin Immunol 2013; 149:534-55; PMID:24263283; http://dx.doi.org/ 10.1016/j.clim.2013.09.006 [DOI] [PubMed] [Google Scholar]
- 28.Marcatili P, Rosi A, Tramontano A. PIGS: automatic prediction of antibody structures. Bioinformatics 2008; 24:1953-4; PMID:18641403; http://dx.doi.org/ 10.1093/bioinformatics/btn341 [DOI] [PubMed] [Google Scholar]
- 29.Shirai H, Ikeda K, Yamashita K, Tsuchiya Y, Sarmiento J, Liang S, Morokata T, Mizuguchi K, Higo J, Standley DM, et al.. High-resolution modeling of antibody structures by a combination of bioinformatics, expert knowledge, and molecular simulations. Proteins 2014; 82:1624-35; PMID:24756852 [DOI] [PubMed] [Google Scholar]
- 30.Sivasubramanian A, Sircar A, Chaudhury S, Gray JJ. Toward high-resolution homology modeling of antibody Fv regions and application to antibody-antigen docking. Proteins 2009; 74:497-514; PMID:19062174; http://dx.doi.org/ 10.1002/prot.22309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu K, Day T, Warshaviak D, Murrett C, Friesner R, Pearlman D. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins 2014; 82:1646-55; PMID:24619874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Choi Y, Deane CM. Predicting antibody complementarity determining region structures without classification. Mol Biosys 2011; 7:3327-34; PMID:22011953; http://dx.doi.org/ 10.1039/c1mb05223c [DOI] [PubMed] [Google Scholar]
- 33.North B, Lehmann A, Dunbrack RL Jr. A new clustering of antibody CDR loop conformations. J Mol Biol 2011; 406:228-56; PMID:21035459; http://dx.doi.org/ 10.1016/j.jmb.2010.10.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weitzner BD, Kuroda D, Marze N, Xu J, Gray JJ. Blind prediction performance of RosettaAntibody 3.0: Grafting, relaxation, kinematic loop modeling, and full CDR optimization. Proteins 2014; 82:1611-23; PMID:24519881; http://dx.doi.org/ 10.1002/prot.24534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Damborsky J, Brezovsky J. Computational tools for designing and engineering enzymes. Curr Opin Chem Biol 2014; 19:8-16; PMID:24780274; http://dx.doi.org/ 10.1016/j.cbpa.2013.12.003 [DOI] [PubMed] [Google Scholar]
- 36.Kazlauskas RJ, Bornscheuer UT. Finding better protein engineering strategies. Nat Chem Biol 2009; 5:526-9; PMID:19620988; http://dx.doi.org/ 10.1038/nchembio0809-526 [DOI] [PubMed] [Google Scholar]
- 37.Kries H, Blomberg R, Hilvert D. De novo enzymes by computational design. Curr Opin Chem Biol 2013; 17:221-8; PMID:23498973; http://dx.doi.org/ 10.1016/j.cbpa.2013.02.012 [DOI] [PubMed] [Google Scholar]
- 38.Dunbar J, Fuchs A, Shi J, Deane C. ABangle: characterising the VH-VL orientation in antibodies. Protein Engineering Design and Selection 2013; 26:611-20; http://dx.doi.org/ 10.1093/protein/gzt020 [DOI] [PubMed] [Google Scholar]
- 39.Kunik V, Ashkenazi S, Ofran Y. Paratome: an online tool for systematic identification of antigen-binding regions in antibodies based on sequence or structure. Nucl Acids Res 2012; 40:W521-W4; PMID:22675071; http://dx.doi.org/ 10.1093/nar/gks480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Olimpieri PP, Chailyan A, Tramontano A, Marcatili P. Prediction of site-specific interactions in antibody-antigen complexes: the proABC method and server. Bioinformatics 2013; 29:2285-91; PMID:23803466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Olimpieri PP, Marcatili P, Tramontano A. Tabhu: Tools for antibody humanization. Bioinformatics 2015; 31:434-5; PMID:25304777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, DeBolt S, Ferguson D, Seibel G, Kollman P.. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules. Comput Phys Commun 1995; 91:1-41; http://dx.doi.org/ 10.1016/0010-4655(95)00041-D [DOI] [Google Scholar]
- 43.Parker AS, Choi Y, Griswold KE, Bailey-Kellogg C. Structure-guided deimmunization of therapeutic proteins. J Comput Biol 2013; 20:152-65; PMID:23384000; http://dx.doi.org/ 10.1089/cmb.2012.0251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang T, Wu M-R, Sentman CL. An NKp30-based chimeric antigen receptor promotes T cell effector functions and antitumor efficacy in vivo. J Immunol 2012; 189:2290-9; PMID:22851709; http://dx.doi.org/ 10.4049/jimmunol.1103495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rammensee H-G, Friede T, Stevanović S. MHC ligands and peptide motifs: first listing. Immunogenetics 1995; 41:178-228; PMID:7890324; http://dx.doi.org/ 10.1007/BF00172063 [DOI] [PubMed] [Google Scholar]
- 46.Ehrenmann F, Lefranc M-P. IMGT/3Dstructure-DB: querying the IMGT database for 3D structures in immunology and immunoinformatics (IG or antibodies, TR, MH, RPI, and FPIA). Cold Spring Harbor Protocols 2011; 2011:750-61 pdb-prot5637 [DOI] [PubMed] [Google Scholar]
- 47.Poiron C, Wu Y, Ginestoux C, Ehrenmann F, Patrice D, Lefranc M-P. IMGT/mAb-DB: the IMGT database for therapeutic monoclonal antibodies. JOBIM 2010; 13 [Google Scholar]
- 48.Giudicelli V, Chaume D, Lefranc M-P. IMGT/V-QUEST, an integrated software program for immunoglobulin and T cell receptor V-J and V-D-J rearrangement analysis. Nucl Acids Res 2004; 32:W435-W40; PMID:15215425; http://dx.doi.org/ 10.1093/nar/gkh412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Retter I, Althaus HH, Munch R, Muller W. VBASE2, an integrative V gene database. Nucl Acids Res 2005; 33:D671-D4; PMID:15608286; http://dx.doi.org/ 10.1093/nar/gki088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tomlinson I, Williams S, Corbett S, Cox J, Winter G. V BASE sequence directory. Cambridge, UK: MRC Centre for protein engineering; 1996. [Google Scholar]
- 51.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol 1990; 215:403-10; PMID:2231712; http://dx.doi.org/ 10.1016/S0022-2836(05)80360-2 [DOI] [PubMed] [Google Scholar]
- 52.Narayanan A, Sellers BD, Jacobson MP. Energy-based analysis and prediction of the orientation between light-and heavy-chain antibody variable domains. J Mol Biol 2009; 388:941-53; PMID:19324053; http://dx.doi.org/ 10.1016/j.jmb.2009.03.043 [DOI] [PubMed] [Google Scholar]
- 53.Foote J, Winter G. Antibody framework residues affecting the conformation of the hypervariable loops. J Mol Biol 1992; 224:487-99; PMID:1560463; http://dx.doi.org/ 10.1016/0022-2836(92)91010-M [DOI] [PubMed] [Google Scholar]
- 54.Safdari Y, Farajnia S, Asgharzadeh M, Khalili M. Antibody humanization methods-a review and update. Biotechnol Genet Eng Rev 2013; 29:175-86; PMID:24568279; http://dx.doi.org/ 10.1080/02648725.2013.801235 [DOI] [PubMed] [Google Scholar]
- 55.Abhinandan K, Martin AC. Analyzing the “degree of humanness” of antibody sequences. J Mol Biol 2007; 369:852-62; PMID:17442342; http://dx.doi.org/ 10.1016/j.jmb.2007.02.100 [DOI] [PubMed] [Google Scholar]
- 56.Gao SH, Huang K, Tu H, Adler AS. Monoclonal antibody humanness score and its applications. BMC Biotechnol 2013; 13:55; PMID:23826749; http://dx.doi.org/ 10.1186/1472-6750-13-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thullier P, Huish O, Pelat T, Martin AC. The humanness of macaque antibody sequences. J Mol Biol 2010; 396:1439-50; PMID:20043919; http://dx.doi.org/ 10.1016/j.jmb.2009.12.041 [DOI] [PubMed] [Google Scholar]
- 58.Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies. MAbs 2010; 2:256-65; PMID:20400861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.King C, Garza EN, Mazor R, Linehan JL, Pastan I, Pepper M, Baker D. Removing T-cell epitopes with computational protein design. Proc Natl Acad Sci 2014; 111:8577-82; PMID:24843166; http://dx.doi.org/ 10.1073/pnas.1321126111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mazor R, Eberle JA, Hu X, Vassall AN, Onda M, Beers R, Lee EC, Kreitman RJ, Lee B, Baker D, et al.. Recombinant immunotoxin for cancer treatment with low immunogenicity by identification and silencing of human T-cell epitopes. Proc Natl Acad Sci 2014; 111:8571-6; PMID:24799704; http://dx.doi.org/ 10.1073/pnas.1405153111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Salvat RS, Choi Y, Bishop A, Bailey-Kellogg C, Griswold KE. Protein deimmunization via structure-based design enables efficient epitope deletion at high mutational loads. Biotechnol Bioeng 2015; PMID:25655032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Salvat RS, Parker AS, Choi Y, Bailey-Kellogg C, Griswold KE. Mapping the Pareto Optimal Design Space for a Functionally Deimmunized Biotherapeutic Candidate. PLoS Comput Biol 2015; 11:e1003988; PMID:25568954; http://dx.doi.org/ 10.1371/journal.pcbi.1003988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Salvat RS, Parker AS, Guilliams A, Choi Y, Bailey-Kellogg C, Griswold KE. Computationally driven deletion of broadly distributed T cell epitopes in a biotherapeutic candidate. Cell Mol Life Sci 2014; 71:4869-80; PMID:24880662; http://dx.doi.org/ 10.1007/s00018-014-1652-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kumar A, Blum KA, Fung HC, Smith MR, Foster PA, Younes A. A phase 1 dose-escalation study of XmAb® 2513 in patients with relapsed or refractory Hodgkin lymphoma. Br J Haematol 2014; PMID:AMBIGUOUS [DOI] [PubMed] [Google Scholar]
- 65.Parham P, Ohta T. Population biology of antigen presentation by MHC class I molecules. Science 1996; 272:67-74; PMID:8600539; http://dx.doi.org/ 10.1126/science.272.5258.67 [DOI] [PubMed] [Google Scholar]
- 66.Reche PA, Reinherz EL. Sequence variability analysis of human class I and class II MHC molecules: functional and structural correlates of amino acid polymorphisms. J Mol Biol 2003; 331:623-41; PMID:12899833; http://dx.doi.org/ 10.1016/S0022-2836(03)00750-2 [DOI] [PubMed] [Google Scholar]
- 67.Lovell SC, Word JM, Richardson JS, Richardson DC. The penultimate rotamer library. Proteins 2000; 40:389-408; PMID:10861930; http://dx.doi.org/ 10.1002/1097-0134(20000815)40:3%3c389::AID-PROT50%3e3.0.CO;2-2 [DOI] [PubMed] [Google Scholar]
- 68.Chen C-Y, Georgiev I, Anderson AC, Donald BR. Computational structure-based redesign of enzyme activity. Proc Natl Acad Sci 2009; 106:3764-9; PMID:19228942; http://dx.doi.org/ 10.1073/pnas.0900266106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gainza P, Roberts KE, Donald BR. Protein design using continuous rotamers. PLoS Comput Biol 2012; 8:e1002335; PMID:22279426; http://dx.doi.org/ 10.1371/journal.pcbi.1002335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Goldstein RF. Efficient rotamer elimination applied to protein side-chains and related spin glasses. Biophys J 1994; 66:1335-40; PMID:8061189; http://dx.doi.org/ 10.1016/S0006-3495(94)80923-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Abhinandan K, Martin AC. Analysis and improvements to Kabat and structurally correct numbering of antibody variable domains. Mol Immunol 2008; 45:3832-9; PMID:18614234; http://dx.doi.org/ 10.1016/j.molimm.2008.05.022 [DOI] [PubMed] [Google Scholar]
- 72.He L, Friedman AM, Bailey-Kellogg C. A divide-and-conquer approach to determine the Pareto frontier for optimization of protein engineering experiments. Proteins 2012; 80:790-806; PMID:22180081; http://dx.doi.org/ 10.1002/prot.23237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Krivov GG, Shapovalov MV, Dunbrack RL. Improved prediction of protein side-chain conformations with SCWRL4. Proteins 2009; 77:778-95; PMID:19603484; http://dx.doi.org/ 10.1002/prot.22488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Choi Y, Deane CM. FREAD revisited: accurate loop structure prediction using a database search algorithm. Proteins 2010; 78:1431-40; PMID:20034110 [DOI] [PubMed] [Google Scholar]
- 75.Kelm S, Vangone A, Choi Y, Ebejer J-P, Shi J, Deane CM. Fragment-based modeling of membrane protein loops: Successes, failures, and prospects for the future. Proteins 2014; 82:175-86; PMID:23589399; http://dx.doi.org/ 10.1002/prot.24299 [DOI] [PubMed] [Google Scholar]
- 76.Ponder JW. TINKER: Software tools for molecular design (Ver. 6.0). Saint Louis, MO: Washington University School of Medicine; 2004; 3. [Google Scholar]
- 77.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins 2006; 65:712-25; PMID:16981200; http://dx.doi.org/ 10.1002/prot.21123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Qiu D, Shenkin PS, Hollinger FP, Still WC. The GB/SA continuum model for solvation. A fast analytical method for the calculation of approximate Born radii. J Phys Chem 1997; 101:3005-14; http://dx.doi.org/ 10.1021/jp961992r [DOI] [Google Scholar]
- 79.Eswar N, Webb B, Marti-Renom MA, Madhusudhan M, Eramian D, Shen M-Y, Pieper U, Sali A. Comparative protein structure modeling using Modeller. Curr Protoc Bioinformat 2006:5-6; PMID:18428767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shen M-y, Sali A. Statistical potential for assessment and prediction of protein structures. Protein Sci 2006; 15:2507-24; PMID:17075131; http://dx.doi.org/ 10.1110/ps.062416606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Al-Lazikani B, Lesk AM, Chothia C. Standard conformations for the canonical structures of immunoglobulins. J Mol Biol 1997; 273:927-48; PMID:9367782; http://dx.doi.org/ 10.1006/jmbi.1997.1354 [DOI] [PubMed] [Google Scholar]
- 82.Chothia C, Lesk AM. Canonical structures for the hypervariable regions of immunoglobulins. J Mol Biol 1987; 196:901-17; PMID:3681981; http://dx.doi.org/ 10.1016/0022-2836(87)90412-8 [DOI] [PubMed] [Google Scholar]
- 83.Chothia C, Lesk AM, Tramontano A, Levitt M, Smith-Gill SJ, Air G, Sheriff S, Padlan EA, Davies D, Tulip WR, et al.. Conformations of immunoglobulin hypervariable regions. Nature 1989; 342:877-83; PMID:2687698; http://dx.doi.org/ 10.1038/342877a0 [DOI] [PubMed] [Google Scholar]
- 84.Deane CM, Blundell TL. CODA: a combined algorithm for predicting the structurally variable regions of protein models. Protein Sci 2001; 10:599-612; PMID:11344328; http://dx.doi.org/ 10.1110/ps.37601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Boesch AW, Brown EP, Cheng HD, Ofori MO, Normandin E, Nigrovic PA, Alter G, Ackerman ME. Highly parallel characterization of IgG Fc binding interactions. mAbs 2014; 6(4):915-27; PMID:24927273; http://dx.doi.org/ 10.4161/mabs.28808 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.