

Abstract
Isoniazid (INH) is highly effective for the management of tuberculosis. However, it can cause liver injury and even liver failure. INH metabolism has been thought to be associated with INH-induced liver injury. This review summarized the metabolic pathways of INH and discussed their associations with INH-induced liver injury.
Abbreviations: AcHz, acetylhydrazine; AcINH, acetylisoniazid; ALP, alkaline phosphatase; ALT, alanine aminotransferase; DiAcHz, diacetylhydrazine; GSH, glutathione; GST, glutathione S-transferase; Hz, hydrazine; INA, isonicotinic acid; INH, isoniazid; MPO, myeloperoxidase; NAD+, nicotinamide adenine dinucleotide; NAT, N-acetyltransferase; P450, cytochrome P450; R.M., reactive metabolite; TB, tuberculosis
Key words: Isoniazid, Metabolism, Hepatotoxicity, Anti-tuberculosis, N-Acetyltransferase 2, Amidase
Graphical abstract
Isoniazid (INH) is highly effective for the management of tuberculosis. However, it can cause liver injury and even liver failure. INH metabolism has been thought to be associated with INH-induced liver injury. This review summarized the metabolic pathways of INH and discussed their associations with INH-induced liver injury.
1. Introduction
Tuberculosis (TB) is a global health issue1. The standard therapies for TB include a combination treatment of isoniazid (INH), rifampicin, pyrazinamide, and ethambutol2. INH can also be used alone for TB prevention3. Despite the beneficial effects of INH, severe adverse effects especially peripheral neuropathy and hepatotoxicity are associated with INH therapies4, 5, 6, 7. About 10%–20% of patients consuming INH have a transient elevation of serum alanine aminotransferase (ALT) level. Most of the patients can adapt to it and their serum ALT levels return to normal without discontinuation, while some patients (less than 1%–3%) develop severe liver injury and even liver failure4, 8, 9, 10. The most current report from the Drug-Induced Liver Injury Network (DILI) indicates that the true incidence of INH-induced liver injury is largely under-reported in the United States, and it is the second-ranking drug that causes liver injury in spite of under-reporting11.
Clinically, INH-associated treatments usually cause a hepatocellular-type of liver injury, as characterized by a marked elevation of ALT levels (>10 times upper limit of normal [ULN]) but minimal increases in alkaline phosphatase (ALP) levels (usually <2 times the ULN)6. Even though INH-induced liver injury has been known and extensively studied, its underlying mechanisms are still poorly understood4, 6, 8, 9, 10, 12, 13, 14, 15. Different experimental animal models have been used to study the hepatotoxicity of INH, including rats13, 16, 17, 18, mice15, 19, 20, 21, and rabbits15, 22, 23, 24. Unfortunately, there is no validated animal model to recapitulate the human patterns of INH-induced liver injury6. Even though 6 doses of 100 mg/kg of INH given to rats hourly can cause necrosis in rats that were pretreated with phenobarbital, the injury and administration patterns in this study were different from those in clinic where chronic administration was used and a late onset of liver injury was observed13. In addition, recent studies suggest that rat is not a good model to replicate the delayed type of INH hepatotoxicity based on comparison of the formation of INH-bound proteins in mice, rat, and human liver microsomes15, 25. Furthermore, INH was found to induce microvesicular steatosis in different animal models, including mice20, rabbits22, 26, and rats15, 23, but these phenotypes are usually not observed in patients with INH-induced liver injury.
INH metabolism is thought to be associated with INH-induced liver injury13, 14, 15, 16, 17, 18, 19, 26, 27, 28, 29, 30, 31, 32, 33. Acetylhydrazine (AcHz), hydrazine (Hz), and acetylisoniazid (AcINH) are the major metabolites of INH. Studies of INH hepatotoxicity in rats showed that AcINH and AcHz can cause hepatic necrosis; however, treatment with INH directly even at high dose and long term did not cause toxicity9, 15. These results suggested INH metabolites are responsible for INH hepatotoxicity. Covalent binding of acetyl group to liver proteins were observed after treating rats with 14C-acetyl-labeled AcINH but not with aromatic ring 14C labeled AcINH, indicating that AcHz is responsible for INH hepatotoxicity in rats13, 16. Studies carried out in mice showed different results. When Hz or AcHz was administrated at a dose of 300 mg/kg to mice, Hz produced hepatic necrosis, macrovesicular degeneration, and steatosis, whereas AcHz did not34, suggesting that Hz is responsible for INH-induced liver injury in mice. In a rabbit model of INH-induced liver injury, the plasma level of Hz is correlated with the extent of INH-induced necrosis and steatosis, but plasma levels of INH and AcHz are not26. In addition, Hz inhibits mitochondrial complex II and affects the function of electron transport chain and ATP production in mouse primary hepatocytes. Co-treatment with Hz and a complex I inhibitor can cause hepatocyte death35. Recent studies also found INH itself can bind to liver proteins and cause immune-mediated hepatotoxicity15, 36.
In summary, despite extensive studies in INH metabolism and its role in INH-induced liver injury, the observations and conclusions are inconsistent and even controversial. This review summarized and updated the pathways of INH metabolism. We also discussed and provided novel insight into the association of INH metabolism with INH-induced liver injury.
2. The metabolic map of INH
INH is a low-molecular weight and water-soluble compound that can be rapidly absorbed from the gastrointestinal tract37. Pharmacokinetic properties of INH are affected by various patient-specific factors, like genetic status, age, comorbidities, and the co-administered food or drugs38, 39, 40, 41, 42, 43, 44. The peak plasma concentration is achieved around 1–3 h after administration of the drug45, 46. Meals containing high fats can decrease absorption of INH as revealed by the reduction of Cmax by 51% and the increasing of Tmax to 2 times47, 48. Hence it is recommended to consume INH on an empty stomach. After absorption, INH diffuses into all tissues and body fluids rapidly, including cerebrospinal fluid, saliva, pleural and peritoneal exudates, bronchi and pulmonary alveoli49, 50, 51, 52. INH also can be excreted into breast milk53, 54.
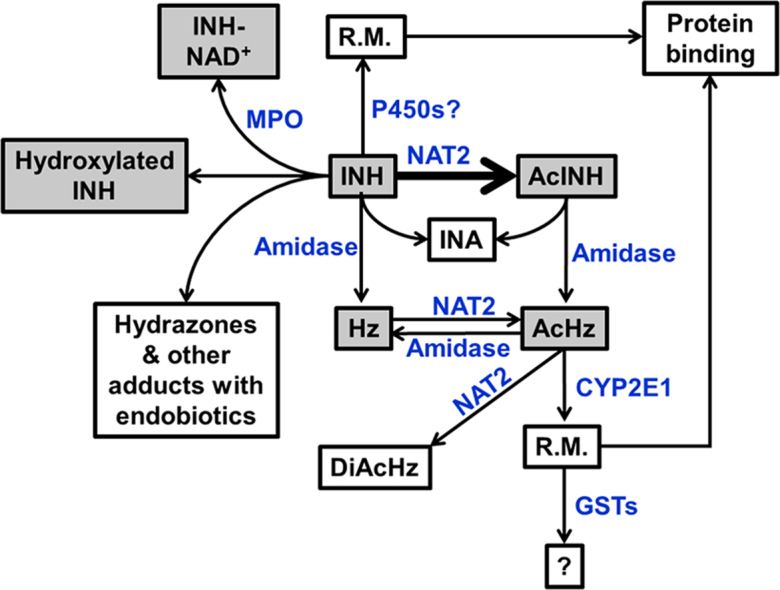
The major pathways of INH metabolism (Fig. 1) include: (1) Acetylation to form AcINH through N-acetyltransferase (NAT) 2; and (2) Hydrolysis to produce isonicotinic acid (INA) and Hz through amidase. AcINH can also be hydrolyzed to form INA and AcHz. In addition, Hz can be acetylated to AcHz and diacetylhydrazine (DiAcHz)55. Hz and AcHz are thought to be further oxidized to reactive metabolites and involved in INH hepatotoxicity13, 16, 28, 56, 57, which was proposed to be mediated by microsomal P450s, especially CYP2E56, 58.
Figure 1.

A schematic representation of isoniazid (INH) metabolism and the enzymes involved in the metabolic pathways of INH. AcHz: acetylhydrazine; AcINH, acetylisoniazid; DiAcHz: diacetylhydrazine; GST: glutathione S-transferases; Hz: Hydrazine; INA: isonicotinic acid; MPO: myeloperoxidase; NAT2: N-acetyltransferase 2; P450: cytochrome P450; R.M.: reactive metabolite.
Besides these major metabolic pathways, INH can also conjugate with several endogenous metabolites59, 60, including ketone acids, vitamin B6 (pyridoxal and pyridoxal 5-phosphate), and NAD+. In addition, INH was found to disturb the homeostasis of endogenous metabolites, such as vitamin B6, bile acids, cholesterol, and triglycerides21, 61, 62. The major metabolic pathways of INH are enzymatic-dependent reactions, including acetylation and hydrolysis of INH by NAT and acyl amidase, respectively6. Catalase-peroxidase (KatG) of mycobacterium tuberculosis (Mtb) and human neutrophil myeloperoxidase can catalyze the formation of INH-NAD+ adducts60, 63. Nevertheless, conjugation of INH with ketone acids and vitamin B6 are non-enzymatic reactions. We illustrated these metabolic pathways of INH in details in the following sections and discussed their associations with INH hepatotoxicity.
3. Role of NATs in INH metabolism and hepatotoxicity
NATs (EC 2.3.1.5, N-acetyltransferase, arylamine N-acetyltransferases) are a class of enzymes that catalyze the acetylation of arylamines from acetyl-CoA. It is widely found in different species, both in eukaryotes and prokaryotes64, 65, 66. NATs are responsible for acetylation of hydrazine drugs and carcinogenic aromatic amines, as well as endogenous molecules, such as serotonin67, 68. NAT1 and NAT2 are the major NATs that are involved in the biotransformation of xenobiotics. The NAT genes are located in close vicinity in the genome and share high sequence identity69, but their expression profiles have distinct tissue distribution patterns and the enzymes have different substrate preferences70, 71. NAT1 is widely expressed in all tissues, including endocrine tissues, blood cells, neural tissue, liver, and the gastrointestinal tract, whereas NAT2 expression is limited to the liver and gastrointestinal tract72. p-Aminobenzoate and p-aminosalicylate, are prefer substrates of NAT1, whereas NAT2 preferentially metabolizes sulfamethazine, procainamide72.
NAT2 is the dominant enzyme that catalyzes the acetylation of INH, Hz, and AcHz46, 73, 74. NAT2 is involved in three steps of INH biotransformation, including deactivation (formation of AcINH), bioactivation (formation of AcHz), and detoxification (formation of DiAcHz)6, 16, 68. The role of NAT2 in INH hepatotoxicity is complicated and still controversial13, 75, 76. NAT2 is highly polymorphic and has been thought to be involved in INH hepatotoxicity68, 76, 77, 78. Rapid acetylators have been proposed to have a higher risk of INH-induced liver injury than slow acetylators, which is based on the proposition of an increased rate of AcHz formation in rapid acetylators75. This proposition is supported by early clinical observations13, 75, 79. In one study, 86% of patients with probable and 60% with possible liver injury were rapid acetylators75. In another study with 143 patients who received INH-containing regimens for anti-TB therapies, 18 patients with abnormal elevated levels and 18 patients with normal levels of serum aminotransferase were investigated. They found out that 14 patients with abnormal serum aminotransferase were rapid acetylators, while only 7 were rapid acetylators in patients with a normal serum aminotransferase level79. These results suggest that rapid acetylators have a higher risk of liver injury with INH therapies.
However, the later clinical studies found that the presence of slow acetylator alleles has a higher risk of INH hepatotoxicity77, 80, 81. In a study of 224 patients that received anti-TB treatment, slow acetylators had a much higher risk of liver toxicity than rapid acetylators (26.4% vs. 11.1%, P=0.013)77. Another study reported the risks of different acetylator status in INH hepatotoxicity in Brazilian patients. The risk of developing hepatitis was 22% for slow acetylators, while only 9.8% for intermediate acetylators and 2.9% for rapid acetylators81. The plasma levels of INH and AcHz are higher in slow acetylators than those in rapid acetylators, which contradicts previous findings75. Even though the acetylation rate of INH is slow in slow acetylators, the acetylation of AcHz is also slow82, thus leading to a higher accumulation of AcHz in slow acetylators37, 46, 83. The clearance rate of INH is also slower in slow acetylators than in rapid acetylators38, which also contributes to the accumulation of INH in slow acetylators. A higher level of free INH might be the cause of the high incidence of liver injury directly as INH can bind to liver proteins and cause immune-mediated liver injury15. Elevation of INH can also lead to an increase in Hz formation, which is supported by an increased rate of hydrolysis process of INH in slow acetylators than in rapid ones27, 73, 84. Furthermore, decreasing the dose of INH in slow acetylators can reduce the incidence of INH hepatotoxicity85. In a multicenter, paralleled, randomized, and controlled clinical trial with Japanese patients, treatment with a lower dose of INH (2.5 mg/kg) used for anti-TB therapy in slow acetylators significantly decreased the incidence of INH-induced liver injury than the standard treatment (5 mg/kg for all patients)85. In addition, NAT2 status also plays an important role in hepatotoxicity caused by INH and rifampicin combination therapies78. In a study with 77 Japanese patients with INH + rifampicin treatment, the risk of liver injury was much higher in slow acetylators than in intermediate and rapid acetylators78.
Even though these clinical reports showed that slow acetylators have a higher risk of INH-induced liver injury with INH treatment in different populations77, 80, 81, 86, several other clinical observations showed modest or no significant difference of incidence of INH hepatotoxicity between different acetylators status87, 88, 89. In addition, the positive prediction value of the NAT2 genotypes for identification of patients with risk of liver injury is low90. Furthermore, the incidence of INH hepatotoxicity did not show significant correlation with NAT2 status in different ethnic populations4, 91. The frequency of slow acetylators in the Asian populations (10%–20%) is much lower than that in Caucasians and Africans (more than 50%)77, 92, but the incidence of INH hepatotoxicity does not show such dramatic ethnic differences4, 91. Overall, the causal role of NAT2 in INH hepatotoxicity remains controversial and the detailed mechanism is still unknown6.
4. Role of amidase in INH metabolism and hepatotoxicity
Amidases (EC 3.5.1.4, acylamidases, acylases, or acylamide amidohydrolases) are a class of enzymes that catalyze the hydrolysis of amides and they usually have carboxylesterase activities that can hydrolyze carboxylic esters93. Carboxylesterases also can hydrolyze amides. Amidases and carboxylesterases have similar catalytic mechanisms and share some substrates. Both enzymes can add water to esters or amides and form the corresponding acids and alcohols or amines, without any co-factors. Amidases and carboxylesterases play important roles in maintaining homeostasis of endobiotics. They are also of great importance in hydrolysis of drugs and environmental toxicants94. Several different types of amidases have been identified in mammals, like trypsin-like acidic arginine amidases95, anandamide amidases (fatty acid amide hydrolase)96, and N-acylethanolamine hydrolyzing acid amidases97.
Amidases can directly hydrolyze INH to INA and Hz, and they also can hydrolyze AcINH and AcHz6, 58, 98 (Fig. 1). Pretreatment with an amidase inhibitor, bis-p-nitrophenyl phosphate (BNPP), can inhibit both the hydrolysis of INH and AcINH and decrease the formation of Hz and AcHz28, 36. Both Hz and AcHz are considered as hepatotoxic metabolites of INH13, 16, 28, 36, 56, 57, thus a higher level of amidase activity can lead to the increasing formation of Hz and AcHz and result in a high incidence of INH hepatotoxicity. Rabbits are more sensitive to INH-induced liver injury as they have higher amidase activities. Around 40% of INH is directly hydrolyzed to INA and Hz in rabbits99, while less than 10% of INH is hydrolyzed in man46. The hydrolysis rate of AcINH is also faster in rabbit than the rate in rats100. A higher amidase activity in rabbit can also be reflected by the formation of higher levels of acetyl-bound proteins than those in rat while treating them with the same dose of AcINH23.
Even though amidases are responsible for the hydrolysis of INH, AcINH, and AcHz, the specific form of amidases that mediate these hydrolysis reactions is elusive. Besides, all previous studies were performed in microsomes, primary hepatocytes, or in animal models rather than in pure enzymatic systems. In addition, BNPP is a non-specific amidase inhibitor and it inhibits both amidases and esterases33, thus the involvement of esterase in INH metabolism cannot be excluded. A recent study suggests that genetic variations in a carboxylesterase gene (CES1) was possibly associated with INH hepatotoxicity; however, the authors also realized that the results are not conclusive and replication in a larger size of population needs to be performed to confirm these correlations101.
5. Role of P450s in INH metabolism and hepatotoxicity
Cytochrome P450 (P450) isoenzymes are a group of heme-containing membrane proteins that are majorly expressed in the endoplasmic reticulum102. In animal cells, P450s are also expressed in the inner membrane of mitochondria103. P450s are major drug metabolizing enzymes102. In addition, they also metabolize endogenous molecules and play important roles in hormone homeostasis (estrogen and testosterone), cholesterol synthesis, and vitamin D metabolism102. CYP3A4, 1A2, 2C9, 2C19, 2D6, 2E1 are the major P450s that are involved in drug metabolism and catalyze the oxidation of almost 90% of human drugs104.
P450s were proposed to be involved in the oxidization of Hz and AcHz to reactive metabolites16, 56, 58. Pretreating rats with phenobarbital increased the covalent binding of AcHz to liver proteins, whereas pretreatment with cobalt chloride plus phenobarbital decreased the formation of covalently bound proteins13, 16. Besides, P450s are also involved in the oxidation of Hz, as well as its toxic effects. The cytotoxic effect of Hz is prevented by 1-aminobenzotriazole, a non-selective P450 inhibitor in rat hepatocyte33. Pretreating rats with phenobarbital increases the extent of hepatic necrosis caused by INH13, whereas pretreatment with cimetidine decreases the toxic effects of INH105. In addition, pretreatment with other P450 inducers, such 3-methylcholanthrene and rifampicin, also increased the production of reactive metabolites and hepatic necrosis in rats28, 98. Furthermore, several studies indicate that rifampicin, a well know human PXR agonist and P450 inducer, can potentiate INH hepatotoxicity in man, especially in slow acetylators106, 107, 108. However, it is still unclear which P450 is responsible for these reactions since rifampicin and phenobarbital are non-specific P450 inducers. Besides, cimetidine and 1-aminobenzotriazole are non-specific P450 inhibitors.
CYP2E1 is well-known to be involved in the formation of reactive oxidative species and other reactive metabolites of hepatotoxins, like acetaminophen and carbon tetrachloride109, 110, 111. Based on the important roles in the formation of reactive metabolites, CYP2E1 was proposed to play important roles in INH hepatotoxicity18, 56, 58, 77. CYP2E1 is highly polymorphic, in which the CYP2E1 c1/c1 genotype had a higher CYP2E1 activity112. Lots of studies investigated the roles of the CYP2E1 polymorphism in INH hepatotoxicity, but the results were inconsistent in different populations86, 87, 90, 112, 113. Some studies suggest a higher CYP2E1 activity is associated with an increasing risk of INH hepatotoxicity. Patients with homozygous wild genotype CYP2E1 c1/c1 have a higher risk of INH hepatotoxicity (20.0%; odds ratio [OR], 2.52) than those with mutant allele c2 (CYP2E1 c1/c2 or c2/c2, 9.0%, P=0.009) in a Chinese population, suggesting the CYP2E1 c1/c1 genotype is an independent risk factor for INH hepatotoxicity after adjustment for acetylator status and age112. Another study showed that the CYP2E1 polymorphism is a useful tool to predict INH hepatotoxicity31. However, there are some reports that showed different results. The CYP2E1 c1/c2 polymorphism did not show a significant association with hepatotoxicity in a study with 175 TB patients who were treated with anti-TB drugs in Argentina86. Another study performed in Chinese patients in the Xinjiang Uyghur autonomous region showed no correlation between the CYP2E1 genotypes with anti-TB drug-induced liver injury90. Involvement of CYP2E1 in INH hepatotoxicity was proposed on the basis of the roles of CYP2E1 in the formation of reactive metabolites, but there was no direct evidence to support it6, 114. Structures of the reactive metabolites are also unclear. However, in animal studies using Cyp2e1 knockout mice to study the roles of CYP2E1 in INH hepatotoxicity, the authors point out that CYP2E1 might not be involved in INH-induced liver injury21. Another study showed that the CYP2E1 inhibitor, diallylsulfide, can potentiate INH-induced oxidative stress rather than decrease the toxic effects in rat primary hepatocytes115. Overall, roles of CYP2E1 in INH metabolism and hepatotoxicity remain controversial and require further studies6.
Besides the oxidation of Hz and AcHz, P450s are also involved in the activation of INH itself15. INH can bind to microsomal proteins in an NADPH-dependent manner, suggesting the key roles of P450s in INH bioactivation, but which P450 is responsible for that remains unclear. Another study carried out by the same group showed the presence of anti-P450 antibodies (anti-CYP2C19, 2E1, and 3A4) in the serum of patients with severe liver injury caused by INH36, suggesting the interaction between CYP2C19, 2E1, and 3A4 with INH. INH is also a mechanism-based inhibitor of CYP1A2, 2A6, 2C19, and 3A4 in human liver microsomes, which suggests that INH interacts with these P450s102, 116, 117, 118. Furthermore, cimetidine administration in man did not decrease the oxidation of AcHz, suggesting that the role of P450s in INH hepatotoxicity in man is different from that in rats105.
6. Role of glutathione S-transferases (GSTs) in INH metabolism and hepatotoxicity
Glutathione S-transferases (GSTs, E.C. 2.5.1.18) comprise a multi-gene family of phase II metabolizing isozymes that are involved in the detoxification of chemicals119. Most GSTs are soluble enzymes and are located in cytosol; a small family of GSTs has been identified in microsome120 and mitochondria121. There are four main classes of mammalian soluble GSTs, alpha (A), mu (M), pi (P), and theta (T)122. GSTs catalyze the conjugation of the reduced form of glutathione (GSH) to electrophilic substrates, thus decreasing their reactivity toward cellular macromolecules122.
In INH metabolism pathways, GSTs are proposed to detoxify the reactive metabolites produced by oxidization of Hz and AcHz6, although the detoxified metabolites by GSTs have not been identified. GST polymorphisms, especially the genetic variants of GSTM1 and GSTT1, have been extensively studied and are reported to associate with INH hepatotoxicity in clinic123, 124. The GSTM1-null genotype in an Asian population and the GSTT1-null genotype in Caucasians have higher risks of liver injury caused by anti-TB drugs83, 125, 126, 127. The null genotypes reduce the catalytic activity of the GST enzymes and hence lead to accumulation of the toxic metabolites that can attack the liver macromolecules. However, studies carried out in Indian and Chinese populations showed no or modest associations between the GST polymorphisms and anti-TB drug induced liver injury87, 90, 128. Hence further investigations are needed to determine the roles of these gene polymorphisms in INH hepatotoxicity in different populations. Further studies are also required to determine the mechanisms of GSTs in INH metabolism.
7. Other enzyme-dependent pathways in INH metabolism
Two minor oxidized metabolites, 2-oxo-1,2-dihydro-pyridine-4-carbohydrazide and isoniazid N-oxide, have been identified in human urine59 (Fig. 1). The enzymes responsible for these two novel oxidized metabolites are still unknown. Formation of 2-oxo-1,2-dihydro-pyridine-4-carbohydrazide was found to be NADPH-independent, suggesting that it was not mediated by P450s. Formation of isoniazid N-oxide is a NADPH-dependent reaction, but it is not mediated by the major P450s, such as CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, 2E1, or 3A459. Further studies are required to determine the enzymes responsible for the formation of these oxidized metabolites and their roles in INH hepatotoxicity.
Besides these two oxidized metabolites of INH, a break-down product of INH-NAD+ was also found in the urine of both TB patients and healthy mice treated with INH129. INH can react with NAD+ to form INH-NAD+ through a neutrophil myeloperoxidase (MPO)60. Oxidation of INH by MPO was proved via a carbon-centered free radical in the presence of NAD+, while the AcINH, Hz, and AcHz cannot go through similar pathways. Another study showed that the interaction of INH with NADP+ and the formation of INH-NADP+ adduct in human liver microsomes were concluded to be mediated by P450s130. INH-NAD+ is well known to be responsible for the action of INH to kill the mycobacteria63, 131, but the role of formation of INH-NAD+ in INH hepatotoxicity is unknown. MPO is most abundantly expressed in neutrophils60. Further studies are required to determine their roles in INH hepatotoxicity.
8. Hydrazones and other adducts of INH
INH is known to interact with ketone acids, which leads to the formation of hydrazones132, 133. INH can condense with pyruvic and α-ketoglutaric acid to form the corresponding hydrazones, isonicotinoylglycine, and 1-isonicotinoyl-2-acetylhydrazine, respectively, in rat urine134. A recent study identified a series of INH-hydrazones in human urine using a LC–MS-based metabolomic approach59. Five novel hydrazones were identified as the condensation of isoniazid with keto acids that are intermediates in the metabolism of leucine and/or isoleucine, lysine, tyrosine, tryptophan, and phenylalanine. The formation of these INH hydrazones is totally a chemical reaction, without the requirement of any enzyme. The formation of these INH hydrazones might affect the metabolism of amino acids, but their role in INH hepatotoxicity still needs further investigation. INH was also found to condense with pyridoxal to form pyridoxal isonicotinoyl hydrazone in human urine59. This is also an enzyme-independent reaction. Since pyridoxal isonicotinoyl hydrazone is a strong iron-chelator135, it might affect iron homeostasis.
9. INH and endobiotic homeostasis
INH has been found to affect the metabolism of bile acids, cholesterols, triglycerides, and free fatty acids in a chronic treatment of INH21. Cheng et al.21 treated WT and Cyp2e1-null mice with INH for 1 month. They found that serum fatty acids were significantly decreased in WT mice, but not in Cyp2e1-null mice. Besides, serum cholesterol and triglycerides, and hepatic bile acids were increased in WT mice after INH treatment. These results suggested INH can also interrupt homeostasis of cholesterol, fatty acids, and bile acids in liver. Besides, conjugation of INH with vitamin B6 (pyridoxal and pyridoxal 5-phosphate) leads to the depletion of pyridoxal 5-phosphate in both humans and lab animals61, 136, INH has also been found to reduce the plasma concentrations of calcium and phosphate ions. These reductions are due to its inhibitory action on the active form of vitamin D137.
10. Conclusions
INH metabolism and its role in INH-induced liver injury have been extensively studied. However, the available data are inconsistent and even controversial. We summarized and updated the pathways of INH metabolism and discussed the association of INH metabolism with INH-induced liver injury. NAT2 is the primary enzyme that contributes to INH metabolism. NAT2 deficiency increases the risk of INH-induced liver injury. However, the detailed mechanism, by which NAT2 deficiency leads to INH hepatotoxicity, remains unknown. Amidases hydrolyze INH and AcINH to produce Hz and AcHz, but the specific isoform of amidases that is involved in this metabolic pathway remains unclear. CYP2E1 and other P450s have been proposed to be associated with INH and AcHz bioactivation. Nevertheless, the products of P450-mediated bioactivation of INH and AcHz have not been identified. GSTs have been proposed to detoxify products of AcHz bioactivation, but the detoxified metabolites have not been determined. In addition to these classical pathways of INH metabolism, INH can form adducts with multiple endogenous metabolites. The significance of the interactions between INH and endobiotics in INH-induced liver injury is understudied. In summary, further studies are needed to explore the field of INH metabolism and hepatotoxicity.
Acknowledgments
This work was supported in part by the U. S. National Institute of Diabetes and Digestive and Kidney Diseases (DK090305) for Xiaochao Ma and the National Institute of General Medical Sciences (GM087376 and GM118367) for Xiao-bo Zhong.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Kim JY, Shakow A, Castro A, Vande C, Farmer P. Tuberculosis Control [Internet]. World Health Organization. 2016: [Cited 2016 Mar 11]. Available from: 〈http://www.who.int/trade/distance_learning/gpgh/gpgh3/en/index4.html〉.
- 2.Hall R.G., Leff R.D., Gumbo T. Treatment of active pulmonary tuberculosis in adults: current standards and recent advances. Insights from the Society of Infectious Diseases Pharmacists. Pharmacotherapy. 2009;29:1468–1481. doi: 10.1592/phco.29.12.1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sia I.G., Wieland M.L. Current concepts in the management of tuberculosis. Mayo Clin Proc. 2011;86:348–361. doi: 10.4065/mcp.2010.0820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nolan C.M., Goldberg S.V., Buskin S.E. Hepatotoxicity associated with isoniazid preventive therapy: a 7-year survey from a public health tuberculosis clinic. JAMA. 1999;281:1014–1018. doi: 10.1001/jama.281.11.1014. [DOI] [PubMed] [Google Scholar]
- 5.Metushi I.G., Cai P., Zhu X., Nakagawa T., Uetrecht J.P. A fresh look at the mechanism of isoniazid-induced hepatotoxicity. Clin Pharmacol Ther. 2011;89:911–914. doi: 10.1038/clpt.2010.355. [DOI] [PubMed] [Google Scholar]
- 6.Boelsterli U.A., Lee K.K. Mechanisms of isoniazid-induced idiosyncratic liver injury: emerging role of mitochondrial stress. J Gastroenterol Hepatol. 2014;29:678–687. doi: 10.1111/jgh.12516. [DOI] [PubMed] [Google Scholar]
- 7.Carlson H.B., Anthony E.M., Russell W.F. Jr., Middlebrook G. Prophylaxis of isoniazid neuropathy with pyridoxine. N Engl J Med. 1956;255:118–122. doi: 10.1056/NEJM195607192550304. [DOI] [PubMed] [Google Scholar]
- 8.Centers for Disease Control and Prevention (CDC) Severe isoniazid-associated liver injuries among persons being treated for latent tuberculosis infection—United States, 2004–2008. MMWR Morb Mortal Wkly Rep. 2010;59:224–229. [PubMed] [Google Scholar]
- 9.Black M., Mitchell J.R., Zimmerman H.J., Ishak K.G., Epler G.R. Isoniazid-associated hepatitis in 114 patients. Gastroenterology. 1975;69:289–302. [PubMed] [Google Scholar]
- 10.Maddrey W.C., Boitnott J.K. Isoniazid hepatitis. Ann Intern Med. 1973;79:1–12. doi: 10.7326/0003-4819-79-1-1. [DOI] [PubMed] [Google Scholar]
- 11.Hayashi P.H., Fontana R.J., Chalasani N.P., Stolz A.A., Talwalker J.A., Navarro V.J. Under-reporting and poor adherence to monitoring guidelines for severe cases of isoniazid hepatotoxicity. Clin Gastroenterol Hepatol. 2015;13(1676-82):e1. doi: 10.1016/j.cgh.2015.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kester N.M. Isoniazid hepatotoxicity—fact of fantasy. JAMA. 1971;217:699. [PubMed] [Google Scholar]
- 13.Mitchell J.R., Zimmerman H.J., Ishak K.G., Thorgeirsson U.P., Timbrell J.A., Snodgrass W.R. Isoniazid liver injury: clinical spectrum, pathology, and probable pathogenesis. Ann Intern Med. 1976;84:181–192. doi: 10.7326/0003-4819-84-2-181. [DOI] [PubMed] [Google Scholar]
- 14.Woo J., Chan C.H., Walubo A., Chan K.K. Hydrazine—a possible cause of isoniazid-induced hepatic necrosis. J Med. 1992;23:51–59. [PubMed] [Google Scholar]
- 15.Metushi I.G., Nakagawa T., Uetrecht J. Direct oxidation and covalent binding of isoniazid to rodent liver and human hepatic microsomes: humans are more like mice than rats. Chem Res Toxicol. 2012;25:2567–2576. doi: 10.1021/tx300341r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nelson S.D., Mitchell J.R., Timbrell J.A., Snodgrass W.R., Corcoran G.B., 3rd Isoniazid and iproniazid: activation of metabolites to toxic intermediates in man and rat. Science. 1976;193:901–903. doi: 10.1126/science.7838. [DOI] [PubMed] [Google Scholar]
- 17.Woodward K.N., Timbrell J.A. Acetylhydrazine hepatotoxicity: the role of covalent binding. Toxicology. 1984;30:65–74. doi: 10.1016/0300-483x(84)90063-5. [DOI] [PubMed] [Google Scholar]
- 18.Yue J., Peng R.X., Yang J., Kong R., Liu J. CYP2E1 mediated isoniazid-induced hepatotoxicity in rats. Acta Pharmacol Sin. 2004;25:699–704. [PubMed] [Google Scholar]
- 19.Chowdhury A., Santra A., Bhattacharjee K., Ghatak S., Saha D.R., Dhali G.K. Mitochondrial oxidative stress and permeability transition in isoniazid and rifampicin induced liver injury in mice. J Hepatol. 2006;45:117–126. doi: 10.1016/j.jhep.2006.01.027. [DOI] [PubMed] [Google Scholar]
- 20.Church R.J., Wu H., Mosedale M., Sumner S.J., Pathmasiri W., Kurtz C.L. A systems biology approach utilizing a mouse diversity panel identifies genetic differences influencing isoniazid-induced microvesicular steatosis. Toxicol Sci. 2014;140:481–492. doi: 10.1093/toxsci/kfu094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cheng J., Krausz K.W., Li F., Ma X., Gonzalez F.J. CYP2E1-dependent elevation of serum cholesterol, triglycerides, and hepatic bile acids by isoniazid. Toxicol Appl Pharmacol. 2013;266:245–253. doi: 10.1016/j.taap.2012.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKennis H., Jr, Yard A.S., Pahnelas E.V. The production of fatty livers in rabbits by isoniazid and other hydrazine derivatives. Am Rev Tuberc. 1956;73:956–959. doi: 10.1164/artpd.1956.73.6.956. [DOI] [PubMed] [Google Scholar]
- 23.Whitehouse L.W., Tryphonas L., Paul C.J., Solomonraj G., Thomas B.H., Wong L.T. Isoniazid-induced hepatic steatosis in rabbits: an explanation for susceptibility and its antagonism by pyridoxine hydrochloride. Can J Physiol Pharmacol. 1983;61:478–487. doi: 10.1139/y83-073. [DOI] [PubMed] [Google Scholar]
- 24.Sarich T.C., Zhou T., Adams S.P., Bain A.I., Wall R.A., Wright J.M. A model of isoniazid-induced hepatotoxicity in rabbits. J Pharmacol Toxicol Methods. 1995;34:109–116. doi: 10.1016/1056-8719(95)00044-i. [DOI] [PubMed] [Google Scholar]
- 25.Metushi I.G., Uetrecht J. Lack of liver injury in Wistar rats treated with the combination of isoniazid and rifampicin. Mol Cell Biochem. 2014;387:9–17. doi: 10.1007/s11010-013-1864-7. [DOI] [PubMed] [Google Scholar]
- 26.Sarich T.C., Youssefi M., Zhou T., Adams S.P., Wall R.A., Wright J.M. Role of hydrazine in the mechanism of isoniazid hepatotoxicity in rabbits. Arch Toxicol. 1996;70:835–840. doi: 10.1007/s002040050347. [DOI] [PubMed] [Google Scholar]
- 27.Lauterburg B.H., Smith C.V., Todd E.L., Mitchell J.R. Pharmacokinetics of the toxic hydrazino metabolites formed from isoniazid in humans. J Pharmacol Exp Ther. 1985;235:566–570. [PubMed] [Google Scholar]
- 28.Timbrell J.A., Mitchell J.R., Snodgrass W.R., Nelson S.D. Isoniazid hepatoxicity: the relationship between covalent binding and metabolism in vivo. J Pharmacol Exp Ther. 1980;213:364–369. [PubMed] [Google Scholar]
- 29.Gent W.L., Seifart H.I., Parkin D.P., Donald P.R., Lamprecht J.H. Factors in hydrazine formation from isoniazid by paediatric and adult tuberculosis patients. Eur J Clin Pharmacol. 1992;43:131–136. doi: 10.1007/BF01740658. [DOI] [PubMed] [Google Scholar]
- 30.Blair I.A., Mansilla Tinoco R., Brodie M.J., Clare R.A., Dollery C.T., Timbrell J.A. Plasma hydrazine concentrations in man after isoniazid and hydralazine administration. Hum Toxicol. 1985;4:195–202. doi: 10.1177/096032718500400210. [DOI] [PubMed] [Google Scholar]
- 31.Vuilleumier N., Rossier M.F., Chiappe A., Degoumois F., Dayer P., Mermillod B. CYP2E1 genotype and isoniazid-induced hepatotoxicity in patients treated for latent tuberculosis. Eur J Clin Pharmacol. 2006;62:423–429. doi: 10.1007/s00228-006-0111-5. [DOI] [PubMed] [Google Scholar]
- 32.Hussain S.M., Frazier J.M. Involvement of apoptosis in hydrazine induced toxicity in rat primary hepatocytes. Toxicol Vitr. 2003;17:343–355. doi: 10.1016/s0887-2333(03)00022-5. [DOI] [PubMed] [Google Scholar]
- 33.Tafazoli S., Mashregi M., O׳Brien P.J. Role of hydrazine in isoniazid-induced hepatotoxicity in a hepatocyte inflammation model. Toxicol Appl Pharmacol. 2008;229:94–101. doi: 10.1016/j.taap.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 34.Richards V.E., Chau B., White M.R., McQueen C.A. Hepatic gene expression and lipid homeostasis in C57BL/6 mice exposed to hydrazine or acetylhydrazine. Toxicol Sci. 2004;82:318–332. doi: 10.1093/toxsci/kfh232. [DOI] [PubMed] [Google Scholar]
- 35.Lee K.K., Fujimoto K., Zhang C., Schwall C.T., Alder N.N., Pinkert C.A. Isoniazid-induced cell death is precipitated by underlying mitochondrial complex I dysfunction in mouse hepatocytes. Free Radic Biol Med. 2013;65:584–594. doi: 10.1016/j.freeradbiomed.2013.07.038. [DOI] [PubMed] [Google Scholar]
- 36.Metushi I.G., Sanders C., Lee W.M., Uetrecht J. Detection of anti-isoniazid and anti-cytochrome P450 antibodies in patients with isoniazid-induced liver failure. Hepatology. 2014;59:1084–1093. doi: 10.1002/hep.26564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Weber W.W., Hein D.W. Clin Pharmacokinet of isoniazid. Clin Pharmacokinet. 1979;4:401–422. doi: 10.2165/00003088-197904060-00001. [DOI] [PubMed] [Google Scholar]
- 38.Seng K.Y., Hee K.H., Soon G.H., Chew N., Khoo S.H., Lee L.S. Population pharmacokinetic analysis of isoniazid, acetylisoniazid, and isonicotinic acid in healthy volunteers. Antimicrob Agents Chemother. 2015;59:6791–6799. doi: 10.1128/AAC.01244-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ramachandran G., Hemanth Kumar A.K., Bhavani P.K., Poorana Gangadevi N., Sekar L., Vijayasekaran D. Age, nutritional status and INH acetylator status affect pharmacokinetics of anti-tuberculosis drugs in children. Int J Tuberc Lung Dis. 2013;17:800–806. doi: 10.5588/ijtld.12.0628. [DOI] [PubMed] [Google Scholar]
- 40.Mach J., Huizer-Pajkos A., Mitchell S.J., McKenzie C., Phillips L., Kane A. The effect of ageing on isoniazid pharmacokinetics and hepatotoxicity in Fischer 344 rats. Fundam Clin Pharmacol. 2016;30:23–34. doi: 10.1111/fcp.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saktiawati A.M., Sturkenboom M.G., Stienstra Y., Subronto Y.W., Sumardi, Kosterink J.G. Impact of food on the pharmacokinetics of first-line anti-TB drugs in treatment-naive TB patients: a randomized cross-over trial. J Antimicrob Chemother. 2016;71:703–710. doi: 10.1093/jac/dkv394. [DOI] [PubMed] [Google Scholar]
- 42.Requena-Méndez A., Davies G., Waterhouse D., Ardrey A., Jave O., López-Romero S.L. Effects of dosage, comorbidities, and food on isoniazid pharmacokinetics in Peruvian tuberculosis patients. Antimicrob Agents Chemother. 2014;58:7164–7170. doi: 10.1128/AAC.03258-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wiltshire C.S., Lamorde M., Scherrer A., Musaazi J., Corti N., Allan B. Low isoniazid and rifampicin concentrations in TB/HIV co-infected patients in Uganda. J Int AIDS Soc. 2014;17(4 Suppl 3):S19585. doi: 10.7448/IAS.17.4.19585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lin M.Y., Lin S.J., Chan L.C., Lu Y.C. Impact of food and antacids on the pharmacokinetics of anti-tuberculosis drugs: systematic review and meta-analysis. Int J Tuberc Lung Dis. 2010;14:806–818. [PubMed] [Google Scholar]
- 45.Mitchison D.A. Plasma concentrations of isoniazid in the treatment of tuberculosis. In: Davies D.S., Prichard B.N., editors. Biological Effects of Drugs in Relation to their Plasma Concentrations. MacMillan; London: 1973. pp. 171–182. [Google Scholar]
- 46.Ellard G.A., Gammon P.T. Pharmacokinetics of isoniazid metabolism in man. J Pharmacokinet Biopharm. 1976;4:83–113. doi: 10.1007/BF01086149. [DOI] [PubMed] [Google Scholar]
- 47.Peloquin C.A., Namdar R., Dodge A.A., Nix D.E. Pharmacokinetics of isoniazid under fasting conditions, with food, and with antacids. Int J Tuberc Lung Dis. 1999;3:703–710. [PubMed] [Google Scholar]
- 48.Melander A., Danielson K., Hanson A., Jansson L., Rerup C., Scherstén B. Reduction of isoniazid bioavailability in normal men by concomitant intake of food. Acta Med Scand. 1976;200:93–97. doi: 10.1111/j.0954-6820.1976.tb08202.x. [DOI] [PubMed] [Google Scholar]
- 49.Bhandari R., Pharmacokinetics Kaur I.P. tissue distribution and relative bioavailability of isoniazid-solid lipid nanoparticles. Int J Pharm. 2013;441:202–212. doi: 10.1016/j.ijpharm.2012.11.042. [DOI] [PubMed] [Google Scholar]
- 50.Hutchings A.D., Monie R.D., Spragg B.P., Routledge P.A. Saliva and plasma concentrations of isoniazid and acetylisoniazid in man. Br J Clin Pharmacol. 1988;25:585–589. doi: 10.1111/j.1365-2125.1988.tb03349.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jutte P.C., Rutgers S.R., Van Altena R., Uges D.R., van Horn J.R. Penetration of isoniazid, rifampicin and pyrazinamide in tuberculous pleural effusion and psoas abscess. Int J Tuberc Lung Dis. 2004;8:1368–1372. [PubMed] [Google Scholar]
- 52.Conte J.E., Jr, Golden J.A., McQuitty M., Kipps J., Duncan S., McKenna E. Effects of gender, AIDS, and acetylator status on intrapulmonary concentrations of isoniazid. Antimicrob Agents Chemother. 2002;46:2358–2364. doi: 10.1128/AAC.46.8.2358-2364.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holdiness M.R. Transplacental pharmacokinetics of the antituberculosis drugs. Clin Pharmacokinet. 1987;13:125–129. doi: 10.2165/00003088-198713020-00005. [DOI] [PubMed] [Google Scholar]
- 54.Singh N., Golani A., Patel Z., Maitra A. Transfer of isoniazid from circulation to breast milk in lactating women on chronic therapy for tuberculosis. Br J Clin Pharmacol. 2008;65:418–422. doi: 10.1111/j.1365-2125.2007.03061.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Preziosi P. Isoniazid: metabolic aspects and toxicological correlates. Curr Drug Metab. 2007;8:839–851. doi: 10.2174/138920007782798216. [DOI] [PubMed] [Google Scholar]
- 56.Delaney J., Timbrell J.A. Role of cytochrome P450 in hydrazine toxicity in isolated hepatocytes in vitro. Xenobiotica. 1995;25:1399–1410. doi: 10.3109/00498259509061927. [DOI] [PubMed] [Google Scholar]
- 57.Lauterburg B.H., Smith C.V., Todd E.L., Mitchell J.R. Oxidation of hydrazine metabolites formed from isoniazid. Clin Pharmacol Ther. 1985;38:566–571. doi: 10.1038/clpt.1985.225. [DOI] [PubMed] [Google Scholar]
- 58.Sarich T.C., Adams S.P., Petricca G., Wright J.M. Inhibition of isoniazid-induced hepatotoxicity in rabbits by pretreatment with an amidase inhibitor. J Pharmacol Exp Ther. 1999;289:695–702. [PubMed] [Google Scholar]
- 59.Li F., Miao Y., Zhang L., Neuenswander S.A., Douglas J.T., Ma X. Metabolomic analysis reveals novel isoniazid metabolites and hydrazones in human urine. Drug Metab Pharmacokinet. 2011;26:569–576. doi: 10.2133/dmpk.DMPK-11-RG-055. [DOI] [PubMed] [Google Scholar]
- 60.Khan S.R., Morgan A.G., Michail K., Srivastava N., Whittal R.M., Aljuhani N. Metabolism of isoniazid by neutrophil myeloperoxidase leads to isoniazid-NAD+ adduct formation: a comparison of the reactivity of isoniazid with its known human metabolites. Biochem Pharmacol. 2016;106:46–55. doi: 10.1016/j.bcp.2016.02.003. [DOI] [PubMed] [Google Scholar]
- 61.Cilliers K., Labadarios D., Schaaf H.S., Willemse M., Maritz J.S., Werely C.J. Pyridoxal-5-phosphate plasma concentrations in children receiving tuberculosis chemotherapy including isoniazid. Acta Paediatr. 2010;99:705–710. doi: 10.1111/j.1651-2227.2010.01696.x. [DOI] [PubMed] [Google Scholar]
- 62.Wason S., Lacouture P.G., Lovejoy F.H., Jr. Single high-dose pyridoxine treatment for isoniazid overdose. JAMA. 1981;246:1102–1104. [PubMed] [Google Scholar]
- 63.Rozwarski D.A., Grant G.A., Barton D.H., Jacobs W.R., Jr., Sacchettini J.C. Modification of the NADH of the isoniazid target (InhA) from Mycobacterium tuberculosis. Science. 1998;279:98–102. doi: 10.1126/science.279.5347.98. [DOI] [PubMed] [Google Scholar]
- 64.Upton A., Johnson N., Sandy J., Sim E. Arylamine N-acetyltransferases—of mice, men and microorganisms. Trends Pharmacol Sci. 2001;22:140–146. doi: 10.1016/s0165-6147(00)01639-4. [DOI] [PubMed] [Google Scholar]
- 65.Payton M., Mushtaq A., Yu T.W., Wu L.J., Sinclair J., Sim E. Eubacterial arylamine N-acetyltransferases—identification and comparison of 18 members of the protein family with conserved active site cysteine, histidine and aspartate residues. Microbiology. 2001;147:1137–1147. doi: 10.1099/00221287-147-5-1137. [DOI] [PubMed] [Google Scholar]
- 66.Sinclair J.C., Sandy J., Delgoda R., Sim E., Noble M.E. Structure of arylamine N-acetyltransferase reveals a catalytic triad. Nat Struct Biol. 2000;7:560–564. doi: 10.1038/76783. [DOI] [PubMed] [Google Scholar]
- 67.Klein D.C., Weller J.L., Moore R.Y. Melatonin metabolism: neural regulation of pineal serotonin: acetyl coenzyme A N-acetyltransferase activity. Proc Natl Acad Sci USA. 1971;68:3107–3110. doi: 10.1073/pnas.68.12.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sim E., Abuhammad A., Ryan A. Arylamine N-acetyltransferases: from drug metabolism and pharmacogenetics to drug discovery. Br J Pharmacol. 2014;171:2705–2725. doi: 10.1111/bph.12598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Blum M., Grant D.M., McBride W., Heim M., Meyer U.A. Human arylamine N-acetyltransferase genes: isolation, chromosomal localization, and functional expression. DNA Cell Biol. 1990;9:193–203. doi: 10.1089/dna.1990.9.193. [DOI] [PubMed] [Google Scholar]
- 70.Grant D.M., Blum M., Beer M., Meyer U.A. Monomorphic and polymorphic human arylamine N-acetyltransferases: a comparison of liver isozymes and expressed products of two cloned genes. Mol Pharmacol. 1991;39:184–191. [PubMed] [Google Scholar]
- 71.Hickman D., Palamanda J.R., Unadkat J.D., Sim E. Enzyme kinetic properties of human recombinant arylamine N-acetyltransferase 2 allotypic variants expressed in Escherichia coli. Biochem Pharmacol. 1995;50:697–703. doi: 10.1016/0006-2952(95)00182-y. [DOI] [PubMed] [Google Scholar]
- 72.Windmill K.F., Gaedigk A., Hall P.M., Samaratunga H., Grant D.M., McManus M.E. Localization of N-acetyltransferases NAT1 and NAT2 in human tissues. Toxicol Sci. 2000;54:19–29. doi: 10.1093/toxsci/54.1.19. [DOI] [PubMed] [Google Scholar]
- 73.Peretti E., Karlaganis G., Lauterburg B.H. Increased urinary excretion of toxic hydrazino metabolites of isoniazid by slow acetylators. Effect of a slow-release preparation of isoniazid. Eur J Clin Pharmacol. 1987;33:283–286. doi: 10.1007/BF00637563. [DOI] [PubMed] [Google Scholar]
- 74.Evans D.A., Manley K.A., McKusick V.A. Genetic control of isoniazid metabolism in man. Br Med J. 1960;2:485–491. doi: 10.1136/bmj.2.5197.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mitchell J.R., Thorgeirsson U.P., Black M., Timbrell J.A., Snodgrass W.R., Potter W.Z. Increased incidence of isoniazid hepatitis in rapid acetylators: possible relation to hydranize metabolites. Clin Pharmacol Ther. 1975;18:70–79. doi: 10.1002/cpt197518170. [DOI] [PubMed] [Google Scholar]
- 76.Huang Y.S., Chern H.D., Su W.J., Wu J.C., Lai S.L., Yang S.Y. Polymorphism of the N-acetyltransferase 2 gene as a susceptibility risk factor for antituberculosis drug-induced hepatitis. Hepatology. 2002;35:883–889. doi: 10.1053/jhep.2002.32102. [DOI] [PubMed] [Google Scholar]
- 77.Walker K., Ginsberg G., Hattis D., Johns D.O., Guyton K.Z., Sonawane B. Genetic polymorphism in N-acetyltransferase (NAT): population distribution of NAT1 and NAT2 activity. J Toxicol Environ Health B Crit Rev. 2009;12:440–472. doi: 10.1080/10937400903158383. [DOI] [PubMed] [Google Scholar]
- 78.Ohno M., Yamaguchi I., Yamamoto I., Fukuda T., Yokota S., Maekura R. Slow N-acetyltransferase 2 genotype affects the incidence of isoniazid and rifampicin-induced hepatotoxicity. Int J Tuberc Lung Dis. 2000;4:256–261. [PubMed] [Google Scholar]
- 79.Yamamoto T., Suou T., Hirayama C. Elevated serum aminotransferase induced by isoniazid in relation to isoniazid acetylator phenotype. Hepatology. 1986;6:295–298. doi: 10.1002/hep.1840060223. [DOI] [PubMed] [Google Scholar]
- 80.Gupta V.H., Amarapurkar D.N., Singh M., Sasi P., Joshi J.M., Baijal R. Association of N-acetyltransferase 2 and cytochrome P450 2E1 gene polymorphisms with antituberculosis drug-induced hepatotoxicity in Western India. J Gastroenterol Hepatol. 2013;28:1368–1374. doi: 10.1111/jgh.12194. [DOI] [PubMed] [Google Scholar]
- 81.Teixeira R.L., Morato R.G., Cabello P.H., Muniz L.M., Moreira Ada S., Kritski A.L. Genetic polymorphisms of NAT2, CYP2E1 and GST enzymes and the occurrence of antituberculosis drug-induced hepatitis in Brazilian TB patients. Mem Inst Oswaldo Cruz. 2011;106:716–724. doi: 10.1590/s0074-02762011000600011. [DOI] [PubMed] [Google Scholar]
- 82.Peretti E., Karlaganis G., Lauterburg B.H. Acetylation of acetylhydrazine, the toxic metabolite of isoniazid, in humans. Inhibition by concomitant administration of isoniazid. J Pharmacol Exp Ther. 1987;243:686–689. [PubMed] [Google Scholar]
- 83.Bing C., Xiaomeia C., Jinhenga L. Gene dose effect of NAT2 variants on the pharmacokinetics of isoniazid and acetylisoniazid in healthy Chinese subjects. Drug Metabol Drug Interact. 2011;26:113–118. doi: 10.1515/DMDI.2011.016. [DOI] [PubMed] [Google Scholar]
- 84.Lauterburg B.H., Smith C.V., Mitchell J.R. Determination of isoniazid and its hydrazino metabolites, acetylisoniazid, acetylhydrazine, and diacetylhydrazine in human plasma by gas chromatography–mass spectrometry. J ChromatogrB Biomed Sci Appl. 1981;224:431–438. [Google Scholar]
- 85.Azuma J., Ohno M., Kubota R., Yokota S., Nagai T., Tsuyuguchi K. NAT2 genotype guided regimen reduces isoniazid-induced liver injury and early treatment failure in the 6-month four-drug standard treatment of tuberculosis: a randomized controlled trial for pharmacogenetics-based therapy. Eur J Clin Pharmacol. 2013;69:1091–1101. doi: 10.1007/s00228-012-1429-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chamorro J.G., Castagnino J.P., Musella R.M., Nogueras M., Aranda F.M., Frías A. Sex, ethnicity, and slow acetylator profile are the major causes of hepatotoxicity induced by antituberculosis drugs. J Gastroenterol Hepatol. 2013;28:323–328. doi: 10.1111/jgh.12069. [DOI] [PubMed] [Google Scholar]
- 87.Cai Y., Yi J., Zhou C., Shen X. Pharmacogenetic study of drug-metabolising enzyme polymorphisms on the risk of anti-tuberculosis drug-induced liver injury: a meta-analysis. PLoS One. 2012;7:e47769. doi: 10.1371/journal.pone.0047769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yamada S., Tang M., Richardson K., Halaschek-Wiener J., Chan M., Cook V.J. Genetic variations of NAT2 and CYP2E1 and isoniazid hepatotoxicity in a diverse population. Pharmacogenomics. 2009;10:1433–1445. doi: 10.2217/pgs.09.66. [DOI] [PubMed] [Google Scholar]
- 89.Leiro-Fernandez V., Valverde D., Vázquez-Gallardo R., Constenla L., Fernández-Villar A. Genetic variations of NAT2 and CYP2E1 and isoniazid hepatotoxicity in a diverse population. Pharmacogenomics. 2010;11:1205–1206. doi: 10.2217/pgs.10.109. (author reply 1207-8) [DOI] [PubMed] [Google Scholar]
- 90.Xiang Y., Ma L., Wu W., Liu W., Li Y., Zhu X. The incidence of liver injury in Uyghur patients treated for TB in Xinjiang Uyghur autonomous region, China, and its association with hepatic enzyme polymorphisms NAT2, CYP2E1, GSTM1 and GSTT1. PLoS One. 2014;9:e85905. doi: 10.1371/journal.pone.0085905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Huang Y.S. Recent progress in genetic variation and risk of antituberculosis drug-induced liver injury. J Chin Med Assoc. 2014;77:169–173. doi: 10.1016/j.jcma.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 92.Chen M., Xia B., Chen B., Guo Q., Li J., Ye M. N-acetyltransferase 2 slow acetylator genotype associated with adverse effects of sulphasalazine in the treatment of inflammatory bowel disease. Can J Gastroenterol. 2007;21:155–158. doi: 10.1155/2007/976804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang C.Y. Microsomal amidases and carboxylesterases. In: Kauffman F.C., editor. Conjugation—deconjugation reactions in drug metabolism and toxicity. Springer; Berlin Heidelberg: 1994. pp. 161–187. [Google Scholar]
- 94.Ross M.K., Crow J.A. Human carboxylesterases and their role in xenobiotic and endobiotic metabolism. J Biochem Mol Toxicol. 2007;21:187–196. doi: 10.1002/jbt.20178. [DOI] [PubMed] [Google Scholar]
- 95.Kobayashi T., Matsuda Y., Park J.Y., Hara I., Kaneko S., Fujimoto Y. Trypsin-like arginine amidases including plasminogen and plasmin in human seminal plasma by affinity adsorption and elution. Arch Androl. 1992;28:165–170. doi: 10.3109/01485019208987694. [DOI] [PubMed] [Google Scholar]
- 96.Bilfinger T.V., Salzet M., Fimiani C., Deutsch D.G., Tramu G., Stefano G.B. Pharmacological evidence for anandamide amidase in human cardiac and vascular tissues. Int J Cardiol. 1998;64:S15–S22. doi: 10.1016/s0167-5273(98)00031-x. [DOI] [PubMed] [Google Scholar]
- 97.Puffenbarger R.A. Molecular biology of the enzymes that degrade endocannabinoids. Curr Drug Targets CNS Neurol Disord. 2005;4:625–631. doi: 10.2174/156800705774933050. [DOI] [PubMed] [Google Scholar]
- 98.Sendo T., Noda A., Noda H., Hsu K.Y., Yamamoto Y. Metabolic hydrolysis of isoniazid by subcellular fractions of rat liver. J UOEH. 1984;6:249–255. doi: 10.7888/juoeh.6.249. [DOI] [PubMed] [Google Scholar]
- 99.Thomas B.H., Wong L.T., Zeitz W., Solomonraj G. Isoniazid metabolism in the rabbit, and the effect of rifampin pretreatment. Res Commun Chem Pathol Pharmacol. 1981;33:235–247. [PubMed] [Google Scholar]
- 100.Thomas B.H., Whitehouse L.W., Zeitz W. Metabolism of [14C]acetylisoniazid and [14C]acetylhydrazine by the rat and rabbit. Fundam Appl Toxicol. 1984;4:646–653. [PubMed] [Google Scholar]
- 101.Yamada S., Richardson K., Tang M., Halaschek-Wiener J., Cook V.J., Fitzgerald J.M. Genetic variation in carboxylesterase genes and susceptibility to isoniazid-induced hepatotoxicity. Pharmacogenomics J. 2010;10:524–536. doi: 10.1038/tpj.2010.5. [DOI] [PubMed] [Google Scholar]
- 102.Gonzalez F.J. The molecular biology of cytochrome P450s. Pharmacol Rev. 1988;40:243–288. [PubMed] [Google Scholar]
- 103.Williams P.A., Cosme J., Sridhar V., Johnson E.F., McRee D.E. Mammalian microsomal cytochrome P450 monooxygenase: structural adaptations for membrane binding and functional diversity. Mol Cell. 2000;5:121–131. doi: 10.1016/s1097-2765(00)80408-6. [DOI] [PubMed] [Google Scholar]
- 104.Rendic S., Di Carlo F.J. Human cytochrome P450 enzymes: a status report summarizing their reactions, substrates, inducers, and inhibitors. Drug Metab Rev. 1997;29:413–580. doi: 10.3109/03602539709037591. [DOI] [PubMed] [Google Scholar]
- 105.Lauterburg B.H., Todd E.L., Smith C.V., Mitchell J.R. Cimetidine inhibits the formation of the reactive, toxic metabolite of isoniazid in rats but not in man. Hepatology. 1985;5:607–609. doi: 10.1002/hep.1840050414. [DOI] [PubMed] [Google Scholar]
- 106.Miguet J.P., Mavier P., Soussy C.J., Dhumeaux D. Induction of hepatic microsomal enzymes after brief administration of rifampicin in man. Gastroenterology. 1977;72(5 Pt 1):924–926. [PubMed] [Google Scholar]
- 107.Shen C., Meng Q., Zhang G., Hu W. Rifampicin exacerbates isoniazid-induced toxicity in human but not in rat hepatocytes in tissue-like cultures. Br J Pharmacol. 2008;153:784–791. doi: 10.1038/sj.bjp.0707611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sarma G.R., Immanuel C., Kailasam S., Narayana A.S., Venkatesan P. Rifampin-induced release of hydrazine from isoniazid. A possible cause of hepatitis during treatment of tuberculosis with regimens containing isoniazid and rifampin. Am Rev Respir Dis. 1986;133:1072–1075. doi: 10.1164/arrd.1986.133.6.1072. [DOI] [PubMed] [Google Scholar]
- 109.Caro A.A., Cederbaum A.I. Oxidative stress, toxicology, and pharmacology of CYP2E1. Annu Rev Pharmacol Toxicol. 2004;44:27–42. doi: 10.1146/annurev.pharmtox.44.101802.121704. [DOI] [PubMed] [Google Scholar]
- 110.Lee S.S., Buters J.T., Pineau T., Fernandez-Salguero P., Gonzalez F.J. Role of CYP2E1 in the hepatotoxicity of acetaminophen. J Biol Chem. 1996;271:12063–12067. doi: 10.1074/jbc.271.20.12063. [DOI] [PubMed] [Google Scholar]
- 111.Wong F.W., Chan W.Y., Lee S.S. Resistance to carbon tetrachloride-induced hepatotoxicity in mice which lack CYP2E1 expression. Toxicol Appl Pharmacol. 1998;153:109–118. doi: 10.1006/taap.1998.8547. [DOI] [PubMed] [Google Scholar]
- 112.Huang Y.S., Chern H.D., Su W.J., Wu J.C., Chang S.C., Chiang C.H. Cytochrome P450 2E1 genotype and the susceptibility to antituberculosis drug-induced hepatitis. Hepatology. 2003;37:924–930. doi: 10.1053/jhep.2003.50144. [DOI] [PubMed] [Google Scholar]
- 113.Cho H.J., Koh W.J., Ryu Y.J., Ki C.S., Nam M.H., Kim J.W. Genetic polymorphisms of NAT2 and CYP2E1 associated with antituberculosis drug-induced hepatotoxicity in Korean patients with pulmonary tuberculosis. Tuberculosis. 2007;87:551–556. doi: 10.1016/j.tube.2007.05.012. [DOI] [PubMed] [Google Scholar]
- 114.Ascenzi P., Coletta A., Cao Y., Trezza V., Leboffe L., Fanali G. Isoniazid inhibits the heme-based reactivity of Mycobacterium tuberculosis truncated hemoglobin N. PLoS One. 2013;8:e69762. doi: 10.1371/journal.pone.0069762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhai Q., Lu S.R., Lin Y., Yang Q.L., Yu B. Oxidative stress potentiated by diallylsulfide, a selective CYP2E1 inhibitor, in isoniazid toxic effect on rat primary hepatocytes. Toxicol Lett. 2008;183:95–98. doi: 10.1016/j.toxlet.2008.10.007. [DOI] [PubMed] [Google Scholar]
- 116.Wen X., Wang J.S., Neuvonen P.J., Backman J.T. Isoniazid is a mechanism-based inhibitor of cytochrome P450 1A2, 2A6, 2C19 and 3A4 isoforms in human liver microsomes. Eur J Clin Pharmacol. 2002;57:799–804. doi: 10.1007/s00228-001-0396-3. [DOI] [PubMed] [Google Scholar]
- 117.Desta Z., Soukhova N.V., Flockhart D.A. Inhibition of cytochrome P450 (CYP450) isoforms by isoniazid: potent inhibition of CYP2C19 and CYP3A. Antimicrob Agents Chemother. 2001;45:382–392. doi: 10.1128/AAC.45.2.382-392.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Feng S., He X. Mechanism-based inhibition of CYP450: an indicator of drug-induced hepatotoxicity. Curr Drug Metab. 2013;14:921–945. doi: 10.2174/138920021131400114. [DOI] [PubMed] [Google Scholar]
- 119.Eaton D.L., Bammler T.K. Concise review of the glutathione S-transferases and their significance to toxicology. Toxicol Sci. 1999;49:156–164. doi: 10.1093/toxsci/49.2.156. [DOI] [PubMed] [Google Scholar]
- 120.Andersson C., Mosialou E., Weinander R., Morgenstern R. Enzymology of microsomal glutathione S-transferase. Adv Pharmacol. 1994;27:19–35. doi: 10.1016/s1054-3589(08)61028-5. [DOI] [PubMed] [Google Scholar]
- 121.Pemble S.E., Wardle A.F., Taylor J.B. Glutathione S-transferase class kappa: characterization by the cloning of rat mitochondrial GST and identification of a human homologue. Biochem J. 1996;319:749–754. doi: 10.1042/bj3190749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Hayes J.D., Pulford D.J. The glutathione S-transferase supergene family: regulation of GST and the contribution of the isoenzymes to cancer chemoprotection and drug resistance. Crit Rev Biochem Mol Biol. 1995;30:445–600. doi: 10.3109/10409239509083491. [DOI] [PubMed] [Google Scholar]
- 123.Lucena M.I., Andrade R.J., Martínez C., Ulzurrun E., García-Martín E., Borraz Y. Glutathione S-transferase M1 and T1 null genotypes increase susceptibility to idiosyncratic drug-induced liver injury. Hepatology. 2008;48:588–596. doi: 10.1002/hep.22370. [DOI] [PubMed] [Google Scholar]
- 124.Monteiro T.P., El-Jaick K.B., Jeovanio-Silva A.L., Brasil P.E., Costa M.J., Rolla V.C. The roles of GSTM1 and GSTT1 null genotypes and other predictors in anti-tuberculosis drug-induced liver injury. J Clin Pharm Ther. 2012;37:712–718. doi: 10.1111/j.1365-2710.2012.01368.x. [DOI] [PubMed] [Google Scholar]
- 125.Tang N., Deng R., Wang Y., Lin M., Li H., Qiu Y. GSTM1 and GSTT1 null polymorphisms and susceptibility to anti-tuberculosis drug-induced liver injury: a meta-analysis. Int J Tuberc Lung Dis. 2013;17:17–25. doi: 10.5588/ijtld.12.0447. [DOI] [PubMed] [Google Scholar]
- 126.Roy B., Chowdhury A., Kundu S., Santra A., Dey B., Chakraborty M. Increased risk of antituberculosis drug-induced hepatotoxicity in individuals with glutathione S-transferase M1 ‘null’ mutation. J Gastroenterol Hepatol. 2001;16:1033–1037. doi: 10.1046/j.1440-1746.2001.02585.x. [DOI] [PubMed] [Google Scholar]
- 127.Huang Y.S., Su W.J., Huang Y.H., Chen C.Y., Chang F.Y., Lin H.C. Genetic polymorphisms of manganese superoxide dismutase, NAD(P)H: quinone oxidoreductase, glutathione S-transferase M1 and T1, and the susceptibility to drug-induced liver injury. J Hepatol. 2007;47:128–134. doi: 10.1016/j.jhep.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 128.Chatterjee S., Lyle N., Mandal A., Kundu S. GSTT1 and GSTM1 gene deletions are not associated with hepatotoxicity caused by antitubercular drugs. J Clin Pharm Ther. 2010;35:465–470. doi: 10.1111/j.1365-2710.2009.01101.x. [DOI] [PubMed] [Google Scholar]
- 129.Mahapatra S., Woolhiser L.K., Lenaerts A.J., Johnson J.L., Eisenach K.D., Joloba M.L. A novel metabolite of antituberculosis therapy demonstrates host activation of isoniazid and formation of the isoniazid-NAD+ adduct. Antimicrob Agents Chemother. 2012;56:28–35. doi: 10.1128/AAC.05486-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Meng X., Maggs J.L., Usui T., Whitaker P., French N.S., Naisbitt D.J. Auto-oxidation of isoniazid leads to isonicotinic–lysine adducts on human serum albumin. Chem Res Toxicol. 2014;28:51–58. doi: 10.1021/tx500285k. [DOI] [PubMed] [Google Scholar]
- 131.Rawat R., Whitty A., Tonge P.J. The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance. Proc Natl Acad Sci USA. 2003;100:13881–13886. doi: 10.1073/pnas.2235848100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Lee H.C. Cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate (NAADP) as messengers for calcium mobilization. J Biol Chem. 2012;287:31633–31640. doi: 10.1074/jbc.R112.349464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Iwainsky H. [Metabolism of isonicotinic acid hydrazide and its derivatives in macroorganisms. IV. Demonstration of isonicotinyl hydrazones of pyruvic acid and α-ketoglutaric acid] Arzneimittelforschung. 1957;7:745–747. (German) [PubMed] [Google Scholar]
- 134.Zamboni V., Defranceschi A. Identification of isonicotinoylhydrazones of pyruvic and α-ketoglutaric acid in rat urine after treatment with isonicotinic acid hydrazide (isoniazid) Biochim Biophys Acta. 1954;14:430–432. doi: 10.1016/0006-3002(54)90203-6. [DOI] [PubMed] [Google Scholar]
- 135.Šimu°nek T., Klimtová I., Kaplanová J., Štêrba M., Mazurová Y., Adamcová M. Study of daunorubicin cardiotoxicity prevention with pyridoxal isonicotinoyl hydrazone in rabbits. Pharmacol Res. 2005;51:223–231. doi: 10.1016/j.phrs.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 136.Sevigny S.J., de J., White S.L., Halsey M.L., Johnston F.A. Effect of isoniazid on the loss of pyridoxal phosphate from, and its distribution in, the body of the rat. J Nutr. 1966;88:45–50. doi: 10.1093/jn/88.1.45. [DOI] [PubMed] [Google Scholar]
- 137.Brodie M.J., Boobis A.R., Hillyard C.J., Abeyasekera G., Maclntyre I., Park B.K. Effect of isoniazid on vitamin D metabolism and hepatic monooxygenase activity. Clin Pharmacol Ther. 1981;30:363–367. doi: 10.1038/clpt.1981.173. [DOI] [PubMed] [Google Scholar]