

Abstract
Polycystic ovary syndrome (PCOS) was hypothesized to result from functional ovarian hyperandrogenism (FOH) due to dysregulation of androgen secretion in 1989–1995. Subsequent studies have supported and amplified this hypothesis. When defined as otherwise unexplained hyperandrogenic oligoanovulation, two-thirds of PCOS cases have functionally typical FOH, characterized by 17-hydroxyprogesterone hyperresponsiveness to gonadotropin stimulation. Two-thirds of the remaining PCOS have FOH detectable by testosterone elevation after suppression of adrenal androgen production. About 3% of PCOS have a related isolated functional adrenal hyperandrogenism. The remaining PCOS cases are mild and lack evidence of steroid secretory abnormalities; most of these are obese, which we postulate to account for their atypical PCOS. Approximately half of normal women with polycystic ovarian morphology (PCOM) have subclinical FOH-related steroidogenic defects. Theca cells from polycystic ovaries of classic PCOS patients in long-term culture have an intrinsic steroidogenic dysregulation that can account for the steroidogenic abnormalities typical of FOH. These cells overexpress most steroidogenic enzymes, particularly cytochrome P450c17. Overexpression of a protein identified by genome-wide association screening, differentially expressed in normal and neoplastic development 1A.V2, in normal theca cells has reproduced this PCOS phenotype in vitro. A metabolic syndrome of obesity-related and/or intrinsic insulin resistance occurs in about half of PCOS patients, and the compensatory hyperinsulinism has tissue-selective effects, which include aggravation of hyperandrogenism. PCOS seems to arise as a complex trait that results from the interaction of diverse genetic and environmental factors. Heritable factors include PCOM, hyperandrogenemia, insulin resistance, and insulin secretory defects. Environmental factors include prenatal androgen exposure and poor fetal growth, whereas acquired obesity is a major postnatal factor. The variety of pathways involved and lack of a common thread attests to the multifactorial nature and heterogeneity of the syndrome. Further research into the fundamental basis of the disorder will be necessary to optimally correct androgen levels, ovulation, and metabolic homeostasis.
Historical Perspective
Definition of Polycystic Ovary Syndrome (PCOS)
-
Normal Androgen Physiology
Biochemical and molecular overview of steroidogenesis
-
Regulation of ovarian function
Regulation of gonadotropin secretion
-
Regulation of ovarian steroidogenesis
Homologous desensitization to LH
Modulation of LH action
Folliculogenesis and its regulation
Regulation of adrenal androgen production
Regulation of peripheral androgen production
Summary of normal androgen physiology
-
Source of Androgen Excess in PCOS
Testing to determine the source of androgen in PCOS
-
Functional Ovarian Hyperandrogenism (FOH) in PCOS
Spectrum of ovarian androgenic function in PCOS: Typical and atypical FOH
Spectrum of ovarian androgenic function in asymptomatic women with polycystic ovarian morphology (PCOM)
Functional adrenal hyperandrogenism (FAH) in PCOS
Other sources of androgen in PCOS
Summary
-
Pathophysiology of PCOS: Abnormal Regulation of Steroidogenesis and Ovarian Function
-
Dysregulation of ovarian function in PCOS
-
Dysregulation of steroidogenesis in PCOS
Intrinsic theca cell dysfunction
Adrenocortical androgenic dysfunction in PCOS
-
Granulosa cell dysfunction and disordered folliculogenesis
Granulosa cell dysfunction contributes to thecal androgen excess
Disordered folliculogenesis and PCOM
Summary
-
-
Relationship of metabolic syndrome to PCOS
-
Role of insulin-resistant hyperinsulinism in PCOS pathogenesis
Insulin resistance and ovarian dysfunction
Insulin resistance and adipose tissue biology
-
Role of obesity in PCOS pathogenesis
Obesity and insulin resistance
Obesity and gonadotropins
-
Relationship of LH excess to PCOS
Modulation of androgen action in PCOS
Summary: Unified model of PCOS pathophysiology
-
-
Etiology of PCOS: A Complex Trait
-
Heritable traits and genetic linkages
Maternal PCOS
Polycystic ovarian morphology
Hyperandrogenemia
Metabolic syndrome and diabetes mellitus (DM)
Gene variants
-
Intrauterine environment
Congenital virilization
Disturbed fetal nutrition
-
Postnatal environment
Insulin resistance
Hyperandrogenism
Other precipitants and risk factors
Implications for evolutionary origin of PCOS
Summary
-
Conclusions and Implications for Future Research
I. Historical Perspective
Polycystic ovary syndrome (PCOS) is the most common endocrine disorder in reproductive aged women, with a prevalence between 5% and 15%, depending on the diagnostic criteria applied (1, 2). PCOS was first described by Stein and Leventhal as a syndrome of oligo-amenorrhea and polycystic ovaries that was variably accompanied by hirsutism, acne, and obesity (3, 4). Demonstration of polycystic ovaries became required for PCOS diagnosis, which required gynecologic expertise, yet polycystic ovaries were found to be variably associated with the signs and symptoms that characterize the disorder (5).
Seminal contributions to our understanding of PCOS pathogenesis began with the 1958 report that urinary LH was elevated by bioassay in the 4 cases studied (6). The 1970 documentation by RIA that serum LH and the ratio of LH to FSH were typically high (7) led both to the adoption of altered gonadotropin secretion as an alternative diagnostic tool and to a focus of research on the putative neuroendocrine genesis of the syndrome. Shortly thereafter, plasma free testosterone was recognized as a marker for hyperandrogenism in hirsute amenorrheic women; subsequent studies suggested the hyperandrogenemia was of ovarian origin (8). During the 1980s, administration of testosterone to female-to-male transsexuals was found to cause polycystic ovaries (9), and ultrasonographic criteria for the identification of polycystic ovarian morphology (PCOM) were developed (10).
Meanwhile, significant insulin resistance was recognized to be related to hyperandrogenism and acanthosis nigricans (11) and to occur independently of obesity in the syndrome (12, 13). In vitro studies subsequently showed that insulin stimulates ovarian androgen production (14), particularly in synergy with LH (15, 16). These studies raised the possibility that hyperinsulinemia contributes to ovarian androgen excess.
In 1989, we published evidence that a GnRH agonist (GnRHag) test, which stimulates the coordinated function of the ovarian follicle in response to endogenous LH and FSH release, disclosed a previously unrecognized form of hyperandrogenism in women with classic PCOS: generalized ovarian steroidogenic hyperresponsiveness (17). 17-Hydroxyprogesterone (17OHP), and to a lesser extent androstenedione, responses were most consistently abnormal, and there was no evidence of a steroidogenic block. This suggested abnormal regulation (dysregulation) of 17-hydroxylase and 17,20-lyase activities, which are 2 activities of the single enzyme cytochrome P450c17 (CYP17) encoded by CYP17A1 (17, 18).
We then found that most hyperandrogenic women (two-thirds of those with oligo-amenorrhea, 30% of eumenorrheic ones) had this type of androgenic ovarian dysfunction and that this was independent of serum LH elevation or PCOM in about half of cases (19, 20). This abnormality was termed functional ovarian hyperandrogenism (FOH), because the steroidogenic disorder is gonadotropin dependent (ie, any treatment that suppresses gonadotropin production suppresses androgen production), and there is not a requisite anatomic basis for the disorder.
This research, suggesting that PCOS was usually a form of FOH due to dysregulation of androgen secretion, generated a paradigm shift in 2 ways. First, the ovary was identified as the source of the disorder rather than the target of a neuroendocrine disturbance. Second, a steroidogenic disorder was attributed to enzyme dysregulation rather than deficient enzyme activity. This research also contributed to the subsequent redefinition of PCOS as otherwise unexplained hyperandrogenic anovulation, irrespective of the presence of polycystic ovaries or LH elevation, ie, National Institutes of Health 1990 conference diagnostic criteria (NIH criteria) (21).
These and subsequent studies by ourselves and others extended and confirmed these pathophysiologic findings and were summarized in a 1995 Endocrine Reviews paper (22). The evidence was consistent with the ovarian hyperandrogenism being a functional abnormality that requires normal adult LH levels but not LH elevation.
Furthermore, despite resistance to the effects of insulin on glucose metabolism in target tissues such as muscle, the ovary seemed to be responsive to the synergistic effect of hyperinsulinemia and LH on ovarian androgen secretion. The FOH of PCOS was ordinarily “primary,” ie, not secondary to any known disorder, although unusual cases can be caused by severe insulin resistance, such as that arising from mutations of the insulin receptor, or by congenital virilizing disorders (22).
Subsequent studies have supported and amplified this hypothesis. Particularly noteworthy is the evidence that the disorder ordinarily is due to an intrinsic disorder of ovarian function (23) and the evidence for ovarian-sparing tissue-specific differences in insulin resistance in PCOS and obesity (24, 25). It is the purpose of the current review to update the evidence regarding the pathogenesis of PCOS and emphasize how new data are providing insights into diagnosis and treatment of the disorder. A review of the literature in English through April 2016 was conducted via PubMed, and data were summarized and integrated from the authors' perspectives.
II. Definition of PCOS
Two international consensus conferences have developed adult diagnostic criteria that widen the definition beyond NIH criteria (21) by incorporating the presence of PCOM, defined by consensus (26), as a diagnostic criterion for PCOS (Table 1). Rotterdam criteria are the broadest and encompass all combinations of otherwise unexplained clinical or biochemical evidence of hyperandrogenism, evidence of oligo-anovulation, and PCOM (27). Androgen Excess-PCOS Society (AE-PCOS) criteria (2006) encompass otherwise unexplained hyperandrogenism with either oligo-anovulation or PCOM (28); this allows a diagnosis of PCOS in women with hyperandrogenism who lack anovulatory symptoms (“ovulatory PCOS”), which comprises about 10% of cases.
Table 1.
Diagnostic Criteria for PCOS
| Adult Diagnostic Criteria (Rotterdam) |
Otherwise unexplained alternative phenotypes:
|
| Adolescent Diagnostic Criteria |
Otherwise unexplained combination of:
|
Modified from Rosenfield, The diagnosis of polycystic ovary syndrome in adolescents. Pediatrics. 2015;136:1154–1165 (50).
AE-PCOS recognizes only hyperandrogenic phenotypes.
An independent panel reviewed the evidence through 2012 in an international workshop and recommended that Rotterdam criteria be adopted with specific identification of phenotype, of which there are 4 (29), listed next in order of decreasing clinical severity, which corresponds to decreasing specificity of the milder phenotypes (Table 1) (30–37). Phenotype 1 is the classic combination of all the reproductive endocrine features of the syndrome, namely evidence of hyperandrogenism, oligo-anovulation, and PCOM. Phenotype 2 is the combination of hyperandrogenism and oligo-anovulation (the essential NIH criteria). Phenotype 3 is the combination of hyperandrogenism and PCOM in the absence of oligo-anovulation (ovulatory PCOS), as endorsed by AE-PCOS. Phenotype 4 is the combination of oligo-anovulation and PCOM (nonhyperandrogenic PCOS). Although insulin resistance and obesity are common in PCOS, they are not recognized as diagnostic criteria. Likewise, alterations in gonadotropin secretion are not included in any of the definitions of the syndrome.
Hyperandrogenism severity decreases in these successive phenotypes, as does, in most populations, the severity of insulin resistance, obesity, and LH elevation, with ethnic and environmental factors playing a role (30, 34–36). The hyperandrogenic phenotypes 1–3 have ovulatory dysfunction ranging successively from severe to nil, whereas phenotype 4 is anovulatory but lacks evidence of hyperandrogenism.
Phenotypes 3 and 4 are successively less specific and successively more contentious. Phenotype 3 permits a PCOS diagnosis in the presence of PCOM in mildly hirsute females with normal serum androgen levels (ie, those with hirsutism scores up to 2-fold above the normal upper limit), who would be considered to have idiopathic hirsutism according to The Endocrine Society Clinical Practice Guidelines (38), or in apparently normal asymptomatic women with subclinical hyperandrogenemia (39), as reviewed below (see section IV.B.2). The lack of hyperandrogenism in phenotype 4 makes it particularly debatable, and it is not considered here; many seem to have functional hypothalamic amenorrhea (40), and it is not recognized as constituting PCOS by AE-PCOS.
Weaknesses of these criteria have emerged. First, the documentation of hyperandrogenemia can be difficult: serum testosterone concentration undergoes episodic, diurnal, and cyclic variation (41) and attains mature levels approximately 1 year after menarche (42). Furthermore, methodologic problems in commercial testosterone assays have emerged (43, 44). Consequently many steroid assays are inaccurate, and the best steroid assays differ from one another modestly but significantly (45–48). For these reasons, hirsutism is often considered the clinical surrogate of hyperandrogenemia although half of mild hirsutism and a small proportion of moderate-severe hirsutism (hirsutism score >2-fold above normal upper limit) are not associated with hyperandrogenemia (38). Second, the consensus sonographic definition of PCOM (26) is prone to lead to overdiagnosis, particularly as it applies to antral follicle count criteria determined by the current generation of high-definition imaging techniques (49) and as it applies to adolescents and young women (30, 50). In adolescents, anovulatory criteria must be age- and pubertal stage-appropriate, and the paucity of normative data obviates the routine use of PCOM as a diagnostic criterion (Table 1) (30, 50, 51). Furthermore, all these criteria overlook the potential presence of the PCOS type of FOH in patients who present with hirsutism, obesity, or insulin-resistance signs such as acanthosis nigricans, but who lack clinical evidence of ovarian dysfunction (19, 52, 53).
Finally, the hyperandrogenism of PCOS improves during middle age, which is sometimes accompanied by normalization of menstrual regularity (54, 55). These changes seem related to the fall in follicle number during the premenopausal transition, which is accompanied by falling serum inhibin-B and rising FSH levels that maintain estradiol secretion (56). Although hyperandrogenism may remit during menopause (54), lifelong metabolic dysfunction persists and may increase postmenopausal cardiovascular disease risk (57). Criteria for the diagnosis of postmenopausal PCOS remain to be defined.
III. Normal Androgen Physiology
Understanding of the normal biochemical and molecular basis of steroidogenesis and of normal androgen physiology is necessary to understand the pathophysiology of PCOS.
Under normal circumstances, the ovaries and adrenal glands contribute about equally to testosterone production (58–60). Approximately half of testosterone originates from direct testosterone secretion by the ovaries and adrenal glands, whereas half is produced by peripheral conversion of circulating androstenedione, which itself arises from approximately equal ovarian and adrenal secretion.
Androgen production is not under direct negative feedback regulation by the neuroendocrine system in females, as is the case for estradiol and cortisol secretion (8, 61). Indeed, modest androgen excess interferes with female sex hormone negative feedback according to recent research (see section V.C).
Androgens are secreted by both the ovaries and adrenal glands in response to their respective tropic hormones, LH and ACTH. Intraglandular paracrine and autocrine mechanisms seem to play a major role in modulating androgen secretion in response to tropic hormone stimulation.
A. Biochemical and molecular overview of steroidogenesis
The rate-determining step for the formation of all steroid hormones in response to tropic hormones in both the gonads and adrenal glands is cholesterol side-chain cleavage, which is mediated by the enzyme cytochrome P450scc (encoded by CYP11A) (62). The ovarian steroidogenic response to LH is slow in the early follicular phase, and is accelerated in the luteinized preovulatory follicle as LH induces the steroidogenic acute regulatory protein, which delivers cholesterol into mitochondria (63).
Cytochrome P450c17 (CYP17A1) is the rate-limiting enzyme for the formation of androgens in the gonads and adrenal cortex (Figure 1) (22, 64). Its expression is absolutely dependent upon tropic hormone stimulation, LH in the ovary (65, 66) and ACTH in the adrenal cortex (67, 68), in a dose-dependent manner. This one enzyme possesses both 17-hydroxylase and 17,20-lyase activities. The first of these activities, 17-hydroxylase, is necessary for the formation of cortisol in the adrenal cortex and the potent sex steroids in the gonads. The second of these activities, 17,20-lyase, less efficiently acts sequentially on enzyme-bound 17-hydroxylated substrate to form 17-ketosteroids, eg, dehydroepiandrosterone (DHEA) or androstenedione. These 17-ketosteroids are, in turn, the precursors of all potent sex steroids in the gonads and adrenal zona reticularis. Specifically, P450c17 mediates conversion of pregnenolone by 17-hydroxylation to form 17-hydroxypregnenolone, which is transformed by 17,20-lyase activity to DHEA. DHEA is then converted by Δ5-isomerase-3β-hydroxysteroid dehydrogenase type 2 (3βHSD2) (HSD3B2) to androstenedione. Progesterone undergoes parallel P450c17 17-hydroxylation to 17OHP. Although 17OHP is a poor substrate for P450c17 17,20-lyase activity in theca cells or cell-free systems, formation of androstenedione has been documented in ovarian and adrenal homogenates and minces (69–71), compatible with the possibility that either paracrine interactions with granulosa cells may up-regulate 17,20-lyase activity for this substrate or that another enzyme accounts for this activity (22).
Figure 1.

Depiction of the organization and regulation of the major steroid biosynthetic pathways in the small antral follicle of the ovary according to the 2-gonadotropin, 2-cell model of ovarian steroidogenesis. LH stimulates androgen formation within theca cells via the steroidogenic pathway common to the gonads and adrenal glands. FSH regulates estradiol biosynthesis from androgen by granulosa cells. Long-loop negative feedback of estradiol on gonadotropin secretion does not readily suppress LH at physiologic levels of estradiol and stimulates LH under certain circumstances. Androgen formation in response to LH appears to be modulated by intraovarian feedback at the levels of 17-hydroxylase and 17,20-lyase, both of which are activities of cytochrome P450c17 that is expressed only in theca cells. The relative quantity of androstenedione formation via 17OHP (dotted arrow) in the intact follicle is probably small, as is the amount of progesterone formed from granulosa cell P450scc activity in response to FSH (data not shown). 17βHSD2 activity is minor in the ovary, and estradiol is primarily formed from androstenedione. Androgens and estradiol inhibit (minus signs) and inhibin, insulin, and IGF-1 (IGF) stimulate (plus signs) 17-hydroxylase and 17,20-lyase activities. Pertinent enzyme activities are italicized: the 17-hydroxylase and 17,20-lyase activities of P450c17 are shown, otherwise enzyme abbreviations are as in the text. Modified with permission from Ehrmann et al, Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion. Endocr Rev. 1995;16:322–353 (22).
The differential regulation of the 2 enzyme activities of P450c17 is incompletely understood. The preferential formation of androgen by P450c17 of the gonads and adrenal zona reticularis is largely dependent on 3 posttranslational factors that up-regulate its 17,20-lyase activity (64). First, this activity is especially sensitive to the molar abundance of the electron-transfer protein cytochrome P450-oxidoreductase (POR). Second, cytochrome b5 (CYB5A) strongly promotes 17,20-lyase activity, principally by acting as an allosteric factor promoting the interaction of P450c17 with POR. Third, the serine/threonine phosphorylation of P450c17 itself by a specific MAPK (p38α) appears to selectively promote 17,20-lyase activity by also promoting the interaction of P450c17 with POR.
Androstenedione is the major precursor for both testosterone and estrogen synthesis in the ovaries (Figure 1) and adrenal cortex (Figure 2) (62). In the ovaries it is in part converted in theca cells by 17βHSD5 (HSD17B5; also termed α-ketoreductase type 1C3 [AKR1C3]) to form testosterone (72) and is in part aromatized in granulosa cells by cytochrome P450aro (CYP19A1) to form estrone. Androstenedione predominates over testosterone as the aromatase substrate, because it is available in 10-fold greater amounts (60, 62, 72–75). Estrone is then converted to estradiol by 17βHSD1 (HSD17B1).
Figure 2.

Depiction of the organization of the major steroid biosynthetic pathways in the adrenal cortex. The top row shows the pathway to aldosterone; the middle row shows the zona fasciculata pathway to cortisol; the lowest, darkly shaded row shows the zona reticularis steps to 17-ketosteroids that are not expressed in the other adrenal zones. Note similarities between the biosynthetic capacities of the zona reticularis and that of ovarian theca cells. Dotted pathways are minor. The zona reticularis is notable for its low 3βHSD2 activity (denoted by small arrow) and unique expression of cytochrome b5, a cofactor which enhances the 17,20-lyase activity of P450c17. Sulfotransferase 2A1 is uniquely expressed in the zona reticularis and rapidly converts DHEA to DHEAS. Compound S (Cpd S), 11-deoxycortisol. Corticosterone and 18-hydroxycorticosterone, the successive intermediates between deoxycorticosterone (DOC) and aldosterone, are not shown. The steroidogenic enzymes are italicized. The clinically relevant electron transfer enzymes also shown are POR and type 1 3′-phosphoadensosine-5′-phosphosulfate synthase (PAPSS). Formation of androstenedione from 17OHP and Cpd S does not seem attributable to CYP450c17. Modified with permission from Rosenfield, Identifying children at risk of polycystic ovary syndrome. J Clin Endocrinol Metab. 2007;92:787–796 (431).
Androgens are preferentially metabolized to dihydrotestosterone rather than estradiol in small ovarian follicles, before follicle selection, because of high steroid 5α-reductase (5αRD) (SRD5A) activity (76). This is carried out by both type 1 and 2 5αRD isozymes in theca, stroma, and granulosa cells, but the predominant reaction is type 1 activity in granulosa cells (77). 17βHSD2 reconversion of testosterone to androstenedione and reconversion of estradiol to estrone are minor pathways.
B. Regulation of ovarian function
Androgens are not only obligate intermediates in the biosynthesis of estradiol. They also have complex effects on follicular growth (78), including up-regulation of aromatase activity (79). It is crucial for the function of the ovary that ovarian androgen secretion be coordinated with the formation of estrogen so that both are optimized for ovulation. Although these processes are critically dependent on LH and FSH concentrations, a variety of intrafollicular modulators are essential to the coordinated function of the ovarian follicle.
1. Regulation of gonadotropin secretion
Androgens are not under tight neuroendocrine negative feedback control by LH, nor is serum LH readily inhibited by modest increases in serum estrogen levels. FSH is reciprocally regulated by estradiol and inhibin in a sensitive, log-dose, negative-feedback loop (Figure 1) (22, 80–82).
It has long been apparent that the mature female neuroendocrine-gonadotropic axis is not very sensitive to negative feedback by androgens (8); only frankly virilizing testosterone levels are clearly gonadotropin suppressive (83). Congenital virilization (due to conditions such as 21-hydroxylase deficiency congenital adrenal hyperplasia), on the other hand, is a positive determinant of LH pulsatility but a negative determinant of the capacity to mount the LH surge necessary for ovulation, masculinizing the pattern of gonadotropin release (83). Congenital androgen effects appear to be mediated in part by permanent impairment of estradiol-induced progesterone receptor gene expression in the hypothalamus (84). Paradoxically, androgen receptor signaling enhances the capacity of females to mount an LH surge in response to estrogen positive feedback (85–87). Recently, it has been discovered that modest increases in serum androgen levels have a stimulatory effect on LH secretion, as discussed in the section on LH excess in PCOS (see section V.C).
2. Regulation of ovarian steroidogenesis
In small antral follicles steroidogenesis is organized as shown in Figure 1 (22). Theca cells produce androgens in response to LH, but granulosa cells do not do so because they do not express CYP17A1 (62). Androgens then diffuse from theca cells to granulosa cells, where they are substrates for estrogen formation in response to FSH because granulosa cells differentially express CYP19A1, which encodes aromatase (62).
a. Homologous desensitization to LH.
The expression of thecal steroidogenic enzymes is absolutely dependent upon LH in a dose-response relationship (22). The normal secretory dose-response curve is asymptotic, however, because as LH rises, desensitization of ovarian responses to LH commences (88–90). Animal models indicate desensitization is in part mediated by down-regulation of LH receptor-binding sites by ligand binding (22, 91), an endocytic process that involves recycling of receptors via membrane raft microdomains or degradation (92–94), and in part by down-regulation of the 17,20-lyase activity of P450c17 (22). Thus, as LH stimulation approaches maximal, 17OHP secretion rises, yet normally androgen production increases very little. Judging from serum 17OHP and steroidogenic responses to the LH analog human chorionic gonadotropin (hCG) in normal women, desensitization normally has commenced by half-maximal stimulation of the LH receptor (Figure 3) (90, 95), whereas maximal stimulation causes about a 5-fold increase in 17OHP and small (2-fold) increases in sex steroids (96). Serum 17OHP responses assessed at 4-hour intervals in response to interim dose changes of pulsatile LH suggest that elevating the serum LH 2-fold affects steroid output in normal women comparably with half-maximal LH receptor stimulation (97); however, it is dubious whether 4 hours are sufficient to ascertain the effect full effect of LH on steroidogenesis.
Figure 3.
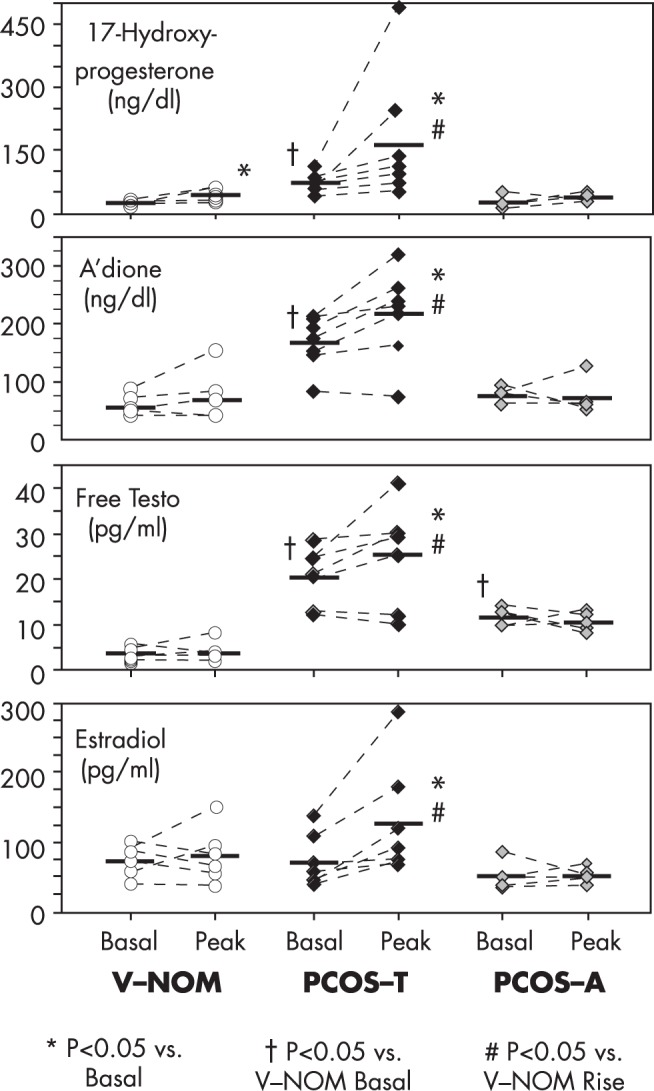
Response to half-maximal hCG stimulation during overnight dexamethasone suppression of normal and PCOS subjects. After bedtime dexamethasone 0.25 mg/m2, 500-IU hCG was administered im at 8 am; a basal blood sample was drawn before hCG, and the peak response to hCG was sampled after a repeat dexamethasone dose 24 hours later. Subjects were healthy volunteers with normal ovarian morphology (V-NOMs), PCOS patients with functionally typical FOH (ie, 17OHP hyperresponsiveness to GnRHag; PCOS-T), and PCOS patients with functionally atypical FOH (ie, normal 17OHP responsiveness to GnRHag; PCOS-A). V-NOM had a small but significant rise in serum 17OHP but not in other steroids. PCOS-T had hyperresponsiveness of all steroids. PCOS-A, a heterogenous group, had elevated basal serum free testosterone, but normal hCG-responses of all steroids. To convert to SI units, multiply 17OHP by 0.0303 (nM), androstenedione (A'dione) by 0.0340 (nM), free testosterone by 3.47 (pM), and estradiol by 3.67 (pM). Regraphed from data of Hirshfeld-Cytron et al, Characterization of functionally typical and atypical types of polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94:1587–1594 (95).
b. Modulation of LH action.
Modulation of ovarian androgenic responsiveness to LH involves a number of hormones and growth factors acting in paracrine, autocrine, and endocrine fashions, as modeled in Figure 1 (22). Substantial evidence indicates that estrogen inhibits P450c17 activity by a short-loop (paracrine) negative feedback mechanism (98–100). Intraovarian androgens inhibit thecal P450c17 activity, but the testosterone effect is not reversed by antiandrogen (101), so it is possible that this androgen effect is estrogen mediated. These inhibiting modulators are counterbalanced by growth factors of granulosa cell origin that are under FSH control and by hormones and cytokines extrinsic to the ovary that amplify P450c17 activities (22, 102–105). Insulin, IGFs, and inhibin are the best recognized of these modulators (Figure 1). Insulin and IGFs up-regulate P450c17 activities (102) and in rat studies have been shown to up-regulate LH receptor sites (16, 91, 106). This counters the normal process of homologous desensitization to LH and thereby potentiates LH-induced androgen synthesis (22, 88). Insulin is approximately equipotent with IGF-1 in this regard, which makes it unlikely that insulin acts through the IGF-1 receptor, because its affinity is about 500-fold less than that for its cognate receptor. More recent studies have indicated that insulin acts through the insulin receptor itself: the effect on human theca cells is specifically neutralized by an antibody to the insulin receptor (105), and selective knockout of theca cell insulin receptors attenuates the androgenic response to hCG in mice (25). Furthermore, the hyperandrogenic anovulation induced by an obesogenic diet in association with 10- to 20-fold elevation of serum insulin in wild-type mice does not occur in transgenic littermates that lack the theca cell insulin receptor (25). Insulin also directly up-regulates 17βHSD5 gene expression and activity, stimulating testosterone formation from androstenedione (107).
Studies in women with PCOS have demonstrated that a previously unrecognized protein variant, differentially expressed in normal and neoplastic development (DENND)1A.V2, is a facilitator of steroidogenesis in androgen-producing cells, as discussed below in the section on etiology of PCOS/genetic traits (see section VI) (108). At this time, its mechanism of action is unclear.
Inhibin-B, a peptide secreted by granulosa cells of small antral follicles in response to FSH, seems essential for androgenic responses to LH and may be modulated by androgens (95, 109). It seems necessary but not sufficient for up-regulation of CYPA1 gene expression (110). Patients with inactivating mutations of FSH lack hyperandrogenism, despite very high levels of LH. Such patients have low inhibin-B levels, and when exogenous FSH is given, inhibin-B rises dramatically, and the response of theca cell steroids to hCG/LH increases markedly (103, 111). Inhibin-B seems to play a permissive role, rather than a stimulatory role, in determining theca cell LH responsiveness because our data indicate that FSH administration normally suppresses testosterone levels, seemingly through the paracrine action of other granulosa cell factors under its control (95).
Numerous other small molecules of granulosa cell and oocyte origin both positively and negatively modulate LH action in theca cells so as to optimize the follicular environment for oocyte maturation (22). These factors include TGF-β superfamily members such as bone morphogenetic proteins (BMPs), other growth and differentiation factors, cytokines such as TNFα, and microRNAs (90, 112–119).
Catecholaminergic overactivity also amplifies the ovarian steroidogenic response to hCG in rodents, with signaling via β2-adrenoreceptors on thecal cells (120–123). In a unique rodent model of PCOM induced by a single injection of estradiol valerate, central sympathetic nervous system (SNS) activity is increased resulting in increased adrenomedullary noradrenergic activity, increased ovarian sympathetic nerve stimulation, and increased intraovarian synthesis of nerve growth factor (NGF), a sympathetic neurotrophin secreted by thecal cells. This is followed by the development of PCOM, anovulation, and increased androgen responses to hCG. These changes are reversed by transection of the superior ovarian nerve. Transgenic mouse NGF overexpression causes increased androgen production in response to pregnant mare serum, a higher prevalence of follicular cysts after sustained increases in LH, enhanced sympathetic outflow, increased body fat with disproportionate hyperinsulinemia, and glucose intolerance (121, 123).
In summary, androgen blood levels are not tightly controlled by direct negative feedback by the pituitary trophic hormones, as is the case for estradiol and cortisol. Rather, intraglandular paracrine and autocrine mechanisms seem to play a major role in the regulation of ovarian androgen secretion. Due to the process of homologous desensitization to LH, once serum LH levels approximate high-normal, intraovarian modulation of LH action seems to be the major factor determining ovarian androgen formation. Insulin counters the desensitization process and sensitizes the ovary to LH.
3. Folliculogenesis and its regulation
The transition from the resting primordial to the growing primary follicle stage (“initial recruitment/activation”) is independent of serum gonadotropins (114, 124, 125). Primordial follicle formation in animal models is directed by the oocyte-specific chemokine S100A8 (126) and BMP2 (127) signaling. Primordial follicles are maintained in a dormant state by mesenchymal-epithelial cell interactions, intraovarian paracrine signals, and oocyte-secreted factors. Among these, oocyte liver kinase b1 (128) and somatic cell forkhead transcription factors inhibit activation (129, 130) and BMP4 promotes apoptosis (131, 132), whereas oocyte kit ligand and phosphatidylinositol-3-kinase are important stimulatory signaling pathways (114). Anti-Müllerian hormone (AMH), of granulosa cell origin, is the major hormonal paracrine inhibitor of primordial follicle progression: Amh-null mice undergo accelerated depletion of the primordial follicle pool, although at a slower rate than do than do Foxo-null mice (129, 133). Estrogen receptor expression is critical for development of the granulosa cell layer (134). Development of primary follicles depends on germ cell/oocyte factors. Growth differentiation factor (GDF)9 successively stimulates granulosa cell differentiation and then initiates theca cell differentiation, in conjunction with kit ligand, BMP6, and BMP15 (114). These primary theca cells express LH receptors and produce androgen. Insulin (114, 135) and androgen (136) promote the primordial-primary follicle transition, although the androgen effect does not occur at normal androgen levels judging from the results of the androgen receptor deletion studies discussed below.
A host of local factors then regulate further follicle growth and development; for example, BMP15 synergizes with GDF9 to stimulate granulosa cell proliferation (137). Only upon reaching the early antral follicle stage does follicle development become strictly dependent on FSH action (103, 125, 138). FSH actions on both growth and steroidogenesis are facilitated by androgen (139), insulin and IGF signaling (89, 138). Inhibin-B, produced by the granulosa cells of small antral follicles at a stage before aromatase becomes highly inducible by FSH (140, 141), seems to be the prime ovarian regulator of FSH secretion via negative feedback on gonadotropes (82).
AMH is an important intrafollicular modulator of follicle growth and FSH action (142). It is a TGF-β superfamily member that is produced by the granulosa cells of small growing follicles. As follicles grow, intrafollicular AMH levels rise sufficiently to inhibit both recruitment of primordial follicles to the primary follicle stage and FSH stimulation of aromatase activity. Because estradiol inhibits AMH production, there exists an intrafollicular short negative feedback loop confining AMH expression to follicles up to about 8 mm in diameter. Thus, AMH appears to act as a follicular gatekeeper, ensuring that each small antral follicle produces little estradiol before selection of the dominant follicle, which allows a direct ovarian-pituitary dialogue regulating the development of the follicle selected to undergo ovulation.
The serum AMH level is an indicator of the number of growing follicles. AMH levels reflect intrafollicular androgenic status (109), probably because androgens stimulates the early phases of follicular growth (136, 143). The AMH level also independently indicates the size of the follicular (“ovarian reserve”) pool (39, 144).
Androgens have complex effects on follicular development that indicate paracrine interactions between theca and granulosa cells (79, 145). As expected from human data indicating that virilization causes polycystic ovaries (9), systemic induction of hypertestosteronemia (4–30 ng/mL for 3–10 d) in nonhuman primates (primates) activates the earliest stages of follicle growth so as to stimulate the growth of small, but not large, antral follicles: granulosa cell androgen receptor and FSH receptor content increase, as do granulosa and theca cell proliferation and cortical thickening (136, 146, 147). Notably, androgen receptor expression precedes FSH receptor expression in human granulosa cells (148). Androgen receptor deletion studies indicate that androgens, in conjunction with gonadotropins (61), normally are important for follicle development from the preantral through the early antral follicle stage and for up-regulating aromatase activity via granulosa cell actions (149, 150). Androgens also synergize with FSH to luteinize follicles by inducing LH receptors (138, 139, 151, 152). In vitro study of primate follicle maturation from the secondary to small antral follicle stage over a 40-day period in serum-containing medium indicates that 10-ng/mL testosterone promotes preantral follicle growth, but 50-ng/mL testosterone or dihydrotestosterone inhibits it (153). In excess, androgens impair selection of the dominant follicle of women (154); this appears likely to result from premature luteinization of follicles (155).
Luteinization of granulosa cells, as indexed by the development of LH receptors, commences as selected follicles reach 5 mm in the midfollicular phase of the menstrual cycle, and in the preovulatory (dominant) follicle LH receptors rise 10-fold more (138, 152). FSH is the primary inducer of these LH receptors. Androgens and estrogens synergize in FSH induction of LH receptors and the subsequent augmentation of progesterone and estradiol formation in luteinized granulosa cells (138, 139, 147, 152). Insulin amplifies these granulosa cell steroidogenic responses to FSH and LH (89, 156). As the dominant follicle emerges, LH signaling comes to predominate, and FSH and androgen receptor expression wane (138, 152, 157). Only the preovulatory follicle continues to grow, seemingly because of its high LH receptor content and an androgen to estrogen ratio that favors estradiol. Reciprocally with dominant follicle emergence, the companion cohort of follicles is growth-inhibited, atresia commences and the androgen to estrogen ratio favors androgen (138). This atresia has been attributed to the combination of relatively low gonadotropin and androgen receptor expression (149, 150, 157).
The biochemical rigidity of the stroma influences follicular development and steroidogenesis (125, 158, 159). The hippopotamus (Hippo) signaling pathway, a serine/threonine kinase signaling cascade that is regulated by the biomechanics of the microenvironment, is growth restrictive and has been postulated to play a role in regulating follicle growth as developing follicles move from the relatively dense cortex, where primordial follicles reside, to softer medullary regions (125). Vascularization of the stroma is a determinant of stromal rigidity and is regulated by the vascular endothelial growth factor (VEGF) family of cytokines (145). They and their receptors are produced by granulosa cells, theca cells, and stroma and are up-regulated by androgens (114, 145, 160). VEGFs not only stimulate endothelial proliferation and permeability, which affect delivery of circulating hormones and cytokines to follicles, they directly restrain primordial follicle growth (160).
In summary, paracrine-acting growth factors originating in germ cells, oocytes, granulosa cells, theca cells, and stroma are the predominant regulators of early follicular development, with FSH becoming essential for follicle growth from the early antral stage. Normal thecal androgen production supports antral follicle growth and development. Androgens do so in part by up-regulating granulosa cell expression of FSH receptors and then augment FSH induction of granulosa cell LH receptors, thus luteinizing granulosa cells and sensitizing them to both gonadotropins. Insulin amplifies the luteinization process. Androgen excess stimulates the excess proliferation of small antral follicles while causing follicle maturation arrest and PCOM. Antral follicle growth is accompanied by increased production of AMH, which normally acts as a gate-keeper that inhibits primordial follicles from entering the growth phase.
C. Regulation of adrenal androgen production
The zona reticularis of the adrenal gland (Figure 2) resembles the theca cell compartment of the ovary in its expression of the core enzymatic pattern for androgen production. Zona reticularis cells become discernable in the central adrenal cortex at 3 years of age. As it begins to develop into a continuous zone at about 5–6 years of age, it becomes discernable as “adrenarche,” the maturational increase in adrenal androgen production that is indexed by increased DHEA sulfate (DHEAS) production (161).
Adrenarche represents a change in the pattern of the adrenocortical secretory response to ACTH. It is characterized over time by disproportionately increasing responsiveness of Δ5-steroid intermediates (17-hydroxypregnenolone and DHEA) compared with Δ4-steroids (eg, 17OHP and androstenedione) in the presence of stable responses of cortisol (162, 163).
A unique zona reticularis enzyme expression profile underlies this adrenarcheal pattern of adrenal secretion. Zona reticularis cells express low 3βHSD2, but high CYB5, and steroid sulfotransferase (SULT2A1) activities (Figure 2) (64, 164, 165). The combination of low 3βHSD2 and high CYB5 activity diverts pregnenolone formation from cortisol to DHEA, and the unique expression of SULT2A1 rapidly sulfates DHEA, forming DHEAS, which accounts for the high secretion of DHEAS by the adrenal cortex (166). SULT2A1 thus acts as a “sump” to direct steroidogenesis to this relatively inert terminal product and prevents adrenal DHEA from being converted into biologically active androgens (165). Seladin-1 (24-dehydrocholesterol RD), expressed in both zona fasciculata and reticularis cells, enhances DHEA, but not cortisol, secretion in vitro, which suggests that it enhances 17,20-lyase activity (167). Enhanced expression of HSD17B5 by this zone accounts for the small but significant adrenal contribution to testosterone secretion (168).
Zona reticularis development requires ACTH, but its determinants are otherwise poorly understood. The stimulus has long been thought to be of pituitary origin (169, 170). Adrenarche is not directly related to the pubertal maturation of the neuroendocrine-gonadotropin-gonadal axis. Adiposity is strongly related to adrenarche, and insulin, IGF-1, and leptin have been suggested as determinants of this relationship (171–177). Adrenarche is severely attenuated in somatotropin- and thyroxine-replaced patients with deficiency of the pituitary-specific transcription factor Pit-1/POU1F: because this gene defect causes a congenital deficiency of somatotropin, thyrotropin, and prolactin, and the former hormones were replaced, a role for prolactin is suggested (178). IL-6 is another candidate because it is strongly expressed in the zona reticularis of the adrenal cortex and stimulates DHEA secretion (179). On the other hand, a BMP4 signaling system has been identified in the zona reticularis that is inhibitory to androgen formation (180). Notably, the theca cell steroidogenesis facilitator protein DENND1A.V2 has been localized to the zona reticularis (see section V.A.1.). Other pathways that potentially modulate adrenal androgen formation have been identified in a human adrenocortical carcinoma cell line (181). We favor the hypothesis that whatever the factor(s) responsible for adrenarche, it affects the growth or differentiation of zona reticularis precursor cells, so that they acquire the enzymatic properties that permit them to respond to ACTH by secreting androgens.
D. Regulation of peripheral androgen production
Peripheral formation of testosterone from androstenedione primarily occurs in liver, skin and fat (107, 182–184). Skin and fat tissue express 3βHSDB1 and 17βHSD5 activities as well as P450aro activity. Although early direct studies of steroid metabolism did not reveal it (185), recent evidence suggests that adipose tissue excess is an important contributor to androgen as well as estrogen excess (47).
No clear picture has emerged of tissue-specific regulation of androgen production in nonendocrine sites. Adipose tissue has become recognized in recent years as an endocrine organ that is an important site of generation of sex steroids and inflammatory cytokines in obesity (186, 187). The ratio of androgenic 17βHSD to aromatase activity in visceral fat is positively correlated with central adiposity, and experimental aromatase deficiency in animals and humans is associated with visceral adiposity (187), implicating increased local androgen production in central adiposity (reviewed in section V.B.1). Notably, insulin up-regulates adipocyte 17βHSD5 gene expression and activity in sc fat (107). Furthermore, 17βHSD5 expression in sc fat correlates with body mass index (BMI) (184). Insulin, glucocorticoids, and inflammatory cytokines also stimulate aromatase activity in adipocytes or preadipocytes (188, 189).
Hepatic 17βHSD5 gene expression, hence testosterone formation from androstenedione, appears to be down-regulated by insulin in liver (107). On the other hand, 5αRD activity, which promotes androgen action, is up-regulated in hyperandrogenic states by mechanisms that involve both insulin resistance (190) and possibly androgen excess itself (191).
Sex hormone-binding globulin (SHBG) is of hepatic origin; it is an important factor in androgen action and metabolism. The SHBG concentration determines the fraction of serum testosterone and other 17β-hydroxysteroid ligands (eg, estradiol, dihydrotestosterone) that are free or bound to albumin. It is thus a major determinant of ligand egress from serum to androgen target tissues and to liver for clearance from the circulation (192). SHBG levels are raised by estrogen and suppressed by androgen, insulin resistance in obesity, and hypothyroidism (192, 193). Although the low SHBG in obese individuals has long been attributed to hyperinsulinemia (194), recent evidence suggests that monosaccharide excess itself, signaling via inflammatory cytokines, mediates the SHBG response to obesity (193, 195). Rarely, mutations cause very low levels of SHBG (196).
E. Summary of normal androgen physiology
Androgen and androgen precursors normally are produced by the ovaries and adrenal cortices in about equal amounts in response to LH and ACTH, respectively. About half of testosterone arises from peripheral metabolism of secreted precursors in liver, skin, and fat, where the factors regulating these conversions are less clear, although insulin stimulates testosterone formation in fat. Androgens are not under tight neuroendocrine negative feedback control. Rather, the ovarian androgenic response to LH appears to be normally modulated by intraovarian mechanisms so as to optimize androgen and estrogen formation so as to promote follicular maturation, because although androgens are essential substrates for estradiol formation, in excess, they hinder ovulation. In part, this modulation seems to be accomplished by homologous desensitization of theca cells to LH, which minimizes the androgenic response to high LH levels commencing with desensitization at the level of the LH receptor. In part, modulation seems to be accomplished by counterbalanced paracrine down-regulatory and up-regulatory mechanisms that primarily act on the rate-limiting step in sex steroid formation, P450c17 activity. Excess insulin is an extraovarian modulator that has the potential to override normal intraovarian down-regulatory mechanisms that control ovarian androgen production.
IV. Source of Androgen Excess in PCOS
A. Testing to determine the source of androgen in PCOS
To attempt to understand PCOS pathophysiology, we have functionally categorized PCOS patients according to whether the source of androgen excess is primarily the ovaries, the adrenal glands, both, or neither (Table 2 and figure 5 below) (47). Test procedures to determine the source of androgen are outlined in Table 3. Our studies reviewed here are based on RIA methods for testosterone, androstenedione, and 17OHP that have a precision of approximately 12% and have been validated against liquid chromatography-tandem mass spectrometry (47).
Table 2.
Functional Classification of PCOS According to Source of Androgen Excess
| PCOS Functional Type | Source of Androgen | GnRHag Test 17OHP Response | DAST Testosterone Response | ACTH test DHEA Response | Prevalence Among PCOS |
|---|---|---|---|---|---|
| PCOS-T | Primary FOH (typical FOH) | Higha | High in 92.5% | High in 28% (associated FAH) | 67%b |
| PCOS-A | Primary FOH (atypical FOH) | Normala | High | High in 30% (associated FAH) | 20% |
| Primary FAH (isolated FAH) | Normal | Normal | High | 5% | |
| PCOS without FOH or FAH (PCOS-A of obesity or idiopathic PCOS-A) | Normal | Normal | Normal | 8% |
Based on data of Rosenfield et al, Determination of the source of androgen excess in functionally atypical polycystic ovary syndrome by a short dexamethasone androgen-suppression test and a low-dose ACTH test. Hum Reprod. 2011;26:3138–3146 (47).
High vs normal denotes defining characteristics; percentages indicate experimentally determined prevalence of abnormality.
Prevalence determined from an age-matched subgroup (n = 60) of an original cohort (n = 99), in which 69% had PCOS-T. Modified with permission from Rosenfield, Polycystic ovary syndrome in adolescents. In: Rose BD, ed. www.uptodate.com. UpToDate. Waltham, MA; 2014.
Table 3.
Test Procedures to Determine Source of Female Androgen Excess
| Test | Rationale | Method | Outcome Measures | Interpretationa |
|---|---|---|---|---|
| GnRHag | Endogenous LH and FSH release stimulates coordinated function of ovarian follicles | Leuprolide acetate 10 μg/kg sc (for maximum stimulation) | Ovarian steroid secretion peaks at 20–24 h | 17OHP >152 ng/dL without steroidogenic block indicates typical FOH (PCOS-T) |
| hCG | Exogenous administration of LH analog stimulates theca-interstitial cells | hCG 3000 IU/m2 (for maximum stimulation) | Ovarian steroid secretion peaks at 24 h | 17OHP >152 ng/dL without steroidogenic block indicates typical FOH (PCOS-T) |
| LDAST | Long DAST: dexamethasone profoundly suppresses adrenal androgens over several days | Dexamethasone 0.5 mg QID per os × 4–5 d | Free testosterone, DHEAS, cortisol: sample early morning d 5 | Free testosterone ≥8 pg/mL with DHEAS <70 and cortisol <1 μg/dL characteristic of FOH |
| SDAST | Short DAST: dexamethasone rapidly suppresses adrenal testosterone and cortisol | Dexamethasone 0.25 mg/m2 per os at 12 noon | Total testosterone, cortisol: sample 4 pm (4 h) | Total testosterone >26 ng/mL, cortisol <5 μg/dL suggests FOH |
| ACTH | Exogenous ACTH stimulates adrenal steroidogenesis | Cosyntropin ≥10 μg/m2 (for maximum stimulation) | DHEA,17OHP, steroid intermediates, cortisol peak at 30–60 min | DHEA 1500–3000 μg/dL without steroidogenic block indicates FAH |
Testing in early follicular phase of menstrual cycle. Conversion multipliers to SI units: 17OHP 0.0303 (nmol/L), cortisol 0.0276 (μmol/L), free testosterone 3.47 (pmol/L), total testosterone 0.0347 (nmol/L), DHEA 0.0347 (nmol/L), and DHEAS 0.0271 (μmol/L). Data from Rosenfield et al, Determination of the source of androgen excess in functionally atypical polycystic ovary syndrome by a short dexamethasone androgen-suppression test and a low-dose ACTH test. Hum Reprod. 2011;26:3138–3146 (47), Levrant et al, A pilot study of the human chorionic gonadotrophin test for ovarian hyperandrogenism. Hum Reprod. 1997;12:1416–1420 (96), and Rosenfield, The diagnosis of polycystic ovary syndrome in adolescents. Pediatrics. 2015;136:1154–1165 (50).
Norms are those in our laboratory; results may differ elsewhere.
The ovarian hyperandrogenism of PCOS is demonstrated directly by the GnRHag test or the hCG test and indirectly by the dexamethasone androgen-suppression test (DAST) (Table 3). The GnRHag test determines the coordinated function of the ovarian follicle. Leuprolide acetate 10 μg/kg sc (or a comparable dose of any other short-acting GnRHag) stimulates endogenous LH and FSH release that peaks at 3–4 hours and persists for 24 hours; this in turn stimulates increased secretion of sex steroids and their precursors, with serum levels peaking at 18–24 hours (17, 22, 197). In the absence of evidence of a steroidogenic block, an elevated 17OHP response is typical of PCOS. Ovarian steroidogenic enzyme deficiency, which is rare, can be detected by an abnormal pattern of steroid intermediates in response to the test (17, 197, 198). hCG is an LH analog: 5000 IU im stimulates steroidogenic responses comparable with those of a GnRHag test at 24 hours (95, 96, 199). We perform the GnRHag test after the DAST so as to blunt coincidental adrenal secretion, which may otherwise occasionally confound the ability to interpret the results.
DAST indirectly tests ovarian androgenic function by suppressing ACTH-dependent adrenal androgen production. In the presence of normal adrenocortical suppression, an inappropriately elevated serum testosterone post-DAST indicates an ACTH-independent source of androgen, which is ordinarily of ovarian origin. In circumstances where an adrenal virilizing disorder is suspected, a “long” (4–5 d) DAST is indicated (200–203), but a “short” (4 h) DAST suffices for most suspected PCOS (Table 3), because the long and short tests yield similar results (47). Notably, the differences in testosterone responses between these 2 tests were greater than expected from assay precision in about 25% of cases, suggesting that biologic variability in ovarian function affects test results.
Adrenal hyperandrogenism is demonstrated by a rapid ACTH test: cosyntropin is administered iv and peak steroid responses occur at 15–60 minutes. Although the test is ordinarily performed using cosyntropin 250 μg, this is a supramaximal dose, and 10 μg/m2 elicits a similar peak response. The low-dose ACTH test (1.0-μg cosyntropin) is more physiologic; it usually elicits nearly as great a peak response that promptly wanes, and in PCOS, does not elicit such a wide spectrum of elevated steroid intermediates as do larger doses (47). DHEAS is a simple correlate of this adrenal androgenic dysfunction (r = 0.708).
B. FOH in PCOS
PCOS is a diagnosis of exclusion by standard criteria (Table 1). Therefore, it is necessary to consider other causes of hyperandrogenism in the differential diagnosis, although they account for only 10%–20% of adults presenting with hyperandrogenic symptoms (Table 4) (50, 204–206).
Table 4.
Differential Diagnosis of Hyperandrogenemia
|
Modified with permission from Rosenfield, The diagnosis of polycystic ovary syndrome in adolescents. Pediatrics. 2015;136:1154–1165 (50).
The unique ovarian dysfunction of PCOS (primary FOH) was first demonstrated by GnRHag testing (22). The pattern of steroidogenesis indicated generalized overactivity of the entire ovarian steroidogenic cascade involved in sex steroid secretion. An elevated 17OHP response was the most consistent abnormality in classic PCOS, which suggested a prominent abnormality at the level of P450c17 activities. Subsequent clinical studies have shown that ovarian steroidogenesis in PCOS is typically similarly hyperresponsive to both the endogenous LH and FSH surge elicited by administration of GnRHag challenge or by hCG challenge (90, 96, 199, 207).
FOH is the common denominator in the vast majority of PCOS. However, as reviewed next, FOH is neither always typical nor always demonstrable in PCOS (Table 2).
1. Spectrum of ovarian androgenic function in PCOS: Typical and atypical FOH
Initially, we reported that the first 7 women with classic PCOS who underwent GnRHag testing had 17OHP hyperresponsiveness in comparison with 16 nonhirsute eumenorrheic women in the midfollicular phase of their menstrual cycle (17). Then, we demonstrated that most (58%) hyperandrogenic women (n = 40) had this PCOS type of 17OHP hyperresponsiveness to GnRHag testing conducted post-DAST in comparison with 13 controls (19). Eighty-seven percent of those with abnormal responses to GnRHag were oligomenorrheic. This abnormality in hyperandrogenic women correlated well (r = 0.75; r2 = 0.56) with elevated plasma free testosterone in response to the 5-day DAST, with concordance in 85% of women, in contrast to the findings that polycystic ovaries or elevated LH levels occurred in only about half.
Although subsequent reports confirmed the presence of a significant difference in 17OHP responses to GnRHag between PCOS and control women, they differed widely in their estimates of the prevalence of this abnormality, with some reporting its presence in only a distinct minority (208, 209). We believe this low prevalence is due to failure to exclude women with PCOM from the reference control group, for reasons discussed in the following section.
From 2000 to 2007, we sought to determine the prevalence of FOH in hyperandrogenic women with anovulatory symptoms (PCOS), controlling for the presence of PCOM (197). We performed a GnRHag test and a DAST in 99 consecutively consenting PCOS patients who presented to our clinics and who met NIH criteria (elevated serum free testosterone and ovulatory dysfunction) and compared them with nonhirsute eumenorrheic volunteers, the reference group being volunteers with ultrasonographically normal ovarian morphology (V-NOM) (n = 21). Volunteers were studied in the midfollicular phase of their menstrual cycles (d 4–10) so as to match them for follicular status as well as possible with PCOS. The study protocol included assessment for PCOM, AMH levels, and glucose tolerance. We found that 69% of PCOS, defined by NIH criteria, had typical 17OHP hyperresponsiveness to GnRHag (197).
To better understand the differences between those PCOS patients with 17OHP hyperresponses (functionally typical PCOS [PCOS-T]) and those who lacked 17OHP hyperresponsiveness (functionally atypical PCOS [PCOS-A]), we then analyzed age-matched subsets of PCOS-T (n = 40), PCOS-A (n = 20), and nonhirsute eumenorrheic volunteers, the reference group being V-NOM. We determined the sources of androgen in these 2 functional types of PCOS and related the findings to glucose intolerance in 1 report (47), related ovarian androgenic function to markers of folliculogenesis (PCOM and serum AMH) in another (39), and then integrated these findings (30). Approximately half of the study subjects were adolescents (>1.0 y postmenarcheal and 11.0–17.9 y of age); adults were 18.0–39.9 years old. Within the volunteer and PCOS groups, adolescents and adults had similar baseline androgen, 17OHP, and LH levels (30). The data are displayed in Figure 4 (30).
Figure 4.

Scatterplots demonstrating relationships among tests for ovarian hyperandrogenism, PCOM, and serum AMH concentrations. Subjects are patients with PCOS identified by NIH criteria (n = 20 PCOS-A, n = 40 PCOS-T) and age-matched healthy eumenorrheic nonhirsute volunteers with normal or PCOM (V-NOM n = 21, V-PCOM n = 32, respectively). Serum for AMH was available in 92% of PCOS and 82% of these volunteers (39). PCOM was defined according to modified Rotterdam criteria: in adults, it was defined as an ovary more than 10.5 cc (in adolescents, >10.8 cc) using the formula for a prolate ellipsoid and/or more than or equal to 10 follicles 2–9 mm in diameter in the maximum plane (27, 39). Dotted lines show normal ranges for the V-NOM reference group; thus (a) quadrant panels show normal ranges. A, PCOS-T is defined by an elevated 17OHP response to the GnRHag test (c and d). SDAST results correlate with GnRHag results (r = 0.671, P < .0001). SDAST is abnormal in 92.5% of PCOS-T. PCOS-A is defined by lack of 17OHP hyperresponse to GnRHag. The SDAST divides PCOS-A into those with (b) and without (a) ovarian androgenic dysfunction. SDAST indicates that 60% of these PCOS-A cases have atypical FOH (b) and 40% of PCOS-A cases have normal ovarian androgenic function (a). B, SDAST relationship to baseline AMH levels. C, 22% (n = 7) of asymptomatic V-PCOM have baseline hyperandrogenemia (hyperandrogenic PCOM [V-PCOMh]). These all proved to have FOH, as indicated by an abnormal SDAST or GnRHag test (b–d). Adolescent V-PCOM tended to have this asymptomatic FOH less often (1/9) than adult (6/23) V-PCOM. Dysregulated PCOM (V-PCOMd), defined by an abnormal 17OHP response to GnRHag in the absence of baseline hyperandrogenemia (d), was found in 25% (n = 8) V-PCOM. D, Mild AMH elevation was found in V-PCOM independently of hyperandrogenemia. So as to best illustrate differences among groups, very high values (post-SDAST testosterone up to 107 ng/dL and post-GnRHag l7OHP up to 1380 ng/dL) are plotted off-scale. To convert to SI units, multiply total testosterone by 0.0347 (nM), 17OHP by 0.0303 (nM), and AMH by 7.125 (pM). Reproduced with permission from Rosenfield, The polycystic ovary morphology-polycystic ovary syndrome spectrum. J Pediatr Adolesc Gynecol. 2015;28:412–419 (30). Since publication of these data, accumulated evidence suggests that in adolescents mean ovarian volume more than 12 cc is a more appropriate criterion for PCOM than more than 10.8 cc (50, 51). Doing so alters the constitution of the V-NOM and V-PCOM groups. With this adjustment and the addition of data on 4 contemporaneously studied but previously overlooked healthy volunteers, our current upper limits for V-NOM (n = 31) are: baseline-free testosterone 9.3 pg/mL and AMH 6.3 ng/mL; SDAST total testosterone 26 ng/dL; and postdexamethasone GnRHag 17OHP 152 ng/dL (50).
Among PCOS-T, those with 17OHP hyperresponsiveness (Table 2 and Figure 5), hyperandrogenism was more severe than in PCOS-A, and the great majority (92.5%) also had an abnormal short DAST (SDAST) and PCOM (Figure 4Ac). Serum AMH was increased in 81%, and the increase was significantly greater than that of any other group. Coincidental FAH was present in 28%. Impaired glucose tolerance (IGT) and frank diabetes were present significantly more often than in PCOS-A or controls; in this series, the only diabetic cases were in PCOS-T. In addition, estradiol secretion was hypersensitive to submaximal hCG stimulation (Figure 3) and FSH administration did not result in the normal inhibition of baseline serum testosterone in PCOS-T (95).
Figure 5.
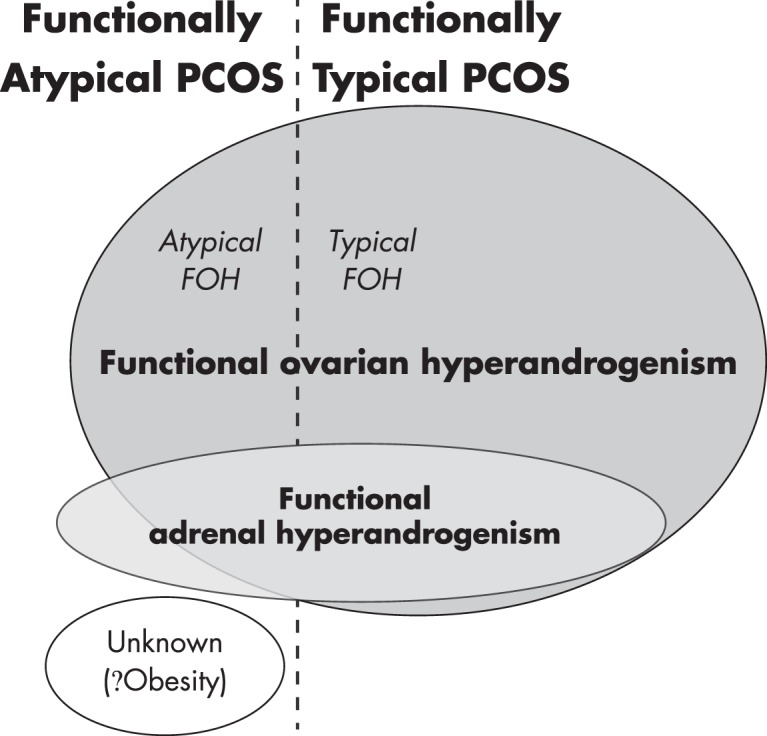
Relationships among sources of androgen in PCOS. About two-thirds of cases have functionally typical PCOS (PCOS-T) that is due to typical FOH, in which there is hypersensitivity to LH, characterized by hyperresponsiveness of 17OHP to a GnRHag or hCG test. The remaining one-third of PCOS is functionally atypical, lacking 17OHP hyperresponsiveness. This is a heterogeneous group, most of which have atypical FOH, in which ovarian androgen excess is indicated only by a DAST. A small number are due to isolated FAH. About one-quarter of FOH also have FAH. In a minority of cases, the source of androgen cannot be identified as ovarian or adrenal; most of these are associated with obesity. Modified and reproduced with permission from Rosenfield, Polycystic ovary syndrome in adolescents. In: Rose BD, ed. www.uptodate.com. Waltham, MA: UpToDate; 2014.
PCOS-A is a functionally heterogeneous group (Table 2 and Figure 5). Sixty percent had an abnormal DAST (Figure 4Ab), which defines an atypical form of FOH (Table 2, atypical FOH). FAH coexisted with FOH in 30% of this subgroup. PCOM (65%) and AMH elevation (39%) were found significantly less frequently than in PCOS-T. The prevalence of glucose intolerance was significantly less than in PCOS-T and did not differ from that in the control subjects.
The other 40% of PCOS-A had a normal DAST (Figure 4Aa), ie, no evidence of an ovarian source of androgen. They had significantly milder hyperandrogenemia. This “nonovarian PCOS” itself seems functionally heterogeneous (Table 2). ACTH testing showed “isolated FAH” (Table 2) in 15% of PCOS-A; two-thirds of these had an elevated baseline DHEAS level. However, 85% of the nonovarian subgroup (25% of PCOS-A) lacked FAH, so evidence for a glandular source of androgen was lacking. All in this small subgroup were obese, and we have attributed their androgen excess to excessive peripheral testosterone formation by excessive adipose tissue (“PCOS-A of obesity” in Table 2) (47).
We conclude that there is a spectrum of pathophysiologic dysfunction in PCOS that generally corresponds to clinical severity (30): the PCOS-T group, which has an ovarian secretory pattern suggestive of dysregulation of steroidogenesis prominent at the level of the 17-hydroxylase/17,20-lyase activities of P450c17 (Figure 1), constitutes two-thirds of PCOS and is significantly more clinically severe than PCOS-A, although considerable clinical overlap exists. Furthermore, these data suggest heterogeneity in the pathophysiologic basis of FOH. It remains to be proven whether these pathophysiologic categorizations have clinical utility beyond possibly identifying a subpopulation of PCOS patients whose androgen excess arises from simple obesity and so would be expected to be reversible by weight loss (210, 211) or distinguishing adolescents with PCOS from those with physiologic anovulation (53). Biochemical categorization is expected to prove useful in developing phenotype-genotype correlations, as discussed later: only when we understand the etiology of PCOS will it be possible to truly assess the sensitivity and specificity of these tests.
2. Spectrum of ovarian androgenic function in asymptomatic women with PCOM
PCOM is a common finding among healthy women. Many of these women have mild PCOS features, ie, irregular menstrual cycles and/or hirsutism (212, 213). When care has been taken to exclude those with such symptoms, groups of apparently normal women with PCOM, including some with documented ovulatory cycles, have been shown to have subclinical androgenic ovarian dysfunction that is intermediate between that of women with normal ovaries and those with PCOS (31, 207, 214).
AMH serum concentrations in normal subjects with PCOM are also intermediate between those of women with NOM and those with PCOS (215–217). Data vary as to whether insulin resistance is associated with PCOM in healthy adult volunteers (31, 214, 218).
We have studied the ovarian androgenic function of normal females with PCOM in some detail. We compared nonhirsute eumenorrheic volunteers with PCOM (V-PCOM) to both the otherwise entirely similar reference group of females with NOM (V-NOM) and to PCOS, all groups age-matched, using the above protocol (39, 197), as shown in Figure 4.
Healthy adolescent and adult volunteers had similar ovarian function, except that V-NOM adolescents by definition had slightly larger ovaries (volume ≤ 10.8 cc) than V-NOM adults and slightly lower FSH and higher AMH levels (30, 197). V-PCOM are heterogeneous, with a spectrum of ovarian function (Figure 4, C and D). At one end of the V-PCOM spectrum was a subgroup of 40% with an extreme variation of normal size and morphology; they had ovarian androgenic function test results and serum AMH like that of V-NOM (Figure 4, Ca–Da). At the other end of the V-PCOM spectrum, was a subgroup of 22% with “hyperandrogenic PCOM” (V-PCOMh in Figure 4C, b–d); although asymptomatic, they had baseline hyperandrogenemia, and so seemed to meet the definition of ovulatory PCOS (Table 1, phenotype 3) since eumenorrheic; all also had a positive SDAST and/or GnRHag test, which indicates subclinical FOH. They also had mildly increased adrenal androgenic function: DHEAS was 159 ± 58, SD, μg/dL, significantly higher than that of V-NOM (80 ± 41 μg/dL), unlike any other V-PCOM subgroup (P < .01); they also tended to have a higher DHEA response to ACTH (P = .15). DHEAS and DHEA peaks were, respectively, elevated (>180 and > 1100 ng/dL) in 42% and 14% of this subgroup.
Between these extremes lay 2 different kinds of ovarian functional variants (39). On the normal side of the spectrum was a V-PCOM subgroup (10% of V-PCOM) with an isolated AMH elevation (V-PCOM in Figure 4Dd) that indicates increased folliculogenesis. On the PCOS side of the spectrum was a V-PCOM subgroup with “dysregulated PCOM,” ie, steroidogenic hyperresponsiveness to GnRHag (17OHP elevation without hyperandrogenemia) (Figure 4Cd); these constituted 28% of V-PCOM, one-third of whom had mildly elevated serum AMH. It seems probable that this dysregulation indicates a very mild and subclinical degree of intraovarian androgen excess. In a multivariate model, AMH correlated independently across all groups (healthy volunteers with or without PCOM and PCOS) with SDAST testosterone (P = .001), but not peak 17OHP response to GnRHag (P = .5). Summary multiple regression analysis across all groups (Figure 4) shows that serum AMH is independently (R2 = 0.3) related to the presence of ovarian hyperandrogenism (P < .001) and to that of PCOM (P = .014).
The picture that emerges from this biochemical spectrum of differences in ovarian function among V-PCOM subjects is that apparently normal women with PCOM occupy a middle position on a spectrum of ovarian function between women with clearly normal ovarian function and those with PCOS (Figure 6). About half have no evidence of steroidogenic dysfunction and, thus, no relation to PCOS. On the other hand, about half of V-PCOM have subclinical evidence of a PCOS-related dysregulation of ovarian steroidogenesis. However, most V-PCOM appear to be ovulatory and at low risk of developing symptomatic PCOS (31, 197, 207, 214, 219). How is this to be interpreted? The subgroup of eumenorrheic young women who have subclinical hyperandrogenemia, which suggests ovulatory PCOS, may well represent a carrier state, or occasionally a risk factor, for PCOS (197), although this remains to be established. The spectrum of ovarian androgenic ovulatory dysfunction may well be wider than ascertained through the presence of PCOM or hyperandrogenemia. Among apparently normal eumenorrheic women, a prospective study indicated that about 8% had sporadic anovulatory cycles and that serum free testosterone and AMH levels were significantly increased in these cycles, although within the normal range (220).
Figure 6.
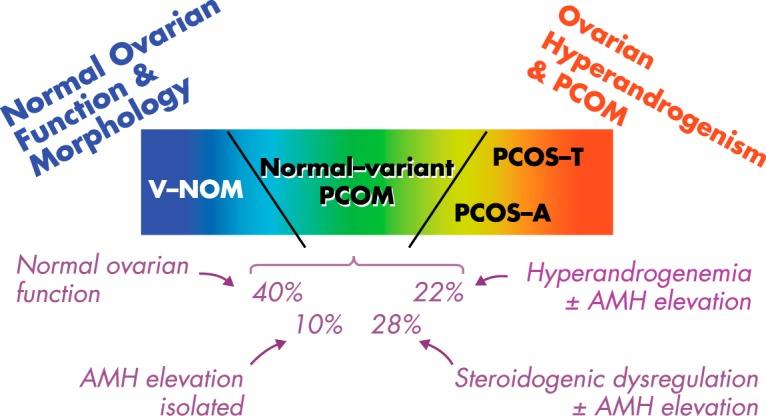
Schematic representation of the spectrum of ovarian function found in eumenorrheic nonhirsute V-PCOM (normal-variant PCOM) in relation to that of normal and PCOS women. Approximately 40% of V-PCOM are functionally variations of normal: this group has ovarian function like that of similar V-NOMs. Another 10% of V-PCOM has elevated AMH in the absence of any evidence of ovarian steroidogenic dysfunction, which suggests an isolated increase in folliculogenesis unrelated to ovarian androgenic dysfunction. The remaining half of V-PCOM have some degree of PCOS-related steroidogenic dysregulation, often with AMH elevation. Of these, nearly half (22% of the V-PCOM group) have biochemically hyperandrogenic PCOM, ie, subclinical FOH that suggests ovulatory PCOS. The remainder have isolated dysregulation of ovarian steroidogenesis (ie, isolated in the sense that 17OHP hyperresponsiveness to GnRHag testing occurs in the absence of hyperandrogenemia). Based on data in Figure 4; percentages are averages derived from different denominators for GnRHag test (n = 32) and AMH (n = 28) determinations in V-PCOM.
On the other hand, a substantial minority of our asymptomatic volunteers have isolated (normoandrogenemic) PCOS-like dysregulated steroidogenic function. Many of this subgroup have elevated AMH levels, which is compatible with a very mild degree of intraovarian androgen excess promoting folliculogenesis (79, 143) without interfering with ovulatory function.
Those V-PCOM with AMH elevation, some of whom have absolutely normal steroidogenic function and some of whom have subclinical evidence of steroidogenic dysregulation (Figure 6), are expected to have an increased population of growing follicles (39, 144). Thus, they can be predicted to have a slightly prolonged reproductive lifespan (221).
In conclusion, about half of asymptomatic V-PCOM have evidence of an ovarian steroidogenic dysfunction related to PCOS, including some who seem to have ovulatory PCOS. They are postulated to be carriers for, or at risk for, PCOS.
C. Functional adrenal hyperandrogenism (FAH) in PCOS
Less than 10% of FAH can be accounted for by well-established pathophysiologic entities, the most common of which is nonclassical virilizing congenital adrenal hyperplasia with a prevalence approximating 5% (Table 4). Most FAH is idiopathic (primary), ie, it cannot be incontrovertibly assigned to any of these well-established disorders.
Primary FAH is defined as 17-ketosteroid hyperresponsiveness to ACTH that is otherwise unexplained, which practically speaking involves ruling out steroidogenic blocks (17). The steroidogenic pattern of response to ACTH resembles an exaggerated adrenarche (22). DHEA is the sole hyperresponsive 17-ketosteroid when testing is performed with low-dose ACTH; it was abnormal in 27% of our PCOS series, with similar prevalence in both PCOS-T and PCOS-A (47). DHEA hyperresponses are accompanied by 17-hydroxypregnenolone hyperresponses (r = 0.773), which suggests a relationship between these two entities. Higher-dose ACTH testing yielded a higher prevalence estimate for FAH in PCOS (46%), and androstenedione hyperresponses accounted for most of the difference (22). The pattern of adrenal secretion is compatible with dysregulation of zona reticularis steroidogenesis prominent at the level of the 17-hydroxylase/17,20-lyase activities of P450c17 (Figure 2). The FAH of PCOS is accompanied by an average 50% increase in adrenal volume that correlates with hyperandrogenemia severity (222).
About one-quarter of FOH have the common (primary) type of FAH (Figure 5). This agrees with most estimates of FAH prevalence in PCOS from serum DHEAS measurements (28). However, the magnitude of the DHEAS correlation with DHEA responsiveness to ACTH (r = 0.7) is such that there is considerable nonspecificity in this estimate, which seems due to the high (∼70%) heritability of DHEAS serum levels (223–225).
D. Other sources of androgen in PCOS
A glandular source of androgen cannot be identified in approximately 8% of hyperandrogenic patients despite thorough testing. Those who present with hirsutism and normal menses, but lack a polycystic ovary, are traditionally given a diagnosis of idiopathic hyperandrogenism.
In our series of PCOS patients who met NIH diagnostic criteria, the subset with no demonstrable ovarian or adrenal dysfunction was obese (Figure 4a and Table 2) (47). We postulated that their excess adipose tissue was both the cause of the testosterone excess (because adipocytes are a site of conversion of circulating androstenedione to testosterone) and the cause of the ovulatory dysfunction (because obesity suppresses LH levels) (83, 226–229), as discussed below (see section V.B.2). These patients with the PCOS-A of obesity were characterized by mild hyperandrogenemia (normal total testosterone, mildly elevated free testosterone, normal DHEAS, low SHBG), and most had normal-size ovaries, normal LH levels, and normal AMH levels; all had insulin resistance that was similar to that of the other PCOS-A subgroups.
The possibility is unexplored that some cases in whom there is no glandular source of androgen are caused by hereditary defects in the peripheral metabolism of steroids, such that testosterone formation from precursors is excessive.
E. Summary
The common denominator of the great majority (87%) of PCOS patients is FOH. Two-thirds of PCOS cases have 17OHP hyperresponsiveness to GnRHag or hCG stimulation (functionally typical FOH). Two-thirds of the remainder have FOH detectable by DAST, in which testosterone remains elevated after suppression of adrenal androgen production. About 3% of PCOS have isolated FAH. The remaining PCOS cases are mild and lack evidence of steroid secretory abnormalities; most of these are obese, and we postulate that their excess adipose tissue accounts for their PCOS. These relationships among the sources of androgen in PCOS are summarized in Table 2 and Figure 5.
V. Pathophysiology of PCOS: Abnormal Regulation of Steroidogenesis and Ovarian Function
The above in vivo data suggest that the fundamental defect in most PCOS is FOH due to an otherwise unexplained (primary) unique type of steroidogenic hyperactivity that seems to disturb the intraovarian processes that normally coordinate ovarian androgen and estrogen secretion (22).
The PCOS ovary is typically hypersensitive to LH stimulation. The initial studies of the responses to GnRHag suggested abnormal steroidogenic dose-response relationships in response to LH that were consistent with partial escape from desensitization (198). Subsequent studies directly demonstrated hypersensitivity to submaximal hCG test doses associated with a similar pattern of increased androgen responsiveness (Figure 3) (90, 95). Initially, the possible causes of this dysregulation of androgen secretion were postulated to include insulin excess, which is known to sensitize the ovary to LH by interfering with the normal process of homologous desensitization to LH as discussed above, or an intrinsic imbalance among intraovarian regulatory systems (198).
Support for an intrinsic theca cell defect has come from both in vivo and in vitro studies. In vivo, ovarian steroidogenic hyperfunction in response to submaximal acute hCG challenge persists after the ovarian quiescence achieved by 1–3 months of gonadotropin suppression (95, 207). In vitro studies have shown the presence of an intrinsic theca cell abnormality that is independent of LH receptor status by demonstrating that an overactive steroidogenic phenotype is constitutively present in isolated theca cells and persists through long-term passage in cell culture, which suggests an inherent defect(s) (23, 72).
These findings support the concept that FOH is usually the essence of PCOS. The intraovarian level of androgens in FOH would seem to be higher than those in FAH and such extraovarian disorders as nonclassical virilizing congenital adrenal hyperplasia in the presence of similar circulating levels of androgen. Only frankly virilizing extraovarian disorders would be expected to boost intraovarian androgen levels to those of FOH. Androgen excess, as noted above, has been demonstrated to enhance the initial recruitment of primordial follicles into the growth pool and thus play a role in initiating the growth of small antral follicles (79, 136, 143). Androgen excess also initiates premature luteinization (138, 139, 147), which hinders ovulation by impairing selection of the dominant follicle (155, 157). Androgen excess has also been shown to cause the classical PCOS histopathologic and gross anatomic changes that constitute PCOM (9, 230).
However, PCOS is a multisystem disorder. A metabolic syndrome, underpinned by insulin resistance and obesity, is common in PCOS, and the insulin resistance is excessive for the degree of adiposity (13, 231–233). PCOS is a state in which tissue-selective resistance to the glucose-metabolic effects of insulin seems to be paradoxically associated with ovarian sensitivity to insulin, such that the compensatory hyperinsulinemia of insulin resistance contributes to ovarian androgen excess (24, 234). Insulin excess appears to do so primarily by leading to a partial “escape” from desensitization of ovarian responsiveness to LH, with the consequence that ovarian steroids are hyperresponsive to LH (see sections III.B.2.b and V.B.1.a) (22, 88, 89).
LH excess is common in the disorder. LH is necessary for the expression of gonadal steroidogenic enzymes and sex hormone secretion. Consequently, PCOS is LH dependent (hence, “functional”), and any treatment or disorder that suppresses LH levels suppresses ovarian steroidogenesis. However, the moderate increase of LH that characterizes PCOS seems unlikely to ordinarily be the primary cause of the ovarian androgen excess, due to the normal process of LH-induced desensitization of theca cells.
A. Dysregulation of ovarian function in PCOS
1. Dysregulation of steroidogenesis in PCOS
a. Intrinsic theca cell dysfunction.
In vitro studies have provided convincing evidence for a thecal cell defect that can account for excess androgen production and the steroidogenic secretory pattern observed in response to gonadotropin stimulation. They show that isolated thecal cells overexpress most steroidogenic enzymes, particularly cytochrome P450c17, and LH receptors (23, 235).
Critical evidence in support of inherent ovarian steroidogenic dysfunction in PCOS came from the demonstration by the McAllister group that “augmented androgen production is a stable steroidogenic phenotype of propagated theca cells from polycystic ovaries” of PCOS patients (23). Theca cells were obtained at hysterectomy from 3- to 5-mm follicles of polycystic ovaries of women meeting NIH criteria for PCOS and asymptomatic fertile women. They were cultured on fibronectin-coated plates in a highly enriched medium including 20% serum; at confluence they were frozen at −70°C. Subsequently, cells were thawed and passaged 3–4 times (22–38 population doublings); then experiments were performed in serum-free medium from age- and follicle-sized matched normal and PCOS subjects. These studies showed that progesterone, 17OHP, and testosterone production per cell were markedly increased in PCOS theca cell cultures. Moreover, basal and cAMP-stimulated pregnenolone, progesterone, and DHEA metabolism were increased dramatically in PCOS theca cells. PCOS theca cells were capable of substantial metabolism of precursors into testosterone, reflecting expression of androgenic 17βHSD activity. cAMP-stimulated CYP11A and CYP17A1 expressions were augmented in PCOS theca cells compared with normal cells, whereas no differences were found in steroidogenic acute regulatory protein mRNA expression. Collectively, these observations establish that increased CYP11A and CYP17A1 mRNA expression, as well as increased P450c17, 3βHSD, and 17βHSD enzyme activity per theca cell, and consequently increased production of progesterone, 17OHP, and testosterone, are stable properties of PCOS theca cells (23). Subsequent study indicated that increased synthesis of testosterone precursors, particularly via increased P450c17 and 3βHSD activity, is the primary factor driving enhanced T secretion in PCOS (72). Stimulated HSD17B5 mRNA was not consistently overexpressed in PCOS, but the statistical power of the study was too low to reach a firm conclusion about the contribution of HSD17B5 gene expression to the increased 17βHSD activity in PCOS.
Recently, drawing upon genome-wide association screening data discussed later, McAllister's group reproduced the PCOS theca phenotype in vitro by forced overexpression of a DENND isoform (A1) that is increased at the mRNA and protein level in PCOS theca cells (108). DENND1A encodes a protein (connecdenn 1) associated with clathrin-coated pits where cell-surface receptors reside that is located in the cytoplasm and nuclei of theca cells. Alternative splicing of DENND1A generates 2 transcripts, and it is the shorter transcript, DENND1A.V2, that is differentially expressed in PCOS and normal theca cells. “Forced overexpression of DENND1A.V2 in normal theca cells resulted in a PCOS phenotype of augmented CYP17A1 and CYP11A1 gene transcription, mRNA abundance, and androgen biosynthesis. Knock-down of DENND1A.V2 in PCOS theca cells reduced androgen biosynthesis and CYP17A1 and CYP11A1 gene expression” (108).
A polyclonal antibody specific to the unique C-terminal sequence of DENND1A.V2 also reduced androgen biosynthesis and CYP17A1 and CYP11A1 mRNA in cultured PCOS theca cells. Additionally, urinary excretion of exosomal DENND1A.V2 RNA in PCOS women was significantly increased in comparison with normal. Thus, DENND1A.V2 potentially plays a key role in the hyperandrogenemia associated with PCOS. It seems likely that excess DENND1A.V2 expression will prove to underlie typical FOH.
The mechanism by which DENND1A.V2 stimulates steroidogenesis is currently unknown. DENND1A is a member of the connecdenn family of proteins, which are clathrin associated adjacent to the inner cytoplasmic membrane and which are involved in protein trafficking, endocytotic processes, and receptor recycling (236). Thus, it is tempting to speculate that it affects LH action by up-regulating LH receptor signaling, much as insulin does in causing escape from LH-induced receptor desensitization.
b. Adrenocortical androgenic dysfunction in PCOS.
FAH often coexists with FOH but may occur in its absence (Table 2). The FAH of PCOS is a unique type of adrenal dysfunction that is characterized by DHEA hyperresponsiveness and hypersensitivity to ACTH of DHEA without evidence of a block in steroidogenesis (22, 47). 17-Hydroxypregnenolone responses to ACTH are highly concordant with DHEA responses in adults and are more often abnormal in adolescents, who have not completed adrenarche (237). Submaximal ACTH stimulation elicits selective DHEA and 17-hydroxypregnenolone hyperresponses in 28% of PCOS patients (47). High-dose ACTH testing demonstrates FAH more often (46%) and elicits additional androstenedione, 17OHP, and/or 11-deoxycortisol hyperresponses; 18% of FAH detected by high-dose ACTH testing is comprised of androstenedione, 17OHP, and/or 11-deoxycortisol hyperresponsiveness in the absence of DHEA hyperresponsiveness (22). The broader spectrum of steroidogenic abnormality in response to high-dose ACTH testing is possibly because PCOS patients have an increased capacity for adrenal androgen secretion (238). FAH is reflected in a high DHEAS level in 20%–30% of patients (239). Adrenal gland volume is reportedly increased about 50% and to correlate positively with age, DHEAS, 17OHP, and testosterone levels and negatively with LH levels (222).
Originally, FAH was attributed to exaggerated adrenarche (22). Subsequently, FAH came to be mistaken for deficiency of 3βHSD2, which is now known to be a rare disorder of steroidogenesis (240, 241). The DHEA responses of FAH are not of the magnitude found in HSD3B2 deficiency (HSD3B2 mutations are not found unless DHEA responses are >5 SD above the mean norm). A functional deficiency of this enzyme activity has been suggested (242).
However, dysregulation of steroidogenesis seems to be the most parsimonious explanation for FAH. The steroidogenic pattern in response to ACTH usually is consistent with 17,20-lyase hyperactivity (22, 240, 243) and overactivity of the early enzymatic steps common to adrenal zona reticularis and ovarian steroidogenesis (eg, P450scc and P450c17 activities). The resultant adrenal secretory pattern differs from the ovarian secretory pattern because of the characteristically different patterns of steroidogenic enzyme expression of these glands: the consequence is that adrenal androgen biosynthesis flows predominantly into DHEA and DHEAS due to the lower 3β-HSD2 and higher sulfotransferase activity of the adrenal zona reticularis than of ovarian theca cells (Figure 1 vs Figure 2). Although no direct evidence exists in support of this hypothesis of adrenal P450c17 activity dysregulation, it is notable that DENND1A is reportedly expressed in the adrenal zona reticularis as well as in ovarian theca cells (236). Furthermore, insulin administered in vitro alters adrenal steroidogenesis much as it does ovarian steroidogenesis (176, 177), and insulin infusion results are consistent with potentiation of 17α-hydroxylase and 17,20-lyase activities in response to ACTH (244). 17-Hydroxypregnenolone responses to ACTH correlate with fasting insulin levels and are lowered by metformin therapy (245). Free fatty acid overload has also been implicated in adrenal hyperandrogenism (246, 247).
Other causes of FAH have been suspected. The unopposed hyperestrogenism of PCOS has been suspected of playing a role in enhancing DHEA secretion by modulating steroidogenic enzyme activity (248); one group reported that steroid ratios indexing 17,20-lyase activity post-CRH stimulation correlated with plasma estradiol in PCOS subjects whose estrogen levels were manipulated. Recent data suggest that intraadrenal end-product inhibition of 3βHSD activity by cortisol might play a role in increasing adrenocortical DHEA secretion (249). Increased cortisol turnover due to elevated peripheral 5αRD activity or decreased peripheral 11β-HSD1 (cortisone RD) activity have been suggested to elicit compensatory ACTH hypersecretion that maintains cortisol levels at the expense of adrenal hyperandrogenism (239, 250). The latter might be related to a common functional polymorphism in the HSD11B1 gene that has been related to FAH (251). On the other hand, obesity may contribute to both pathways (239), and androgens may inhibit 11β-HSD1 (252). The possible role of 5αRD activity in PCOS is discussed in more detail below (see section V.D).
In summary, dysregulation of zona reticularis steroidogenesis akin to that of theca cells seems to be the most parsimonious explanation for the FAH of PCOS, but there is a paucity of direct evidence.
2. Granulosa cell dysfunction and disordered folliculogenesis
a. Granulosa cell dysfunction contributes to thecal androgen excess.
A large body of clinical observations and animal studies suggests that theca cell function is modulated by paracrine factors produced by granulosa cells in response to FSH (Figure 1). Inhibin-B, a peptide that is reciprocally regulated by FSH in a negative feedback loop, is essential and permissive for thecal androgen production, as noted earlier (95). The fall in inhibin-B as follicle number diminishes during menopause may well account for the waning hyperandrogenism of PCOS patients during middle age, as noted above (54, 56). In typical FOH, serum inhibin-B levels are hyperresponsive to FSH, even after prolonged gonadotropin suppression (95). Ovarian inhibin-B betaglycan receptor expression is also increased in PCOS (253). Whether inhibin-B excess plays a role in the hyperandrogenemia of PCOS is unclear because normally FSH predominantly seems to reduce testosterone levels (95). This normal FSH effect is not found in typical FOH. Although this lack of testosterone suppression by FSH might be ascribed to the inhibin-B hyperresponsiveness to FSH, in vitro observations suggest that normal granulosa cells exert a restraining effect on thecal androgen secretion in vivo that is lacking in PCOS; isolated thecal cells from clinically normoandrogenic ovulatory subjects with polycystic ovaries have been reported to secrete as much 17OHP and androgen in culture as did those from hyperandrogenic anovulatory PCOS patients (254). Other granulosa cell peptides are known to induce expression of LH receptors and steroidogenic enzymes during early theca cell development (255, 256) and may mediate the granulosa cell effects on theca cells in PCOS.
b. Disordered folliculogenesis and PCOM.
The ovaries of PCOS patients classically show an excessive number of small antral follicles and various degrees of theca cell hyperplasia and hypertrophy (“hyperthecosis” and “luteinization” in histologic terms), stromal hyperplasia and hypertrophy, and cortical thickening (257). The increased number of small (2–9 mm) antral follicles results from an increased proportion of follicles leaving the resting (primordial follicle) phase to become growing (primary) follicles and eventually small antral follicles that have a prolonged lifespan when the follicle maturation arrest occurs that hinders dominant follicle development (258, 259). Androgen excess can account for all of these morphologic changes (9, 136) and insulin excess likely contributes, but it is unclear whether intrinsic disturbances exist in the ovarian follicle itself.
Enhanced follicle recruitment in PCOS has been attributed to androgen excess promoting the number of small follicles (143). In the rhesus monkey, testosterone implants that raised serum levels 5- to 10-fold rapidly increased the number of primary follicles as well as healthy preantral and small antral follicles by 3- to 5-fold (136). Cystic follicle development may be promoted by androgenic inhibition of follicular 11β-HSD1 increasing cortisol locally (252). Due to their roles in folliculogenesis, GDF9 and BMP15 have been examined in PCOS, but aside from reduced expression of GDF9 in the early stages of follicle development, no convincing evidence has emerged to support their involvement in the abnormal folliculogenesis (137, 260).
Increased folliculogenesis is responsible for the increased AMH production by polycystic ovaries; the increase in AMH is due in part to the increase in follicle count (142), and in part due to increased AMH production per granulosa cell (261, 262). The androgen-induced increase in folliculogenesis has been postulated to cause the AMH elevation of PCOS (143).
Premature luteinization of granulosa cells is indicated biochemically by their acquisition of LH responsiveness at an inappropriately early stage (156). Premature luteinization seems attributable to both androgen excess (9, 136) and insulin excess (156). Both granulosa and theca cells overexpress LH receptors (235), which increases LH responsiveness. Androgen increases LH responsiveness by an indirect mechanism; LH-dependent excessive thecal androgen production causes increased granulosa cell FSH receptor expression (147), which in turn increases thecal LH receptor expression (138, 139, 152). Insulin augmentation of gonadotropin action (89) can account for follicular LH responsiveness to develop at a premature stage (156). The large pool of prematurely luteinized follicles is particularly hyperresponsive to LH. Consequently, unlike midfollicular phase normal women, PCOS women inappropriately secrete estradiol as well as excessive androgen in response to LH/hCG (Figure 3) (95).
Prematurely luteinized granulosa cells are also hypersensitive to FSH. Estradiol is overproduced in response to FSH (95, 263, 264) and seems important for further stimulation of granulosa cell and follicular growth (265, 266). Inhibin-B is hyperresponsive to FSH in typical FOH (95), despite low follicular fluid levels and normal baseline serum levels (140), and combines with the inappropriate estradiol secretion to inhibit FSH secretion and lower FSH levels, and thereby prevent frank hyperestrogenism. However, if challenged with exogenous FSH, as during fertility treatment, PCOS women are at risk of developing ovarian hyperstimulation syndrome (267).
The mechanism of follicular maturation arrest is a subject of debate. Granulosa cell IGF receptor and IGF-binding protein gene expression, which are, respectively, enhanced and inhibited by FSH, are consistent with follicular maturation arrest (268). Premature luteinization may also play a role in follicle maturation arrest by inhibiting further follicle proliferation and so hinder dominant follicle emergence (156). Follicle maturation arrest may be an indirect consequence of the increase in early folliculogenesis via the AMH inhibitory effect on the response of antral follicles to FSH (142).
VEGF serum levels and ovarian gene expression are increased in PCOS, as is ovarian vascularity (114, 145). It has been postulated that this hypervascularity contributes to the relatively dense cortex of the PCOS ovary (159) and inappropriately delivers inflammatory cytokines, and that these factors aggravate the disorder and contribute to ovarian hyperstimulation risk (114, 145). The sclerotic cortex has also been postulated to inhibit the growth-restraining Hippo signaling pathway and contribute to proliferation of granulosa, theca, and stromal cells (125). Local factors, such as VEGF and Hippo, have been postulated to be important mediators of the ovulation induced by ovarian surgery (125, 269).
Differential expression profiling of granulosa cell cDNA is being used as a means to detect the nature of signaling pathway abnormalities in PCOS (270–272). These data suggest that there are multiple and complex disturbances of biological functions: regulation of fatty acid metabolism, insulin and kinase signaling, cell-cell signal transduction, immune, oxidative metabolism, oxidative stress, and inflammatory responses.
Oocyte gene expression is also dysregulated in PCOS and the possibility has been raised that this might be related to the increased risk for pregnancy loss of some PCOS patients (273). A comparison of high quality oocytes from 9 PCOS and 10 unmatched controls undergoing in vitro fertilization showed the PCOS oocytes to have abnormal gene expression profiles that are associated with defective meiosis or early embryonic development. A high proportion of the differentially expressed genes contain androgen and peroxisome proliferator-activated receptors, which suggests that excessive androgen and epigenetic metabolic signals contribute to the reduced developmental competency of PCOS oocytes. However, in vitro oocyte maturation rate, fertilization rate, and grade 1 embryo rate were reported to be significantly higher for PCOS than in controls, and the pregnancy, miscarriage, and live birth rate outcomes were reportedly normal for the number and quality of transferred embryos (274, 275). Factors other than oocyte quality, such as obesity and placental compromise, seem to determine pregnancy outcomes of PCOS women.
3. Summary
Theca cells from the polycystic ovaries of PCOS patients (“classic PCOS”) have provided convincing in vitro evidence for an intrinsic defect that can account for excess androgen production and the steroidogenic secretory pattern observed in vivo. This defect is independent of normal endocrine and paracrine regulatory processes. After long-term passage, these cells in culture overexpress most steroidogenic enzymes, particularly cytochrome P450c17. Overexpression of DENND1A.V2, a protein identified via genome-wide association screening data discussed later, has reproduced this PCOS phenotype in normal theca cells in vitro. FAH is likely to have a similar basis, but no direct evidence exists.
Granulosa cell dysfunction appears to contribute to theca cell overproduction of androgens. The data suggest paracrine defects in FSH inhibition of theca cell function that involve inhibin-B and likely other as-yet-unidentified factors. Granulosa cells prematurely luteinize primarily as a result of androgen and insulin excess. Folliculogenesis is excessive as indicated by PCOM and AMH elevation; although this may be secondary to androgen excess, the possibility cannot be excluded that these abnormalities may be a manifestation of an intrinsic defect in the intraovarian regulation of folliculogenesis (155).
B. Relationship of metabolic syndrome to PCOS
Metabolic syndrome is defined by a cluster of hyperglycemia, central obesity, hypertension, and dyslipidemia. Expression of these individual components can vary between individuals. It ordinarily results from the interactions of insulin resistance and obesity with age (276, 277). Metabolic syndrome occurs in up to one-third of PCOS adolescents and nearly half of PCOS adults (278–282). Among PCOS patients, an abnormal degree of insulin resistance is reported in about one- to two-thirds (although insulin resistance per se is not a defining feature) (13, 233, 283–285); obesity prevalence is similar, with considerable variability among populations (1). A number of observations suggest that among obese women with PCOS, metabolic abnormalities related to insulin resistance and obesity are in many instances more important in the mechanism of anovulation in PCOS than androgen excess (286–288).
Type 2 diabetes mellitus (DM) itself is related to PCOS (289, 290), and deterioration of glucose tolerance can be accelerated in PCOS (291). Glucose intolerance and diabetes arise when β-cell failure compromises the ability of insulin secretion to compensate for insulin resistance. There is a strong heritable component to β-cell dysfunction in women with PCOS (292), and glucose intolerance, ie, β-cell failure, seems to be specifically associated with PCOS-T rather than PCOS-A (47).
1. Role of insulin-resistant hyperinsulinism in PCOS pathogenesis
Insulin resistance is not only common in PCOS, it is often excessive for the degree of adiposity and is found in nonobese PCOS women (13, 24, 231–234). Insulin resistance in PCOS is characterized by reduced sensitivity and responsiveness to insulin-mediated glucose utilization primarily in skeletal muscle and adipose tissue, although the nature of these defects differs (24, 293). The insulin resistance of PCOS typically has a prominent intrinsic element, although in some cases, it may simply be acquired because of exogenous obesity. The mechanism characteristically involves constitutive, tissue specific, postbinding defects in receptor signaling that selectively affect metabolic pathways but not mitogenic or steroidogenic, actions (22, 232, 234, 294). Intracellular serine kinases account for phosphorylation of the insulin receptor and insulin receptor substrate-1 that decreases insulin activation of the phosphatidylinositol-3-kinase signaling pathway that activates glucose transport; serine kinase phosphorylation also activates mitogenic pathways mediated by ERK/MAPK (232). PCOS-related commonalities in insulin signaling in the ovary and other tissues have been noted. Constitutive activation of serine kinases that contribute to resistance to insulin's metabolic actions in skeletal muscle has been proposed to contribute to the increased 17,20-lyase activity of P450c17 in steroidogenic tissue, but commonalities in kinase signaling pathways involving both effects has been elusive (64). Postreceptor insulin signaling through the transcriptional coregulator protein Kruppel-like factor 15 in steroidogenic tissues and adipose tissue up-regulates testosterone formation via increased HSD17B5 gene expression and also stimulates adipogenesis in fat depots (107); this signaling appears to be intact in the face of insulin resistance (25).
A paradox is created by the tissue-selective resistance to the metabolic effects of insulin: the hyperinsulinemia compensatory for resistance to the glucose-metabolic effect of insulin elicits excess insulin action in some tissues in the presence of resistance to insulin action in others. This insulin-resistant hyperinsulinism (hyperinsulinism) is a major extraovarian factor in the steroidogenic dysregulation and DM-related comorbidities of PCOS (231, 232, 295). Mitogenic signaling and protein metabolism also remain sensitive to insulin, according to studies in cultured skin fibroblasts from PCOS patients (232, 296).
a. Insulin resistance and ovarian dysfunction.
Insulin has been shown to stimulate PCOS theca cell (297) and normal granulosa cell (105, 298) steroidogenesis through the insulin receptor rather than through the IGF-1 receptor. The initial report of antibody blockade of the insulin receptor showing an IGF-1 receptor mediated effect of insulin on PCOS thecal steroidogenesis (297) was of dubious significance because supraphysiologic doses of insulin were used in PCOS theca cells. However, selective knockout of theca cell insulin receptors in transgenic mice has been shown to ameliorate the hyperandrogenic anovulation of insulin resistance induced by an obesogenic diet; this indicates that hyperandrogenic anovulation was caused by insulin signaling through the theca cell insulin receptor (25). Although this mouse model differs from human PCOS in some regards (eg, LH levels are high in obese mice, normal in obese PCOS), this constitutes proof of principle that insulin signaling in the ovary is preserved in a state of resistance to the metabolic effects of insulin. Insulin signaling in the GnRH neuron similarly seems to be preserved (299).
Hyperinsulinemia augments LH stimulation of ovarian androgen production by up-regulating LH-binding sites and enhancing androgen production in response to LH at the level of cytochrome P450c17, as discussed earlier (Figure 1). Insulin and IGF-1 also stimulate expression of adrenal P450c17 and 3βHSD2 activities (176, 177), with lesser effects on other steroidogenic steps (300). Insulin potentially augments LH-stimulated androgen production through several other mechanisms. Insulin may also act via the IGF-1 receptor, atypical IGF-1 receptors, or a hybrid receptor that contains a combination of α- and β-subunits of both receptors (22). It has also been postulated that, by lowering levels of IGF-binding protein 1, insulin may raise the fraction of IGF-1 that is bioavailable (22).
All known forms of insulin resistance are associated with PCOS (22). PCOS is evident in patients with the extreme insulin resistance of hereditary insulin receptor mutations or lipodystrophy (22, 301). The PCOS of such extreme generalized insulin resistance may well be the result of extreme hyperinsulinemia promiscuously signaling through the IGF-1 receptor in the ovarian thecal cell. The IGF-1 excess state of acromegaly also is associated with PCOS (302).
The severe insulin resistance of pseudoacromegaly is characterized by a selective defect in postreceptor insulin signaling that only affects the glucose metabolic pathway mediated by phosphoinositide 3-kinase, whereas mitogenic signaling via the MAPK system remains intact (52, 303). In this syndrome, as in ordinary PCOS where the mechanisms are less clear (232), hyperinsulinemia appears to cause clinical symptoms (in this case, acromegaloid overgrowth) and to augment ovarian androgen production by signaling through one arm of the insulin signaling cascade via the insulin receptor itself.
More modest forms of insulin resistance also are associated with PCOS, including both major forms of DM. As an example, premenopausal women with type 2 DM have a PCOS prevalence of 30%–40% (304, 305). Type 1 DM also is associated with PCOS; this association is thought to be due to the supraphysiologic systemic doses of insulin required to control glycemia in the absence of the efficient hepatic glucose utilization that normally results from insulin secretion into the portal system (306). In both types of diabetes, insulin levels are not sufficiently high to act via the IGF-1 receptor
All treatments that cause a reduction in serum insulin levels, whether by weight loss, bariatric surgery, or by administration of somatostatin, metformin, or insulin-sensitizing thiazolidinediones, significantly improve ovulation and hyperandrogenemia in PCOS (1, 22, 210, 287, 288, 307, 308). The extent to which hyperinsulinemia is fundamental to PCOS pathophysiology has been a subject of debate. About half of PCOS patients experience improvement in the PCOS symptoms when they lose weight, and patients with the most severe ovarian dysfunction are those least likely to benefit symptomatically from weight loss (309). This is consistent with the concept that it is the patients with the atypical FOH of obesity, discussed earlier, that can expect the greatest symptomatic benefits from weight loss (47). Metformin, an antidiabetic drug, is well established to transiently reduce BMI, improve menstrual frequency, and lower testosterone levels by about 20%, which has a limited, if any, efficacy for hirsutism (41, 310, 311). Well-controlled studies indicate that metformin therapy offers no advantage over lifestyle modification in regards to weight, insulin levels, menstrual frequency, or ovulation (57, 211, 312, 313). The efficacy of thiazolidinedione therapy appears to be similar (313), but these agents are seldom used because they promote adipogenesis. Dietary supplementation with the insulin sensitizers myo- or D-chiro-inositol has shown anecdotal (314) or inconsistent effects on PCOS ovarian function (315).
Ibáñez et al have issued a series of positive reports of a prolonged randomized nonblinded trial comparing combined metformin-thiazolidenedione-antiandrogen therapy with estrogen-progestin therapy in hyperinsulinemic PCOS adolescents (316). They found these treatments to similarly lower serum total testosterone levels (but probably not free testosterone because marked differences in SHBG responses exist) with 94% normalization of menstrual irregularity while conferring markedly superior metabolic outcomes. Their experience has led them to equate PCOS with the hyperinsulinemia of insulin resistance (“Hyperinsulinaemic androgen excess is the most common cause of hirsutism, acne and menstrual irregularity in adolescent girls”; see Ref. 317). However, their Catalan PCOS population is both nonobese and hyperinsulinemic, which does not seem representative of the type of PCOS described in the United States and Europe. To illustrate this, we have applied their hyperinsulinemia criteria post hoc to a comparable subset of our study population that was studied by similar methodology (197). We compared the 14 nonobese PCOS adolescents (15.9 ± 1.4, SD, years old, BMI 24.7 ± 3.0), which represents 26% of our cohort of 53 adolescent PCOS, with our 19 nonobese control adolescents (15.0 ± 1.6 y, BMI 22.9 ± 2.4). Four of this PCOS subset, 1 of whom had asymptomatic diabetes by glucose tolerance test (GTT) criteria, had greater insulin resistance by homeostatic model assessment (HOMA-IR) (318) than the controls (P < .05). However, this PCOS subset was not significantly hyperinsulinemic, neither compared with our controls nor by the criteria of Ibáñez et al, which were met by 36%–37% of both our groups (316, 319).
Another group has reported that metformin and rosiglitazone, alone and in combination, similarly improve ovulation rates and testosterone levels in nonobese women with PCOS and normal indices of insulin sensitivity (320). These data are compatible with insulin-promoting hyperandrogenism independently of hyperinsulinemic insulin resistance and are consistent with the concept that levels of insulin in the normal range modulate the androgenic response to LH (88). This possibility is consistent with experimental data in which selective knockout of theca cell insulin receptors attenuated the androgenic response to hCG in both lean and obese transgenic mice (25).
On the other hand, caution must be exercised in attributing metformin actions to specific targeting of insulin resistance. Metformin's “insulin-sensitizing effect” is more accurately an insulin-lowering effect that appears to be a consequence of the reduction of hepatic glucose output and inhibition of gastrointestinal glucose absorption. Increasing glucose effectiveness occurs via effects on mitochondrial complex I adenosine triphosphate generation and activation of cAMP-activated protein kinase (321–326). These biochemical processes have the potential to exert a broad spectrum of effects, among which are known to be direct inhibition of steroidogenesis (327–329) and rarely lactic acidosis (330).
Liraglutide, a glucagon-like peptide receptor 1 agonist, in a 12-week trial was recently reported to be as efficacious as metformin in treating PCOS (331). Liraglutide improves glucose homeostasis by enhancing the early insulin response to meal ingestion and inhibiting glucagon secretion; it also inhibits appetite. Weight loss, HOMA-IR, glucose tolerance, and serum free testosterone were similar after liraglutide and metformin. Indeed, monotherapy outcomes in the most obese and insulin-resistant subset of their study group were significantly better than with metformin. Furthermore, the liraglutide effect was additive with that of metformin (332). The possibility that these effects were due in part to direct effects on the hypothalamic-pituitary-ovarian axis (333) cannot be excluded.
b. Insulin resistance and adipose tissue biology.
Insulin signaling is of major importance to the size and function of the adipose tissue depot: it stimulates adipogenesis (development of preadipocytes into adipocytes) and lipogenesis while inhibiting lipolysis (334, 335). The critical periods for establishment of the adipocyte population are fetal life and adolescence, after which lipid accumulation occurs primarily by cell hypertrophy (335). The overexpansion of PCOS fat depots suggests that insulin signaling in fat is intact in PCOS.
Studies on adipocytes from women with PCOS have revealed impaired insulin sensitivity (293, 336–338) and reduced glucose transport (24, 293, 336). In some studies this has been shown to be related to lower expression of the insulin-sensitive glucose transporter-4 (GLUT-4) (339, 340). Defects in tyrosine phosphorylation in the insulin signaling pathway have been identified, although not consistently (24, 340). Epigenetic changes such as microRNA overexpression have also been reported in association with reduced glucose transport in abdominal adipocytes of women with PCOS (340).
However, in contrast to skeletal muscle, in PCOS, the abdominal adipocyte cell lineage has no intrinsic defect in insulin-stimulated glucose transport, the first step of lipogenesis, and glycogen formation. Testosterone causes an acquired defect in lipogenesis by impairing sensitivity of glucose transport to insulin in both abdominal and visceral adipocytes (24, 341–344). Androgen excess may also contribute to adipocyte insulin resistance by suppressing production of the insulin-sensitizing adipokine adiponectin (345–347).
Androgens have been shown to inhibit differentiation of human and murine preadipocytes at a point before peroxisome-proliferator activated receptor expression (345, 348), although adipocyte stem cell commitment to abdominal preadipocytes is increased according to the prenatally androgenized monkey model of PCOS (349). These data suggest the potential for a local negative feedback regulatory loop for adipogenesis whereby androgen generated by sc adipocytes, as they differentiate in response to insulin (107), in turn impairs insulin-stimulated adipocyte lipogenesis (342, 348, 349). This loop remains to be documented, as does the extent to which these effects occur in visceral fat.
Although the relationship between hyperandrogenism and insulin resistance appears to primarily result from the effect of the compensatory insulin secretion on steroidogenesis, excess androgen may modestly reduce insulin sensitivity in vivo. Long-term testosterone-induced virilization of female-to-male transsexuals and female nonhuman primates fed a high-fat diet reduced insulin sensitivity of glucose uptake (343, 350, 351). On the other hand, androgen action via 5αRD type 1 promotes insulin sensitivity in males (352, 353). In PCOS, reducing androgen levels by antiandrogen or GnRHag treatment has improved insulin sensitivity in only a minority of studies (343).
The central fat accumulation in PCOS and the occasional presentation of pseudo-Cushing's syndrome as a manifestation of severely hyperinsulinemic PCOS (52) are consistent with insulin adipocyte actions being intact in PCOS and suggest a relation of hyperinsulinemia to glucocorticoid action (354, 355). Glucocorticoids augment insulin stimulation of adipogenesis and lipogenesis and redistribute fat to visceral stores (335, 354) while causing insulin resistance and attenuating secretion of insulin by the pancreatic β-cell (356). Local cortisol generation by adipocyte HSD11B1 is up-regulated in obesity and PCOS; a role for insulin in this process is unclear (357). On the other hand, in contrast to fat-specific insulin receptor knockout, which reduces fat stores by half (334), fat-specific glucocorticoid receptor deletion (358) does not affect baseline fat stores or fat distribution on either standard or high-fat diets, although it protects against cortisol-induced obesity. Thus, the mechanisms of glucocorticoid-insulin interactions in the regulation of adipogenesis and adipocyte function are unclear.
In summary, about half of PCOS women have an abnormal degree of insulin resistance, and this usually has a constitutive basis in which insulin resistance selectively affects tissue-specific metabolic, but not mitogenic or steroidogenic, insulin actions. The compensatory insulin-resistant hyperinsulinemia sensitizes ovarian theca cells to secrete androgen in response to LH and seems to have a similar effect on the adrenal androgenic response to ACTH. Parsimony suggests that hyperinsulinemia is unlikely to play a primary pathogenetic role in the pathogenesis of most PCOS, because it is an inconsistent and usually mild feature of the syndrome, particularly in the developmental (adolescent) phase of the syndrome. Furthermore, it does not account for the intrinsic theca cell dysfunction characteristic of classic PCOS. However, it may play a primary pathogenetic role in those functionally PCOS-A patients in whom it is a prominent clinical feature (52, 53). Regardless, it is an important aggravating factor in PCOS pathogenesis in about half of cases., and any treatment that lowers insulin levels improves hyperandrogenism. The available data are consistent with the concept that levels of insulin in the normal range modulate androgen production, most notably those in response to LH. Stimulation of adipogenesis and lipogenesis and inhibition of lipolysis by insulin excess also appear to contribute to the obesity of PCOS.
2. Role of obesity in PCOS pathogenesis
PCOS is the most common obesity-related endocrine syndrome in females. The prevalence of obesity in PCOS case series is influenced by ethnicity (359) and by referral patterns (360). Most evidence indicates that body fat content is excessive for BMI (118, 346, 361–364). One-third or more of normal-weight women with PCOS have abdominal obesity, whereas obese PCOS women accumulate fat globally. Obesity plays roles in PCOS via insulin resistance and generating testosterone from circulating androstenedione while suppressing gonadotropin production.
a. Obesity and insulin resistance.
As noted in the preceding section, insulin-resistant hyperinsulinemism likely is a major factor in the excessive adipogenesis and lipogenesis of PCOS, and obesity in turn seems to aggravate the hyperandrogenism of PCOS by exaggerating insulin resistance.
Regional differences in adipose tissue lipolysis have been reported in PCOS that contribute to the enlarged adipocyte size of the sc depot and promote later development of obesity in PCOS (365). Visceral fat contributes more to the insulin resistance of PCOS than does abdominal fat because of its enhanced lipolytic response to catecholamines, the major lipolytic stimulus in man (335, 366–369). The enhanced visceral fat lipolysis of PCOS appears to cause hepatic insulin resistance by the lipotoxicity of the excess free fatty acids released into the portal circulation (368, 369). The nature of the independent relationship of visceral fat to muscle insulin resistance is unclear, but a potential hepato-muscular endocrine role for fibroblast growth factor 19 (FGF19) has been suggested (370).
The relationship of androgens to the excess visceral fat lipolysis of PCOS has been unclear. This excessive lipolysis is not simply explicable by androgens. In human sc fat, but not visceral fat, androgens directly inhibit lipolysis in vitro (365, 368). In nonhuman primates, androgen administered in vivo appears to indirectly inhibit visceral fat lipolysis via suppression of luteal phase progesterone levels (351). A preliminary report in this primate model indicates that the coadministration of a high-fat/calorie diet with testosterone stimulated visceral fat lipolysis and increased insulin resistance while suppressing luteal phase progesterone (371). Thus, although the mechanism remains unclear, testosterone in conjunction with a high-calorie diet seems to promote visceral fat accumulation and insulin resistance in females by a combination of inhibiting lipolysis and promoting lipogenesis.
Proinflammatory cytokines arising from the mononuclear cells (MNCs) of adipose tissue are another mediator of the insulin resistance of PCOS. Abdominal adipocyte hypertrophy triggers an inflammatory response (335), which is aggravated in PCOS by hyperandrogenism priming MNCs to secrete proinflammatory cytokines in response to glucose and saturated fat ingestion (118, 372–374). Glucose ingestion by PCOS women stimulates a MNC prooxidant inflammatory response that promotes insulin resistance and atherogenesis and may be deleterious to pancreatic β-cell function.
Although inflammation from excess abdominal adipose tissue is an important element in the pathogenesis of insulin resistance, it seems insufficient to promote insulin resistance in the absence of increased body weight, as indicated by the following observations (118, 372). Normal-weight women with PCOS who lack excessive abdominal adiposity are insulin resistant in the presence of normal baseline serum levels of proinflammatory cytokines such as TNF-α, although their MNCs have an abnormal proinflammatory cytokine response to glucose ingestion. However, the third of normal-weight PCOS women with excess abdominal adiposity have elevated fasting TNF-α, and TNF-α levels of PCOS women rise as their BMI increases, in parallel with insulin resistance.
Obstructive sleep apnea (OSA) is characterized by sleep fragmentation and hypoxemia, and both obesity and male sex are risk factors for it. Women with PCOS have at least a 5-fold higher risk for OSA than similarly obese women without PCOS (375, 376). In some studies, insulin resistance has been found to be a stronger predictor of OSA than age, BMI, or circulating testosterone concentrations. Its effects seem to be additive with androgen; in one study, women with PCOS taking oral contraceptives may be less likely to have sleep disordered breathing, analogous to the lower likelihood of sleep disordered breathing among postmenopausal women on hormone replacement therapy (377). Conversely, the severity of OSA is a highly significant predictor of the fasting concentrations of glucose and insulin as well as the 2-hour glucose concentration during an oral GTT and HOMA-IR. Several well-controlled experimental studies in healthy human subjects involving sleep restriction as a model of OSA and assessments of glucose metabolism by iv GTT or euglycemic-hyperinsulinemic clamp have shown that sleep restriction cause a reduction of insulin sensitivity ranging from 18% to 24% without simultaneous increases in insulin levels, thus resulting in reduced glucose tolerance and an increased risk of diabetes (378). Thus, there appears to be a bidirectional cause-and-effect relationship between insulin resistance and OSA.
An important link between OSA and insulin resistance may be its relationship to overactivity of the SNS (379), which is related to PCOS independently of its relationship to adiposity (373). PCOS women with OSA have elevated norepinephrine levels over a 24-hour sleep-wake cycle compared with PCOS women without OSA (376). The higher muscle sympathetic nerve activity of PCOS compared with weight-matched controls likewise seems related to OSA (380–382). Improvement of OSA by treatment with continuous positive airway pressure has been shown to significantly reduce insulin resistance and norepinephrine levels (376).
SNS overactivity promotes inflammatory cytokine production and is associated with cardiovascular instability (122, 373). It has been proposed that the SNS may also contribute to the PCOM/PCOS phenotype via the sympathetic neurotropin NGF. Ovarian catecholaminergic nerve fibers and NGF production are excessive in women with PCOS and PCOM (121).
b. Obesity and gonadotropins.
Evidence is accumulating that obesity is associated with suppression of serum gonadotropin levels independently of insulin resistance, at least in part. Inverse associations between BMI and LH production have been reported in about half of studies of healthy eumenorrheic women with BMIs up to 40 kg/m2 (83, 383–385). A series of studies have shown consistently blunted follicular phase LH levels, suppressed LH pulse amplitude, and subtle FSH suppression in morbidly obese eumenorrheic women who are clinically normoandrogenic (226, 229, 386). These lowered gonadotropins had a subtle but important effect on follicular function: estrogen production, normal during the follicular phase of the menstrual cycle, was significantly depressed at the time of the midcycle ovulatory surge, after which corpus luteum insufficiency ensued. These studies further suggested that obesity-related inflammatory cytokine excess mediates suppression of pituitary gonadotropin release and that both are improved by transdermal estradiol administration to slightly boost estradiol levels (229).
In PCOS, BMI is likewise inversely related to baseline mean LH levels (83). The fall in LH levels with obesity is attributable to a fall in pulse amplitude (384, 385, 387). Although the early LH response to GnRHag is significantly elevated in PCOS, it is significantly lower in the obese than in nonobese subset (83, 385, 387). Baseline and stimulated LH are normal in most PCOS patients with BMI more than 40.
The blunting of LH pulsations in obese PCOS is at least in part due to accelerated metabolism of LH (388). Clearance of gonadotropins from the circulation is related to the sulfonation and sialylation patterns of the component isoforms: sulfonated isoforms are cleared more rapidly than sialylated ones (389). Although sialylation of LH molecules is increased in PCOS, the percent of sulfonated LH isoforms is proportional to BMI in PCOS (390). Thus, the increase in sulfonated LH isoforms in obese PCOS seems likely to account for their accelerated LH turnover. Because LH turnover seems to be the major determinant of LH bioavailability, this change would be expected to decrease LH in vivo bioactivity (391).
The mechanism by which obesity suppresses LH pulse amplitude is unclear. Because obesity causes insulin resistance and because PCOS patients are more insulin-resistant than BMI-matched controls (13), insulin resistance is a major candidate mediator. However, studies of the effects of insulin on gonadotropin production by diverse methods have not yielded a consistent picture (392–394). The possibility also exists that estrogen production in excess adipose tissue plays a role in suppressing women's LH pulse amplitude, as reported in men (395), and LH bioactivity (396), perhaps by affecting LH sialylation.
Obese women with hyperandrogenic anovulation without FOH or FAH constituted 8% of women in our PCOS series. We have proposed that simple obesity explains their mild hyperandrogenic anovulation (the PCOS-A of obesity) (47). As noted in the preceding paragraph, obesity itself is associated with suppressed ovulation and LH levels (83, 227, 228). Furthermore, obesity itself can account for excess peripheral formation of testosterone independently of PCOS (184, 397, 398). Adipocytes convert circulating androstenedione to testosterone via type 5 17βHSD, which is up-regulated by insulin (107, 184). The expression of this enzyme in sc fat correlates with BMI and falls with weight loss in simple obesity. The extent to which the hyperandrogenic anovulation of obese women is due to simple obesity remains to be determined in a prospective study. PCOS symptoms may improve with 5%–10% weight loss (399). However, 25%–50% weight loss may be required in the very obese, and correction of anovulation may require correction of both hyperandrogenism and metabolic syndrome (288). The hyperandrogenism of most morbidly obese PCOS patients can be corrected upon the substantial weight loss achieved by bariatric surgery (210, 288, 400). We postulate that it is those who lack an ovarian source of androgen excess that achieve resolution of hyperandrogenism with weight loss.
On the other hand, in most PCOS cases, whose hyperandrogenism is significantly greater, adipose tissue excess seems to contribute negligibly to testosterone excess, judging from androstenedione to testosterone ratios as an index of peripheral conversion of secreted androstenedione (401).
In summary, the major role of obesity in PCOS pathogenesis seems to be related to an increase in insulin resistance, thereby aggravating FOH. However, simple obesity may sometimes cause a mild PCOS picture that is atypical in that anovulation results from LH suppression rather than FOH.
C. Relationship of LH excess to PCOS
Increased LH relative to FSH was the first laboratory abnormality identified in classic PCOS. Mean LH levels correlate positively with LH pulse frequency (384, 385). Patients with PCOS have an increased LH pulse frequency, which is particularly striking in overnight studies, because these women typically lack the normal nocturnal slowing of pulse frequency that is the residual effect of ovulation in the previous cycle on the subsequent early follicular phase (384, 402). PCOS LH pulse amplitude is also increased, which is reflected in the size of the pulse induced promptly by GnRH or GnRHag administration (83). Elevated LH levels occur in about half of PCOS patients (197, 384).
Elevated LH has been thought to play a role in the pathogenesis of PCOS by increasing androgen production and secretion by ovarian theca cells (Figure 1). LH is necessary for the expression of gonadal steroidogenic enzymes and sex hormone secretion (Figure 1). Consequently, PCOS is LH dependent (hence, functional), and any treatment or disorder that suppresses LH levels suppresses ovarian steroidogenesis.
However, there are reasons to doubt that serum LH elevation is often the primary cause of FOH. For one, about half of PCOS subjects, particularly obese cases, with a documented ovarian source of hyperandrogenism were demonstrated to have normal baseline and GnRH-stimulated LH levels, as discussed in the previous section (83). For another, normal homologous desensitization of theca cells begins limiting the androgenic response to LH/hCG at submaximal stimulation, as discussed above. Nevertheless, LH is capable of overstimulating steroidogenesis in the presence of the escape from desensitization that occurs with hyperinsulinism. The hyperinsulinism common in PCOS would thus seem to contribute to the frequent ovarian steroidogenic hypersensitivity to LH stimulation (Figure 3). LH excess also seems to play an important role in the PCOS that follows congenital virilization (see section VI).
Recent research also supports the concept that the increase in LH is the result of abnormal sex steroid feedback rather than the cause of androgen excess (83). The elevated mean LH and, particularly, LH pulse frequency, of PCOS are less sensitive to negative feedback by combined estrogen-progestin administration than are those of controls (80, 403). In particular, higher concentrations of progesterone are required to suppress LH pulse frequency in the presence of luteal phase estradiol levels in PCOS (403). Furthermore, sensitivity to estrogen-progestin negative feedback is restored by antiandrogen treatment of PCOS (404). These data indicate that androgen excess interferes with the hypothalamic inhibitory feedback of female hormones, particularly progesterone. This conclusion is supported by a recent reexamination of the effects of testosterone infusion using deconvolution analysis to quantify the pulsatile properties of LH secretion in a 12-hour overnight study, in which 7 normal postmenarcheal and 7 PCOS adolescent girls were tested (405). In normals, when testosterone levels were increased 3-fold (to 120 ng/dL = 4.1nM), LH pulsatile secretion increased 50% (P < .05), whereas basal LH secretion over the 12-hour period did not change. When testosterone levels were raised 6-fold (to 245 ng/dL = 8.5nM), mean serum LH fell 22% (P < .05) due to a fall in basal LH secretion and a return of pulsatile secretion to normal. PCOS adolescents' LH levels were resistant to testosterone: only when testosterone was raised 4-fold to 300 ng/dL (10.4nM) were effects seen: testosterone significantly further increased LH pulsatile secretion and reduced basal LH secretion. Thus, induction of modest hyperandrogenemia appears to stimulate increased LH pulsatility in both normal and PCOS females. It is unclear why the dose-response relationship is shifted in PCOS; it may be due to the different antecedent progesterone or testosterone milieu, or other factors may be involved, for example, in the prenatally androgenized mouse model of PCOS AMH has been shown to directly activate GnRH neurons and increase LH secretion (406).
The androgen effect can be accounted for by increased gonadotrope responsiveness to GnRH: the testosterone elevation of PCOS is associated with enhanced GnRH-stimulated early LH secretion (17, 95, 197, 407). In PCOS, as in normal men, GnRHag administration elicits exaggerated early LH release (Figure 7), reflecting a large gonadotrope pool of readily releasable LH, consistent with this being an effect of the ambient testosterone level (95, 407). However, unlike normal men, and like normal women, GnRHag elicits a subsequent LH surge (Figure 7), which seems to reflect the capacity of female gonadotropes to form the large pool of newly synthesized LH that men are incapable of forming. Notably, GnRHag elicits a lower FSH surge (seemingly secondary to negative feedback by the excessive estrogen and inhibin production), which may contribute to ovulatory inefficiency.
Figure 7.
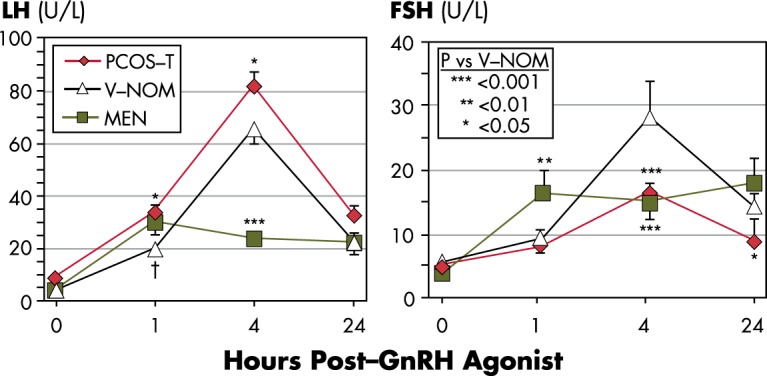
Effects of PCOS and sex on response to GnRHag. The early LH responses (1 h after 10-μg/kg GnRHag) of PCOS-T subjects resemble those of normal men (P < .05 vs V-NOM), whereas their surge responses (4 h after GnRHag) resemble those of normal women (P < .001 vs men). The FSH responses of PCOS-T subjects are significantly less than those of V-NOM from 4–24 hours and below those of normal men at 1 and 24 hours after GnRHag (P < .05). PCOS-T and V-NOM are previously reported age-matched groups (47), except NOM in postmenarcheal adolescents has been redefined as mean ovarian volume up to 12.0 cc, consistent with current consensus (30, 51). Adult male data shown for comparison were previously reported using a slightly different GnRHag sampling protocol (407); †, all head-to-head comparisons of normal male and female responses have shown a significant sex difference in the early releasable pool of LH (17, 407).
In summary, the data suggests that the response of LH to androgen is biphasic: mild testosterone excess is stimulatory, severe excess is inhibitory. LH is necessary for the expression of steroidogenic enzymes, and any treatment that suppresses LH levels improves ovarian hyperandrogenism. On the other hand, the phenomenon of homologous desensitization normally limits the role of LH in causing ovarian androgen excess. Thus, the data are compatible with LH excess in PCOS being the result rather than the cause of FOH. However, in the presence of escape from desensitization, such as is induced by hyperinsulinism, ovarian theca cells become more sensitive to LH, which aggravates ovarian hyperandrogenism. For these reasons, and because LH excess is an inconstant feature of PCOS, it is difficult to attribute a primary role to LH excess in the pathogenesis of most PCOS. However, LH excess seems to play a prominent role in the PCOS that follows congenital virilization.
D. Modulation of androgen action in PCOS
Although testosterone appears to be the main circulating androgen (38), the possibility has been raised that atypical adrenal androgens such as 11-ketotestosterone, which has 25% of testosterone's potency, might contribute to androgen action (408). Although serum bioactivity can essentially be accounted for by testosterone in normal women (409), the extent to which such atypical androgens contribute in hyperandrogenic remains to be systematically determined.
The formation of DHT from testosterone by 5αRD is a major determinant of androgen action. Evidence of increased global peripheral 5αRD activity in PCOS has been consistently obtained from evaluation of urinary androgen and cortisol metabolites (250). Indexes of increased 5αRD activity were increased in both nonobese and obese PCOS and significantly greater in the obese than nonobese subjects. The cause is unknown. The possibility of differential expression of “backdoor pathway” enzymes for the formation of DHT, such as 5αRD1/2, α-ketoreductase type 1C2, and 17β-HSD6 (62), in PCOS ovarian theca cells and adrenal zona reticularis cells has been raised by a preliminary report (410). It is possible that the increased 5αRD activity is a consequence of hyperandrogenism because 5αRD activity is up-regulated by androgen action (191, 411). Insulin appears to up-regulate 5αRD activity (250); this effect may be exerted on the type 1 isozyme (412), which in turn up-regulates insulin sensitivity (352, 353). Genetics may play a role, because 5αRD gene variants are associated with PCOS prevalence (413). The increased 5αRD activity has been postulated to play a role in adrenal hyperandrogenism (250). It could also potentiate androgen actions in other organs or tissues, such as the pilosebaceous unit and granulosa cells (77, 412, 413). Serum allopregnanolone, a 5α-reduced progesterone metabolite that is a γ-amino butyric acid receptor agonist, has been reported to be significantly higher in PCOS than in BMI-matched controls (25 vs 17 ng/dL; 0.8 vs 0.5nM) and proposed to play a role in appetite dysregulation (414).
Androgen receptor alternate splice variant heterozygosity has recently been reported in the luteinized granulosa cells of 62% of Han Chinese with PCOS undergoing in vitro fertilization; it is absent in controls (415). Either an insertion or a deletion isoform were coexpressed with wild-type receptor and accounted for the androgen receptor overexpression in these PCOS cells. They reduced nuclear translocation of wild-type receptor, which has an overall negative effect on androgen action. In isolation, neither transduced the normal up-regulation of aromatase by dihydrotestosterone, whereas the insertion variant transduced up-regulation of CYP17A1 by dihydrotestosterone. The expression pattern was tissue specific, not being found in peripheral lymphocytes, where wild-type receptor was found to be up-regulated. The variants were associated with more severe hyperandrogenemia, which was attributed to deficiency of aromatase activity secondary to deficient up-regulation by androgen receptor signaling. They were also associated with lowered expression of genes related to folliculogenesis and ovulation. The mechanism by which these gene variants arise appears to be locally regulated and in part may be epigenetically determined. It has been postulated that these changes are an adaptation of the PCOS ovary to the abnormally hyperandrogenic environment rather than a contributor to the hyperandrogenism (416).
The length of androgen receptor CAG trinucleotide repeats is known to be inversely related to the efficacy of androgen receptor activity. Many studies have examined the associations of polymorphic CAG repeats in the androgen receptor gene, or of X-inactivation favoring short repeats, with PCOS risk, but with inconsistent results (417). A 2013 metaanalysis demonstrated no evident association between the CAG length variations in the AR gene and PCOS risk, whereas the CAG length appeared to be positively associated with testosterone levels in PCOS patients (418).
In summary, although such clinical observations as hirsutism in the apparent absence of hyperandrogenemia and anovulation in association with isolated mild FAH suggest that androgen action may be excessive at the target tissue level in some individuals, information is scarce about the mechanism of such putative effects. Improved knowledge about the role of atypical androgens and postreceptor mechanisms of androgen action will be necessary to understand the role of tissue responsiveness to androgen in PCOS pathogenesis.
E. Summary: Unified model of PCOS pathophysiology
Our working model of PCOS pathogenesis is shown in Figure 8. Approximately 90% of PCOS have an abnormal SDAST or GnRHag test (the most specific of current diagnostic tools for testing ovarian androgenic function), whereas only about half of these have abnormally elevated insulin and/or LH. Thus, the common denominator in PCOS appears to be FOH. FOH typically has a steroidogenic abnormality suggestive of the constitutive biochemical dysfunction that is characteristic of classic PCOS theca cells. (A similar adrenal dysfunction seems to account for the commonly associated FAH.) FOH can account for all the clinical features that characterize PCOS: hirsutism, anovulation, and polycystic ovaries or, in severe cases, hyperthecosis. In about half of cases, tissue-specific resistance to the metabolic effects of insulin causes compensatory hyperinsulinemia. This aggravates anovulation and the development of PCOM by up-regulating thecal androgen production in response to LH and synergizing with androgen to cause premature luteinization of ovarian follicles; it also stimulates adipogenesis. Then, 2 vicious cycles of feed-forward effects occur. The modest hyperandrogenemia causes secondary LH elevation by interfering with female hormone negative feedback; in the presence of hyperinsulinemia, this LH excess aggravates the ovarian dysfunction. Hyperinsulinemia also promotes adiposity, which in turn aggravates the insulin-resistant state.
Figure 8.

Unified minimal model of PCOS pathophysiology. A, Ovarian hyperandrogenism is nearly universal in PCOS and can account for all the cardinal clinical features of the syndrome: hyperandrogenemia, oligo-anovulation, and polycystic ovaries (1). Pituitary LH secretion is necessary to sustain the ovarian androgen excess but is not sufficient to cause it. B, About half of patients with FOH have insulin-resistant hyperinsulinism (2). Insulin-resistant hyperinsulinism acts on theca cells to aggravate hyperandrogenism, synergizes with androgen to prematurely luteinize granulosa cells, and stimulates adipogenesis. The increased hyperandrogenemia provokes LH excess (3), which then acts on both theca and luteinized granulosa cells to worsen hyperandrogenism. LH also stimulates luteinized granulosa cells to secrete estradiol (4), which suppresses FSH secretion. These hyperinsulinism-initiated changes in granulosa cell function further exacerbate PCOM and further hinder ovulation. Obesity increases insulin resistance, and the resultant increased hyperinsulinism further aggravates hyperandrogenism. Heaviness of lines and fonts represents severity. Both FOH and insulin resistance typically have an intrinsic basis. This model does not exclude the possibility that the unknown intrinsic ovarian defects that underpin the ovarian steroidogenic dysfunction also involve granulosa cell folliculogenesis as well. The figure also does not depict other associated defects, such as the FAH that often accompanies the ovarian hyperandrogenism and the contribution of excess adiposity to peripheral androgen production and gonadotropin suppression.
In most PCOS, the cause of ovarian hyperandrogenism seems to be intrinsic, and there is evidence for a constitutive basis for much of the insulin resistance as well. In the absence of intrinsic ovarian dysfunction, modest hyperandrogenemia of extraovarian origin (adrenal or peripheral sources) or severe insulin resistance are unusual causes of hyperandrogenic anovulation and polycystic ovaries.
VI. Etiology of PCOS: A Complex Trait
A number of hereditary and environmental factors contribute to ovarian hyperandrogenism and/or insulin resistance. Polycystic ovaries, androgen levels, and insulin resistance have hereditary components. Environmental factors may be congenital or acquired and include intrauterine factors such as androgen exposure and prenatal nutrition, whereas acquired obesity is a major postnatal factor influencing the phenotype. The complex interactions generally mimic an autosomal dominant trait with variable penetrance: the disorder is correlated in identical twins (r = 0.7) (419); about half of sisters are hyperandrogenic, and half of these also have oligo-amenorrhea and thus PCOS (420, 421); and polycystic ovaries appear to be inherited as an autosomal dominant trait (421, 422). Although estimates vary widely, 3%–35% of mothers of women with PCOS also have PCOS, as do about 25% of sisters (423, 424), and metabolic syndrome prevalence is high in parents and siblings (280, 425, 426). The syndrome's phenotypic diversity is affected by ethnic diversity (427–429).
A. Heritable traits and genetic linkages
Familial cases of PCOS that appeared to be inherited as an autosomal dominant trait with variable penetrance were recognized long ago (430). Twin studies suggest that there is a strong contribution of familial factors to PCOS pathogenesis (419, 431). In a Dutch twin-family study, the correlation between monozygotic twins for PCOS was 0.71, and for dizygotic twin or nontwin sister pairs, the correlation was 0.38. Heritable traits that have been identified as PCOS risk factors are maternal PCOS, PCOM, hyperandrogenemia, and metabolic syndrome.
1. Maternal PCOS
Maternal PCOS is a risk factor for PCOS in daughters. One set of studies compared the singleton daughters, about two-thirds of whom were pubertal, of 99 Chilean women with PCOS (defined by NIH criteria) with those of 88 control women (432, 433). A study in subsets of these groups showed that in infancy and early childhood the daughters of women with PCOS had higher serum AMH (434); however, these findings were not replicated in a Nordic PCOS population in which an unknown proportion had nonhyperandrogenic or ovulatory PCOS phenotypes (435). From midchildhood onwards, the daughters of the Chilean women with PCOS had larger ovaries and higher insulin responses to an oral GTT than the daughters of women without PCOS (ethnic or environmental influences may play a role because evidence of insulin resistance did not emerge until late puberty in an American study of PCOS daughters) (436). Peripubertally, higher DHEAS levels emerged. In late puberty, higher basal testosterone and higher 17OHP responses to GnRHag testing emerged during the late stages of puberty. Half of the daughters of women with PCOS who were postmenarcheal had higher testosterone levels than any control daughters, which is consistent with the familial distribution of hyperandrogenemia found by Legro et al (420).
2. Polycystic ovarian morphology
Two studies suggest that the PCOM of PCOS is inherited in an autosomal dominant fashion (422, 437). Most PCOS adolescents with PCOM have either a mother with a polycystic ovary, which is usually asymptomatic, or a father with metabolic syndrome (280).
A presumed second factor, in concert with the intrinsic genetic susceptibility defect in the ovary, would appear necessary to result in the clinical phenotype of PCOS (hyperandrogenemic anovulation) in the affected daughter. This was suggested by the results of a study of 214 sisters of 125 PCOS probands, defined by NIH criteria (80%) or by broader Rotterdam criteria (20%), which showed that 70.5% had a polycystic ovary (421). Of the sisters with a PCOM, 25% had clinically hyperandrogenic anovulation, 42% had hirsutism or oligomenorrhea, and 33% were asymptomatic. The group of sisters with a polycystic ovary resembled their proband sisters in both androgenic and glucose-metabolic traits, indicating significant heritability and cosegregation of these traits.
Genetic studies have not taken into account the heterogeneity of steroidogenic function of the polycystic ovary (30). On one hand, about half of asymptomatic young women with PCOM have no biochemical evidence of ovarian androgenic dysfunction and thus represent a variation of normal. On the other hand, half of asymptomatic women with PCOM have biochemical evidence of ovarian androgenic dysfunction (Figure 6), and nearly half of these have biochemical hyperandrogenemia and thus seem to have ovulatory PCOS although eumenorrheic and normal to all appearances, as discussed above. We have postulated that such women are PCOS carriers and may be at risk of PCOS with excessive weight gain, although the latter outcome seems to be uncommon (219). However, oligomenorrheic women with PCOM and functional hypothalamic amenorrhea and a very elevated AMH level are at high risk for hyperandrogenic PCOS when hypothalamic function normalizes with weight gain (438, 439).
3. Hyperandrogenemia
Approximately one-half of sisters of PCOS probands have an elevated serum testosterone level (420). However, only one-half of these sisters with excess androgen have menstrual irregularity, and the other half are asymptomatic. This suggests that these asymptomatic patients with excess androgen only become symptomatic in the presence of another precipitating factor(s).
4. Metabolic syndrome and DM
Metabolic syndrome and its core pathogenetic constituents, insulin resistance and obesity, have heritable components (421, 440, 441). Perhaps less appreciated is the evidence that defective insulin secretion is also highly heritable in PCOS (289, 290, 292) and is closely associated with PCOS-T (47).
Parental factors related to metabolic syndrome (ie, insulin resistance and/or obesity) are strongly associated with the pathogenesis of PCOS. There is a high prevalence of these features in first-degree relatives of PCOS patients (421, 425, 426, 442). In our series of 35 families in which an adolescent girl had PCOS, 70% of the probands had a parent with metabolic syndrome; using National Cholesterol Education Program Adult Treatment Panel III (ATP-III) criteria for defining metabolic syndrome, 53% of fathers and 34% of mothers were affected (280). Using 2004 criteria for metabolic syndrome (which include assessment of glucose tolerance), 79% of fathers and 37% of mothers were affected. Excess adiposity was found in most parents; 94% of fathers and 66% of mothers were obese or overweight. Abnormal glucose tolerance was present in 84% of fathers and 49% of mothers: of these, half were diabetic (half occult) and half had IGT (“prediabetes”). Notably, this case series showed complete concordance between paternal metabolic syndrome and a polycystic ovary in affected daughters but no relationship to metabolic syndrome in these daughters, which suggested a fundamental relationship of paternal metabolic syndrome to the PCOM of PCOS. In a study that defined metabolic syndrome by ATP-III criteria and did not include GTTs, fathers were confirmed to have an increased prevalence of excess adiposity (about 85%) and metabolic syndrome (42%) (426). In another study of fasting blood sugar, fathers were also found to have a significantly higher prevalence of fasting dysglycemia than mothers; however, only maternal fasting dysglycemia was demonstrably heritable, which suggests genetic or epigenetic parent-of-origin effects (443).
5. Gene variants
Many attempts are being made to identify specific genes that underlie the intrinsic cause of PCOS. A wide variety of gene variants with linkage and/or association with PCOS have been identified by candidate gene studies (1, 417, 444, 445). Polymorphisms, linkages, and differential expression have been reported for genes encoding steroidogenic enzymes, SHBG, the androgen receptor, and gonadotropin receptors; genetic loci associated with insulin sensitivity and susceptibility to obesity, and congenital adrenal hyperplasia (418, 446–450). Dysregulation of genes involved in cell growth is suggested by the reports of up-regulation of proto-oncogenic genes in endometrium (451) and of telomere shortening in leukocytes (452).
More recently, there has been widespread interest in the identification of linked genes through genome-wide association studies (GWAS), commencing with a series of studies conducted in large populations of ethnic Han Chinese in 2011 (453). The major findings in this population have been generally replicated in most other populations studied (427, 454–457). Linkages for polymorphisms in the fibrillin 3, proopiomelanocortin, and diverse signaling pathway genes have been robust in many populations (115, 453, 458). How the various linkages are functionally related to PCOS is often unclear. Variations in fibrillin 3 have been proposed to dysregulate TGF-β signaling (115) and account for ovarian stroma hyperplasia (117). The most recent and largest of the GWAS as of this writing is the report from a multicenter study of 984 PCOS cases and 2964 population controls followed by replication in 1799 PCOS cases and 1231 phenotyped reproductively normal control women (457). This was followed by a metaanalysis of the top 24 associations detected in the first and second case-control groups. Three loci reached genome-wide significance in the case-control metaanalysis of all 3 strata; 2 novel loci, chr 8p32.1 in the region of the GATA4 transcription factor and the NEIL2 endonuclease-encoding gene, and chr 11p14.1 in the region encoding the FSH beta subunit (FSHB), and one previously found in Chinese PCOS, chr 9q22.32 in the region of c9orf3/FANCC which encodes an aminopeptidase. The same chr 11p14.1 SNP, rs11031006, in the region of the FSHB gene also reached genome-wide significance in the metaanalysis of the quantitative LH levels. These findings implicate gonadotropin levels in the pathogenesis of PCOS. The GWAS approach has also been applied to ovary-specific methylation, and the findings support a role for epigenetic gene modifications in the pathogenesis of PCOS (459).
Among the most striking findings from GWAS to date has been the recent discovery of DENND1A (MIM *613633) as a highly significant intronic locus linked to PCOS in many populations (108). Even though the odds ratio for this association was modest (odds ratio < 2.0), a previously unsuspected protein, DENND1A, was found to play an unanticipated role in ovarian androgen formation. DENND1A.V2 isoform expression was then demonstrated to be increased in PCOS theca cells, to up-regulate cytochrome P450c17 and side chain cleavage activities, and its mRNA to be excreted in excess in urine of PCOS women, as discussed in the section V.A. Details of DENND1A pathophysiology remain to be worked out. Likewise, the genetic mechanism(s) accounting for DENND1A.V2 overexpression have yet to be determined.
There are many pieces to the puzzle of how the components of PCOS are interrelated, and the puzzle remains to be solved. It seems increasingly likely that the mechanistic pathways underlying the diverse manifestations of PCOS may involve transcription factors or other intrinsic cellular processes common to the many tissues involved. The variety of pathways involved and lack of a common thread attests to the multifactorial nature and heterogeneity of the syndrome and suggests that the syndrome may have evolved heterogeneously as diverse means of preserving anabolism and reproductive capacity in times of nutritional deprivation (460).
B. Intrauterine environment
It is becoming increasingly apparent that environmental insults during development induce persistent changes in the epigenome that lead to altered gene expression and adult disease (461, 462). Congenital virilization and intrauterine nutrition have been incriminated as risk factors for PCOS.
1. Congenital virilization
PCOS is common in females with congenital virilizing disorders, of which congenital adrenal hyperplasia is the most frequent, but not exclusive, cause (201, 463). Congenital androgenization has now been well established to lead to PCOS features in several experimental animal models (84, 431, 464, 465). Studies in the rhesus monkey exposed to androgen excess early in gestation have been particularly informative, showing characteristic PCOS features. These animals have ovarian and adrenal hyperandrogenism, oligomenorrhea, polyfollicular ovaries, and elevated LH levels associated with resistance to negative feedback inhibition of LH release by progesterone. They also have abdominal obesity, insulin resistance, IGT, and dyslipidemia. Prenatally androgenization of sheep causes increased antral follicle AMH expression similar to that in PCOS (466).
Mechanistic studies in the rat indicate that prenatal androgen programs for pubertal LH elevation and absence of the capacity to mount an LH surge via suppression of estrogen induction of hypothalamic progesterone receptors (84, 467). Studies in sheep indicate that prenatal virilization causes tissue-specific differential changes in genes that determine insulin sensitivity, with liver and muscle being insulin resistant and adipose tissue being insulin sensitive (468).
The PCOS secondary to congenital virilization indicates the potential for in utero epigenetic programming of postnatal development (462). However, the relevance of this to ordinary PCOS is unclear in view of what is known about structure-function relationships of normal human fetal ovary development.
By the beginning of the second trimester, human fetal testes have differentiated, and their secretions have begun to differentiate male from female genitalia. At this time, the normal human fetal ovary, although histologically undifferentiated, has the capacity to form and respond to androgen and estrogen, and this increases thereafter (469–473). Histologic studies indicate that primordial follicles commence organizing and growing at about 16 weeks of gestation and the oocyte population peaks at 20 weeks, at about the time that primary follicles begin appearing (472); meanwhile, follicle growth is active, and preantral follicles become developed at 24–26 weeks (471, 474). Antral follicles are first detected near term both histologically (471, 474) and ultrasonographically: pelvic ultrasonography has detected antral follicles on the seventh postnatal day in no premature infants less than 34 weeks of gestation, 5% of preterm infants (34- to 37-wk gestation), and 30% of term infants (P ≤ .001) (475). Corresponding to antral follicle status, premature infants have high gonadotropin levels, of the magnitude seen in ovarian insufficiency, yet do not begin estrogen and AMH production until they approach term gestational age (475, 476). This timing of ovarian acquisition of estrogen responsiveness to gonadotropins resembles that in the nonhuman primate (477) and corresponds to the late temporal appearance of FSH receptors in the primate ovary, where the developmental increase in FSH receptors reflects the estrogen-dependent increase in folliculogenesis (265). Thus, despite having the capacity for estrogen formation from the time of ovarian differentiation, ovarian estrogen production appears to be virtually unresponsive to gonadotropins until follicles become capable of responding to FSH with antral follicle growth near term gestational age.
Androgen production by the primate fetal ovary seems to parallel this quiescent fetal ovarian pattern of estrogen production and contrasts with that of testosterone in the male fetus (472, 477, 478); fetal male serum testosterone peaks above female levels at 16–20 weeks of gestation as fetal pituitary LH secretion peaks and CG levels are waning (471, 479, 480). Therefore, it would be surprising if the PCOS ovary were found to be hyperfunctioning in utero. Nevertheless, some have postulated that to be the case (462).
The most direct way of demonstrating fetal hyperandrogenism, if it exists, would seem to be measurement of amniotic fluid testosterone levels at 12–24 weeks. (A methodological note: direct assays of androgens are often problematic [45]; the steroid output of the fetal adrenal-placental unit generates a steroid milieu that is unique to pregnancy [481]; this pregnancy-related steroid pattern interferes with direct testosterone assays that are accurate for children and adults, so high-specificity assays after preliminary chromatography are necessary for the accurate assay of pregnancy-related samples [482]; whether any specific postchromatographic RIA or mass spectrometry method is superior to another in this setting remains to be directly tested.) In normal pregnancies a distinct sex difference in amniotic fluid testosterone has been shown by postchromatographic RIA and mass spectrometry (483–485). This contrasts with the absence of fetal sex-dependent differences in normal maternal serum androgen levels (484, 486) or umbilical cord androgen levels among normal and PCOS offspring (486, 487).
In view of all the reciprocal paracrine interactions that seem necessary for normal postnatal theca cell function, it seems unlikely that PCOS theca cell dysfunction could emerge and initiate developmental programming during the window of time in midgestation (16–24 wk) after sexual differentiation is complete and before fetal gonadal endocrine production is suppressed by high adrenoplacental unit estrogen production and the coincident fall in fetal pituitary and CG levels (471, 479, 480). Furthermore, it is unlikely that endogenous ovarian androgen overproduction could exceed the high capacity of placental aromatase to protect the fetus from androgen excess (488, 489), as is the case in different-sex twin pregnancies. Nevertheless, it has been proposed that the midgestational uterine environment causes excessive fetal androgenization (462). This notion has received some support that is of questionable significance. Term PCOS placentas have changes that favor increased androgen production: they possess significantly more (40%) 3βHSD1 activity and less (30%) aromatase activity than controls (489); the relationship of this biochemical change to the significant histoanatomic evidence of hypoxic change of such placentas is unclear (490). However, no clear picture of placental aromatase deficiency has emerged from measurement of umbilical cord blood sex steroid concentrations: estriol has been reported to be higher (489), and estradiol, estrone, and androstenedione have variably been reported to be lower (486, 487). PCOS women bearing a girl had second trimester amniotic fluid testosterone concentrations that were intermediate between those of controls bearing a girl (P = .019) and controls bearing a boy (P = .10) (491); however, these data have a substantial element of nonspecificity, because they were obtained by a direct RIA that yielded absolute values averaging 80%–500% higher for amniotic fluid obtained from female-bearing women than reported for the postchromatographic assays noted above (483–485). PCOS women bearing a girl had significantly higher serum testosterone and androstenedione levels by postchromatographic mass spectrometry at 20 weeks of gestation and at delivery than did comparable controls, with a similar trend seen in PCOS women bearing a boy (486). This is both intriguing and unexpected; if confirmed, this would seem more likely to arise from maternal ovaries than from PCOS fetal ovaries secreting more androgen into the maternal circulation than normal fetal testes.
Because of the difficulty of safely measuring indexes of fetal hyperandrogenism, postnatal markers of in utero androgen action have been explored. Congenital androgen excess of a degree sufficient to affect genital differentiation is known to be significantly disruptive to the development of normal female gender satisfaction (492, 493). Reversed sexual orientation is unusual in PCOS (494), and there is no evidence of excess gender dissatisfaction aside from that resulting from body image perception. The ratio of second and fourth digit lengths has been evaluated because it is slightly but significantly lower in men than women and masculinized (lengthened) in prenatally androgenized vs control primates (462, 495); however, in 3/4 reports, this ratio was not supportive of a fetal masculinizing effect in PCOS (462). This ratio in normal females, but not males, at birth was found to correlate with amniotic fluid testosterone (484). An alternative approach has been to search for biochemical markers of prenatal programming. A preliminary report has appeared of infant daughters of women with PCOS having urinary steroid metabolite evidence of increased global 5αRD activity compared with control infants, a finding compatible with congenital androgen programming (496).
2. Disturbed fetal nutrition
There is good evidence that fetal undernutrition programs for metabolic syndrome and related cardiovascular disease in adulthood (497, 498). It has been proposed that low birth weight is likewise a marker for a fetal origin of PCOS (499); this proposal is more controversial.
Several studies support an association between inadequate fetal nutrition and subsequent development of PCOS. First, a retrospective study evaluating Catalan girls with premature pubarche found that it was associated with intrauterine growth restriction and postmenarcheal PCOS and insulin resistance (499): this led to the hypothesis that low birth weight was a marker for a fetal origin of PCOS (and an even more sensitive marker of premature adrenarche). Probes of birth records have supported the concept that low birth weight is associated with PCOS. An Italian neonatal registry-based study showed low birth weight, irrespective of gestational age, to be associated with PCOS features and insulin resistance (500). A similar Brazilian birth cohort follow-up (43 small for gestational age, 122 appropriate for gestational age) examined at an average age of 29 years showed small for gestational age to confer a 2.4-fold risk for PCOS but none for hyperinsulinemia (501). An Australian follow-up of a birth cohort of 2199 women showed that the weight-for-length at birth was significantly inversely associated with increased risk for PCOS (502). A converse association between birth weight and PCOS was suggested by 2 studies of adults in which PCOM and other PCOS features were associated with a relatively high birth weight (503, 504).
In contrast to these studies, larger longitudinal or retrospective studies in Northern Europe demonstrated no relationship between birth weight and PCOS symptoms (505, 506). Similarly, birth weight by recall was similar to that of controls and/or population data among 854 United States and 170 Italian adults and adolescents with PCOS (507, 508), only 9%–10% recalled low birth weight.
C. Postnatal environment
Postnatal environmental risk factors for PCOS can be viewed as precipitating latent, congenitally programmed susceptibility traits to become manifest.
1. Insulin resistance
All extreme insulin-resistant states are associated with PCOS. The compensatory hyperinsulinemia of insulin resistance is closely associated with the anovulation of PCOS. Ovulatory patients with PCOS are less insulin-resistant than anovulatory patients with PCOS (509). All treatments that lower insulin levels improve ovarian dysfunction and ovulation (510–512).
Ordinary obesity is the most common cause of insulin resistance, and we are in the midst of a worldwide obesity epidemic (513), which is one reason why PCOS may be recognized more often than in decades past. Weight loss sufficient to improve indexes of insulin sensitivity in PCOS improves menstrual cyclicity and ovulation (210, 288, 308, 514–516).
Two syndromes of intractable obesity in childhood, pseudo-Cushing's syndrome and pseudoacromegaly, herald PCOS in adolescence (52, 303). These syndromes are characterized by moderately severe insulin resistance.
It is possible that the transient physiologic insulin resistance of puberty may contribute to physiologic anovulation or the development of PCOS during adolescence. Insulin resistance and compensatory hyperinsulinemia normally peak in midpuberty (517–520). The waning insulin resistance as puberty progresses generally parallels improvement in menstrual regularity, but the nature of this association remains to be investigated.
2. Hyperandrogenism
Postnatal androgen excess causes ovarian hyperandrogenism in some animal models (461), as does androgen excess in girls with poorly controlled congenital adrenal virilizing disorders (201). In some species, adipogenesis is also stimulated and glucose metabolism deteriorates (461, 521).
3. Other precipitants and risk factors
Excessive LH stimulation at puberty may play a role in the pathogenesis of PCOS (“hyperpuberty”) (522). The best support for this theory is in the setting of congenital virilization, in which there is prenatal programming for LH excess at puberty (see section VI.B.1).
Most uncontrolled follow-up studies of idiopathic central precocious puberty have not been consistent with an increased incidence of PCOS (523). However, a controlled study of girls with early puberty reported a higher prevalence of PCOS in 25 adolescents who chose treatment with GnRHag, as compared with a similar group of 55 girls who declined treatment (36% vs 15%) (524). The treated group had significant elevation of androstenedione in association with menstrual dysfunction or polycystic ovaries. In this study, PCOS was defined using AE-PCOS criteria (phenotypes 1 through 3), which are broader and less specific than the modified NIH criteria now accepted for the diagnosis of adolescent PCOS (phenotypes 1 and 2) (Table 1). Confirmation in a randomized trial using highly specific androgen assays and more stringent PCOS diagnostic criteria is needed before considering GnRHag treatment to be a risk factor for PCOS.
Premature adrenarche may pose a moderately increased risk for PCOS/FOH (431, 525). These individuals overall appear to carry approximately a 2-fold (15%–20% risk of developing PCOS), although the risk may vary with ethnic group. Some studies suggest that this risk is related to low birth weight (499), others do not (431). The association of premature adenarche with the subsequent development of PCOS may indicate that premature adrenarche is sometimes an early manifestation of steroidogenic dysregulation (431).
Adolescents with epilepsy appear particularly susceptible to develop PCOS when treated with valproic acid, an antiepileptic drug that augments the transcription of P450c17 and other steroidogenic enzymes (526, 527).
Endocrine disruptors have been suspected of aggravating PCOS (445, 528, 529). Serum bisphenol A levels are elevated in PCOS, giving reason to believe that it may play a role in the pathogenesis of the syndrome.
D. Implications for evolutionary origin of PCOS
PCOS presents an evolutionary paradox; it is very common across populations, although it is an infertility disorder. The classical theories postulate that PCOS developed through natural selection as a spectrum of independent, diverse genetic adaptations that evolved to preserve anabolism and reproductive capacity via increased androgen and insulin production in ancient times of nutritional deprivation, although in current times of plenty this phenotype is disadvantageous (460, 530, 531). An alternate mechanism may be “intralocus sexual conflict,” that is, some PCOS-related genotypes may not disappear because they improve the reproductive fitness of the human male (eg, by promoting male hyperandrogenism) and thus compensate for reduced female fertility (531).
E. Summary
PCOS seems to arise as a complex trait that results from the interaction of diverse genetic and environmental factors that usually first becomes manifest at puberty. At its simplest, this is a “2-hit” hypothesis that can be thought of because of a congenitally programmed predisposition (“first hit”) that becomes manifest upon exposure to a provocative environmental factor (“second hit”) (Table 5). There is evidence for the congenital hit being genetic or acquired, with diverse causes of each. The provocative factor is postnatal and usually seems to be insulin-resistant hyperinsulinemism, which may have been programmed congenitally either on a hereditary or acquired basis or acquired postnatally due to simple (exogenous) obesity.
Table 5.
PCOS Etiology as a Complex Trait Involving 2 Hits
|
VII. Conclusions and Implications for Future Research
This review indicates that research to date is consistent with the concept that most PCOS is due to FOH that arises from dysregulation of steroidogenesis that sensitizes ovarian steroidogenesis to LH. In our experience, FOH is the common denominator in approximately 90% of the hyperandrogenic anovulation cases. Typical FOH accounts for two-thirds of FOH: it is characterized by 17OHP hyperresponsiveness to gonadotropin stimulation. This secretory abnormality resembles the biochemical dysfunction that is a constitutive characteristic of theca cells of classic PCOS and seems to indicate an intrinsic abnormality in the normal mechanism for down-regulation of the steroidogenic response to LH. Similar dysregulation of adrenocortical steroidogenesis seems to account for the associated FAH found in about one-quarter of cases. The pathophysiologic and biochemical basis of functionally atypical FOH is unclear. Typical FOH has more severe hyperandrogenism and a higher prevalence of PCOM than atypical FOH. It is also associated with a significantly higher prevalence of glucose intolerance in the presence of similarly increased insulin resistance, which suggests a relationship of the intrinsic ovarian abnormality to pancreatic β-cell failure.
Insulin-resistant hyperinsulinism is often an important aggravating factor in PCOS pathogenesis. About half of PCOS women have an abnormal degree of insulin resistance for BMI. The insulin resistance of PCOS is independent of obesity and thus to some extent constitutive. It selectively affects tissue-specific metabolic actions of insulin with the result that the compensatory hyperinsulinemia sensitizes ovarian theca cells to LH stimulation. Stimulation of adipogenesis and lipogenesis and inhibition of lipolysis by insulin excess also appear to contribute to the obesity of PCOS.
Currently, specific testing for FOH has little clinical utility beyond possibly identifying a subpopulation of PCOS patients whose androgen excess arises from simple obesity, and so would be expected to be reversible by weight loss, or distinguishing adolescents with PCOS from those with physiologic anovulation. The main place of biochemical phenotyping of PCOS would seem to lie in the investigation of phenotype-genotype correlations. Ultimately, the goal of research into PCOS pathophysiology is to understand the biochemical and genetic subtypes of PCOS as well as we currently understand the biochemical and genetic subtypes of congenital adrenal hyperplasia.
The etiology of FOH is multifactorial. FOH usually seems to develop as a complex trait from interactions between predisposing congenital factor(s) and provocative environmental factor(s). The most common provocative factors seem to be obesity and insulin resistance, which occur in about half of cases and which themselves have heritable components. Obesity up-regulates ovarian androgen production primarily via insulin-resistant hyperinsulinemia and to some extent via inflammatory cytokines.
Improved comprehension of the cause of PCOS will be necessary to facilitate diagnosis and treatment of the disorder. The current diagnostic criteria are very broad. Although this approach facilitates diagnosis, it obscures the recognition that obesity alone appears capable of causing hyperandrogenic anovulation. Thus, the elucidation of simple specific biochemical markers for the syndrome as distinct from such features as PCOM would be desirable. Accomplishing this would also enable the identification of specific genetic predisposing factors, the search for which is hampered by the somewhat nonspecific nature of the phenotypic criteria in current use.
Knowledge about the nature of the congenital predisposing factors is in in its infancy. Although congenital androgenization is the most well-established mechanism for experimentally reproducing the PCOS phenotype, it is unlikely to cause ordinary PCOS. In the search for causes of PCOS, this seems most likely to serve as a model for exploring epigenetic programming. The promise of molecular genetic approaches to understanding the cause of PCOS is illustrated by the recent identification by genome-wide association screening of a previously unrecognized protein variant in androgen-producing cells, DENND1A.V2, as a facilitator of steroidogenesis.
A complementary approach to understanding the pathogenesis of PCOS would involve developing untransformed human cell lines for all the tissues that are dysfunctional in PCOS: theca, granulosa, adrenocortical zona reticularis, and preadipocyte cells. This would facilitate knowledge about the normal and abnormal regulation of ovarian steroidogenesis, homologous desensitization to LH, and folliculogenesis as well as potentially identify commonality among the mechanisms underpinning the associated abnormalities seen in PCOS. One of the great mysteries about PCOS is what the common denominator may be that links ovarian hyperandrogenism, obesity, and insulin resistance. Considerable basic research will be necessary to discern this.
Acknowledgments
We thank Susan Sam, MD and Annie Newell-Fugate, DVM, PhD, for guidance about the role of insulin and androgen in adipocyte biology.
This research was supported in part by the Eunice Kennedy Shriver National Institute of Child Health and Human Development/National Institutes of Health through Cooperative Agreement U54–041859 as part of the Specialized Cooperative Centers Program in Reproduction and Infertility Research and by National Center For Research Resources Grants RR-00055 and UL1RR024999. Additional support was provided by The University of Chicago Grants DRTC P60-DK20595 and P50 HD057796 (“Sex Steroids, Sleep, and Metabolic Dysfunction in Women”) (to D.A.E.).
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AE-PCOS
- Androgen Excess-PCOS Society
- AMH
- anti-Müllerian hormone
- BMI
- body mass index
- BMP
- bone morphogenetic protein
- CYP17
- cytochrome P450c17
- DAST
- dexamethasone androgen-suppression test
- DENND
- differentially expressed in normal and neoplastic development
- DHEA
- dehydroepiandrosterone
- DHEAS
- DHEA sulfate
- DM
- diabetes mellitus
- FAH
- functional adrenal hyperandrogenism
- FOH
- functional ovarian hyperandrogenism
- GDF
- growth differentiation factor
- GnRHag
- GnRH agonist
- GTT
- glucose tolerance test
- GWAS
- genome-wide association studies
- hCG
- human chorionic gonadotropin
- Hippo
- hippopotamus
- HOMA-IR
- insulin resistance by homeostatic model assessment
- 3βHSD2
- Δ5-isomerase-3β-hydroxysteroid dehydrogenase type 2
- IGT
- impaired glucose tolerance
- MNC
- mononuclear cell
- NGF
- nerve growth factor
- NIH criteria
- National Institutes of Health 1990 conference diagnostic criteria
- NOM
- normal ovarian morphology
- 17OHP
- 17-hydroxyprogesterone
- OSA
- obstructive sleep apnea
- PCOM
- polycystic ovarian morphology
- PCOS
- polycystic ovary syndrome
- PCOS-A
- functionally atypical PCOS
- PCOS-T
- functionally typical PCOS
- POR
- cytochrome P450-oxidoreductase
- RD
- reductase
- SDAST
- short DAST
- SHBG
- sex hormone-binding globulin
- SNS
- sympathetic nervous system
- SULT2A1
- steroid sulfotransferase
- VEGF
- vascular endothelial growth factor
- V-NOM
- volunteer with NOM.
Reference
- 1. Ehrmann DA. Polycystic ovary syndrome. N Engl J Med. 2005;352:1223–1236. [DOI] [PubMed] [Google Scholar]
- 2. Lauritsen MP, Bentzen JG, Pinborg A, et al. The prevalence of polycystic ovary syndrome in a normal population according to the Rotterdam criteria versus revised criteria including anti-Mullerian hormone. Hum Reprod. 2014;29:791–801. [DOI] [PubMed] [Google Scholar]
- 3. Stein IF, Leventhal ML. Amenorrhea associated with bilateral polycystic ovaries. Am J Obstet Gynecol. 1935;29:181–191. [Google Scholar]
- 4. Azziz R, Adashi EY. Stein and Leventhal: 80 years on. Am J Obstet Gynecol. 2016;214:247.e1–247.e11. [DOI] [PubMed] [Google Scholar]
- 5. Goldzieher MW, Green JA. The polycystic ovary. I. Clinical and histologic features. J Clin Endocrinol Metab. 1962;22:325–338. [DOI] [PubMed] [Google Scholar]
- 6. McArthur JW, Ingersoll FM, Worcester J. The urinary excretion of interstitial-cell and follicle-stimulating hormone activity by women with diseases of the reproductive system. J Clin Endocrinol Metab. 1958;18:1202–1215. [DOI] [PubMed] [Google Scholar]
- 7. Yen SS, Vela P, Rankin J. Inappropriate secretion of follicle-stimulating hormone and luteinizing hormone in polycystic ovarian disease. J Clin Endocrinol Metab. 1970;30:435–442. [DOI] [PubMed] [Google Scholar]
- 8. Rosenfield RL, Ehrlich EN, Cleary R. Adrenal and ovarian contributions to the elevated free plasma androgen levels in hirsute women. J Clin Endocrinol Metab. 1972;34:92–98. [DOI] [PubMed] [Google Scholar]
- 9. Futterweit W, Deligdisch L. Histopathological effects of exogenously administered testosterone in 19 female to male transsexuals. J Clin Endocrinol Metab. 1986;62:16–21. [DOI] [PubMed] [Google Scholar]
- 10. Franks S. Polycystic ovary syndrome: a changing perspective. Clin Endocrinol. 1989;31:87–120. [DOI] [PubMed] [Google Scholar]
- 11. Barbieri RL, Ryan KJ. Hyperandrogenism, insulin resistance, and acanthosis nigricans syndrome: a common endocrinopathy with distinct pathophysiologic features. Am J Obstet Gynecol. 1983;147:90–101. [DOI] [PubMed] [Google Scholar]
- 12. Chang RJ, Nakamura RM, Judd HL, Kaplan SA. Insulin resistance in nonobese patients with polycystic ovary disease. J Clin Endocrinol Metab. 1983;57:356–359. [DOI] [PubMed] [Google Scholar]
- 13. Dunaif A, Segal KR, Futterweit W, Dobrjansky A. Profound peripheral insulin resistance, independent of obesity, in polycystic ovary syndrome. Diabetes. 1989;38:1165–1174. [DOI] [PubMed] [Google Scholar]
- 14. Barbieri R, Makris A, Randall R, Daniels G, Kistner R, Ryan K. Insulin stimulates androgen accumulation in incubations of ovarian stroma obtained from women with hyperandrogenism. J Clin Endocrinol Metab. 1986;904–910. [DOI] [PubMed] [Google Scholar]
- 15. Hernandez ER, Resnick CE, Holtzclaw WD, Payne DW, Adashi EY. Insulin as a regulator of androgen biosynthesis by cultured rat ovarian cells: cellular mechanism (s) underlying physiological and pharmacological hormonal actions. Endocrinology. 1988;122:2034. [DOI] [PubMed] [Google Scholar]
- 16. Cara JF, Rosenfield RL. Insulin-like growth factor I and insulin potentiate luteinizing hormone-induced androgen synthesis by rat ovarian theca-interstitial cells. Endocrinology. 1988;123:733–739. [DOI] [PubMed] [Google Scholar]
- 17. Barnes RB, Rosenfield RL, Burstein S, Ehrmann DA. Pituitary-ovarian responses to nafarelin testing in the polycystic ovary syndrome. N Engl J Med. 1989;320:559–565. [DOI] [PubMed] [Google Scholar]
- 18. Rosenfield RL, Barnes RB, Cara JF, Lucky AW. Dysregulation of cytochrome P450c 17α as the cause of polycystic ovary syndrome. Fertil Steril. 1990;53:785–791. [PubMed] [Google Scholar]
- 19. Ehrmann DA, Rosenfield RL, Barnes RB, Brigell DF, Sheikh Z. Detection of functional ovarian hyperandrogenism in women with androgen excess. N Engl J Med. 1992;327:157–162. [DOI] [PubMed] [Google Scholar]
- 20. Rosenfield RL, Ehrmann DA, Barnes RB, Brigell DF, Chandler DW. Ovarian steroidogenic abnormalities in polycystic ovary syndrome: evidence for abnormal coordinate regulation of androgen and estrogen secretion. In: Dunaif A, Givens J, Haseltine F, Merriam G, eds. Polycystic Ovary Syndrome. Cambridge, MA: Blackwell Scientific Publications; 1992:83–110. [Google Scholar]
- 21. Zawadzki J, Dunaif A. Diagnostic criteria for polycystic ovary syndrome: towards a rational approach. In: Dunaif A, Givens J, Haseltine F, Merriam G, eds. Polycystic Ovary Syndrome. Cambridge, MA: Blackwell Scientific Publications; 1992:377–384. [Google Scholar]
- 22. Ehrmann DA, Barnes RB, Rosenfield RL. Polycystic ovary syndrome as a form of functional ovarian hyperandrogenism due to dysregulation of androgen secretion. Endocr Rev. 1995;16:322–353. [DOI] [PubMed] [Google Scholar]
- 23. Nelson VL, Legro RS, Strauss JF, 3rd, McAllister JM. Augmented androgen production is a stable steroidogenic phenotype of propagated theca cells from polycystic ovaries. Mol Endocrinol. 1999;13:946–957. [DOI] [PubMed] [Google Scholar]
- 24. Ciaraldi TP, Aroda V, Mudaliar S, Chang RJ, Henry RR. Polycystic ovary syndrome is associated with tissue-specific differences in insulin resistance. J Clin Endocrinol Metab. 2009;94:157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wu S, Divall S, Nwaopara A, et al. Obesity-induced infertility and hyperandrogenism are corrected by deletion of the insulin receptor in the ovarian theca cell. Diabetes. 2014;63:1270–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Balen AH, Laven JS, Tan SL, Dewailly D. Ultrasound assessment of the polycystic ovary: international consensus definitions. Hum Reprod Update. 2003;9:505–514. [DOI] [PubMed] [Google Scholar]
- 27. Rotterdam ESHRE/ASRM-Sponsored PCOS Consensus Workshop Group. Revised 2003 consensus on diagnostic criteria and long-term health risks related to polycystic ovary syndrome. Fertil Steril. 2004;81:19–25. [DOI] [PubMed] [Google Scholar]
- 28. Azziz R, Carmina E, Dewailly D, et al. The Androgen Excess and PCOS Society criteria for the polycystic ovary syndrome: the complete task force report. Fertil Steril. 2009;91:456–488. [DOI] [PubMed] [Google Scholar]
- 29. Johnson T, Kaplan L, Ouyang P, Rizza R. National Institutes of Health evidence-based methodology workshop on polycystic ovary syndrome (PCOS). NIH EbMW Report. 2013. Available from https://prevention.nih.gov/programs-events/pathways-to-prevention/previous-workshops/pcos/workshop-resources Bethesda, MD: National Institutes of Health; 1–14. [Google Scholar]
- 30. Rosenfield RL. The polycystic ovary morphology-polycystic ovary syndrome spectrum. J Pediatr Adolesc Gynecol. 2015;28:412–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adams JM, Taylor AE, Crowley WF, Jr, Hall JE. Polycystic ovarian morphology with regular ovulatory cycles: insights into the pathophysiology of polycystic ovarian syndrome. J Clin Endocrinol Metab. 2004;89:4343–4350. [DOI] [PubMed] [Google Scholar]
- 32. Barber TM, Wass JA, McCarthy MI, Franks S. Metabolic characteristics of women with polycystic ovaries and oligo-amenorrhoea but normal androgen levels: implications for the management of polycystic ovary syndrome. Clin Endocrinol. 2007;66:513–517. [DOI] [PubMed] [Google Scholar]
- 33. Guastella E, Longo RA, Carmina E. Clinical and endocrine characteristics of the main polycystic ovary syndrome phenotypes. Fertil Steril. 2010;94:2197–2201. [DOI] [PubMed] [Google Scholar]
- 34. Clark NM, Podolski AJ, Brooks ED, et al. Prevalence of polycystic ovary syndrome phenotypes using updated criteria for polycystic ovarian morphology: an assessment of over 100 consecutive women self-reporting features of polycystic ovary syndrome. Reprod Sci. 2014;21:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ladron de Guevara A, Fux-Otta C, et al. Metabolic profile of the different phenotypes of polycystic ovary syndrome in two Latin American populations. Fertil Steril. 2014;101:1732–1739.e1731–e1732. [DOI] [PubMed] [Google Scholar]
- 36. Christ JP, Vanden Brink H, et al. Ultrasound features of polycystic ovaries relate to degree of reproductive and metabolic disturbance in polycystic ovary syndrome. Fertil Steril. 2015;103:787–794. [DOI] [PubMed] [Google Scholar]
- 37. Romualdi D, Di Florio C, Tagliaferri V, et al. The role of anti-Müllerian hormone in the characterization of the different polycystic ovary syndrome phenotypes. Reprod Sci. 2015;23:655–661. [DOI] [PubMed] [Google Scholar]
- 38. Martin KA, Chang RJ, Ehrmann DA, et al. Evaluation and treatment of hirsutism in premenopausal women: an Endocrine Society Clinical Practice Guideline. J Clin Endocrin Metab. 2008;93:1105–1120. [DOI] [PubMed] [Google Scholar]
- 39. Rosenfield RL, Wroblewski K, Padmanabhan V, Littlejohn E, Mortensen M, Ehrmann DA. Antimüllerian hormone levels are independently related to ovarian hyperandrogenism and polycystic ovaries. Fertil Steril. 2012;98:242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lauritsen MP, Pinborg A, Loft A, et al. Revised criteria for PCOS in WHO Group II anovulatory infertility - a revival of hypothalamic amenorrhoea? Clin Endocrinol. 2015;82:584–591. [DOI] [PubMed] [Google Scholar]
- 41. Rosenfield RL. Clinical practice. Hirsutism [comment appears in N Engl J Med. 2006;354(14):1533–1535; author reply 1533–1535]. N Engl J Med. 2005;353:2578–2588. [DOI] [PubMed] [Google Scholar]
- 42. Rosenfield RL. Clinical review: adolescent anovulation: maturational mechanisms and implications. J Clin Endocrinol Metab. 2013;98:3572–3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rosner W, Vesper H. Toward excellence in testosterone testing: a consensus statement. J Clin Endocrinol Metab. 2010;95:4542–4548. [DOI] [PubMed] [Google Scholar]
- 44. Auchus RJ. Steroid assays and endocrinology: best practices for basic scientists. Endocrinology. 2014;155:2049–2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Rosner W, Auchus RJ, Azziz R, Sluss PM, Raff H. Position statement: utility, limitations, and pitfalls in measuring testosterone: an Endocrine Society position statement. J Clin Endocrinol Metab. 2007;92:405–413. [DOI] [PubMed] [Google Scholar]
- 46. Legro RS, Schlaff WD, Diamond MP, et al. Total testosterone assays in women with polycystic ovary syndrome: precision and correlation with hirsutism. J Clin Endocrinol Metab. 2010;95:5305–5313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rosenfield RL, Mortensen M, Wroblewski K, Littlejohn E, Ehrmann DA. Determination of the source of androgen excess in functionally atypical polycystic ovary syndrome by a short dexamethasone androgen-suppression test and a low-dose ACTH test. Hum Reprod. 2011;26:3138–3146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Salameh WA, Redor-Goldman MM, Clarke NJ, Mathur R, Azziz R, Reitz RE. Specificity and predictive value of circulating testosterone assessed by tandem mass spectrometry for the diagnosis of polycystic ovary syndrome by the National Institutes of Health 1990 criteria. Fertil Steril. 2014;101:1135–1141 e1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dewailly D, Lujan ME, Carmina E, et al. Definition and significance of polycystic ovarian morphology: a task force report from the Androgen Excess and Polycystic Ovary Syndrome Society. Hum Reprod Update. 2014;20:334–352. [DOI] [PubMed] [Google Scholar]
- 50. Rosenfield RL. The diagnosis of polycystic ovary syndrome in adolescents. Pediatrics. 2015;136:1154–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Witchel SF, Oberfield S, Rosenfield RL, et al. The diagnosis of polycystic ovary syndrome during adolescence. Horm Res Paediatr. 2015;83:376–389. [DOI] [PubMed] [Google Scholar]
- 52. Littlejohn EE, Weiss RE, Deplewski D, Edidin DV, Rosenfield R. Intractable early childhood obesity as the initial sign of insulin resistant hyperinsulinism and precursor of polycystic ovary syndrome. J Pediatr Endocrinol Metab. 2007;20:41–51. [DOI] [PubMed] [Google Scholar]
- 53. Rosenfield RL, Ehrmann DA, Littlejohn E. Adolescent polycystic ovary syndrome due to functional ovarian hyperandrogenism persists into adulthood. J Clin Endocrinol Metab. 2015;100:1537–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Winters SJ, Talbott E, Guzick DS, Zborowski J, McHugh KP. Serum testosterone levels decrease in middle age in women with the polycystic ovary syndrome. Fertil Steril. 2000;73:724–729. [DOI] [PubMed] [Google Scholar]
- 55. Elting MW, Korsen TJ, Schoemaker J. Obesity, rather than menstrual cycle pattern or follicle cohort size, determines hyperinsulinaemia, dyslipidaemia and hypertension in ageing women with polycystic ovary syndrome. Clin Endocrinol. 2001;55:767–776. [DOI] [PubMed] [Google Scholar]
- 56. Burger HG, Hale GE, Robertson DM, Dennerstein L. A review of hormonal changes during the menopausal transition: focus on findings from the Melbourne Women's Midlife Health Project. Hum Reprod Update. 2007;13:559–565. [DOI] [PubMed] [Google Scholar]
- 57. Fauser BC, Tarlatzis BC, Rebar RW, et al. Consensus on women's health aspects of polycystic ovary syndrome (PCOS): the Amsterdam ESHRE/ASRM-Sponsored 3rd PCOS Consensus Workshop Group. Fertil Steril. 2012;97:28–38.e25. [DOI] [PubMed] [Google Scholar]
- 58. Horton R, Romanoff E, Walker J. Androstenedione and testosterone in ovarian venous and peripheral plasma during ovariectomy for breast cancer. J Clin Endocrinol Metab. 1966;26:1267–1269. [DOI] [PubMed] [Google Scholar]
- 59. Horton R, Tait JF. Androstenedione production and interconversion rates measured in peripheral blood and studies on the possible site of its conversion to testosterone. J Clin Invest. 1966;45:301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kirschner MA, Bardin CW. Androgen production and metabolism in normal and virilized women. Metabolism. 1972;21:667–688. [DOI] [PubMed] [Google Scholar]
- 61. Walters KA, Allan CM, Jimenez M, et al. Female mice haploinsufficient for an inactivated androgen receptor (AR) exhibit age-dependent defects that resemble the AR null phenotype of dysfunctional late follicle development, ovulation, and fertility. Endocrinology. 2007;148:3674–3684. [DOI] [PubMed] [Google Scholar]
- 62. Miller WL, Auchus RJ. The molecular biology, biochemistry, and physiology of human steroidogenesis and its disorders. Endocr Rev. 2011;32:81–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kiriakidou M, McAllister JM, Sugawara T, Strauss JF., 3rd Expression of steroidogenic acute regulatory protein (StAR) in the human ovary. J Clin Endocrinol Metab. 1996;81:4122–4128. [DOI] [PubMed] [Google Scholar]
- 64. Miller WL, Tee MK. The post-translational regulation of 17,20 lyase activity. Mol Cell Endocrinol. 2015;408:99–106. [DOI] [PubMed] [Google Scholar]
- 65. Anakwe OO, Payne AH. Noncoordinate regulation of de novo synthesis of cytochrome P-450 cholesterol side-chain cleavage and cytochrome P-450 17α-hydroxylase/C17–20 lyase in mouse Leydig cell cultures: relation to steroid production. Mol Endocrinol. 1987;1:595–603. [DOI] [PubMed] [Google Scholar]
- 66. Magoffin D. Evidence that luteinizing hormone-stimulated differentiation of purified ovarian thecal-interstitial cells is mediated by both type I and type II adenosine 3′,5′-monophosphate-dependent protein kinases. Endocrinology. 1989;125:1464–1473. [DOI] [PubMed] [Google Scholar]
- 67. John ME, John MC, Boggaram V, Simpson ER, Waterman MR. Transcriptional regulation of steroid hydroxylase genes by corticotropin. Proc Natl Acad Sci USA. 1986;83:4715–4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Di Blasio A, Voutilainen R, Jaffe RB, Miller WL. Hormonal regulation of messenger ribonucleic acids for P450scc (cholesterol side-chain cleavage enzyme) and P450c17 (17 α-hydroxylase/17,20 lyase) in cultured human fetal adrenal cells. J Clin Endocrinol Metab. 1987;65:170–175. [DOI] [PubMed] [Google Scholar]
- 69. Kase N, Forchielli E, Dorfman R. In vitro production of testosterone and androst-4-ene-3,17-dione in a human ovarian homogenate. Acta Endocrinol (Copenh). 1961;37:19–23. [DOI] [PubMed] [Google Scholar]
- 70. Axelrod LR, Goldzieher JW. The polycystic ovary. III. Steroid biosynthesis in normal and polycystic ovarian tissue. J Clin Endocrinol. 1962;22:431–440. [DOI] [PubMed] [Google Scholar]
- 71. Dorfman RI, Forchielli E, Gut M. Androgen biosynthesis and related studies. Rec Prog Horm Res. 1963;19:251–273. [PubMed] [Google Scholar]
- 72. Nelson VL, Qin KN, Rosenfield RL, et al. The biochemical basis for increased testosterone production in theca cells propagated from patients with polycystic ovary syndrome. J Clin Endocrinol Metab. 2001;86:5925–5933. [DOI] [PubMed] [Google Scholar]
- 73. Longcope C, Johnston CC., Jr Androgen and estrogen dynamics in pre- and postmenopausal women: a comparison between smokers and nonsmokers. J Clin Endocrinol Metab. 1988;67:379–383. [DOI] [PubMed] [Google Scholar]
- 74. Gibb W, Lavoie JC. Substrate specificity of the placental microsomal aromatase. Steroids. 1980;36:507–519. [DOI] [PubMed] [Google Scholar]
- 75. Simpson ER, Mahendroo MS, Means GD, et al. Aromatase cytochrome P450, the enzyme responsible for estrogen biosynthesis. Endocr Rev. 1994;15:342–355. [DOI] [PubMed] [Google Scholar]
- 76. McNatty KP, Makris A, Reinhold VN, De Grazia C, Osathanondh R, Ryan KJ. Metabolism of androstenedione by human ovarian tissues in vitro with particular reference to reductase and aromatase activity. Steroids. 1979;34:429–443. [DOI] [PubMed] [Google Scholar]
- 77. Jakimiuk AJ, Weitsman SR, Magoffin DA. 5α-reductase activity in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1999;84:2414–2418. [DOI] [PubMed] [Google Scholar]
- 78. Prizant H, Gleicher N, Sen A. Androgen actions in the ovary: balance is key. J Endocrinol. 2014;222:R141–R151. [DOI] [PubMed] [Google Scholar]
- 79. Walters KA, Allan CM, Handelsman DJ. Androgen actions and the ovary. Biol Reprod. 2008;78:380–389. [DOI] [PubMed] [Google Scholar]
- 80. Daniels TL, Berga SL. Resistance of gonadotropin releasing hormone drive to sex steroid-induced suppression in hyperandrogenic anovulation. J Clin Endocrinol Metab. 1997;82:4179–4183. [DOI] [PubMed] [Google Scholar]
- 81. Bogumil RJ, Ferin M, Rootenberg J, Speroff L, Vande Wiele RL. Mathematical studies of the human menstrual cycle. I. Formation of a mathematical model. J Clin Endocrinol Metab. 1972;35:126–143. [DOI] [PubMed] [Google Scholar]
- 82. Welt CK, Smith ZA, Pauler DK, Hall JE. Differential regulation of inhibin A and inhibin B by luteinizing hormone, follicle-stimulating hormone, and stage of follicle development. J Clin Endocrinol Metab. 2001;86:2531–2537. [DOI] [PubMed] [Google Scholar]
- 83. Rosenfield RL, Bordini B. Evidence that obesity and androgens have independent and opposing effects on gonadotropin production from puberty to maturity. Brain Res. 2010;1364:186–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Foecking EM, Szabo M, Schwartz NB, Levine JE. Neuroendocrine consequences of prenatal androgen exposure in the female rat: absence of luteinizing hormone surges, suppression of progesterone receptor gene expression, and acceleration of the gonadotropin-releasing hormone pulse generator. Biol Reprod. 2005;72:1475–1483. [DOI] [PubMed] [Google Scholar]
- 85. Aono T, Miyake A, Kinugasa T, Kurachi K, Matsumoto K. Absence of positive feedback effect of oestrogen on LH release in patients with testicular feminization syndrome. Acta Endocrinol (Copenh). 1978;87:259–267. [DOI] [PubMed] [Google Scholar]
- 86. Nicoletti I, Filipponi P, Fedeli L, Santori PA, Santeusanio F. Effect of estrogens and progesterone on gonadotropin and prolactin release in a patient with androgen insensitivity. Obstet Gynecol. 1981;58:527–532. [PubMed] [Google Scholar]
- 87. Wu S, Chen Y, Fajobi T, et al. Conditional knockout of the androgen receptor in gonadotropes reveals crucial roles for androgen in gonadotropin synthesis and surge in female mice. Mol Endocrinol. 2014;28:1670–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Nahum R, Thong KJ, Hillier SG. Metabolic regulation of androgen production by human thecal cells in vitro. Hum Reprod. 1995;10:75–81. [DOI] [PubMed] [Google Scholar]
- 89. Willis D, Mason H, Gilling-Smith C, Franks S. Modulation by insulin of follicle-stimulating hormone and luteinizing hormone actions in human granulosa cells of normal and polycystic ovaries. J Clin Endocrinol Metab. 1996;81:302–309. [DOI] [PubMed] [Google Scholar]
- 90. Chang RJ, Cook-Andersen H. Disordered follicle development. Mol Cell Endocrinol. 2013;373:51–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Cara JF, Fan J, Azzarello J, Rosenfield RL. Insulin-like growth factor-I enhances luteinizing hormone binding to rat ovarian theca-interstitial cells. J Clin Invest. 1990;86:560–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Bhaskaran RS, Ascoli M. The post-endocytotic fate of the gonadotropin receptors is an important determinant of the desensitization of gonadotropin responses. J Mol Endocrinol. 2005;34:447–457. [DOI] [PubMed] [Google Scholar]
- 93. Menon B, Gulappa T, Menon KM. Eukaryotic initiation factor 5A plays an essential role in luteinizing hormone receptor regulation. Mol Endocrinol. 2014;28:1796–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Wolf-Ringwall AL, Winter PW, Roess DA, George Barisas B. Luteinizing hormone receptors are confined in mesoscale plasma membrane microdomains throughout recovery from receptor desensitization. Cell Biochem Biophys. 2014;68:561–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Hirshfeld-Cytron J, Barnes RB, Ehrmann DA, Caruso A, Mortensen MM, Rosenfield RL. Characterization of functionally typical and atypical types of polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94:1587–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Levrant SG, Barnes RB, Rosenfield RL. A pilot study of the human chorionic gonadotrophin test for ovarian hyperandrogenism. Hum Reprod. 1997;12:1416–1420. [DOI] [PubMed] [Google Scholar]
- 97. McCartney CR, Bellows AB, Gingrich MB, et al. Exaggerated 17-hydroxyprogesterone response to intravenous infusions of recombinant human LH in women with polycystic ovary syndrome. Am J Physiol Endocrinol Metab. 2004;286:E902–E908. [DOI] [PubMed] [Google Scholar]
- 98. Couse JF, Korach KS. Estrogen receptor null mice: what have we learned and where will they lead us? [erratum appears in Endocr Rev. 1999;20(4):459]. Endocr Rev. 1999;20:358–417. [DOI] [PubMed] [Google Scholar]
- 99. Britt KL, Findlay JK. Estrogen actions in the ovary revisited. J Endocrinol. 2002;175:269–276. [DOI] [PubMed] [Google Scholar]
- 100. Taniguchi F, Couse JF, Rodriguez KF, Emmen JM, Poirier D, Korach KS. Estrogen receptor-α mediates an intraovarian negative feedback loop on thecal cell steroidogenesis via modulation of Cyp17a1 (cytochrome P450, steroid 17α-hydroxylase/17,20 lyase) expression. FASEB. 2007;21:586–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Duda M, Grzesiak M, Knet M, et al. The impact of antiandrogen 2-hydroxyflutamide on the expression of steroidogenic enzymes in cultured porcine ovarian follicles. Mol Biol Rep. 2014;41:4213–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. McAllister JM, Byrd W, Simpson ER. The effects of growth factors and phorbol esters on steroid biosynthesis in isolated human theca interna and granulosa-lutein cells in long term culture. J Clin Endocrinol Metab. 1994;79:106–112. [DOI] [PubMed] [Google Scholar]
- 103. Barnes RB, Namnoum AB, Rosenfield RL, Layman LC. The role of LH and FSH in ovarian androgen secretion and ovarian follicular development: clinical studies in a patient with isolated FSH deficiency and multicystic ovaries. Hum Reprod. 2002;17:88–91. [DOI] [PubMed] [Google Scholar]
- 104. Barnes RB, Rosenfield RL, Namnoum A, Layman LC. Effect of follicle-stimulating hormone on ovarian androgen production in a woman with isolated follicle-stimulating hormone deficiency. N Engl J Med. 2000;343:1197–1198. [DOI] [PubMed] [Google Scholar]
- 105. Munir I, Yen HW, Geller DH, et al. Insulin augmentation of 17α-hydroxylase activity is mediated by phosphatidyl inositol 3-kinase but not extracellular signal-regulated kinase-1/2 in human ovarian theca cells. Endocrinology. 2004;145:175–183. [DOI] [PubMed] [Google Scholar]
- 106. Magoffin DA, Weitsman SR. Insulin-like growth factor-I regulation of luteinizing hormone (LH) receptor messenger ribonucleic acid expression and LH-stimulated signal transduction in rat ovarian theca-interstitial cells. Biol Reprod. 1994;51:766–775. [DOI] [PubMed] [Google Scholar]
- 107. Du X, Rosenfield RL, Qin K. KLF15 is a transcriptional regulator of the human 17β-hydroxysteroid dehydrogenase type 5 gene. A potential link between regulation of testosterone production and fat stores in women. J Clin Endocrinol Metab. 2009;94:2594–2601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. McAllister JM, Modi B, Miller BA, et al. Overexpression of a DENND1A isoform produces a polycystic ovary syndrome theca phenotype. Proc Natl Acad Sci USA. 2014;111:E1519–E1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Andersen CY, Lossl K. Increased intrafollicular androgen levels affect human granulosa cell secretion of anti-Müllerian hormone and inhibin-B. Fertil Steril. 2008;89:1760–1765. [DOI] [PubMed] [Google Scholar]
- 110. Hoang YD, McTavish KJ, Chang RJ, Shimasaki S. Paracrine regulation of theca androgen production by granulosa cells in the ovary. Fertil Steril. 2013;100:561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Kottler ML, Chou YY, Chabre O, et al. A new FSHβ mutation in a 29-year-old woman with primary amenorrhea and isolated FSH deficiency: functional characterization and ovarian response to human recombinant FSH. Eur J Endocrinol. 2010;162:633–641. [DOI] [PubMed] [Google Scholar]
- 112. Dooley CA, Attia GR, Rainey WE, Moore DR, Carr BR. Bone morphogenetic protein inhibits ovarian androgen production. J Clin Endocrinol Metab. 2000;85:3331–3337. [DOI] [PubMed] [Google Scholar]
- 113. Young JM, McNeilly AS. Theca: the forgotten cell of the ovarian follicle. Reproduction. 2010;140:489–504. [DOI] [PubMed] [Google Scholar]
- 114. Dumesic DA, Richards JS. Ontogeny of the ovary in polycystic ovary syndrome. Fertil Steril. 2013;100:23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Raja-Khan N, Urbanek M, Rodgers RJ, Legro RS. The role of TGF-β in polycystic ovary syndrome. Reprod Sci. 2014;21:20–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Glister C, Satchell L, Bathgate RA, et al. Functional link between bone morphogenetic proteins and insulin-like peptide 3 signaling in modulating ovarian androgen production. Proc Natl Acad Sci USA. 2013;110:E1426–E1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Hatzirodos N, Bayne RA, Irving-Rodgers HF, et al. Linkage of regulators of TGF-β activity in the fetal ovary to polycystic ovary syndrome. FASEB. 2011;25:2256–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. González F. Nutrient-induced inflammation in polycystic ovary syndrome: role in the development of metabolic aberration and ovarian dysfunction. Semin Reprod Med. 2015;33:276–286. [DOI] [PubMed] [Google Scholar]
- 119. Jiang L, Huang J, Li L, et al. MicroRNA-93 promotes ovarian granulosa cells proliferation through targeting CDKN1A in polycystic ovarian syndrome. J Clin Endocrinol Metab. 2015;100:E729–E738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Heider U, Pedal I, Spanel-Borowski K. Increase in nerve fibers and loss of mast cells in polycystic and postmenopausal ovaries. Fertil Steril. 2001;75:1141–1147. [DOI] [PubMed] [Google Scholar]
- 121. Dissen GA, Garcia-Rudaz C, Paredes A, Mayer C, Mayerhofer A, Ojeda SR. Excessive ovarian production of nerve growth factor facilitates development of cystic ovarian morphology in mice and is a feature of polycystic ovarian syndrome in humans. Endocrinology. 2009;150:2906–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Lansdown A, Rees DA. The sympathetic nervous system in polycystic ovary syndrome: a novel therapeutic target? Clin Endocrinol. 2012;77:791–801. [DOI] [PubMed] [Google Scholar]
- 123. Wilson JL, Chen W, Dissen GA, et al. Excess of nerve growth factor in the ovary causes a polycystic ovary-like syndrome in mice, which closely resembles both reproductive and metabolic aspects of the human syndrome. Endocrinology. 2014;155:4494–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Fauser BC, Van Heusden AM. Manipulation of human ovarian function: physiological concepts and clinical consequences. Endocr Rev. 1997;18:71–106. [DOI] [PubMed] [Google Scholar]
- 125. Hsueh AJ, Kawamura K, Cheng Y, Fauser BC. Intraovarian control of early folliculogenesis. Endocr Rev. 2015;36:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Teng Z, Wang C, Wang Y, et al. S100A8, an oocyte-specific chemokine, directs the migration of ovarian somatic cells during mouse primordial follicle assembly. J Cell Physiol. 2015;230:2998–3008. [DOI] [PubMed] [Google Scholar]
- 127. Chakraborty P, Roy SK. Bone morphogenetic protein 2 promotes primordial follicle formation in the ovary. Sci Rep. 2015;5:12664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Jiang ZZ, Hu MW, Ma XS, et al. LKB1 acts as a critical gatekeeper of ovarian primordial follicle pool. Oncotarget. 2016;7:5738–5753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Castrillon DH, Miao L, Kollipara R, Horner JW, DePinho RA. Suppression of ovarian follicle activation in mice by the transcription factor Foxo3a. Science. 2003;301:215–218. [DOI] [PubMed] [Google Scholar]
- 130. Schmidt D, Ovitt CE, Anlag K, et al. The murine winged-helix transcription factor Foxl2 is required for granulosa cell differentiation and ovary maintenance. Development. 2004;131:933–942. [DOI] [PubMed] [Google Scholar]
- 131. Childs AJ, Kinnell HL, Collins CS, et al. BMP signaling in the human fetal ovary is developmentally regulated and promotes primordial germ cell apoptosis. Stem Cells. 2010;28:1368–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Glister C, Satchell L, Knight PG. Changes in expression of bone morphogenetic proteins (BMPs), their receptors and inhibin co-receptor betaglycan during bovine antral follicle development: inhibin can antagonize the suppressive effect of BMPs on thecal androgen production. Reproduction. 2010;140:699–712. [DOI] [PubMed] [Google Scholar]
- 133. Durlinger AL, Kramer P, Karels B, et al. Control of primordial follicle recruitment by anti-Müllerian hormone in the mouse ovary. Endocrinology. 1999;140:5789–5796. [DOI] [PubMed] [Google Scholar]
- 134. Couse JF, Hewitt SC, Bunch DO, et al. Postnatal sex reversal of the ovaries in mice lacking estrogen receptors α and β. Science. 1999;286:2328–2331. [DOI] [PubMed] [Google Scholar]
- 135. Kezele PR, Nilsson EE, Skinner MK. Insulin but not insulin-like growth factor-1 promotes the primordial to primary follicle transition. Mol Cell Endocrinol. 2002;192:37–43. [DOI] [PubMed] [Google Scholar]
- 136. Vendola KA, Zhou J, Adesanya OO, Weil SJ, Bondy CA. Androgens stimulate early stages of follicular growth in the primate ovary. J Clin Invest. 1998;101:2622–2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Persani L, Rossetti R, Di Pasquale E, Cacciatore C, Fabre S. The fundamental role of bone morphogenetic protein 15 in ovarian function and its involvement in female fertility disorders. Hum Reprod Update. 2014;20:869–883. [DOI] [PubMed] [Google Scholar]
- 138. Gougeon A. Regulation of ovarian follicular development in primates: facts and hypotheses. Endocrin Rev. 1996;17:121–155. [DOI] [PubMed] [Google Scholar]
- 139. Rani CS, Salhanick AR, Armstrong DT. Follicle-stimulating hormone induction of luteinizing hormone receptor in cultured rat granulosa cells: an examination of the need for steroids in the induction process. Endocrinology. 1981;108:1379–1385. [DOI] [PubMed] [Google Scholar]
- 140. Welt CK, Taylor AE, Fox J, Messerlian GM, Adams JM, Schneyer AL. Follicular arrest in polycystic ovary syndrome is associated with deficient inhibin A and B biosynthesis. J Clin Endocrinol Metab. 2005;90:5582–5587. [DOI] [PubMed] [Google Scholar]
- 141. Jakimiuk AJ, Weitsman SR, Brzechffa PR, Magoffin DA. Aromatase mRNA expression in individual follicles from polycystic ovaries. Mol Hum Reprod. 1998;4:1–8. [DOI] [PubMed] [Google Scholar]
- 142. Dewailly D, Andersen CY, Balen A, et al. 2014 The physiology and clinical utility of anti-Mullerian hormone in women [erratum appears in Hum Reprod Update. 2014;2020(2015):2804]. Hum Reprod Update. 2014;20:370–385. [DOI] [PubMed] [Google Scholar]
- 143. Jonard S, Dewailly D. The follicular excess in polycystic ovaries, due to intra-ovarian hyperandrogenism, may be the main culprit for the follicular arrest. Hum Reprod Update. 2004;10:107–117. [DOI] [PubMed] [Google Scholar]
- 144. Eldar-Geva T, Margalioth EJ, Gal M, et al. Serum anti-Mullerian hormone levels during controlled ovarian hyperstimulation in women with polycystic ovaries with and without hyperandrogenism. Hum Reprod. 2005;20:1814–1819. [DOI] [PubMed] [Google Scholar]
- 145. Lebbe M, Woodruff TK. Involvement of androgens in ovarian health and disease. Mol Hum Reprod. 2013;19:828–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Weil SJ, Vendola K, Zhou J, et al. Androgen receptor gene expression in the primate ovary: cellular localization, regulation, and functional correlations. J Clin Endocrinol Metab. 1998;83:2479–2485. [DOI] [PubMed] [Google Scholar]
- 147. Weil S, Vendola K, Zhou J, Bondy CA. Androgen and follicle-stimulating hormone interactions in primate ovarian follicle development. J Clin Endocrinol Metab. 1999;84:2951–2956. [DOI] [PubMed] [Google Scholar]
- 148. Rice S, Ojha K, Whitehead S, Mason H. Stage-specific expression of androgen receptor, follicle-stimulating hormone receptor, and anti-Müllerian hormone type II receptor in single, isolated, human preantral follicles: relevance to polycystic ovaries. J Clin Endocrinol Metab. 2007;92:1034–1040. [DOI] [PubMed] [Google Scholar]
- 149. Sen A, Hammes SR. Granulosa cell-specific androgen receptors are critical regulators of ovarian development and function. Mol Endocrinol. 2010;24:1393–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Walters KA, Middleton LJ, Joseph SR, et al. Targeted loss of androgen receptor signaling in murine granulosa cells of preantral and antral follicles causes female subfertility. Biol Reprod. 2012;87:151. [DOI] [PubMed] [Google Scholar]
- 151. Hillier SG. Gonadotropic control of ovarian follicular growth and development. Mol Cell Endocrinol. 2001;179:39–46. [DOI] [PubMed] [Google Scholar]
- 152. Jeppesen JV, Kristensen SG, Nielsen ME, et al. LH-receptor gene expression in human granulosa and cumulus cells from antral and preovulatory follicles. J Clin Endocrinol Metab. 2012;97:E1524–E1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Rodrigues JK, Navarro PA, Zelinski MB, Stouffer RL, Xu J. Direct actions of androgens on the survival, growth and secretion of steroids and anti-Müllerian hormone by individual macaque follicles during three-dimensional culture. Hum Reprod. 2015;30:664–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Hoffman F, Meger RC. [On the action of intraovarian injection of androgen on follicle and corpus luteum maturation in women]. Geburtshilfe Frauenheilklinic. 1965;25:1132–1137. [PubMed] [Google Scholar]
- 155. Franks S, Stark J, Hardy K. Follicle dynamics and anovulation in polycystic ovary syndrome. Hum Reprod Update. 2008;14:367–378. [DOI] [PubMed] [Google Scholar]
- 156. Willis DS, Watson H, Mason HD, Galea R, Brincat M, Franks S. Premature response to luteinizing hormone of granulosa cells from anovulatory women with polycystic ovary syndrome: relevance to mechanism of anovulation. J Clin Endocrinol Metab. 1998;83:3984–3991. [DOI] [PubMed] [Google Scholar]
- 157. Hillier SG, Tetsuka M. Role of androgens in follicle maturation and atresia. Baillieres Clin Obstet Gynaecol. 1997;11:249–260. [DOI] [PubMed] [Google Scholar]
- 158. Tingen CM, Kiesewetter SE, Jozefik J, et al. A macrophage and theca cell-enriched stromal cell population influences growth and survival of immature murine follicles in vitro. Reproduction. 2011;141:809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Woodruff TK, Shea LD. A new hypothesis regarding ovarian follicle development: ovarian rigidity as a regulator of selection and health. J Assist Reprod Genet. 2011;28:3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Roberts AE, Arbogast LK, Friedman CI, Cohn DE, Kaumaya PT, Danforth DR. Neutralization of endogenous vascular endothelial growth factor depletes primordial follicles in the mouse ovary. Biol Reprod. 2007;76:218–223. [DOI] [PubMed] [Google Scholar]
- 161. Nakamura Y, Gang HX, Suzuki T, Sasano H, Rainey WE. Adrenal changes associated with adrenarche. Rev Endocr Metab Disord. 2009;10:19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Rosenfield RL. Plasma 17-ketosteroids and 17-β hydroxysteroids in girls with premature development of sexual hair. J Pediatr. 1971;79:260–266. [DOI] [PubMed] [Google Scholar]
- 163. Rich BH, Rosenfield RL, Lucky AW, Helke JC, Otto P. Adrenarche: changing adrenal response to adrenocorticotropin. J Clin Endocrinol Metab. 1981;52:1129–1136. [DOI] [PubMed] [Google Scholar]
- 164. Rainey WE, Carr BR, Sasano H, Suzuki T, Mason JI. Dissecting human adrenal androgen production. Trends Endocrinol Metab. 2002;13:234–239. [DOI] [PubMed] [Google Scholar]
- 165. Noordam C, Dhir V, McNelis JC, et al. Inactivating PAPSS2 mutations in a patient with premature pubarche. N Engl J Med. 2009;360:2310–2318. [DOI] [PubMed] [Google Scholar]
- 166. Baulieu EE, Corpechot C, Dray F, et al. An adrenal-secreted “androgen”: dehydroisoandrosterone sulfate. Its metabolism and a tentative generalization on the metabolism of other steroid conjugates in man. Rec Prog Horm Res. 1965;21:411–500. [PubMed] [Google Scholar]
- 167. Battista MC, Roberge C, Martinez A, Gallo-Payet N. 24-dehydrocholesterol reductase/seladin-1: a key protein differentially involved in adrenocorticotropin effects observed in human and rat adrenal cortex. Endocrinology. 2009;150:4180–4190. [DOI] [PubMed] [Google Scholar]
- 168. Nakamura Y, Hornsby PJ, Casson P, et al. Type 5 17β-hydroxysteroid dehydrogenase (AKR1C3) contributes to testosterone production in the adrenal reticularis. J Clin Endocrinol Metab. 2009;94:2192–2198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. Mills I, Brooks R, Prunty F. The relationship between the production of cortisol and androgen by the human adrenal. In: Currie A, Symington T, Grant J, eds. The Human Adrenal Cortex. Baltimore, MD: Williams, Wilkins; 1962. [Google Scholar]
- 170. Grumbach M, Richards C, Conte F, Kaplan S. Clinical disorders of adrenal function and puberty: an assessment of the role of the adrenal cortex in normal and abnormal puberty in man and evidence for an ACTH-like pituitary adrenal androgen stimulating hormone. In: James V, Serio M, Giusti C, Martini L, eds. The Endocrine Function of the Human Adrenal Cortex. London, United Kingdom: Academic Press; 1978;583. [Google Scholar]
- 171. Biason-Lauber A, Zachmann M, Schoenle EJ. Effect of leptin on CYP17 enzymatic activities in human adrenal cells: new insight in the onset of adrenarche. Endocrinology. 2000;141:1446–1454. [DOI] [PubMed] [Google Scholar]
- 172. Guercio G, Rivarola MA, Chaler E, Maceiras M, Belgorosky A. Relationship between the growth hormone/insulin-like growth factor-I axis, insulin sensitivity, and adrenal androgens in normal prepubertal and pubertal girls. J Clin Endocrinol Metab. 2003;88:1389–1393. [DOI] [PubMed] [Google Scholar]
- 173. Baquedano MS, Berensztein E, Saraco N, et al. Expression of the IGF system in human adrenal tissues from early infancy to late puberty: implications for the development of adrenarche. Pediatr Res. 2005;58:451–458. [DOI] [PubMed] [Google Scholar]
- 174. Smith CP, Dunger DB, Williams AJ, et al. Relationship between insulin, insulin-like growth factor I, and dehydroepiandrosterone sulfate concentrations during childhood, puberty, and adult life. J Clin Endocrinol Metab. 1989;68:932–937. [DOI] [PubMed] [Google Scholar]
- 175. Palmert MR, Hayden DL, Mansfield MJ, et al. The longitudinal study of adrenal maturation during gonadal suppression: evidence that adrenarche is a gradual process. J Clin Endocrinol Metab. 2001;86:4536–4542. [DOI] [PubMed] [Google Scholar]
- 176. Endoh A, Kristiansen SB, Casson PR, Buster JE, Hornsby PJ. The zona reticularis is the site of biosynthesis of dehydroepiandrosterone and dehydroepiandrosterone sulfate in the adult human adrenal cortex resulting from its low expression of 3β-hydroxysteroid dehydrogenase. J Clin Endocrinol Metab. 1996;81:3558–3565. [DOI] [PubMed] [Google Scholar]
- 177. l'Allemand D, Penhoat A, Lebrethon MC, et al. Insulin-like growth factors enhance steroidogenic enzyme and corticotropin receptor messenger ribonucleic acid levels and corticotropin steroidogenic responsiveness in cultured human adrenocortical cells. J Clin Endocrinol Metab. 1996;81:3892–3897. [DOI] [PubMed] [Google Scholar]
- 178. Taha D, Mullis PE, Ibáñez L, de Zegher F. Absent or delayed adrenarche in Pit-1/POU1F1 deficiency. Horm Res. 2005;64:175–179. [DOI] [PubMed] [Google Scholar]
- 179. Ehrhart-Bornstein M, Hinson JP, Bornstein SR, Scherbaum WA, Vinson GP. Intraadrenal interactions in the regulation of adrenocortical steroidogenesis. Endocr Rev. 1998;19:101–143. [DOI] [PubMed] [Google Scholar]
- 180. Rege J, Nishimoto HK, Nishimoto K, Rodgers RJ, Auchus RJ, Rainey WE. Bone morphogenetic protein-4 (BMP4): a paracrine regulator of human adrenal C19 steroid synthesis. Endocrinology. 2015;156:2530–2540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Udhane SS, Pandey AV, Hofer G, Mullis PE, Fluck CE. Retinoic acid receptor β and angiopoietin-like protein 1 are involved in the regulation of human androgen biosynthesis. Sci Rep. 2015;5:10132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Rosenfield RL. Role of androgens in growth and development of the fetus, child, and adolescent. Adv Pediatr. 1972;19:171–213. [Google Scholar]
- 183. Zouboulis CC, Chen WC, Thornton MJ, Qin K, Rosenfield R. Sexual hormones in human skin. Horm Metab Res. 2007;39:85–95. [DOI] [PubMed] [Google Scholar]
- 184. Quinkler M, Sinha B, Tomlinson JW, Bujalska IJ, Stewart PM, Arlt W. Androgen generation in adipose tissue in women with simple obesity–a site-specific role for 17β-hydroxysteroid dehydrogenase type 5. J Endocrinol. 2004;183:331–342. [DOI] [PubMed] [Google Scholar]
- 185. Longcope C, Baker R, Johnston CC., Jr Androgen and estrogen metabolism: relationship to obesity. Metabolism. 1986;35:235–237. [DOI] [PubMed] [Google Scholar]
- 186. Bélanger C, Luu-The V, Dupont P, Tchernof A. Adipose tissue intracrinology: potential importance of local androgen/estrogen metabolism in the regulation of adiposity. Horm Metab Res. 2002;34:737–745. [DOI] [PubMed] [Google Scholar]
- 187. Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89:2548–2556. [DOI] [PubMed] [Google Scholar]
- 188. McTernan PG, Anwar A, Eggo MC, Barnett AH, Stewart PM, Kumar S. Gender differences in the regulation of P450 aromatase expression and activity in human adipose tissue. Int J Obes Rel Metab Disord. 2000;24:875–881. [DOI] [PubMed] [Google Scholar]
- 189. Subbaramaiah K, Howe LR, Bhardwaj P, et al. Obesity is associated with inflammation and elevated aromatase expression in the mouse mammary gland. Cancer Prev Res (Phila). 2011;4:329–346. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 190. Tsilchorozidou T, Honour JW, Conway GS. Altered cortisol metabolism in polycystic ovary syndrome: insulin enhances 5α-reduction but not the elevated adrenal steroid production rates. J Clin Endocrinol Metab. 2003;88:5907–5913. [DOI] [PubMed] [Google Scholar]
- 191. Mowszowicz I, Melanitou E, Kirchhoffer M, Mauvais J, P Dihydrotestosterone stimulates 5α-reductase activity in public skin fibroblasts. J Clin Endocrinol Metab. 1983;53:320. [DOI] [PubMed] [Google Scholar]
- 192. Rosenfield RL, Moll GW. The role of proteins in the distribution of plasma androgens and estradiol. In: Molinatti G, Martini L, James V, eds. Androgenization in Women. New York, NY: Raven Press; 1983;25–45. [Google Scholar]
- 193. Pugeat M, Nader N, Hogeveen K, Raverot G, Déchaud H, Grenot C. Sex hormone-binding globulin gene expression in the liver: drugs and the metabolic syndrome. Mol Cell Endocrinol. 2010;316:53–59. [DOI] [PubMed] [Google Scholar]
- 194. Nestler JE, Powers LP, Matt DW, et al. A direct effect of hyperinsulinemia on serum sex hormone-binding globulin levels in obese women with the polycystic ovary syndrome. J Clin Endocrinol Metab. 1991;72:83–89. [DOI] [PubMed] [Google Scholar]
- 195. Simó R, Barbosa-Desongles A, Lecube A, Hernandez C, Selva DM. Potential role of tumor necrosis factor-α in downregulating sex hormone-binding globulin. Diabetes. 2012;61:372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Hogeveen KN, Cousin P, Pugeat M, Dewailly D, Soudan B, Hammond GL. Human sex hormone-binding globulin variants associated with hyperandrogenism and ovarian dysfunction. J Clin Invest. 2002;109:973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197. Mortensen M, Ehrmann DA, Littlejohn E, Rosenfield RL. Asymptomatic volunteers with a polycystic ovary are a functionally distinct but heterogeneous population. J Clin Endocrinol Metab. 2009;94:1579–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198. Rosenfield RL, Barnes RB, Ehrmann DA. Studies of the nature of 17-hydroxyprogesterone hyperresponsiveness to gonadotropin-releasing hormone agonist challenge in functional ovarian hyperandrogenism. J Clin Endocrinol Metab. 1994;79:1686–1692. [DOI] [PubMed] [Google Scholar]
- 199. Ibañez L, Hall JE, Potau N, Carrascosa A, Prat N, Taylor AE. Ovarian 17-hydroxyprogesterone hyperresponsiveness to gonadotropin-releasing hormone (GnRH) agonist challenge in women with polycystic ovary syndrome is not mediated by luteinizing hormone hypersecretion: evidence from GnRH agonist and human chorionic gonadotropin stimulation testing. J Clin Endocrinol Metab. 1996;81:4103–4107. [DOI] [PubMed] [Google Scholar]
- 200. Rosenfield RL, Rich BH, Wolfsdorf JI, et al. Pubertal presentation of congenital Δ5–3β-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab. 1980;51:345–353. [DOI] [PubMed] [Google Scholar]
- 201. Barnes RB, Rosenfield RL, Ehrmann DA, et al. Ovarian hyperandrogynism as a result of congenital adrenal virilizing disorders: evidence for perinatal masculinization of neuroendocrine function in women. J Clin Endocrinol Metab. 1994;79:1328–1333. [DOI] [PubMed] [Google Scholar]
- 202. Derksen J, Nagesser SK, Meinders AE, Haak HR, van de Velde CJ. Identification of virilizing adrenal tumors in hirsute women. N Engl J Med. 1994;331:968–973. [DOI] [PubMed] [Google Scholar]
- 203. Kaltsas GA, Isidori AM, Kola BP, et al. The value of the low-dose dexamethasone suppression test in the differential diagnosis of hyperandrogenism in women. J Clin Endocrinol Metab. 2003;88:2634–2643. [DOI] [PubMed] [Google Scholar]
- 204. Azziz R, Sanchez LA, Knochenhauer ES, et al. Androgen excess in women: experience with over 1000 consecutive patients. J Clin Endocrinol Metab. 2004;89:453–462. [DOI] [PubMed] [Google Scholar]
- 205. Carmina E, Rosato F, Jannì A, Rizzo M, Longo RA. Extensive clinical experience: relative prevalence of different androgen excess disorders in 950 women referred because of clinical hyperandrogenism. J Clin Endocrinol Metab. 2006;91:2–6. [DOI] [PubMed] [Google Scholar]
- 206. Rosenfield RL, Barnes RB, Ehrmann DA. Hyperandrogenism, hirsutism, and the polycystic ovary syndrome. In: Jameson JL, DeGroot LJ, eds. Endocrinology. 7th ed Philadelphia, PA: Elsevier; 2015;2275–2296. [Google Scholar]
- 207. Gilling-Smith C, Story H, Rogers V, Franks S. Evidence for a primary abnormality of thecal cell steroidogenesis in the polycystic ovary syndrome. Clin Endocrinol. 1997;47:93–99. [DOI] [PubMed] [Google Scholar]
- 208. Pasquali R, Patton L, Pocognoli P, Cognigni GE, Gambineri A. 17-Hydroxyprogesterone responses to gonadotropin-releasing hormone disclose distinct phenotypes of functional ovarian hyperandrogenism and polycystic ovary syndrome. J Clin Endocrinol Metab. 2007;92:4208–4217. [DOI] [PubMed] [Google Scholar]
- 209. Maas KH, Chuan SS, Cook-Andersen H, Su HI, Duleba A, Chang RJ. Relationship between 17-hydroxyprogesterone responses to human chorionic gonadotropin and markers of ovarian follicle morphology in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2015;100:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Escobar-Morreale HF, Botella-Carretero JI, Alvarez-Blasco F, Sancho J, San Millán JL. The polycystic ovary syndrome associated with morbid obesity may resolve after weight loss induced by bariatric surgery. J Clin Endocrinol Metab. 2005;90:6364–6369. [DOI] [PubMed] [Google Scholar]
- 211. Domecq JP, Prutsky G, Mullan RJ, et al. Lifestyle modification programs in polycystic ovary syndrome: systematic review and meta-analysis. J Clin Endocrinol Metab. 2013;98:4655–4663. [DOI] [PubMed] [Google Scholar]
- 212. Polson D, Adams J, Wadsworth J, Franks S. Polycystic ovaries–a common finding in normal women. Lancet. 1988;331:870–872. [DOI] [PubMed] [Google Scholar]
- 213. Farquhar CM, Birdsall M, Manning P, Mitchell JM, France JT. The prevalence of polycystic ovaries on ultrasound scanning in a population of randomly selected women. Aust N Z J Obstet Gynaecol. 1994;34:67–72. [DOI] [PubMed] [Google Scholar]
- 214. Chang PL, Lindheim SR, Lowre C, et al. Normal ovulatory women with polycystic ovaries have hyperandrogenic pituitary-ovarian responses to gonadotropin-releasing hormone-agonist testing. J Clin Endocrinol Metab. 2000;85:995–1000. [DOI] [PubMed] [Google Scholar]
- 215. Johnstone EB, Rosen MP, Neril R, et al. The polycystic ovary post-Rotterdam: a common, age-dependent finding in ovulatory women without metabolic significance. J Clin Endocrinol Metab. 2010;95:4965–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216. Hart R, Doherty DA, Norman RJ, et al. Serum antimullerian hormone (AMH) levels are elevated in adolescent girls with polycystic ovaries and the polycystic ovarian syndrome (PCOS). Fertil Steril. 2010;94:1118–1121. [DOI] [PubMed] [Google Scholar]
- 217. Villarroel C, Merino PM, López P, et al. Polycystic ovarian morphology in adolescents with regular menstrual cycles is associated with elevated anti-Mullerian hormone. Hum Reprod. 2011;26:2861–2868. [DOI] [PubMed] [Google Scholar]
- 218. Legro RS, Chiu P, Kunselman AR, Bentley CM, Dodson WC, Dunaif A. Polycystic ovaries are common in women with hyperandrogenic chronic anovulation but do not predict metabolic or reproductive phenotype. J Clin Endocrinol Metab. 2005;90:2571–2579. [DOI] [PubMed] [Google Scholar]
- 219. Murphy MK, Hall JE, Adams JM, Lee H, Welt CK. Polycystic ovarian morphology in normal women does not predict the development of polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91:3878–3884. [DOI] [PubMed] [Google Scholar]
- 220. Sjaarda LA, Mumford SL, Kissell K, et al. Increased androgen, anti-Müllerian hormone, and sporadic anovulation in healthy, eumenorrheic women: a mild PCOS-like phenotype? J Clin Endocrinol Metab. 2014;99:2208–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221. Depmann M, Faddy MJ, van der Schouw YT, et al. The relationship between variation in size of the primordial follicle pool and age at natural menopause. J Clin Endocrinol Metab. 2015;100:E845–E851. [DOI] [PubMed] [Google Scholar]
- 222. Unlu E, Unlu BS, Yildiz Y, et al. Adrenal gland volume assessed by magnetic resonance imaging in women with polycystic ovary syndrome. Diagn Interv Imaging. 2016;97:57–63. [DOI] [PubMed] [Google Scholar]
- 223. Rotter JI, Wong FL, Lifrak ET, Parker LN. A genetic component to the variation of dehydroepiandrosterone sulfate. Metabolism. 1985;34:731–736. [DOI] [PubMed] [Google Scholar]
- 224. Rice T, Sprecher DL, Borecki IB, Mitchell LE, Laskarzewski PM, Rao DC. The Cincinnati Myocardial Infarction and Hormone Family Study: family resemblance for dehydroepiandrosterone sulfate in control and myocardial infarction families. Metabolism. 1993;42:1284–1290. [DOI] [PubMed] [Google Scholar]
- 225. Louwers YV, de Jong FH, van Herwaarden NA, et al. Variants in SULT2A1 affect the DHEA sulphate to DHEA ratio in patients with polycystic ovary syndrome but not the hyperandrogenic phenotype. J Clin Endocrinol Metab. 2013;98:3848–3855. [DOI] [PubMed] [Google Scholar]
- 226. Jain A, Polotsky AJ, Rochester D, et al. Pulsatile luteinizing hormone amplitude and progesterone metabolite excretion are reduced in obese women. J Clin Endocrinol Metab. 2007;92:2468–2473. [DOI] [PubMed] [Google Scholar]
- 227. Yeung EH, Zhang C, Albert PS, et al. Adiposity and sex hormones across the menstrual cycle: the BioCycle Study. Int J Obes. 2013;37:237–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228. Roth LW, Allshouse AA, Bradshaw-Pierce EL, et al. Luteal phase dynamics of follicle-stimulating and luteinizing hormones in obese and normal weight women. Clin Endocrinol. 2014;81:418–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229. Al-Safi ZA, Liu H, Carlson NE, et al. Estradiol priming improves gonadotrope sensitivity and pro-inflammatory cytokines in obese women. J Clin Endocrinol Metab. 2015;100:4372–4381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230. Pache TD, Chadha S, Gooren LJ, et al. Ovarian morphology in long-term androgen-treated female to male transsexuals. A human model for the study of polycystic ovarian syndrome? Histopathology. 1991;19:445–452. [DOI] [PubMed] [Google Scholar]
- 231. Rosenfield RL. Polycystic ovary syndrome and insulin-resistant hyperinsulinemia. J Am Acad Dermatol. 2001;45:S095–S104. [DOI] [PubMed] [Google Scholar]
- 232. Diamanti-Kandarakis E, Dunaif A. Insulin resistance and the polycystic ovary syndrome revisited: an update on mechanisms and implications. Endocr Rev. 2012;33:981–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233. Lewy VD, Danadian K, Witchel SF, Arslanian S. Early metabolic abnormalities in adolescent girls with polycystic ovarian syndrome. J Pediatr. 2001;138:38–44. [DOI] [PubMed] [Google Scholar]
- 234. Poretsky L, Seto-Young D, Shrestha A, et al. Phosphatidyl-inositol-3 kinase-independent insulin action pathway(s) in the human ovary. J Clin Endocrinol Metab. 2001;86:3115–3119. [DOI] [PubMed] [Google Scholar]
- 235. Jakimiuk AJ, Weitsman SR, Navab A, Magoffin DA. Luteinizing hormone receptor, steroidogenesis acute regulatory protein, and steroidogenic enzyme messenger ribonucleic acids are overexpressed in thecal and granulosa cells from polycystic ovaries. J Clin Endocrinol Metab. 2001;86:1318–1323. [DOI] [PubMed] [Google Scholar]
- 236. McAllister JM, Legro RS, Modi BP, Strauss JF., 3rd Functional genomics of PCOS: from GWAS to molecular mechanisms. Trends Endocrinol Metab. 2015;26:118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237. Rosenfield RL, Ghai K, Ehrmann DA, Barnes RB. Diagnosis of polycystic ovary syndrome in adolescence: comparison of adolescent and adult hyperandrogenism. J Pediatr Endocrinol Metab. 2000;13:1285–1289. [PubMed] [Google Scholar]
- 238. Azziz R, Black V, Hines GA, Fox LM, Boots LR. Adrenal androgen excess in the polycystic ovary syndrome: sensitivity and responsivity of the hypothalamic-pituitary-adrenal axis. J Clin Endocrinol Metab. 1998;83:2317–2323. [DOI] [PubMed] [Google Scholar]
- 239. Goodarzi MO, Carmina E, Azziz R. DHEA, DHEAS and PCOS. J Ster Biochem Mol Biol. 2015;145:213–225. [DOI] [PubMed] [Google Scholar]
- 240. Barnes RB, Ehrmann DA, Brigell DF, Rosenfield RL. Ovarian steroidogenic responses to the gonadotropin-releasing hormone agonist nafarelin in hirsute women thought to have 3β-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab. 1993;76:450–455. [DOI] [PubMed] [Google Scholar]
- 241. Lutfallah C, Wang W, Mason JI, et al. Newly proposed hormonal criteria via genotypic proof for type II 3β-hydroxysteroid dehydrogenase deficiency. J Clin Endocrinol Metab. 2002;87:2611–2622. [DOI] [PubMed] [Google Scholar]
- 242. Carbunaru G, Prasad P, Scoccia B, et al. The hormonal phenotype of nonclassic 3 β-hydroxysteroid dehydrogenase (HSD3B) deficiency in hyperandrogenic females is associated with insulin-resistant polycystic ovary syndrome and is not a variant of inherited HSD3B2 deficiency. J Clin Endocrinol Metab. 2004;89:783–794. [DOI] [PubMed] [Google Scholar]
- 243. Gonzalez F, Chang L, Horab T, Lobo RA. Evidence for heterogeneous etiologies of adrenal dysfunction in polycystic ovary syndrome. Fertil Steril. 1996;66:354–361. [PubMed] [Google Scholar]
- 244. Rosenfield RL. Evidence that idiopathic functional adrenal hyperandrogenism is caused by dysregulation of adrenal steroidogenesis and that hyperinsulinemia may be involved. J Clin Endocrinol Metab. 1996;81:878–880. [DOI] [PubMed] [Google Scholar]
- 245. Arslanian SA, Lewy V, Danadian K, Saad R. Metformin therapy in obese adolescents with polycystic ovary syndrome and impaired glucose tolerance: amelioration of exaggerated adrenal response to adrenocorticotropin with reduction of insulinemia/insulin resistance. J Clin Endocrinol Metab. 2002;87:1555–1559. [DOI] [PubMed] [Google Scholar]
- 246. Mai K, Bobbert T, Reinecke F, et al. Intravenous lipid and heparin infusion-induced elevation in free fatty acids and triglycerides modifies circulating androgen levels in women: a randomized, controlled trial. J Clin Endocrinol Metab. 2008;93:3900–3906. [DOI] [PubMed] [Google Scholar]
- 247. Bellanger S, Battista MC, Fink GD, Baillargeon JP. Saturated fatty acid exposure induces androgen overproduction in bovine adrenal cells. Steroids. 2012;77:347–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248. Ditkoff EC, Fruzzetti F, Chang L, Stancyzk FZ, Lobo RA. The impact of estrogen on adrenal androgen sensitivity and secretion in polycystic ovary syndrome. J Clin Endocrinol Metab. 1995;80:603–607. [DOI] [PubMed] [Google Scholar]
- 249. Topor LS, Asai M, Dunn J, Majzoub JA. Cortisol stimulates secretion of dehydroepiandrosterone in human adrenocortical cells through inhibition of 3βHSD2. J Clin Endocrinol Metab. 2011;96:E31–E39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250. Vassiliadi DA, Barber TM, Hughes BA, et al. Increased 5 α-reductase activity and adrenocortical drive in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94:3558–3566. [DOI] [PubMed] [Google Scholar]
- 251. Gambineri A, Vicennati V, Genghini S, et al. Genetic variation in 11β-hydroxysteroid dehydrogenase type 1 predicts adrenal hyperandrogenism among lean women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91:2295–2302. [DOI] [PubMed] [Google Scholar]
- 252. Michael AE, Glenn C, Wood PJ, Webb RJ, Pellatt L, Mason HD. Ovarian 11β-hydroxysteroid dehydrogenase (11βHSD) activity is suppressed in women with anovulatory polycystic ovary syndrome (PCOS): apparent role for ovarian androgens. J Clin Endocrinol Metab. 2013;98:3375–3383. [DOI] [PubMed] [Google Scholar]
- 253. Zhu R, Zhou X, Chen Y, Qiu C, Xu W, Shen Z. Aberrantly increased mRNA expression of betaglycan, an inhibin co-receptor in the ovarian tissues in women with polycystic ovary syndrome. J Obstet Gynaecol Res. 2010;36:138–146. [DOI] [PubMed] [Google Scholar]
- 254. Gilling-Smith C, Willis DS, Beard RW, Franks S. Hypersecretion of androstenedione by isolated theca cells from polycystic ovaries. J Clin Endocrinol Metab. 1994;79:1158–1165. [DOI] [PubMed] [Google Scholar]
- 255. Magarelli PC, Zachow RJ, Magoffin DA. Developmental and hormonal regulation of rat theca-cell differentiation factor secretion in ovarian follicles. Biol Reprod. 1996;55:416–420. [DOI] [PubMed] [Google Scholar]
- 256. Liu C, Peng J, Matzuk MM, Yao HH. Lineage specification of ovarian theca cells requires multicellular interactions via oocyte and granulosa cells. Nat Commun. 2015;6:6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257. Hughesdon PE. Morphology and morphogenesis of the Stein-Leventhal ovary and of so-called “hyperthecosis.” Obstet Gynecol Surv. 1982;37:59–77. [DOI] [PubMed] [Google Scholar]
- 258. Webber LJ, Stubbs S, Stark J, et al. Formation and early development of follicles in the polycystic ovary. Lancet. 2003;362:1017–1021. [DOI] [PubMed] [Google Scholar]
- 259. Webber LJ, Stubbs SA, Stark J, et al. Prolonged survival in culture of preantral follicles from polycystic ovaries. J Clin Endocrinol Metab. 2007;92:1975–1978. [DOI] [PubMed] [Google Scholar]
- 260. Wei LN, Huang R, Li LL, Fang C, Li Y, Liang XY. Reduced and delayed expression of GDF9 and BMP15 in ovarian tissues from women with polycystic ovary syndrome. J Assist Reprod Genet. 2014;31:1483–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261. Pellatt L, Hanna L, Brincat M, et al. Granulosa cell production of anti-Mullerian hormone is increased in polycystic ovaries. J Clin Endocrinol Metab. 2007;92:240–245. [DOI] [PubMed] [Google Scholar]
- 262. Alebić MŠ, Stojanović N, Duhamel A, Dewailly D. The phenotypic diversity in per-follicle anti-Mullerian hormone production in polycystic ovary syndrome. Hum Reprod. 2015;30:1927–1933. [DOI] [PubMed] [Google Scholar]
- 263. Mason HD, Willis DS, Beard RW, Winston RM, Margara R, Franks S. Estradiol production by granulosa cells of normal and polycystic ovaries: relationship to menstrual cycle history and concentrations of gonadotropins and sex steroids in follicular fluid. J Clin Endocrinol Metab. 1994;79:1355–1360. [DOI] [PubMed] [Google Scholar]
- 264. Coffler MS, Patel K, Dahan MH, et al. Evidence for abnormal granulosa cell responsiveness to follicle-stimulating hormone in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88:1742–1747. [DOI] [PubMed] [Google Scholar]
- 265. Zachos NC, Billiar RB, Albrecht ED, Pepe GJ. Developmental regulation of follicle-stimulating hormone receptor messenger RNA expression in the baboon fetal ovary. Biol Reprod. 2003;68:1911–1917. [DOI] [PubMed] [Google Scholar]
- 266. Lofrano-Porto A, Barra GB, Giacomini LA, et al. Luteinizing hormone β mutation and hypogonadism in men and women. N Engl J Med. 2007;357:897–904. [DOI] [PubMed] [Google Scholar]
- 267. Scheele F, Hompes PG, van der Meer M, Schoute E, Schoemaker J. The effects of a gonadotrophin-releasing hormone agonist on treatment with low dose follicle stimulating hormone in polycystic ovary syndrome. Hum Reprod. 1993;8:699–704. [DOI] [PubMed] [Google Scholar]
- 268. el-Roeiy A, Chen X, Roberts VJ, et al. Expression of the genes encoding the insulin-like growth factors (IGF-I and II), the IGF and insulin receptors, and IGF- binding proteins-1–6 and the localization of their gene products in normal and polycystic ovary syndrome ovaries. J Clin Endocrinol Metab. 1994;78:1488–1496. [DOI] [PubMed] [Google Scholar]
- 269. Hendriks ML, Ket JC, Hompes PG, Homburg R, Lambalk CB. Why does ovarian surgery in PCOS help? Insight into the endocrine implications of ovarian surgery for ovulation induction in polycystic ovary syndrome. Hum Reprod Update. 2007;13:249–264. [DOI] [PubMed] [Google Scholar]
- 270. Hu ZX, Qiao J, Li MZ, et al. [The differential expression profile of polycystic ovary syndrome associated genes]. Beijing Da Xue Xue Bao. 2004;36:600–604. [PubMed] [Google Scholar]
- 271. Kaur S, Archer KJ, Devi MG, Kriplani A, Strauss JF, 3rd, Singh R. Differential gene expression in granulosa cells from polycystic ovary syndrome patients with and without insulin resistance: identification of susceptibility gene sets through network analysis. J Clin Endocrinol Metab. 2012;97:E2016–E2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272. Lan CW, Chen MJ, Tai KY, et al. Functional microarray analysis of differentially expressed genes in granulosa cells from women with polycystic ovary syndrome related to MAPK/ERK signaling. Sci Rep. 2015;5:14994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273. Wood JR, Dumesic DA, Abbott DH, Strauss JF., 3rd Molecular abnormalities in oocytes from women with polycystic ovary syndrome revealed by microarray analysis. J Clin Endocrinol Metab. 2007;92:705–713. [DOI] [PubMed] [Google Scholar]
- 274. Ludwig DS, Currie J. The association between pregnancy weight gain and birthweight: a within-family comparison. Lancet. 2010;376:984–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275. Kdous M, Chaker A, Zhioua A, Zhioua F. [Oocyte and embryo quality and outcome of ICSI cycles in patients with polycystic ovary syndrome (PCOS) versus normo-ovulatory]. J Gynecol Obstet Biol Reprod (Paris). 2009;38:133–143. [DOI] [PubMed] [Google Scholar]
- 276. Grundy SM, Brewer HB, Jr, Cleeman JI, Smith SC, Jr, Lenfant C. Definition of metabolic syndrome: report of the National Heart, Lung, and Blood Institute/American Heart Association conference on scientific issues related to definition. Circulation. 2004;109:433–438. [DOI] [PubMed] [Google Scholar]
- 277. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005;365:1415–1428. [DOI] [PubMed] [Google Scholar]
- 278. Apridonidze T, Essah PA, Iuorno MJ, Nestler JE. Prevalence and characteristics of the metabolic syndrome in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2005;90:1929–1935. [DOI] [PubMed] [Google Scholar]
- 279. Coviello AD, Legro RS, Dunaif A. Adolescent girls with polycystic ovary syndrome have an increased risk of the metabolic syndrome associated with increasing androgen levels independent of obesity and insulin resistance. J Clin Endocrinol Metab. 2006;91:492–497. [DOI] [PubMed] [Google Scholar]
- 280. Leibel NI, Baumann EE, Kocherginsky M, Rosenfield RL. Relationship of adolescent polycystic ovary syndrome to parental metabolic syndrome. J Clin Endocrinol Metab. 2006;91:1275–1283. [DOI] [PubMed] [Google Scholar]
- 281. Nandalike K, Strauss T, Agarwal C, et al. Screening for sleep-disordered breathing and excessive daytime sleepiness in adolescent girls with polycystic ovarian syndrome. J Pediatr. 2011;159:591–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282. Rossi B, Sukalich S, Droz J, et al. Prevalence of metabolic syndrome and related characteristics in obese adolescents with and without polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93:4780–4786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. DeUgarte CM, Bartolucci AA, Azziz R. Prevalence of insulin resistance in the polycystic ovary syndrome using the homeostasis model assessment. Fertil Steril. 2005;83:1454–1460. [DOI] [PubMed] [Google Scholar]
- 284. Kahn SE. Clinical review 135: the importance of β-cell failure in the development and progression of type 2 diabetes. J Clin Endocrinol Metab. 2001;86:4047–4058. [DOI] [PubMed] [Google Scholar]
- 285. Stepto NK, Cassar S, Joham AE, et al. Women with polycystic ovary syndrome have intrinsic insulin resistance on euglycaemic-hyperinsulaemic clamp. Hum Reprod. 2013;28:777–784. [DOI] [PubMed] [Google Scholar]
- 286. Robinson S, Kiddy D, Gelding SV, et al. The relationship of insulin insensitivity to menstrual pattern in women with hyperandrogenism and polycystic ovaries. Clin Endocrinol. 1993;39:351–355. [DOI] [PubMed] [Google Scholar]
- 287. Legro RS, Dodson WC, Kris-Etherton PM, et al. Randomized controlled trial of preconception interventions in infertile women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2015;100:4048–4058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288. Turkmen S, Ahangari A, Bäckstrom T. Roux-en-Y gastric bypass surgery in patients with polycystic ovary syndrome and metabolic syndrome. Obes Surg. 2016;26:111–118. [DOI] [PubMed] [Google Scholar]
- 289. Ehrmann DA, Sturis J, Byrne MM, Karrison T, Rosenfield RL, Polonsky KS. Insulin secretory defects in polycystic ovary syndrome. Relationship to insulin sensitivity and family history of non-insulin-dependent diabetes mellitus. J Clin Invest. 1995;96:520–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290. Dunaif A, Finegood DT. β-Cell dysfunction independent of obesity and glucose intolerance in the polycystic ovary syndrome. J Clin Endocrinol Metab. 1996;81:942–947. [DOI] [PubMed] [Google Scholar]
- 291. Ehrmann DA, Barnes RB, Rosenfield RL, Cavaghan MK, Imperial J. Prevalence of impaired glucose tolerance and diabetes in women with polycystic ovary syndrome. Diabetes Care. 1999;22:141–146. [DOI] [PubMed] [Google Scholar]
- 292. Colilla S, Cox NJ, Ehrmann DA. Heritability of insulin secretion and insulin action in women with polycystic ovary syndrome and their first degree relatives. J Clin Endocrinol Metaab. 2001;86:2027–2031. [DOI] [PubMed] [Google Scholar]
- 293. Dunaif A, Segal KR, Shelley DR, Green G, Dobrjansky A, Licholai T. Evidence for distinctive and intrinsic defects in insulin action in polycystic ovary syndrome. Diabetes. 1992;41:1257–1266. [DOI] [PubMed] [Google Scholar]
- 294. Corbould A, Kim YB, Youngren JF, et al. Insulin resistance in the skeletal muscle of women with PCOS involves intrinsic and acquired defects in insulin signaling. Am J Physiol Endocrinol Metab. 2005;288:E1047–E1054. [DOI] [PubMed] [Google Scholar]
- 295. Tosi F, Di Sarra D, Kaufman JM, et al. Total body fat and central fat mass independently predict insulin resistance but not hyperandrogenemia in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2015;100:661–669. [DOI] [PubMed] [Google Scholar]
- 296. Book CB, Dunaif A. Selective insulin resistance in the polycystic ovary syndrome. J Clin Endocrinol Metab. 1999;84:3110–3116. [DOI] [PubMed] [Google Scholar]
- 297. Nestler JE, Jakubowicz DJ, de Vargas AF, Brik C, Quintero N, Medina F. Insulin stimulates testosterone biosynthesis by human thecal cells from women with polycystic ovary syndrome by activating its own receptor and using inositolglycan mediators as the signal transduction system. J Clin Endocrinol Metab. 1998;83:2001–2005. [DOI] [PubMed] [Google Scholar]
- 298. Willis D, Franks S. Insulin action in human granulosa cells from normal and polycystic ovaries is mediated by the insulin receptor and not the type-I insulin-like growth factor receptor. J Clin Endocrinol Metab. 1995;80:3788–3790. [DOI] [PubMed] [Google Scholar]
- 299. DiVall SA, Herrera D, Sklar B, et al. Insulin receptor signaling in the GnRH neuron plays a role in the abnormal GnRH pulsatility of obese female mice. PLoS One. 2015;10:e0119995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300. Kristiansen SB, Endoh A, Casson PR, Buster JE, Hornsby PJ. Induction of steroidogenic enzyme genes by insulin and IGF-I in cultured adult human adrenocortical cells. Steroids. 1997;62:258–265. [DOI] [PubMed] [Google Scholar]
- 301. Lungu AO, Zadeh ES, Goodling A, Cochran E, Gorden P. Insulin resistance is a sufficient basis for hyperandrogenism in lipodystrophic women with polycystic ovarian syndrome. J Clin Endocrinol Metab. 2012;97:563–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302. Kaltsas GA, Androulakis II, Tziveriotis K, et al. Polycystic ovaries and the polycystic ovary syndrome phenotype in women with active acromegaly. Clin Endocrinol. 2007;67:917–922. [DOI] [PubMed] [Google Scholar]
- 303. Flier JS, Moller DE, Moses AC, et al. Insulin-mediated pseudoacromegaly: clinical and biochemical characterization of a syndrome of selective insulin resisitance. J Clin Endocrinol Metab. 1993;76:1533–1541. [DOI] [PubMed] [Google Scholar]
- 304. Conn JJ, Jacobs HS, Conway GS. The prevalence of polycystic ovaries in women with type 2 diabetes mellitus. Clin Endocrinol. 2000;52:81–86. [DOI] [PubMed] [Google Scholar]
- 305. Peppard HR, Marfori J, Iuorno MJ, Nestler JE. Prevalence of polycystic ovary syndrome among premenopausal women with type 2 diabetes. Diabetes Care. 2001;24:1050–1052. [DOI] [PubMed] [Google Scholar]
- 306. Codner E, Escobar-Morreale HF. Clinical review: hyperandrogenism and polycystic ovary syndrome in women with type 1 diabetes mellitus. J Clin Endocrinol Metab. 2007;92:1209–1216. [DOI] [PubMed] [Google Scholar]
- 307. Nestler JE, Jakubowicz DJ. Decreases in ovarian cytochrome P450c17a activity and serum free testosterone after reduction of insulin secretion in women with polycystic ovary syndrome. N Engl J Med. 1996;335:617–623. [DOI] [PubMed] [Google Scholar]
- 308. Eid GM, Cottam DR, Velcu LM, et al. Effective treatment of polycystic ovarian syndrome with Roux-en-Y gastric bypass. Surg Obes Relat Dis. 2005;1:77–80. [DOI] [PubMed] [Google Scholar]
- 309. Moran LJ, Noakes M, Clifton PM, Norman RJ. The use of anti-mullerian hormone in predicting menstrual response after weight loss in overweight women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2007;92:3796–3802. [DOI] [PubMed] [Google Scholar]
- 310. Harborne L, Fleming R, Lyall H, Norman J, Sattar N. Descriptive review of the evidence for the use of metformin in polycystic ovary syndrome. Lancet. 2003;361:1894–1901. [DOI] [PubMed] [Google Scholar]
- 311. Cosma M, Swiglo BA, Flynn DN, et al. Clinical review: insulin sensitizers for the treatment of hirsutism: a systematic review and metaanalyses of randomized controlled trials. J Clin Endocrinol Metab. 2008;93:1135–1142. [DOI] [PubMed] [Google Scholar]
- 312. Tang T, Glanville J, Hayden CJ, White D, Barth JH, Balen AH. Combined lifestyle modification and metformin in obese patients with polycystic ovary syndrome. A randomized, placebo-controlled, double-blind multicentre study. Hum Reprod. 2006;21:80–89. [DOI] [PubMed] [Google Scholar]
- 313. Tang T, Lord JM, Norman RJ, Yasmin E, Balen AH. Insulin-sensitising drugs (metformin, rosiglitazone, pioglitazone, D-chiro-inositol) for women with polycystic ovary syndrome, oligo amenorrhoea and subfertility. Cochrane Database Syst Rev. 2012;(5):CD003053. [DOI] [PubMed] [Google Scholar]
- 314. Papaleo E, Unfer V, Baillargeon JP, et al. Myo-inositol in patients with polycystic ovary syndrome: a novel method for ovulation induction. Gynecol Endocrinol. 2007;23:700–703. [DOI] [PubMed] [Google Scholar]
- 315. Facchinetti F, Bizzarri M, Benvenga S, et al. Results from the International Consensus Conference on Myo-inositol and d-chiro-inositol in Obstetrics and Gynecology: the link between metabolic syndrome and PCOS. Europ J Obstet Gynecol Reprod Biol. 2015;195:72–76. [DOI] [PubMed] [Google Scholar]
- 316. Ibáñez L, Díaz M, Sebastiani G, Marcos MV, López-Bermejo A, de Zegher F. Oral contraception vs insulin sensitization for 18 months in nonobese adolescents with androgen excess: posttreatment differences in C-reactive protein, intima-media thickness, visceral adiposity, insulin sensitivity, and menstrual regularity. J Clin Endocrinol Metab. 2013;98:E902–E907. [DOI] [PubMed] [Google Scholar]
- 317. Ibáñez L, Ong KK, López-Bermejo A, Dunger DB, de Zegher F. Hyperinsulinaemic androgen excess in adolescent girls. Nat Rev Endocrinol. 2014;10:499–508. [DOI] [PubMed] [Google Scholar]
- 318. Matthews DR, Hosker JP, Rudenski AS, Naylor BA, Treacher DF, Turner RC. Homeostasis model assessment: insulin resistance and β-cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia. 1985;28:412–419. [DOI] [PubMed] [Google Scholar]
- 319. Ibanez L, Potau N, Zampolli M, et al. Hyperinsulinemia in postpubertal girls with a history of premature pubarche and functional ovarian hyperandrogenism. J Clin Endocrinol Metab. 1996;81:1237–1243. [DOI] [PubMed] [Google Scholar]
- 320. Baillargeon JP, Jakubowicz DJ, Iuorno MJ, Jakubowicz S, Nestler JE. Effects of metformin and rosiglitazone, alone and in combination, in nonobese women with polycystic ovary syndrome and normal indices of insulin sensitivity. Fertil Steril. 2004;82:893–902. [DOI] [PubMed] [Google Scholar]
- 321. Pau CT, Keefe C, Duran J, Welt C. Metformin improves glucose effectiveness, not insulin sensitivity: predicting treatment response in women with polycystic ovary syndrome in an open-label, interventional study. J Clin Endocrinol Metab. 2014;99:1870–1878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322. Miller RA, Chu Q, Xie J, Foretz M, Viollet B, Birnbaum MJ. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature. 2013;494:256–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 323. Pernicova I, Korbonits M. Metformin–mode of action and clinical implications for diabetes and cancer. Nat Rev Endocrinol. 2014;10:143–156. [DOI] [PubMed] [Google Scholar]
- 324. Madiraju AK, Erion DM, Rahimi Y, et al. Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014;510:542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325. Buse JB, DeFronzo RA, Rosenstock J, et al. The primary glucose-lowering effect of metformin resides in the gut, not the circulation: results from short-term pharmacokinetic and 12-week dose-ranging studies. Diabetes Care. 2015;39:198–205. [DOI] [PubMed] [Google Scholar]
- 326. Legro RS, Barnhart HX, Schlaff WD, et al. Ovulatory response to treatment of polycystic ovary syndrome is associated with a polymorphism in the STK11 gene. J Clin Endocrinol Metab. 2008;93:792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327. Kurzthaler D, Hadziomerovic-Pekic D, Wildt L, Seeber BE. Metformin induces a prompt decrease in LH-stimulated testosterone response in women with PCOS independent of its insulin-sensitizing effects. Reprod Biol Endocrinol. 2014;12:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 328. Di Pietro M, Parborell F, Irusta G, et al. Metformin regulates ovarian angiogenesis and follicular development in a female polycystic ovary syndrome rat model. Endocrinology. 2015;156:1453–1463. [DOI] [PubMed] [Google Scholar]
- 329. Rice S, Elia A, Jawad Z, Pellatt L, Mason HD. Metformin inhibits follicle-stimulating hormone (FSH) action in human granulosa cells: relevance to polycystic ovary syndrome. J Clin Endocrinol Metab. 2013;98:E1491–E1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330. Orban JC, Fontaine E, Ichai C. Metformin overdose: time to move on. Crit Care. 2012;16:164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331. Jensterle M, Kravos NA, Pfeifer M, Kocjan T, Janez A. A 12-week treatment with the long-acting glucagon-like peptide 1 receptor agonist liraglutide leads to significant weight loss in a subset of obese women with newly diagnosed polycystic ovary syndrome. Hormones. 2015;14:81–90. [DOI] [PubMed] [Google Scholar]
- 332. Jensterle Sever M, Kocjan T, Pfeifer M, Kravos NA, Janez A. Short-term combined treatment with liraglutide and metformin leads to significant weight loss in obese women with polycystic ovary syndrome and previous poor response to metformin. Eur J Endocrinol. 2014;170:451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Outeiriño-Iglesias V, Romaní-Pérez M, González-Matías LC, Vigo E, Mallo F. GLP-1 increases preovulatory LH source and the number of mature follicles, as well as synchronizing the onset of puberty in female rats. Endocrinology. 2015;156:4226–4237. [DOI] [PubMed] [Google Scholar]
- 334. Kahn CR. Knockout mice challenge our concepts of glucose homeostasis and the pathogenesis of diabetes. Exp Diabesity Res. 2003;4:169–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335. Rosen ED, Spiegelman BM. What we talk about when we talk about fat. Cell. 2014;156:20–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336. Ciaraldi TP, el-Roeiy A, Madar Z, Reichart D, Olefsky JM, Yen SS. Cellular mechanisms of insulin resistance in polycystic ovarian syndrome. J Clin Endocrinol Metab. 1992;75:577–583. [DOI] [PubMed] [Google Scholar]
- 337. Ciaraldi TP, Morales AJ, Hickman MG, Odom-Ford R, Olefsky JM, Yen SS. Cellular insulin resistance in adipocytes from obese polycystic ovary syndrome subjects involves adenosine modulation of insulin sensitivity. J Clin Endocrinol Metab. 1997;82:1421–1425. [DOI] [PubMed] [Google Scholar]
- 338. Marsden PJ, Murdoch A, Taylor R. Severe impairment of insulin action in adipocytes from amenorrheic subjects with polycystic ovary syndrome. Metabolism. 1994;43:1536–1542. [DOI] [PubMed] [Google Scholar]
- 339. Rosenbaum D, Haber RS, Dunaif A. Insulin resistance in polycystic ovary syndrome: decreased expression of GLUT-4 glucose transporters in adipocytes. Am J Physiol. 1993;264:E197–E202. [DOI] [PubMed] [Google Scholar]
- 340. Chen YH, Heneidi S, Lee JM, et al. miRNA-93 inhibits GLUT4 and is overexpressed in adipose tissue of polycystic ovary syndrome patients and women with insulin resistance. Diabetes. 2013;62:2278–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341. Corbould A, Dunaif A. The adipose cell lineage is not intrinsically insulin resistant in polycystic ovary syndrome. Metabolism. 2007;56:716–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342. Corbould A. Chronic testosterone treatment induces selective insulin resistance in subcutaneous adipocytes of women. J Endocrinol. 2007;192:585–594. [DOI] [PubMed] [Google Scholar]
- 343. Corbould A. Effects of androgens on insulin action in women: is androgen excess a component of female metabolic syndrome? Diabetes Metab Res Rev. 2008;24:520–532. [DOI] [PubMed] [Google Scholar]
- 344. Varlamov O, White AE, Carroll JM, et al. Androgen effects on adipose tissue architecture and function in nonhuman primates. Endocrinology. 2012;153:3100–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345. Capllonch-Amer G, Lladó I, Proenza AM, García-Palmer FJ, Gianotti M. Opposite effects of 17-β estradiol and testosterone on mitochondrial biogenesis and adiponectin synthesis in white adipocytes. J Mol Endocrinol. 2014;52:203–214. [DOI] [PubMed] [Google Scholar]
- 346. Huang ZH, Manickam B, Ryvkin V, et al. PCOS is associated with increased CD11c expression and crown-like structures in adipose tissue and increased central abdominal fat depots independent of obesity. J Clin Endocrinol Metab. 2013;98:E17–E24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347. Li S, Huang X, Zhong H, et al. Low circulating adiponectin levels in women with polycystic ovary syndrome: an updated meta-analysis. Tumour Biol. 2014;35:3961–3973. [DOI] [PubMed] [Google Scholar]
- 348. Chazenbalk G, Singh P, Irge D, Shah A, Abbott DH, Dumesic DA. Androgens inhibit adipogenesis during human adipose stem cell commitment to preadipocyte formation. Steroids. 2013;78:920–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349. Keller E, Chazenbalk GD, Aguilera P, et al. Impaired preadipocyte differentiation into adipocytes in subcutaneous abdominal adipose of PCOS-like female rhesus monkeys. Endocrinology. 2014;155:2696–2703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350. Polderman KH, Gooren LJ, Asscheman H, Bakker A, Heine RJ. Induction of insulin resistance by androgens and estrogens. J Clin Endocrinol Metab. 1994;79:265–271. [DOI] [PubMed] [Google Scholar]
- 351. Varlamov O, Chu MP, McGee WK, et al. Ovarian cycle-specific regulation of adipose tissue lipid storage by testosterone in female nonhuman primates. Endocrinology. 2013;154:4126–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352. Upreti R, Hughes KA, Livingstone DE, et al. 5α-reductase type 1 modulates insulin sensitivity in men. J Clin Endocrinol Metab. 2014;99:E1397–E1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353. Livingstone DE, Barat P, Di Rollo EM, et al. 5α-Reductase type 1 deficiency or inhibition predisposes to insulin resistance, hepatic steatosis, and liver fibrosis in rodents. Diabetes. 2015;64:447–458. [DOI] [PubMed] [Google Scholar]
- 354. Lee MJ, Pramyothin P, Karastergiou K, Fried SK. Deconstructing the roles of glucocorticoids in adipose tissue biology and the development of central obesity. Biochim Biophys Acta. 2014;1842:473–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355. Morgan SA, McCabe EL, Gathercole LL, et al. 11β-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc Natl Acad Sci USA. 2014;111:E2482–E2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356. Ehrmann DA, Breda E, Corcoran MC, et al. Impaired β-cell compensation to dexamethasone-induced hyperglycemia in women with polycystic ovary syndrome. Am J Physiol Endocrinol Metab. 2004;287:E241–E246. [DOI] [PubMed] [Google Scholar]
- 357. Chapman K, Holmes M, Seckl J. 11β-hydroxysteroid dehydrogenases: intracellular gate-keepers of tissue glucocorticoid action. Physiol Rev. 2013;93:1139–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358. Desarzens S, Faresse N. Adipocyte glucocorticoid receptor has a minor contribution in adipose tissue growth. J Endocrinol. 2016;230:1–11. [DOI] [PubMed] [Google Scholar]
- 359. Carmina E, Koyama T, Chang L, Stanczyk FZ, Lobo RA. Does ethnicity influence the prevalence of adrenal hyperandrogenism and insulin resistance in polycystic ovary syndrome? Am J Obstet Gynecol. 1992;167:1807–1812. [DOI] [PubMed] [Google Scholar]
- 360. Ezeh U, Yildiz BO, Azziz R. Referral bias in defining the phenotype and prevalence of obesity in polycystic ovary syndrome. J Clin Endocrinol Metab. 2013;98:E1088–E1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361. Kirchengast S, Huber J. Body composition characteristics and body fat distribution in lean women with polycystic ovary syndrome. Hum Reprod. 2001;16:1255–1260. [DOI] [PubMed] [Google Scholar]
- 362. Barber TM, Golding SJ, Alvey C, et al. Global adiposity rather than abnormal regional fat distribution characterises women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93:999–1004. [DOI] [PubMed] [Google Scholar]
- 363. Mannerås-Holm L, Leonhardt H, Kullberg J, et al. Adipose tissue has aberrant morphology and function in PCOS: enlarged adipocytes and low serum adiponectin, but not circulating sex steroids, are strongly associated with insulin resistance. J Clin Endocrinol Metab. 2011;96:E304–E311. [DOI] [PubMed] [Google Scholar]
- 364. Borruel S, Fernández-Durán E, Alpañés M, et al. Global adiposity and thickness of intraperitoneal and mesenteric adipose tissue depots are increased in women with polycystic ovary syndrome (PCOS). J Clin Endocrinol Metab. 2013;98:1254–1263. [DOI] [PubMed] [Google Scholar]
- 365. Arner P. Effects of testosterone on fat cell lipolysis. Species differences and possible role in polycystic ovarian syndrome. Biochimie. 2005;87:39–43. [DOI] [PubMed] [Google Scholar]
- 366. Ek I, Arner P, Rydén M, et al. A unique defect in the regulation of visceral fat cell lipolysis in the polycystic ovary syndrome as an early link to insulin resistance. Diabetes. 2002;51:484–492. [DOI] [PubMed] [Google Scholar]
- 367. Faulds G, Rydén M, Ek I, Wahrenberg H, Arner P. Mechanisms behind lipolytic catecholamine resistance of subcutaneous fat cells in the polycystic ovarian syndrome. J Clin Endocrinol Metab. 2003;88:2269–2273. [DOI] [PubMed] [Google Scholar]
- 368. Dicker A, Rydén M, Näslund E, et al. Effect of testosterone on lipolysis in human pre-adipocytes from different fat depots. Diabetologia. 2004;47:420–428. [DOI] [PubMed] [Google Scholar]
- 369. Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. Lancet. 2010;375:2267–2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370. Chen DL, Liess C, Poljak A, et al. Phenotypic characterization of insulin-resistant and insulin-sensitive obesity. J Clin Endocrinol Metab. 2015;100:4082–4091. [DOI] [PubMed] [Google Scholar]
- 371. Varlamov O, White A, Takahashi DL, McCurdy CE, Roberts CT., Jr Hyperandrogenemia induces adipose tissue dysfunction in adolescent female rhesus macaques consuming a western-style diet. Endocr Rev. 2016;37:Abst SUN–167. [Google Scholar]
- 372. González F, Sia CL, Shepard MK, Rote NS, Minium J. The altered mononuclear cell-derived cytokine response to glucose ingestion is not regulated by excess adiposity in polycystic ovary syndrome. J Clin Endocrinol Metab. 2014;99:E2244–E2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373. Shorakae S, Teede H, de Courten B, Lambert G, Boyle J, Moran LJ. The emerging role of chronic low-grade inflammation in the pathophysiology of polycystic ovary syndrome. Semin Reprod Med. 2015;33:257–269. [DOI] [PubMed] [Google Scholar]
- 374. González F, Sia CL, Bearson DM, Blair HE. Hyperandrogenism induces a proinflammatory TNFα response to glucose ingestion in a receptor-dependent fashion. J Clin Endocrinol Metab. 2014;99:E848–E854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375. Tasali E, Van Cauter E, Ehrmann DA. Relationships between sleep disordered breathing and glucose metabolism in polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91:36–42. [DOI] [PubMed] [Google Scholar]
- 376. Tasali E, Chapotot F, Leproult R, Whitmore H, Ehrmann DA. Treatment of obstructive sleep apnea improves cardiometabolic function in young obese women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2011;96:365–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 377. Vgontzas AN, Legro RS, Bixler EO, Grayev A, Kales A, Chrousos GP. Polycystic ovary syndrome is associated with obstructive sleep apnea and daytime sleepiness: role of insulin resistance. J Clin Endocrinol Metab. 2001;86:517–520. [DOI] [PubMed] [Google Scholar]
- 378. Broussard JL, Ehrmann DA, Van Cauter E, Tasali E, Brady MJ. Impaired insulin signaling in human adipocytes after experimental sleep restriction: a randomized, crossover study. Ann Intern Med. 2012;157:549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379. Ehrmann DA. Metabolic dysfunction in PCOS: relationship to obstructive sleep apnea. Steroids. 2012;77:290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380. Narkiewicz K, Montano N, Cogliati C, van de Borne PJ, Dyken ME, Somers VK. Altered cardiovascular variability in obstructive sleep apnea. Circulation. 1998;98:1071–1077. [DOI] [PubMed] [Google Scholar]
- 381. Sverrisdóttir YB, Mogren T, Kataoka J, Janson PO, Stener-Victorin E. Is polycystic ovary syndrome associated with high sympathetic nerve activity and size at birth? Am J Physiol Endocrinol Metab. 2008;294:E576–E581. [DOI] [PubMed] [Google Scholar]
- 382. Lambert EA, Teede H, Sari CI, et al. Sympathetic activation and endothelial dysfunction in polycystic ovary syndrome are not explained by either obesity or insulin resistance. Clin Endocrinol. 2015;83:812–819. [DOI] [PubMed] [Google Scholar]
- 383. Holte J, Bergh T, Gennarelli G, Wide L. The independent effects of polycystic ovary syndrome and obesity on serum concentrations of gonadotrophins and sex steroids in premenopausal women. Clin Endocrinol. 1994;41:473–481. [DOI] [PubMed] [Google Scholar]
- 384. Taylor AE, McCourt B, Martin KA, et al. Determinants of abnormal gonadotropin secretion in clinically defined women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1997;82:2248–2256. [DOI] [PubMed] [Google Scholar]
- 385. Arroyo A, Laughlin GA, Morales AJ, Yen SS. Inappropriate gonadotropin secretion in polycystic ovary syndrome: influence of adiposity. J Clin Endocrinol Metab. 1997;82:3728–3733. [DOI] [PubMed] [Google Scholar]
- 386. Rochester D, Jain A, Polotsky AJ, et al. Partial recovery of luteal function after bariatric surgery in obese women. Fertil Steril. 2009;92:1410–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387. Pagán YL, Srouji SS, Jimenez Y, Emerson A, Gill S, Hall JE. Inverse relationship between luteinizing hormone and body mass index in polycystic ovarian syndrome: investigation of hypothalamic and pituitary contributions. J Clin Endocrinol Metab. 2006;91:1309–1316. [DOI] [PubMed] [Google Scholar]
- 388. Srouji SS, Pagán YL, D'Amato F, et al. Pharmacokinetic factors contribute to the inverse relationship between luteinizing hormone and body mass index in polycystic ovarian syndrome. J Clin Endocrinol Metab. 2007;92:1347–1352. [DOI] [PubMed] [Google Scholar]
- 389. Wide L, Eriksson K, Sluss PM, Hall JE. Serum half-life of pituitary gonadotropins is decreased by sulfonation and increased by sialylation in women. J Clin Endocrinol Metab. 2009;94:958–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390. Wide L, Naessén T, Sundström-Poromaa I, Eriksson K. Sulfonation and sialylation of gonadotropins in women during the menstrual cycle, after menopause, and with polycystic ovarian syndrome and in men. J Clin Endocrinol Metab. 2007;92:4410–4417. [DOI] [PubMed] [Google Scholar]
- 391. Mi Y, Fiete D, Baenziger JU. Ablation of GalNAc-4-sulfotransferase-1 enhances reproduction by altering the carbohydrate structures of luteinizing hormone in mice. J Clin Invest. 2008;118:1815–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392. Eagleson CA, Bellows AB, Hu K, Gingrich MB, Marshall JC. Obese patients with polycystic ovary syndrome: evidence that metformin does not restore sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by ovarian steroids. J Clin Endocrinol Metab. 2003;88:5158–5162. [DOI] [PubMed] [Google Scholar]
- 393. Lawson MA, Jain S, Sun S, Patel K, Malcolm PJ, Chang RJ. Evidence for insulin suppression of baseline luteinizing hormone in women with polycystic ovarian syndrome and normal women. J Clin Endocrinol Metab. 2008;93:2089–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394. Mehta RV, Patel KS, Coffler MS, et al. Luteinizing hormone secretion is not influenced by insulin infusion in women with polycystic ovary syndrome despite improved insulin sensitivity during pioglitazone treatment. J Clin Endocrinol Metab. 2005;90:2136–2141. [DOI] [PubMed] [Google Scholar]
- 395. Zumoff B, Miller LK, Strain GW. Reversal of the hypogonadotropic hypogonadism of obese men by administration of the aromatase inhibitor testolactone. Metabolism. 2003;52:1126–1128. [DOI] [PubMed] [Google Scholar]
- 396. Lucky AW, Rebar RW, Rosenfield RL, Roche-Bender N, Helke J. Reduction of the potency of luteinizing hormone by estrogen. N Engl J Med. 1979;300:1034–1036. [DOI] [PubMed] [Google Scholar]
- 397. Taponen S, Martikainen H, Järvelin MR, et al. Hormonal profile of women with self-reported symptoms of oligomenorrhea and/or hirsutism: Northern Finland birth cohort 1966 study. J Clin Endocrinol Metab. 2003;88:141–147. [DOI] [PubMed] [Google Scholar]
- 398. Strain GW, Zumoff B, Miller LK, Rosner W. Sex difference in the effect of obesity on 24-hour mean serum gonadotropin levels. Horm Metab Res. 2003;35:362–366. [DOI] [PubMed] [Google Scholar]
- 399. Teede HJ, Misso ML, Deeks AA, et al. Assessment and management of polycystic ovary syndrome: summary of an evidence-based guideline. Med J Austral. 2011;195:S65–S112. [DOI] [PubMed] [Google Scholar]
- 400. Jamal M, Gunay Y, Capper A, Eid A, Heitshusen D, Samuel I. Roux-en-Y gastric bypass ameliorates polycystic ovary syndrome and dramatically improves conception rates: a 9-year analysis. Surg Obes Relat Dis. 2012;8:440–444. [DOI] [PubMed] [Google Scholar]
- 401. O'Reilly MW, Taylor AE, Crabtree NJ, et al. Hyperandrogenemia predicts metabolic phenotype in polycystic ovary syndrome: the utility of serum androstenedione. J Clin Endocrinol Metab. 2014;99:1027–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402. McCartney CR, Gingrich MB, Hu Y, Evans WS, Marshall JC. Hypothalamic regulation of cyclic ovulation: evidence that the increase in gonadotropin-releasing hormone pulse frequency during the follicular phase reflects the gradual loss of the restraining effects of progesterone. J Clin Endocrinol Metab. 2002;87:2194–2200. [DOI] [PubMed] [Google Scholar]
- 403. Pastor CL, Griffin-Korf ML, Aloi JA, Evans WS, Marshall JC. Polycystic ovary syndrome: evidence for reduced sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 1998;83:582–590. [DOI] [PubMed] [Google Scholar]
- 404. Eagleson CA, Gingrich MB, Pastor CL, et al. Polycystic ovarian syndrome: evidence that flutamide restores sensitivity of the gonadotropin-releasing hormone pulse generator to inhibition by estradiol and progesterone. J Clin Endocrinol Metab. 2000;85:4047–4052. [DOI] [PubMed] [Google Scholar]
- 405. Ropelato MG, Rudaz MC, Escobar ME, et al. Acute effects of testosterone infusion on the serum luteinizing hormone profile in eumenorrheic and polycystic ovary syndrome adolescents. J Clin Endocrinol Metab. 2009;94:3602–3610. [DOI] [PubMed] [Google Scholar]
- 406. Cimino I, Casoni F, Liu X, et al. Novel role for anti-Mullerian hormone in the regulation of GnRH neuron excitability and hormone secretion. Nat Commun. 2016;7:10055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407. Zimmer CA, Ehrmann DA, Rosenfield RL. Potential diagnostic utility of intermittent administration of short-acting GnRH agonist administration in gonadotropin deficiency. Fertil Steril. 2010;94:2697–2702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408. Rege J, Nakamura Y, Satoh F, et al. Liquid chromatography-tandem mass spectrometry analysis of human adrenal vein 19-carbon steroids before and after ACTH stimulation. J Clin Endocrinol Metab. 2013;98:1182–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409. Campana C, Rege J, Turcu AF, et al. Development of a novel cell based androgen screening model. J Ster Biochem Mol Biol. 2016;156:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410. Marti N, Galván J, Pandey AV, et al. The backdoor androgen biosynthesis pathway exists in the human ovary and seems altered in PCOS. Endocr Rev. 2016;37:Abst SUN–224. [Google Scholar]
- 411. Rosenfield RL, Lawrence AM, Liao S, Landau RL. Androgens and androgen responsiveness in the feminizing testis syndrome. Comparison of complete and “incomplete” forms. J Clin Endocrinol Metab. 1971;32:625–632. [DOI] [PubMed] [Google Scholar]
- 412. Kayampilly PP, Wanamaker BL, Stewart JA, Wagner CL, Menon KM. Stimulatory effect of insulin on 5α-reductase type 1 (SRD5A1) expression through an Akt-dependent pathway in ovarian granulosa cells. Endocrinology. 2010;151:5030–5037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413. Goodarzi MO, Shah NA, Antoine HJ, Pall M, Guo X, Azziz R. Variants in the 5α-reductase type 1 and type 2 genes are associated with polycystic ovary syndrome and the severity of hirsutism in affected women. J Clin Endocrinol Metab. 2006;91:4085–4091. [DOI] [PubMed] [Google Scholar]
- 414. Hedstrom H, Backstrom T, Bixo M, et al. Women with polycystic ovary syndrome have elevated serum concentrations of and altered GABA(A) receptor sensitivity to allopregnanolone. Clin Endocrinol. 2015;83:643–650. [DOI] [PubMed] [Google Scholar]
- 415. Wang F, Pan J, Liu Y, et al. Alternative splicing of the androgen receptor in polycystic ovary syndrome. Proc Natl Acad Sci USA. 2015;112:4743–4748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416. Walters KA, Handelsman DJ. Androgen receptor splice variants and polycystic ovary syndrome: cause or effect? Asian J Androl. 2016;18:442–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417. Escobar-Morreale HF, Luque-Ramírez M, San Millán JL. The molecular-genetic basis of functional hyperandrogenism and the polycystic ovary syndrome. Endocr Rev. 2005;26:251–282. [DOI] [PubMed] [Google Scholar]
- 418. Zhang T, Liang W, Fang M, Yu J, Ni Y, Li Z. Association of the CAG repeat polymorphisms in androgen receptor gene with polycystic ovary syndrome: a systemic review and meta-analysis. Gene. 2013;524:161–167. [DOI] [PubMed] [Google Scholar]
- 419. Vink JM, Sadrzadeh S, Lambalk CB, Boomsma DI. Heritability of polycystic ovary syndrome in a Dutch twin-family study. J Clin Endocrinol Metab. 2006;91:2100–2104. [DOI] [PubMed] [Google Scholar]
- 420. Legro RS, Driscoll D, Strauss JF, 3rd, Fox J, Dunaif A. Evidence for a genetic basis for hyperandrogenemia in polycystic ovary syndrome. Proc Natl Acad Sci USA. 1998;95:14956–14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 421. Franks S, Webber LJ, Goh M, et al. Ovarian morphology is a marker of heritable biochemical traits in sisters with polycystic ovaries. J Clin Endocrinol Metab. 2008;93:3396–3402. [DOI] [PubMed] [Google Scholar]
- 422. Govind A, Obhrai MS, Clayton RN. Polycystic ovaries are inherited as an autosomal dominant trait: analysis of 29 polycystic ovary syndrome and 10 control families. J Clin Endocrinol Metab. 1999;84:38–43. [DOI] [PubMed] [Google Scholar]
- 423. Kahsar-Miller MD, Nixon C, Boots LR, Go RC, Azziz R. Prevalence of polycystic ovary syndrome (PCOS) in first-degree relatives of patients with PCOS. Fertil Steril. 2001;75:53–58. [DOI] [PubMed] [Google Scholar]
- 424. Sam S, Legro RS, Essah PA, Apridonidze T, Dunaif A. Evidence for metabolic and reproductive phenotypes in mothers of women with polycystic ovary syndrome. Proc Natl Acad Sci USA. 2006;103:7030–7035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425. Sam S, Legro RS, Bentley-Lewis R, Dunaif A. Dyslipidemia and metabolic syndrome in the sisters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2005;90:4797–4802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426. Coviello AD, Sam S, Legro RS, Dunaif A. High prevalence of metabolic syndrome in first-degree male relatives of women with polycystic ovary syndrome is related to high rates of obesity. J Clin Endocrinol Metab. 2009;94:4361–4366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427. Louwers YV, Stolk L, Uitterlinden AG, Laven JS. Cross-ethnic meta-analysis of genetic variants for polycystic ovary syndrome. J Clin Endocrinol Metab. 2013;98:E2006–E2012. [DOI] [PubMed] [Google Scholar]
- 428. Williamson K, Gunn AJ, Johnson N, Milsom SR. The impact of ethnicity on the presentation of polycystic ovarian syndrome. Aust N Z J Obstet Gynaecol. 2001;41:202–206. [DOI] [PubMed] [Google Scholar]
- 429. Glintborg D, Mumm H, Hougaard D, Ravn P, Andersen M. Ethnic differences in Rotterdam criteria and metabolic risk factors in a multiethnic group of women with PCOS studied in Denmark. Clin Endocrinol. 2010;73:732–738. [DOI] [PubMed] [Google Scholar]
- 430. Givens JR. Familial polycystic ovarian disease. Endocrinol Metab Clin N Am. 1988;17:771–783. [PubMed] [Google Scholar]
- 431. Rosenfield RL. Identifying children at risk of polycystic ovary syndrome. J Clin Endocrinol Metab. 2007;92:787–796. [DOI] [PubMed] [Google Scholar]
- 432. Sir-Petermann T, Codner E, Pérez V, et al. Metabolic and reproductive features before and during puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94:1923–1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433. Maliqueo M, Sir-Petermann T, Pérez V, et al. Adrenal function during childhood and puberty in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2009;94:3282–3288. [DOI] [PubMed] [Google Scholar]
- 434. Sir-Petermann T, Codner E, Maliqueo M, et al. Increased anti-mullerian hormone serum concentrations in prepubertal daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2006;91:3105–3109. [DOI] [PubMed] [Google Scholar]
- 435. Vanky E, Carlsen SM. Androgens and antimullerian hormone in mothers with polycystic ovary syndrome and their newborns. Fertil Steril. 2012;97:509–515. [DOI] [PubMed] [Google Scholar]
- 436. Kent SC, Gnatuk CL, Kunselman AR, Demers LM, Lee PA, Legro RS. Hyperandrogenism and hyperinsulinism in children of women with polycystic ovary syndrome: a controlled study. J Clin Endocrinol Metab. 2008;93:1662–1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437. Carey AH, Chan KL, Short F, White D, Williamson R, Franks S. Evidence for a single gene effect causing polycystic ovaries and male pattern baldness. Clin Endocrinol. 1993;38:653–658. [DOI] [PubMed] [Google Scholar]
- 438. Wang JG, Lobo RA. The complex relationship between hypothalamic amenorrhea and polycystic ovary syndrome. J Clin Endocrinol Metab. 2008;93:1394–1397. [DOI] [PubMed] [Google Scholar]
- 439. Robin G, Gallo C, Catteau-Jonard S, et al. Polycystic ovary-like abnormalities (PCO-L) in women with functional hypothalamic amenorrhea. J Clin Endocrinol Metab. 2012;97:4236–4243. [DOI] [PubMed] [Google Scholar]
- 440. Povel CM, Boer JM, Feskens EJ. Shared genetic variance between the features of the metabolic syndrome: heritability studies. Mol Genet Metab. 2011;104:666–669. [DOI] [PubMed] [Google Scholar]
- 441. Waalen J. The genetics of human obesity. Transl Res. 2014;164:293–301. [DOI] [PubMed] [Google Scholar]
- 442. Yildiz BO, Yarali H, Oguz H, Bayraktar M. Glucose intolerance, insulin resistance, and hyperandrogenemia in first degree relatives of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2003;88:2031–2036. [DOI] [PubMed] [Google Scholar]
- 443. Kobaly K, Vellanki P, Sisk RK, et al. Parent-of-origin effects on glucose homeostasis in polycystic ovary syndrome. J Clin Endocrinol Metab. 2014;99:2961–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444. Urbanek M, Legro RS, Driscoll DA, et al. Thirty-seven candidate genes for polycystic ovary syndrome: strongest evidence for linkage is with follistatin. Proc Natl Acad Sci USA. 1999;96:8573–8578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 445. Goodarzi MO, Dumesic DA, Chazenbalk G, Azziz R. Polycystic ovary syndrome: etiology, pathogenesis and diagnosis. Nat Rev Endocrinol. 2011;7:219–231. [DOI] [PubMed] [Google Scholar]
- 446. Urbanek M. The genetics of the polycystic ovary syndrome. Nat Clin Pract Endocrinol Metab. 2007;3:103–111. [DOI] [PubMed] [Google Scholar]
- 447. Witchel SF, Lee PA, Suda-Hartman M, Hoffman EP. Hyperandrogenism and manifesting heterozygotes for 21-hydroxylase deficiency. Biochem Mol Med. 1997;62:151–158. [DOI] [PubMed] [Google Scholar]
- 448. Ewens KG, Stewart DR, Ankener W, et al. Family-based analysis of candidate genes for polycystic ovary syndrome. J Clin Endocrinol Metab. 2010;95:2306–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 449. Jones MR, Chazenbalk G, Xu N, et al. Steroidogenic regulatory factor FOS is underexpressed in polycystic ovary syndrome (PCOS) adipose tissue and genetically associated with PCOS susceptibility. J Clin Endocrinol Metab. 2012;97:E1750–E1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450. Boqun X, Xiaonan D, Yugui C, et al. Expression of SET protein in the ovaries of patients with polycystic ovary syndrome. Int J Endocrinol. 2013;2013:367956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451. Piltonen TT, Chen J, Erikson DW, et al. Mesenchymal stem/progenitors and other endometrial Cell types From women With polycystic ovary syndrome (PCOS) display inflammatory and oncogenic potential. J Clin Endocrinol Metab. 2013;98:3765–3775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452. Li Q, Du J, Feng R, et al. A possible new mechanism in the pathophysiology of polycystic ovary syndrome (PCOS): the discovery that leukocyte telomere length is strongly associated with PCOS. J Clin Endocrinol Metab. 2014;99:E234–E240. [DOI] [PubMed] [Google Scholar]
- 453. Shi Y, Zhao H, Shi Y, et al. Genome-wide association study identifies eight new risk loci for polycystic ovary syndrome. Nat Genet. 2012;44:1020–1025. [DOI] [PubMed] [Google Scholar]
- 454. Kosova G, Urbanek M. Genetics of the polycystic ovary syndrome. Mol Cell Endocrinol. 2013;373:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 455. Brower MA, Jones MR, Rotter JI, et al. Further investigation in Europeans of susceptibility variants for polycystic ovary syndrome discovered in genome-wide association studies of Chinese individuals. J Clin Endocrinol Metab. 2015;100:E182–E186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 456. Ha L, Shi Y, Zhao J, Li T, Chen ZJ. Association study between polycystic ovarian syndrome and the susceptibility genes polymorphisms in Hui Chinese women. PLoS One. 2015;10:e0126505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 457. Hayes MG, Urbanek M, Ehrmann DA, et al. A genome-wide association of polycystic ovary syndrome implicates alterations in gonadotropin secretion in European ancestry populations. Nat Commun. 2015;6:7502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 458. Welt CK, Styrkarsdottir U, Ehrmann DA, et al. Variants in DENND1A are associated with polycystic ovary syndrome in women of european ancestry. J Clin Endocrinol Metab. 2012;97:E1342–E1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459. Yu YY, Sun CX, Liu YK, Li Y, Wang L, Zhang W. Genome-wide screen of ovary-specific DNA methylation in polycystic ovary syndrome. Fertil Steril. 2015;104:145–153.e146. [DOI] [PubMed] [Google Scholar]
- 460. Corbett S, Morin-Papunen L. The polycystic ovary syndrome and recent human evolution. Mol Cell Endocrinol. 2013;373:39–50. [DOI] [PubMed] [Google Scholar]
- 461. Padmanabhan V, Veiga-Lopez A. Developmental origin of reproductive and metabolic dysfunctions: androgenic versus estrogenic reprogramming. Semin Reprod Med. 2011;29:173–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 462. Dumesic DA, Goodarzi MO, Chazenbalk GD, Abbott DH. Intrauterine environment and polycystic ovary syndrome. Semin Reprod Med. 2014;32:159–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463. Ghizzoni L, Virdis R, Vottero A, et al. Pituitary-ovarian responses to leuprolide acetate testing in patients with congenital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab. 1996;81:601–606. [DOI] [PubMed] [Google Scholar]
- 464. Abbott DH, Bacha F. Ontogeny of polycystic ovary syndrome and insulin resistance in utero and early childhood. Fertil Steril. 2013;100:2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465. Nicol LE, O'Brien TD, Dumesic DA, Grogan T, Tarantal AF, Abbott DH. Abnormal infant islet morphology precedes insulin resistance in PCOS-Like monkeys. PLoS One. 2014;9:e106527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466. Veiga-Lopez A, Ye W, Padmanabhan V. Developmental programming: prenatal testosterone excess disrupts anti-Mullerian hormone expression in preantral and antral follicles. Fertil Steril. 2012;97:748–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467. Caldwell AS, Eid S, Kay CR, et al. Haplosufficient genomic androgen receptor signaling is adequate to protect female mice from induction of polycystic ovary syndrome features by prenatal hyperandrogenization. Endocrinology. 2015;156:1441–1452. [DOI] [PubMed] [Google Scholar]
- 468. Nada SE, Thompson RC, Padmanabhan V. Developmental programming: differential effects of prenatal testosterone excess on insulin target tissues. Endocrinology. 2010;151:5165–5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469. Payne AH, Jaffe RB. Androgen formation from pregnenolone sulfate by the human fetal ovary. J Clin Endocrinol Metab. 1974;39:300–304. [DOI] [PubMed] [Google Scholar]
- 470. George FW, Wilson JD. Conversion of androgen to estrogen by the human fetal ovary. J Clin Endocrinol Metab. 1978;47:550–555. [DOI] [PubMed] [Google Scholar]
- 471. Rabinovici J, Jaffe R. Development and regulation of growth and differentiated function in human and subhuman primate fetal gonads. Endocr Rev. 1990;11:532. [DOI] [PubMed] [Google Scholar]
- 472. Cole B, Hensinger K, Maciel GA, Chang RJ, Erickson GF. Human fetal ovary development involves the spatiotemporal expression of p450c17 protein. J Clin Endocrinol Metab. 2006;91:3654–3661. [DOI] [PubMed] [Google Scholar]
- 473. Fowler PA, Anderson RA, Saunders PT, et al. Development of steroid signaling pathways during primordial follicle formation in the human fetal ovary. J Clin Endocrinol Metab. 2011;96:1754–1762. [DOI] [PubMed] [Google Scholar]
- 474. Peters H, Byskov A, Grinsted J. Follicular growth in fetal and prepubertal ovaries of humans and other primates. Clin Endocrinol Metab. 1978;7:469. [DOI] [PubMed] [Google Scholar]
- 475. Kuiri-Hänninen T, Kallio S, Seuri R, et al. Postnatal developmental changes in the pituitary-ovarian axis in preterm and term infant girls. J Clin Endocrinol Metab. 2011;96:3432–3439. [DOI] [PubMed] [Google Scholar]
- 476. Kuiri-Hänninen T, Haanpää M, Turpeinen U, et al. Postnatal ovarian activation has effects in estrogen target tissues in infant girls. J Clin Endocrinol Metab. 2013;98:4709–4716. [DOI] [PubMed] [Google Scholar]
- 477. Ellinwood WE, McClellan MC, Brenner RM, Resko JA. Estradiol synthesis by fetal monkey ovaries correlates with antral follicle formation. Biol Reprod. 1983;28:505–516. [DOI] [PubMed] [Google Scholar]
- 478. Wilson EA, Jawad MJ. The effect of trophic agents on fetal ovarian steroidogenesis in organ culture. Fertil Steril. 1979;32:73–79. [DOI] [PubMed] [Google Scholar]
- 479. Reyes FI, Boroditsky RS, Winter JS, Faiman C. Studies on human sexual development. II. Fetal and maternal serum gonadotropin and sex steroid concentrations. J Clin Endocrinol Metab. 1974;612–617. [DOI] [PubMed] [Google Scholar]
- 480. Beck-Peccoz P, Padmanabhan V, Baggiani AM, et al. Maturation of hypothalamic-pituitary-gonadal function in normal human fetuses: circulating levels of gonadotropins, their common α-subunit and free testosterone, and discrepancy between immunological and biological activities of circulating follicle-stimulating hormone. J Clin Endocrinol Metab. 1991;73:525–532. [DOI] [PubMed] [Google Scholar]
- 481. Eberlein WR. Steroids and sterols in umbilical cord blood. J Clin Endocrinol Metab. 1965;25:1101–1118. [DOI] [PubMed] [Google Scholar]
- 482. Wudy SA, Wachter UA, Homoki J, Teller WM. 17 α-hydroxyprogesterone, 4-androstenedione, and testosterone profiled by routine stable isotope dilution/gas chromatography-mass spectrometry in plasma of children. Pediatr Res. 1995;38:76–80. [DOI] [PubMed] [Google Scholar]
- 483. Forest MG, de Peretti E, Lecoq A, Cadillon E, Zabot MT, Thoulon JM. Concentration of 14 steroid hormones in human amniotic fluid of midpregnancy. J Clin Endocrinol Metab. 1980;51:816–822. [DOI] [PubMed] [Google Scholar]
- 484. Ventura T, Gomes MC, Pita A, Neto MT, Taylor A. Digit ratio (2D:4D) in newborns: influences of prenatal testosterone and maternal environment. Early Hum Dev. 2013;89:107–112. [DOI] [PubMed] [Google Scholar]
- 485. Fahlbusch FB, Heussner K, Schmid M, et al. Measurement of amniotic fluid steroids of midgestation via LC-MS/MS. J Ster Biochem Mol Biol. 2015;152:155–160. [DOI] [PubMed] [Google Scholar]
- 486. Caanen MR, Kuijper EA, Hompes PG, et al. Mass spectrometry methods measured androgen and estrogen concentrations during pregnancy and in newborns of mothers with polycystic ovary syndrome. Eur J Endocrinol. 2016;174:25–32. [DOI] [PubMed] [Google Scholar]
- 487. Anderson H, Fogel N, Grebe SK, Singh RJ, Taylor RL, Dunaif A. Infants of women with polycystic ovary syndrome have lower cord blood androstenedione and estradiol levels. J Clin Endocrinol Metab. 2010;95:2180–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488. Hickey M, Sloboda DM, Atkinson HC, et al. The relationship between maternal and umbilical cord androgen levels and polycystic ovary syndrome in adolescence: a prospective cohort study. J Clin Endocrinol Metab. 2009;94:3714–3720. [DOI] [PubMed] [Google Scholar]
- 489. Maliqueo M, Lara HE, Sanchez F, Echiburú B, Crisosto N, Sir-Petermann T. Placental steroidogenesis in pregnant women with polycystic ovary syndrome. Eur J Obstet Gynecol Reprod Biol. 2013;166:151–155. [DOI] [PubMed] [Google Scholar]
- 490. Koster MP, de Wilde MA, Veltman-Verhulst SM, et al. Placental characteristics in women with polycystic ovary syndrome. Hum Reprod. 2015;30:2829–2837. [DOI] [PubMed] [Google Scholar]
- 491. Palomba S, Marotta R, Di Cello A, et al. Pervasive developmental disorders in children of hyperandrogenic women with polycystic ovary syndrome: a longitudinal case-control study. Clin Endocrinol. 2012;77:898–904. [DOI] [PubMed] [Google Scholar]
- 492. Wilson JD. Androgens, androgen receptors, and male gender role behavior. Horm Behav. 2001;40:358–366. [DOI] [PubMed] [Google Scholar]
- 493. Lee PA, Houk CP, Ahmed SF, Hughes IA. Consensus statement on management of intersex disorders. International Consensus Conference on Intersex. Pediatrics. 2006;118:e488–e500. [DOI] [PubMed] [Google Scholar]
- 494. Brunner F, Fliegner M, Krupp K, Rall K, Brucker S, Richter-Appelt H. Gender role, gender identity and sexual orientation in CAIS (“XY-women”) compared with subfertile and infertile 46,XX women. J Sex Res. 2016;53:109–124. [DOI] [PubMed] [Google Scholar]
- 495. Abbott AD, Colman RJ, Tiefenthaler R, Dumesic DA, Abbott DH. Early-to-mid gestation fetal testosterone increases right hand 2D:4D finger length ratio in polycystic ovary syndrome-like monkeys. PLoS One. 2012;7:e42372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 496. Torchen LC, Idkowiak J, Fogel NR, et al. Evidence for increased 5α-reductase activity during early childhood in daughters of women with polycystic ovary syndrome. J Clin Endocrinol Metab. 2016;101:2069–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 497. Barker DJ, Eriksson JG, Forsén T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol. 2002;31:1235–1239. [DOI] [PubMed] [Google Scholar]
- 498. Heijmans BT, Tobi EW, Stein AD, et al. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci USA. 2008;105:17046–17049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 499. Ibáñez L, Potau N, Francois I, de Zegher F. Precocious pubarche, hyperinsulinism, and ovarian hyperandrogenism: relation to reduced fetal growth. J Clin Endocrinol Metab. 1998;83:3558–3562. [DOI] [PubMed] [Google Scholar]
- 500. Pandolfi C, Zugaro A, Lattanzio F, et al. Low birth weight and later development of insulin resistance and biochemical/clinical features of polycystic ovary syndrome. Metabolism. 2008;57:999–1004. [DOI] [PubMed] [Google Scholar]
- 501. Melo AS, Vieira CS, Barbieri MA, et al. High prevalence of polycystic ovary syndrome in women born small for gestational age. Hum Reprod. 2010;25:2124–2131. [DOI] [PubMed] [Google Scholar]
- 502. Davies MJ, March WA, Willson KJ, Giles LC, Moore VM. Birthweight and thinness at birth independently predict symptoms of polycystic ovary syndrome in adulthood. Hum Reprod. 2012;27:1475–1480. [DOI] [PubMed] [Google Scholar]
- 503. Cresswell JL, Barker DJ, Osmond C, Egger P, Phillips DI, Fraser RB. Fetal growth, length of gestation, and polycystic ovaries in adult life. Lancet. 1997;350:1131–1135. [DOI] [PubMed] [Google Scholar]
- 504. Michelmore K, Ong K, Mason S, et al. Clinical features in women with polycystic ovaries: relationships to insulin sensitivity, insulin gene VNTR and birth weight. Clin Endocrinol. 2001;55:439–446. [DOI] [PubMed] [Google Scholar]
- 505. Sadrzadeh S, Klip WA, Broekmans FJ, et al. Birth weight and age at menarche in patients with polycystic ovary syndrome or diminished ovarian reserve, in a retrospective cohort. Hum Reprod. 2003;18:2225–2230. [DOI] [PubMed] [Google Scholar]
- 506. Laitinen J, Taponen S, Martikainen H, et al. Body size from birth to adulthood as a predictor of self-reported polycystic ovary syndrome symptoms. Int J Obes. 2003;27:710–715. [DOI] [PubMed] [Google Scholar]
- 507. Legro RS, Roller RL, Dodson WC, Stetter CM, Kunselman AR, Dunaif A. Associations of birthweight and gestational age with reproductive and metabolic phenotypes in women with polycystic ovarian syndrome and their first-degree relatives. J Clin Endocrinol Metab. 2010;95:789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508. Fulghesu AM, Manca R, Loi S, Fruzzetti F. Insulin resistance and hyperandrogenism have no substantive association with birth weight in adolescents with polycystic ovary syndrome. Fertil Steril. 2015;103:808–814. [DOI] [PubMed] [Google Scholar]
- 509. Franks S. Controversy in Clin Endocrinol: diagnosis of polycystic ovarian syndrome: in defense of the Rotterdam criteria. J Clin Endocrinol Metab. 2006;91:786–789. [DOI] [PubMed] [Google Scholar]
- 510. Ehrmann DA, Schneider DJ, Sobel BE, et al. Troglitazone improves defects in insulin action, insulin secretion, ovarian steroidogenesis, and fibrinolysis in women with polycystic ovary syndrome. J Clin Endocrinol Metab. 1997;82:2108–2116. [DOI] [PubMed] [Google Scholar]
- 511. Nestler JE, Jakubowicz DJ, Evans WS, Pasquali R. Effects of metformin on spontaneous and clomiphene-induced ovulation in the polycystic ovary syndrome. N Engl J Med. 1998;338:1876–1880. [DOI] [PubMed] [Google Scholar]
- 512. Lord JM, Flight IH, Norman RJ. Insulin-sensitising drugs (metformin, troglitazone, rosiglitazone, pioglitazone, D-chiro-inositol) for polycystic ovary syndrome. Cochrane Database Syst Rev. 2003;CD003053. [DOI] [PubMed] [Google Scholar]
- 513. James WP. WHO recognition of the global obesity epidemic. Int J Obes (Lond). 2008;32(suppl 7):S120–S126. [DOI] [PubMed] [Google Scholar]
- 514. Pasquali R, Antenucci D, Casimirri F, et al. Clinical and hormonal characteristics of obese amenorrheic hyperandrogenic women before and after weight loss. J Clin Endocrinol Metab. 1989;68:173–179. [DOI] [PubMed] [Google Scholar]
- 515. Huber-Buchholz MM, Carey DG, Norman RJ. Restoration of reproductive potential by lifestyle modification in obese polycystic ovary syndrome: role of insulin sensitivity and luteinizing hormone. J Clin Endocrinol Metab. 1999;84:1470–1474. [DOI] [PubMed] [Google Scholar]
- 516. Kiddy DS, Hamilton-Fairley D, Bush A, et al. Improvement in endocrine and ovarian function during dietary treatment of obese women with polycystic ovary syndrome. Clin Endocrinol. 1992;36:105–111. [DOI] [PubMed] [Google Scholar]
- 517. Bloch CA, Clemons P, Sperling MA. Puberty decreases insulin sensitivity. J Pediatr. 1987;110:481–487. [DOI] [PubMed] [Google Scholar]
- 518. Caprio S, Jones T, Tamborlane W. Developmental changes in insulin action and secretion in childhood health and disease. Adv Endocrinol Metab. 1994;5:171–201. [Google Scholar]
- 519. Caprio S. Insulin: the other anabolic hormone of puberty. Acta Paediatr Suppl. 1999;88:84–87. [DOI] [PubMed] [Google Scholar]
- 520. Moran A, Jacobs DR, Jr, Steinberger J, et al. Insulin resistance during puberty: results from clamp studies in 357 children. Diabetes. 1999;48:2039–2044. [DOI] [PubMed] [Google Scholar]
- 521. van Houten EL, Kramer P, McLuskey A, Karels B, Themmen AP, Visser JA. Reproductive and metabolic phenotype of a mouse model of PCOS. Endocrinology. 2012;153:2861–2869. [DOI] [PubMed] [Google Scholar]
- 522. Nobels F, Dewailly D. Puberty and polycystic ovarian syndrome: the insulin/insulin-like growth factor I hypothesis. Fertil Steril. 1992;58:655–666. [DOI] [PubMed] [Google Scholar]
- 523. Carel JC, Eugster EA, Rogol A, et al. Consensus statement on the use of gonadotropin-releasing hormone analogs in children. Pediatrics. 2009;123:e752–e762. [DOI] [PubMed] [Google Scholar]
- 524. Chiavaroli V, Liberati M, D'Antonio F, et al. GNRH analog therapy in girls with early puberty is associated with the achievement of predicted final height but also with increased risk of polycystic ovary syndrome. Eur J Endocrinol. 2010;163:55–62. [DOI] [PubMed] [Google Scholar]
- 525. Ibañez L, Potau N, Virdis R, et al. Postpubertal outcome in girls diagnosed of premature pubarche during childhood: increased frequency of functional ovarian hyperandrogenism. J Clin Endocrinol Metab. 1993;76:1599–1603. [DOI] [PubMed] [Google Scholar]
- 526. Nelson-DeGrave VL, Wickenheisser JK, Cockrell JE, et al. Valproate potentiates androgen biosynthesis in human ovarian theca cells. Endocrinology. 2004;145:799–808. [DOI] [PubMed] [Google Scholar]
- 527. Herzog AG. Menstrual disorders in women with epilepsy. Neurology. 2006;66:S23–S28. [DOI] [PubMed] [Google Scholar]
- 528. Kandaraki E, Chatzigeorgiou A, Livadas S, et al. Endocrine disruptors and polycystic ovary syndrome (PCOS): elevated serum levels of bisphenol A in women with PCOS. J Clin Endocrinol Metab. 2011;96:E480–E484. [DOI] [PubMed] [Google Scholar]
- 529. Yang Q, Zhao Y, Qiu X, Zhang C, Li R, Qiao J. Association of serum levels of typical organic pollutants with polycystic ovary syndrome (PCOS): a case-control study. Hum Reprod. 2015;30:1964–1973. [DOI] [PubMed] [Google Scholar]
- 530. Azziz R, Dumesic DA, Goodarzi MO. Polycystic ovary syndrome: an ancient disorder? Fertil Steril. 2011;95:1544–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531. Casarini L, Brigante G. The polycystic ovary syndrome evolutionary paradox: a GWAS-based, in silico, evolutionary explanation. J Clin Endocrinol Metab. 2014;99:E2412–E2420. [DOI] [PubMed] [Google Scholar]