

Abstract
The neuropeptide kisspeptin, encoded by Kiss1, regulates reproduction by stimulating GnRH secretion. Neurons synthesizing kisspeptin are predominantly located in the hypothalamic anteroventral periventricular (AVPV) and arcuate nuclei, but smaller kisspeptin neuronal populations also reside in extrahypothalamic brain regions, such as the medial amygdala (MeA). In adult rodents, estradiol (E2) increases Kiss1 expression in the MeA, as in the AVPV. However, unlike AVPV and arcuate nuclei kisspeptin neurons, little else is currently known about the development, regulation, and function of MeA Kiss1 neurons. We first assessed the developmental onset of MeA Kiss1 expression in males and found that MeA Kiss1 expression is absent at juvenile ages but significantly increases during the late pubertal period, around postnatal day 35, coincident with increases in circulating sex steroids. We next tested whether developmental MeA Kiss1 expression could be induced early by E2 exposure prior to puberty. We found that juvenile mice given short-term E2 had greatly increased MeA Kiss1 expression at postnatal day 18. Although MeA Kiss1 neurons are known to be E2 up-regulated, the specific estrogen receptor (ER) pathway(s) mediating this stimulation are unknown. Using adult ERα knockout and ERβ knockout mice, we next determined that ERα, but not ERβ, is required for maximal E2-induced MeA Kiss1 expression in both sexes. These results delineate both the developmental time course of MeA Kiss1 expression and the specific ER signaling pathway required for E2-induced up-regulation of Kiss1 in this extrahypothalamic brain region. These findings will help drive future studies ascertaining the potential functions of this understudied kisspeptin population.
Kisspeptin, a neuropeptide encoded by the Kiss1 gene, is essential for reproduction, as demonstrated by the fact that humans and mice harboring mutations in the Kiss1r or Kiss1 genes show striking deficits in puberty, reproductive hormone release, and fertility (1–4). Kiss1 neurons are located primarily in the hypothalamic anteroventral periventricular (AVPV) and arcuate (ARC) nuclei, but a smaller population of Kiss1 neurons is also present outside the hypothalamus in the medial amygdala (MeA) (5–12). Most kisspeptin research has focused on the regulation and reproductive role of the two hypothalamic Kiss1 populations, and very little is currently known about the development, regulation, and function of MeA Kiss1 neurons (8, 10, 11, 13). The MeA has numerous behavioral and physiological functions, including but not limited to effects on reproductive physiology and behavior (14–19). Thus, understanding the developmental and regulatory aspects of MeA Kiss1 neurons may provide important insight into potential functions of kisspeptin signaling arising from the MeA.
Kiss1 gene expression in the two hypothalamic regions is differentially regulated by sex steroids (T and estradiol [E2]) (8–10), and these differential effects most likely reflect the different roles of these two Kiss1 populations in positive and negative feedback. The ARC is likely involved in mediating negative feedback of sex steroids because removal of sex steroids via gonadectomy greatly increases ARC Kiss1 expression, and exogenous T or E2 treatment suppresses ARC Kiss1 levels (8–10, 20). In contrast to the ARC, in the AVPV, gonadectomy significantly decreases Kiss1 expression, whereas exogenous E2 treatment robustly increases AVPV Kiss1 expression, potentially linking the AVPV kisspeptin population to a role in E2-positive feedback in females (8–10). As in the AVPV, we have previously reported that Kiss1 expression in the MeA is greatly reduced after gonadectomy (11). Moreover, like in the AVPV, treatment with either T or E2, but not DHT, upregulates Kiss1 levels in the MeA (11). The lack of DHT effect indicates that sex steroid stimulation of Kiss1 in the MeA occurs specifically through estrogen receptor (ER)-mediated pathways, rather than androgen receptor pathways (11). However, exactly which specific ER pathway is responsible for mediating E2's up-regulation of Kiss1 in the MeA is not currently known. Previous studies indicate that E2 regulation of ARC and AVPV Kiss1 occurs primarily via ERα (8, 10, 21). However, unlike the ARC and AVPV, which both express ERα at much higher levels than ERβ (10), the MeA highly expresses both ERα and ERβ (22, 23), suggesting either (or both) ERs may regulate MeA Kiss1 expression.
We previously demonstrated that Kiss1 is expressed in the MeA of adults (11) but is not readily detectable at postnatal day (PND) 14 in juvenile mice (24). Likewise, MeA Kiss1 is not readily detectable at the prepubertal age of PND 19 in rats (25). This is in stark contrast to Kiss1 in the hypothalamus, which is readily expressed in the ARC before birth and in the AVPV starting around the second to third week of postnatal life (26–29). It is currently unknown when Kiss1 first becomes expressed in the MeA or whether the developmental onset of MeA Kiss1 expression relates to pubertal increases in gonadal steroids because MeA Kiss1 is up-regulated by sex steroids.
This study had several main goals to fill in gaps in our knowledge about the development and regulation of MeA Kiss1 neurons. First, we examined the developmental expression of MeA Kiss1 in juvenile, prepubertal, and peripubertal males to determine when MeA Kiss1 is first expressed, how this expression changes with pubertal development, and whether any changes in Kiss1 expression coincide with pubertal changes in circulating sex steroid levels. Next, we determined whether MeA Kiss1 expression in juvenile male mice could be enhanced after exogenous short-term E2 treatment, thereby assessing whether MeA Kiss1 expression could be developmentally induced by early sex steroid exposure or whether the developmental onset of MeA Kiss1 expression is guided by nonsex steroid factors. In the final set of experiments, we used adult ERα knockout (KO) and ERβKO mice of both sexes to determine which specific ER, ERα, and/or ERβ (both of which are very highly expressed in the MeA), is necessary for E2's ability to up-regulate Kiss1 in the MeA.
Materials and Methods
Animals
Experiments used either male C57BL6 mice (experiments 1 and 2) or mice of both sexes from the ERαKO or ERβKO lines (experiments 3 and 4). Male and female ERαKO mice and WT littermate controls were generated from heterozygous breeder ERαKO mice (obtained from Dr James L. Jameson, Philadelphia, Pennsylvania, and originally created by the Chambon laboratory) (30). Male and female ERβKO and WT control mice were similarly generated from heterozygous ERβKO breeders (Jackson Labs). All experimental animals were housed in a 12-hour light, 12-hour dark cycle at approximately 22°C. Mice were weaned at 21 days of age and housed two to three mice/cage (mixed WTs and KOs), unless noted otherwise, with ad libitum access to food and water. All surgeries occurred under isoflurane anesthesia. All experimental procedures were approved by the University of California, San Diego, Institutional Animal Care and Use Committee in accordance with the National Institute of Health policies.
Tissue collection
All blood sampling and tissue collection was performed on mice that were briefly anesthetized with isoflurane and then rapidly decapitated. Blood samples were collected retroorbitally just prior to decapitation and centrifuged (15 min at 5000 rpm) 90 minutes after collection and the serum stored at −20°C. Serum samples were assayed in singlet by the University of Virginia's Center for Research in Reproduction Ligand Assay and Analysis Core to measure the LH, E2, and/or T levels. Brains were collected immediately after decapitation, frozen on dry ice, and stored at −80°C until sectioning. Brains were sectioned on a cryostat into five coronal sets of 20 μm sections spanning the entire hypothalamus and amygdala regions. Brain slices were mounted on to SuperFrost Plus slides (VWR Scientific) and stored at −80°C until in situ hybridization (ISH).
Single-label ISH
Single-label ISH for Kiss1 expression was performed as previously described (26, 31–35). Briefly, one of the five coronal sets of slides containing the entire AVPV (experiments 2–4) or ARC and MeA (experiments 1–4) was assayed to examine Kiss1 expression. Slides were first fixed in 4% paraformaldehyde and then pretreated with acetic anhydride and rinsed in a 2× sodium citrate/sodium chloride (SSC) solution. Slides were then delipidated in chloroform, dehydrated in ethanol washes, and air dried for 90 minutes. Radiolabeled (33P) Kiss1 (0.04 pmol/mL) antisense riboprobes were added to tRNA, heat denatured, and combined with hybridization buffer, and then 100 μL of this probe mixture was applied to each slide before hybridizing overnight in a 55°C humidity chamber. The next day, slides were washed in 4× SSC at room temperature and treated with ribonuclease (RNase) A for 30 minutes at 37°C. Slides were then washed in RNase buffer (no RNase A) at 37°C and 2× SSC at room temperature, each for 30 minutes. After washes in 0.1× SSC at 62°C for 1 hour, the slides were dehydrated in ethanols, air dried for 90 minutes, dipped in Kodak nitroblue tetrazolium salt emulsion, air dried for 90 minutes, and then stored at 4°C until being developed. AVPV slides were developed 3–5 days later, depending on the experiment, whereas ARC and MeA slides were developed 9–12 days later, depending on the experiment. Due to the large size of the number of slides, male and female assays were run separately for the ERαKO and ERβKO experiments.
As in previous studies (26, 31–34), we used an automated computer image processing system (Dr Don Clifton, University of Washington, Seattle, Washington) to quantify Kiss1 expression for each brain slice. The custom software takes into account background staining levels and counts the number of silver grain clusters, representing cells expressing Kiss1 mRNA. We analyzed both the number of Kiss1 cells and grains per cell during data analysis. However, for most assays of the MeA and AVPV, there was at least one of the four treatment groups that had either no cells or very few Kiss1 cells (eg, none to two cells). As such, the grains per cell for those groups would not be calculable or would be based on just one or two cells, essentially eliminating that group from any proper statistical analyses. Thus, we do not report data for grains per cell, but we note that the data for grains per cell showed a comparable pattern as was seen with the number of Kiss1 cells. All ISH slides were analyzed by a person blinded to treatment groups.
Experiment 1: when are MeA Kiss1 neurons initially expressed in development and does this coincide with developmental increases in gonadal steroids?
Hypothalamic Kiss1 expression has been the focus of most kisspeptin research, but very little is known about the regulation and development of Kiss1 neurons in the MeA. Although MeA Kiss1 expression is readily found in adult mice (8, 10, 11), it was not previously observed in juvenile (PND 14) mice (24), and at present, nothing is known about the developmental pattern of MeA Kiss1 expression. Because MeA Kiss1 neurons are strongly up-regulated by E2 (11) and gonadal steroid levels are not elevated until puberty, we hypothesized that MeA Kiss1 would first be detected around the age of puberty. In this experiment, we therefore examined the developmental time course of MeA Kiss1 expression in male mice and assessed how that relates to pubertal changes in sex steroid levels. Because MeA Kiss1 expression is notably greater in gonad-intact adult males than females, we therefore used males in this experiment to assess developmental MeA Kiss1 expression and to ensure detection of MeA Kiss1 at early ages in gonad-intact animals. Developing C57BL6 male mice were briefly anesthetized with isoflurane and killed via rapid decapitation at PND 15, 20, 25, 30, 35, or 40. In male mice, the first two ages are juvenile, the next two are prepubertal, PND 35 is peripubertal, and PND 40 is around the onset of adulthood. At the time the animals were killed, blood and brains were collected to analyze serum T levels and MeA Kiss1 expression, respectively (n = 4–6 mice per group).
Experiment 2: is MeA Kiss1 expression capable of being induced prior to puberty with exogenous E2 treatment?
Experiment 1 determined that MeA Kiss1 expression was essentially absent at juvenile and prepubertal ages but became progressively more abundant during the pubertal ages and beyond, correlating with simultaneous increases in circulating sex steroid levels. Because MeA Kiss1 expression in adults is strongly up-regulated with E2 (11), we hypothesized that MeA Kiss1 expression is not observed at young juvenile/prepubertal ages because circulating gonadal steroids are too low. However, it is also possible that MeA Kiss1 expression is not observed at juvenile ages because MeA Kiss1 neurons are not fully developed at this age. In this experiment, we gave short-term exogenous E2 capsules to PND 14 male mice to determine whether early sex steroid exposure could induce a precocious rise in the MeA Kiss1 levels. Juvenile male C57BL6 mice (PND 14) received either a small SILASTIC brand capsule (Dow Corning) with 1 mm of 1:25 E2 to cholesterol mix (based on doses used in experiments 3 and 4) or received no exogenous hormonal treatment. All mice were then killed 4 days later on PND 18, and brains were collected to measure Kiss1 expression in the MeA (n = 8–10 mice per group).
Experiment 3: is ERβ necessary for the E2-induced increase in Kiss1 in the MeA?
It has been shown that short-term T or E2 treatment, but not DHT, robustly increases Kiss1 levels in the MeA of adult mice and rats of both sexes (11), but the specific ER pathway(s) mediating this stimulatory effect is currently unknown. ERα appears to be the key ER pathway for regulating hypothalamic Kiss1 neurons (8, 10). However, unlike the Kiss1 neurons in the hypothalamus, which expresses ERα at much higher levels than ERβ (10), the MeA contains very high concentrated levels of both ERβ and ERα (22, 23), indicating that either (or both) of these ER pathways might mediate the stimulatory effects of E2 on MeA Kiss1 neurons. In experiment 3, we used ERβKO mice (lacking ERβ globally) to determine whether functional ERβ is necessary for E2-induced Kiss1 expression in the MeA. Adult ERβKO mice of both sexes, and their WT littermates, were gonadectomized (GDX) at 7 weeks of age. One week after GDX, all mice were individually housed and received either a SILASTIC brand capsule (Dow Corning) containing 2 mm of a 1:25 mixture of E2 to cholesterol (previously shown to provide constant high elevated E2 levels in adult mice) or no hormonal treatment. Five days later, all mice were killed, and blood and brains were collected to measure hormone levels and to examine Kiss1 expression in the MeA and hypothalamus (n = 5–8 mice per group).
Experiment 4: is ERα necessary for the E2-induced increase in Kiss1 in the MeA?
Expression of Kiss1 mRNA in the MeA is robustly upregulated by E2 (11), but the specific ER pathway mediating this increase in MeA Kiss1 expression is unknown. ERα is the primary receptor regulating both AVPV and ARC Kiss1 neurons (8, 10), and the MeA contains high levels of ERα (22, 23, 36). As a complementary experiment to experiment 3 above, experiment 4 used ERαKO mice (global ERα knockouts) to determine whether ERα is specifically required for the E2 up-regulation of MeA Kiss1. As was done for ERβKO mice, ERαKO and WT mice of both sexes were GDX at 7 weeks of age, received no hormonal treatment or an exogenous E2 SILASTIC brand capsule (Dow Corning) at 8 weeks of age, and the brains and blood were collected 5 days later (n = 5–10 mice per group).
Statistical analysis
All data are expressed as mean ± SEM. For experiment 1, MeA Kiss1 expression in each developmental age group was compared with MeA Kiss1 expression in the PND15 group using a one-way ANOVA and Bonferroni correction post hoc tests. When examining Kiss1 expression in the AVPV and MeA (experiments 2–4), at least one of the treatment groups had low Kiss1 expression (eg, < 10 cells) and one treatment group had much higher Kiss1 expression (eg, ≥ 50 cells), so the variances were not equal between groups and required the appropriate use of Mann-Whitney U (experiment 2; AVPV analysis) or Kruskal-Wallis (experiments 3 and 4) tests, using Dunn's multiple comparison tests for post hoc analysis. In experiment 2, there were no Kiss1 cells found in the MeA of non-E2-treated PND18 mice, and thus, a one-sample Student's t test was used to determine whether the number of MeA Kiss1 cells induced by E2 treatment at PND 18 was significantly greater than 0. When examining ARC Kiss1 expression (experiments 3 and 4), group differences were examined using ANOVAs and Bonferroni post hoc tests. Statistical significance was set at P < .05 for all experiments.
Results
Experiment 1: MeA Kiss1 and T levels show a similar developmental increase around PND 35
This experiment examined MeA Kiss1 expression in male mice during development to identify when MeA Kiss1 is initially expressed, how this expression changes during puberty, and whether the expression coincides with developmental increases in gonadal T secretion. MeA Kiss1 expression was virtually absent at juvenile ages PND 15 and 20 but then gradually increased throughout peripubertal development, with a significant increase in MeA Kiss1 first observed at PND 35 (P < .05, relative to PND 15, Figure 1). The highest MeA Kiss1 expression was detected at PND 40. Circulating T levels in the same developing males followed a similar pattern to that of MeA Kiss1 expression, with T levels being low at juvenile and prepubertal ages and then significantly increasing at PND 35 and PND 40 (P < .05 relative to PND 15, Figure 1).
Figure 1.
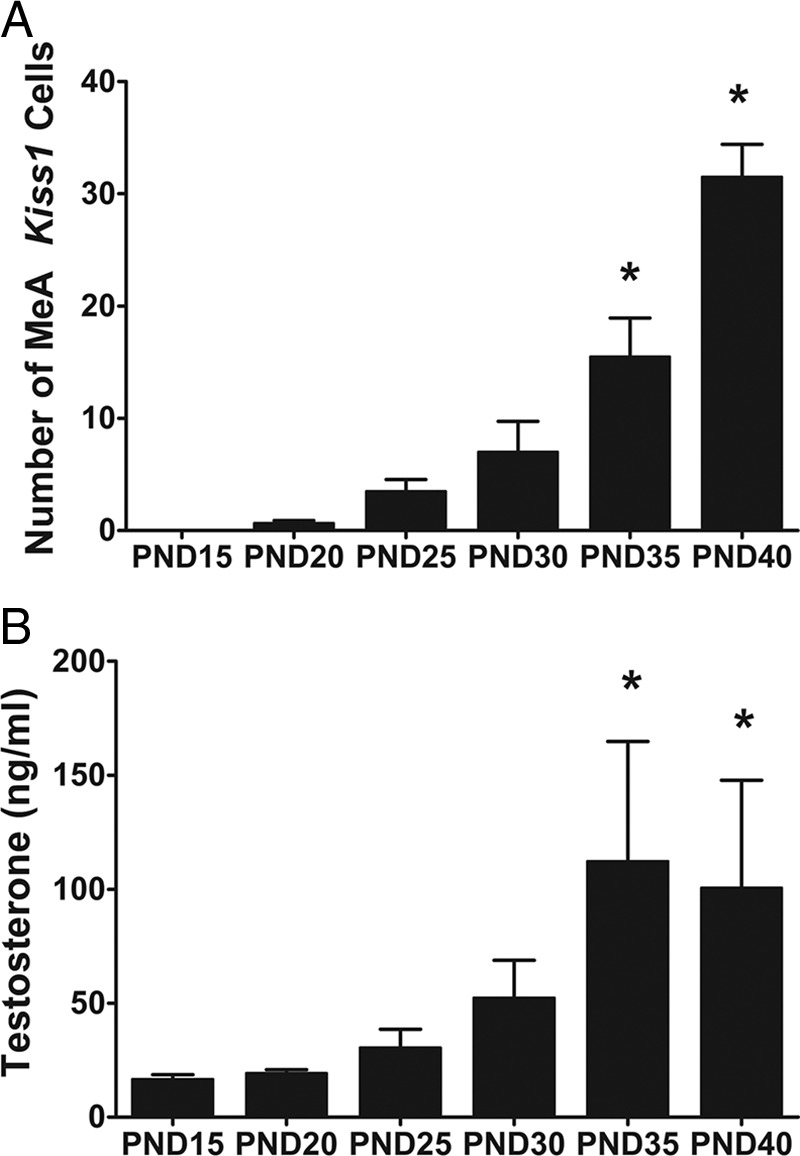
Developmental expression of MeA Kiss1 in male mice. MeA Kiss1 expression (A) and serum T (B) increase throughout development in male C57BL6 mice, with both measures demonstrating a significant increase around PND 35. *, P < .05, relative to PND 15.
Experiment 2: juvenile E2 treatment significantly increases MeA Kiss1 expression
The previous experiment showed that Kiss1 expression in the MeA is absent or very low at juvenile and prepubertal ages when circulating sex steroids are also low but then starts to increase at pubertal ages in synchrony with increased sex steroid levels. This suggests that the absence of MeA Kiss1 expression at young juvenile ages may reflect a lack of stimulatory sex steroid signaling. To test this possibility, juvenile males (PND 14) were treated short term with a high dose of E2 to determine whether early sex steroid exposure could prematurely increase MeA Kiss1 expression at a young age. As a control comparison, Kiss1 levels were also measured in the AVPV (where Kiss1 is similarly stimulated by E2 exposure). Similar to what has previously been observed in adult mice, E2 treatment significantly increased Kiss1 in the AVPV in PND 18 males in comparison with nontreated males, indicating the early E2 treatment is sufficient to induce Kiss1 in the AVPV (Figure 2; P < .05). As in the AVPV, early E2 treatment also induced significantly more Kiss1 expression in the amygdala on PND 18 (Figure 2; P < .05). Of note, the heightened Kiss1 expression in the amygdala at this age appeared more scattered throughout the greater MeA region than normally observed in adults, in which the predominance of Kiss1 cells are in the posterior dorsal region of the MeA along with additional cells scattered in other MeA regions.
Figure 2.
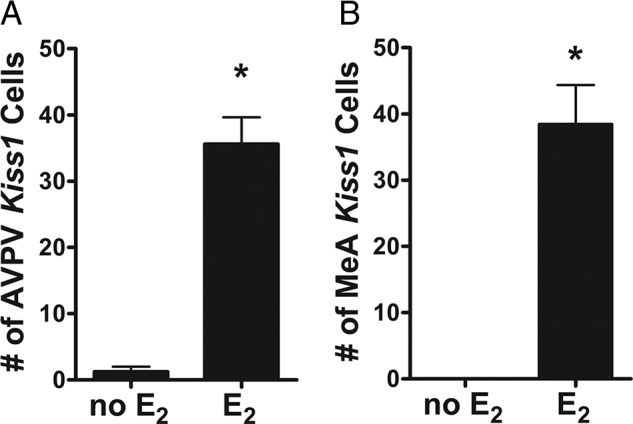
Kiss1 mRNA expression in the AVPV (A) and MeA (B) of PND 18 male mice with or without short-term E2 treatment. E2 exposure significantly increased Kiss1 levels in both AVPV and MeA of juvenile males, though Kiss1 cells were more scattered throughout the MeA region than typically seen in adults. *, P < .05.
Experiment 3: ERβ is not required for maximal Kiss1 expression in the MeA
Kiss1 in the MeA is markedly increased with short-term E2 treatment in adults, but the ER pathway(s) regulating this increase are unknown. This experiment determined whether ERβ, which is highly expressed in the MeA, is required for E2 up-regulation of MeA Kiss1. In adult ERβKO mice, serum E2 levels were significantly increased with 5-day E2 treatment, with no significant differences in E2 levels between WT and ERβKO mice (Figure 3; P < .05). Likewise, serum levels of LH (which are typically reduced by exogenous E2 negative feedback) were elevated in GDX mice of both genotypes and significantly suppressed with E2 treatment in both WT and ERβKO mice of both sexes (Figure 3; P < .05).
Figure 3.

Serum E2 and LH levels for male and female ERβKO mice with or without 5-day E2 implants. E2 levels were elevated in both WT and ERβKO E2-treated female (A) and male (B) mice. LH levels were suppressed by E2 in both WT and ERβKO female (C) and male (D) mice. Different letters denote significant group differences within each sex (P < .05).
Because Kiss1 expression in the AVPV is up-regulated by E2, AVPV Kiss1 expression served as a positive control measure for the E2 treatment. Both WT and ERβKO GDX mice of both sexes showed low levels of Kiss1 in the AVPV, which was increased similarly in both genotypes with E2 treatment (Figures 4 and 5; P < .05). In contrast to the AVPV, in the ARC, both WT and ERβKO GDX mice had significantly more Kiss1 cells than non-E2-treated mice (Figures 4 and 5; P < .05). As was seen in the AVPV, in the MeA, Kiss1 expression significantly increased with E2 treatment in both WT and ERβKO mice (Figures 4 and 5; P < .05). This increased MeA Kiss1 expression was present in both male and female ERβKOs, demonstrating that ERβ is not required for the E2 up-regulation of Kiss1 in the MeA, similar to Kiss1 in the AVPV.
Figure 4.

Representative images of Kiss1 mRNA expression in the AVPV (female), ARC (male), and MeA (male) of ERβKO mice with or without 5-day E2 treatment. opt, optic tract; 3V, third ventricle.
Figure 5.

Kiss1 mRNA expression in ERβKO mice. Mean number of Kiss1 cells in the AVPV of female (A) and male (B), ARC of female (C) and male (D), and MeA of female (E) and male (F) ERβKO and WT littermates. Different letters denote significant group differences within each sex (P < .05).
Experiment 4: ERα is required for maximal Kiss1 expression in the MeA
This experiment determined whether ERα, which is also highly expressed in the MeA, is required for E2 induction of MeA Kiss1 expression. In ERαKO and WT mice, circulating E2 levels were significantly elevated in 5-day E2-treated mice in comparison with GDX mice (Figure 6; P < .05) and were comparable between genotypes (Figure 6; P < .05). Serum LH levels were significantly elevated after GDX in both WT and ERαKO mice and were significantly suppressed by E2 treatment in WTs but not in ERαKOs of either sex (as expected, given the known necessary role for ERα in E2 negative feedback) (Figure 6; P < .05).
Figure 6.

Serum E2 and LH levels for female and male ERαKO mice. E2 levels were elevated in both female (A) and male (B) ERαKO and WT mice treated with E2. However, LH was suppressed by E2 in female (C) and male (D) WT littermate control, but not in ERαKO females (C) and males (D). Different letters denote significant group differences within each sex (P < .05).
GDX WT and ERαKO mice both showed low AVPV Kiss1 levels, and E2 treatment significantly increased AVPV Kiss1 expression in WTs but not ERαKOs of either sex (Figure 7 and Supplemental Figure 1; P < .05), indicating that ERα is required for the E2 up-regulation of Kiss1 in the AVPV, as previously reported for just female mice. In contrast to the AVPV, in the ARC, GDX WT mice of both sexes had significantly more Kiss1 cells than E2-treated WT mice (Figure 7 and Supplemental Figure 1; P < .05). However, in ERαKOs, there was no E2-induced suppression of ARC Kiss1, with all ERαKO mice, regardless of treatment, showing high ARC Kiss1 levels comparable with those in GDX WT mice (Figure 7 and Supplemental Figure 1; P < .05). In the MeA, WT mice of both sexes showed the expected E2 up-regulation of Kiss1 expression. Interestingly, both male and female ERαKO mice did exhibit a small but significant increase in MeA Kiss1 levels with E2 treatment relative to GDX ERαKOs; however, this E2-induced increase in MeA Kiss1 expression in ERαKO mice was of lower magnitude than the increase observed in their E2-treated WT littermates (Figure 7 and Supplemental Figure 1; P < .05). This demonstrates that ERα is required for maximal MeA Kiss1 expression, but another ER may also be involved in regulating MeA Kiss1 to a lesser degree.
Figure 7.

Kiss1 mRNA expression in ERαKO mice. Mean number of Kiss1 cells in the AVPV of female (A) and male (B), ARC of female (C) and male (D), and MeA of female (E) and male (F) ERαKO and WT littermates. Different letters denote significant group differences within each sex (P < .05).
Discussion
Kisspeptin synthesized in the hypothalamus stimulates GnRH secretion and the reproductive axis (1, 5, 6, 37–41), and as a result, the vast majority of kisspeptin research has focused on the role of hypothalamic Kiss1 neurons in reproduction and sex steroid feedback. However, Kiss1 is also located at moderate levels in extrahypothalamic areas, such as the MeA (8, 10, 11, 24), a region of the limbic system involved in regulating numerous physiological and behavioral responses, including aspects of reproduction (14–19). Yet, at present, little is known about the development, regulation, and function of Kiss1 neurons in the MeA. In the present study, we filled in several gaps in our knowledge about the development and regulation of MeA Kiss1 neurons. First, we delineated the developmental time course of MeA Kiss1 expression and linked this onset of expression, which occurs around puberty, to developmental sex steroid exposure. Next, using multiple global knockout mouse lines, we determined the specific ER signaling pathway that is required for E2-induced up-regulation of Kiss1 in this MeA region, identifying ERα, but not ERβ, as a critical mediator of E2 signaling for MeA Kiss1 stimulation.
Kiss1 is readily expressed in the MeA in adult rodents of both sexes (8, 10, 11, 24), but this is not the case at younger ages. Previous research demonstrated that, although Kiss1 is already being expressed in the AVPV and ARC at young ages (26–29), Kiss1 expression is absent in the MeA in juvenile (PND 14) mice (24) and prepubertal (PND 19) rats (25). However, it remained unknown when in development MeA Kiss1 begins to be expressed between juvenile and adult stages. Moreover, given that MeA Kiss1 is up-regulated by sex steroids (11), it was also unknown how or whether this initial Kiss1 expression related to developmental changes in circulating sex steroid hormones, which themselves increase at puberty. We therefore examined the developmental expression of Kiss1 in the MeA in male mice every 5 days between PND 15 and PND 40. Kiss1 mRNA in the MeA was essentially absent at both PND 15 and 20 (juvenile ages) but showed a small, gradual increase throughout prepubertal and peripubertal development, with a low number of cells present around PND 25–30 and then substantially more cells detected at subsequent later ages (PND 35 and 40). Interestingly, serum T levels mimicked a similar developmental pattern, with T levels being low initially and then gradually increasing but not showing a significant increase until PND 35 and 40. Given the synchrony in the developmental timing of the Kiss1 and T increases, it suggests that MeA Kiss1 expression onset is likely guided by gonadal sex steroid levels and first becomes notably expressed when T levels first begin to rise substantially around puberty.
Although MeA Kiss1 neurons showed a significant increase in expression at PND 35, coinciding with an increase in serum T, this developmental pattern does not preclude the possibility that earlier Kiss1 expression is absent because either Kiss1 neurons are not fully developed yet or some additional stimulatory (or lessening of an inhibitory factor) is also required and not yet present. In experiment 2, we addressed this issue by testing whether MeA Kiss1 expression could in fact be elicited earlier in development if higher circulating sex steroids were present. Indeed, as in adults, in juvenile (PND 18) male mice, short-term E2 treatment significantly increased Kiss1 levels in both the AVPV and the MeA in comparison with non-E2-treated mice. This indicates that the lack of detectable MeA Kiss1 expression at juvenile and prepubertal ages is most likely due to a lack of sufficient sex steroid exposure to stimulate the Kiss1 gene at this time, rather than due to absent Kiss1 neurons or an absent nonsteroidal stimulatory signal. Interestingly, we also note that Kiss1 expression in these E2-treated juveniles was less abundant in the posterodorsal subdivision of the MeA, in which Kiss1 expression in adults is most predominant, but was observed more scattered throughout multiple parts of the greater medial amygdala area. It is not currently clear whether the abundant yet more scattered MeA Kiss1 cells observed with E2 treatment at PND 18 are the same cells expressing Kiss1 in adulthood, which have migrated closer to the MePD subregion during puberty.
MeA Kiss1 is up-regulated by E2 (11), suggesting that a functional role of MeA Kiss1 neurons involving kisspeptin signaling might occur primarily when sex steroid levels are elevated. However, many behaviors and physiological functions are modulated by either ERα or ERβ. Thus, understanding which specific ER(s) pathway regulates MeA Kiss1 and how this compares with the regulation of hypothalamic Kiss1 populations exclusively by ERα could provide an important framework for future investigations of potential functions of MeA Kiss1 neurons. Based on this rationale, we used ER knockout mice to examine whether ERα and/or ERβ were necessary for the E2-induced Kiss1 expression in the MeA. Previous research in female mice has shown that ERβ is not required for hypothalamic Kiss1 expression (10), and our present data in both females and males support this, with ERβKO females and males each showing normal E2-regulated AVPV and ARC Kiss1 expression patterns comparable with WT mice. Similarly, we found that in the MeA, despite very high ERβ expression in this region, ERβKO mice of both sexes showed a marked increase in Kiss1 levels with E2 treatment, comparable with the increase observed in WT mice, indicating that ERβ is also not necessary for E2-induced MeA Kiss1 expression.
In contrast to ERβ, ERα is known to be required for the E2 regulation of AVPV and ARC Kiss1 (8, 10). Our present data in both male and female ERαKO mice support this previous conclusion. Indeed, in both sexes, we found that despite comparable E2 levels between E2-treated WT and ERαKO mice, E2-treated ERαKOs fail to show any significant increase in Kiss1 in the AVPV or suppression of Kiss1 in the ARC. ERα also appears to be required for normal E2 induction of Kiss1 in the MeA. We found that in the MeA, as in the hypothalamus, E2-treated ERαKO mice had significantly fewer Kiss1 cells than E2-treated WTs, indicating that ERα is required for the maximal expression of Kiss1 in the MeA. However, it was interesting to note that E2-treated ERαKOs had significantly more MeA Kiss1 neurons than GDX ERαKOs, indicative of a small degree of E2 up-regulation, perhaps by some other ER, possibly ERβ or G protein-coupled receptor (GPR)-30, that may compensate in the absence of ERα or be sufficient to induce MeA Kiss1. This minor up-regulation with E2 treatment was limited to the MeA Kiss1 population and not observed in either the AVPV or ARC Kiss1 populations. ERβ is highly expressed in the MeA, and thus, it is possible that ERβ is sufficient, but not required, for E2-induced MeA Kiss1 expression. At this time, there is not a consistently suitable and well-established positive control for ERβ action in the brain, making it difficult to assess this possibility at present. However, the role of ERβ in this minimal up-regulation of MeA Kiss1 could be examined in future research by examining E2-up-regulation in double-ERα/ERβ KO mice. It is also possible that MeA Kiss1 in WT mice is normally regulated solely via ERα but that the developmental compensation by another ER is induced in the MeA of ERαKO mice, allowing for their minor E2-stimulated increase in MeA Kiss1 expression. Lastly, the current experiments indicate that ERα, but not ERβ, is required for MeA Kiss1 expression, but future research is needed to know whether ERα has a direct or indirect effect on Kiss1 neurons in the MeA.
The primary known functions of hypothalamic kisspeptin are its roles in stimulating reproduction and mediating sex steroid feedback signaling. Although the MeA has many ascribed functions in multiple diverse physiological and behavioral systems, this region has also been implicated in modulating some aspects of reproduction. Lesions of the MeA disrupt ovarian cycles (15–17), and electrical stimulation of the MeA results in increases in LH (14), mimicking an LH surge, suggesting that in adulthood, some factor in the MeA facilitates reproduction. Thus, it is possible that the mechanism by which the MeA influences reproduction is via kisspeptin signaling arising from this region. ERα is the primary ER regulating both hypothalamic Kiss1 and reproductive sex steroid feedback loops (8, 10), and our data demonstrate that ERα is also necessary for the complete E2-induced increase in MeA Kiss1. Thus, it is possible that Kiss1 neurons in the MeA modulate reproductive parameters under the influence of ERα, as occurs for Kiss1 neurons in the AVPV and ARC. However, the small degree of partial modulation of MeA Kiss1 by another ER other than ERα may indicate an additional estrogen-mediated role(s) of MeA Kiss1 neurons beyond reproduction. Recent data demonstrating projections from the accessory olfactory bulb to the MeA Kiss1 neurons (42) suggest that these neurons may relay chemosensory information, which may indicate a role of MeA Kiss1 neurons in modulating reproductive and/or nonreproductive behaviors or physiology that are modulated by olfactory cues.
In summary, very little is known about the development and regulation of Kiss1 neurons in the MeA. Our data are the first to demonstrate that MeA Kiss1 expression increases around puberty, coincident with developmental increases in sex steroids. We also provide evidence to suggest that the developmental onset and increase in MeA Kiss1 are likely driven primarily by exposure to rising circulating sex steroids, although we cannot currently rule out that MeA Kiss1 neurons also undergo additional yet-to-be identified changes during pubertal development that prevent earlier expression. Finally, our data indicate that Kiss1 in the MeA is regulated primarily by ERα, and not ERβ, and therefore, these neurons are likely involved in mediating ERα-regulated behaviors or physiology, beginning around or after puberty.
Acknowledgments
This work was supported by National Science Foundation Grant IOS-1457226 and Eunice Kennedy Shriver National Institute of Child Health and Human Development Specialized Cooperative Centers Program in Reproduction and Infertility Research Grants U54 HD012303 (to the University of California, San Diego) and U54 HD28934 (to the University of Virginia). S.B.Z.S. is supported by Grants F32 HD088060 and T32 HD007203.
Disclosure Summary: The authors have nothing to disclose.
Footnotes
- ARC
- arcuate nucleus
- AVPV
- anteroventral periventricular nucleus
- E2
- estradiol
- ER
- estrogen receptor
- GDX
- gonadectomized
- GPR
- G protein-coupled receptor
- ISH
- in situ hybridization
- KO
- knockout
- MeA
- medial amygdala
- PND
- postnatal day
- RNase
- ribonuclease
- SSC
- sodium citrate/sodium chloride
- WT
- wild type.
References
- 1. Seminara SB, Messager S, Chatzidaki EE, et al. The GPR54 gene as a regulator of puberty. N Engl J Med. 2003;349:1614–1627. [DOI] [PubMed] [Google Scholar]
- 2. Topaloglu AK, Tello JA, Kotan LD, et al. Inactivating KISS1 mutation and hypogonadotropic hypogonadism. N Engl J Med. 2012;366:629–635. [DOI] [PubMed] [Google Scholar]
- 3. Lapatto R, Pallais JC, Zhang D, et al. Kiss1−/− mice exhibit more variable hypogonadism than Gpr54−/− mice. Endocrinology. 2007;148:4927–4936. [DOI] [PubMed] [Google Scholar]
- 4. de Roux N, Genin E, Carel JC, Matsuda F, Chaussain JL, Milgrom E. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci USA. 2003;100:10972–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Shahab M, Mastronardi C, Seminara SB, Crowley WF, Ojeda SR, Plant TM. Increased hypothalamic GPR54 signaling: a potential mechanism for initiation of puberty in primates. Proc Natl Acad Sci USA. 2005;102:2129–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gottsch ML, Cunningham MJ, Smith JT, et al. A role for kisspeptins in the regulation of gonadotropin secretion in the mouse. Endocrinology. 2004;145:4073–4077. [DOI] [PubMed] [Google Scholar]
- 7. Franceschini I, Lomet D, Cateau M, Delsol G, Tillet Y, Caraty A. Kisspeptin immunoreactive cells of the ovine preoptic area and arcuate nucleus co-express estrogen receptor α. Neurosci Lett. 2006;401:225–230. [DOI] [PubMed] [Google Scholar]
- 8. Smith JT, Dungan HM, Stoll EA, et al. Differential regulation of KiSS-1 mRNA expression by sex steroids in the brain of the male mouse. Endocrinology. 2005;146:2976–2984. [DOI] [PubMed] [Google Scholar]
- 9. Kauffman AS, Gottsch ML, Roa J, et al. Sexual differentiation of Kiss1 gene expression in the brain of the rat. Endocrinology. 2007;148:1774–1783. [DOI] [PubMed] [Google Scholar]
- 10. Smith JT, Cunningham MJ, Rissman EF, Clifton DK, Steiner RA. Regulation of Kiss1 gene expression in the brain of the female mouse. Endocrinology. 2005;146:3686–3692. [DOI] [PubMed] [Google Scholar]
- 11. Kim J, Semaan SJ, Clifton DK, Steiner RA, Dhamija S, Kauffman AS. Regulation of Kiss1 expression by sex steroids in the amygdala of the rat and mouse. Endocrinology. 2011;152:2020–2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim W, Jessen HM, Auger AP, Terasawa E. Postmenopausal increase in KiSS-1, GPR54, and luteinizing hormone releasing hormone (LHRH-1) mRNA in the basal hypothalamus of female rhesus monkeys. Peptides. 2009;30:103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Xu Z, Kaga S, Mochiduki A, et al. Immunocytochemical localization of kisspeptin neurons in the rat forebrain with special reference to sexual dimorphism and interaction with GnRH neurons. Endocr J. 2012;59:161–171. [DOI] [PubMed] [Google Scholar]
- 14. Beltramino C, Taleisnik S. Facilitatory and inhibitory effects of electrochemical stimulation of the amygdala on the release of luteinizing hormone. Brain Res. 1978;144:95–107. [DOI] [PubMed] [Google Scholar]
- 15. Chateau D, Kauffmann MT, Aron C. Are the amygdaloid projections to the hypothalamic ventromedial nucleus involved in estrous rhythm regulation in the female rat? Exp Clin Endocrinol. 1984;83:303–309. [DOI] [PubMed] [Google Scholar]
- 16. Velasco ME, Taleisnik S. Effects of the interruption of amygdaloid and hippocampal afferents to the medial hypothalamus on gonadotrophin release. J Endocrinol. 1971;51:41–55. [DOI] [PubMed] [Google Scholar]
- 17. Tyler JL, Gorski RA. Effects of corticomedial amydgala lesions or olfactory bulbectomy on LH responses to ovarian steroids in the female rat. Biol Reprod. 1980;22:927–934. [DOI] [PubMed] [Google Scholar]
- 18. DiBenedictis BT, Ingraham KL, Baum MJ, Cherry JA. Disruption of urinary odor preference and lordosis behavior in female mice given lesions of the medial amygdala. Physiol Behav. 2012;105:554–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maras PM, Petrulis A. Chemosensory and steroid-responsive regions of the medial amygdala regulate distinct aspects of opposite-sex odor preference in male Syrian hamsters. Eur J Neurosci. 2006;24:3541–3552. [DOI] [PubMed] [Google Scholar]
- 20. Kauffman AS, Navarro VM, Kim J, Clifton DK, Steiner RA. Sex differences in the regulation of Kiss1/NKB neurons in juvenile mice: implications for the timing of puberty. Am J Physiol Endocrinol Metab. 2009;297:E1212–E1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roa J, Vigo E, Castellano JM, et al. Opposite roles of estrogen receptor (ER)-α and ERβ in the modulation of luteinizing hormone responses to kisspeptin in the female rat: implications for the generation of the preovulatory surge. Endocrinology. 2008;149:1627–1637. [DOI] [PubMed] [Google Scholar]
- 22. Shughrue PJ, Scrimo PJ, Merchenthaler I. Evidence for the colocalization of estrogen receptor-β mRNA and estrogen receptor-α immunoreactivity in neurons of the rat forebrain. Endocrinology. 1998;139:5267–5270. [DOI] [PubMed] [Google Scholar]
- 23. Osterlund M, Kuiper GG, Gustafsson JA, Hurd YL. Differential distribution and regulation of estrogen receptor-α and -β mRNA within the female rat brain. Brain Res Mol Brain Res. 1998;54:175–180. [DOI] [PubMed] [Google Scholar]
- 24. Di Giorgio NP, Semaan SJ, Kim J, et al. Impaired GABAB receptor signaling dramatically up-regulates Kiss1 expression selectively in nonhypothalamic brain regions of adult but not prepubertal mice. Endocrinology. 2014;155:1033–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cao J, Patisaul HB. Sex-specific expression of estrogen receptors α and β and Kiss1 in the postnatal rat amygdala. J Comp Neurol. 2013;521:465–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Semaan SJ, Kauffman AS. Daily successive changes in reproductive gene expression and neuronal activation in the brains of pubertal female mice. Mol Cell Endocrinol. 2015;401:84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Clarkson J, Herbison AE. Postnatal development of kisspeptin neurons in mouse hypothalamus; sexual dimorphism and projections to gonadotropin-releasing hormone neurons. Endocrinology. 2006;147:5817–5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Desroziers E, Droguerre M, Bentsen AH, et al. Embryonic development of kisspeptin neurones in rat. J Neuroendocrinol. 2012;24:1284–1295. [DOI] [PubMed] [Google Scholar]
- 29. Kumar D, Periasamy V, Freese M, Voigt A, Boehm U. In utero development of kisspeptin/GnRH neural circuitry in male mice. Endocrinology. 2015;156:3084–3090. [DOI] [PubMed] [Google Scholar]
- 30. Dupont S, Krust A, Gansmuller A, Dierich A, Chambon P, Mark M. Effect of single and compound knockouts of estrogen receptors α (ERα) and β (ERβ) on mouse reproductive phenotypes. Development. 2000;127:4277–4291. [DOI] [PubMed] [Google Scholar]
- 31. Stephens SB, Tolson KP, Rouse ML, Jr, et al. Absent progesterone signaling in kisspeptin neurons disrupts the LH surge and impairs fertility in female mice. Endocrinology. 2015;156:3091–3097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kim J, Tolson KP, Dhamija S, Kauffman AS. Developmental GnRH signaling is not required for sexual differentiation of kisspeptin neurons but is needed for maximal Kiss1 gene expression in adult females. Endocrinology. 2013;154:3273–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Russo KA, LA JL, Stephens SB, et al. Circadian control of the female reproductive axis through gated responsiveness of the RFRP-3 system to VIP signaling. Endocrinology. 2015;156:2608–2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luo E, Stephens SB, Chaing S, Munaganuru N, Kauffman AS, Breen KM. Corticosterone blocks ovarian cyclicity and the LH surge via decreased kisspeptin neuron activation in female mice. Endocrinology. 2016;157:1187–1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dror T, Franks J, Kauffman AS. Analysis of multiple positive feedback paradigms demonstrates a complete absence of LH surges and GnRH activation in mice lacking kisspeptin signaling. Biol Reprod. 2013;88:146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shughrue PJ, Lane MV, Merchenthaler I. Comparative distribution of estrogen receptor-α and -β mRNA in the rat central nervous system. J Comp Neurol. 1997;388:507–525. [DOI] [PubMed] [Google Scholar]
- 37. Dhillo WS, Chaudhri OB, Patterson M, et al. Kisspeptin-54 stimulates the hypothalamic-pituitary gonadal axis in human males. J Clin Endocrinol Metab. 2005;90:6609–6615. [DOI] [PubMed] [Google Scholar]
- 38. Navarro VM, Castellano JM, Fernandez-Fernandez R, et al. Characterization of the potent luteinizing hormone-releasing activity of KiSS-1 peptide, the natural ligand of GPR54. Endocrinology. 2005;146:156–163. [DOI] [PubMed] [Google Scholar]
- 39. Navarro VM, Castellano JM, Fernandez-Fernandez R, et al. Effects of KiSS-1 peptide, the natural ligand of GPR54, on follicle-stimulating hormone secretion in the rat. Endocrinology. 2005;146:1689–1697. [DOI] [PubMed] [Google Scholar]
- 40. Irwig MS, Fraley GS, Smith JT, et al. Kisspeptin activation of gonadotropin releasing hormone neurons and regulation of KiSS-1 mRNA in the male rat. Neuroendocrinology. 2004;80:264–272. [DOI] [PubMed] [Google Scholar]
- 41. Messager S, Chatzidaki EE, Ma D, et al. Kisspeptin directly stimulates gonadotropin-releasing hormone release via G protein-coupled receptor 54. Proc Natl Acad Sci USA. 2005;102:1761–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Pineda R, Plaisier F, Millar RP, Ludwig M. Amygdala kisspeptin neurons: putative mediators of olfactory control of the gonadotropic axis. Neuroendocrinology. In press. [DOI] [PubMed] [Google Scholar]