

Abstract
Intestine is responsible for the biotransformation of many orally-exposed chemicals. The constitutive androstane receptor (CAR/Nr1i3) is known to up-regulate many genes encoding drug-metabolizing enzymes and transporters (drug-processing genes/DPGs) in liver, but less is known regarding its effect in intestine. Sixty-day-old wild-type and Car−/− mice were administered the CAR-ligand TCPOBOP or vehicle once daily for 4 days. In wild-type mice, Car mRNA was down-regulated by TCPOBOP in liver and duodenum. Car−/− mice had altered basal intestinal expression of many DPGs in a section-specific manner. Consistent with the liver data (Aleksunes and Klaassen, 2012), TCPOBOP up-regulated many DPGs (Cyp2b10, Cyp3a11, Aldh1a1, Aldh1a7, Gsta1, Gsta4, Gstm1-m4, Gstt1, Ugt1a1, Ugt2b34, Ugt2b36, and Mrp2–4) in specific sections of small intestine in a CAR-dependent manner. However, the mRNAs of Nqo1 and Papss2 were previously known to be up-regulated by TCPOBOP in liver but were not altered in intestine. Interestingly, many known CAR-target genes were highest expressed in colon where CAR is minimally expressed, suggesting that additional regulators are involved in regulating their expression. In conclusion, CAR regulates the basal expression of many DPGs in intestine, and although many hepatic CAR-targeted DPGs were bona fide CAR-targets in intestine, pharmacological activation of CAR in liver and intestine are not identical.
Abbreviations: Aldh, aldehyde dehydrogenase; Asbt, solute carrier family 10, member 2 (apical sodium/bile acid cotransporter); CAR, constitutive androstane receptor; cDNA, complementary DNA; CITCO, 6-(4-chlorophenyl)imidazo [2,1-b](1,3)thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime; Cq, quantification cycle; Cyp, cytochrome P450; ddCq, delta delta Cq; DPGs, drug-processing genes (genes that encodes drug metabolizing enzymes or transporters); Gst, glutathione S-trasnferase; H3, Histone 3; hCAR, human constitutive androstane receptor; HRP, horseradish peroxidase; Mrp, multi-drug resistance-associated protein (ABC transporter family C member); Nqo1, NAD(P)H dehydrogenase quinone 1; Nrf2, nuclear factor erythroid 2-related factor 2; Oatp, organic anion transporting polypeptide (solute carrier organic anion transporter family member); Papss2, 3ʹ-phosphoadenosine 5ʹ-phosphosulfate synthase 2; PBS, phosphate-buffered saline; PBST, phosphate-buffered saline with 0.05% tween 20; PPARα, peroxisome proliferator activated receptor alpha; PVDF, polyvinylidene difluoride; qPCR, quantitative polymerase chain reaction; ST buffer, sucrose Tris buffer; Sult, sulfotransferase; TCPOBOP, 3,3ʹ,5,5ʹ-tetrachloro-1,4-bis(pyridyloxy)benzene; Ugt, UDP glucuronosyltransferase; WT, wild-type
KEY WORDS: Drug-processing genes, Intestine, Mice, CAR, Drug-metabolizing enzymes, Transporters
Graphical abstract
Intestine is responsible for the biotransformation of many orally-exposed chemicals. The constitutive androstane receptor (CAR) is known as an important xenobiotic-sensing transcription factor in liver. The present study used wild-type and CAR-null mice and showed that many critical drug-metabolizing enzymes and transporters are also bona fide CAR-target genes in intestine.

1. Introduction
Oral administration is the preferred route by patients due to its convenience, price, comfort, and handling1. The orally-administered drugs may undergo extensive first-pass metabolism in the gastro-intestinal tract, and this may result in limited systematic bioavailability, and decreased therapeutic effects2, 3. Absorption of orally administered drugs takes place primarily in the small intestine, followed by delivery to the liver via the portal blood. The small intestine is efficient in the absorption of a wide-spectrum of chemicals due to the high concentration of villi and microvilli in the order of duodenum, jejunum, and ileum4, abundant epithelial transporters, optimal pH for absorption, high peristalsis, high blood flow, as well as contact for a long time. Alteration or failure to maintain one of these conditions may result in lower bioavailability of the drug3, 5, 6. In addition to the small intestine, the large intestine may also be important for the absorption of xenobiotics, especially oral drugs formulated for sustained release6. The bacteria in the large intestine contain various enzymes that metabolize xenobioitcs as well as endogenous chemicals such as bile acids and dietary constituents7, 8. In addition, colon-specific oral drug-delivery systems have been utilized recently to administer a variety of therapeutic agents9. Therefore, it is important to determine the regulation of xenobiotic biotransformation in the intestine.
The drug-processing genes involved in the xenobiotic biotransformation include various phase-I and phase-II drug metabolizing enzymes, as well as uptake and efflux transporters. In general, the content of DPGs is lower in intestine than that in liver10. DPGs play a critical role in the absorption, metabolism, disposition, elimination and detoxification of xenobiotics and other drugs11. Phase-I enzymes catalyze hydrolysis, reduction, and oxidation of drugs. The cytochrome P450s (CYPs) in the first 3 families are among the most important phase-I enzymes that contribute to the biotransformation of the majority of xenobiotics, whereas the CYP4 family members are important for fatty acid metabolism. The NAD(P)H dehydrogenase, quinone 1 (Nqo1) is involved in reduction reactions, and it is a prototypical target gene of the oxidative stress sensor nuclear factor erythroid 2-related factor 2 (NRF2). Aldehyde dehydrogenases (ALDHs) are important phase-I enzymes for the detoxification of aldehydes, which are the metabolites of alcohols. Phase-II metabolism refers to the conjugation reactions that generally increase the water-solubility of substrates to form more polar compounds with exceptions. The three major classes of phase-II enzymes include UDP-glucuronosyltransferases (UGTs), sulfotransferases (SULTs), as well as glutathione S-transferases12. Whereas some drugs diffuse into the intestinal cells, there are two major classes of transporters, namely the solute carriers (SLC) and ATP-binding cassette (ABC) transporters, that are important in the disposition of many large and/or polar chemicals. Intestinal transporters mediate the translocation of chemicals in and out of enterocytes, and this process is important for drug disposition in the body13.
DPGs can be trans-activated by various nuclear receptors following xenobiotic exposure. The constitutive androstane receptor (CAR/Nr1i3) is one of the important xenobiotic-sensing nuclear receptors that regulate the transcription of DPGs. CAR is activated by various chemicals including steroid hormones, bile acids, pharmaceuticals, as well as environmental, dietary, and occupational chemicals14, via direct or indirect mechanisms. Direct activation of CAR refers to ligand-binding to the CAR protein, and the prototypical CAR ligands include TCPOBOP for the mouse CAR and CITCO for the human CAR. The indirect activation of CAR by chemicals such as phenobarbital disassociates CAR from its cytosolic repressor protein. CAR activation leads to its nuclear translocation and binding to the targeted response elements of genes, and this usually leads to the transcriptional up-regulation of DPGs. Chronic activation of CAR is known to cause liver tumor in rodents but to a much lesser extent in humans15, 16. Pharmacological activation of CAR by TCPOBOP has also been shown to reduce obesity in mice17. Car−/− mice have been engineered to determine the necessity of CAR in xenobiotic metabolism and liver physiology18. Phenobarbital-mediated up-regulation of the prototypical CAR-target gene Cyp2b10 does not occur in livers of Car−/− mice, and there is also decreased metabolism of the classic CYP substrate zoxazolamine, as well as a complete loss of the liver hypertrophic and hyperplastic responses to CAR-inducers.
CAR is highly expressed in liver, but it can also be detected at high amounts in the small intestine, and a lower amount in the large intestine19, 20, 21. Extensive studies have been done regarding the effect of CAR-activation on the hepatic DPG expression18, 22, 23, 24. Despite the important role of the intestine in xenobiotic biotransformation, relatively less is known regarding the effect of genetic depletion of Car and the pharmacological activation of CAR on the basal and inducibility of DPGs in different sections of intestine. Therefore, the goal of this study was to determine whether the well-known CAR-targeted DPGs in liver are also regulated by CAR in duodenum, jejunum, ileum, and colon.
2. Materials and methods
2.1. Chemicals and reagents
The mouse CAR ligand 1,4-bis-[2-(3,5-dichloropyridyloxy)]benzene (3,3′,5,5′-tetrachloro-1,4-bis(pyridyloxy)benzene, TCPOBOP) and corn oil were purchased from Sigma–Aldrich (St. Louis, MO, USA).
2.2. Animal procedures
C57BL/6 wild-type (WT) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). Breeder pairs of the Car−/− mice in the C57BL/6 background were obtained from Amgen (Thousand Oaks, CA, USA). Mice were housed according to the American Animal Association Laboratory Animal Care Guidelines, and were bred under standard conditions at the University of Washington (WA, USA). All animals were given ad libitum access to water and irradiated Picolab Rodent Diet 20 number 5053 (PMI Nutrition International, Brentwood, MO, USA). Sixty-day-old wild type and Car−/− male mice were administered the CAR-ligand TCPOBOP (3 mg/kg, i.p.), or vehicle, once daily for 4 days (n=4–5 per group). Various sections of intestine (duodenum, jejunum, ileum, and colon) were collected on the 5th day. The tissues were frozen immediately in liquid nitrogen and then stored in a –80 °C freezer prior further analysis. All animal procedures were approved by the Institutional Animal Care and Use Committee at the University of Washington.
2.3. RNA isolation
Total RNA was isolated from each section of frozen intestine tissues using RNA zol Bee reagent (Tel-Test Inc., Friendswood, TX, USA) directed by the manufacturer׳s protocol. The total RNA concentration of each sample was quantified spectrophotometrically at 260 nm using a NanoDrop 1000 Sectrophotometer (Thermo Scientific, Waltham, MA, USA). The RNA integrity of each sample was evaluated by formaldehyde agarose gel electrophoresis, and assured by appearance of 18S and 28S rRNA bands.
2.4. Reverse transcription and quantitative polymerase chain reaction (RT-qPCR)
Total RNA was reverse-transcribed to cDNA by High-Capacity cDNA Reverse Transcription Kits 1001073 (Applied Biosystems, Foster, CA, USA) in a final volume 10 µL containing 5 µL of RNA sample and 5 µL of 2×RT master mix directed by the manufacturer׳s protocol. The cDNAs were diluted 10 times and amplified by PCR, using the SsoAdvanced Universal SYBR Green Supermix in a Bio-Rad CFX384 Real-Time PCR Detection System (Bio-rad, Hercules, CA, USA). Two µL of cDNA were added to 8 µL of PCR mix, containing SsoAdvanced Universal SYBR Green Supermix (2×), forward and reverse primers, and nuclear-free water. The primers for PCR were designed using the NCBI Primer Design Tool as shown in Table 1, and were purchased from the Integrated DNA Technologies (Coralville, IA, USA). Data were expressed as percentage of the housekeeping gene β-actin.
Table 1.
RT-qPCR primer sequences
 |
2.5. Western blotting analysis
Each section of intestinal homogenate was prepared using 250 mL ST buffer (250 mmol/L sucrose, 10 mmol/L Tris base, pH 7.5) with protease inhibitors. The crude membranes were prepared from each section of frozen intestine samples as described previously25. The protein concentrations in each section of intestines were determined by a Qubit Protein Assay Kit (Thermo Fisher Scientific, Grand Island, NY, USA) as directed by the manufacturer׳s instructions. The samples were subjected to polyacrylamide gel electrophoresis and transferred onto a polyvinylidene difluoride (PVDF) membrane for 3 h on the ice. After transfer, the membranes were blocked by 5% nonfat dry milk in phosphate-buffered saline with 0.05% Tween 20 (PBST) for 1 h and incubated overnight with the following primary antibodies diluted in 1% dry milk in PBST: rabbit anti-mouse CYP2B10 polyclonal antibody (AB9916, 1:5000, EMD Millipore); or goat anti-mouse H3 polyclonal antibody (ab12079, 1:500, Abcam). After washing with 1% dry milk in PBST, the membranes were incubated for 1 h with a 1:2000 HRP-linked species-appropriate secondary antibody (Sigma Aldrich, St. Louis, MO, USA) diluted in 1% dry milk in PBST. After incubation, the membranes were washed again with 1% dry milk in PBST and then with 1% PBS, followed by incubation in Novex ECL Chemiluminescent Substrate Reagent Kit (Life Technologies, Carlsbad, CA, USA).
2.6. Statistical analysis
For RT-qPCR analysis, data among multiple groups were analyzed using analysis of variance (ANOVA) followed by the Duncan׳s post hoc test (P<0.05) using the SPSS software (IBM Cooperation, Armonk, North Castle, NY, USA). Asterisk (*) indicates statistically significant differences between control and TCPOBOP-treated wild-type mice. Pound sign (#) indicates statistically significant differences (P<0.05) between control and TCPOBOP-treated Car−/− mice. Dollar sign ($) indicates significant differences (P<0.05) in the basal mRNA expression of DPGs between control WT and control Car−/− mice. Protein density was quantified using Image J Software (National Institutes of Health, Bethesda, MD, USA).
3. Results
3.1. DPG expressions in intestine
BioGPS26 data were examined to determine which DPSs are highly expressed in the small and large intestine (Fig. 1). Those DPGs that were expressed in the intestine were selected (Table 2) to examine their mRNA expression in response to the CAR-ligand TCPOBOP in intestine, based on the following selection criteria: (1) the DPGs have been shown to be bona fide CAR-target genes in mouse liver24, or (2) the DPGs that were not examined in liver24 but are highly expressed in small or large intestine (http://biogps.org/)26, 27.
Figure 1.

Expression of drug processing genes (DPGs) in small and large intestine of mice. The mRNA abundance of DPGs was retrieved from BioGPS (Biogps.org, Su et al.26). The abundance of DPGs was expressed as average probe intensity from the microarray data in BioGPS. DPGs that were highly expressed in at least one section of intestine and/or are known as CAR-target genes (Aleksunes and Klaassen24) in liver were selected for the analysis (n=2 per tissue). For each graph, black bar represents DPG expression in the small intestine whereas red bar represents DPG expression in the large intestine.
Table 2.
Liver and Intestine regulation difference in Car-null control mice compared to WT control mice.
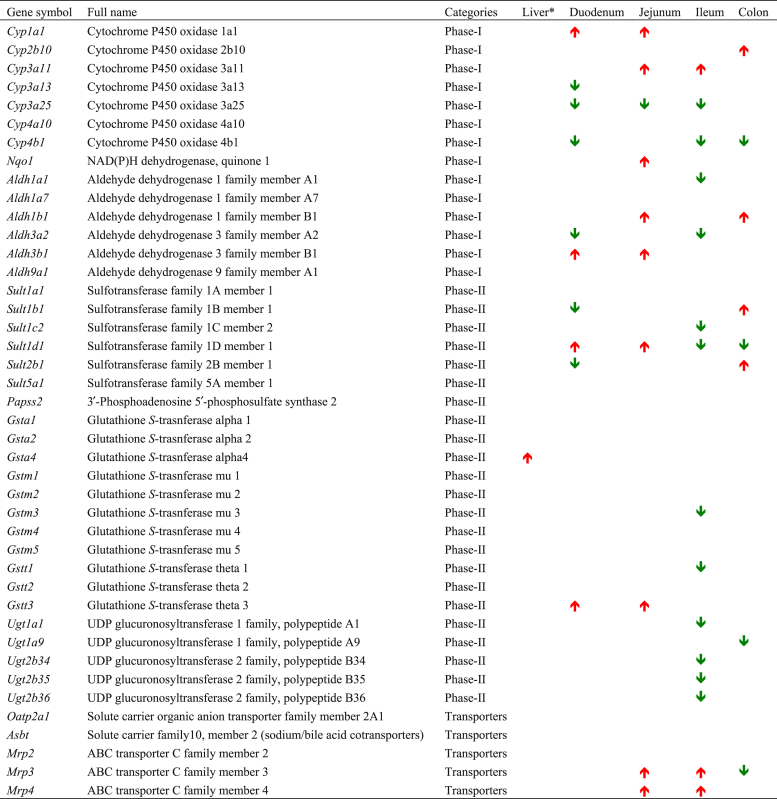 |
Basal expression of genes is shown as increased or decreased relative to that in Car-null mice. Up-regulation suggests CAR suppresses the basal expression of the gene and down-regulation suggests CAR is necessary in maintaining the constitutive expression of the gene.
⁎ Note: The liver data were obtained from Aleksunes and Klaassen24, 2012.
The relative mRNA abundance of the major DPG families in the small and large intestine are shown in Fig. 1. Data were retrieved from BioGPS as described above. For the Cyp1 family, Cyp1a1 appeared to be the major intestinal Cyp1 isoform and its mRNA was much higher in small intestine than that in large intestine. Cyp1a2 mRNA was minimally expressed in intestine (Fig. 1a), although it was shown to be the major Cyp1 isoform in liver28. Cyp1b1 was expressed at low levels and it was also small intestine-enriched. For the Cyp2b family, Cyp2b10 was the only isoform that was expressed in intestine, and its mRNA was predominantly found in the small intestine but not in large intestine (Fig. 1b). For the Cyp3a family, the mRNAs of Cyp3a11, 3a13, and 3a25 were highly expressed in small intestine; Cyp3a13 mRNA was also detected at low levels in the large intestine, whereas other Cyp3a isoforms (including Cyp3a16, 3a41a, and 3a44) were not expressed in intestine (Fig. 1c). For the Cyp4 family, Cyp4b1 was the major intestinal isoform in both small and large intestine. Cyp4a10 and 4a31 mRNAs were also detected at low levels in the large intestine, whereas they were minimally expressed in the small intestine. Other Cyp4 isoforms (including Cyp4a12a and 4a14) were minimally expressed in intestine (Fig. 1d). The Nqo1 mRNA appeared to be higher than Nqo2 mRNA in both small and large intestines (Fig. 1e). For the Aldh family, Aldh1a1, 1b1, and 9a1 were the predominant isoforms and in general, they were more highly expressed in small intestine than in large intestine. Aldh1a7, 1l1, 3a2, and 6a1 were also expressed at low levels in both the small and large intestine, Aldh16a1 and 18a1 were lowly expressed in small intestine but minimally expressed in large intestine, whereas other Aldh isoforms (including Aldh1a2, 1a3, 1l2, 2, 3a1, 3b1, 3b2, 4a1, 5a1, 7a1, and 8a1) were minimally expressed in small and large intestine (Fig. 1f).
For the Gsta family, Gsta1 appeared to be the major intestinal isoform and its mRNA was much higher in small intestine than in large intestine. Gsta4 mRNA was also detected at low levels in both small and large intestine. Gsta2 and Gsta3 were minimally expressed in intestine (Fig. 1g). For the Gstm family, Gstm1, 2, and 5 were highly expressed in large intestine; Gstm1, 2, 3, and 5 were also detected at low levels in small intestine, whereas other Gstm isoforms (including Gstm4, m6, and m7) were minimally expressed in small and large intestine (Fig. 1h). For the Gstt family, Gstt1, 2, and 3 mRNAs were the predominant isoforms in both small and large intestine, whereas Gstt4 was minimally expressed in small and large intestine (Fig. 1i). For the Sult family, Sult1b1, 1d1, and 2b1 appeared to be highly expressed in small intestine with the mRNA abundance in descending order; Sult1b1 and 1d1 were also expressed at high levels in the large intestine. Sult1a1 was only expressed in large intestine but not in small intestine, and its mRNA was highly enriched in the large intestine. Sult1c2 and 2b1 were also lowly expressed in the large intestine. Other Sult isoforms (including Sult1e1, 2a2, 3a1, 4a1, 5a1, and 6b1) were minimally expressed in small and large intestine (Fig. 1j). For the Ugt family, Ugt1a transcripts (note: probes were not specific to differentiate various Ugt1a isoforms) and Ugt2b34 mRNA appeared to be the major Ugt genes expressed in small and large intestine. Ugt1a transcripts were more highly expressed in large intestine than small intestine. Ugt2b5 and 2b35 were lowly expressed in both small and large intestines. Ugt2a3 was detected at low levels only in the small intestine, whereas Ugt2b38 and 3a2 were expressed at low levels only in large intestine. Other Ugt isoforms were minimally expressed in intestine (Fig. 1k).
For the Solute carrier organic anion (Slco) transporter family (also known as the Oatps), Slco2a1, 2b1, 3a1, and 4a1 were the intestine-enriched Slco isoforms and they were more highly expressed in small than in large intestine. Other Slco isoforms (including Slco1a1, 1a4, 1a5, 1a6, 1b2, 1c1, 5a1, 6b1, 6c1, and 6d1) were not expressed in intestine (Fig. 1I). For the Abcc family (Mrp), Abcc3 appeared to be the major intestinal isoform and its mRNA was approximately twice in large intestine as that in small intestine. Abcc1 was detected at low levels in large intestine, whereas Abcc2 was detected at low levels in small intestine. Abcc4, 5, 9, and 10 were also expressed at low levels in both small and large intestine. Other Abcc isoforms (including Abcc6, c8, and c12) were not detected in the intestine (Fig. 1m).
In summary, BioGPS has identified distinct DPG isoforms that are expressed in the intestine. Based on this information, as well as the previous findings regarding known CAR-targeted DPGs in liver24, DPGs listed in Table 2 were selected for the induction studies in WT and Car−/− mice.
Previously, it has been shown that CAR is highly expressed in liver24, and the basal expression of CAR was also found at relatively high levels in various sections of intestine19, 21. To further determine the distribution of CAR in intestine as compared to liver in control and TCPOBOP-treated conditions, and to confirm the depletion of Car in intestine of the Car−/− mice, the Car mRNA was quantified in various sections of intestine and liver from WT and Car−/− mice treated with corn oil or TCPOBOP (Fig. 2). CAR was most highly expressed in liver of the WT mice, followed by duodenum and jejunum, whereas ileum and colon had very low levels of Car expression. TCPOBOP down-regulated the Car mRNA in liver and duodenum of WT mice, and tended to decrease in jejunum, although a statistically significant difference was not achieved. As expected, the Car mRNA was not expressed in any of the tissues examined in the Car−/− mice.
Figure 2.

Messenger RNA expression of Car in mice liver and intestine. The Car mRNA in liver and various sections of the intestine was quantified using RT-qPCR as described in Section 2 (WT mice only). Data are expressed as percentage of the housekeeping gene β-actin (n=4–5 per group). Data between control and TCPOBOP-treated groups were analyzed using a Student׳s t-test. Asterisks (*) indicate statistically significant differences (P<0.05) between control and TCPOBOP-treated WT mice in the same tissue.
3.2. Regulation of phase-I drug metabolizing enzymes (Cyps and Aldh) in intestine by CAR
The expressions of selected phase-I enzymes are shown in Figure 3, Figure 4. For Cyps (Fig. 3a–g), the basal expression of Cyp1a1 was highest in duodenum, followed by jejunum and ileum, and was minimal in colon. Interestingly, in control Car−/− mice, the basal Cyp1a1 mRNA increased 11.97-fold in duodenum and 6.02-fold in jejunum, suggesting that CAR suppresses the basal expression of Cyp1a1 in duodenum and jejunum of WT mice. TCPOBOP did not alter Cyp1a1 mRNA in any portions of the intestine in WT mice; however, it down-regulated Cyp1a1 89.3% in duodenum of the Car−/− mice, suggesting an off-target effect of TCPOBOP independent of CAR (Fig. 3a). Regarding the prototypical CAR-target gene Cyp2b10, as shown in Fig. 3b, the basal expression of Cyp2b10 was the highest in duodenum followed by jejunum, but was minimally expressed in ileum and colon. In Car−/− mice, the basal Cyp2b10 mRNA increased in colon (22.1-fold) but the expression was still minimal. TCPOBOP up-regulated Cyp2b10 mRNA 4.98-fold in duodenum, 5.72-fold in jejunum, and 20.2-fold in ileum of WT mice in a CAR-dependent manner. As shown in Fig. 3c, the basal expression of Cyp3a11 was highest in duodenum, followed by jejunum, ileum, and colon. In Car−/− mice, basal Cyp3a11 mRNA decreased 55% in jejunum and 75% in ileum, suggesting that CAR is necessary for the basal expression of Cyp3a11 in these sections. TCPOBOP up-regulated Cyp3a11 expression 70% in duodenum in a CAR-dependent manner. However, it did not alter the Cyp3a11 mRNAs in other sections of the intestine. As shown in Fig. 3d, the basal expression of Cyp3a13 was highest in duodenum, followed by jejunum, and was much lower in ileum and colon. In Car−/− mice, the basal Cyp3a13 mRNA expression decreased 60% in duodenum, suggesting that CAR is necessary in maintaining the constitutive expression of Cyp3a13 in the duodenum. TCPOBOP up-regulated Cyp3a13 mRNA expression 38% in duodenum and 44% in jejunum in a CAR-dependent manner; however, it did not alter Cyp3a13 mRNA in ileum or colon. As shown in Fig. 3e, the basal expression of Cyp3a25 was highest in duodenum, followed by jejunum and ileum but was minimal in colon. In Car−/− mice, the basal Cyp3a25 mRNA decreased in duodenum (77.8%), jejunum (58.4%), and ileum (59.6%), suggesting that CAR is necessary for the basal expression of Cyp3a25 in the small intestine. TCPOBOP in general did not alter the Cyp3a25 mRNA expression in any sections of intestine. As shown in Fig. 3f, the basal expression of Cyp4a10 was the highest in duodenum, followed by colon, jejunum, and ileum. TCPOBOP in general did not alter the expression of Cyp4a10, except for a down-regulation (63.5%) in ileum in a CAR-dependent manner. As shown in Fig. 3g, the basal expression of Cyp4b1 was similarly expressed in all portions of the intestine. In Car−/− mice, the basal Cyp4b1 mRNA decreased 34% in duodenum, 71% in ileum and 81% in colon, suggesting that CAR is necessary in maintaining the basal expression of Cyp4b1 in these sections. TCPOBOP down-regulated Cyp4b1 mRNA expression 27.4% in ileum of WT mice. As shown in Fig. 3h, the basal expression of Nqo1 was highest in colon, followed by duodenum, ileum, and jejunum. In Car−/− mice, the basal Nqo1 mRNA increased 1.09-fold in jejunum, suggesting that CAR suppresses the basal expression of Nqo1 in WT mice intestine. TCPOBOP moderately down-regulated Nqo1 27.1% in ileum of the Car−/− mice. In summary, CAR suppresses the basal expression of Cyp1a1 and Nqo1, but maintains the constitutive expression of Cyp3a11, Cyp3a13, Cyp3a25, and Cyp4b1, in distinct sections of intestine; whereas pharmacological activation of CAR by TCPOBOP up-regulates Cyp2b10, Cyp3a13, Cyp3a13, but down-regulates Cyp4a10 and Cyp4b1 in distinct sections of intestine, in a CAR-dependent manner.
Figure 3.

Messenger RNA expression of phase-I drug-metabolizing enzymes, including Cyp1–4 and Nqo1 in various sections of intestine (duodenum, jejunum, ileum, and colon) of WT and Car−/− male mice treated with vehicle (corn oil) or TCPOBOP as described in Section 2. Data are expressed as percentage of the housekeeping gene β-actin (n=4–5 per group). Data among multiple groups were analyzed using ANOVA followed by Duncan׳s post hoc test. Asterisks (*) indicate statistically significant differences (P<0.05) between control WT and TCPOBOP-treated WT mice at the same section of intestines. Pound signs (#) indicate statistically significant differences (P<0.05) between control Car−/− and with TCPOBOP-treated Car−/− mice. Dollar signs ($) indicate statistically significant differences (P<0.05) of the basal mRNA expression between control WT and control Car−/− mice.
Figure 4.

Messenger RNA expression of the phase-I drug-metabolizing enzymes Aldhs in various sections of intestine (duodenum, jejunum, ileum, and colon) of WT and Car−/− male mice treated with vehicle (corn oil) or TCPOBOP as described in Section 2. Data are expressed as percentage of the housekeeping gene β-actin (n=4–5 per group). Data among multiple groups were analyzed using ANOVA followed by Duncan׳s post hoc test. Asterisks (*) indicate statistically significant differences (P<0.05) between control WT and TCPOBOP-treated WT mice at the same section of intestines. Pound signs (#) indicate statistically significant differences (P<0.05) between control Car−/− and with TCPOBOP-treated Car−/− mice. Dollar signs ($) indicate statistically significant differences (P<0.05) of the basal mRNA expression between control WT and control Car−/− mice.
Regarding the regulation of the aldehyde dehydrogenases (Aldhs), as shown in Fig. 4a, the basal expression of Aldh1a1 was highest in colon and duodenum, followed by jejunum, and ileum. In Car−/− mice, the basal Aldh1a1 mRNA moderately decreased 54% in ileum. TCPOBOP up-regulated Aldh1a1 mRNA 5.37-fold in duodenum and 87% in jejunum in a CAR-dependent manner. However, TCPOBOP did not alter the Aldh1a1 mRNA expression in ileum and colon. As shown in Fig. 4b, the basal expression of Aldh1a7 was highest in ileum, followed by colon, jejunum, and minimal in duodenum. The absence of CAR leads to an apparent increase in Aldh1a7 in duodenum and jejunum, although a statistical significance was not achieved. TCPOBOP up-regulated Aldh1a7 mRNA 51.4-fold in duodenum and 5.00-fold in jejunum in a CAR-dependent manner, but it slightly decreased Aldh1a7 mRNA in colon. As shown in Fig. 4c, the basal expression of Aldh1b1 was relatively similar in all portions of the intestine. In Car−/− mice, the basal expression of Aldh1b1 mRNA was 70.9% higher in jejunum and 57.4% in colon than in WT mice, and TCPOBOP did not alter Aldh1b1 mRNA in any intestinal sections. As shown in Fig. 4d, the basal expression of Aldh3a2 was relatively similar in all portions of intestine. In Car−/− mice, basal Aldh3a2 mRNA decreased 41.6% in duodenum and 65.6% in ileum. TCPOBOP did not have any effect on the Aldh3a2 mRNA expression in intestine. As shown in Fig. 4e, the basal expression of Aldh3b1 expression was highest in colon and ileum, and was minimal in duodenum and jejunum. Interestingly, In Car−/− mice, the basal Aldh3b1 mRNA markedly increased 124.7-fold in duodenum and 26.8-fold in jejunum, which suggest that CAR suppresses the basal expression of Aldh3b1 in WT mice intestine. TCPOBOP up-regulated Aldh3b1 expression 160.4-fold in duodenum, 20.9-fold in jejunum, and 41.9% in ileum in a CAR-dependent manner. As shown in Fig. 4f, the basal expression of Aldh9a1 decreased from duodenum to colon. TCPOBOP did not alter the Aldh9a1 mRNA expression in any portions of intestine. In summary, CAR suppresses the basal expression of Aldh3b1 but maintains the constitutive expression of Aldh1a1, Aldh1b1, and Aldh3a2 in distinct sections of intestine, whereas pharmacological activation of CAR by TCOBOP up-regulates Aldh1a7 and Aldh3b1 in a CAR-dependent manner in distinct sections of small intestine.
3.3. Regulation of phase-II drug metabolizing enzymes (Sult, Gsta, Gstt, Gstm, Ugt) in intestine by CAR
Figure 5, Figure 6, Figure 7, Figure 8 illustrate the regulation of various phase-II enzymes by CAR in intestine. Regarding the regulation of Sult, as shown in Fig. 5, the basal Sult1a1 mRNA expression was predominantly expressed in colon, and was very lowly expressed in all portions of the small intestine. TCPOBOP did not alter the expression of Sult1a1 mRNA in any portions of intestine (Fig. 5a). As shown in Fig. 5b, the basal expression of Sult1b1 mRNA was highest in duodenum, followed by jejunum, colon, and ileum. In Car−/− mice, there was a decrease in the basal Sultb1 mRNA in duodenum (47.1%), but an increase in the basal Sult1b1 mRNA in colon (61.2%). TCPOBOP did not alter the Sult1b1 mRNA expression in any portions of intestine. As shown in Fig. 5c, the basal mRNA expression of Sult1c2 is highest in colon, followed by ileum and jejunum, and was minimally expressed in duodenum. In Car−/− mice, the basal Sult1c2 mRNA expression was 60% lower in ileum, suggesting that CAR is necessary in maintaining constitutive expression of Sult1c2 in ileum of WT mice. TCPOBOP up-regulated Sult1c2 mRNA 73.6-fold in duodenum and 4.8-fold in jejunum in a CAR-dependent manner. However, TCPOBOP did not alter Sult1c2 mRNA expression in ileum or colon. As shown in Fig. 5d, the basal mRNA expression of Sult1d1 increased from duodenum to colon. In Car−/− mice, the basal Sult1d1 mRNA increased 23.9-fold and 6.21-fold in duodenum and jejunum; however, it decreased 75.8% in ileum and 74.4% in colon. TCPOBOP up-regulated Sult1d1 mRNA 52.4-fold in duodenum and 12.5-fold in jejunum in a CAR-dependent manner, whereas it had no effect on the Sult1d1 mRNA in ileum, and down-regulated 47.5% in colon of WT mice. As shown in Fig. 5e, the basal expression of Sult2b1 mRNA decreased from duodenum, to colon. In Car−/− mice, the basal Sult2b1 mRNA decreased in duodenum (53.1%) but increased in colon (1.31-fold). TCPOBOP down-regulated Sult2b1 mRNA 61.4% in duodenum, but had no effect in other portions of the intestine. As shown in Fig. 5f, the basal expression of Sult5a1 was highest in colon and duodenum, and lower in ileum, and jejunum. TCPOBOP had minimal effect on Sult5a1 mRNA expression, except for a moderate increase (57.4%) in jejunum in a CAR-dependent manner. As shown in Fig. 5g, the basal mRNA expression of Papss2 was highest in duodenum and colon, but was much lower in jejunum and ileum. TCPOBOP did not have any effect on Papss2 mRNA expression in any portions of intestine of either genotype.
Figure 5.

Messenger RNA expression of the phase-II drug-metabolizing enzymes Sults in various sections of intestine (duodenum, jejunum, ileum, and colon) of WT and Car−/− male mice treated with vehicle (corn oil) or TCPOBOP as described in Section 2. Data are expressed as percentage of the housekeeping gene β-actin (n=4–5 per group). Data among multiple groups were analyzed using ANOVA followed by Duncan׳s post hoc test. Asterisks (*) indicate statistically significant differences (P<0.05) between control WT and TCPOBOP-treated WT mice at the same section of intestines. Pound signs (#) indicate statistically significant differences (P<0.05) between control Car−/− and with TCPOBOP-treated Car−/− mice. Dollar signs ($) indicate statistically significant differences (P<0.05) of the basal mRNA expression between control WT and control Car−/− mice.
Figure 6.

Messenger RNA expression of the phase-II drug-metabolizing enzymes Gstas and Gstts in various sections of intestine (duodenum, jejunum, ileum, and colon) of WT and Car−/− male mice treated with vehicle (corn oil) or TCPOBOP as described in Section 2. Data are expressed as percentage of the housekeeping gene β-actin (n=4–5 per group). Data among multiple groups were analyzed using ANOVA followed by Duncan׳s post hoc test. Asterisks (*) indicate statistically significant differences (P<0.05) between control WT and TCPOBOP-treated WT mice at the same section of intestines. Pound signs (#) indicate statistically significant differences (P<0.05) between control Car−/− and with TCPOBOP-treated Car−/− mice. Dollar signs ($) indicate statistically significant differences (P<0.05) of the basal mRNA expression between control WT and control Car−/− mice.
Figure 7.

Messenger RNA expression of the phase-II drug-metabolizing enzymes Gstms in various sections of intestine (duodenum, jejunum, ileum, and colon) of WT and Car−/− male mice treated with vehicle (corn oil) or TCPOBOP as described in Section 2. Data are expressed as percentage of the housekeeping gene β-actin (n=4–5 per group). Data among multiple groups were analyzed using ANOVA followed by Duncan׳s post hoc test. Asterisks (*) indicate statistically significant differences (P<0.05) between control WT and TCPOBOP-treated WT mice at the same section of intestines. Pound signs (#) indicate statistically significant differences (P<0.05) between control Car−/− and with TCPOBOP-treated Car−/− mice. Dollar signs ($) indicate statistically significant differences (P<0.05) of the basal mRNA expression between control WT and control Car−/− mice.
Figure 8.

Messenger RNA expression of the phase-II drug-metabolizing enzymes Ugts in various sections of intestine (duodenum, jejunum, ileum, and colon) of WT and Car−/− male mice treated with vehicle (corn oil) or TCPOBOP as described in Section 2. Data are expressed as percentage of the housekeeping gene β-actin (n=4–5 per group). Data among multiple groups were analyzed using ANOVA followed by Duncan׳s post hoc test. Asterisks (*) indicate statistically significant differences (P<0.05) between control WT and TCPOBOP-treated WT mice at the same section of intestines. Pound signs (#) indicate statistically significant differences (P<0.05) between control Car−/− and with TCPOBOP-treated Car−/− mice. Dollar signs ($) indicate statistically significant differences (P<0.05) of the basal mRNA expression between control WT and control Car−/− mice.
Regarding the Gsta, as shown in Fig. 6a–c, the basal mRNAs of multiple Gsta family members (Gsta1, 2, and 4) followed a similar expression pattern, which was highest expression in duodenum, followed by jejunum, and minimal expression in ileum and colon. In duodenum, TCPOBOP up-regulated the mRNAs of Gsta1 (1.46-fold), Gsta2 (4.56-fold), and Gsta4 (2.45-fold) in a CAR-dependent manner (Fig. 6a–c). In jejunum, TCPOBOP also up-regulated Gsta2 mRNA 1.72-fold in a CAR-dependent manner. However, TCPOBOP had no effect on the Gsta expression in other portions of intestine in either WT or Car−/− mice (Fig. 6a–c). Regarding the Gstt, as shown in Fig. 6d, Gstt1 mRNA basal expression was highest in duodenum, followed by colon, jejunum, and ileum, and was not readily altered by TCPOBOP in any portions of intestine, except for a moderate increase by TCPOBOP in ileum of WT mice. CAR deficiency moderately decreased the basal Gstt1 mRNA in ileum (27.8%). As shown in Fig. 6e, the basal expression of Gstt2 mRNA was highest in colon, followed by duodenum, ileum, and jejunum, and was not altered by TCPOBOP in any intestinal sections. As shown in Fig. 6f, the basal expression of Gstt3 was low in duodenum and jejunum, but was much higher in ileum and highest in colon. CAR deficiency resulted in a 34.5-fold up-regulation of Gstt3 mRNA in duodenum and a 10.2-fold in jejunum of WT mice. Interestingly, TCPOBOP also up-regulated Gstt3 mRNA in these two sections (47.4-fold and 9.34-fold, respectively) in a CAR-dependent manner, whereas it did not have any effect in ileum and colon. Regarding the Gstm family members (Fig. 7a–e), the basal mRNA expression of Gstm1 (Fig. 7a), Gstm4 (Fig. 7d), and Gstm5 (Fig. 7e) was highest in duodenum, followed by colon, jejunum, and ileum; the basal mRNA expression of Gstm2 was highest in colon, followed by duodenum, ileum, and jejunum; whereas the basal mRNA expression of Gstm3 was highest in duodenum, followed by jejunum, colon, and ileum (Fig. 6d–g). In Car−/− mice, the basal Gstm3 mRNA decreased 67.0% in ileum, but Gstm5 mRNA increased 58.4% in colon. TCPOBOP up-regulated all Gstm genes, except for Gstm5, in duodenum (15.8-fold, 6.28-fold, 5.27-fold, and 5.07-fold, respectively) and jejunum (6.20-fold, 3.65-fold, 3.17-fold, and 3.16-fold, respectively), in a CAR-dependent manner. In ileum, TCPOBOP also moderately up-regulated certain Gstm genes, such as Gstm1 (58%) and Gstm3 (74.7%) in a CAR dependent manner. TCPOBOP in general had minimal effect on the Gstm genes in colon of either genotype (Fig. 7a–e). TCPOBOP moderately down-regulated the Gstm1 and Gstm2 mRNAs in ileum, and up-regulated Gstm4 in colon of Car−/− mice, likely due to off-target effect of the chemical.
Regarding the regulation of the Ugt family, as shown in Fig. 8a, the basal mRNA expression of Ugt1a1 was expressed at comparable levels in various sections of the intestine. In Car−/− mice, there was a moderate decrease in the basal Ugt1a1 mRNA only in ileum (46.3%). TCPOBOP up-regulated Ugt1a1 mRNA 3.95-fold in duodenum and 1.46-fold in jejunum in a CAR-dependent manner. However, TCPOBOP had no effect on the Ugt1a1 mRNA in ileum or colon. As shown in Fig. 8b, the basal expression of Ugt1a9 was highest in duodenum, and was lowly expressed in the other sections of the intestine. The basal Ugt1a9 mRNA was further decreased in colon of the Car−/− mice. TCPOBOP in general did not alter the Ugt1a9 mRNA expression in any sections of intestine, although it tended to increase the Ugt1a9 mRNA in duodenum (a statistical significance was not achieved). As shown in Fig. 8c and d, the basal mRNAs of Ugt2b34 and Ugt2b35 were both highest in duodenum, followed by colon, jejunum, and ileum. In Car−/− mice, the basal Ugt2b34 and 2b35 mRNAs decreased in ileum (50.5% and 40.7%, respectively), suggesting that CAR is necessary in maintaining constitutive expression of both Ugt2b34 and 2b35. TCPOBOP up-regulated Ugt2b34 mRNA 5.09-fold in duodenum in a CAR-dependent manner; however, it did not alter the Ugt2b34 mRNA in other sections of intestine. TCPOBOP had a similar effect on the Ugt2b35, except that the mRNA increased in duodenum was not statistically significant. As shown in Fig. 8e, the basal mRNA expression of Ugt2b36 was comparable in various sections of intestine. In Car−/− mice, the basal Ugt2b36 mRNA was down-regulated 61.4% in ileum. TCPOBOP up-regulated Ugt2b36 mRNA 1.65-fold in duodenum and 73.1% in jejunum in a CAR-dependent manner, however, it did not alter the Ugt2b36 mRNA in ileum or colon.
In summary, CAR regulates the basal expression of many phase-II enzymes in distinct sections of intestine, whereas pharmacological activation of CAR up-regulates Sult1c2, Sult1d1, Sult5a1, Gsta1, Gsta2, Gsta4, Gstt3, Gstm1-4, Ugt1a1, Ugt2b34, Ugt2b36, and tends to up-regulate Ugt1a9 and Ugt2b35, in distinct sections of intestine in a CAR-dependent manner. In contrast, pharmacological activation of CAR down-regulates Sult1d1 in colon and Sult2b1 in duodenum in a CAR-dependent manner; however, because Car is lowly expressed in colon (Fig. 2), the TCPOBOP-mediated effects in colon may be due to the involvement of other regulatory factors.
3.4. Regulation of the transporters in intestine by CAR
Fig. 9 shows the mRNA expression of uptake and efflux transporters in various sections of intestine of WT and Car−/− mice treated with corn oil or TCPOBOP. Regarding the uptake transporters (Fig. 9a–c), the basal mRNA expression of Oatp2a1 was higher in colon than in the three sections of the small intestine, and TCPOBOP did not alter the Oatp2a1 mRNA expression in any sections of intestine, except for a moderate increase in ileum of Car−/− mice (46.5%, Fig. 9a). The basal mRNA expression of Asbt was highest in ileum and colon, but was minimally expressed in duodenum and jejunum, and TCPOBOP had no effect on the Asbt mRNA expression in any sections of intestine (Fig. 9b).
Figure 9.

Messenger RNA expression of the transporters in various sections of intestine (duodenum, jejunum, ileum, and colon) of WT and Car−/− male mice treated with vehicle (corn oil) or TCPOBOP as described in the Section of materials and methods. Data are expressed as percentage of the housekeeping gene β-actin (n=4–5 per group). Data among multiple groups were analyzed using ANOVA followed by Duncan׳s post hoc test. Asterisks (*) indicate statistically significant differences (P<0.05) between control WT and TCPOBOP-treated WT mice at the same section of intestines. Pound signs (#) indicate statistically significant differences (P<0.05) between control Car−/− and with TCPOBOP-treated Car−/− mice. Dollar signs ($) indicate statistically significant differences (P<0.05) of the basal mRNA expression between control WT and control Car−/− mice.
Regarding the efflux transporters Mrp2–4 (Fig. 9c–e), the basal mRNA expression of Mrp2 was highest in duodenum, followed by jejunum, and ileum, but was minimally expressed in colon. TCPOBOP up-regulated Mrp2 1.93-fold in duodenum, 1.16-fold in jejunum, and 40.3% in ileum in a CAR-dependent manner. In Car−/− mice, TCPOBOP down-regulated Mrp2 mRNA 96.5% in ileum, but up-regulated Mrp2 mRNA 9.03-fold in colon, which may be due to off-target effect of the chemical (Fig. 9c). The basal mRNA expression of Mrp3 was highest in colon, followed by duodenum, jejunum, and ileum. In Car−/− mice, the basal Mrp3 mRNA expression was up-regulated 1.18-fold in jejunum and 1.05-fold in ileum, but was down-regulated 48.4% in colon. TCPOBOP up-regulated Mrp3 mRNA 2.11-fold in duodenum and 71.9% in ileum in a CAR-dependent manner (it also tended to increase Mrp3 mRNA in jejunum although a statistically significant difference was not achieved). In contrast, TCPOBOP down-regulated Mrp3 mRNA 50.7% in colon of WT mice. Considering that Car is minimally expressed in colon (Fig. 2), the TCPOBOP-mediated down-regulation of Mrp3 may be due to off-target effect of the chemical (Fig. 9d). The basal mRNA expression of Mrp4 was highest in colon followed by ileum, but was minimally expressed in duodenum and jejunum. Interestingly, in Car−/− mice, there was a marked increase in the basal Mrp4 mRNA in both duodenum and jejunum (81.1-fold and 15.6-fold, respectively), suggesting CAR suppresses Mrp4 basal expression in these two sections. Conversely, pharmacological activation of CAR by TCPOBOP also increased Mrp4 mRNA in duodenum and jejunum (74.2-fold and 15.4-fold, respectively), in a CAR-dependent manner. However, TCPOBOP did not alter the Mrp4 mRNA in ileum and colon (Fig. 9e).
In summary, CAR suppresses the basal expression of Mrp3 in jejunum and ileum, as well as Mrp4 in duodenum and jejunum, whereas pharmacological activation of CAR by TCPOBOP has minimal effect on the uptake transporters but markedly increases the efflux transporters Mrp2–4 in small intestine, but decreases the Mrp3 mRNA in colon, in a CAR-dependent manner. Even though Mrp3 and Mrp4 are well known CAR-target genes in liver24 and intestine (Fig. 9c–e), their basal expressions are highest in colon where Car is lowly expressed, suggesting that other regulatory factors are involved in the basal expression of these transporters.
3.5. Regulation of CYP2B10 protein in duodenum by CAR
Because duodenum has the highest Car and Cyp2b10 mRNA expression (Figs. 2 and 3b), the protein for the prototypical CAR-target gene Cyp2b10 was quantified in duodenum of WT and Car−/− mice by Western blotting analysis (Fig. 10). Following TCPOBOP treatment, consistent with the mRNA data, CYP2B10 protein was also increased (5.11-fold) in the duodenum of WT mice (Fig. 10b). However, such TCPOBOP-mediated induction in the CYP2B10 protein expression was completely abolished in the duodenum of Car−/− mice, suggesting that TCPOBOP-mediated up-regulation of CYP2B10 protein in duodenum is CAR-dependent.
Figure 10.

Western blot analysis of CYP2B10 protein and H3 in duodenum (small intestine) of wild-type and Car−/− mice treated with vehicle (corn oil) or TCPOBOP. Asterisks (*) indicate statistically significant differences (P<0.05) between control WT and TCPOBOP-treated WT mice in duodenum.
4. Discussion
In conclusion, the present study has demonstrated that in addition to its important roles in liver24, CAR is also critical in both maintaining the basal expression of certain DPGs and the pharmacological regulation of certain DPGs in a section-specific manner of the intestine. A systematic comparison between liver (previous studies) and intestine (present study) has shown that CAR activation in liver and intestine produces overlapping but not identical results. The present study has also compared the section-specific CAR-mediated effect on the DPG expression, and has demonstrated that in general, duodenum appears to be the most responsive section following exposure to the CAR-ligand TCPOBOP, likely because CAR is highest expressed in duodenum as compared to other sections of the intestine. TCPOBOP not only has inducible but also suppressive effect on the DPG expression in intestine. In addition, the CAR-independent off-target effect of TCPOBOP has also been observed in the present study, evidenced by TCPOBOP-mediated changes in DPG expression in Car−/− mice, and TCPOBOP-mediated changes in DPG expression in WT colon where CAR is minimally expressed. Many bona fide CAR-target genes in small intestine were highest expressed in colon where CAR is minimally expressed, suggesting that additional regulatory factors are involved in the basal expression of these genes.
A systematic comparison of the CAR effect on DPG expression in between liver and various sections of intestine is shown in Table 2 (CAR-mediated basal expression of DPGs) and Table 3 (effect of pharmacological activation of CAR on the expression of DPGs). In general, the basal CAR expression is more important for the constitutive expression of DPGs in intestine rather than in liver24 (Table 2). Regarding the effect of the pharmacological activation of CAR, consistent with liver data24, TCPOBOP up-regulated many DPGs (Cyp2b10, Cyp3a11, Aldh1a1, Aldh1a7, Gsta1, Gsta4, Gstm1-m4, Ugt1a1, Ugt2b34, Ugt2b36, and Mrp2–4) in certain portions of the small intestine in a CAR-dependent manner, with duodenum generally being the most inducible section. In contrast, Nqo1, Papss2, Ugt1a9, and Ugt2b35 were up-regulated by TCPOBOP in liver but were not changed in intestine, therefore the pharmacological activation of CAR in liver and intestine are not identical. Such tissue-specific effects may be due to tissue-specific chromatin epigenetic environment, such as different signatures for DNA methylation (suppressive signal for gene transcription) and/or histone modification patterns, which prevent the CAR-mediated trans-activation of certain DPGs in intestine. The epigenetic signatures within the enhancers and promoters of certain DPGs will need to be examined in future studies. Using the epithelial cells scraped from the whole small intestine, another study in the literature has also shown liver- vs. small intestine–specific regulation of some DPGs by TCPOBOP29. Our finding is consistent with that study regarding the regulation of Cyp2b10, Gsta1, Gstm2, Mrp2, and Mrp3, and in addition, the present study has investigated the intestinal-specific CAR-mediated regulation of many other genes, including Cyp3a13, 3a25, Cyp4a10, Cyp4b1, Nqo1, Gsta2, Gsta4, Gstt2, Gstt3, Gstm3, Gstm4, Mrp4, and this has added new information to the existing knowledge. Certain discrepancies are also observed between the present study and the Maglich et al.29 study, in that Aldh1a1, Aldh1a7, and Cyp3a11 were not changed in the whole small intestine by TCPOBOP in the previous study, but were up-regulated by TCPOBOP in the present study in a CAR-dependent manner. In addition, the basal Cyp1a1 expression was decreased in the previous study but increased in the present study. Such discrepancy may be due to difference doses of TCPOBOP (a single dose of TCPOBOP at 0.3 mg/kg in corn oil with 5% DMSO of the previous study vs. 3 mg/kg of TCPOBOP in corn oil once daily for 4-days in the present study), or different sample preparation procedures (epithelial cells scraped from whole small intestine in the previous study vs. various sections of small intestine in the present study).
Table 3.
Liver and Intestine regulation difference in WT TCPOBOP-treated mice compared to WT control mice.
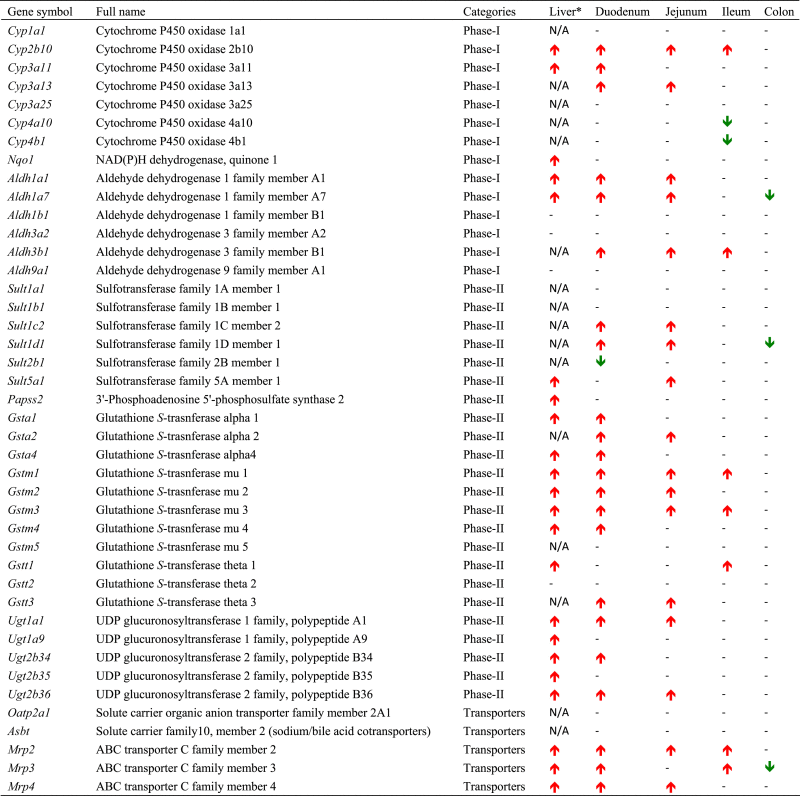 |
Basal expression of genes is shown as increased or decreased relative to that in wild type mice. (–) denotes none change. N/A: not available.
⁎ Note: The liver data were obtained from Aleksunes and Klaassen24, 2012.
Previous studies have demonstrated that the basal expression of Car is high in liver and small but is lower in the large intestine19, 21, 30, whereas the present study has confirmed the basal tissue distribution of CAR, and is among the first to show that pharmacological activation of CAR by TCPOBOP actually down-regulates the Car expression in liver, duodenum, and jejunum (Fig. 2), and this is likely due to a negative feedback mechanism to prevent excessive CAR-signaling through decreasing the CAR synthesis. Regarding the regulation of DPGs by CAR, a previous study in the literature has demonstrated the CAR-dependent up-regulation of Cyp2b10 in duodenum30. The present study on Cyp2b10 in duodenum is consistent with that study, and our study has also examined the expression of Cyp2b10 and other DPGs in other sections of intestine. Another previous study has performed a preliminary survey in WT male mice regarding the regulation of a few Ugts by TCPOBOP in duodenum, jejunum, ileum, and colon using pooled samples (i.e., 1 pooled sample from n=5 biological replicates)31. The apparent TCPOBOP-mediated increase in the mRNAs of Ugt1a1 and Ugt2b35 in that previous study is consistent with the present study, but Ugt2b34 mRNA is only up-regulated by TCPOBOP in the present study. Such differences are likely due to different method of detection (branched DNA amplification technology vs. RT-qPCR), and/or pooled vs. individual samples. Our finding on CAR-dependent up-regulation of Ugt1a1 mRNA by TCPOBOP in duodenum is also consistent with a previous study using Northern blot of Ugt1a1 in duodenum of WT and Car−/− mice29. The expression of Car is gender-divergent (i.e. higher in females than in males) in liver but not in any sections of intestine19. Therefore, the present study has only tested the effect of CAR activation in intestines of male mice.
Although many orally administered drug absorption and delivery are known to take place mostly in small intestine and liver, the present study has shown that many bona fide CAR-target DPGs in liver and small intestine are highest expressed in colon, where Car is lowly expressed. Examples of these genes include Aldh3b1, Sult1c2, Sult1d1, Gstt3, Mrp3 and Mrp4. Functionally speaking, the high expression of Aldh3b1 may be important in detoxifying the microbial aldehyde produced from ethanol by the intestinal bacteria, and this may be critical in reducing the risk of colon cancer derived from microbial aldehyde32. The high expression of certain phase-II enzymes such as Sults and Gst in colon correlate with its critical function to conjugate and thus detoxify various substances in large intestine. GSTs are also involved in the metabolism of endogenous and exogenous carcinogenic substances, which are implicated in the risks of colorectal cancer33, 34. The colon-specific efflux transporters Mrp3 and Mrp4 may also favor the elimination of various potentially toxic chemicals into feces. Another functional significant of colon-specific expression of certain DPGs is that it is associated with the critical roles of DPGs in metabolizing colon-targeted drugs or prodrugs. In addition to the microbial enzymes capability to bio-activate and/or detoxify xenobiotics, the colon tissue derived host enzymes may also contribute to the biotransformation of certain chemicals.
One critical question that arises is in regard of the potential species differences in the CAR-mediated regulation of intestinal DPGs. CAR is highly expressed in liver and small intestine of both mice and humans21, 35. The species differences of CAR and its tumorigenesis potential have been well characterized in liver, in that pharmacological activation of mouse CAR leads hepatomegaly followed by hepatocarcinogenesis in a CAR-dependent manner36. Although human CAR activation is not a risk to cause liver tumor in human, it may cause liver hypertrophy without hyperplasia in response to the human CAR activators phenobarbital and chlordane, suggesting that hCAR is able to induce hypertrophic responses in response to xenobiotic stress37. However, both the mouse and human CAR proteins appear to share high similarities in regulating the genes involved in xenobiotic biotransformation in liver. For example, the mouse CAR activation by TCPOBOP up-regulates the expression of Cyp1a2, Cyp2b10, Cyp3a11, and Ugt1a1 in a CAR-dependent manner in liver24, whereas the human CAR activation by the human CAR activators also up-regulates the expression of the human orthologs CYP1A2, CYP2B6, and UGT1A129. In human intestine-derived Caco2 cells phenobarbital up-regulates CYP2B6 and CYP3A438, 39, and our finding regarding the CAR-mediated up-regulation of the mouse orthologs Cyp2b10 and Cyp3a11 in duodenum is consistent with the previous studies. However, relatively less is known regarding the intestinal effect of pharmacological activation or genetic depletion of Car in the regulation of many other DPGs in vivo, thus the present study has filled this critical knowledge gap. Identification of the xenobiotic responses to CAR activation in mouse and human intestines is critical for understanding certain adverse drug reactions for orally exposed chemicals.
Acknowledgment
The authors would like to thank all members in Dr. Cui׳s laboratory for help in tissue collection, sample preparation, laboratory procedures, and bioinformatics. We also would like to thank Dr. Curtis Klaassen for proof-reading this manuscript. This study is supported by U. S. National Institute of Health R-01 grants ES019487, ES025708, and GM11138, as well as start-up funds from University of Washington Center for Ecogenetics and Environmental Health (P30ES007033).
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Liu G., Franssen E., Fitch M.I., Warner E. Patient preferences for oral versus intravenous palliative chemotherapy. J Clin Oncol. 1997;15:110–115. doi: 10.1200/JCO.1997.15.1.110. [DOI] [PubMed] [Google Scholar]
- 2.Alqahtani S., Mohamed L.A., Kaddoumi A. Experimental models for predicting drug absorption and metabolism. Expert Opin Drug Metab Toxicol. 2013;9:1241–1254. doi: 10.1517/17425255.2013.802772. [DOI] [PubMed] [Google Scholar]
- 3.Ward N. The impact of intestinal failure on oral drug absorption: a review. J Gastrointest Surg. 2010;14:1045–1051. doi: 10.1007/s11605-009-1151-9. [DOI] [PubMed] [Google Scholar]
- 4.Pang K.S. Modeling of intestinal drug absorption: roles of transporters and metabolic enzymes (for the Gillette Review Series) Drug Metab Dispos. 2003;31:1507–1519. doi: 10.1124/dmd.31.12.1507. [DOI] [PubMed] [Google Scholar]
- 5.Nauli A.M., Nauli S.M. Intestinal transport as a potential determinant of drug bioavailability. Curr Clin Pharmacol. 2013;8:247–255. doi: 10.2174/1574884711308030012. [DOI] [PubMed] [Google Scholar]
- 6.Doherty M.M., Charman W.N. The mucosa of the small intestine: how clinically relevant as an organ of drug metabolism? Clin Pharmacokinet. 2002;41:235–253. doi: 10.2165/00003088-200241040-00001. [DOI] [PubMed] [Google Scholar]
- 7.Selwyn F.P., Csanaky I.L., Zhang Y., Klaassen C.D. Importance of large intestine in regulating bile acids and glucagon-like peptide-1 in germ-free mice. Drug Metab Dispos. 2015;43:1544–1556. doi: 10.1124/dmd.115.065276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Klaassen C.D., Cui J.Y. Review: mechanisms of how the intestinal microbiota alters the effects of drugs and bile acids. Drug Metab Dispos. 2015;43:1505–1521. doi: 10.1124/dmd.115.065698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Patel M., Shah T., Amin A. Therapeutic opportunities in colon-specific drug-delivery systems. Crit Rev Ther Drug Carr Syst. 2007;24:147–202. doi: 10.1615/critrevtherdrugcarriersyst.v24.i2.20. [DOI] [PubMed] [Google Scholar]
- 10.Doherty M.M., Pang K.S. First-pass effect: significance of the intestine for absorption and metabolism. Drug Chem Toxicol. 1997;20:329–344. doi: 10.3109/01480549709003891. [DOI] [PubMed] [Google Scholar]
- 11.Xu C., Li C.Y., Kong A.N. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res. 2005;28:249–268. doi: 10.1007/BF02977789. [DOI] [PubMed] [Google Scholar]
- 12.Jancova P., Anzenbacher P., Anzenbacherova E. Phase II drug metabolizing enzymes. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2010;154:103–116. doi: 10.5507/bp.2010.017. [DOI] [PubMed] [Google Scholar]
- 13.Zakeri-Milani P., Valizadeh H. Intestinal transporters: enhanced absorption through P-glycoprotein–related drug interactions. Expert Opin Drug Metab Toxicol. 2014;10:859–871. doi: 10.1517/17425255.2014.905543. [DOI] [PubMed] [Google Scholar]
- 14.Hernandez J.P., Mota L.C., Baldwin W.S. Activation of CAR and PXR by dietary, environmental and occupational chemicals alters drug metabolism, intermediary metabolism, and cell proliferation. Curr Pharmacogenom Person Med. 2009;7:81–105. doi: 10.2174/187569209788654005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamamoto Y., Moore R., Goldsworthy T.L., Negishi M., Maronpot R.R. The orphan nuclear receptor constitutive active/androstane receptor is essential for liver tumor promotion by phenobarbital in mice. Cancer Res. 2004;64:7197–7200. doi: 10.1158/0008-5472.CAN-04-1459. [DOI] [PubMed] [Google Scholar]
- 16.Braeuning A., Gavrilov A., Brown S., Wolf C.R., Henderson C.J., Schwarz M. Phenobarbital-mediated tumor promotion in transgenic mice with humanized CAR and PXR. Toxicol Sci. 2014;140:259–270. doi: 10.1093/toxsci/kfu099. [DOI] [PubMed] [Google Scholar]
- 17.Gao J., He J., Zhai Y., Wada T., Xie W. The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity. J Biol Chem. 2009;284:25984–25992. doi: 10.1074/jbc.M109.016808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wei P., Zhang J., Egan-Hafley M., Liang S., Moore D.D. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature. 2000;407:920–923. doi: 10.1038/35038112. [DOI] [PubMed] [Google Scholar]
- 19.Petrick J.S., Klaassen C.D. Importance of hepatic induction of constitutive androstane receptor and other transcription factors that regulate xenobiotic metabolism and transport. Drug Metab Dispos. 2007;35:1806–1815. doi: 10.1124/dmd.107.015974. [DOI] [PubMed] [Google Scholar]
- 20.Wei P., Zhang J., Dowhan D.H., Han Y., Moore D.D. Specific and overlapping functions of the nuclear hormone receptors CAR and PXR in xenobiotic response. Pharmacogenom J. 2002;2:117–126. doi: 10.1038/sj.tpj.6500087. [DOI] [PubMed] [Google Scholar]
- 21.Bookout A.L., Jeong Y., Downes M., Yu R.T., Evans R.M., Mangelsdorf D.J. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126:789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shah P., Guo T., Moore D.D., Ghose R. Role of constitutive androstane receptor in Toll-like receptor-mediated regulation of gene expression of hepatic drug-metabolizing enzymes and transporters. Drug Metab Dispos. 2014;42:172–181. doi: 10.1124/dmd.113.053850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tojima H., Kakizaki S., Yamazaki Y., Takizawa D., Horiguchi N., Sato K. Ligand dependent hepatic gene expression profiles of nuclear receptors CAR and PXR. Toxicol Lett. 2012;212:288–297. doi: 10.1016/j.toxlet.2012.06.001. [DOI] [PubMed] [Google Scholar]
- 24.Aleksunes L.M., Klaassen C.D. Coordinated regulation of hepatic phase I and II drug-metabolizing genes and transporters using AhR-, CAR-, PXR-, PPARα-, and Nrf2-null mice. Drug Metab Dispos. 2012;40:1366–1379. doi: 10.1124/dmd.112.045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li C.Y., Renaud H.J., Klaassen C.D., Cui J.Y. Age-specific regulation of drug-processing genes in mouse liver by ligands of xenobiotic-sensing transcription factors. Drug Metab Dispos. 2016;44:1038–1049. doi: 10.1124/dmd.115.066639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Su A.I., Cooke M.P., Ching K.A., Hakak Y., Walker J.R., Wiltshire T. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci U S A. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Su A.I., Wiltshire T., Batalov S., Lapp H., Ching K.A., Block D. A gene atlas of the mouse and human protein-encoding transcriptomes. Proc Natl Acad Sci U S A. 2004;101:6062–6067. doi: 10.1073/pnas.0400782101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Renaud H.J., Cui J.Y., Khan M., Klaassen C.D. Tissue distribution and gender-divergent expression of 78 cytochrome P450 mRNAs in mice. Toxicol Sci. 2011;124:261–277. doi: 10.1093/toxsci/kfr240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maglich J.M., Stoltz C.M., Goodwin B., Hawkins-Brown D., Moore J.T., Kliewer S.A. Nuclear pregnane X receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol Pharmacol. 2002;62:638–646. doi: 10.1124/mol.62.3.638. [DOI] [PubMed] [Google Scholar]
- 30.Xu C., Wang X., Staudinger J.L. Regulation of tissue-specific carboxylesterase expression by pregnane X receptor and constitutive androstane receptor. Drug Metab Dispos. 2009;37:1539–1547. doi: 10.1124/dmd.109.026989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Buckley D.B., Klaassen C.D. Induction of mouse UDP-glucuronosyltransferase mRNA expression in liver and intestine by activators of aryl-hydrocarbon receptor, constitutive androstane receptor, pregnane X receptor, peroxisome proliferator-activated receptor α, and nuclear factor erythroid 2-related factor 2. Drug Metab Dispos. 2009;37:847–856. doi: 10.1124/dmd.108.024190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh S., Arcaroli J., Thompson D.C., Messersmith W., Vasiliou V. Acetaldehyde and retinaldehyde-metabolizing enzymes in colon and pancreatic cancers. Adv Exp Med Biol. 2015;815:281–294. doi: 10.1007/978-3-319-09614-8_16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Klusek J., Głuszek S., Klusek J. GST gene polymorphisms and the risk of colorectal cancer development. Contemp Oncol (Pozn) 2014;18:219–221. doi: 10.5114/wo.2014.41388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koh W.P., Nelson H.H., Yuan J.M., van den Berg D., Jin A.Z., Wang R.W. Glutathione S-transferase (GST) gene polymorphisms, cigarette smoking and colorectal cancer risk among Chinese in Singapore. Carcinogenesis. 2011;32:1507–1511. doi: 10.1093/carcin/bgr175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lamba J.K., Lamba V., Yasuda K., Lin Y.S., Assem M., Thompson E. Expression of constitutive androstane receptor splice variants in human tissues and their functional consequences. J Pharmacol Exp Ther. 2004;311:811–821. doi: 10.1124/jpet.104.069310. [DOI] [PubMed] [Google Scholar]
- 36.Huang W., Zhang J., Washington M., Liu J., Parant J.M., Lozano G. Xenobiotic stress induces hepatomegaly and liver tumors via the nuclear receptor constitutive androstane receptor. Mol Endocrinol. 2005;19:1646–1653. doi: 10.1210/me.2004-0520. [DOI] [PubMed] [Google Scholar]
- 37.Ross J., Plummer S.M., Rode A., Scheer N., Bower C.C., Vogel O. Human constitutive androstane receptor (CAR) and pregnane X receptor (PXR) support the hypertrophic but not the hyperplastic response to the murine nongenotoxic hepatocarcinogens phenobarbital and chlordane in vivo. Toxicol Sci. 2010;116:452–466. doi: 10.1093/toxsci/kfq118. [DOI] [PubMed] [Google Scholar]
- 38.Martin P., Riley R., Back D.J., Owen A. Comparison of the induction profile for drug disposition proteins by typical nuclear receptor activators in human hepatic and intestinal cells. Br J Pharmacol. 2008;153:805–819. doi: 10.1038/sj.bjp.0707601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Al-Salman F., Plant N. Non-coplanar polychlorinated biphenyls (PCBs) are direct agonists for the human pregnane-X receptor and constitutive androstane receptor, and activate target gene expression in a tissue-specific manner. Toxicol Appl Pharmacol. 2012;263:7–13. doi: 10.1016/j.taap.2012.05.016. [DOI] [PubMed] [Google Scholar]