

Abstract
Increasing evidence has highlighted the important roles of dysregulated long non-coding RNA (lncRNA) expression in tumorigenesis, tumor progression and metastasis. However, lncRNA expression patterns and their prognostic value for tumor relapse in lung adenocarcinoma (LUAD) patients have not been systematically elucidated. In this study, we evaluated lncRNA expression profiles by repurposing the publicly available microarray expression profiles from a large cohort of LUAD patients and identified specific lncRNA signature closely associated with tumor relapse in LUAD from significantly altered lncRNAs using the weighted voting algorithm and cross-validation strategy, which was able to discriminate between relapsed and non-relapsed LUAD patients with sensitivity of 90.9% and specificity of 81.8%. From the discovery dataset, we developed a risk score model represented by the nine relapse-related lncRNAs for prognosis prediction, which classified patients into high-risk and low-risk subgroups with significantly different recurrence-free survival (HR=45.728, 95% CI=6.241-335.1; p=1.69e-04). The prognostic value of this relapse-related lncRNA signature was confirmed in the testing dataset and other two independent datasets. Multivariable Cox regression analysis and stratified analysis showed that the relapse-related lncRNA signature was independent of other clinical variables. Integrative in silico functional analysis suggested that these nine relapse-related lncRNAs revealed biological relevance to disease relapse, such as cell cycle, DNA repair and damage and cell death. Our study demonstrated that the relapse-related lncRNA signature may not only help to identify LUAD patients at high risk of relapse benefiting from adjuvant therapy but also could provide novel insights into the understanding of molecular mechanism of recurrent disease.
Keywords: long non-coding RNA, lung adenocarcinoma, prognosis, relapse, recurrence-free survival
INTRODUCTION
Lung cancer, including small cell lung cancer (SCC) and non-small cell lung cancer (NSCLC), is one of the most common cancers that severely threaten human health. The number of deaths from lung cancer is increasing, and it is estimated that nearly one in four cancer-related deaths is due to lung cancer [1]. Lung adenocarcinoma (LUAD) is the most frequent histological subtype of NSCLC and its incidence remains a rapidly increasing trend over the past few decades in China [2]. Despite improvement in diagnosis and treatment, the overall five-year survival rate for LUAD patients is only about 15% [3]. Moreover, more than 30% of patients treated with surgical resection will have a relapse within five years after surgery [4].
Recent advances in large-scale genomic analysis and high-throughput sequencing technologies have greatly increased our understanding of non-coding RNA (ncRNA) world. It has become increasingly apparent that a large proportion of human genome can be transcribed and produced a huge number of ncRNA molecules [5]. NcRNAs are briefly divided into two broad categories on the basis of their size: short ncRNAs and long ncRNAs. MicroRNA (miRNAs) is very important short ncRNAs, recently several predictors have been proposed to accurately predict miRNAs from other RNAs [6–8], which are very useful for the studies of the ncRNAs. Recently a web server called repRNA [9] was established to extract various features from RNA sequences, which will benefit the studies of RNAs. Long non-coding RNAs (lncRNAs), representing the largest class of ncRNAs, are mRNA-like transcripts and defined arbitrarily as ncRNAs of greater than 200 nucleotides in length [10, 11]. Evidence from growing publications has demonstrated that lncRNAs play important roles in various fundamental biological processes including development, differentiation and metabolism by executing functions as signals, decoys, guides and scaffolds [12, 13]. Furthermore, there is increasing evidence that lncRNAs are emerging as crucial components in the cancer paradigm [14]. The findings from transcriptome profiling analysis have shown highly aberrant lncRNA expression pattern in various types of human cancer [15, 16]. These differently expressed lncRNAs may be associated with tumorigenesis, tumor progression and metastasis [17, 18]. Moreover, lncRNA expression tended to be cell-, tissue- and cancer-type specific, thus making them attractive as independent biomarkers for diagnosis and prognosis [19]. Some well-known lncRNAs, such as HOTAIR, MALAT1, H19, Xist, HULC and PTENP1, have been found to possess oncogenic or/and tumor suppressor properties in various types of cancer [20, 21]. Several combinations of multiple lncRNAs were proposed as potential prognostic signature associated with overall survival in some cancers, including glioblastoma multiforme, colorectal cancer, breast cancer, oesophageal squamous cell carcinoma, non-small cell lung cancer and multiple myeloma [22–29]. Recent studies have shown the close relationship between cancer metastasis/relapse and dysregulated lncRNA expression [28, 30–34], implying the potential of lncRNAs as biomarkers to predict the risk of cancer metastasis/relapse. However, lncRNA expression patterns and their prognostic value for LUAD relapse have not been systematically elucidated.
In this study, we performed a systematic analysis of lncRNA expression profiles across 403 LUAD patients who did or did not relapse by repurposing the publicly available microarray expression profiles to determine whether there is significantly altered lncRNA expression pattern that could distinguish LUAD with relapse and without relapse. We aimed to detect potential lncRNA biomarkers closely correlated with LUAD relapse, and to develop novel lncRNA signature to identify LUAD patients who are at the higher risk for developing relapse.
RESULTS
Identification of altered lncRNA expression associated with tumor relapse
Here, the Okayama dataset, which is the largest dataset in our study, contains 64 LUAD patients who developed relapse and 162 relapse-free LUAD patients [35]. To identify relapse-related lncRNAs, we selected 88 favorable patients (alive > 5 years without any evidence of relapse) and 33 fatal samples (dead in 5 years with evidence of relapse) in the Okayama dataset to form a discovery dataset (n=121). The remaining patients in the Okayama dataset was considered as the internal testing dataset (n=105). Analysis of lncRNA expression profiles for LUAD patients in the discovery dataset revealed obvious differences and identified a total of 25 differentially expressed lncRNAs (adjusted p-value <0.01 after Bonferroni correction) between LUAD patients who developed relapse and relapse-free LUAD patients (Supplementary File S1). Unsupervised hierarchical clustering of 121 LUAD patients in the discovery dataset according to the expression patterns of these 25 differentially expressed lncRNAs showed two distant patient clusters, which were highly correlated with the tumor relapse status (p=7.557e-12, chi-square test; Figure 1A). Indeed, cluster I contained close to the majority of relapsed patients (n=30; 90.9%). Conversely, cluster II contained the majority of non-relapsed patients (n=70; 79.5%). Moreover, the Kaplan-Meier analysis and log-rank test revealed significant difference in recurrence-free survival (RFS) between these two patients subgroups (p=4.93-e14, log-rank test; Figure 1B). At three and five years, the RFS rates of LUAD patients in cluster II were 95.9% and 95.9%, respectively, whereas the corresponding rates in the cluster I were 45.8% and 37.5%, respectively. The above results demonstrated that dysregulated lncRNAs might have a predictive power in the prognosis of LUAD patients.
Figure 1. LncRNA expression patterns can distinguish patients who developed relapse from those who did not relapse in the discovery dataset.
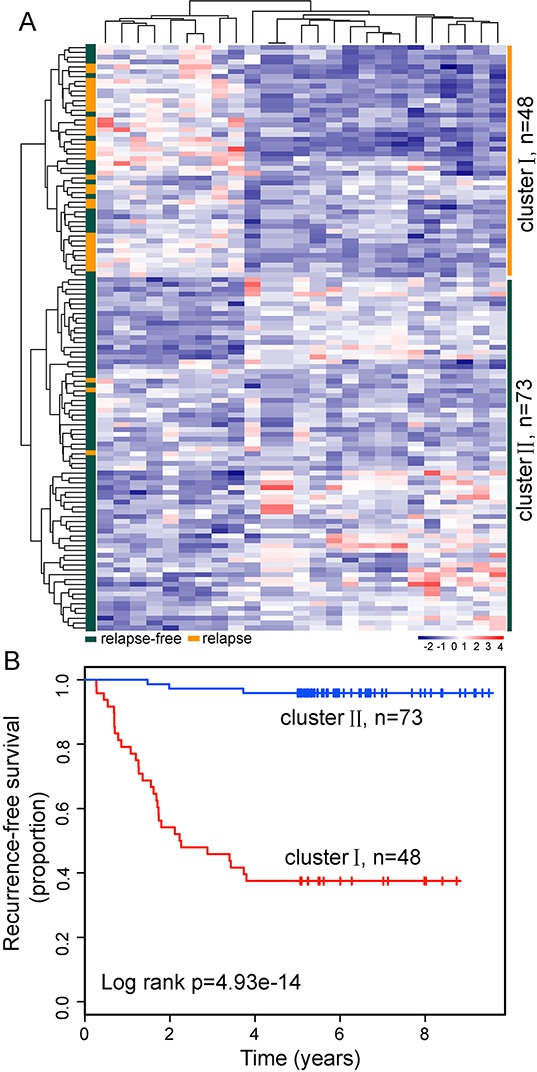
A. The unsupervised hierarchical clustering heatmap of 121 patients based on the 25 differentially expressed lncRNAs in the discovery dataset. B. Kaplan-Meier survival curve for RFS in the two lncRNA transcriptomic classifications.
Identification of optimal relapse-related lncRNA set
To identify relapse-related lncRNA signature, we used the weighted voting classification algorithm to predict outcome with the expression levels of these 25 differentially expressed lncRNAs as described in Materials and methods. These 25 differentially expressed lncRNAs were firstly ranked according to signal-to-noise metric. Then the average number of misclassified patients of the 5-fold cross-validation in 100 permutations was calculated when increasing numbers of top ranked predictive lncRNAs (Figure 2A). As a result, nine lncRNAs were found to yield a balance between accuracy and the number of lnRNAs, and were identified as optimal relapse-related lncRNA set. When choosing more than nine lncRNAs, there is a very slight increase in prediction accuracy (Figure 2B). With the selected nine lncRNAs and relapse status taken together, 121 LUAD patients were assigned as either relapse or relapse-free with accuracy of 84.3%. The classification of 121 LUAD patients produced a receiver operating characteristic (ROC) curve with AUC of 0.923, sensitivity of 90.9%, and specificity of 81.8% (Figure 2C). Furthermore, the Kaplan-Meier analysis for RFS demonstrated a significant difference between the groups predicted to be relapse or relapse-free (p=3.44e-15, log-rank test; Figure 2D). At three and five years, the RFS rates of LUAD patients in the predicted relapse-free group were 96% and 96%, respectively, whereas the corresponding rates in the predicted relapse group were 45.7% and 34.8%, respectively. We clustered LUAD patients in the discovery dataset according to the expression levels of nine relapse-related lncRNAs by hierarchical clustering analysis and obtained two distinctive patient groups with significantly different RFS (p=2.56e-07, log-rank test; Figure 3A and 3B). These results revealed better performance of nine relapse-related lncRNAs in prognosis prediction.
Figure 2. Identification of the relapse-related lncRNA signature in the discovery dataset.
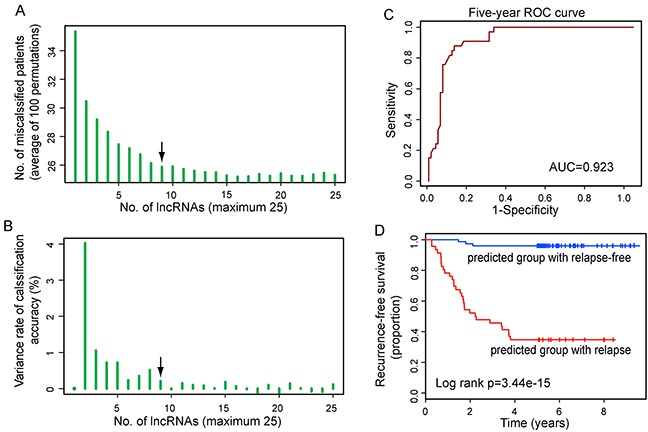
A. The learning errors for top N-lncRNA model using 5-fold cross-validation procedures with 100 random partitions of the discovery dataset. B. The variance rate of classification accuracy when increasing numbers of the predictive lncRNAs. C. ROC analysis of the relapse-related lncRNA signature for relapse prediction within five years as the defining point. D. Kaplan-Meier survival curve for RFS of patients with relapse or relapse-free according to nine-lncRNA model-based prediction.
Figure 3. The heatmap and survival analysis of hierarchical clustering based on relapse-related lncRNA signature in the discovery dataset.
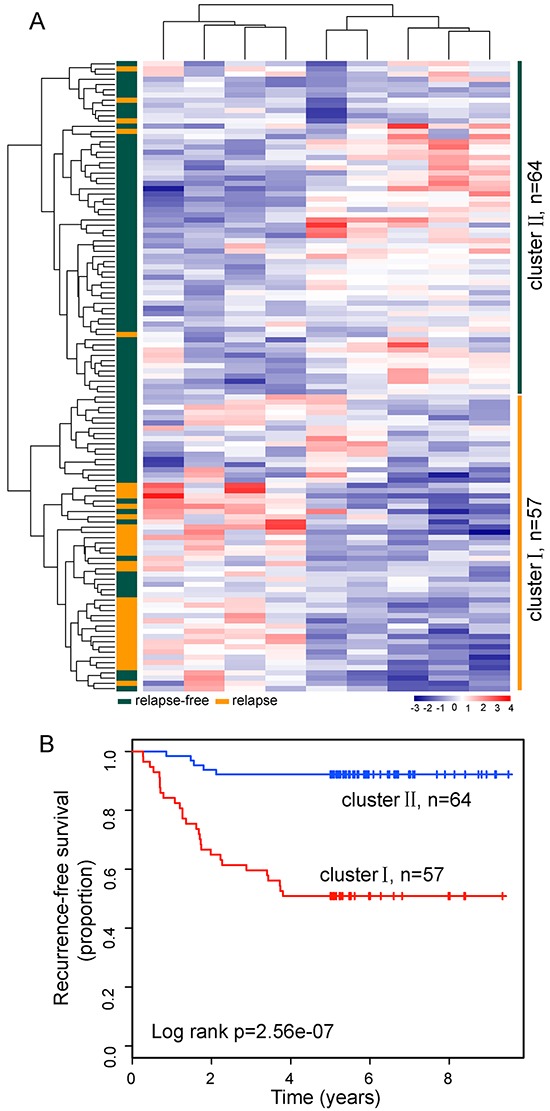
A. The unsupervised hierarchical clustering heatmap of 121 patients based on selected optimal nine lncRNAs in the discovery dataset. B. Kaplan-Meier survival curve for RFS in the two lncRNA transcriptomic classifications.
Construction of relapse-related lncRNA signature from the discovery dataset
We applied univariate Cox proportional hazard regression to each of these nine relapse-related lncRNAs and found all of them significantly correlated with patient's RFS (Table 1). We then used these nine relapse-related lncRNAs to construct a signature by the risk score method as the classifier for prognosis prediction. This risk score model was defined as a linear combination of the expression levels of the nine relapse-related lncRNAs and the multivariate Cox regression coefficient as the weight as follows: (−1.049×expression value of DACT3-AS1) + (0.027×expression value of CTD-2524L6.3) + (0.485×expression value of EFCAB14-AS1) + (0.93×expression value of AP000679.2) + (0.439×expression value of CTB-129P6.4) + (0.091× expression value of LRRC2-AS1) + (−0.357×expression value of RP11-517O13.3) + (0.489×expression value of AL133249.1)+(0.015×expression value of CTC-366B18.2). Each LUAD patient in the discovery dataset was assigned a risk score and was classified into different prognostic groups (the high-risk group and low-risk group) according to the threshold of the median risk score (−0.054). The Kaplan-Meier analysis demonstrated a significant difference in RFS between two patient groups predicted to have good or poor prognosis (median RFS > 8 years vs. 3.73 years, p=1.01e-10, log-rank test; Figure 4A). The three-year and five-year RFS rates of the high-risk group were 55% and 46.7%, respectively, whereas the corresponding rates in the low-risk group were 98.4% and 98.4%, respectively. The univariate analysis revealed a significant association between the risk score and RFS, in which the hazard ratio (HR) of high-risk group versus low-risk group for RFS is 45.728 (95% confidence interval (CI) 6.241-335.1; p=1.69e-04; Table 2). The time-dependent ROC curves analysis for the relapse-related lncRNA signature prognostic model achieved an AUC of 0.932 at five years of RFS (Figure 4B). These results demonstrated that the relapse-related lncRNA signature has better performance in prognosis prediction of LUAD.
Table 1. Relapse-related lncRNAs significantly associated with RFS in patients with LUAD.
| Ensembl ID | Gene name | Chromosome | Coef | HR | 95% CI of HR | P-value |
|---|---|---|---|---|---|---|
| ENSG00000245598 | DACT3-AS1 | Chr19: 46,660,364-46,677,447(+) | −0.964 | 0.382 | 0.26-0.559 | 7.52e-07 |
| ENSG00000260037 | CTD-2524L6.3 | Chr15: 71,818,396-71,823,384(+) | −0.783 | 0.457 | 0.334-0.626 | 1.09e-06 |
| ENSG00000228237 | EFCAB14-AS1 | Chr1: 46,674,036-46,692,098(+) | −1.226 | 0.294 | 0.182-0.474 | 5.13e-07 |
| ENSG00000176984 | AP000679.2 | Chr11: 120,168,977-120,171,679(+) | 1.058 | 2.881 | 1.976-4.202 | 3.83e-08 |
| ENSG00000267282 | CTB-129P6.4 | Chr19: 44,882,027-44,890,876(−) | 0.835 | 2.304 | 1.682-3.157 | 2.04e-07 |
| ENSG00000268324 | LRRC2-AS1 | Chr3: 46,557,398-46,559,694 (+) | 0.711 | 2.037 | 1.574-2.637 | 6.52e-08 |
| ENSG00000258658 | RP11-517O13.3 | Chr14: 58,370,023-58,395,641(−) | −0.843 | 0.43 | 0.299-0.619 | 5.44e-06 |
| ENSG00000223647 | AL133249.1 | Chr2: 31,793,823-31,803,980(−) | 0.843 | 2.323 | 1.636-3.3 | 2.51e-06 |
| ENSG00000248881 | CTC-366B18.2 | Chr5: 75,598,482-75,599,380(−) | −0.841 | 0.431 | 0.305-0.609 | 1.79e-06 |
Abbreviations: Coef, Coefficient; HR, hazard ratio; CI, confidence interval.
Figure 4. The relapse-related signature by risk score method in prognosis of RFS of patients with LUAD in the discovery dataset.
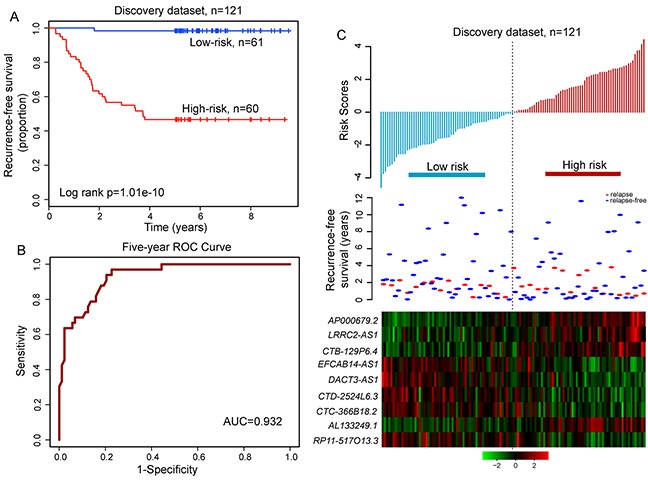
A. Kaplan-Meier survival curve for RFS of patients with high-risk or low-risk scores. B. ROC analysis of the risk score model for prognosis prediction within five years as the defining point. C. The distribution of risk scores, patients' relapse status and the heatmap of lncRNA expression profiles.
Table 2. Univariate and multivariate Cox regression analysis of RFS in each dataset.
| Variables | Univariate analysis | Multivariate analysis | |||||
|---|---|---|---|---|---|---|---|
| HR | 95% CI of HR | P-value | HR | 95% CI of HR | P-value | ||
| Discovery dataset, n=121 | |||||||
| lncRNA signature | High risk vs. Low risk | 45.728 | 6.241-335.1 | 1.69e-04 | 31.259 | 4.158-235.021 | 8.25e-04 |
| Age | >65 vs. <=65 | 2.876 | 1.44-5.744 | 2.75e-03 | 2.771 | 1.314-5.845 | 0.007 |
| Gender | Male vs. Female | 1.623 | 0.818-3.222 | 0.166 | 1.23 | 0.505-2.995 | 0.649 |
| Stage | II vs. I | 4.761 | 2.397-9.455 | 8.31e-06 | 2.243 | 1.102-4.566 | 0.026 |
| Smoking status | Yes vs. No | 1.574 | 0.789-3.14 | 0.198 | 1.099 | 0.43-2.809 | 0.843 |
| Testing dataset, n=105 | |||||||
| lncRNA signature | High risk vs. Low risk | 11.02 | 3.346-36.3 | 7.96e-05 | 11.5 | 3.344-39.57 | 1.06e-04 |
| Age | >65 vs. <=65 | 1.015 | 0.439-2.348 | 0.972 | 1.213 | 0.516-2.856 | 0.658 |
| Gender | Male vs. Female | 1.02 | 0.509-2.044 | 0.955 | 0.481 | 0.165-1.402 | 0.18 |
| Stage | II vs. I | 2.52 | 1.234-5.145 | 0.011 | 1.147 | 0.546-2.408 | 0.717 |
| Smoking status | Yes vs. No | 1.197 | 0.597-2.399 | 0.612 | 1.695 | 0.585-4.913 | 0.331 |
| Okayama dataset, n=226 | |||||||
| lncRNA signature | High risk vs. Low risk | 20.66 | 7.5-56.91 | 4.7e-09 | 17.619 | 6.266-49.543 | 5.36e-08 |
| Age | >65 vs. <=65 | 1.801 | 1.72-3.025 | 0.026 | 1.949 | 1.145-3.315 | 0.014 |
| Gender | Male vs. Female | 1.268 | 0.783-2.055 | 0.335 | 0.905 | 0.471-1.738 | 0.765 |
| Stage | II vs. I | 3.37 | 2.066-5.497 | 1.13e-06 | 1.609 | 0.969-2.67 | 0.066 |
| Smoking status | Yes vs. No | 1.324 | 0.816-2.148 | 0.256 | 1.09 | 0.565-2.102 | 0.797 |
| Der dataset, n=124 | |||||||
| lncRNA signature | High risk vs. Low risk | 1.829 | 1.074-3.115 | 0.026 | 1.938 | 1.135-3.31 | 0.015 |
| Age | >65 vs. <=65 | 1.327 | 0.756-2.328 | 0.324 | 1.397 | 0.788-2.477 | 0.252 |
| Gender | Male vs. Female | 1.267 | 0.761-2.109 | 0.364 | 1.312 | 0.778-2.213 | 0.308 |
| Stage | II vs. I | 2.64 | 1.559-4.469 | 3.02e-04 | 2.746 | 1.615-4.67 | 1.92e-04 |
| Smoking status | Yes vs. No | 1.219 | 0.628-2.366 | 0.559 | 0.968 | 0.49-1.914 | 0.926 |
| Botling dataset, n=53 | |||||||
| lncRNA signature | High risk vs. Low risk | ç1.86 | 0.939-3.685 | 0.075 | 1.997 | 0.976-4.084 | 0.058 |
| Age | >65 vs. <=65 | 1.231 | 0.653-2.321 | 0.521 | 1.118 | 0.58-2.155 | 0.74 |
| Gender | Male vs. Female | 1.123 | 0.59-2.138 | 0.725 | 1.152 | 0.552-2.404 | 0.706 |
| Stage | I | 1.000 (reference) | 1.000 (reference) | ||||
| II | 1.823 | 0.796-4.176 | 0.156 | 2.074 | 0.878-4.898 | 0.096 | |
| III | 2.338 | 1.027-5.322 | 0.043 | 2.624 | 1.066-6.459 | 0.036 | |
| IV | 4.61 | 0.588-36.143 | 0.146 | 3.866 | 0.46-32.479 | 0.213 | |
Abbreviations: HR, hazard ratio; CI, confidence interval.
The distribution of prognostic risk scores, the relapse status and expression pattern of lncRNA signature of 121 LUAD patients in the discovery dataset was shown in Figure 4C. Of these nine relapse-related lncRNAs, five were protective lncRNAs whose high expression were associated with low risk, and the remaining four were risky lncRNAs whose high expression were associated with high risk.
Validation of relapse-related lncRNA signature in the testing and entire okayama dataset
To confirm our findings, the predictive ability of relapse-related lncRNA signature was validated in LUAD patients from the testing dataset and entire Okayama dataset. With the same risk score formula and cutoff value derived from the discovery dataset, patients of the testing dataset were classified into the high-risk group (n=58) and low-risk group (n=47). As in the discovery dataset, the RFS time of patients in the high-risk group was significantly shorter than that in the low-risk group (median RFS 4.01 years vs. > 5 years, p=8.14e-07, log-rank test) (Figure 5A). The risk stratification of the entire Okayama dataset (i.e. combined the discovery and testing series) also yielded similar result. This relapse-related lncRNA signature was able to separate 226 patients in the entire Okayama dataset into two groups with significantly different RFS (median 3.73 years vs. > 8 years, p=1.11e-16, log-rank test) (Figure 5B). A significant association between the relapse-related lncRNA signature and RFS in the univariate Cox regression analysis was observed both in the testing and entire Okayama datasets. The hazard ratios of the high-risk group versus the low-risk group for RFS was 11.02 (p=7.96e-05; 95% CI 3.346-36.3) in the testing dataset, and 20.66 (p=4.7e-09; 95% CI 7.5-56.91) in the entire Okayama dataset (Table 2). The distribution of prognostic risk scores, the relapse status and expression pattern of lncRNA signature of LUAD patients in the testing and entire Okayama datasets were shown in Figure 5C and 5D. Patients with high prognostic scores tended to express risky lncRNAs, whereas patients with low prognostic scores tended to express protective lncRNAs.
Figure 5. The relapse-related signature predicts RFS of patients with LUAD in the testing dataset and entire Okayama dataset.
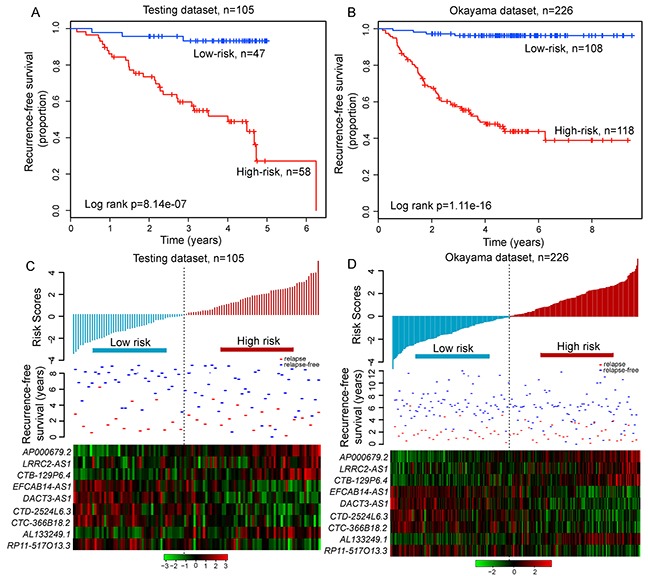
Kaplan-Meier survival curves of RFS between high-risk and low-risk patients in A. the testing dataset and B. entire Okayama dataset. The distribution of risk scores, patients' relapse status and the heatmap of lncRNA expression profiles in C. the testing dataset and D. entire Okayama dataset.
Further validation of relapse-related lncRNA signature with two additional independent datasets of LUAD patients
Further validation of the prognostic power of relapse-related lncRNA signature in LUAD patients was conducted using two additional completely independent cohorts of 124 and 53 LUAD patients obtained from Der's study [36] and Botling's study [37], which will be further referred to as the Der dataset and Botling dataset. The median cutoff value of risk score obtained from the discovery dataset was used for the Der dataset and Botling dataset to classify patients into either high-risk or low-risk groups. For the Der dataset, patients with high-risk scores had significantly shorter RFS than those with low-risk scores (median 3.74 years vs. 7.61 years, p=2.42e-02, log-rank test) (Figure 6A). Among patients in the Botling dataset, the high-risk group and low-risk group were marginally significantly different in their RFS (median 1.38 years vs. 7.96 years, p=7.07e-02, log-rank test) (Figure 6B). At three and five years, the respective absolute differences in RFS between the high-risk group and low-risk group were 15.6% (56.6% vs.72.2%) and 18% (43.3% vs. 61.3%) for Der dataset, and 22.8% (42.2% vs. 65%) and 26.1% (38.9% vs. 65%) for Botling dataset, respectively. In the univariate analysis, the hazard ratios of high-risk scores versus low-risk scores for RFS were 1.829 (p=2.62e-02; 95% CI 1.074-3.115) for Der dataset and 1.86 (p=7.52e-02; 95% CI 0.939-3.685) for Botling dataset, respectively (Table 2). The distribution of prognostic risk scores, the relapse status and expression pattern of lncRNA signature of LUAD patients in the two independent cohorts were consistent with those observed in the discovery, testing and entire Okayama datasets (Figure 6C and 6D).
Figure 6. Independent validation of relapse-related signature for prognosis prediction in two additional independent datasets.
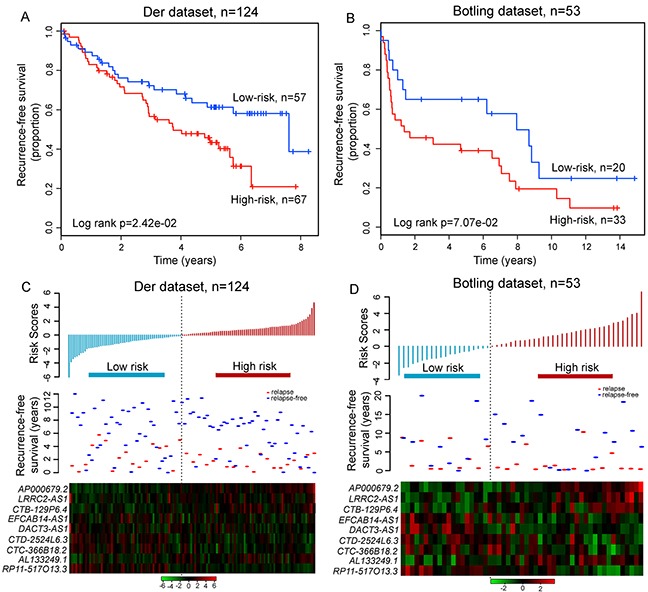
Kaplan-Meier survival curves of RFS between high-risk and low-risk patients in A. the Der dataset and B. the Botling dataset. The distribution of risk scores, patients' relapse status and the heatmap of lncRNA expression profiles in C. the Der dataset and D. the Botling dataset.
Independence of prognostic value of relapse-related lncRNA signature from other clinical variables
To determine whether the prognostic value of the relapse-related lncRNA signature was independent of other clinical variables, we conducted a multivariate Cox regression analysis including lncRNA signature, age, gender, smoking status and tumor stage as covariates. The results of multivariable Cox regression analysis from five LUAD patient datasets showed that the relapse-related lncRNA signature was still significantly associated with RFS after adjusted by these clinical variables in each dataset (Table 2). We also found that age and tumor stage was significant in the multivariate analysis in some datasets. So we performed data stratification analysis according to the age and tumor stage. All LUAD patients enrolled in this study were first stratified into either the younger stratum (age≤65) or the elder stratum (age>65). This analysis showed that within each age stratum, the relapse-related lncRNA signature could further classified patients into the high-risk group and low-risk group with significantly different RFS (median 4.73 years vs. > 8.68 years, p=2.55e-12 for younger stratum; median 2.94 years vs. 8.83 years, p=3.39e-05 for elder stratum; log-rank test) (Figure 7A and 7B). Next, the stratified analysis was carried out in tumor stage, which stratified patients into the stage I stratum and stage II stratum. For patients within stage I stratum, significant differences for RFS between high-risk group and low-risk group were observed (median 4.68 years vs. >10 years, p=2.8e-13, log-rank test) (Figure 7C). Among stage II patients, RFS was also marginally significantly different between the groups with high-risk and low-risk scores (median 2.26 years vs. 4.88 years, p=9.38e-02, log-rank test) (Figure 7D). Because of limited patient size, the stratified analysis was not conducted for stage III (n=8) and IV (n=1) patients. Taken together, the results of multivariate Cox regression and stratification analyses suggested that the relapse-related lncRNA signature is independent of other clinical features for prognosis prediction of LUAD patients. Moreover, the discrimination performance of relapse-related lncRNA signature measured by the C-index was much higher than that of other clinical variables in each dataset (Table 3), demonstrating the better predictive ability to discriminate between LUAD patients who are or not likely to develop relapse.
Figure 7. Prognosis prediction in patients stratified by age and tumor stage.
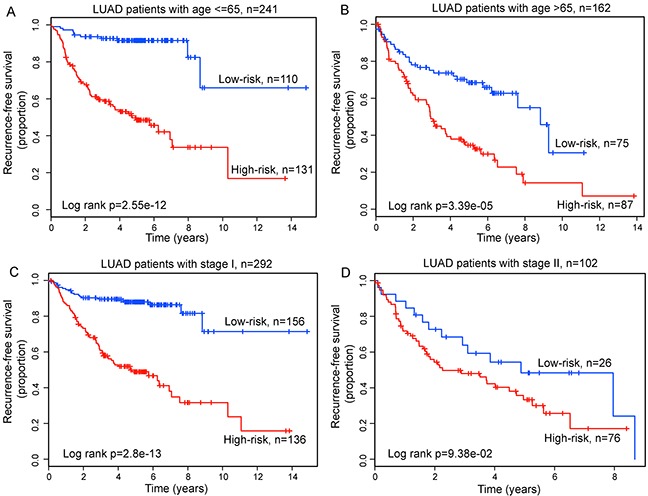
Kaplan-Meier survival curves for younger patients A. and elder patients B. Kaplan-Meier survival curves for stage I patients C. and stage II patients D.
Table 3. Concordance index values of the relapse-related lncRNA signature and other clinical feature for prognosis prediction.
| C-index | |||||
|---|---|---|---|---|---|
| Variables | Discovery dataset | Testing dataset | Okayama dataset | Der dataset | Botling dataset |
| lncRNA signature | 0.876 | 0.761 | 0.822 | 0.677 | 0.657 |
| Age | 0.608 | 0.498 | 0.556 | 0.528 | 0.514 |
| Gender | 0.567 | 0.490 | 0.536 | 0.536 | 0.534 |
| Stage | 0.671 | 0.590 | 0.635 | 0.591 | 0.585 |
| Smoking | 0.561 | 0.525 | 0.544 | 0.531 | - |
“-” means no corresponding information available in this dataset.
Identification of associated biological functions of relapse-related lncRNA signature
As an initial step toward gaining insights into the biological functions of relapse-related lncRNA signature, we first applied GSEA to identify associated biological pathways and processes from gene expression profiles of LUAD patients in the high-risk and low-risk groups classified by the relapse-related lncRNA signature in the Okayama dataset. The high-risk scores were associated with coordinated transcriptional up-regulation of multiple gene sets (Figure 8A and 8B) (Supplementary File S2), mainly involved in glucose metabolism and proteasome that have been reported to be involved in lung cancer [38, 39]. The low-risk score was accompanied with up-regulation of circadian clock and JNK-MAPK pathway (Figure 8A and 8B) (Supplementary File S2), both of which have inhibitory effects on the growth of lung cancer [40, 41]. Then we measured the co-expressed relationships between nine relapse-related lncRNAs and mRNAs by calculating the Pearson correlation coefficient of paired lncRNA and mRNA expression profiles and identified 1414 mRNAs positively correlated (ranked top 1%) with at least one of nine relapse-related lncRNAs. The functional enrichment analysis of GO and KEGG pathway revealed that the co-expressed mRNAs were most significantly enriched in 15 GO functional annotation clusters (mainly involved in cell cycle, DNA repair and damage, macromolecular complex assembly, RNA splicing and cell death) (Figure 8C) (Supplementary File S3), and 10 KEGG pathways including cell cycle, oocyte meiosis, progesterone-mediated oocyte maturation, DNA replication, insulin signaling pathway, spliceosome, p53 signaling pathway, One carbon pool by folate, gap junction and ErbB signaling pathway (p<0.05 and Fold Enrichment>2) (Figure 8D) (Supplementary File S4). This integrative functional analysis suggested that the dysregulated expression of relapse-related lncRNAs might affect the critical biological pathways and processes involved in tumor progression and recurrence.
Figure 8. Functional analysis of the prognostic lncRNAs.
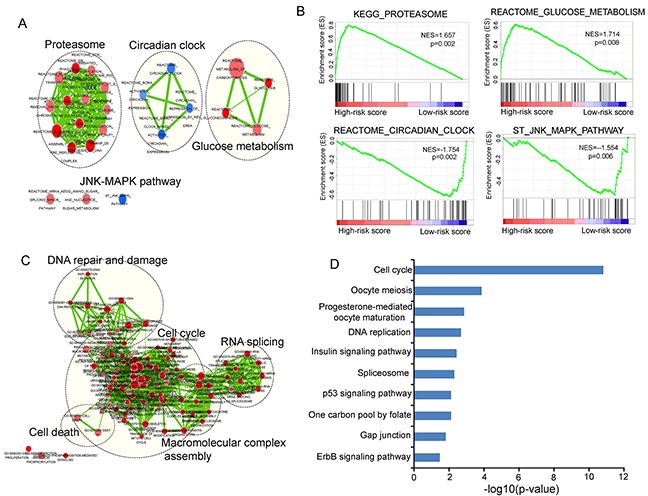
A. The enrichment map of gene sets with each node represents a gene set and an edge represents the proportion of shared genes between connecting gene sets. B. The enriched biological pathways and processes associated with risk score. C. The functional enrichment map of GO terms with each node represents a GO term and an edge represents the proportion of shared genes between connecting GO terms. D. The enriched KEGG pathways ranked by −log10 (p-value).
DISCUSSION
LUAD, the most frequent type of NSCLC, remains to be the leading cause of cancer-related deaths in women and men. LUAD is a recurrent disease, and more than 30% of patients still faced relapse after surgical resection and treatment and ultimately die of relapse [4]. In the past years, great efforts have been made to improve our understanding of the possible molecular mechanism of relapse process at protein, mRNA and microRNA (miRNA) levels, and some protein/mRNA/miRNA-based predictive signature were reported to identify patients at the high risk of relapse, which will enable them to benefit from adjuvant therapy [35, 42–50]. LncRNAs is a novel layer of gene regulation network and their aberrant expression has been demonstrated to be associated with tumorigenesis, tumor progression and metastasis [14, 17, 18, 30]. Until now, several lncRNAs, including MALAT-1, CCAT2, HOTAIR, and ZXF1, have been found to contribute to LUAD [51–54]. More recently, differentially expressed lncRNAs were observed between LUAD tissues and normal tissues by microarray analysis [55]. However, lncRNA expression patterns and their prognostic value for LUAD relapse have not been systematically investigated.
In the present study, we obtained lncRNA expression profiles in 403 LUAD patients by repurposing the publicly available microarray expression profiles, and performed comparison analysis between the groups of 121 LUAD patients that dead in 5 years with evidence of relapse and those that alive > 5 years without any evidence of relapse. Although Okayama et al. analyzed ALK-positive and triple negative LUAD patients [35], current study did not differentiate them. We found that two patient groups have significantly different lncRNA expression patterns and identified 25 differentially expressed lncRNAs. Hierarchical clustering analysis revealed that these differentially expressed lncRNAs were significantly correlated with LUAD relapse. By using the lncRNA expression data and a weighted voting algorithm, we were able to generate an optimal set of nine lncRNAs that could clearly distinguish patients who developed relapse from those who did not relapse with high accuracy, demonstrating their potential clinically application to identify patients with higher risk of relapse and improve prognosis prediction of LUAD. In statistical prediction, the following three cross-validation methods are often used to examine a predictor for its effectiveness in practical application: independent dataset test, subsampling test, and jackknife test. However, of the three test methods, the jackknife test is deemed the least arbitrary that can always yield a unique result for a given benchmark dataset as elaborated in [56]. Accordingly, the jackknife test has been widely recognized and increasingly used by investigators to examine the quality of various predictors [57–61]. However, to reduce the computational time, we adopted the 5-fold cross-validation in this study as done by many investigators with SVM as the prediction engine. Furthermore, the small subset of predictive lncRNAs perhaps not only has better transferability in the clinics but also reduce the possibilities of false positives in the selection of predictive lncRNAs. By focusing on relapse-related lncRNAs, we constructed a nine-lncRNA signature for prognosis by the risk score method based on the linear combination of expression data of relapse-related lncRNAs and weighted by the regression coefficients from multivariate Cox regression analysis, which effectively classified patients of the discovery dataset into high-risk group and low-risk group with significantly different RFS. Moreover, the prognostic power of this relapse-related lncRNA signature was further validated by the testing dataset and other two independent non-overlapping datasets, indicating good reproducibility and robustness of relapse-related lncRNA signature in patient prognosis.
The conventional indicators for making adjuvant treatment decisions for patients after surgical resection are based on some clinical factors, such as tumor stage, tumor size, margin status and so on [45]. Therefore, we performed multivariate Cox regression analysis to assess the independence of the relapse-related lncRNA signature in prognosis prediction, and in these datasets, the signature maintained an independent correlation with RFS after adjusting for age, gender, stage and smoking status (Table 2). However, it can be found that age and stage also were two important factors affecting RFS when accessed in the multivariate Cox regression analysis. So, we further stratified patients according age and stage, and applied this lncRNA signature to classify patients within the same age stratum or the same stage into two subgroups with good and poor prognosis. The stratification analysis showed that patients in the predicted good prognosis group tended to have longer RFS than those in the predicted poor prognosis group across these stratified patient datasets, demonstrating the age- and stage-independent prognostic value of this lncRNA signature. Moreover, the discriminatory power of relapse-related lncRNA signature measured by the C-index value was better than the discrimination provided by the clinical variables in different datasets. Taken together, this relapse-related lncRNA signature was a significant and independent prognostic marker in predicting relapse risk of patients with LUAD.
Although the number of identified lncRNAs in human is continuously increasing, only a small proportion have been well functionally characterized to date and the functional study of lncRNAs remains to be in its infancy. For example, only 181 lncRNAs with functional evidence were recorded in lncRNAdb v2.0 database by manually literature mining [62]. To gain functional insight into these nine relapse-related lncRNAs, we performed an integrative bioinformatics analysis to predict lncRNA function which could overcome the bias derived from the single prediction method. We found that the GO composition of co-expressed mRNAs with these lncRNAs revealed biological relevance to disease relapse, such as cell cycle, DNA repair and damage and cell death. Moreover, the pathway analysis also revealed a significant enrichment of co-expressed mRNAs with these lncRNAs in lung cancer-related pathways. These in silico functional analysis based on the co-expressed mRNAs with lncRNAs suggested that expression variation of these relapse-related lncRNAs might affect some critical biological pathways and processes involved in tumor progression and recurrence. However, biological significance of these relapse-related lncRNAs should be validated using wet experiments on cell lines and clinical samples in the future. As shown in a series of recent publications [63–68] in developing or reporting new methods or findings, user-friendly and publicly accessible web-servers will significantly enhance their impacts [69], we shall make efforts in our future work to provide a web-server for the method presented in this article.
In summary, we investigated lncRNA expression patterns in LUAD relapse and their effects on patient outcome for the first time. We identified an optimal small set of nine lncRNAs whose expression patterns were able to discriminate between relapsed and non-relapsed LUAD patients with sensitivity of 90.9% and specificity of 81.8%. A relapse-related lncRNA signature was developed by the risk model method that effectively classified patients into good and poor prognosis groups across different datasets. Moreover, the prognostic power of this signature was independent of other clinical variables. The relapse-related lncRNA signature may not only help to identify LUAD patients at high risk of relapse benefiting from adjuvant therapy but also could provide novel insights into the molecular mechanism of recurrent disease.
MATERIALS AND METHODS
LUAD patient datasets
LUAD patients with whole-genome gene expression profiles (generated from the Affymetrix Human Genome U133 Plus 2.0 Array) and corresponding clinical information were collected from the publicly available Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). A total of 403 LUAD patients were enrolled in this study, including 226 patients from Okayama's study (the accession number is GSE31210, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE31210) [35], 124 patients from Der's study (the accession number is GSE50081, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE50081) [36] and 53 patients from Botling's study (the accession number is GSE37745, http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE37745) [37]. LUAD patients and tumor features are detailed in Table 4.
Table 4. Clinical features of LUAD patients enrolled in this study.
| Covariates | Discovery dataset N=121 | Testing dataset N=105 | Okayama dataset N=226 | Der dataset N=124 | Botling dataset N=53 | |
|---|---|---|---|---|---|---|
| Age (years), no (%) | <=65 | 93(76.9) | 83(79.0) | 176(77.9) | 40(32.3) | 25(47.2) |
| >65 | 28(23.1) | 22(21.0) | 50(22.1) | 84(67.7) | 28(52.8) | |
| Gender, no (%) | Male | 55(45.6) | 50(47.6) | 105(46.5) | 63(50.8) | 20(37.7) |
| Female | 66(54.4) | 55(52.4) | 121(53.5) | 61(49.2) | 33(62.3) | |
| Vital status, no (%) | Alive | 88(72.7) | 103(98.1) | 191(84.5) | 75(60.5) | 16(30.2) |
| Dead | 33(27.3) | 2(1.90) | 35(15.5) | 49(39.5) | 37(69.8) | |
| Relapse status, no (%) | Relapse | 33(27.3) | 31(29.5) | 64(28.3) | 37(29.8) | 26(49.1) |
| Not relapse | 88(72.7) | 74(70.5) | 162(71.7) | 87(70.2) | 27(50.9) | |
| Tumor stage, no (%) | I | 93(76.9) | 75(71.4) | 168(74.3) | 90(72.6) | 34(64.2) |
| II | 28(23.1) | 30(28.6) | 58(25.7) | 34(27.4) | 10(18.9) | |
| III | - | - | - | - | 8(15.1) | |
| IV | - | - | - | - | 1(1.8) | |
| Smoking status, no (%) | Never-smoker | 62(51.2) | 53(50.5) | 115(50.9) | 23(18.5) | - |
| Ever-smoker | 59(48.8) | 52(49.5) | 111(49.1) | 90(72.6) | - | |
| Undetermined | - | - | - | 11(8.9) | - | |
“-” means no corresponding information available in the dataset.
Acquisition and analysis of lncRNA expression profiles of LUAD patients
The raw array data (.CEL files) of 403 LUAD patients were retrieved from the GEO database and were uniformly pre-processed using the Robust Multichip Average (RMA) algorithm for background correction, quantile normalization and log2-transformation [70]. To account for the heterogeneity of multiple microarray datasets in systematic measurement, each dataset was standardized independently by the Z-score transformation to scale expression intensities of each probe as follows [71]:
Where ei the raw intensity data of probei, is the overall average intensity of probes in a single experiment and δ is the standard deviation (SD) of all of the measured intensities.
The probe sequences of Affymetrix HG-U133 Plus 2.0 array were obtained from the Affymetrix website (http://www.affymetrix.com). LncRNA expression data of 403 LUAD patients were obtained by repurposing Affymetrix array probes as previous described [72, 73]. Briefly, probe sets for Affymetrix HG-U133 Plus 2.0 array were re-annotated to the human genome (GRCh38) and lncRNA genes based on the annotations from GENCODE (release 21) using SeqMap tool [74]. Then those probes (or probe sets) that were uniquely mapped to the human genome and lncRNA genes with no mismatch were generated to represent the lncRNAs. For each lncRNA, all corresponding probe set signals were averaged to produce a single expression value. Finally, the expression data of 2313 lncRNAs was obtained.
The expression profiles of lncRNAs between relapse-free LUAD patients (alive > 5 years without any evidence of relapse) and LUAD patients who developed relapse (dead in 5 years with evidence of relapse) were compared and the differentially expressed lncRNAs were identified using two-tailed T-test. Those lncRNAs with an adjusted p-value <0.01 after Bonferroni correction were considered as differentially expressed lncRNAs. The unsupervised hierarchical clustering of both LUAD patients and lncRNAs was performed with R software using the euclidean distance and complete linkage method.
Identification of relapse-related lncRNA set
To identify optimal lncRNA set associated with relapse of LUAD patients, we used the weighted voting algorithm to develop a supervised classification model, assessed using 5-fold cross-validation with 100 randomized permutations in the discovery dataset as follows: (i) patients of discovery dataset were divided into five non-overlapping sets with equal quantity. (ii) With four of five sample sets, the signal-to-noise statistic (Slnc-i) of each lncRNA was calculated as
where and is the mean value and standard deviation (SD) of expression level of lncRNA i in LUAD patients who developed relapse (relapse-free). In addition, the classification boundary of two classes (relapse or non-relapse) for each lncRNA i was also calculated as
(iii) lncRNAs were ranked according to its signal-to-noise statistic with absolute value, and top N ranked lncRNA were selected to develop a supervised classification model. The top N was initially set to top 1, and increase one lncRNA each time until N was equal to the number of candidate lncRNAs. (iv) The above classification model was used to classify patients in the remaining one set into relapse or non-relapse based on voting rules: each lncRNA i in the top N model has a vote Vln c−i (, where eln c–i is the expression level of lncRNA i in the corresponding patient. The votes of top N ranked lncRNAs were summed to determine the relapse or relapse-free class of sample. (v) repeat steps i-iii for each of the five non-overlapping sets. (vi) The 5-fold cross-validation process was repeated 100 times. The average number of misclassified patients of 100 randomized permutations for top N model as follows:
Finally, the top N which yielded the optimal numbers of learning errors was selected as the optimal number (OPN) of predictive lncRNAs. The frequencies of lncRNAs in 500 candidate lncRNAs ranking list according to their signal-to-noise statistic were ranked and top OPN of lncRNAs were identified as optimal relapse-related lncRNA signature.
Statistical analysis for classification and prediction
The association between expression levels of relapse-related lncRNAs and patients' RFS was assessed using the univariate Cox regression analysis. RFS was calculated as the time to tumor recurrence or death due to any cause, and was censored at the time of last following-up when recurrence has happened. To construct a prognostic model, the relapse-related lncRNAs were fitted in the multivariate Cox regression model in the discovery dataset. Then we applied these relapse-related lncRNAs to build an expression signature by risk score method as follows [75]:
Where N is the number of relapse-related lncRNAs, Expi is the expression levels of lncRNAi, and wi is the estimated regression coefficient of lncRNAi derived from the above multivariable Cox regression analysis in the discovery dataset. This relapse-related lncRNA expression signature was established by taking into account the contribution of independent relapse-related lncRNA to patient's RFS. Finally, LUAD patients were assigned a risk score according to the relapse-related lncRNA expression signature, and were divided into high-risk and low-risk groups using the median of the risk score generated from the discovery dataset as the cutoff value. The LUAD patients with higher scores were considered to have high risk of poor outcome. The difference in RFS between high-risk group and low-risk group was demonstrated by Kaplan-Meier survival plots, and the statistical significance was assessed by two-sided log-rank test. Univariate and multivariate analyses with Cox proportional hazards regression were performed with RFS as the dependent variable and relapse-related lncRNA risk score and clinical features as explanatory variables in each dataset. Hazard ratio (HR) and 95% confidence intervals (CI) was estimated by Cox proportional hazards regression model. The sensitivity and specificity of relapse-related lncRNA risk score in RFS prediction was evaluated by analysis of the time-dependent ROC curve within 5 years as the defining point. The Harrell's concordance index (C-index) was calculated to quantify the discriminatory power of relapse-related lncRNA risk score [76]. A C-index of 1.0 indicates perfect prediction accuracy, whereas a C-index of 0.5 represents prognosis prediction is equivalent to random guessing. All statistical analyses were performed using R software and Bio-conductor.
Integrative prediction analysis of lncRNA function
In order to explore the potential biological roles of lncRNA, we performed function enrichment analysis by the integration of gene sets, Gene Ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG). Gene set enrichment analysis (GSEA) was performed by the JAVA program using MSigDB (c2.cp.v5.0, 1330 gene sets) to rank gene set associated with risk score by enrichment score [77]. The gene sets with positive enrichment score (or negative enrichment score) and p-value <0.01 after performing 1000 permutations of the risk-phenotype labels were considered as significant enriched gene sets in which most of the genes are up-regulated accompanied with high-risk scores (or low-risk scores). GO and KEGG enrichment analysis were carried out to assess over-representation of functional categories among a gene set of interest using DAVID Bioinformatics Tool (version 6.7) limited to GO terms in the “Biological Process”(GOTERM-BP-FAT) and KEGG pathway categories [78]. Functional categories with p-value of <0.05 and an enrichment score of >2 using the whole human genome as background were considered significant. Significant functional annotations of GSEA and GO analysis were organized into an interaction network with similar functions using the Enrichment Map [79] plugin in Cytoscape 3.2.1 [80].
SUPPLEMENTARY FILES
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 61403111), China Postdoctoral Science Foundation (Grant No. 2014M551268) and Postdoctoral Foundation of Heilongjiang Province (Grant No. LBH-Z14212).
Footnotes
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
REFERENCES
- 1.Siegel R, Ma J, Zou Z, Jemal A. Cancer statistics, 2014. CA. 2014;64:9–29. doi: 10.3322/caac.21208. [DOI] [PubMed] [Google Scholar]
- 2.Zhou C. Lung cancer molecular epidemiology in China: recent trends. Translational Lung Cancer Research. 2014;3:270. doi: 10.3978/j.issn.2218-6751.2014.09.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A. Mapping the hallmarks of lung adenocarcinoma with massively parallel sequencing. Cell. 2012;150:1107–1120. doi: 10.1016/j.cell.2012.08.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goodgame B, Viswanathan A, Miller CR, Gao F, Meyers B, Battafarano RJ, Patterson A, Cooper J, Guthrie TJ, Bradley J. A clinical model to estimate recurrence risk in resected stage I non-small cell lung cancer. American journal of clinical oncology. 2008;31:22–28. doi: 10.1097/COC.0b013e3180ca77d1. [DOI] [PubMed] [Google Scholar]
- 5.Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, Thurman RE. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 2007;447:799–816. doi: 10.1038/nature05874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu B, Fang L, Wang S, Wang X, Li H, Chou KC. Identification of microRNA precursor with the degenerate K-tuple or Kmer strategy. J Theor Biol. 2015;385:153–159. doi: 10.1016/j.jtbi.2015.08.025. [DOI] [PubMed] [Google Scholar]
- 7.Liu B, Fang L, Liu F, Wang X, Chou KC. iMiRNA-PseDPC: microRNA precursor identification with a pseudo distance-pair composition approach. J Biomol Struct Dyn. 2016;34:223–235. doi: 10.1080/07391102.2015.1014422. [DOI] [PubMed] [Google Scholar]
- 8.Liu B, Fang L, Liu F, Wang X, Chen J, Chou KC. Identification of real microRNA precursors with a pseudo structure status composition approach. PLoS One. 2015;10:e0121501. doi: 10.1371/journal.pone.0121501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu B, Liu F, Fang L, Wang X, Chou KC. repRNA: a web server for generating various feature vectors of RNA sequences. Mol Genet Genomics. 2016;291:473–481. doi: 10.1007/s00438-015-1078-7. [DOI] [PubMed] [Google Scholar]
- 10.Ponting CP, Oliver PL, Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 11.Sun J, Zhou M, Mao ZT, Hao DP, Wang ZZ, Li CX. Systematic analysis of genomic organization and structure of long non-coding RNAs in the human genome. FEBS Lett. 2013;587:976–982. doi: 10.1016/j.febslet.2013.02.036. [DOI] [PubMed] [Google Scholar]
- 12.Mercer TR, Dinger ME, Mattick JS. Long non-coding RNAs: insights into functions. Nature Reviews Genetics. 2009;10:155–159. doi: 10.1038/nrg2521. [DOI] [PubMed] [Google Scholar]
- 13.Wang KC, Chang HY. Molecular mechanisms of long noncoding RNAs. Molecular cell. 2011;43:904–914. doi: 10.1016/j.molcel.2011.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Qiu MT, Hu JW, Yin R, Xu L. Long noncoding RNA: an emerging paradigm of cancer research. Tumour Biol. 2013;34:613–620. doi: 10.1007/s13277-013-0658-6. [DOI] [PubMed] [Google Scholar]
- 15.Gibb EA, Vucic EA, Enfield K, Stewart GL, Lonergan KM, Kennett JY, Becker-Santos DD, MacAulay CE, Lam S, Brown CJ. Human cancer long non-coding RNA transcriptomes. PloS one. 2011;6:e25915. doi: 10.1371/journal.pone.0025915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dong R, Jia D, Xue P, Cui X, Li K, Zheng S, He X, Dong K. Genome-wide analysis of long noncoding RNA (lncRNA) expression in hepatoblastoma tissues. PloS one. 2014:9. doi: 10.1371/journal.pone.0085599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang H, Chen Z, Wang X, Huang Z, He Z, Chen Y. Long non-coding RNA: a new player in cancer. J Hematol Oncol. 2013;6:1–7. doi: 10.1186/1756-8722-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qiu M-T, Hu J-W, Yin R, Xu L. Long noncoding RNA: an emerging paradigm of cancer research. Tumor Biology. 2013;34:613–620. doi: 10.1007/s13277-013-0658-6. [DOI] [PubMed] [Google Scholar]
- 19.Cheetham S, Gruhl F, Mattick J, Dinger M. Long noncoding RNAs and the genetics of cancer. British journal of cancer. 2013;108:2419–2425. doi: 10.1038/bjc.2013.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hauptman N, Glavač D. Long non-coding RNA in cancer. International journal of molecular sciences. 2013;14:4655–4669. doi: 10.3390/ijms14034655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fatima R, Akhade VS, Pal D, Rao SM. Long noncoding RNAs in development and cancer: potential biomarkers and therapeutic targets. Mol Cell Ther. 2015;3:5. doi: 10.1186/s40591-015-0042-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou M, Zhao H, Wang Z, Cheng L, Yang L, Shi H, Yang H, Sun J. Identification and validation of potential prognostic lncRNA biomarkers for predicting survival in patients with multiple myeloma. J Exp Clin Cancer Res. 2015;34:102. doi: 10.1186/s13046-015-0219-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu Y, Chen HY, Yu CY, Xu J, Wang JL, Qian J, Zhang X, Fang JY. A long non-coding RNA signature to improve prognosis prediction of colorectal cancer. Oncotarget. 2014;5:2230–2242. doi: 10.18632/oncotarget.1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Meng J, Li P, Zhang Q, Yang Z, Fu S. A four-long non-coding RNA signature in predicting breast cancer survival. J Exp Clin Cancer Res. 2014;33:84. doi: 10.1186/s13046-014-0084-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang XQ, Sun S, Lam KF, Kiang KM, Pu JK, Ho AS, Lui WM, Fung CF, Wong TS, Leung GK. A long non-coding RNA signature in glioblastoma multiforme predicts survival. Neurobiol Dis. 2013;58:123–131. doi: 10.1016/j.nbd.2013.05.011. [DOI] [PubMed] [Google Scholar]
- 26.Zhou M, Guo M, He D, Wang X, Cui Y, Yang H, Hao D, Sun J. A potential signature of eight long non-coding RNAs predicts survival in patients with non-small cell lung cancer. J Transl Med. 2015;13:231. doi: 10.1186/s12967-015-0556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li J, Chen Z, Tian L, Zhou C, He MY, Gao Y, Wang S, Zhou F, Shi S, Feng X, Sun N, Liu Z, Skogerboe G, Dong J, Yao R, Zhao Y, et al. LncRNA profile study reveals a three-lncRNA signature associated with the survival of patients with oesophageal squamous cell carcinoma. Gut. 2014;63:1700–1710. doi: 10.1136/gutjnl-2013-305806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Crea F, Watahiki A, Quagliata L, Xue H, Pikor L, Parolia A, Wang Y, Lin D, Lam WL, Farrar WL. Identification of a long non-coding RNA as a novel biomarker and potential therapeutic target for metastatic prostate cancer. Oncotarget. 2014;5:764–774. doi: 10.18632/oncotarget.1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou M, Wang X, Shi H, Cheng L, Wang Z, Zhao H, Yang L, Sun J. Characterization of long non-coding RNA-associated ceRNA network to reveal potential prognostic lncRNA biomarkers in human ovarian cancer. Oncotarget. 2016 doi: 10.18632/oncotarget.7181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun J, Chen X, Wang Z, Guo M, Shi H, Wang X, Cheng L, Zhou M. A potential prognostic long non-coding RNA signature to predict metastasis-free survival of breast cancer patients. Scientific reports. 2015:5. doi: 10.1038/srep16553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Malek E, Jagannathan S, Driscoll JJ. Correlation of long non-coding RNA expression with metastasis, drug resistance and clinical outcome in cancer. Oncotarget. 2014;5:8027–8038. doi: 10.18632/oncotarget.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu Y, Liu H, Shi X, Yao Y, Yang W, Song Y. The long non-coding RNA HNF1A-AS1 regulates proliferation and metastasis in lung adenocarcinoma. Oncotarget. 2015;6:9160. doi: 10.18632/oncotarget.3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang Z, Zhou L, Wu L-M, Lai M-C, Xie H-Y, Zhang F, Zheng S-S. Overexpression of long non-coding RNA HOTAIR predicts tumor recurrence in hepatocellular carcinoma patients following liver transplantation. Annals of surgical oncology. 2011;18:1243–1250. doi: 10.1245/s10434-011-1581-y. [DOI] [PubMed] [Google Scholar]
- 34.Lai M-c, Yang Z, Zhou L, Zhu Q-q, Xie H-y, Zhang F, Wu L-m, Chen L-m, Zheng S-s. Long non-coding RNA MALAT-1 overexpression predicts tumor recurrence of hepatocellular carcinoma after liver transplantation. Medical oncology. 2012;29:1810–1816. doi: 10.1007/s12032-011-0004-z. [DOI] [PubMed] [Google Scholar]
- 35.Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer research. 2012;72:100–111. doi: 10.1158/0008-5472.CAN-11-1403. [DOI] [PubMed] [Google Scholar]
- 36.Der SD, Sykes J, Pintilie M, Zhu C-Q, Strumpf D, Liu N, Jurisica I, Shepherd FA, Tsao M-S. Validation of a Histology-Independent Prognostic Gene Signature for Early-Stage, Non–Small-Cell Lung Cancer Including Stage IA Patients. Journal of Thoracic Oncology. 2014;9:59–64. doi: 10.1097/JTO.0000000000000042. [DOI] [PubMed] [Google Scholar]
- 37.Botling J, Edlund K, Lohr M, Hellwig B, Holmberg L, Lambe M, Berglund A, Ekman S, Bergqvist M, Pontén F. Biomarker discovery in Non–Small cell lung cancer: Integrating gene expression profiling, meta-analysis, and tissue microarray validation. Clinical Cancer Research. 2013;19:194–204. doi: 10.1158/1078-0432.CCR-12-1139. [DOI] [PubMed] [Google Scholar]
- 38.Duhaylongsod FG, Lowe VJ, Patz EF, Vaughn AL, Coleman RE, Wolfe WG. Lung tumor growth correlates with glucose metabolism measured by fluoride-18 fluorodeoxyglucose positron emission tomography. The Annals of thoracic surgery. 1995;60:1348–1352. doi: 10.1016/0003-4975(95)00754-9. [DOI] [PubMed] [Google Scholar]
- 39.Escobar M, Velez M, Belalcazar A, Santos ES, Raez LE. The role of proteasome inhibition in nonsmall cell lung cancer. BioMed Research International. 2011:2011. doi: 10.1155/2011/806506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu N, Gu C, Gu H, Hu H, Han Y, Li Q. Metformin induces apoptosis of lung cancer cells through activating JNK/p38 MAPK pathway and GADD153. Neoplasma. 2010;58:482–490. doi: 10.4149/neo_2011_06_482. [DOI] [PubMed] [Google Scholar]
- 41.Gery S, Komatsu N, Baldjyan L, Yu A, Koo D, Koeffler HP. The circadian gene per1 plays an important role in cell growth and DNA damage control in human cancer cells. Molecular cell. 2006;22:375–382. doi: 10.1016/j.molcel.2006.03.038. [DOI] [PubMed] [Google Scholar]
- 42.Tomida S, Takeuchi T, Shimada Y, Arima C, Matsuo K, Mitsudomi T, Yatabe Y, Takahashi T. Relapse-related molecular signature in lung adenocarcinomas identifies patients with dismal prognosis. Journal of Clinical Oncology. 2009;27:2793–2799. doi: 10.1200/JCO.2008.19.7053. [DOI] [PubMed] [Google Scholar]
- 43.Raponi M, Zhang Y, Yu J, Chen G, Lee G, Taylor JM, MacDonald J, Thomas D, Moskaluk C, Wang Y. Gene expression signatures for predicting prognosis of squamous cell and adenocarcinomas of the lung. Cancer research. 2006;66:7466–7472. doi: 10.1158/0008-5472.CAN-06-1191. [DOI] [PubMed] [Google Scholar]
- 44.Yanagisawa K, Tomida S, Shimada Y, Yatabe Y, Mitsudomi T, Takahashi T. A 25-signal proteomic signature and outcome for patients with resected non–small-cell lung cancer. Journal of the National Cancer Institute. 2007;99:858–867. doi: 10.1093/jnci/djk197. [DOI] [PubMed] [Google Scholar]
- 45.Subramanian J, Simon R. Gene expression–based prognostic signatures in lung cancer: ready for clinical use? Journal of the National Cancer Institute. 2010;102:464–474. doi: 10.1093/jnci/djq025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Del Vescovo V, Grasso M, Barbareschi M, Denti MA. MicroRNAs as lung cancer biomarkers. World journal of clinical oncology. 2014;5:604. doi: 10.5306/wjco.v5.i4.604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Larsen JE, Pavey SJ, Passmore LH, Bowman RV, Hayward NK, Fong KM. Gene expression signature predicts recurrence in lung adenocarcinoma. Clinical Cancer Research. 2007;13:2946–2954. doi: 10.1158/1078-0432.CCR-06-2525. [DOI] [PubMed] [Google Scholar]
- 48.Boutros PC, Lau SK, Pintilie M, Liu N, Shepherd FA, Der SD, Tsao MS, Penn LZ, Jurisica I. Prognostic gene signatures for non-small-cell lung cancer. Proc Natl Acad Sci U S A. 2009;106:2824–2828. doi: 10.1073/pnas.0809444106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu CQ, Ding K, Strumpf D, Weir BA, Meyerson M, Pennell N, Thomas RK, Naoki K, Ladd-Acosta C, Liu N, Pintilie M, Der S, Seymour L, Jurisica I, Shepherd FA, Tsao MS. Prognostic and predictive gene signature for adjuvant chemotherapy in resected non-small-cell lung cancer. J Clin Oncol. 2010;28:4417–4424. doi: 10.1200/JCO.2009.26.4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen HY, Yu SL, Chen CH, Chang GC, Chen CY, Yuan A, Cheng CL, Wang CH, Terng HJ, Kao SF, Chan WK, Li HN, Liu CC, Singh S, Chen WJ, Chen JJ, et al. A five-gene signature and clinical outcome in non-small-cell lung cancer. N Engl J Med. 2007;356:11–20. doi: 10.1056/NEJMoa060096. [DOI] [PubMed] [Google Scholar]
- 51.Tano K, Mizuno R, Okada T, Rakwal R, Shibato J, Masuo Y, Ijiri K, Akimitsu N. MALAT-1 enhances cell motility of lung adenocarcinoma cells by influencing the expression of motility-related genes. FEBS letters. 2010;584:4575–4580. doi: 10.1016/j.febslet.2010.10.008. [DOI] [PubMed] [Google Scholar]
- 52.Qiu M, Xu Y, Yang X, Wang J, Hu J, Xu L, Yin R. CCAT2 is a lung adenocarcinoma-specific long non-coding RNA and promotes invasion of non-small cell lung cancer. Tumor Biology. 2014;35:5375–5380. doi: 10.1007/s13277-014-1700-z. [DOI] [PubMed] [Google Scholar]
- 53.Liu Z, Sun M, Lu K, Liu J, Zhang M, Wu W, De W, Wang Z, Wang R. The long noncoding RNA HOTAIR contributes to cisplatin resistance of human lung adenocarcinoma cells via downregualtion of p21 (WAF1/CIP1) expression. PLoS One. 2013;8:e77293. doi: 10.1371/journal.pone.0077293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang L, Zhou X-F, Pan G-F, Zhao J-P. Enhanced expression of long non-coding RNA ZXF1 promoted the invasion and metastasis in lung adenocarcinoma. Biomedicine & Pharmacotherapy. 2014;68:401–407. doi: 10.1016/j.biopha.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 55.Xu G, Chen J, Pan Q, Huang K, Pan J, Zhang W, Chen J, Yu F, Zhou T, Wang Y. Long noncoding RNA expression profiles of lung adenocarcinoma ascertained by microarray analysis. PloS one. 2014;9:e104044. doi: 10.1371/journal.pone.0104044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Chou K. Some remarks on protein attribute prediction and pseudo amino acid composition. Journal of Theoretical Biology. 2011;273:236–247. doi: 10.1016/j.jtbi.2010.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hajisharifi Z, Piryaiee M, Beigi M, Behbahani M, Mohabatkar H. Predicting anticancer peptides with Chou's pseudo amino acid composition and investigating their mutagenicity via Ames test. Journal of Theoretical Biology. 2014;341:34–40. doi: 10.1016/j.jtbi.2013.08.037. [DOI] [PubMed] [Google Scholar]
- 58.Nanni L, Brahnam S, Lumini A. Prediction of protein structure classes by incorporating different protein descriptors into general Chou's pseudo amino acid composition. Journal of Theoretical Biology. 2014;360:109–116. doi: 10.1016/j.jtbi.2014.07.003. [DOI] [PubMed] [Google Scholar]
- 59.Ahmad S, Kabir M, Hayat M. Identification of Heat Shock Protein families and J-protein types by incorporating Dipeptide Composition into Chou's general PseAAC. Computer Methods and Programs in Biomedicine. 2015;122:165–174. doi: 10.1016/j.cmpb.2015.07.005. [DOI] [PubMed] [Google Scholar]
- 60.Liu B, Fang L, Long R, Lan X, Chou K. iEnhancer-2L: a two-layer predictor for identifying enhancers and their strength by pseudo k-tuple nucleotide composition. Bioinformatics. 2016;32:362–9. doi: 10.1093/bioinformatics/btv604. [DOI] [PubMed] [Google Scholar]
- 61.Chen W, Feng P, Ding H, Lin H, Chou K. iRNA-Methyl: Identifying N 6 -methyladenosine sites using pseudo nucleotide composition. Analytical Biochemistry. 2015;490:26–33. doi: 10.1016/j.ab.2015.08.021. [DOI] [PubMed] [Google Scholar]
- 62.Quek XC, Thomson DW, Maag JL, Bartonicek N, Signal B, Clark MB, Gloss BS, Dinger ME. lncRNAdb v2. 0: expanding the reference database for functional long noncoding RNAs. Nucleic acids research. 2014:gku988. doi: 10.1093/nar/gku988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lin H, Deng E, Ding H, Chen W, Chou K. iPro54-PseKNC: a sequence-based predictor for identifying sigma-54 promoters in prokaryote with pseudo k-tuple nucleotide composition. Nucleic Acids Research. 2014;42:12961–12972. doi: 10.1093/nar/gku1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen W, Feng P, Deng E, Lin H, Chou K. iTIS-PseTNC: A sequence-based predictor for identifying translation initiation site in human genes using pseudo trinucleotide composition. Analytical Biochemistry. 2014;462:76–83. doi: 10.1016/j.ab.2014.06.022. [DOI] [PubMed] [Google Scholar]
- 65.Liu B, Xu J, Lan X, Xu R, Zhou J, Wang X, Chou K. iDNA-Prot|dis: Identifying DNA-Binding Proteins by Incorporating Amino Acid Distance-Pairs and Reduced Alphabet Profile into the General Pseudo Amino Acid Composition. PLOS ONE. 2014;9:e106691–e106691. doi: 10.1371/journal.pone.0106691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Liu B, Zhang D, Xu R, Xu J, Wang X, Chen Q, Dong Q, Chou K. Combining evolutionary information extracted from frequency profiles with sequence-based kernels for protein remote homology detection. Bioinformatics. 2014;30:472–9. doi: 10.1093/bioinformatics/btt709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Qiu W, Xiao X, Chou K. iRSpot-TNCPseAAC: Identify Recombination Spots with Trinucleotide Composition and Pseudo Amino Acid Components. International Journal of Molecular Sciences. 2014;15:1746–1766. doi: 10.3390/ijms15021746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu B, Liu F, Wang X, Chen J, Fang L, Chou K. Pse-in-One: a web server for generating various modes of pseudo components of DNA, RNA, and protein sequences. Nucleic Acids Research. 2015;43:W65–W71. doi: 10.1093/nar/gkv458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chou K. Impacts of Bioinformatics to Medicinal Chemistry. Med Chem. 2015;11:218–34. doi: 10.2174/1573406411666141229162834. [DOI] [PubMed] [Google Scholar]
- 70.Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- 71.Cheadle C, Vawter MP, Freed WJ, Becker KG. Analysis of microarray data using Z score transformation. The Journal of molecular diagnostics. 2003;5:73–81. doi: 10.1016/S1525-1578(10)60455-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Du Z, Fei T, Verhaak RG, Su Z, Zhang Y, Brown M, Chen Y, Liu XS. Integrative genomic analyses reveal clinically relevant long noncoding RNAs in human cancer. Nature structural & molecular biology. 2013;20:908–913. doi: 10.1038/nsmb.2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhou M, Guo M, He D, Wang X, Cui Y, Yang H, Hao D, Sun J. A potential signature of eight long non-coding RNAs predicts survival in patients with non-small cell lung cancer. Journal of translational medicine. 2015;13:231. doi: 10.1186/s12967-015-0556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang H, Wong WH. SeqMap: mapping massive amount of oligonucleotides to the genome. Bioinformatics. 2008;24:2395–2396. doi: 10.1093/bioinformatics/btn429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lossos IS, Czerwinski DK, Alizadeh AA, Wechser MA, Tibshirani R, Botstein D, Levy R. Prediction of survival in diffuse large-B-cell lymphoma based on the expression of six genes. New England Journal of Medicine. 2004;350:1828–1837. doi: 10.1056/NEJMoa032520. [DOI] [PubMed] [Google Scholar]
- 76.Harrell FE, Califf RM, Pryor DB, Lee KL, Rosati RA. Evaluating the yield of medical tests. Jama. 1982;247:2543–2546. [PubMed] [Google Scholar]
- 77.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America; 2005; pp. 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Merico D, Isserlin R, Stueker O, Emili A, Bader GD. Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PloS one. 2010;5:e13984. doi: 10.1371/journal.pone.0013984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome research. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.