

Abstract
Comparison of 11 human nuclear receptor amino acid sequences revealed a conserved phosphorylation motif within their DNA-binding domains as an intramolecular signal that regulates proteolytic degradation. Nuclear receptors use this signal to either degrade or proscribe degradation through either the proteasome or nonproteasome pathways. A phosphomimetic farnesoid X receptor (FXR) S154D mutant neither bound to nor trans-activated an FXR-response element-driven reporter gene and was rapidly degraded in COS-1 cells. Ectopically expressed FXR had increased Ser154 phosphorylation in COS-1 cells after ligand treatment, and knock-down of the nuclear vaccinia-related kinase 1 (VRK1) greatly reduced this phosphorylation. FXR was phosphorylated at Ser154 in the nucleus of centrilobular hepatocytes only in ligand-treated mice. Thus, FXR Ser154 phosphorylation is a rheostat for activation and subsequent degradation that controls receptor levels and activity.
Phosphorylation of nuclear receptors results in diverse biological functions that can be distinct from transcriptional control (1). Antibodies against phospho-peptides were used to detect phosphorylation of endogenous nuclear receptors in tissues. For example, the androgen receptor (AR) is phosphorylated at Ser210 in prostate cancer cells or in spinal and bulbar muscular atrophy tissue (2, 3). Estrogen receptor (ER)α Ser118 or Ser305 are phosphorylated in human breast cancers (4, 5). The phosphorylation status of AR and ERα has been used as diagnostic markers for cancer progression (1). However, the general significance of phosphorylation at these sites in other nuclear receptors is questionable, because the modified amino acid at AR Ser210 and ERα Ser118 or Ser305 are not conserved. It was first suggested with Ser51 of vitamin D receptor (VDR) that a protein kinase C (PKC) phosphorylation site was conserved between the 2 zinc fingers within the DNA-binding domain (DBD) of nuclear receptors (6). In fact, sequence alignments revealed that this motif is conserved in 41 out of the total 46 human nuclear receptors (Supplemental Figure 1). Mutation studies of this motif revealed that some nuclear receptors, such as VDR Ser51, hepatocyte nuclear receptor (HNF)4α Ser78, and farnesoid X receptor (FXR) Ser154, could affect transactivation activity or proteolytic degradation (6–9). Thus, this conserved phosphorylation site provides an experimental opportunity to determine whether it elicits a common regulatory signal that can be implicated in most nuclear receptors.
Phosphorylation of this motif was previously confirmed only with 2 nuclear receptors constitutive androstane receptor (CAR) (at Thr38) and ERα (at Ser216) in tissues in vivo (10, 11). Notably, Thr38 is involved in the molecular mechanism of CAR activation in the liver (12, 13), whereas Ser216 was implicated in the ERα-mediated regulation of neutrophils in mouse uterus (11). However, the regulatory functions observed in CAR and ERα are unique to each nuclear receptor and not conserved in other receptors. In an attempt to investigate a common regulatory role for phosphorylation, phosphomimetic and nonphosphomimetic mutants were produced at Ser or Thr residues in the conserved motif of 11 nuclear receptors and the properties of these mutants determined. Wild-type and mutated nuclear receptors were expressed in COS-1 cells with or without proteasome inhibitor MG132 to examine whether or not phosphorylation is able to regulate their expression levels. Domain-based deletion mutants were generated to define the domains regulating the phosphorylation-based expressions of nuclear receptors. With the finding that phosphorylation of the DBD at the conserved motif is a protein degradation signal, FXR (Ser154) was selected for further characterization of the mechanism through which phosphorylation regulates FXR function as a transcription factor. Moreover, a phospho-Ser154 peptide antibody was employed to demonstrate that FXR can, in fact, be phosphorylated in COS-1 cells as well as in mouse livers in a ligand-dependent manner. The phosphorylation-based mechanism by which ligand enables FXR to link its transactivation and inactivation and subsequent degradation will be discussed.
Materials and Methods
Materials
The antibodies against FXR (PP-A9033A-00), HNF4α (PP-K9218-00), PPARα (PP-H0723-00), TRα (PP-H2804-00), and Rev-erbα (PP-A8740A-00) were obtained from Perseus Proteomics, Inc. Antibody against FXR (ab28676) recognizing the C-terminal region and antibody to GFP-HRP (ab6663) were purchased from Abcam. An antibody against GFP (632381) was obtained from Clontech Laboratories, Inc, and antibodies against VDR (sc-1009), retinoid X receptor (RXR)α (sc-553), retinoic acid receptor-related orphan receptor (ROR)α (sc-6062), ERα (sc-7207), β-actin (sc-47778), ubiquitin (sc-9133), and TBP (sc-273) were from Santa Cruz Biotechnology, Inc. Antibody to V5 (46-0705), TOPO TA cloning kit with pCR2.1 TOPO, pcDNA3.1/V5-His TOPO TA Expression kit, Dynabeads Protein G, and Lipofectamine 2000 were from Invitrogen. Antibody against HSP90 (610419) was purchased from BD Transduction Laboratories. Anti-P-S154 peptide antibody against human FXR was generated by GeneScript. PrimeSTAR Max DNA Polymerase and LA-taq DNA polymerase were obtained from Takara. The human VDR cDNA (clone ID, 30343975) was from GE Healthcare, COS-1 cells (CRL-1650) were from the American Type Culture Collection, and FuGENE 6 was from Roche Diagnostics. The Bio-Rad Protein Assay was obtained from Bio-Rad. WesternBright ECL and Sirius HRP substrate were purchased from Advansta. The Protein kinase C and Dual Luciferase Assay System were from Promega. The phosphatase inhibitor cocktail 2 and 3 and chenodeoxycholic acid (CDCA) were obtained from Sigma-Aldrich. The ligand GW4064 was from Tocris Bioscience. DNA polymerase Klenow fragment and restriction enzymes were purchased from New England Biolabs, and the pHACE vector harboring constitutively active human PKCα (Plasmid 21234) was from Addgene. ON-TARGETplus SMART pool human VRK1 siRNA and ON-TARGETplus Non-Targeting pool were from Dharmacon Research.
Animal maintenance and treatment
Fxr-null mice (14) on a C57BL/6N background were housed in a pathogen-free animal facility under a standard 12-hour light, 12-hour dark cycle and given pelleted NIH-31 chow diet and water ad libitum. Male mice and their wild-type counterparts age matched were received ip injections of either DMSO/corn oil or GW4064 (30 mg/kg) once per day for 4 days. Four hours after the final injection, the mice were killed and liver tissues harvested and divided into 2 parts: one part was immediately frozen in liquid nitrogen and the other treated with 10% neutral formalin for histological evaluation. Liver tissues were kept at −80°C until use. All mouse studies were carried out in accordance with Institute of Laboratory Animal Resources guidelines and protocols approved by the National Cancer Institute Animal Care and Use Committee.
Plasmids
A given DNA was amplified and cloned into pCR2.1 or pcDNA3.1. GFP-hCAR/pEGFP-C1, hERα/pcDNA3.1, and hRXRα/pcDNA3.1 were previously constructed (10, 15, 16). TRα, PPARα, and Rev-erbα were amplified from human brain cDNA and human liver cDNA libraries, respectively, and cloned into pcDNA3.1. Human VDR cDNA (clone ID, 30343975) was cloned into pCR3. Proper primer sets were used to introduce deletions or base mutations or to construct chimeric mutants using a QuikChange Site-Directed Mutagenesis system (Stratagene). All constructs were verified by nucleotide sequencing. In addition, plasmids used were Nr0b2-#UTR/pGL4-tk, phRL-tk (Promega), hFXR WT/pCMV-ICIS, hRORα WT/pcDNA3.1, hHNF4α/pcDNA3.1, hSF1(Ad4BP)/pcDNA3.1, pCW7-Myc-Ub, and FLAG-hVRK1/pcDNA3.1.
Cell culture, transfection, and fractionation
COS-1 cells were grown and maintained in DMEM supplemented with 10% fetal bovine serum, 100-U/mL penicillin, and 100-μg/mL streptomycin at 37°C in a humidified atmosphere containing 5% CO2. FuGENE 6 or Lipofectamine 2000 was used to transfect cells according to a manufacturer's instruction. For cell fractionations, COS-1 cells (5.0 × 105 cells/6-cm dish) were washed and scraped into PBS. The harvested cells were dissolved in 10mM HEPES-NaOH buffer (pH7.5), 10mM KCl, 1.5mM MgCl2, protease inhibitors, and 0.3% NP-40 to prepare whole-cell extracts. Whole-cell extracts were homogenized using glass homogenizer and centrifuged, from which supernatants were used as cytosolic extracts. The resulting precipitates were washed with the above-mentioned buffer but without NP-40 and resuspended in 10mM HEPES-NaOH (pH 7.5) buffer containing 400mM NaCl, 100mM KCl, 3mM MgCl2, 20% glycerol, and protease inhibitor at 4°C for 30 minutes. The suspensions were centrifuged at 100 000g for 30 minutes at 4°C to prepare nuclear extracts. Protein concentrations were determined by the Bradford assay.
Western blottings
Ten micrograms of proteins were electrophoresed on a 10% or 12.5% SDS-PAGE gel and transferred onto polyvinylidene difluoride membranes at 10 V for 1 hour. The membranes were probed with a given antibody overnight at 4°C, reacted with horseradish peroxidase-conjugated goat antirabbit, monkey antimouse, or donkey antigoat IgG secondary antibody using WesternBright ECL or Sirius kit, exposed to a x-ray film, or visualized by a C-DiGit Chemiluminescent Western Blot Scanner (LI-COR, Inc).
In vitro phosphorylation assays
Four micrograms of purified bacterially expressed GST-hFXR WT or GST-hFXR S154A were incubated with 50 mU of PKC in 30 μL of TBS buffer containing 2mM CaCl2, 2mM dithiothreitol, 17mM MgCl2, 0.6-mg/mL phosphatidyl serine, and 170μM ATP in the presence or absence of PKC at 30°C for 1 hour and were subjected to Western blot analysis using the anti-P-S154 peptide antibody in 5% BSA solution (1:500 dilution).
Ubiquitination and Co-IP assays
For detection of ubiquitinated proteins, assays were performed as previously reported with minor modifications (17). COS-1 cells transfected with expression plasmids for 24 hours were homogenized in lysis buffer 2% SDS, 150mM NaCl, 10mM Tris-HCl (pH 8.0), protease inhibitors, boiled to inactivate the deubiquitinating enzyme, sonicated, and diluted in 10mM Tris-HCl buffer (pH 8.0) containing 150mM NaCl, 1% Triton X-100, and protease inhibitors. These homogenates were incubated at 4°C for 30 minutes with rotation and centrifuged to obtain supernatants for subsequent analyses. The supernatants (500-μg protein) were incubated with FXR antibody-conjugated Dynabeads Protein G at 4°C for 12 hours, and the beads were recovered and washed with PBS containing 0.02% Tween 20. The beads were treated in 1× SDS sample buffer (25 μL) and loaded onto a SDS-PAGE gel for Western blot analysis with an anti-ubiquitin antibody. For the Co-IP assays, the cells were lysed in IP buffer containing 50mM Tris-HCl (pH 8.0), 150mM NaCl, 1% NP-40, protease inhibitors, and phosphatase inhibitor cocktails. After IP with anti-GFP antibody conjugated agarose resin at 4°C for 3 hours, the resin was washed with the above IP buffer, subjected to SDS-PAGE for Western blotting.
Luciferase reporter assays
COS-1 cells were cotransfected with a given FXR expression vector and Nr0b2-#UTR/pGL4-TK (18) and control Renilla luciferase plasmids for 24 hours before ligand treatments for an additional 24 hours. The treatments were 50μM CDCA or 5μM GW4064 for FXR. Cell lysates were prepared to measure firefly luciferase activities using Dual Luciferase Assay System and normalized with Renilla luciferase activities.
Gel-shift assays
Gel mobility shift assays were conducted as described in a previous study (10). Briefly, a 5′-overhanging double stranded oligonucleotides were filled in with DNA polymerase Klenow fragment in the presence of [α-32P] dATP to produce radioactive probes; 5′-GATCTCAAGAGGTCATTGACCTTTTTG for FXR response element (FXRE). For supershifts, the mixtures were incubated with 1 μg of anti-FXR at room temperature for 10 minutes.
cDNA microarrays
COS-1 cells were transfected with hFXR S154A or hFXR S154D for 24 hours and then were treated with 50μM CDCA or DMSO for control for an additional 24 hours. RNA purification, cDNA synthesis, microarrays, and data analysis were performed as previously described (16). The GEO accession for the analysis was assigned GSE82278.
Immunohistochemistry
Paraffin-embedded liver sections (6 μm) from DMSO- or GW4064-treated mice were deparaffinized and hydrated. For antigen unmasking, the sections were heated at 120°C for 3 minutes and then blocked with 3% H2O2 for 15 minutes. Sections were incubated in anti-P-S154 peptide antibody for FXR at a 1:50 dilution and biotinylated secondary antibody according to the Vectastain Elite ABC kit (Vector laboratories). The sections were treated with ExtrAvidin-peroxidase (Sigma) for 20 minutes, incubated with 3,3′-diaminobenzidine (Agilent Technologies) to visualize a color signal, dehydrated, and cover slipped.
Results
Phosphorylation motif and degradation
Specific Thr or Ser residues that constitute the conserved phosphorylation motifs within the DBDs of selected human nuclear receptors were investigated in the present study (Figure 1A). These residues include FXR Ser154. In vitro kinase assays, using bacterially expressed glutathione S-transferase (GST)-tagged proteins as substrates and pure PKC as enzyme, confirmed that Ser154 could be phosphorylated and detected by Western blottings with anti-P-S154 peptide antibody (Figure 1B). Wild-type FXR, FXR S154A, and FXR S154D were ectopically expressed in COS-1 cells, and whole-cell extracts were prepared for Western blot analysis. The phosphomimetic FXR S154D mutant was poorly expressed compared with the wild-type and the nonphosphomimetic FXR S154A mutant (Figure 1C). Moreover, MG132 treatment of cells expressing FXR S154D restored FXR protein to levels observed with the wild-type FXR and FXR S154A mutant. These results indicated that the phosphomimetic mutation is correlated with enhanced FXR degradation through proteasomes. As expected, the FXR S154D mutant markedly decreased expression compared with the wild-type FXR and FXR S154A in the presence of cycloheximide in COS-1 cells (Figure 1D).
Figure 1. Conserved phosphorylation of FXR.

A, Schematic diagram of the conserved phosphorylation site between 2 zinc fingers within the DBD among nuclear receptors. B, In vitro phosphorylation of the Ser154 in hFXR by recombinant PKCs. GST-hFXRs were purified and used as the substrate. After phosphorylation, the proteins were subjected to Western blotting using anti-P-S154 peptide antibody. C, Transient expression of hFXR WT, S154A, and S154D in COS-1 cells. Twenty-four hours after transfection, the cells were treated with either DMSO (0.1%) or 10μM MG132 for 3 hours and then were harvested for Western blot analysis. D, Protein stabilities. COS-1 cells were transfected with expression plasmids for hFXR WT, S154A, or S154D for 24 hours and then were treated with 10μM MG132 for 3 hours to make at equal the protein levels. Subsequently, 50-μg/mL cycloheximide (CHX) was added to media for given time, from which cells were collected for Western blot analysis.
Phosphorylation as a protein degradation signal
A mutation study with FXR suggested that phosphorylation of this conserved motif within the DBD functioned as a protein degradation signal. Subsequently, a similar mutation study was extended to 10 other nuclear receptors by expressing them in COS-1 cells. RORα, PPARα, and HNF4α accumulated when their corresponding Ser100, Thr129, and Ser87 were mutated to Asp, respectively (Figure 2). MG132 treatment did not affect the degradation patterns (data not shown), suggesting those Asp mutants are resistant to proteasome-independent degradation. In the absence of their ligand binding domains (LBDs), both RORα and PPARα lost Asp mutation-dependent accumulation and were degraded in the manner observed with their wild-types and Ala mutants. On the other hand, HNF4α was degraded in an Asp mutation-dependent manner when its LBD was removed (Figure 2). With CAR, VDR, TRα, and Rev-erbα, any mutation dependencies were not observed when the full-length protein were expressed (Figure 2). However, when the LBD was removed, all 4 nuclear receptors were degraded in an Asp mutation-dependent manner as seen with the full-length FXR. In contrast, ERα, SF1/Ad4BP, and RXRα remained unchanged with or without the presence of the LBD. Table 1 summarizes these findings observed with 11 nuclear receptors.
Figure 2. The protein expressions of selected nuclear receptors.

The expression plasmids for a full length of nuclear receptors were transfected into COS-1 cells for 24 hours, and then the cells were harvested for Western blot analysis. The LBD domain deletion mutants for each nuclear receptor were constructed and ectopically expressed in COS-1 cells for 24 hours followed by treatment with 10μM MG132 for 3 hours. The samples were used for Western blot analysis as described in Materials and Methods.
Table 1.
DBD Phosphorylation as a Degradation Signal for Nuclear Receptors
| NR | Phosphorylation Dependent |
Proteasome Dependent |
||
|---|---|---|---|---|
| Full | ΔLBD | Full | ΔLBD | |
| Degradation | ||||
| FXR | + | + | + | + |
| CAR | − | + | − | + |
| VDR | − | + | − | + |
| TRα | − | + | − | + |
| Rev-erbα | − | + | − | + |
| Stabilization | ||||
| RORα | + | − | − | − |
| HNF4α | + | + (Degrade) | − | + (Degrade) |
| PPARα | + | − | − | − |
| None | ||||
| RXRα | − | − | − | − |
| ERα | − | − | − | − |
| SF1 | − | − | − | − |
The effects of phosphomimetic mutation on nuclear receptors were summarized. The marks mean as follows: +, degradation; −, no degradation; + (Degrade), degradation.
Activation function 1 (AF1)-directed proteasomal degradation of phosphorylated FXR
Phosphorylation at the conserved motif within the DBD appeared to be a general protein degradation signal of nuclear receptors. Given the fact that this signal was differently regulated by the LBD in a given nuclear receptor, we further investigated how FXR phosphorylation regulated its susceptibility to degradation and promoter transactivitation potential. The LBD was deleted from wild type, FXR S154A, and FXR S154D, which were then ectopically expressed in COS-1 cells. These LBD-deleted FXRs exhibited the same expression as observed with their full-length counterparts, FXR S154D (Figure 3A). Following an additional deletion of the hinge region, FXR S154D ΔHinge/LBD was degraded similar to FXR S154D ΔLBD (Figure 3B). On the other hand, when the AF1 was deleted from FXR S154D, the FXR S154D ΔAF1 was also degraded, but MG132 did not inhibit this degradation (Figure 3C). Moreover, MG132 did not inhibit the degradation of FXR S154D DBD (Figure 3D). These results indicated that the AF1 directed FXR S154D DBD to degrade through MG132-dependent pathway. To confirm that this degradation was catalyzed by the proteasome, coimmunoprecipitation assays and Western blotting with anti-ubiquitin antibody were performed to examine ubiquitination of FXR. Ubiquitinated FXR S154D was increased after MG132 treatment (Figure 4A); the LBD deletion did not affect this ubiquitination, suggesting that potential ubiquitination occurred at residues within the AF1/DBD/hinge region (Figure 4B). All of possible Lys residues within this region were singularly or combinatorically mutated, but none of these mutations affected ubiquitination (data not shown).
Figure 3. Domain deletion assays for FXR.

The domain deletion mutants were constructed as described in Materials and Methods. The protein expressions of hFXRΔLBD (A), hFXRΔHinge/LBD (B), hFXRΔAF1 (C), and hFXR DBD (D). COS-1 cells were transfected with each plasmid for 24 hours and then were treated with 10μM MG132 for 3 hours. The protein extracts were subjected to Western blot analysis. For detection, the hFXR DBD was expressed as C-terminal V5-tagged protein.
Figure 4. Coimmunoprecipitations for ubiquitination assays.

COS-1 cells were transfected with full-length form of hFXR S154D (A) or its ΔLBD mutant (B) along with ubiquitin for 24 hours and then were treated with 10μM MG132 for 6 hours. The protein extracts were immunoprecipitated with anti-FXR antibody followed by Western blotting with anti-ubiquitin antibody.
Chimeras of FXR and RXRα were constructed to investigate the functionality of the AF1 domain as a structural motif that directs phosphorylation-dependent degradation of nuclear receptors. The RXRα T162D mutant was neither degraded nor accumulated in COS-1 cells compared with RXRα WT and the RXRα T162A mutant (Figure 2). However, when its AF1 was replaced by the AF1 of FXR, RXRα degraded in an Asp mutation-dependent manner and accumulated in the presence of MG132 as observed with FXR (Figure 5). Thus, FXR AF1 retained its function to direct degradation. On the other hand, the degradation of FXR became independent from the Asp mutation when the AF1 was replaced with the AF1 of RXRα (Figure 5). These results indicate that phosphorylated DBD enables these receptors to degrade, whereas AF1 dictates to the DBD to determine how they degrade.
Figure 5. The protein expressions of chimeric proteins consisting of hFXR and hRXRα.

The plasmids which express the hRXRα AF1 and hFXRΔAF1/LBD mutants or hFXR AF1 and hRXRαΔAF1/LBD were transfected into COS-1 cells for 24 hours. The protein expressions were examined after 10μM MG132 treatment for 3 hours using anti-RXRα or anti-FXR for each chimera molecule.
Functionalities of phosphomimetic mutants
Cell-based luciferase reporter and gel-shift assays were performed to examine the influence of the phosphomimetic mutations on promoter activation by FXR. Both wild-type FXR and the FXR S154A mutant effectively activated the FXRE-Luc reporter gene after ligand treatment (CDCA or GW4064) of COS-1 cells (Figure 6A). On the other hand, the FXR S154D mutant exhibited neither basal nor ligand-mediated transactivation (Figure 6A). Consistent with the differences in their transactivation activities, the FXR S154D mutant did not bind to FXRE oligonucleotides in gel mobility shift assays (Figure 6B). Unlike previous reports (8), neither phorbol-12-myristate-13-acetate (PMA) treatments nor cotransfection of constitutively active PKC increased FXR-mediated FXRE transactivation activities in COS-1 cells (Supplemental Figure 2).
Figure 6. Functional characterization of hFXR mutants.

A, Cell-based reporter assay. Expression plasmids were cotransfected with a luciferase reporter construct for 24 hours. COS-1 cells were treated with 50μM CDCA and 5μM GW4064 for 24 hours. Empty expression plasmid pcDNA3.1 was used for mock transfection. Results are presented as mean ± SD of triplicate experiments. B, Gel-shift assays. The in vitro transcribed/translated proteins of the indicated hFXRs was incubated with 32P-labeled probe, electrophoresed on a polyacrylamide gel, and detected by autoradiography using x-ray film. C, Intracellular localization of hFXR mutants. COS-1 cells were transfected with expression vectors for 24 hours and then treated with the ligands for 24 hours. The cell extracts were fractionated into cytosol and nucleus as described in Materials and Methods. Western blotting was performed with anti-β-actin antibody for whole, anti-HSP90 antibody for cytosol, and anti-TBP antibody for nucleus fraction as positive control.
How the phosphomimetic mutation alters intracellular localization was next examined in COS-1 cells. Transfected cells were treated with a given ligand and fractionated into whole, nuclear, and cytosolic extracts for subsequent Western blot analysis. Wild-type FXR and FXR S154A were largely expressed in the nuclear fractions but also in the cytosolic fractions (Figure 6C). FXR S154D was equally distributed in both the nuclear and cytosolic fractions. Ligand treatments did not affect these localizations.
cDNA microarrays were employed to examine genes that were regulated by ectopically expressed FXR S154D in COS-1 cells in response to CDCA. The FXR S154D was unable to regulate genes that are regulated by FXR, including FABP6 and FGF19. Ten genes that were regulated by FXR S154A- vs FXR S154D-transfected COS-1 cells are listed in Supplemental Table 1. It appears that not all FXR target genes were regulated by this phosphorylation-mediated mechanism, because the representative target genes such as SHP and CYP7A1 gene were not significantly changed at their mRNA levels. The CYP8B1 gene was increased by the FXR S154D mutant, suggesting the possibility that phosphorylated FXR directly activates this gene and not indirectly via small heterodimer partner (SHP).
Phosphorylation of Ser154
Wild-type FXR or FXR S154A were ectopically expressed in COS-1 cells with or without a ligand CDCA or GW4064 and extracts subjected to Western blot analysis with P-S154 antibody. The wild-type FXR, but not the FXR S154A, was phosphorylated (Figure 7A). Moreover, treatment with CDCA or GW4064 increased this phosphorylation. When nuclear VRK1 was knocked down by siRNA in COS-1 cells, wild-type FXR dramatically reduced its phosphorylation levels (Figure 7B). Conversely, when VRK1 was overexpressed in COS-1 cells, wild-type FXR increased its phosphorylation, and coimmunoprecipitation between VRK1 and FXR increased (Figure 7C). Although recombinant active VRK1 and GST-tagged FXRs were used for in vitro phosphorylation assay (Supplemental Figure 3A, a and b), GST FXRs were not phosphorylated. Transient expression of VRK1 into COS-1 cells hardly affected FXRE-driven reporter activity (Supplemental Figure 3B). Phosphorylation was further examined with endogenous FXR in mouse livers. Whole-liver extracts and liver nuclear extracts were prepared from wild-type and Fxr-null mice treated with GW4064, and Western blot analysis was carried out. In the wild-type livers, FXR was decreased in both extracts after GW4064 treatment (Figure 8A). Phosphorylated FXR was detected in the nuclear extracts obtained from FXR WT mice and GW4064 treatment induced its phosphorylation. Immunohistochemistry of liver sections revealed that the P-S154 antibody stained nuclei of centrilobular hepatocytes in the only in wild-type mice after GW4064 treatment (Figure 8B). These results indicated that FXR was phosphorylated in the nucleus in response to GW4064 treatment in mouse liver.
Figure 7. The signaling for phosphorylation of FXR.

A, GFP-FXR WT/pEGFP-C1 or S154A/pEGFP-C1 were transfected into COS-1 cells for 24 hours. The cells were treated with 50μM CDCA and 5μM GW4064 for 3 hours in fetal bovine serum (FBS)-free media. The harvested samples were immunoprecipitated with anti-GFP-conjugated agarose resin. Western blot analysis was performed with anti-P-S154 peptide antibody for FXR or anti-GFP antibody. B, GFP-FXR WT/pEGFP-C1 and siRNA for VRK1 were cotransfected into COS-1 cells for 24 hours. The cells were subsequently treated with 5μM GW4064 for 3 hours in FBS-free media. IP assay was performed with the anti-GFP resin, followed by Western blotting with anti-P-S154 peptide antibody and anti-GFP antibody. C, FLAG-hVRK1/pcDNA3.1 was transfected into COS-1 cells along with GFP-FXRs/pEGFP-C1 for 24 hours. After Co-IP assay with the anti-GFP resin, Western blotting was performed with anti-P-S154 peptide antibody, anti-GFP, and anti-FLAG antibodies.
Figure 8. Phosphorylated FXR at Ser154 in mouse livers.

A, Mouse livers treated with either DMSO/corn oil or GW4064 (30 mg/kg) for 72 hours were harvested, and the protein extracts for whole or nuclear fraction were subjected to Western blot analysis using anti-FXR, anti-β-actin, anti-P-S154 peptide antibody for FXR, or anti-HDAC1. Relative phosphorylated FXR level was quantified using Image Studio software. The intensity was normalized to HDAC1 followed by FXR and calculated with respect to DMSO/corn oil treatment set to 1. B, Immunohistochemistry of the mouse liver sections with anti-P-S154 peptide antibody for FXR. The experimental procedure was described in Materials and Methods. PV, portal vein; CV, central vein.
Discussion
Nuclear receptors have a conserved phosphorylation motif between 2 zinc fingers of their DBDs. The present work with 11 nuclear receptors has now defined phosphorylation of this motif as a general protein degradation signal. Various nuclear receptors utilized phosphorylation differently to regulate their degradation. Notably, FXR used this signal to stimulate degradation via proteasomes as described herein, whereas RORα, PPARα, and HNF4α proscribed nonproteasomal degradation. Although no phosphorylation-dependent regulation was observed with ERα, SF1/Ad4BP, and RXRα, phosphorylation may regulate the first 2 nuclear receptors in endocrine organ-derived cells. Because RXRα is a heterodimer partner of numerous nuclear receptors including those used in the present study, regulation by phosphorylation may be more delicate and complex. How phosphorylation elicited signals to degrade nuclear receptors was determined by the function of AF1 or LBD. AF1 directed FXR to degrade, whereas LBD prevented CAR, VDR, TRα, and Rev-erbα from undergoing phosphorylation-dependent degradation. As such, phosphorylation through either the AF1 or LBD domains was capable of regulating degradation of 8 out of the 11 total nuclear receptors examined.
FXR is the third nuclear receptor, after CAR and ERα, for which phosphorylation of the conserved motif was confirmed in tissues in vivo (10, 11). VRK1, a nuclear protein kinase, originally cloned from mouse liver and named 51PK (19), is a potential candidate kinase that phosphorylates FXR in the nucleus. Coexpression and siRNA knockdown of VRK1 greatly increased and decreased phosphorylation of FXR at Ser154, respectively, in COS-1 cells. However, phosphorylation by a bacterially expressed active recombinant VRK1 in an in vitro kinase assay failed to demonstrate phosphorylation. Although it remains elusive as to whether or not VRK1 directly phosphorylated FXR at Ser154, VRK1 was essential for this phosphorylation to occur in response to ligand treatment. Alternatively, VRK1 may recruit a protein kinase to a ligand-activated FXR for direct phosphorylation of Ser154. A unique feature of this phosphorylation is that agonist ligands facilitated phosphorylation, from which a scenario of phosphorylation-mediated regulation could be envisioned (Figure 9). For example, FXR spontaneously accumulates in the nucleus and after binding to ligand, transactivates target gene promoters coincident with recruitment of VRK1 to stimulate FXR phosphorylation. As indicated by the FXR S154D mutant that was unable to bind FXRE and be degraded, this phosphorylated FXR should dissociate from the promoter for subsequent ubiquitination and degradation. By undergoing these processes, phosphorylation enables FXR to link ligand activation with inactivation and degradation. It was noted in our experiments that FXR S154A reproducibly exhibited higher ligand-activated transactivation activity compared with wild-type FXR. This higher activation could be explained if FXR WT, but not the FXR S154A mutant, increased phosphorylation after ligand treatment. However, the transactivation activity of FXR WT remained constant even after VRK1 overexpression. Apparently, FXR WT was not sufficiently phosphorylated to reflect the functional features of the FXR S154D mutant under these conditions.
Figure 9.
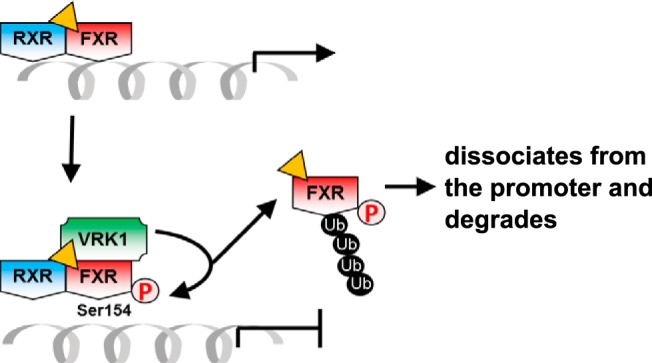
Schematic representation of ligand-dependent activation and phosphorylation-dependent deactivation for FXR.
With ERα, phosphorylation linking transactivation, ubiquitination, and degradation was previously documented; estrogen stimulated phosphorylation of ERα at residues such as Ser294 and/or Ser341, thus linking the abilities of ERα to activating promoters and degrading through proteasomes (20, 21). Through phosphorylation, AR was ubiquitinated and degraded (22), although this phosphorylated residue has not been identified. These studies with AR and ERα are of great interest, because they likely do not extend to other nuclear receptors because their phosphorylated residues and motifs are not conserved. On the other hand, information obtained with FXR could be applied to investigate a regulatory role played by the corresponding residue in a given nuclear receptor, because Ser154 is conserved as a phosphorylation motif in most nuclear receptors. Although there is a still possibility that VRK1 recruits PKC as the direct kinase that phosphorylates FXR, our recent study revealed that purified recombinant p38 MAPK could phosphorylate CAR Thr38 (23). This result implies that the conserved phosphorylation motif could be phosphorylated by various types of kinases, thereby responding to the diverse cell signals. Nuclear receptors can cross-talk with each other or other transcription factors as reported in a number of studies (24–26). The highly conserved phosphorylation motif might be critical switch to initiate or terminate cross talk of nuclear receptors because their protein stability could be controlled via DBD phosphorylation as shown in the present work. Further studies using specific knock in mouse models would be worthwhile to elucidate how nuclear receptors utilize the phosphorylation motif within DBD in vivo.
Acknowledgments
Human FXR/pCMV-ICIS and Nr0b2-#UTR/pGL4-TK were provided by Grace L. Guo (Rutgers University); pCMX-hRORα1 was from Jetten Anton (NIEHS); the expression plasmid and antiserum for hSF1/Ad4BP were from Ken-ichirou Morohashi (Kyushu University); and pCW7-Myc-Ub vector was from Andrew Wallace (North Carolina State University). S.A. was on leave with pay from Daiichi Sankyo Co., Ltd., Medicinal Safety Research Laboratories, Japan. T.H. was supported by the Strategic Young Researcher Overseas Visits Program for Accelerating Brain Circulation, Japan Society for the Promotion of Science (JSPS).
This work was supported by the National Institutes of Health Intramural Research Program: Z01ES1005-01.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- AR
- androgen receptor
- CAR
- constitutive androstane receptor
- CDCA
- chenodeoxycholic acid
- DBD
- DNA-binding domain
- ER
- estrogen receptor
- FXR
- farnesoid X receptor
- HNF
- hepatocyte nuclear receptor
- PKC
- protein kinase C
- VDR
- vitamin D receptor
- VRK1
- vaccinia-related kinase 1.
References
- 1. Anbalagan M, Huderson B, Murphy L, Rowan BG. Post-translational modifications of nuclear receptors and human disease. Nucl Recept Signal. 2012;10:e001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. McCall P, Gemmell LK, Mukherjee R, Bartlett JM, Edwards J. Phosphorylation of the androgen receptor is associated with reduced survival in hormone-refractory prostate cancer patients. Brit J Cancer. 2008;98:1094–1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Palazzolo I, Burnett BG, Young JE, et al. Akt blocks ligand binding and protects against expanded polyglutamine androgen receptor toxicity. Hum Mol Genet. 2007;16:1593–1603. [DOI] [PubMed] [Google Scholar]
- 4. Skliris GP, Rowan BG, Al-Dhaheri M, et al. Immunohistochemical validation of multiple phospho-specific epitopes for estrogen receptor α (ERα) in tissue microarrays of ERα positive human breast carcinomas. Breast Cancer Res Tr. 2009;118:443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Holm C, Kok M, Michalides R, et al. Phosphorylation of the oestrogen receptor α at serine 305 and prediction of tamoxifen resistance in breast cancer. J Pathology. 2009;217:372–379. [DOI] [PubMed] [Google Scholar]
- 6. Hsieh JC, Jurutka PW, Nakajima S, et al. Phosphorylation of the human vitamin D receptor by protein kinase C. Biochemical and functional evaluation of the serine 51 recognition site. J Biol Chem. 1993;268:15118–15126. [PubMed] [Google Scholar]
- 7. Hsieh JC, Jurutka PW, Galligan MA, et al. Human vitamin D receptor is selectively phosphorylated by protein kinase C on serine 51, a residue crucial to its trans-activation function. Proc Natl Acad Sci USA. 1991;88:9315–9319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Gineste R, Sirvent A, Paumelle R, et al. Phosphorylation of farnesoid X receptor by protein kinase C promotes its transcriptional activity. Mol Endocrinol. 2008;22:2433–2447. [DOI] [PubMed] [Google Scholar]
- 9. Sun K, Montana V, Chellappa K, et al. Phosphorylation of a conserved serine in the deoxyribonucleic acid binding domain of nuclear receptors alters intracellular localization. Mol Endocrinol. 2007;21:1297–1311. [DOI] [PubMed] [Google Scholar]
- 10. Mutoh S, Osabe M, Inoue K, et al. Dephosphorylation of threonine 38 is required for nuclear translocation and activation of human xenobiotic receptor CAR (NR1I3). J Biol Chem. 2009;284:34785–34792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shindo S, Moore R, Flake G, Negishi M. Serine 216 phosphorylation of estrogen receptor α in neutrophils: migration and infiltration into the mouse uterus. PLoS One. 2013;8:e84462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mutoh S, Sobhany M, Moore R, et al. Phenobarbital indirectly activates the constitutive active androstane receptor (CAR) by inhibition of epidermal growth factor receptor signaling. Sci Signal. 2013;6:ra31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang H, Garzel B, Heyward S, Moeller T, Shapiro P, Wang H. Metformin represses drug-induced expression of CYP2B6 by modulating the constitutive androstane receptor signaling. Mol Pharmacol. 2014;85:249–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–744. [DOI] [PubMed] [Google Scholar]
- 15. Osabe M, Negishi M. Active ERK1/2 protein interacts with the phosphorylated nuclear constitutive active/androstane receptor (CAR; NR1I3), repressing dephosphorylation and sequestering CAR in the cytoplasm. J Biol Chem. 2011;286:35763–35769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shindo S, Sakuma T, Negishi M, Squires J. Phosphorylation of serine 212 confers novel activity to human estrogen receptor α. Steroids. 2012;77:448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Choo YS, Zhang Z. Detection of protein ubiquitination. J Vis Exp. 2009;30:e1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li G, Thomas AM, Hart SN, et al. receptor activation mediates head-to-tail chromatin looping in the Nr0b2 gene encoding small heterodimer partner. Mol Endocrinol. 2010;24:1404–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zelko I, Kobayashi R, Honkakoski P, Negishi M. Molecular cloning and characterization of a novel nuclear protein kinase in mice. Arch Biochem Biophys. 1998;352:31–36. [DOI] [PubMed] [Google Scholar]
- 20. Williams CC, Basu A, El-Gharbawy A, Carrier LM, Smith CL, Rowan BG. Identification of four novel phosphorylation sites in estrogen receptor α: impact on receptor-dependent gene expression and phosphorylation by protein kinase CK2. BMC Biochem. 2009;10:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhou W, Srinivasan S, Nawaz Z, Slingerland JM. ERα, SKP2 and E2F-1 form a feed forward loop driving late ERα targets and G1 cell cycle progression. Oncogene. 2014;33:2341–2353. [DOI] [PubMed] [Google Scholar]
- 22. Lin HK, Wang L, Hu YC, Altuwaijri S, Chang C. Phosphorylation-dependent ubiquitylation and degradation of androgen receptor by Akt require Mdm2 E3 ligase. EMBO J. 2002;21:4037–4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hori T, Moore R, Negishi M. p38 MAP kinase links CAR activation and inactivation in the nucleus via phosphorylation at threonine 38. Drug Metab Dispos. 2016;44:871–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kodama S, Koike C, Negishi M, Yamamoto Y. Nuclear receptors CAR and PXR cross talk with FOXO1 to regulate genes that encode drug-metabolizing and gluconeogenic enzymes. Mol Cell Biol. 2004;24:7931–7940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. De Bosscher K, Vanden Berghe W, Haegeman G. Cross-talk between nuclear receptors and nuclear factor κB. Oncogene. 2006;25:6868–6886. [DOI] [PubMed] [Google Scholar]
- 26. Watanabe K, Sakurai K, Tsuchiya Y, Yamazoe Y, Yoshinari K. Dual roles of nuclear receptor liver X receptor α (LXRα) in the CYP3A4 expression in human hepatocytes as a positive and negative regulator. Biochem Pharmacol. 2013;86:428–436. [DOI] [PubMed] [Google Scholar]