

Abstract
Ligand-dependent actions of the vitamin D receptor (VDR) play a pleiotropic role in the regulation of innate and adaptive immunity. The liganded VDR is required for recruitment of macrophages during the inflammatory phase of cutaneous wound healing. Although the number of macrophages in the granulation tissue 2 days after wounding is markedly reduced in VDR knockout (KO) compared with wild-type mice, VDR ablation does not alter macrophage polarization. Parabiosis studies demonstrate that circulatory chimerism with wild-type mice is unable to rescue the macrophage defect in the wounds of VDR KO mice and reveal that wound macrophages are of local origin, regardless of VDR status. Wound cytokine analyses demonstrated a decrease in macrophage colony-stimulating factor (M-CSF) protein levels in VDR KO mice. Consistent with this, induction of M-CSF gene expression by TGFβ and 1,25-dihydroxyvitamin D was impaired in dermal fibroblasts isolated from VDR KO mice. Because M-CSF is important for macrophage self-renewal, studies were performed to evaluate the response of tissue resident macrophages to this cytokine. A decrease in M-CSF induced proliferation and cyclin D1 expression was observed in peritoneal resident macrophages isolated from VDR KO mice, suggesting an intrinsic macrophage abnormality. Consistent with this, wound-healing assays in mice with macrophage-specific VDR ablation demonstrate that a normal wound microenvironment cannot compensate for the absence of the VDR in macrophages and thus confirm a critical role for the macrophage VDR in the inflammatory response to injury.
Vitamin D is produced in the skin in response to UV radiation. Sequential hydroxylation steps result in formation of the active hormone, 1,25-dihydroxyvitamin D, which regulates gene expression by binding to the vitamin D receptor (VDR), a member of the nuclear receptor superfamily of ligand-dependent transcription factors (1). In addition to the classic actions of 1,25-dihydroxyvitamin D that contribute to the regulation of mineral ion hemostasis, the receptor-dependent actions of 1,25-dihydroxyvitamin D play an important role in several nontraditional target tissues such as the heart, the skin and cells of the immune system, including macrophages (2–4).
Macrophages play a critical role in innate and adaptive immune responses. In addition to their role in host defense against pathogens, they contribute to tissue remodeling under homeostatic conditions and in response to injury (5). The first link between vitamin D and macrophage function can be traced back more than a century to the time when increasing vitamin D levels by exposure to solar radiation or ingestion of cod liver oil was used to treat tuberculosis (6). Subsequent investigations demonstrated that macrophages associated with granulomata (7) and nonpathological states (8) were able to convert 25-hydroxyvitamin D to its active metabolite, 1,25-dihydroxyvitamin D, due to inducible expression of Cyp27B1, the vitamin D 1α-hydroxylase (9). These findings led to numerous investigations examining the effect of the liganded VDR on innate and adaptive immunity,
Activation of the innate immune system by microbial pathogens promotes an inflammatory process that leads to clearance of the offending pathogens. Notably, activation of the Toll-like receptor 2/1 pattern recognition receptors by Mycobacterium tuberculosis induces the expression of the VDR and Cyp27B1 in macrophages, resulting in induction of antimicrobial peptides and accelerated mycobacterial clearance (10). Similarly, induction of Cyp27B1 by activation of Toll-like receptor 2/6 triggers VDR ligand-dependent immune responses in keratinocytes, including the expression of antimicrobial peptides (11).
The VDR and its ligand also have pleiotropic roles in adaptive immunity. The most extensively investigated role of vitamin D in the adaptive immune system is inhibition of T-cell proliferation and modulation of the T-cell phenotype (6). Although the liganded VDR promotes immune tolerance, paradoxically, the absence of the VDR in mice impairs cytokine production by Th2 and iNKT cells, protecting against airway hyperreactivity (12). Ligand-dependent actions of the VDR inhibit dendritic cell maturation (13, 14) and cytokine production as well as suppress the ability of macrophages to present antigens and produce inflammatory cytokines (15).
Macrophages comprise a heterogeneous population of cells that either promote or inhibit host responses to pathogens and injury. Macrophages are categorized into classically (M1) and alternatively (M2) activated groups (16). M1 macrophages produce inflammatory cytokines and reactive oxygen species that contribute to host defense against pathogens, whereas M2 macrophages attenuate inflammation and participate in tissue remodeling and parasite clearance. Whereas this paradigm is an oversimplification of the complexity and plasticity of macrophage phenotypes (17), based on this classification, tissue-resident macrophages are M2-like and play an important role in tissue homeostasis and resolution of inflammation (18).
Our previous investigations demonstrated that the ligand-dependent actions of the VDR are required for the increase in macrophage number and granulation tissue formation in response to cutaneous injury (19). Investigations were therefore undertaken to characterize the activation state of macrophages in the wounds of VDR null mice and to address the pathophysiologic basis for the impaired macrophage response observed.
Materials and Methods
Animal studies
Studies were approved by the institutional animal care committee. With the exception of mice expressing the K14-VDR transgene, which are on a mixed C57BL6/J;FVB/N background (20), all mice were on the C57BL6/J background. Both males and females were studied; no sexual dimorphism was observed. Mice were maintained in a virus- and parasite-free animal facility under a 12-hour light, 12-hour dark cycle. To prevent abnormalities in mineral ion homeostasis, which are observed by the third week of life, VDR knockout (KO) mice were weaned at day 18 onto a diet that maintains normal mineral ion homeostasis in the absence of VDR signaling (TD96348; Harlan Teklad) (21). Vitamin D-deficient mice were bred and maintained in a UV-free environment and were weaned onto a similar diet lacking vitamin D metabolites (TD97340; Harlan Teklad), which results in undetectable circulating 25-hydroxyvitamin D and 1,25-dihydroxyvitamin D levels after three generations (22). To evaluate the consequences of vitamin D deficiency, studies were performed in five mixed-gender mice raised under these conditions for four or more generations. Under general anesthesia, mice were subjected to dorsal full-thickness wounds using a 3.5-mm biopsy punch (Unipunch; Premier). Parabiosis studies were performed in five pairs of age- and sex-matched mice differing in the expression of a β-actin/enhanced green fluorescent protein (GFP) transgene (23). Four weeks after the surgery, circulatory chimerism was confirmed by flow cytometry. Parabiotic pairs were subjected to wound-healing assays 6 weeks after the surgery. Mice with floxed VDR alleles (24) were mated to LysM-cre mice (Jackson Labs) to obtain mice lacking the VDR in macrophages. Wounds were harvested from six mice to determine the effects of macrophage-specific VDR ablation on macrophage number.
Histology and immunohistochemistry
Tissue samples were fixed in 4% paraformaldehyde in PBS for 3 hours at room temperature, processed, and embedded in paraffin. For immunofluorescent immunohistochemistry, antigen retrieval was performed using 1 mg/mL trypsin (T7168; Sigma) for 10 minutes at 37°C. Sections were blocked and permeabilized in 10% heat-inactivated fetal bovine serum/0.25% Triton X-100 in PBS. Endogenous avidin and biotin were blocked (SP-2001; Vector Labs) prior to incubation with secondary antibodies. Primary antibodies were used at the following dilutions: α-mouse F4/80, 1:100 (14–4801; Affymetrix eBioscience); α-arginase I, 1:100 (sc-18351; Santa Cruz Biotechnology); resistin-like molecule-α (RELM-α), 1:100 (ab39626; Abcam); and α-TNF-α, 1:100 (ab9739; Abcam). The Tyramide Signal Amplification biotin kit (PerkinElmer) was used for signal amplification. Streptavidin-conjugated Alexa Fluor 594 and 488 (S-11227 and S-11223; Life Technologies) were used to detect secondary antibodies. The total number of positive cells per high-power field (hpf; ×20) was manually quantified using digitally captured images. Masson's Trichrome staining was used to evaluate fibrosis.
Monocyte depletion by liposomal clodronate
Liposomal Clodronate suspension (200μcl; Encapsula Nano Sciences) was injected iv 24 hours before and immediately prior to wounding. Wounds were harvested at 48 hours as was the peripheral blood. Macrophages were evaluated in six wounds from clodronate-injected and control mice. Red blood cells were lysed (lysis buffer, R7757; Sigma) prior to flow cytometry for allophycocyanin (APC)-CD11b+ (553312, BD Biosciences) and Phycoerythrin-F4/80+ (FAB5580P, R&D Systems) cells using an eight-laser SORP LSR II flow cytometer (Becton Dickinson).
Evaluation of in vivo cytokine expression
ELISA cytokine arrays (RayBiotech) were performed according to the manufacturer's instructions. Wounds were harvested after 24 hours and 900 μg of wound protein was incubated with the antibody-coated wells. Fluorescent intensity was analyzed with an Axon GenePix 4000B scanner. Cytokine concentration was calculated based on cytokine-specific standard curves.
Dermal fibroblast culture and treatment
Dermal fibroblasts were isolated from neonatal mice as previously described (19). Cells were treated with 10−8 M 1,25-dihydroxyvitamin D or 10 ng/mL recombinant human TGF-β1 (R&D Systems) prior to the isolation of RNA. Data were obtained using three replicates per genotype.
Peritoneal macrophage culture and treatment
Immediately after euthanasia, the peritoneum of mice was lavaged with 10 mL ice-cold Dulbecco's PBS without calcium and magnesium (17–512Q; Lonza). Pelleted cells were incubated with APC-conjugated α-CD11b and phycoerythrin conjugated α-F4/80 prior to sorting. Double-positive cells were cultured with cytokines (20 ng/mL of macrophage colony stimulating factor (M-CSF), IL-9, or IL-13) in DMEM/F12 (Invitrogen) supplemented with 10% fetal bovine serum (AXG44975; Hyclone), 10 mM L-glutamine (Gibco), penicillin, and streptomycin. Media and growth factors were replenished every 24 hours. Proliferation was evaluated by flow cytometry of seven samples per genotype 3 hours after the addition of 5-ethynyl-2'-deoxyuridine (EdU) to cells cultured with M-CSF for 24 hours. RNA was isolated after 72 hours from five samples per genotype. For the evaluation of M-CSF-induced ERK1/2 phosphorylation, cells were plated in the absence of growth factors for 16 hours and treated with M-CSF for 10 minutes. Quantitation of phosphorylated ERK1/2 was performed on four samples per genotype using an AlphaLISA SureFire Ultra kit (PerkinElmer).
Real-time PCR
RNA was extracted from CD11b;F4/80 double-positive peritoneal cells using the RNeasy Plus minikit (QIAGEN) immediately after sorting or after 72 hours of in vitro cytokine treatment. RNA was reversed transcribed using Superscript II reverse transcriptase (Invitrogen). Quantitative real-time PCR was performed using primers designed to span introns. Target gene expression was normalized to actin mRNA in the same sample, and relative gene expression was calculated using the method of Livak and Schmittgen (25).
Statistics
Statistical significance between data obtained from wild-type (WT) and VDR KO mice was evaluated by a Student's t test. Statistical differences between three groups (WT, VDRKO, and vitamin D deficient or LysM-Cre VDR floxed mice) were determined by an ANOVA and a Tukey's multiple comparison test. A value of P < .05 was considered significant.
Results
Ligand-dependent actions of the VDR regulate M2 macrophage response to cutaneous injury.
Ligand-dependent actions of the VDR are required for recruitment of macrophages in response to cutaneous injury in mice (19). Because 1,25-dihydroxyvitamin D and TGF-β signaling (19), both of which suppress the M1 macrophage phenotype (26–28), are impaired in VDR KO mice, investigations were undertaken to determine whether the observed decrease in macrophage number reflects absence of ligand-dependent actions of the VDR on wound macrophage polarization. Based on the absence of sexual dimorphism in macrophage recruitment to cutaneous wounds, 8-week-old WT, VDR-KO, and vitamin D-deficient male and female mice were subjected to full-thickness skin wounds. Due to the small size of the wound, which precludes characterization of macrophages by flow cytometry, immunofluorescence studies were performed to quantitate and characterize macrophages. Consistent with previous data (19), the number of F4/80+ macrophages was markedly reduced in the wounds of VDR-KO and vitamin D-deficient mice relative to WT controls at 48 hours after injury (Figure 1). Immunohistochemical (IHC) analyses performed to evaluate macrophage polarization revealed that fewer than 10% of the F4/80+ macrophages in the granulation tissue of WT, VDR-KO, and vitamin D-deficient mice expressed TNF-α, a marker of classically activated macrophages (M1) (data not shown). Consistent with previous findings that macrophages in aseptic cutaneous wounds express an M2 phenotype (29), approximately 80% of the F4/80+ macrophages in the wounds of WT mice expressed arginase-1 and RELM-α, markers of alternatively activated M2 macrophages (Figure 1). Whereas the total number of F4/80+ macrophages was significantly decreased in the wounds of the VDR-KO and vitamin D-deficient mice, the percentage of alternatively activated (M2) macrophages was not significantly different from that observed in WT mice.
Figure 1.
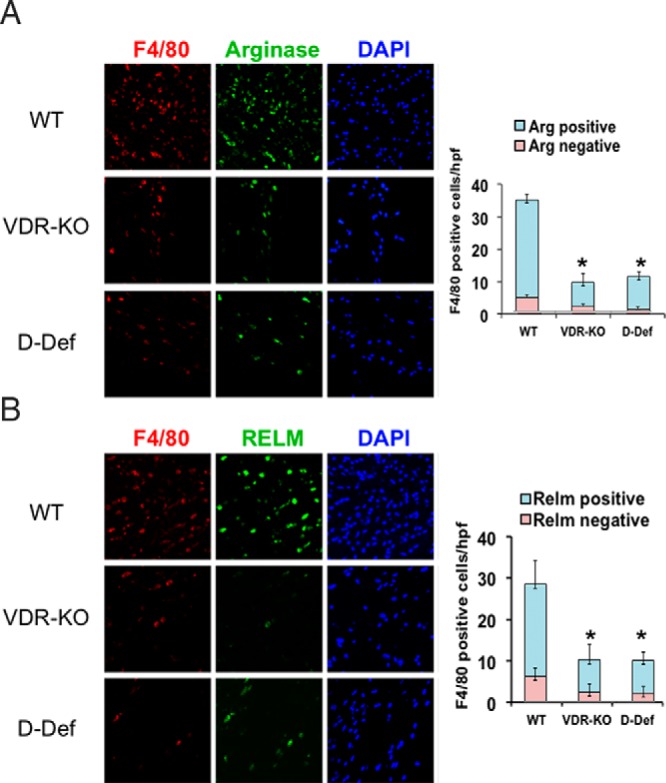
Ligand-dependent actions of the VDR regulate M2 macrophage response to cutaneous injury. IHC analyses for F4/80 were performed on 2-day wounds of WT, VDR KO, and vitamin D-deficient (D-Def) mice to evaluate M2 macrophages. Markers of M2 polarization, arginase (A) and RELMα (B), were also evaluated. Graphs represent the number of F4/80+ cells per high-power field that were found to be arginase (Arg) and RELM positive or negative. Data represent those obtained from five wounds per condition. *, P < .0001 vs WT by ANOVA (VDR KO vs D-Def, P = NS). DAPI, 4′,6′-diamino-2-phenylindole.
Cutaneous wound granulation tissue macrophages are of local tissue origin
Investigations in a murine model of atopic dermatitis demonstrate that keratinocyte VDR expression is required for the proliferation of epidermal macrophage-like Langerhans cells (30). However, restoring VDR signaling in keratinocytes, by transgenic overexpression of the VDR under a keratinocyte-specific K14-VDR transgene (20), does not normalize the VDR KO wound macrophage phenotype (Figure 2A). This suggests that an intrinsic macrophage defect or abnormalities in the local wound microenvironment lead to the decreased macrophage number observed in the VDR KO wounds. To address this, studies were performed in parabiotic mice, a model in which surgically conjoining mice results in circulatory chimerism within 2 weeks (23). Age- and sex-matched WT and VDR KO mice were conjoined with mice differing in the presence of an actin-enhanced GFP transgene. Cross-circulation was shown to be established 4 weeks after surgery by flow cytometric analyses of peripheral blood. Parabionts were wounded 6 weeks after surgery, and wounds were harvested 48 hours later. Although the wound clots of all mice exhibited marked GFP fluorescence, confirming circulatory chimerism (Figure 2B), approximately 70% of the nucleated cells in the granulation tissue were of autologous origin, regardless of the VDR status of the parabionts (WTGFP+/WTGFP−, WTGFP+/VDRKOGFP− or VDRKOGFP+/VDRKOGFP−) (Figure 2B and Table 1). Conjoining WT and VDR KO circulations did not normalize macrophage numbers in the wounds of the VDR KO mice. Moreover, the majority (>84.6%) of wound macrophages were of autologous origin, regardless of VDR status (Figure 2B and Table 1), demonstrating that most macrophages present in the granulation tissue 48 hours after wounding are not derived from the circulation.
Figure 2.

Cutaneous wound granulation tissue macrophages are of local tissue origin. A, Restoring VDR expression in keratinocytes of VDR KO (VDR KOK14VDR) mice does not normalize cutaneous wound macrophage number. Data represent the mean and SD of the number of F4/80-positive macrophages per high-power field in five wounds per genotype. *, P < .0001 vs WT by ANOVA (VDR KO vs VDR KOK14-VDR, P = NS). B, Parabiosis studies were performed in WT and VDR KO mice differing in GFP status. GFP fluorescence and DAPI nuclear staining of the clot and granulation tissue of day 2 wounds are shown under low magnification (×10). GFP and F4/80 dual immunofluorescence is shown under high power (×20). Data are representative of those obtained with five parabiotic pairs per condition. C, Flow cytometric analyses demonstrate depletion of circulating F4/80-positive cells by clodronate liposomes. D, F4/80 IHC assays of wound macrophages in clodronate liposome- and PBS-injected mice. Graph represents the mean and SD of the number of F4/80-positive cells/hpf. Data are representative of those obtained in six wounds per genotype. DAPI, 4′,6′-diamino-2-phenylindole.
Table 1.
Quantification of GFP+ and F4/80+ Cells in Parabiotic Mice
| Genotype | Parabiont | %GFP+ Cells | F4/80+/hpf | %GFP+ F4/80+ Cells |
|---|---|---|---|---|
| WTGFP+/WTGFP− | WT GFP+ | 71.1 ± 4.1 | 43 ± 13 | 86.6 ± 7.3 |
| WT GFP− | 29.0 ± 2.4* | 44 ± 11 | 14.1 ± 8.1*** | |
| WTGFP+/KOGFP− | WT GFP+ | 69.4 ± 4.5 | 55 ± 23 | 84.6 ± 4.0 |
| KO GFP− | 22.1 ± 5.1* | 15 ± 6** | 17.6 ± 6.9*** | |
| KOGFP+/KOGFP− | KO GFP+ | 69.5 ± 5.9 | 10 ± 7** | 94.0 ± 8.9 |
| KO GFP− | 28.5 ± 3.2* | 17 ± 2** | 20.6 ± 4.2*** |
Results are based on data from 5 mice per condition. Statistical significance is indicated as follows *p < 0.05 and ***p < 0.001 vs the parabiotic partner for GFP+ and %GFP+F4/80+ cells; **p < 0.01 vs WT for F4/80+ cells/hpf.
To confirm the contribution of tissue resident macrophages to cutaneous wound healing, circulating cells were depleted by injection of clodronate liposomes 24 hours prior to, and immediately before, wounding. Clodronate liposomes resulted in a greater than 90% decrease in circulating macrophages 24 hours after injection (Figure 2C and data not shown) but did not alter the number of wound macrophages present (Figure 2D).
To determine whether the reduced macrophage number observed in the wounds of VDR KO mice represents a delay, rather than an block in macrophage expansion, wounds of single (data not shown) or parabiotic mice were examined 7 days after wounding. These studies demonstrated a persistent reduction in the number of macrophages in the wounds of the VDR KO mice and continued predominance of autologous macrophages in the parabionts (Supplemental Figure 1A). Because M2 macrophages promote matrix deposition during this tissue remodeling phase of wound healing, the matrix was evaluated by Masson's trichrome staining. A dramatic decrease in connective tissue was observed in the wounds of VDR KO mice, regardless of the VDR status of its parabiotic partner, suggesting impaired macrophage-mediated granulation tissue maturation (Supplemental Figure 1B).
Impaired cytokine production in VDR-KO wounds
Numerous cytokines have been shown to regulate the proliferation of tissue resident macrophages in response to infection, inflammation, and injury (31). To determine whether local cytokine production is impaired in the wounds of VDR KO mice, protein lysates from the granulation tissue of 24-hour wounds were quantitated using a cytokine array ELISA. Although 1,25-dihydroxyvitamin D induces expression of interferon-γ, IL-6, TNFα, and granulocyte macrophage colony-stimulating factor (32), the levels of IL-6, TNFα, and granulocyte macrophage colony-stimulating factor were not altered in the wounds of VDR KO mice and interferon-γ was not detected in the wound lysates of either control or VDR KO mice. However, the levels of IL-13 and M-CSF were found to be lower in the wound lysates of VDR KO mice compared with those of WT mice (respectively, 139.8 ± 21.0 pg/mg vs 36.4 ± 18.1 pg/mg, P = .02; 184.2 ± 17.7 pg/mg vs 69.6 ± 20.5 pg/mg, P = .01; and 17.6 ± 4.1 pg/mg vs 3.1 ± 2.8 pg/mg, P = .06) (Figure 3A). As reported by others (29), IL-4 was undetectable in the skin wounds.
Figure 3.

Wound cytokine production and M-CSF-induced tissue resident macrophage proliferation is impaired in VDR KO mice. A, Concentration of IL-9, IL-13, and M-CSF in the wounds of VDR KO mice was determined by a cytokine array ELISA using six wounds per genotype. *, P < .05 vs WT by Student's paired t test. B, Induction of M-CSF mRNA in dermal fibroblasts from WT and VDR KO mice in response to a 4-hour treatment with 10 ng/mL TGFβ or 10−8 M 1,25-dihydroxyvitamin. Data are representative of three replicates per genotype. *, P < .003, **, P < .03 by Student's paired t test. C, Peritoneal tissue resident macrophage proliferation was evaluated after 27 hours of treatment with M-CSF in vitro. Data are representative of seven samples per genotype. *, P < .0003 vs WT by Student's paired t test. D, Basal and M-CSF (10 min treatment) induced ERK1/2 phosphorylation in cultured peritoneal macrophages. Data are representative of four samples per genotype. *, P < .04 vs WT by Student's paired t test. E, Fold induction of colony stimulating factor 1 receptor, cyclin D1, and GATA6 mRNA expression in peritoneal macrophages after 72 hours of M-CSF treatment in vitro. Data represent the mean and SEM of those obtained with five samples per group. *, P < .03 vs WT by Student's paired t test.
Because M-CSF is important for the self-renewal of tissue resident macrophages (33) and both TGFβ and 1,25-dihydroxyvitamin D signaling are impaired in the wounds of VDR KO mice, studies were performed to determine whether these factors regulate M-CSF production by dermal fibroblasts. Primary dermal fibroblasts, isolated from WT and VDR KO mice, were treated with 10 ng/mL TGFβ or 10−8 M 1,25-dihydroxyvitamin D for 3 hours prior to the isolation of RNA. Although basal M-CSF expression was not affected by the VDR status, TGFβ induction of M-CSF was attenuated in the VDR KO fibroblasts, whereas induction by 1,25-dihydroxyvitamin D was abolished, suggesting that abnormalities in the local wound microenvironment may contribute to the decreased macrophage number observed in the wounds of VDR KO mice (Figure 3B).
To determine whether impaired macrophage proliferation contributes to the phenotype of the VDR KO skin wounds, investigations were undertaken to evaluate whether the proliferation of tissue resident VDR KO macrophages could be normalized by exogenous cytokines. Because normal skin and the 3.5-mm wounds provide insufficient numbers of macrophages for analyses, based on their accessibility, peritoneal macrophages were isolated by flow cytometry (CD11b+;F4/80+) and cultured in the presence of M-CSF, IL-9, or IL-13. IL-9 was unable to induce proliferation of WT or VDR-KO peritoneal macrophages in vitro. IL-13 treatment resulted in rare EdU+ cells in WT but not VDR KO macrophages after 24 hours. M-CSF induced proliferation of VDR KO macrophages was attenuated relative to that observed in the macrophages isolated from WT mice, based on flow cytometry of EdU+ macrophages (Figure 3C).
M-CSF signaling through its receptor induces ERK1/2 phosphorylation in macrophages (34). To evaluate this signaling pathway, M-CSF receptor mRNA levels and ERK1/2 phosphorylation in response to M-CSF were examined. As demonstrated in Figure 3D, basal ERK1/2 phosphorylation was not affected by VDR status. However, M-CSF-induction of ERK1/2 phosphorylation was paradoxically enhanced in the VDR KO macrophages. Consistent with this, expression of the M-CSF receptor, colony stimulating factor 1 receptor, was not decreased in the VDR KO macrophages (Figure 3E). Evaluation of M-CSF-regulated genes known to play a role in macrophage proliferation demonstrated that neither GATA6 (35) nor c-myc expression was impaired in the VDR KO macrophages under basal conditions or after exposure to CSF-1. However, a reduction in both basal (44.4% ± 4.7% of WT; P = .001) and M-CSF induced cyclin D1 expression (Figure 3E) was observed in the in VDR KO macrophages, consistent with the decrease in M-CSF-induced proliferation observed in culture.
VDR expression in macrophages is essential for the inflammatory response to wound injury
To determine the relative contributions of the VDR in macrophages and in dermal fibroblasts to the impaired inflammatory response to wounding, mice lacking the VDR in macrophages were generated. LysM-cre-mediated VDR deletion resulted in a phenotype indistinguishable from that observed in mice with global VDR ablation, demonstrating that the expression of the VDR in macrophages is essential for the normal expansion of tissue-resident macrophages in response to cutaneous wounding (Figure 4).
Figure 4.
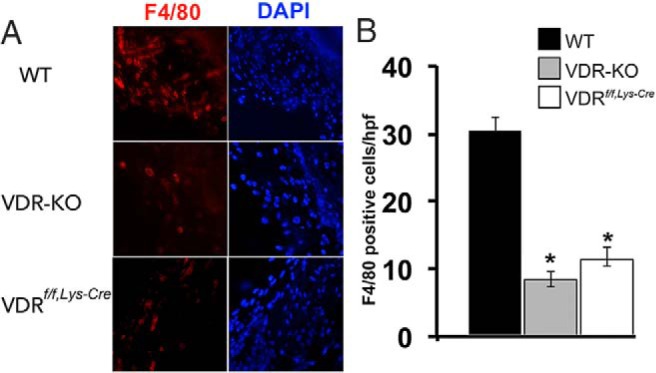
Macrophage VDR expression is required for the inflammatory response to tissue injury. A, IHC analyses for F4/80 were performed on 2-day wounds of WT, VDR KO, and Lys-Cre VDR f/f mice. B, Graphs represent the number of F4/80-positive cells per high-power field. Data are based on five to six wounds per genotype. *, P < .0001 vs WT by ANOVA (VDR KO vs VDRf/f,Lys-Cre, P = NS). DAPI, 4′,6′-diamino-2-phenylindole.
Discussion
Tissue repair is characterized by several overlapping phases: hemostasis, inflammation, granulation tissue formation, and tissue remodeling (36). During the inflammatory stage of cutaneous wound healing, neutrophils and macrophages are recruited to the wound area (37), participating in host defense and debridement of damaged tissue. Studies in transgenic models of inducible macrophage depletion demonstrate that macrophages are critical for clearance of pathogens and necrotic debris as well as angiogenesis and tissue repair (38, 39).
Whereas circulating macrophages are derived from bone marrow-resident adult hematopoietic stem and progenitor cells, tissue-resident macrophages originate largely from embryonic yolk sac progenitors (40). In many pathological settings, the relative roles of tissue-resident vs circulating macrophages have not been established. Although it was originally postulated that macrophages recruited to sites of injury and inflammation were derived from circulating monocytes of bone marrow origin, more recently a critical role for tissue resident macrophages in this processes has been established. Studies of microglia, the brain-resident macrophages, and Langerhans cells, the epidermal resident macrophages, support the hypothesis that under homeostatic conditions, macrophages in many organ systems are repopulated by tissue-resident cells (41, 42). Whereas steady-state Langerhans cell proliferation is independent of the VDR, induction of proliferation in a model of atopic dermatitis requires keratinocyte VDR expression (30). However, our investigations in VDR KO mice demonstrate that expressing a VDR transgene in epidermal keratinocytes does not restore a normal wound macrophage phenotype. Consistent with the hypothesis that the increase in macrophage number observed with different stimuli may be due to the proliferation of local cells vs recruitment from the bone marrow, the expansion of Langerhans cells observed in response to UV irradiation injury and graft-vs-host disease is dependent on bone marrow-derived macrophages.
Studies performed to address the relative roles of circulating vs resident macrophages in injury repair have taken advantage of bone marrow transplantation strategies performed with lineage traceable donor bone marrow-derived cells. These studies demonstrate a significant contribution of bone marrow-derived macrophages to the inflammatory response; however, the radiation doses used in these studies may ablate or damage endogenous tissue resident macrophages as well as significantly disrupt vascular permeability, increasing cellular extravasation and thereby the contribution of circulating cells. Furthermore, intravascular injection of donor cells derived from whole bone marrow may result in the persistence of macrophage/monocyte progenitors in the circulation of the recipient that have the potential to recapitulate the developmental establishment of a tissue resident macrophage network. In contrast to these studies, parabiosis studies demonstrate that microglial response to neurological injury involves cells of local origin (41, 43). Unlike the central nervous system (CNS), the skin and the intestine are constantly exposed to potential pathogenic stimuli. Intestinal resident macrophages are continuously repopulated by circulating precursors (44, 45). Parabiosis studies in WT mice demonstrate that lung macrophages are of autologous origin (33), whereas approximately 10%–20% of skin macrophages originate from circulating cells (46, 47). This is consistent with our data demonstrating that approximately 15%–20% of macrophages in the skin wounds of VDR KO or WT parabionts originate from the parabiotic partner.
Our investigations demonstrate that the macrophages present during the early stages of cutaneous wound repair are predominantly M2 macrophages of local, rather than circulatory, origin and suggest that induction of local macrophage proliferation is critical for the increase in cell number observed in response to cutaneous injury. These findings are consistent with the origin of macrophages that contribute to host response to nematode infection (48). However, our results contrast with those of a previous study demonstrating a significant contribution of circulating (23) and proinflammatory M1 (49) macrophages to the early inflammatory response to cutaneous injury. However, those studies examined the response to more extensive injuries, which would be expected to elicit a more significant inflammatory phenotype and be associated with a greater pathogen burden. In addition, our observations of local expansion of M2 polarized macrophages in the cutaneous wound model are consistent with prior results using parabiosis between young and old mice to track the contributions of circulation-derived macrophages to the repair of demyelinating lesions in the aging mouse spinal cord. These studies demonstrated that, whereas the recruitment of young circulating monocytes into the demyelinated lesions of older mice is associated with an increased density of M2 microglia and macrophages (50) and a restoration of remyelinating activity (51), the recruited cells contribute only minimally to the M2 macrophage pool; instead, these circulation-derived macrophages act indirectly to promote M2 polarization of endogenous macrophages, which in turn promote the differentiation of remyelinating oligodendrocytes (50).
Our investigations, which reveal that absence of the VDR attenuates macrophage number and fibrotic matrix deposition in a cutaneous wound-healing model, also contrast with previous studies demonstrating that the liganded VDR suppresses inflammation and fibrosis in models of liver (52) and renal (28, 53, 54) injury. Although none of these preclinical models is able to address the effects of vitamin D insufficiency or mild vitamin D deficiency, the profound differences in the phenotypes observed may be a reflection of models investigated. Both the liver and renal injury models result in significant cellular necrosis. Similar to muscle injury, in which chemically induced injury but not laceration induces an M1 macrophage response (55), necrotic tissue in these renal and liver injury models would be predicted to elicit an inflammatory response characterized by classical activation of macrophages (M1), a phenotype that is profibrotic. Thus, these latter studies are consistent with observations that the liganded VDR attenuates M1 macrophage activity (15, 28), including suppression of interferon-γ expression (56). In our skin punch biopsy model, tissue is removed rather than damaged, leaving little necrotic tissue behind. This difference may be responsible for the observed predominance of alternatively activated M2 macrophages that are known to play an important role in tissue remodeling and matrix deposition in addition to immunoregulation.
Whereas ligand-dependent effects of the VDR attenuate expression of classic M1 macrophage markers and promote the M2 phenotype in models of inflammation (15, 28), our studies did not demonstrate a significant effect of VDR ablation on macrophage polarization. Although the wounds in our model were exposed to commensal organisms commonly present in pathogen-free barrier facilities, our findings are similar to investigations demonstrating that sterile incisional peritoneal wound healing is characterized by resident M2 macrophage proliferation (48). Taken together, these findings suggest that tissue injury induces local M2 macrophages involved in tissue repair and that only in the presence of significant necrosis or pathogen burden are bone marrow derived macrophages recruited. These data also demonstrate that the absence of the VDR leads to impaired cytokine production in response to injury and resistance to M-CSF induction of macrophage proliferation and cyclin D1 expression, independently of ERK1/2 phosphorylation. Thus, in addition to decreased M-CSF in the VDR KO wounds, our studies define a role for the VDR in regulating tissue-resident M2 macrophages, which are critical for tissue remodeling after an injury. Future investigations will be required to determine whether the effects of the VDR on postinjury repair are dependent on circulating 1,25-dihydroxyvitamin D or autocrine activation of 25-hydroxyvitamin D by tissue resident macrophages (10).
Acknowledgments
This work was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases P30 Core facility (Grant AR066261) and by grants from the National Institutes of Health (Grant R01DK46974) (to M.B.D.) and (Grant UO1 HL100402) (to A.J.W.). H.Z. was supported by a Research Fellowship from the Department of Dermatology, the First Affiliated Hospital of Chongqing Medical University. A.J.W. is an Early Career Scientist of Howard Hughes Medical Institute.
Disclosure Summary: The authors have nothing to disclose.
Appendix
Table 2.
Antibody Table
| Epitope | Company | Product Number |
|---|---|---|
| f4/80 | eBioscience | 14-4801 |
| Arginase | Santa Cruz Biotechnology | sc-18351 |
| RELM-a | Abcam | ab39626 |
| TNF-a | Abcam | ab9739 |
| CD11b-APC | BD Biosciences | 553312 |
| f4/80-PE | R&D Systems | FAB5580P |
Footnotes
- APC
- allophycocyanin
- CNS
- central nervous system
- EdU
- 5-ethynyl-2'-deoxyuridine
- GFP
- green fluorescent protein
- hpf
- high-power field
- IHC
- immunohistochemical
- KO
- knockout
- M-CSF
- macrophage colony-stimulating factor
- RELM-α
- resistin-like molecule-α
- VDR
- vitamin D receptor
- WT
- wild type.
References
- 1. Haussler MR, Whitfield GK, Haussler CA, et al. The nuclear vitamin D receptor: biological and molecular regulatory properties revealed. J Bone Miner Res. 1998;13(3):325–349. [DOI] [PubMed] [Google Scholar]
- 2. Bouillon R, Carmeliet G, Verlinden L, et al. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev. 2008;29(6):726–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baeke F, Takiishi T, Korf H, Gysemans C, Mathieu C. Vitamin D: modulator of the immune system. Curr Opin Pharmacol. 2010;10(4):482–496. [DOI] [PubMed] [Google Scholar]
- 4. Hewison M. Vitamin D and the immune system: new perspectives on an old theme. Rheum Dis Clin North Am. 2012;38(1):125–139. [DOI] [PubMed] [Google Scholar]
- 5. Murray PJ, Wynn TA. Protective and pathogenic functions of macrophage subsets. Nat Rev Immunol. 2011. November;11(11):723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chun RF, Liu PT, Modlin RL, Adams JS, Hewison M. Impact of vitamin D on immune function: lessons learned from genome-wide analysis. Front Physiol. 2014;5:151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Adams JS, Sharma OP, Gacad MA, Singer FR. Metabolism of 25-hydroxyvitamin D3 by cultured pulmonary alveolar macrophages in sarcoidosis. J Clin Invest. 1983;72(5):1856–1860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Koeffler HP, Reichel H, Bishop JE, Norman AW. γ-Interferon stimulates production of 1,25-dihydroxyvitamin D3 by normal human macrophages. Biochem Biophys Res Commun. 1985;127(2):596–603. [DOI] [PubMed] [Google Scholar]
- 9. Fu GK, Lin D, Zhang MY, et al. Cloning of human 25-hydroxyvitamin D-1α-hydroxylase and mutations causing vitamin D-dependent rickets type 1. Mol Endocrinol. 1997;11(13):1961–1970. [DOI] [PubMed] [Google Scholar]
- 10. Liu PT, Stenger S, Li H, et al. Toll-like receptor triggering of a vitamin D-mediated human antimicrobial response. Science. 2006;311(5768):1770–1773. [DOI] [PubMed] [Google Scholar]
- 11. Schauber J, Dorschner RA, Coda AB, et al. Injury enhances TLR2 function and antimicrobial peptide expression through a vitamin D-dependent mechanism. J Clin Invest. 2007;117(3):803–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu S, Zhao J, Cantorna MT. Invariant NKT cell defects in vitamin D receptor knockout mice prevents experimental lung inflammation. J Immunol. 2011;187(9):4907–4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Griffin MD, Lutz W, Phan VA, Bachman LA, McKean DJ, Kumar R. Dendritic cell modulation by 1α,25 dihydroxyvitamin D3 and its analogs: a vitamin D receptor-dependent pathway that promotes a persistent state of immaturity in vitro and in vivo. Proc Natl Acad Sci USA. 2001;98(12):6800–6805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Penna G, Adorini L. 1α,25-Dihydroxyvitamin D3 inhibits differentiation, maturation, activation, and survival of dendritic cells leading to impaired alloreactive T cell activation. J Immunol. 2000;164(5):2405–2411. [DOI] [PubMed] [Google Scholar]
- 15. Korf H, Wenes M, Stijlemans B, et al. 1,25-Dihydroxyvitamin D3 curtails the inflammatory and T cell stimulatory capacity of macrophages through an IL-10-dependent mechanism. Immunobiology. 2012;217(12):1292–1300. [DOI] [PubMed] [Google Scholar]
- 16. Martinez FO, Gordon S. The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep. 2014;6:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Murray PJ, Allen JE, Biswas SK, et al. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity. 2014;41(1):14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mantovani A, Biswas SK, Galdiero MR, Sica A, Locati M. Macrophage plasticity and polarization in tissue repair and remodelling. J Pathol. 2013;229(2):176–185. [DOI] [PubMed] [Google Scholar]
- 19. Luderer HF, Nazarian RM, Zhu ED, Demay MB. Ligand-dependent actions of the vitamin D receptor are required for activation of TGF-β signaling during the inflammatory response to cutaneous injury. Endocrinology. 2013;154(1):16–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen C, Sakai Y, Demay M. Targeting expression of the human vitamin D receptor to the keratinocytes of vitamin D receptor null mice prevents alopecia. Endocrinology. 2001;142:5386–5389. [DOI] [PubMed] [Google Scholar]
- 21. Li YC, Amling M, Pirro AE, et al. Normalization of mineral ion homeostasis by dietary means prevents hyperparathyroidism, rickets, and osteomalacia, but not alopecia in vitamin D receptor-ablated mice. Endocrinology. 1998;139(10):4391–4396. [DOI] [PubMed] [Google Scholar]
- 22. Sakai Y, Kishimoto J, Demay M. Metabolic and cellular analysis of alopecia in vitamin D receptor knockout mice. J Clin Invest. 2001;107:961–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Song G, Nguyen DT, Pietramaggiori G, et al. Use of the parabiotic model in studies of cutaneous wound healing to define the participation of circulating cells. Wound Repair Regen. 2010;18(4):426–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chen S, Law CS, Grigsby CL, et al. Cardiomyocyte-specific deletion of the vitamin D receptor gene results in cardiac hypertrophy. Circulation. 2011;124(17):1838–1847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-δδC([T)]) method. Methods. 2001;25(4):402–408. [DOI] [PubMed] [Google Scholar]
- 26. Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 macrophages and the Th1/Th2 paradigm. J Immunol. 2000;164(12):6166–6173. [DOI] [PubMed] [Google Scholar]
- 27. Mathieu C, Waer M, Laureys J, Rutgeerts O, Bouillon R. Prevention of autoimmune diabetes in NOD mice by 1,25 dihydroxyvitamin D3. Diabetologia. 1994;37(6):552–558. [DOI] [PubMed] [Google Scholar]
- 28. Zhang XL, Guo YF, Song ZX, Zhou M. Vitamin D prevents podocyte injury via regulation of macrophage M1/M2 phenotype in diabetic nephropathy rats. Endocrinology. 2014;155(12):4939–4950. [DOI] [PubMed] [Google Scholar]
- 29. Daley JM, Brancato SK, Thomay AA, Reichner JS, Albina JE. The phenotype of murine wound macrophages. J Leukocyte Biol. 2010;87(1):59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chorro L, Sarde A, Li M, et al. Langerhans cell (LC) proliferation mediates neonatal development, homeostasis, and inflammation-associated expansion of the epidermal LC network. J Exp Med. 2009;206(13):3089–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gentek R, Molawi K, Sieweke MH. Tissue macrophage identity and self-renewal. Immunol Rev. 2014;262(1):56–73. [DOI] [PubMed] [Google Scholar]
- 32. Tobler A, Gasson J, Reichel H, Norman AW, Koeffler HP. Granulocyte-macrophage colony-stimulating factor. Sensitive and receptor-mediated regulation by 1,25-dihydroxyvitamin D3 in normal human peripheral blood lymphocytes. J Clin Invest. 1987;79(6):1700–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hashimoto D, Chow A, Noizat C, et al. Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 2013;38(4):792–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Valledor AF, Comalada M, Xaus J, Celada A. The differential time-course of extracellular-regulated kinase activity correlates with the macrophage response toward proliferation or activation. J Biol Chem. 2000;275(10):7403–7409. [DOI] [PubMed] [Google Scholar]
- 35. Rosas M, Davies LC, Giles PJ, et al. The transcription factor Gata6 links tissue macrophage phenotype and proliferative renewal. Science. 2014;344(6184):645–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Singer AJ, Clark RA. Cutaneous wound healing. N Engl J Med. 1999;341(10):738–746. [DOI] [PubMed] [Google Scholar]
- 37. Li J, Chen J, Kirsner R. Pathophysiology of acute wound healing. Clin Dermatol. 2007;25(1):9–18. [DOI] [PubMed] [Google Scholar]
- 38. Mirza R, DiPietro LA, Koh TJ. Selective and specific macrophage ablation is detrimental to wound healing in mice. Am J Pathol. 2009;175(6):2454–2462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lucas T, Waisman A, Ranjan R, et al. Differential roles of macrophages in diverse phases of skin repair. J Immunol. 2010;184(7):3964–3977. [DOI] [PubMed] [Google Scholar]
- 40. Gomez Perdiguero E, Klapproth K, Schulz C, et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature. 2015;518(7540):547–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM. Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci. 2007;10(12):1538–1543. [DOI] [PubMed] [Google Scholar]
- 42. Merad M, Manz MG, Karsunky H, et al. Langerhans cells renew in the skin throughout life under steady-state conditions. Na Immunol. 2002;3(12):1135–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Massengale M, Wagers AJ, Vogel H, Weissman IL. Hematopoietic cells maintain hematopoietic fates upon entering the brain. J Exp Med. 2005;201(10):1579–1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zigmond E, Jung S. Intestinal macrophages: well educated exceptions from the rule. Trends Immunol. 2013;34(4):162–168. [DOI] [PubMed] [Google Scholar]
- 45. Sieweke MH, Allen JE. Beyond stem cells: self-renewal of differentiated macrophages. Science. 2013;342(6161):1242974. [DOI] [PubMed] [Google Scholar]
- 46. Jakubzick C, Gautier EL, Gibbings SL, et al. Minimal differentiation of classical monocytes as they survey steady-state tissues and transport antigen to lymph nodes. Immunity. 2013;39(3):599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Tamoutounour S, Guilliams M, Montanana Sanchis F, et al. Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity. 2013;39(5):925–938. [DOI] [PubMed] [Google Scholar]
- 48. Jenkins SJ, Ruckerl D, Cook PC, et al. Local macrophage proliferation, rather than recruitment from the blood, is a signature of TH2 inflammation. Science. 2011;332(6035):1284–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Willenborg S, Lucas T, van Loo G, et al. CCR2 recruits an inflammatory macrophage subpopulation critical for angiogenesis in tissue repair. Blood. 2012;120(3):613–625. [DOI] [PubMed] [Google Scholar]
- 50. Miron VE, Boyd A, Zhao JW, et al. M2 microglia and macrophages drive oligodendrocyte differentiation during CNS remyelination. Nat Neurosci. 2013;16(9):1211–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ruckh JM, Zhao JW, Shadrach JL, et al. Rejuvenation of regeneration in the aging central nervous system. Cell Stem Cell. 2012;10(1):96–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ding N, Yu RT, Subramaniam N, et al. A vitamin D receptor/SMAD genomic circuit gates hepatic fibrotic response. Cell. 2013;153(3):601–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Ito I, Waku T, Aoki M, Abe R, et al. A nonclassical vitamin D receptor pathway suppresses renal fibrosis. J Clin Invest. 2013;123(11):4579–4594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Park JW, Bae EH, Kim IJ, et al. Renoprotective effects of paricalcitol on gentamicin-induced kidney injury in rats. Am J Physiol Renal Physiol. 2010;298(2):F301–F313. [DOI] [PubMed] [Google Scholar]
- 55. Novak ML, Koh TJ. Macrophage phenotypes during tissue repair. J Leukocyte Biol. 2013;93(6):875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Helming L, Bose J, Ehrchen J, et al. 1α,25-Dihydroxyvitamin D3 is a potent suppressor of interferon γ-mediated macrophage activation. Blood. 2005;106(13):4351–4358. [DOI] [PubMed] [Google Scholar]