

Abstract
Elevated basal insulin secretion under fasting conditions together with insufficient stimulated insulin release is an important hallmark of type 2 diabetes, but the mechanisms controlling basal insulin secretion remain unclear. Membrane rafts exist in pancreatic islet cells and spatially organize membrane ion channels and proteins controlling exocytosis, which may contribute to the regulation of insulin secretion. Membrane rafts (cholesterol and sphingolipid containing microdomains) were dramatically reduced in human type 2 diabetic and diabetic Goto-Kakizaki (GK) rat islets when compared with healthy islets. Oxidation of membrane cholesterol markedly reduced microdomain staining intensity in healthy human islets, but was without effect in type 2 diabetic islets. Intriguingly, oxidation of cholesterol affected glucose-stimulated insulin secretion only modestly, whereas basal insulin release was elevated. This was accompanied by increased intracellular Ca2+ spike frequency and Ca2+ influx and explained by enhanced single Ca2+ channel activity. These results suggest that the reduced presence of membrane rafts could contribute to the elevated basal insulin secretion seen in type 2 diabetes.
Type-2 diabetes (T2D) develops when the pancreatic β-cells fail to increase insulin secretion to compensate for systemic insulin resistance. This condition is also characterized by increased levels of plasma cholesterol, a general disorder of lipid metabolism and increased basal insulin secretion. However, it remains incompletely elucidated how this affects cholesterol levels in the endocrine pancreatic islets from individuals with diabetes and its consequences for insulin section and pathophysiology of T2D.
Membrane rafts are specialized platforms enriched in cholesterol and glycosphingolipids. Several important biological functions have been associated with membrane raft integrity including hormone release in pancreatic β-cells (1–5). Membrane raft microdomains in neuroendocrine and epithelial cells determine the spatial organization of important exocytotic regulatory proteins such as soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor (SNARE) protein, syntaxin 1, vesicle-associated membrane protein, and the Ca2+ sensor synaptotagmin 1 (6–8). Important β-cell ion channels like L-type Cav1.2 Ca2+ channels and Kv2.1 potassium channels are also suggested to colocalize with membrane rafts in both clonal pancreatic β-cells and rodent islets (9–11).
Membrane rafts can be manipulated in terms of their cholesterol content by different compounds (12). Methyl β-cyclodextrin (MβCD) extracts membrane cholesterol using its hydrophobic core (13). Longer incubations with MβCD suppress the genes that control the endogenous cholesterol efflux ability of the cells, which could pose potential problems with cell viability (13–16). MβCD treatment is also known to cause selective release of some important membrane proteins such as Glycosylphosphatidylinositol (GPI)-linked proteins and caveolin (17). As an enzyme-based regulator, cholesterol oxidase (CO) is more accurate to target cholesterol more selectively and is capable of oxidizing cholesterol also in less accessible, tightly packed areas of cell membrane resulting from high lateral surface pressure (18, 19). CO has the added advantage of not causing cell breakage and is also capable of oxidizing unesterified cholesterol in both high-density and low-density lipoproteins and, furthermore, does not oxidize intracellular cholesterol pools (20), and negligible amounts of lactate dehydrogenase are released from CO-treated cells (19). Further evidence also shows that CO causes oxidation of the plasma membrane cholesterol content without disturbing the native plasma membrane composition (21–23).
Several investigations suggest a role for membrane rafts in insulin secretion (9–11). Exocytosis of insulin secretory granules is tightly regulated by the regulatory SNARE complex (11) that is activated by a rise in intracellular calcium ([Ca2+]i) to trigger insulin exocytosis (24). Normal blood glucose, or euglycemia, is maintained by glucose-stimulated and constitutive release of insulin via exocytosis (25). Type 2 diabetes patients show not only impaired response to glucose stimulation but also elevated basal insulin levels (26, 27). Constitutive or basal insulin secretion occurs during the fasting or resting state to inhibit hepatic gluconeogenesis, ketogenesis, and hepatic glycogenolysis (28). Basal release may occur both with or without activation of Ca2+ channels (29).
In this study we have aimed at studying the impact of membrane rafts using different modifying compounds such as CO and MβCD in different diabetic models to carefully scrutinize the role of membrane rafts for human pancreatic β-cell function and its involvement in type 2 diabetes.
Materials and Methods
Reagents
All reagents were from Sigma, except ATTO 647N-Sphingomyelin (ATTO-SM; ATTO-tec) (30), Fluo5F, and BODIPY FL C5-ganglioside GM1 (Invitrogen).
Cell culture and islet isolation
INS-1 832/13 cells were cultured as described elsewhere (31). Islets from Wistar and Goto-Kakizaki (GK) rats were isolated by collagenase digestion and hand picked. Islets from human donors were donated by the Nordic Islet Transplantation Programme. Islets were obtained from six nondiabetic (five males, one female, body mass index [BMI] 26.4 ± 2 kg/m2, glycated hemoglobin [HbA1c] 5.7 ± 0.23) and three diabetic donors (three males, BMI 26.9 ± 2.2 kg/m2, HbA1c 5.8 ± 0.06).
Membrane raft staining and immunohistochemistry
Human islets, INS-1 832/13, Wistar, and GK rat islet cells were seeded on 35-mm glass-bottom dishes cultured with or without 8 U CO in Krebs for 2 hours before staining with ATTO 647N-Sphingomyelin (0.1 mM) or BODIPY FL C5-ganglioside GM1 (0.1 mM) for 20 minutes on ice.
Wistar rat islet cells were seeded on gridded dishes (IBIDI μ-Dish 35 mm, Grid-50) and stained with ATTO-SM after 24 hours, followed by immunocytochemistry as described elsewhere (31). Primary antibodies were against rat insulin (guinea pig anti-insulin, 1:400) or rat glucagon (mouse anti-glucagon, 1:200) and secondary antibodies anti-guinea pig-Cy3, 1:400, and antimouse Cy2, 1:400, respectively.
Rat experiments were approved by the Malmö/Lund Ethical Committee for Animal Research (Sweden). The use of human samples in the study was approved by the local ethical committee (Regional Ethical Review Board in Lund). All experiments were performed in accordance with the approved guidelines.
Insulin secretion assay
Insulin secretion in islets and cells was measured as previously described (31) and assayed by the Coat-a-Count kit (Siemens).
Electrophysiology
INS-1 832/13 cells were seeded on 35-mm plastic petri dishes used for electrophysiology measurements. The cells were treated with 2 U CO or not for 2 hours in Krebs at 37°C. Whole-cell currents were measured as previously detailed (31). Single Ca2+ channel activity was recorded in extracellular solution containing (in millimoles) 125 KCl, 1 MgCl2, 10 EGTA, and 5 HEPES (pH 7.2 with KOH) in the cell-attached configuration. The pipette solution contained (in millimoles) 120 BaCl2, 5 HEPES, and 0.1 μM BAY K8644. Single-channel sweeps were analyzed with programs TAC and TACfit (version 4.3.0; Bruxton). Sweeps not containing any channel activity were averaged and subtracted from the active sweeps.
Intracellular Ca2+ imaging
Krebs buffer supplemented with 5 mM glucose was used throughout this experiment. INS-1 832/13 cells were incubated in Krebs buffer with or without 8 U CO for 2 hours. Changes in [Ca2+]i were visualized by confocal imaging using 1 μM Fluo5F as detailed elsewhere (47). The cells were perfused with 70 mM K+ Krebs or treated with 0.2 μM thapsigargin for 10 minutes to empty the internal stores. The mean intensity in membrane and cytosol were measured. A ratio was calculated by taking the experimental fluorescence intensity divided by the average fluorescence intensity under nonstimulatory conditions.
Live cell imaging
Cells seeded onto coverslips and mounted in the experimental chamber were perifused and temperature controlled during the entire experiment. Confocal imaging was performed using enhanced green fluorescent protein (EGFP)-Cav1.2 (excitation 488 nm, emission 500–530 nm bandpass filter); GM1 and ATTO-SM (excitation 488 nm and 633 nm, emission 500–530 nm and 650–710 nm bandpass). Three dimensional (3D) reconstruction and calculation of colocalization coefficients were done using the ZEN software (Zeiss). The ratio of fluorescence intensity was calculated using the formula: where i1, i2, and i3 represent the measured intensity of whole cell, cytosol, and nucleus, respectively. a1, a2, and a3 represent the area of the whole cell, cytosol, and nucleus, respectively.
Plasmid purification and transfection
A plasmid encoding the N terminus of Cav1.2 fused with EGFP (pEGFP-N1 vector) was kindly provided by Jörg Striessnig (Innsbruck, Austria). The cells were transfected with 0.3 μg of pEGFP-Cav1.2 plasmid and two different concentrations (6–8 μL) of QIAGEN transfection reagent were used as control. The next day, cells were transferred to 96-well plates used for imaging. After 24 hours, cells were washed twice with PBS before use and treated with control buffer, 2 U CO, or 10 μM MβCD.
Fluorescence correlation spectroscopy (FCS)
Membrane rafts were identified using FCS (32, 33) using cells labeled with Bodipy-GM1 (excitation/emission 502/511 nm). INS-1 832/13 cells were incubated with Bodipy-GM1 for 20 minutes at 37°C with or without 2 U CO treatment. FCS measurements were carried out on a Zeiss 780 laser scanning microscope with FCS capabilities, using the Zen software. FCS curves were analyzed using a homemade Matlab based software and Microcal Origin.
Filipin staining of Wistar and GK rat islets
Isolated Wistar and GK rat islets were washed and fixed with 3% paraformaldehyde in PBS for 30 minutes at 37°C. Unbound paraformaldehyde was quenched (1.5 mg/mL glycine for 10 min at room temperature), followed by incubation with 50 μg/mL of filipin for 30 minutes at room temperature, 3× wash with PBS, and air drying for 30 minutes, and finally the islets were imaged in a two-photon imaging system.
Laurdan imaging and analysis
The INS-1 832/13 cells were treated with CO or MβCD and then stained by 2.5 μM Laurdan for 1 hour in a 37°C incubator. Laurdan was excited by two-photon imaging with 780 nm wavelength excitation and the emission fluorescence was collected by two filter sets in which CH1 was 395–465 nm and CH2 was 500–530 nm. The generalized polarization (GP) images of Laurdan and GP value calculation were created by simFCS software (34).
Statistics
Values are expressed as mean ± SEM. Group means were compared with a Student's unpaired two-tailed t test and a one-way ANOVA test. The level of significance was set as follows: *, P < .05; **, P < .01; ***, P < .001.
Results
Type 2 diabetic islets show decreased plasma membrane cholesterol content and membrane rafts
To assess the plasma membrane cholesterol content of the islets, we stained the Wistar and GK rat islets with free cholesterol binding dye filipin (Figure 1a). We observed that GK rat islets exhibited lower filipin fluorescence intensity compared with healthy Wistar islets (P < .01, n = 3, Figure 1b). Cholesterol forms the integral constituent of membrane rafts. These specialized structures were here visualized using ATTO-SM, a sphingomyelin labeling lipophilic dye (30). Interestingly, the diabetic GK rat islets displayed less membrane raft area than islets from healthy Wistar rats (P < .01, n = 3 rats/condition; Figure 1, c and d). To study the distribution of membrane rafts in insulin- and glucagon-secreting β- and α-cells, dispersed Wistar rat islet cells were stained with ATTO-SM and then fixed and immunostained for insulin and glucagon. We observed that glucagon-secreting α-cells displayed significantly lower ATTO-SM fluorescence intensity as compared with the β-cells (P < .05, Figure 1e). Decreased levels of plasma membrane cholesterol staining in diabetic GK rat islets as measured by filipin (Figure 1a) can be physiologically mimicked by treatment with cholesterol-oxidizing agents like CO, which was optimized in terms of dosage and time, for activity as well as cell toxicity in INS-1 832/13 cells before treatment in islets (Supplemental Figure 1). In agreement with the observations in rat islets, human islets from healthy human donors exhibited almost twice as intense ATTO-SM fluorescence, when compared with islets from donors with diabetes (Figure 1, f and g). Furthermore, healthy islets treated with CO showed clearly decreased ATTO-SM intensity (Figure 1F). By contrast, islets from diabetic donors were largely resistant to CO treatment, and ATTO-SM fluorescence intensity remained unaffected by CO. In summary, these experiments clearly indicate that diabetic islets exhibit a decreased presence of membrane rafts.
Figure 1. Disruption of membrane rafts of pancreatic islet cells in type 2 diabetes.
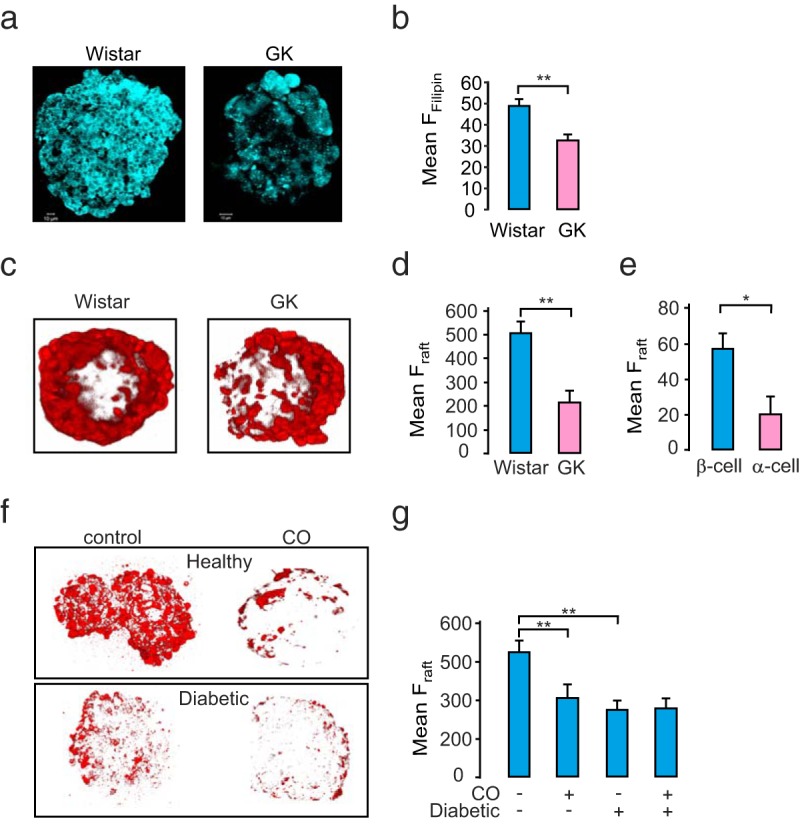
a, Representative images indicating decreased cholesterol in GK rat islets as compared with healthy Wistar rat islets as visualized by filipin fluorescence. b, Mean filipin fluorescence ± SEM (n = 3 in both groups). **, P < .01. c, 3D-rendered view of single Wistar and GK rat islets stained with membrane raft dye ATTO-SM. d, Mean ATTO-SM fluorescence ± SEM (n = 3 in both groups). **, P < .01. e, Mean ATTO-SM fluorescence intensity ± SEM in dispersed α- and β-cells from Wistar rat islets (n = 3 in both groups, P < .01). f, 3D-rendered view of a healthy human islet (Hba1c < 6.0) and a diabetic human islet (Hba1c > 6.0), treated or not with CO for 2 hours. g, Mean ATTO-SM fluorescence ± SEM per islet (n = 3 in both groups; P < .01 by ANOVA).
Disruption of membrane rafts causes increased basal insulin secretion
To assess the consequences of CO treatment in β-cells, insulin secretion was measured in rodent and human islet batch incubations. Preceding this procedure, we first tested the time dependence (0.5, 1, and 2 h) of CO pretreatment on insulin secretion in INS-1 832/13 cells (Figure 2a). For further experiments, pretreatment for 1 hour was chosen because it stimulated both basal and stimulated insulin secretion. Glucose stimulated insulin secretion in untreated control INS-1 832/13 cells by 4-fold. However, CO treatment significantly elevated basal insulin release in 2.8 mM glucose, whereas at 16.7 mM glucose, it showed only a tendency for increase (Figure 2b). Consequently, when assessing the fold stimulation by glucose at 16.7 over that at 2.8 mM glucose, we observed a clear reduction in the CO-treated group (Figure 2b, right panel). To verify that this effect on insulin release is related to the cholesterol oxidation effect of CO, we next used MβCD (35, 36), which complexes with cholesterol. Remarkably, the cells treated with MβCD also exhibited a marked greater than 245% increase in basal insulin secretion, whereas glucose stimulation calculated as fold change was reduced by 41% (Figure 2b). Next, in Figure 2c we investigated insulin secretion in freshly isolated rat islets.
Figure 2. Dispersion of membrane rafts causes increased basal insulin secretion.
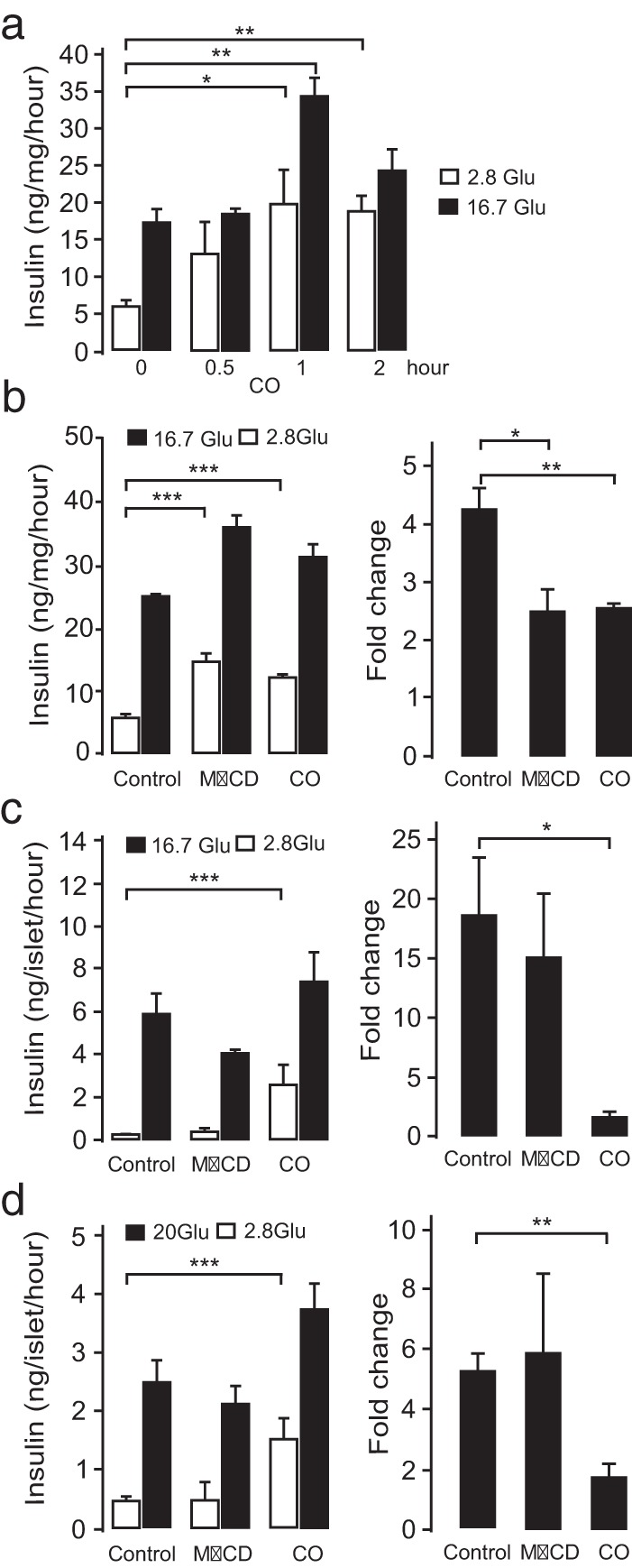
a, Insulin secretion in INS-1 832/13 cells in response to 2.8 and 16.7 mM glucose (Glu) pretreated or not with 2 U CO for 0 hours, 0.5 hours, 1 hour, and 2 hours. b, Insulin secretion in human islets in response to stimulation with 2.8 and 16.7 mM glucose, pretreated or not with 2 U CO or 10 μM MβCD. To the right, histograms show the respective fold changes in insulin secretion at 16.7 mM glucose vs 2.8 mM glucose between the control and CO- and MβCD-treated cells, as indicated. Panels c and d are as in panel B but using rat islets and INS-1 832/13 cells, respectively. Data represent mean values ± SEM from three independent experiments in all groups. *, P < .05, **, P = 0.01, ***, P = 0.001 by ANOVA.
These findings validated the CO results in INS-1 832/13 cells. Rat islets treated with CO also showed a significant 8-fold increase in basal insulin secretion at 2.8 mM glucose when compared with control. Surprisingly, rat islets treated with MβCD showed no significant change with respect to the basal insulin levels. Human islets (Figure 2D) were next investigated and with respect to CO, the results were in perfect line with other two model systems tested. However, in agreement with the rat islet data MβCD exerted no changes in either basal or stimulated insulin secretion. Because SNARE proteins, eg, synatxin 1a, are involved in the regulation of insulin secretion, we studied the effects of CO on syntaxin 1a clustering. We found that the number of syntaxin 1a clusters was much reduced upon incubation with CO (Supplemental Figure 2). The ratio of plasma to cytosol localization decreased from 3.2 ± 0.8 to 1.0 ± 0.2. This reduction was also apparent in 3D reconstructed membrane images (Supplemental Figure 3). In summary, the disruption of the membrane raft by CO reproducibly induced an increase in the basal insulin release.
Activation of [Ca2+]i oscillations in CO-treated cells under depolarizing and resting conditions
The entry of Ca2+ via the voltage-gated Ca2+ channels (Cav channels) and the resulting increase in the [Ca2+]i concentration triggers insulin exocytosis (24). It is hence essential to study the effect of membrane rafts on Ca2+ influx and [Ca2+]i. To this end, we tested the effects of CO on [Ca2+]i measured as fluo-5F fluorescence. The CO-treated or control cells were subjected to depolarizing buffer (70 mM K+) for 2 minutes (Figure 3a). The peak fluo-5F ratio (Figure 3b) and integrated [Ca2+]i (area under the curve; Figure 3c) were measured. It was observed that the CO-treated cells showed higher increases in [Ca2+]i compared with controls by both measurements.
Figure 3. Increased [Ca2+]i oscillations in stimulated cells upon dispersion of membrane rafts.

a, Increases in [Ca2+]i during a 120-second exposure to depolarizing K+ (HK) concentrations in control (in gray) or CO-treated (in black) cells. b, Mean peak fluorescence (Fi) over initial fluorescence (Fo) ratios (n = 3 in both conditions). c, Area under the curve (AUC) in control and CO-treated cells (n = 3 in both groups). d, [Ca2+]i measured in control (in gray) and CO (in black)-treated cells under resting conditions (5 mM glucose) and when stimulated with 0.2 mM thapsigargin. Mean spike frequency (number of spikes per minute) in control and CO-treated cells at resting e and TG-stimulated states f, respectively, are shown (n = 3 in both groups). g, L-type Cav1.2 channel declustering by membrane raft dispersion. INS-1 832/13 cells were transfected with phosphorylated EGFP-Cav1.2 treated either with control buffer or CO for 2 hours, respectively. h, Mean Cav1.2 cluster number per cell in control or CO-treated cells (n = 3 in both). i, Ratio of plasma membrane Cav1.2 immunoreactivity over that of the cell interior in control or CO-treated cells. All data are represented as mean ± SEM. ***, P < .001; *, P < .05. ns, not significant.
The increased Ca2+ concentration in response to high K+ could be caused by either influx of extracellular Ca2+ or by augmented release from internal stores. To address this, in Figure 3d we first measured [Ca2+]i at the resting state and after exposure to thapsigargin (TG). For the resting-state condition, the cells were modestly stimulated by 5 mM glucose, which resulted in some oscillatory activity. Interestingly, CO-treated cells (black trace) displayed more frequent [Ca2+]i oscillations assessed as spike frequency, which increased by 85% (Figure 3e). To assess whether [Ca2+]i stores also may contribute to the CO-induced oscillations, we tested the effects of the inhibitor TG that prevents pumping of Ca2+ ions into internal stores by blocking the sarco/endoplamatic Ca2+ ATPase or sarcoendoplasmic reticulum Ca2+-ATPase pump. Spikes in [Ca2+]i were observed during TG treatment. Interestingly, also after pretreatment with CO, thapsigargin retained its capacity to accelerate Ca2+ spiking (Figure 3f). This observation suggests that sarcoendoplasmic reticulum Ca2+-ATPase-regulated intracellular Ca2+ mobilization is unrelated to CO-stimulated Ca2+ oscillations observed at resting conditions. Taken together, cholesterol oxidation causes increased Ca2+ influx at both depolarized and resting states, and intracellular Ca2+ stores do not seem to contribute to this increase.
We next set out to determine the mechanisms underlying for the increased Ca2+ influx in response to CO. It has previously been reported that cholesterol-rich regions of the plasma membrane anchor Cav channels including the L-type Cav1.2 (37). We therefore checked whether cholesterol oxidation had any effect on Cav1.2 channel distribution in β-cells. To this end, INS-1 832/13 cells were transfected with pEGFP-Cav1.2 (Figure 3g). Treatment with CO decreased the Cav1.2 cluster number (Figure 3h) when compared with control. To evaluate whether membrane cholesterol oxidation results in Cav channel redistribution within the cell, we measured the ratio of the fluorescence intensity of Cav1.2 at the plasma membrane over that located intracellularly (ratio; Figure 3i). Interestingly, CO-treated cells displayed a clear reduction in Cav1.2 clusters, but the total intensity of EGFP-Cav1.2 was not significantly changed after CO treatment, and Ca2+ currents were the same in the enhanced EGFP-Cav1.2- and control green fluorescent protein-overexpressing cells (Supplemental Figure 4). Taken together, the disruption of membrane rafts by CO leads to an apparent declustering of Cav1.2 channels in the plasma membrane, which is associated with increased Ca2+ entry in both resting and depolarized states.
CO treatment increases Ca2+ influx via Cav channels
Given the finding that cholesterol oxidation leads to increased Ca2+ oscillations, we hypothesized that this may be due to increased Cav channel activity. To determine the consequences of cholesterol oxidation on Ca2+ currents, whole-cell Ca2+ currents were measured by patch clamp recordings in INS-1 832/13 cells. To analyze the voltage dependence of the currents, we measured the Ca2+-carried charge (Q) in response to voltage depolarizations from the holding −70 mV to membrane potentials between −50 and 40 mV. In line with the CO-increased, depolarization-induced Ca2+ influx (Figure 3), the carried Ca2+ charge (black; Figure 4a) was significantly stimulated by CO at potentials of −30 mV or greater. Isradipine inhibited Ca2+ influx in CO-treated cells showing their involvement in this process (Figure 4b).
Figure 4. Increased single Ca2+ channel activity after dispersion of membrane rafts.

a, Effect on charge-voltage (V) relationship of the voltage-dependent currents in control (in gray, n = 22) and CO (in black, n = 21)-treated cells. *, P < .05, **, P < .01, and ***, P < .001, tested by one-way ANOVA. b, Same conditions as in panel A, but cells were perfused with or without 2 mM isradipine (ISR) after 2 hours of preincubation with 2 U CO. *, P < .05, **, P < .01, one-way ANOVA. c, Single Ca2+ channel activity evoked by 100-millisecond (frequency 0.5 Hz) voltage-clamp depolarizations from −70 to 0 mV. The recordings were made using the cell-attached configuration under control conditions (in gray) or after incubation of CO for 2 hours (in black). d, Distribution of Ca2+ channel open times under control conditions (gray bars) and in the presence of CO treatment (black bars). Two exponentials were required to fit the distribution of the open times. e, Measurement of membrane potential with the voltage-sensitive dye FLIPR Red in INS-1 832/13 cells pretreated or not with 2 U CO and stimulated by 70 mM K+.
These results clearly suggest that Ca2+ channel activity was increased. To confirm this finding, we performed single Ca2+ channel experiments. Figure 4c shows single Ca2+ channel currents activated by a depolarization from −70 mV to 0 mV with or without CO pretreatment. Cells treated with CO showed a clearly increased number of channel openings in comparison with control cells. The effect of CO on single Ca2+ channel open time distribution is summarized in Figure 4d. The distribution of the single-event open times was not significantly different between the CO and the control cells. However, the number of single-channel events was nearly doubled by treatment with CO. We then further assessed possible changes in the regulation of membrane potential caused by CO treatment. Interestingly CO-treated cells depolarized significantly faster in response to high potassium (70 mM K+) compared with untreated cells (Figure 4e). In summary, the increased basal insulin release (Figure 2) coincides with enhanced intracellular Ca2+ signaling at both basal and stimulated conditions. This is caused by increased voltage-gated Ca2+ influx, resulting from increased single-Ca2+ channel open probability and rapid membrane depolarization.
Impairment of membrane rafts assessed by alternative approaches
The functions of membrane rafts can be studied using different approaches for impairing their structure and organization, which in turn can be assessed by alternative methods to visualize the plasma membrane qualities. Using cholesterol oxidation as a means to impair membrane raft structure, we next compared in INS-1 832/13 cells results when labeling with ATTO-SM (Figure 5a), membrane raft marker ganglioside Bodipy-GM1 (Figure 5c), or the organic polarity sensitive dye Laurdan (Figure 5e). First, CO treatment (as in Figure 1), markedly reduced ATTO-SM fluorescence as quantified in Figure 5b. Membrane rafts can alternatively be labeled using the ganglioside Bodipy-GM1 (Figure 5c). Also, when using this marker, CO treatment resulted in an apparent loss of membrane rafts, as evidenced by the reduction in Bodipy-GM1 fluorescence (Figure 5d). Laurdan fluorescence reports the insolubility of the plasma membrane and changes in membrane phase behavior as a shift in spectral emission. This may occur in response to alterations in membrane cholesterol. To address this, we performed two-photon imaging to obtain both gel- and liquid-phase information and reconstructed GP images of Laurdan. Surprisingly both CO and MβCD increased intensities of both phases in the plasma membrane (Supplemental Figure 5). However, the GP value was dramatically decreased after incubation with CO and MβCD in INS-1 832/13 cells (Figure 5e). To further confirm the effects of CO on membrane rafts by a biochemical approach, we next performed sucrose gradient centrifugation comparing cells treated or not with CO. For this purpose, we blotted the sucrose fractions with the established membrane raft maker flotillin. We observed a clear shift of flotillin from lipid raft fraction (5% sucrose) into the 30% fractions after CO treatment for 2 hours. The nonraft fractions (45% sucrose) were not significantly changed by the treatment (Figure 5f).
Figure 5. Validation of membrane raft disruption by cholesterol oxidase.
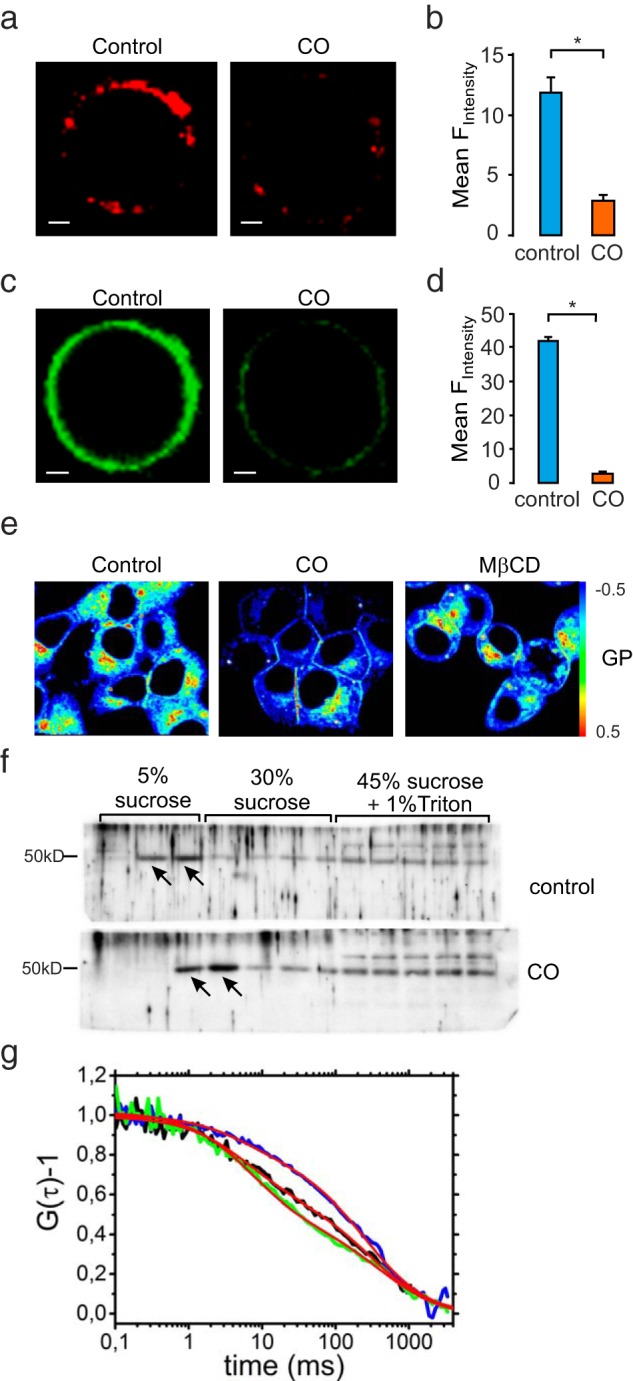
a, Confocal imaging of membrane raft markers ATTO-SM in INS-1 832/13 cells treated with CO or not (control). Scale bar, 2 μm. b, Mean ATTO-SM fluorescence intensity ± SEM (n = 3 in both; P < .05). c, Confocal imaging of Bodipy-GM1-labeled cells treated with CO or not (control). Scale bar, 2 μm. d, Data presented as mean ± SEM (n = 3; P < .05). e, GP images of Laurdan in INS-1 832/13 cells with treatment of CO or MβCD. The pseudocolored GP image showed the GP value, which can be calculated by the color scale bar. The images are presented from three independent experiments. f, Sucrose gradient fractionations of cells pretreated or not with CO blotted for membrane raft marker flotillin-1. Note the CO-induced shift of flotillin-1 out of the raft fraction (indicated by arrows). g, Normalized FCS curves from measurements in untreated (blue), CO-treated (black), and MβCD-treated (green) cells, all containing the fluorescent raft marker Bodipy-GM1.
Finally, another way to verify the disruption of membrane rafts is by analyzing their diffusion pattern in the plasma membrane. To allow this, we used FCS to quantify the diffusion coefficient before and after membrane raft disruption. In this experiment Bodipy-GM1-labeled cells provided technically superior results and were analyzed without treatment, after treatment with CO, and after treatment with MβCD (Figure 5g). In all three cases, the FCS curves required fitting with models containing two components: one faster component with a characteristic diffusion time through the detection focus of τD1 = 7–10 milliseconds and one slower component with τD2 = 300–340 milliseconds. Although the two components remained qualitatively similar in all three preparations, a marked difference was observed for the relative amplitudes of these components. For the untreated cells, the amplitudes were 28% ± 14% and 72% ± 14% for the fast and the slow components, respectively, ie, the slower component was dominating. In contrast, for the CO-treated cells, the corresponding amplitudes were 45% ± 7% and 55% ± 7% and for the MβCD treated cells 56% ± 22% and 44% ± 22%. This indicates that CO and MβCD both increase the fraction of freely diffusing Bodipy-GM1 and reduce the fraction of Bodipy-GM1 bound in clusters or rafts. A very similar effect of CO has previously been reported in neuronal cells (38).
Taking the data from these different approaches in Figure 5 together, there is solid support for concluding that treatment with CO changes the membrane phase characteristics and disperses membrane microdomains in insulin-secreting cells.
A prolonged higher concentration of glucose disperses membrane rafts
We next tested the effects of glucose on membrane rafts in a time series in INS-1 832/13 cells for 24, 48, or 72 hours (Figure 6a). Under normal glucose culture conditions, the membrane rafts staining intensity remained unchanged. In contrast, during high-glucose culture, membrane raft staining intensity decreased by 90% within 48 hours (Figure 6b). This result indicates that high-glucose culture decreases the presence of membrane rafts and that their integrity may be compromised in type 2 diabetes. We also measured membrane raft intensity after incubation with 1 mM palmitate for 24 hours to see whether a lipotoxicity stimulus regulates these structures. Intriguingly, palmitate had opposite effects and enhanced raft intensity staining, even in the simultaneous presence of 20 mM glucose (Supplemental Figure 6). These results suggest that glucose and palmitate have opposite effects on the plasma membrane lipid microdomains.
Figure 6. Reduced numbers of membrane rafts under glucotoxic conditions.

a, Confocal images of ATTO-SM in INS-1 832/13 cells cultured in normal glucose (9.7 mM) RPMI 1640 medium or high glucose (21.1 mM) for 24 hours, 48 hours, or 72 hours, as indicated. b, Mean ATTO-SM fluorescence intensity ± SEM (n = 3 in all groups). *, P < .05, by ANOVA.
Discussion
Membrane rafts anchor several important cell signaling receptors and associated proteins, and in pancreatic β-cells, they harbor the major exocytotic proteins. An imbalance to the native raft structure poses potential problem to the basic function of pancreatic β-cells. In this study, we have addressed this issue by dispersing membrane rafts using CO, a treatment that effectively alters the organization of the membrane rafts yet without extracting important plasma membrane proteins.
The principal findings of the current study are as follows: 1) membrane raft dispersion leads to hyperactivation of voltage-gated Ca2+ channels under basal conditions; 2) membrane raft integrity is a prerequisite for controlling basal insulin secretion; and 3) impaired membrane raft organization is observed in human type 2 diabetic islets. This suggests that impaired membrane rafts may contribute to the elevated basal insulin release in type 2 diabetes. In the following text, we aim to address the physiological importance of membrane rafts pertaining to the results observed.
In the literature, the consequences of manipulating the membrane cholesterol content on insulin secretion are somewhat contradictory. First, elevating cholesterol levels in vivo, as well as in vitro, decrease insulin secretion in mice (39), an effect that recently has been ascribed to cholesterol accumulation in insulin granules and impaired granule trafficking (40). These secretory defects could be rescued by MβCD treatment, which restored normal insulin release. In line with these reports, another study demonstrated a stimulatory effect of MβCD on insulin granule exocytosis and insulin secretion in clonal HIT-T15 cells and rat islet cells (9). Seemingly contrasting these reports, it has been reported that cholesterol extraction by MβCD decreases insulin granule docking and exocytosis as well as insulin secretion in mouse beta cells (11). These diverging effects of MβCD could probably largely be explained by different starting concentrations of cholesterol and different MβCD concentrations used. However, novel alternatives based on specific enzymatic digestion are offered by, for example, CO. Whereas the effects of CO are strikingly similar in islets from humans or rats and in clonal INS-1 832/13 cells, the effects of MβCD vary and in primary cell models differ markedly from the enzymatic alternative. Another way to interfere with membrane raft properties is silencing caveolin-1. Caveolin-1 is the principal component of the specialized form of rafts, called caveolae. It has been shown that mouse pancreatic islets treated with small interfering RNA against Cav-1 showed increased basal insulin secretion (41). Our study in conjunction with previous studies project that the overall function of membrane rafts in β-cell function lies in regulating basal insulin secretion. Furthermore, in addition to enhancing basal insulin secretion, membrane raft dispersion also causes increased basal glucagon secretion (10). This evidence adds further impetus to the hypothesis that membrane rafts are important for the hormone regulation of glucose homeostasis.
Early reports suggested that some degree of basal insulin is necessary for maintaining normal secretory capacity of the β-cell; however, the factors controlling basal secretion are not fully known. Several reports suggest that, for example, palmitate treatment causes a similar increase in basal secretion as observed here (42, 43); whether this also involves changes in membrane rafts remains to be determined. Furthermore, the mechanisms behind the increased basal insulin levels after membrane raft dispersion have not been investigated previously. Increased Ca2+ oscillations observed after membrane raft dispersion could contribute to the increased basal insulin levels, as suggested by a previous study (44), in which they observed increased Ca2+ levels accompanied by abnormal elevations in the basal insulin levels in mouse MIN6B1 clonal cells. Our data show that despite CO treatment resulting in declustering of Cav1.2 channels in the plasma membrane, the activity of these channels is increased. In fact, Ca2+ influx is accelerated after raft dispersion. This is in line with other reports indicating that the cell membrane cholesterol content influences the gating of inwardly rectifying Kir channels and also the current density of N-type Ca2+ channels by increasing the number of active channels (30, 45, 46). Likewise, our present results show that with the oxidation of the cholesterol content within the rafts, the apparent single-channel activity is markedly increased. The resulting increase in Ca2+ influx could contribute to the mixed Ca2+ oscillations observed after membrane raft dispersion in the resting state.
Oxidation of cholesterol in healthy islets exhibited decreased proportion of membrane rafts. type 2 diabetic islets have been reported to express lower amounts of SNARE-regulating proteins (47). In agreement with this, our data demonstrate that type 2 diabetic human islets exhibit reduced membrane rafts that harbor the SNARE proteins, thereby resembling the CO-treated healthy human islets. Interestingly, there has been a previous report showing that INS-1 cells cultured in high glucose exhibit decreased cholesterol content and dispersed membrane rafts (48), which is in perfect agreement with our observations (Figure 6A). It could be speculated that a lower expression of membrane rafts in diabetic islets could contribute to the decreased expression of exocytosis-regulating SNARE proteins in T2D and also that T2D islets have aberrant cholesterol metabolism or content. Further support for this view is provided by reports on the increased risk of incident diabetes in individuals receiving cholesterol-lowering statin treatment, although the capacity of statins to reduce cardiovascular mortality by far outdistance this negative effect (49, 50).
The factor governing the regulation of cholesterol levels in T2D remains unknown. Does cholesterol synthesis and absorption account for decreased membrane rafts in T2D? This remains to be studied in greater detail. In conclusion, we propose that membrane raft integrity is of prime importance for the proper functioning of pancreatic islets, wherein the Ca2+ channels within the rafts appear to play a major role in regulating the mode of insulin secretion.
Acknowledgments
We thank Britt-Marie Nilsson and Anna-Maria Veljanovska-Ramsay for their expert technical assistance.
Present address for S.B.: Department of Biotechnology, University of North Bengal, Siliguri, West Bengal, India.
Present address for T.R.: Institute of Neuroscience and Physiology, Department of Physiology, Gothenburg University, Gothenburg, Sweden.
Author contributions included the following: E.Z. and E.R. conceived and designed the experiments. V.N., S.F., J.H., C.L., S.B., P.B., E.Z., A.K., and T.R. performed the experiments. V.N. and E.Z. analyzed the data. E.Z. contributed the reagents/materials/analysis tools; V.N. and E.Z. wrote the manuscript, and E.R. reviewed the manuscript.
E.R. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
This work was supported by grants from the Swedish Research Council, the Knut and Alice Wallenbergs Stiftelse, the NovoNordisk Foundation, the Ragnar Söderbergs Stiftelse, the Region Skåne and the Albert Påhlsson Foundation (to E.R.). The fellowships of E.Z. was initially partially supported by the Integrated project BetaBat supported by the European Union and later by the KAW Foundation. This project is also supported by grants from the Juvenile Diabetes Research Foundation and the Swedish Strategic Research area in diabetes EXODIAB. This work was done using imaging and other equipment financed by the Linnaeus grant to the Lund University Diabetes Center and the Knut and Alice Wallenberg Foundation.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ATTO-SM
- ATTO 647N-Sphingomyelin
- BMI
- body mass index
- [Ca2+]i
- intracellular calcium concentration
- Cav channels
- voltage-gated Ca2+ channels
- CO
- cholesterol oxidase
- 3D
- three dimensional
- EGFP
- enhanced green fluorescent protein
- FCS
- fluorescence correlation spectroscopy
- GK
- Goto-Kakizaki
- GP
- generalized polarization
- HbA1c
- glycated hemoglobin
- MβCD
- methyl β-cyclodextrin
- SNARE
- soluble N-ethylmaleimide-sensitive fusion protein attachment protein receptor
- T2D
- type 2 diabetes
- TG
- thapsigargin.
References
- 1. Bickel PE. Lipid rafts and insulin signaling. Am J Physiol Endocrinol Metab. 2002;282:E1–E10. [DOI] [PubMed] [Google Scholar]
- 2. Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997;387:569–572. [DOI] [PubMed] [Google Scholar]
- 3. Simons K, van Meer G. Lipid sorting in epithelial cells. Biochemistry. 1988;27:6197–6202. [DOI] [PubMed] [Google Scholar]
- 4. Simons K, Toomre D. Lipid rafts and signal transduction. Nat Rev Mol Cell Biol. 2000;1:31–39. [DOI] [PubMed] [Google Scholar]
- 5. Salaun C, James DJ, Chamberlain LH. Lipid rafts and the regulation of exocytosis. Traffic. 2004;5:255–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chamberlain LH, Burgoyne RD, Gould GW. SNARE proteins are highly enriched in lipid rafts in PC12 cells: implications for the spatial control of exocytosis. Proc Natl Acad Sci USA. 2001;98:5619–5624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lang T. SNARE proteins and 'membrane rafts.' J Physiol. 2007;585:693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lang T, Bruns D, Wenzel D, et al. SNAREs are concentrated in cholesterol-dependent clusters that define docking and fusion sites for exocytosis. EMBO J. 2001;20:2202–2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Xia F, Gao X, Kwan E, et al. Disruption of pancreatic β-cell lipid rafts modifies Kv2.1 channel gating and insulin exocytosis. J Biol Chem. 2004;279:24685–24691. [DOI] [PubMed] [Google Scholar]
- 10. Xia F, Leung YM, Gaisano G, et al. Targeting of voltage-gated K+ and Ca2+ channels and soluble N-ethylmaleimide-sensitive factor attachment protein receptor proteins to cholesterol-rich lipid rafts in pancreatic α-cells: effects on glucagon stimulus-secretion coupling. Endocrinology. 2007;148:2157–2167. [DOI] [PubMed] [Google Scholar]
- 11. Vikman J, Jimenez-Feltstrom J, Nyman P, Thelin J, Eliasson L. Insulin secretion is highly sensitive to desorption of plasma membrane cholesterol. FASEB J. 2009;23:58–67. [DOI] [PubMed] [Google Scholar]
- 12. Allen JA, Halverson-Tamboli RA, Rasenick MM. Lipid raft microdomains and neurotransmitter signalling. Nat Rev Neurosci. 2007;8:128–140. [DOI] [PubMed] [Google Scholar]
- 13. Ohtani Y, Irie T, Uekama K, Fukunaga K, Pitha J. Differential effects of α-, β- and γ-cyclodextrins on human erythrocytes. Eur J Biochem. 1989;186:17–22. [DOI] [PubMed] [Google Scholar]
- 14. Neufeld EB, Cooney AM, Pitha J, et al. Intracellular trafficking of cholesterol monitored with a cyclodextrin. J Biol Chem. 1996;271:21604–21613. [DOI] [PubMed] [Google Scholar]
- 15. Yancey PG, Rodrigueza WV, Kilsdonk EP, et al. Cellular cholesterol efflux mediated by cyclodextrins. Demonstration of kinetic pools and mechanism of efflux. J Biol Chem. 1996;271:16026–16034. [DOI] [PubMed] [Google Scholar]
- 16. Smart EJ, Anderson RG. Alterations in membrane cholesterol that affect structure and function of caveolae. Methods Enzymol. 2002;353:131–139. [DOI] [PubMed] [Google Scholar]
- 17. Oncul S, Klymchenko AS, Kucherak OA, et al. Liquid ordered phase in cell membranes evidenced by a hydration-sensitive probe: effects of cholesterol depletion and apoptosis. Biochim Biophys Acta. 2010;1798:1436–1443. [DOI] [PubMed] [Google Scholar]
- 18. MacLachlan J, Wotherspoon AT, Ansell RO, Brooks CJ. Cholesterol oxidase: sources, physical properties and analytical applications. J Steroid Biochem Mol Biol. 2000;72:169–195. [DOI] [PubMed] [Google Scholar]
- 19. Lange Y. Tracking cell cholesterol with cholesterol oxidase. J Lipid Res. 1992;33:315–321. [PubMed] [Google Scholar]
- 20. Slotte JP, Gronberg L. Oxidation of cholesterol in low density and high density lipoproteins by cholesterol oxidase. J Lipid Res. 1990;31:2235–2242. [PubMed] [Google Scholar]
- 21. Harder T, Scheiffele P, Verkade P, Simons K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J Cell Biol. 1998;141:929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Keller P, Simons K. Cholesterol is required for surface transport of influenza virus hemagglutinin. J Cell Biol. 1998;140:1357–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Scheiffele P, Roth MG, Simons K. Interaction of influenza virus haemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. EMBO J. 1997;16:5501–5508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lang J. Molecular mechanisms and regulation of insulin exocytosis as a paradigm of endocrine secretion. Eur J Biochem. 1999;259:3–17. [DOI] [PubMed] [Google Scholar]
- 25. Del Prato S, Marchetti P, Bonadonna RC. Phasic insulin release and metabolic regulation in type 2 diabetes. Diabetes. 2002;51(suppl 1):S109–S116. [DOI] [PubMed] [Google Scholar]
- 26. Weyer C, Hanson RL, Tataranni PA, Bogardus C, Pratley RE. A high fasting plasma insulin concentration predicts type 2 diabetes independent of insulin resistance: evidence for a pathogenic role of relative hyperinsulinemia. Diabetes. 2000;49:2094–2101. [DOI] [PubMed] [Google Scholar]
- 27. Dankner R, Chetrit A, Shanik MH, Raz I, Roth J. Basal-state hyperinsulinemia in healthy normoglycemic adults is predictive of type 2 diabetes over a 24-year follow-up: a preliminary report. Diabetes Care. 2009;32:1464–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Borgono CA, Zinman B. Insulins: past, present, and future. Endocrinol Metab Clin North Am. 2012;41:1–24. [DOI] [PubMed] [Google Scholar]
- 29. Gerber SH, Sudhof TC. Molecular determinants of regulated exocytosis. Diabetes. 2002;51(suppl 1):S3–S11. [DOI] [PubMed] [Google Scholar]
- 30. Romanenko VG, Rothblat GH, Levitan I. Modulation of endothelial inward-rectifier K+ current by optical isomers of cholesterol. Biophys J. 2002;83:3211–3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Buda P, Reinbothe T, Nagaraj V, et al. Eukaryotic translation initiation factor 3 subunit e controls intracellular calcium homeostasis by regulation of cav1.2 surface expression. PLoS One. 2013;8:e64462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bergstrand J, Ronnlund D, Widengren J, Wennmalm S. Scanning inverse fluorescence correlation spectroscopy. Optics Express. 2014;22:13073–13090. [DOI] [PubMed] [Google Scholar]
- 33. Wennmalm S, Thyberg P, Xu L, Widengren J. Inverse-fluorescence correlation spectroscopy. Anal Chem. 2009;81:9209–9215. [DOI] [PubMed] [Google Scholar]
- 34. Parasassi T, De Stasio G, Ravagnan G, Rusch RM, Gratton E. Quantitation of lipid phases in phospholipid vesicles by the generalized polarization of Laurdan fluorescence. Biophys J. 1991;60:179–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Yu C, Alterman M, Dobrowsky RT. Ceramide displaces cholesterol from lipid rafts and decreases the association of the cholesterol binding protein caveolin-1. J Lipid Res. 2005;46:1678–1691. [DOI] [PubMed] [Google Scholar]
- 36. Colon-Saez JO, Yakel JL. The α7 nicotinic acetylcholine receptor function in hippocampal neurons is regulated by the lipid composition of the plasma membrane. J Physiol. 2011;589:3163–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Jacobo SM, Guerra ML, Hockerman GH. Cav1.2 and Cav1.3 are differentially coupled to glucagon-like peptide-1 potentiation of glucose-stimulated insulin secretion in the pancreatic β-cell line INS-1. J Pharmacol Exp Ther. 2009;331:724–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Eggeling C, Ringemann C, Medda R, et al. Direct observation of the nanoscale dynamics of membrane lipids in a living cell. Nature. 2009;457(7233):1159–1162. [DOI] [PubMed] [Google Scholar]
- 39. Hao M, Head WS, Gunawardana SC, Hasty AH, Piston DW. Direct effect of cholesterol on insulin secretion: a novel mechanism for pancreatic β-cell dysfunction. Diabetes. 2007;56:2328–2338. [DOI] [PubMed] [Google Scholar]
- 40. Bogan JS, Xu Y, Hao M. Cholesterol accumulation increases insulin granule size and impairs membrane trafficking. Traffic. 2012;13(11):1466–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nevins AK, Thurmond DC. Caveolin-1 functions as a novel Cdc42 guanine nucleotide dissociation inhibitor in pancreatic β-cells. J Biol Chem. 2006;281:18961–18972. [DOI] [PubMed] [Google Scholar]
- 42. Hoppa MB, Collins S, Ramracheya R, et al. Chronic palmitate exposure inhibits insulin secretion by dissociation of Ca(2+) channels from secretory granules. Cell Metab. 2009;10:455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Olofsson CS, Collins S, Bengtsson M, et al. Long-term exposure to glucose and lipids inhibits glucose-induced insulin secretion downstream of granule fusion with plasma membrane. Diabetes. 2007;56:1888–1897. [DOI] [PubMed] [Google Scholar]
- 44. Jaques F, Jousset H, Tomas A, et al. Dual effect of cell-cell contact disruption on cytosolic calcium and insulin secretion. Endocrinology. 2008;149:2494–2505. [DOI] [PubMed] [Google Scholar]
- 45. Lundbaek JA, Birn P, Girshman J, Hansen AJ, Andersen OS. Membrane stiffness and channel function. Biochemistry. 1996;35:3825–3830. [DOI] [PubMed] [Google Scholar]
- 46. Jennings LJ, Xu QW, Firth TA, Nelson MT, Mawe GM. Cholesterol inhibits spontaneous action potentials and calcium currents in guinea pig gallbladder smooth muscle. Am J Physiol. 1999;277:G1017–G1026. [DOI] [PubMed] [Google Scholar]
- 47. Ostenson CG, Gaisano H, Sheu L, Tibell A, Bartfai T. Impaired gene and protein expression of exocytotic soluble N-ethylmaleimide attachment protein receptor complex proteins in pancreatic islets of type 2 diabetic patients. Diabetes. 2006;55:435–440. [DOI] [PubMed] [Google Scholar]
- 48. Somanath S, Barg S, Marshall C, Silwood CJ, Turner MD. High extracellular glucose inhibits exocytosis through disruption of syntaxin 1A-containing lipid rafts. Biochem Biophys Res Commun. 2009;389:241–246. [DOI] [PubMed] [Google Scholar]
- 49. Sattar S, Preiss N, Murray D, et al. Statins and risk of incident diabetes: a collaborative meta-analysis of randomised statin trials. Lancet. 2010;375:735–742. [DOI] [PubMed] [Google Scholar]
- 50. Simsek S, Schalkwijk CG, Wolffenbuttel BH. Effects of rosuvastatin and atorvastatin on glycaemic control in type 2 diabetes—the CORALL study. Diabet Med. 2012;29:628–631. [DOI] [PubMed] [Google Scholar]