

Supplemental Digital Content is available in the text.
Keywords: electrophysiology, heart failure, L-type calcium channel, membrane microdomains, myocardial infarction
Abstract
Rationale:
Disruption in subcellular targeting of Ca2+ signaling complexes secondary to changes in cardiac myocyte structure may contribute to the pathophysiology of a variety of cardiac diseases, including heart failure (HF) and certain arrhythmias.
Objective:
To explore microdomain-targeted remodeling of ventricular L-type Ca2+ channels (LTCCs) in HF.
Methods and Results:
Super-resolution scanning patch-clamp, confocal and fluorescence microscopy were used to explore the distribution of single LTCCs in different membrane microdomains of nonfailing and failing human and rat ventricular myocytes. Disruption of membrane structure in both species led to the redistribution of functional LTCCs from their canonical location in transversal tubules (T-tubules) to the non-native crest of the sarcolemma, where their open probability was dramatically increased (0.034±0.011 versus 0.154±0.027, P<0.001). High open probability was linked to enhance calcium–calmodulin kinase II–mediated phosphorylation in non-native microdomains and resulted in an elevated ICa,L window current, which contributed to the development of early afterdepolarizations. A novel model of LTCC function in HF was developed; after its validation with experimental data, the model was used to ascertain how HF-induced T-tubule loss led to altered LTCC function and early afterdepolarizations. The HF myocyte model was then implemented in a 3-dimensional left ventricle model, demonstrating that such early afterdepolarizations can propagate and initiate reentrant arrhythmias.
Conclusions:
Microdomain-targeted remodeling of LTCC properties is an important event in pathways that may contribute to ventricular arrhythmogenesis in the settings of HF-associated remodeling. This extends beyond the classical concept of electric remodeling in HF and adds a new dimension to cardiovascular disease.
Heart failure (HF) is a major cause of morbidity and mortality, contributing significantly to global health expenditure. Sudden death due to arrhythmia is responsible for >50% of deaths among patients with HF and therefore preventing arrhythmia and ameliorating the risk of sudden cardiac death secondary to HF is a paramount clinical need.1,2 Pathophysiological remodeling of cardiac function in HF occurs at multiple levels and includes the alterations in ion channel profile, Ca2+-handling proteins, and proteins mediating cell–cell coupling, predisposing to arrhythmias.3–7 Recent studies have demonstrated that disruption of proper cellular organization accompanied by a progressive loss of transverse tubule (T-tubule [TT]) microdomains in HF may also have an impact on calcium cycling, thus, promoting the development of arrhythmogenic triggers.8 Specifically, it has been hypothesized9,10 that the communication between L-type Ca2+ channels (LTCCs) and ryanodine receptors (RyRs) is impaired in HF, perhaps indicating that LTCCs are less strictly confined to TTs. In addition, LTCC protein phosphorylation defects have been identified in HF.3,11 Therefore, we hypothesized that there is altered distribution of single LTCCs in cellular microdomains in HF after TT degradation, which results in channel dysfunction critically contributing to the development of arrhythmogenic triggers.
Here, we used the super-resolution scanning patch-clamp technique12 to probe the microdomain-specific localization of functional LTCCs with nanospatial resolution in normal and failing ventricular cardiac myocytes. We showed, in failing cells, dislocation of functional LTCCs to the sarcolemma surface (or crest membrane, a term based on micrograph topography in contrast to TT), where they are rarely present in healthy cardiac myocytes. Obtaining evidence from multiple imaging modalities, electrophysiology and biochemistry, we discovered that these relocated channels exhibit higher open probability (Po) and phosphorylation status, which we found to be linked to enhanced activity of calcium–calmodulin kinase II (CaMKII). The experiments were complemented by the development and use of a novel accurate HF computational model, which also includes single-channel behavior to ascertain that LTCC relocation to the cell crest combined with enhanced CaMKII activity gives rise to the measured Po values. The model then demonstrated how this abnormal behavior leads to cell-level oscillations in membrane voltage and development of arrhythmogenic triggers, and how these propagate to become arrhythmias at the organ level. The combined experimental/simulation approach presented here provides a comprehensive understanding of how disease-induced remodeling at the microdomain level is manifested into dysfunction at the organ level.
Methods
For full details of Methods section, please see the Online Data Supplement.
Study Approval
All animal experiments were carried out in accordance with the United Kingdom Home Office Animals (Scientific Procedures) Act 1986 Amendment Regulations 2012, incorporating the EU Directive 2010/63/EU, which conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH publication No. 85-23, revised 1996). Experiments on isolated human cardiac myocytes were approved by the Imperial College Institutional Review Board.
Myocytes Isolation and TT Characterization
Failing ventricular myocytes were isolated from a 16 weeks post myocardial infarction rat model of HF (Online Figure I) and transplanted human hearts from patients with dilated cardiomyopathy (Online Table I). Age-matched sham-operated rats and human tissue biopsy samples (Online Table II) were used to isolate control, nonfailing cardiac myocytes. The subcellular TT system was visualized by confocal imaging of Di-8-ANEPPS–stained cells.6 Surface topography was characterized by scanning ion conductance microscopy, which uses a glass nanopipette as sensitive probe as described elsewhere.13
Super-Resolution Scanning Patch-Clamp With Pipette Clipping Modification
After generating a topographical image of the cell surface by scanning ion conductance microscopy, the tip diameter of the pipette was widened by clipping13 to increase the area of attachment. The pipette was then lowered to a specific location until it touched the membrane and a high resistance seal was established. Single LTCC recordings were then performed in a cell-attached mode.12 Controlled widening of the scanning nanopipette tip is described in detail in the Online Data Supplement (Online Figure II and Online Figure III).
Optical Mapping of Calcium Activity
Optical mapping of cells loaded with the Ca2+-sensitive fluorescent dye Fluo-4AM via CMOS camera ULTIMA-L (SciMedia, USA Ltd, CA; 1000 fps, 1.5–2 μm/pixel) was used to monitor localized changes in [Ca2+]i.14
Western Blot
Western blotting was done using monoclonal anti-phospho-CaMKII (Thermo Scientific, MA1-047) α-tubulin (Sigma, T9026), and GAPDH (Santa Cruz Biotechnology, FL-335) primary antibodies, followed by secondary anti-mouse antibodies Amersham ECL detection (GE Healthcare). Western blots were analyzed by using ImageJ software.
Statistical Analysis
All graphs and statistical analysis were performed using either GraphPad prism 5 or Origin version 6.1. Normality was tested using the Kolmogorov–Smirnov test. In cases where data failed the normality test, the nonparametric Mann–Whitney test was used instead of the unpaired Student t test. Statistical differences were assessed with Student t test, Mann–Whitney test, Kruskal–Wallis test, and Fisher exact test as appropriate. All data are expressed as mean±SEM. A value of P<0.05 was considered statistically significant.
Computational Simulations
Single-Channel Human L-Type Ca2+ Current (ICa,L)
Human ventricular cell electrophysiological behavior was represented by the O’Hara-Rudy model.15 To model stochastic single-channel behavior and determine the channel Po for comparison with human experimental data, for ICa,L, we used its Markov-equivalent representation (Online Figure IV). To evolve the channel gating in response to a 1-s voltage change step from resting state to −6.7 mV, we used the Gillespie Exact Algorithm. Barium simulations matched barium experiments; with this as validation, we extrapolated to physiological calcium simulations. Example single-channel sweeps are shown in Online Figure V.
Once single-channel current simulation results were generated and validated with experimental Po measurements, the ICa,L model was reverted back to a Hodgkin–Huxley formulation, retaining the CaMKII mode definitions and the behavior of the equivalent Markov version. Failing crest LTCCs were assumed to operate in CaMKII-phosphorylated mode, based on experimental findings (see Results section of this article); their inactivation was via the slow-gating mode. Failing LTCCs in TTs and control LTCCs were sensitive to standard CaMKII, and so inactivation was both fast and slow. Ensemble current computed by summation of single-channel sweeps matched the deterministic Hodgkin–Huxley current (Online Figure VI).
Models of Human Control and Failing Myocytes
The original O’Hara-Rudy myocyte model did not include a subsarcolemmal volume and, thus, did not allow for Ca2+ accumulation near the intracellular mouth of Crest LTCCs. We, thus, incorporated subsarcolemmal volume and related fluxes in the O’Hara-Rudy myocyte model based on the work of Grandi et al16 and Shannon et al.17 Description of how this was done can be found in the Online Data Supplement.
The original O’Hara-Rudy model included LTCCs exclusively at TT sites with dyadic intracellular face. Here, LTCCs in TTs sensed and contributed to dyadic Ca2+; channels newly added to the crest sensed and contributed to subsarcolemmal Ca2+. In each of TT and crest locations, LTCC permeability, PCa, representing whole-cell current density in TTs (PCaTT) and crest (PCaCrest) needed to be defined. We utilized experimental data obtained in this study to assign values to PCaTT and PCaCrest in control and failing human cells. The data used were % occurrence of LTCCs, LTCC single-channel current amplitude, and in failing cells, the degree of TT loss. Specific values for PCaTT and PCaCrest in control and failing human cells and the methodology by which they were assigned can be found in the Online Data Supplement.
Outside of ICa,L several factors were included to recreate an accurate HF model. Descriptions of the formulations for Na+/Ca2+ exchanger, representation of orphaned RyRs, and additional parameters representing others HF ion channel remodeling in the myocyte model can be also be found in the Online Data Supplement.
Whole Heart Model
Organ-level simulations were performed using an MRI-based, anatomically realistic human left ventricular model as described previously.18 Fiber orientation was assigned using a rule-based approach.19 A computational mesh at a resolution of <300 μm was generated using a validated approach.20 Transmural cell types and conductivity were specified according to experimental data from human left ventricles7 (Online Figure VII). All parameters defining differences between the endocardial and epicardial cell variations of the O’Hara-Rudy model were scaled linearly across the ventricular walls to generate smoothly varying intermediate transmural types (10 segments defined using a Laplace-Dirichlet technique).19 Pacing was delivered to the apex using a 2-cm diameter electrode. Electrophysiological simulation and numeric methods were identical to previous work.18 Simulations were executed in monodomain mode using the CARP software package.21,22
Results
HF is associated with the loss of TTs with consequent alterations in LTCC spatial distribution and their functional properties.
In both rat and human failing myocytes, we observed a significant decrease in internal TT density when compared with nonfailing controls (Figure 1A and 1B; ≈50% decrease in human, P<0.01), consistent with our previous findings.6 Using scanning ion conductance microscopy,23 we found that the loss of TT correlates with the loss of surface structures as well (Figure 1C). Using Z-groove index as metric of surface integrity,24 we found that in both human and rat failing myocytes, surface structure was impaired (Figure 1D, ≈40% reduction in human, P<0.01). We also detected reduction in the number of TT openings on the surface of failing cardiac myocytes, identified as dark circles in scanning ion conductance microscopy images (≈35% reduction in rat compared with control, P<0.001; Online Figure VIII).
Figure 1.
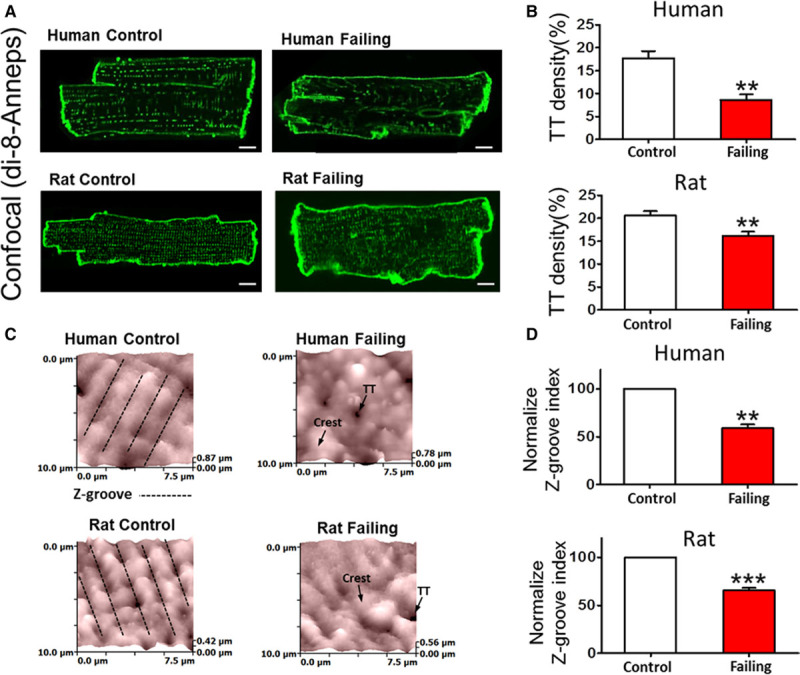
T-tubule (TT) loss in ventricular myocytes from failing human and rat hearts. A, Example confocal images of human control and failing (top) and rat control and failing (bottom) cardiac myocytes showing membranes stained with Di-8-ANEPPS; scale bar, 10 μm. B, TT density in control and failing cells (human control n=20, failing n=8; rat control n=20, failing n=20; **P<0.01 by unpaired Student t test). C, Example scanning ion conductance microscopy scans from a 10×10 μm portion of cell membrane shows regular undulations, indicating spatially alternating TT invaginations and surface membrane crests in human (top left) and rat (bottom left) control cardiac myocytes that are relatively absent in failing cells (right). D, Z-groove index in human and rat failing cells normalized to control average value (human control n=30, failing n=59; top, P<0.01; rat control n=91, failing n= 122; bottom, P<0.001 by unpaired Student t test).
The loss of TT microdomains in failing myocytes was accompanied by altered spatial distribution of LTCCs. In control rat and human cardiac myocytes, LTCC activity was predominantly recorded in TTs (26.7% of 86 successful patches in rat and 28.6% of 21 successful patches in human cardiac myocytes showed LTCC activity) as opposed to the crest, where LTCC activity was rarely recorded (only 7.02% of 57 successful patches in rat and 9.1% of 11 successful patches in human cells showed LTCC activity, Figure 2A and 2B, control). This confirmed our previous observation that the majority of functional LTCCs reside in the TTs.12 Interestingly, LTCC occurrence along the Z-groove in rat cardiac myocytes was found to be intermediate between that in TT and crest areas (15% of 20 successful patches), suggesting a density gradient of channels throughout the membrane.
Figure 2.
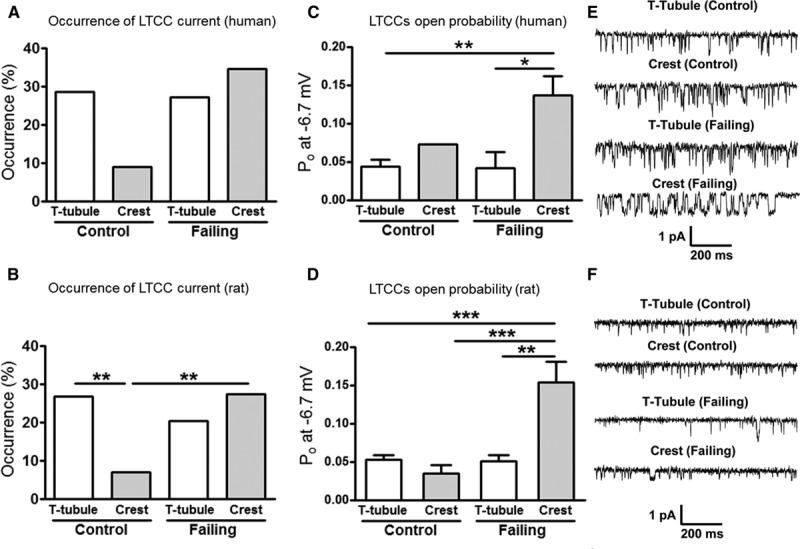
Abnormal L-type Ca2+ channel (LTCC) localization and function in failing cardiac myocytes. Chance of obtaining an LTCC current (% occurrence) in human (A) and rat (B) control and failing cells (**P<0.01 by Fisher exact test). Open probability (Po) during a maximum activating voltage step (to −6.7 mV) in human (C) and rat (D) failing cells (human control, T-tubule [TT] n=6, crest n=1; failing, TT n=6, crest n=9, * P<0.05, ** P<0.01 by Mann–Whitney test; rat control, TT n=19, crest n=4; failing, TT n=11, crest n=13, ** P<0.01, ***P<0.001 by Mann–Whitney test). Representative single-channel traces at −6.7 mV showing LTCC activity in human (E) and rat (F) cardiac myocytes.
In contrast, in failing rat cardiac myocytes, LTCC current was recorded with similar frequency from both TTs and crests (20.4% of 54 successful patches in the TTs versus 27.3% of 55 successful patches in the crest showed LTCC activity). In failing human cardiac myocytes, LTCC activity was even higher in the crest (27.3% of 22 successful patches in the TTs versus 34.6% of 26 successful patches in the crest showed LTCC activity). This indicates that the distribution of functional LTCCs in the cardiac myocyte membrane was significantly altered in HF (Figure 2A and 2B, failing).
Our experiments demonstrated that the HF-associated increase in the number of functional LTCCs outside of their native microdomains is accompanied by the changes in their behavior. The LTCC Po was significantly elevated at the crest of failing myocytes when compared with that in crest of control myocytes in rat (Po at −6.7 mV: 0.034±0.011 for control crest LTCCs versus 0.153±0.026 for failing crest LTCCs, P<0.001; Figure 2D) and to that in TTs (0.053±0.005 for control TT LTCCs, P<0.001; 0.051±0.008 for failing TT LTCCs, P<0.01; Figure 2D and 2F). No changes in LTCC amplitude were observed in rat HF (Online Figure IX). In failing humans myocytes, the Po was also significantly elevated at the crest when compared with that in TT of control and failing myocytes (Po at −6.7 mV: 0.136±0.025 for failing crest LTCCs versus 0.043±0.01 for control TT LTCCs, P<0.01 and versus 0.042±0.021 for failing TT LTCCs, P<0.05; Figure 2C and 2E). We next endeavored to determine the mechanisms for LTCC functional changes in HF.
Constitutive Phosphorylation of LTCCs by Cytoplasmic CaMKII Leads to an Increase in LTCC Po
Elevated phosphorylation can lead to increased Po of LTCCs.25 Also, CaMKII activity is elevated in HF,26 and CaMKII can phosphorylate LTCCs at specific sites.27 Therefore, we tested the hypothesis that the elevated activity of CaMKII in failing cardiac myocytes is responsible for phosphorylation and, thus, for the high Po of LTCCs in crest.
Western blots indicated that the phosphorylated CaMKII T286 was higher in rat and human failing cells (Figure 3A and 3B); this p-CaMKII could be reduced after the application of the CaMKII inhibitor KN-93 (Figure 3C; only rat myocytes were used in these experiments because of the paucity of human cells). Because this residue is critical for the association and phosphorylation of the LTCC β2a subunit,28 these data indicate that LTCC phosphorylation should be increased. Besides, as we have shown in Figure 3D and Online Figure X, local Po measurements demonstrate that LTCC Po in the crest of failing myocytes was reduced to a value similar to control after the application of KN-93 (from 0.153±0.026 for failing crest LTCCs to 0.061±0.018 for failing crest LTCCs with KN-93, P<0.01). LTCCs from control cells or from TT failing cells were not affected by the inhibitor (Figure 3D), suggesting that the increase of CaMKII in failing cells had only effect on the failing myocyte crest, confirming our hypothesis. It has been shown that KN-93 can block LTCC directly under certain conditions29; to strengthen our conclusion, autocamptide-2–related inhibitor peptide was used on failing crest channels (Figure 3B) and the results showed the reduction of Po to control values (0.031±0.003, P<0.05 versus failing crest values), confirming our initial findings.
Figure 3.
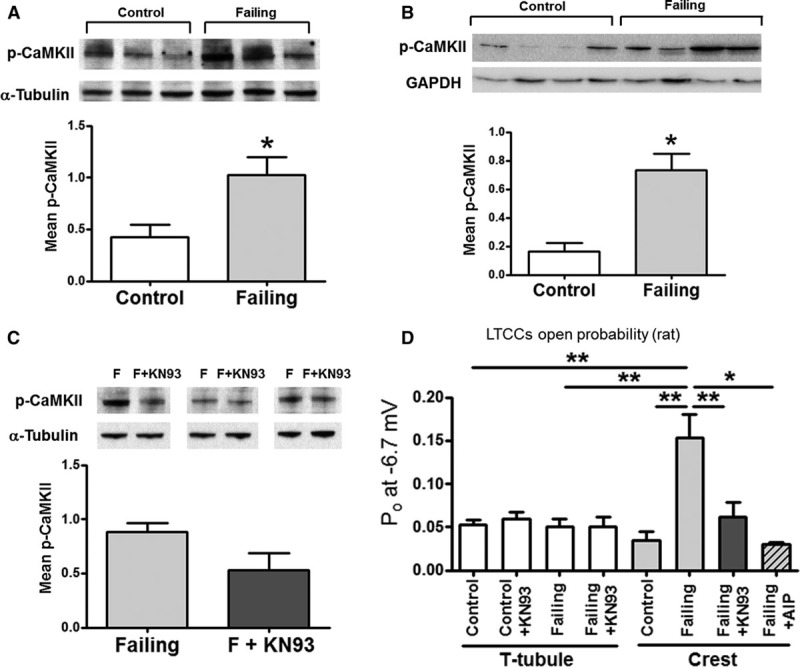
L-type Ca2+ channels (LTCCs) at crests in rat failing myocytes are hyperphosphorylated by Ca2+ calmodulin-dependent protein kinase II (CaMKII). A, Mean phosphorylated CaMKII (Thr286) from whole-cell lysate in control and failing cardiac myocytes. A representative blot is shown above the graph. Results are normalized to α-tubulin (B) Mean p-CaMKII from whole tissue from control and failing human samples. A representative blot is shown above the graph. Results are normalized to GAPDH. C, Mean p-CaMKII from whole-cell lysate in failing cardiac myocytes with or without CaMKII inhibition. A representative blot is shown above the graph. Results are normalized to α-tubulin. D, LTCC open probability (Po) in control and failing rat myocytes, under control conditions and after CaMKII inhibition with 5-μmol/L KN-93 or with 5-μmol/L autocamptide-2 related inhibitor peptide (AIP; control T-tubule [TT] n=19; control TT+KN-93 n=5; failing TT n=11; failing TT+KN-93 n=4; control crest n=4; failing crest n=13; failing crest+KN-93 n=9; failing crest+AIP n=3. * P<0.05, **P<0.01 by Kruskal–Wallis test).
Abnormal [Ca2+]i Behavior in Failing Cells
Because HF remodeling altered LTCC function in failing myocytes, we investigated whether there were concomitant changes in Ca2+ transients. Optical mapping of Ca2+ transients at pacing rates of 0.5 to 1 Hz revealed spontaneous [Ca2+]i oscillations, which occurred during the decay phase in a greater proportion of failing cardiac myocytes compared with control (0.5 Hz, ≈45% failing versus ≈4% control P<0.05; Figure 4A). These Ca2+ oscillations were completely suppressed after treatment with KN-93 (Figure 4B) but not by KN-92 (Online Figure XI), an inactive analog of KN-93. This suggests that the phosphorylation inhibition by KN-93 reduces the late LTCC current resulting from the increased LTCC Po at the cell crest and prevents [Ca2+]i oscillations.
Figure 4.
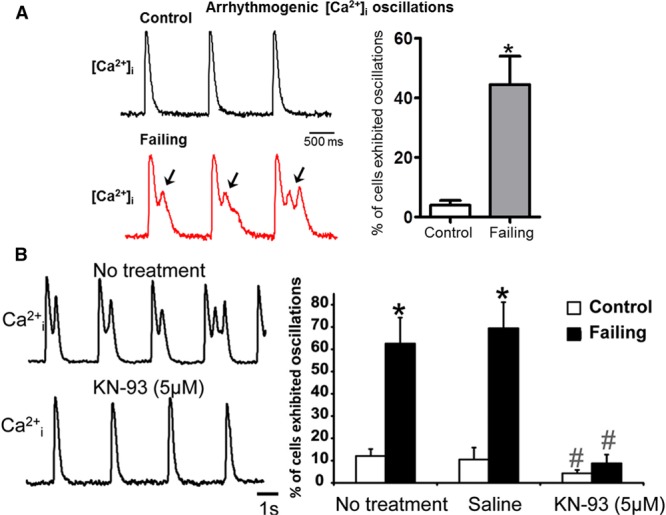
Ca2+ oscillations that develop in failing but not in control rat cardiac myocytes are Ca2+ calmodulin-dependent protein kinase II dependent. A, Left, Spontaneous [Ca2+]i oscillation occurred at a pacing rate of 0.5 Hz during the decay phase of calcium transients in failing rat cardiac myocytes only. Right, Percentage of cells exhibiting Ca2+ oscillations (control n=106, failing=73, * P<0.05 by Mann–Whitney test). B, Left, Example of Ca2+ transients recorded from the same failing cardiac myocytes before and after treatment with KN-93. Right, Number of cells (in %) exhibiting Ca2+ oscillations (n=2 rats, 32–114 cardiac myocytes in each group; * P<0.05 for failing vs control; #P<0.05 for KN93 effect within the same group by Mann–Whitney test). Saline mimics no treatment.
In rat failing cardiac myocytes, whole-cell patch-clamp recordings (Online Figure XII) show prolonged action potential (AP) duration similar to what had been previously described in HF.30,31 It has been shown that such AP duration prolongation is accompanied by spontaneous occurrence of single and multiple early afterdepolarizations (EADs) in failing ventricular myocytes.31 Oscillations in the Ca2+ transient such as shown in Figure 4, occurring in the settings of HF-associated ion channel remodeling, are also widely associated with EADs, a class of possible arrhythmogenic triggers in the heart.32 However, linking Ca2+ transient oscillations to the development of HF-related arrhythmias at the organ level is a challenging task, both in terms of demonstrating by experimentation, across the spatial scales of structural hierarchy, and also causally, as electrotonic influences at the tissue/organ level could suppress cell-level triggers.33 To prove that HF-induced changes in microdomain localization of LTCCs and their functional consequences at the cellular level could result in arrhythmia in the failing human heart, we developed and utilized a novel computational model of human HF that incorporated the experimental findings described above.
Computational Model Based on Single-Channel Kinetics Predicts the Development of Cellular-Level Triggers of Arrhythmias in Human HF
The human HF model was based on the Monte Carlo simulations of the kinetics of a single human LTCC (Online Data Supplement). Incorporating the experimental finding that in failing cardiac myocytes LTCCs at crests are CaMKII-phosphorylated, we calculated Po values in TTs and crests in control and failing human myocytes (Figure 5A, example sweeps in Online Figure V). Simulations were able to relate LTCC phosphorylation at crests to the elevated local Po value. The close match between simulation and experimental results served as a validation of the model, allowing us to use simulations to explore the downstream effect of LTCC functional changes.
Figure 5.
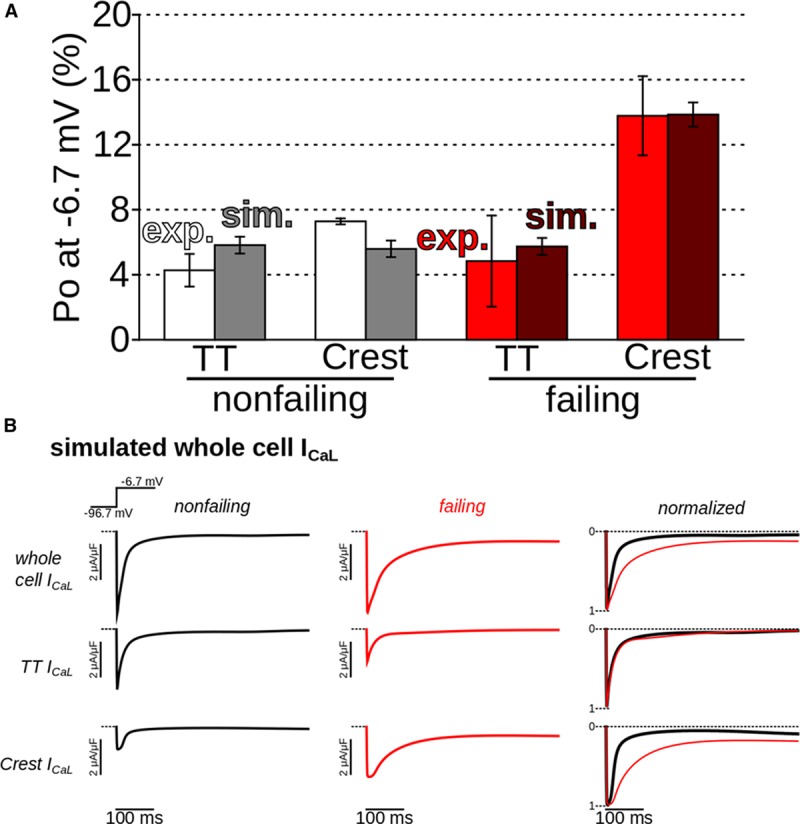
Simulations demonstrated increased open probability (Po) of L-type Ca2+ channels (LTCCs) at failing crest sites, as in experiments, and slowed inactivation. A, Comparison between simulated and experimental Po values for 1-s voltage steps to −6.7 mV (average of 500 stochastic sweeps in simulations). B, Simulated whole-cell T-tubule (TT) and crest LTCC current (ICa,L) in control (black) and failing (red) cells. Traces in right column were normalized to allow visual comparison of decay rates.
Simulated human whole-cell Ca2+current is of similar magnitude in failing and control cells, consistent with previous findings34–37 and our results on failing rat cardiac myocytes (Online Figure XIII). Current decay is slower in failing myocytes compared with control (Figure 5B), suggesting a potential for destabilization of repolarization. Slower decay is because of enhanced CaMKII phosphorylation of LTCC channels in the crest. Traces corresponding to TTs were nearly identical for failing and control cells with regard to decay rate; however, TTs in failing myocytes were depopulated and thus current magnitude in TT was reduced relative to control.
The LTCC kinetics model was incorporated, together with other HF electrophysiological alterations, in an AP model of the human (endocardial) ventricular myocyte to determine the cell-level consequences of LTCC dysfunction. AP simulation results showed that EADs developed because of late L-type Ca2+ current appearance in failing myocytes only (Figure 6). Here, CaMKII was set to be maximal in the failing crest (validated by experimental data in Figure 5A), causing phosphorylation of all LTCCs there. CaMKII block eliminated L-type Ca2+ current appearance and EADs. These simulation results were supported by optical [Ca2+] recordings (Figure 4; Ca2+ transient oscillations in failing human cells were simulated at a pacing rate of 0.25 Hz, which could be considered similar to 0.5 Hz pacing in the rat15).
Figure 6.
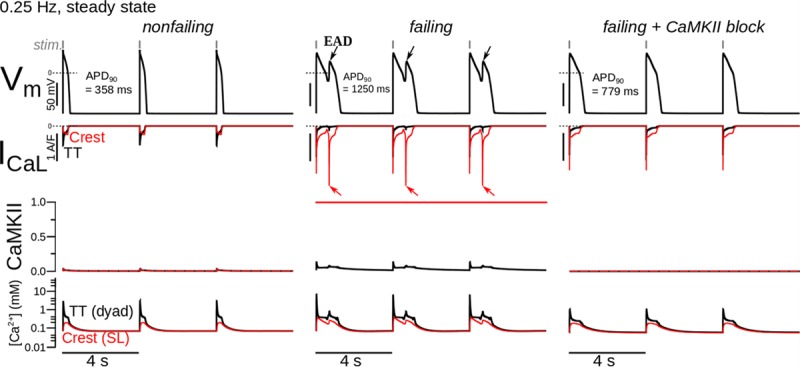
Simulations demonstrating the development of proarrhythmic triggers (early afterdepolarizations [EADs]) in failing human ventricular (endocardial) myocytes. Rows from top to bottom show simulated action potentials, T-tubule (TT, black) and crest (red) ICa,L, TT and crest Ca2+ calmodulin-dependent protein kinase II (CaMKII) activity (0.0–none, no L-type Ca2+ channels [LTCCs] phosphorylated; 1.0–maximal, all LTCCs phosphorylated), and TT and crest Ca2+ concentrations (ie, concentrations in the dyadic and subsarcolemmal volumes). Columns from left to right show cases of control, failing, and failing cells with CaMKII block.
Although EADs in isolated cells are potential ectopic triggers, their formation does not guarantee the occurrence of arrhythmogenic triggers or arrhythmias in the whole heart. We, therefore, conducted organ-level simulations to test whether EADs resulting from HF-induced LTCC relocalization could form propagating triggers and result in arrhythmia in the heart using more physiological human heart rates, such as 1 Hz.
Computational Model Predicts the Development of Arrhythmias in the Failing Human Ventricles
Simulation results in Online Figure XIV demonstrated that epicardial cells did not develop EADs for any of the simulated conditions, indicating that arrhythmia triggers in the failing heart were expected to develop in the endocardial layer.
The formation of arrhythmogenic triggers and reentrant arrhythmia in the failing human heart is shown in Figure 7 (1 Hz pacing with a single-skipped beat; arrhythmia did not take place without a skipped beat at this pacing rate) and in the Online Movie I. AP traces at 3 different endocardial locations are shown in Figure 7A; site “i” was the closest to the pacing location at the apex. In the control ventricles (Online Movie II), wavefronts propagated in an organized fashion in response to each pacing stimulus (short horizontal gray lines relate stimuli, one-to-one, to resulting APs at sites throughout the ventricles). In the failing ventricles, EAD triggers appeared near site “ii” and propagated (dashed gray lines from EAD triggers toward triggered APs). Activation in the control ventricles was completed within 250 ms, undisturbed by the skipped beat (Figure 7B1 and 7B2). In contrast, activation took twice as long to excite the failing ventricles. After the skipped beat pause, cells near site “ii” failed to repolarize, and an endocardial EAD trigger formed (Figure 7B1 and 7B2). As shown in Figure 7C, triggered activity propagated from that location, resulting in reentrant arrhythmia. The skipped beat pause had no discernable effect on subsequent activity in the control model (results not shown). Simulations also showed that triggered activity and arrhythmias did not develop in the failing ventricles when CaMKII was blocked or when crest LTCCs were made to sense dyadic [Ca2+], where Ca-dependent inactivation was enhanced. The whole-heart simulations, thus, demonstrated a causal link between HF-induced microdomain localization of LTCCs and arrhythmias in the failing human heart.
Figure 7.
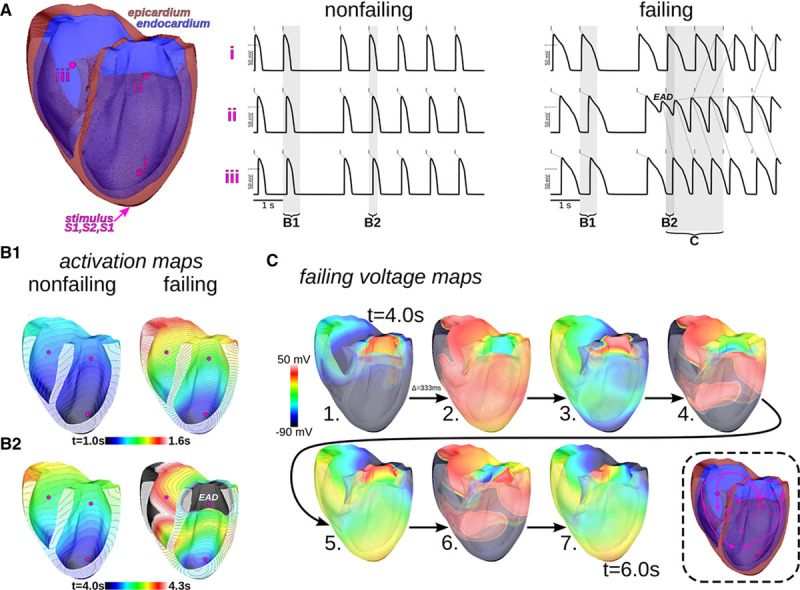
Whole-heart simulations predict the formation of arrhythmogenic triggers and reentrant arrhythmia in human heart failure. 1 Hz pacing with a single-skipped beat was from the apex. A, Action potential traces at endocardial sites “i–iii” for control and failing hearts. Small dashed gray lines relate stimuli (solid gray tick marks) to result action potentials at the sites. In the failing heart, early afterdepolarization (EAD) triggers (red, arrows) appeared near site ii and propagated outwardly to other sites (gray lines emanating from EADs and triggered action potentials). Vertical semitransparent bands denote time windows over which activation and voltage maps are shown in remaining panels. B1 and B2, Activation maps, showing the time at which membrane voltage first crossed activation threshold, in the control and failing ventricles during the respective time windows B1 and B2 shown in A. C, Progression of activation recovery during the time window C shown in A (from 4.0 to 6.0 s, time points separated by 1/3 s). An island of EAD generating tissue can be seen near site iii in snapshot 1. Reentry developed after the propagation of triggered activity emanating from the EAD site (path summarized in the cartoon at bottom right; arrows show general direction of propagation and should not be taken as belonging to specific wavefronts).
Discussion
This study adds a new dimension to the understanding of cardiovascular disease, highlighting microdomain-specific changes in LTCC function, which acts in concert with well-established changes in protein expression. The major discovery of this study is that a disruption in the delicately balanced dynamic interactions between LTCCs and their cellular microenvironment can lead to pathological changes in cellular physiology and to a downstream dysfunction at the organ level. This novel concept may help to explain the molecular mechanisms of HF and other human diseases.
Relocalization of LTCCs in HF
Here, for the first time, we provide direct evidence of the presence, in HF, of abnormally functioning LTCCs in the extradyadic space (crests) of ventricular cardiac myocytes, concurrent with changes in microdomain structure. These extradyadic LTCCs may lose the communication with the RyRs, as previous work has shown that RyR regularity and distribution do not change during HF.38 Only LTCCs localized in the crest had abnormally high Po, which contributed to the pathophysiology of HF suggesting that nanoscale changes in the location of proteins can be detrimental to their function. In fact, it has been proposed that the long open states of the LTCCs are particularly proarrhythmic in the setting of AP prolongation,39 suggesting that the channels that we found in the crest of failing cells can be a source of arrhythmias.
Several studies in animal models of HF40,41 have demonstrated a reduction in whole-cell Ca2+ current and in the average LTCC density, which seems to be a consequence of the profound loss of TTs, but other studies did not find changes in whole-cell Ca2+.42 Interestingly, Bryant et al38 showed a decreased ICa density in the TT and increased ICa density on the cell surface in rat HF ventricular myocytes, which supports our findings at the single-channel level. In human failing cardiac myocytes, no significant changes in LTCC density have been observed34,36,37; however, an impaired Cav1.2 trafficking to the TT cell membrane has been suggested.43 Taking into account the increased Po of LTCC observed in our study (Figure 3) and in previous reports,25 one would expect a reduction of the number of functional channels,5 although we cannot exclude that a more complex mechanism could be involved. Impaired communication between LTCCs and RyRs, together with an increase in LTCC Po, may slow down the inactivation time, as demonstrated experimentally.3
It remains an open question what is the precise location of functional LTCC on the crests of sarcolemma. Are they sparsely localized on the cell surface or do they organize in special membrane microdomains forming complexes with others proteins? It is plausible that these channels are located in caveolae domains on the plasma membrane of the crest, as we recently demonstrated for extratubular LTCCs in atrial myocytes.44 Through the function of caveolae-based LTCCs is unknown, Makarewich et al45 recently demonstrated that Ca2+ influx through LTCCs within caveolae signaling domains can activate pathological cardiac hypertrophic signaling, and this Ca2+ influx can be selectively blocked without reducing cardiac contractility. Whether these channels are associated with the hyperphosphorylated LTCCs observed in our study and the extent to which they may contribute to EADs remain an open question.
Microdomain-Dependent Changes in CaMKII Signaling
For the first time, our study reports microdomain-dependent changes in CaMKII-mediated Ca2+ signaling in HF (Figures 3 and 4). CaMKII, a well-known modulator of LTCCs, is upregulated under pathological conditions,26,46 resulting in increased LTCC Po.42 We extended these finding to show that CaMKII-dependent phosphorylation of LTCCs is increased specifically in crest microdomains (Figures 3 and 4) without affecting TT domains. CaMKII also contributed to the occurrence of abnormal calcium oscillations and lethal arrhythmias (Figures 4, 6, and 7). CaMKII is a therapeutic target, and CaMKII inhibition provides cardioprotection.26,47 Using experiments and simulations, we elucidated an additional mechanism for the success of CaMKII inhibition in ameliorating HF, namely the inhibition of phosphorylation of dislocated LTCCs.48 It has been shown that in HF, the ICa,L current peak density is not changed, whereas LTCCs density is decreased, suggesting an increase in the activity of the channels.42 We propose that the dislocated LTCCs found on the crest can be modulated by CaMKII, which could represent a new mechanism explaining this discrepancy.
CaMKII-mediated phosphorylation is an essential signaling event in triggering Ca2+/CaM-dependent LTCC facilitation, which requires the presence of LTCC β-subunits that can also be directly phosphorylated by CaMKII.49 β-subunits are upregulated in human failing myocardium and their overexpression correlates with an increase in the Po of LTCCs.50 Thus, we suggest that in failing myocytes, where the loss of TT structure is associated with dislocation of LTCCs to the crests, increase in CaMKII activity would phosphorylate and trigger facilitation of the dislocated LTCCs via 2 factors: the weak CDI in this microdomain and direct phosphorylation of LTCC β-subunits, leading to an abnormal LTCC activity.
Linking Subcellular Changes to Arrhythmia Propensity in HF
Using a new modeling approach spanning from stochastic LTCC gating to arrhythmogenesis at the organ level, we were able to understand how the subcellular changes are able to influence the development of arrhythmias. We found that the increase in LTCC Po, although CaMKII hyperactivity is specific to the crest microdomain (Figure 2), and this produces an increase of the slow inactivation of the ICa,L in failing cells (Figure 5B), as described previously in a stochastic model.51 Recently, Morotti et al52 using a mathematical model of rabbit AP linked a decreased CDI to an increase of the slow inactivation of the ICa,L and the occurrence of EADs, which was also observed in this study.
We demonstrated that in the failing ventricle, microdomain, cell- and tissue-level abnormalities act in synergy to produce whole-organ arrhythmia. It is important to note that HF is a systemic disorder in which it affects all of these hierarchical biological levels. For accurate representation of a human HF remodeling process, the membrane model of HF presented in this work used the descriptions provided in the study by Elshrif et al53 to define the rest of HF ion channel remodeling, outside of ICa,L. Our study confirms that CaMKII is an important node in this network of changes in the link between HF and arrhythmias. It is involved in disease pathways although the phosphorylation of multiple key proteins, modulating ion channel functioning and affecting gene transcription, metabolism and cell survival.54,55 It has also been linked to the HF-associated upregulation of the late sodium current, an important contributor to EAD development.56,57 Although many ionic currents can contribute to EAD formation in the settings of HF-associated remodeling, ICa,L late appearance plays a central role in providing a regenerative inward current required for EADs to propagate,58,59 thereby causing triggered activity in multicellular tissue.60 It has been recently shown that reducing the amplitude of the noninactivating pedestal component of ICa,L (ie, late or window ICa,L) effectively suppressed both H2O2− and hypokalemia-induced EADs.61 In nonfailing cardiac myocytes, in the settings of potassium current blockade, ICa,L was shown to be the main contributor to EADs formation.62 Conversely in HF, enhanced late sodium current may also have an important role.57,63 It, thus, may indicate their additive role in the enhancement of these net inward currents during the plateau phase of the AP, contributing to EAD development.
Clinical Implications
Conventional calcium channel blockers are generally felt to be contraindicated in HF because of their negatively inotropic effects via inhibiting both peak and window ICa,L.59 However, such adverse effects may relate to nonspecific targeting of LTCCs. The findings from this study can, therefore, facilitate the development of targeted and effective molecular therapies for preventing sudden cardiac death, without harmful side effects, and to steer the development of new and improved approaches to arrhythmia risk stratification of patients with HF. In the future, our assay could be used as a development platform for improved therapeutic approaches in combating HF based on the subcellular distribution of their targets. For example, selective block of the non-native LTCC pathway—directly or through subsequent regulatory proteins, including CaMKII—by novel reagents might provide an effective strategy for predicting and ameliorating the risk of sudden cardiac death in patients with cardiac disease.
Limitation
Failing cardiac myocytes were isolated from human tissue provided with the support of NIHR Cardiovascular BRU at Royal Brompton and Harefield from explanted failing hearts only. Control cardiac myocytes were isolated from human tissue provided with the support of Hammersmith Hospital from patients who underwent mitral valve replacement surgery, with normal ejection fraction values. This indeed may introduce a difference between samples because of the regional heterogeneity within the ventricles; however, the differences in human cells between control and failing cases are consistent with the results on rat cells, which are isolated from the same region of the posterior lateral left ventricle free wall. Control human tissue for protein studies was obtained from the left ventricle free wall with the support of University of Sydney.
Acknowledgments
We thank Dr Steven Houser for helpful discussion and critical comments on the article. We thank Peter O’Gara for cardiac myocyte isolation and Karina Zimmermann for Western blots. We are grateful to Prof Cristobal dos Remedios (University Sydney) and Prof Steve Marston for human heart muscle samples for Western blots; they were sourced through and with approval from the Australian Red Cross Blood Service and are covered by Human Research Ethics Approval from the University of Sydney (#2012/2814). Human heart failure tissue provided with the support of NIHR Cardiovascular BRU at Royal Brompton and Harefield.
Sources of Funding
This work was supported by Wellcome Trust (J. Gorelik-WT090594; M.B. Sikkel-WT092852), British Heart Foundation (J. Gorelik-12/18/30088, A.R. Lyon-FS/11/67/28954), MRC grant (J. Gorelik-MR/L006855/1), Imperial College London Rector Award (J. Gorelik), National Institutes of Health (NIH) grants (N.A. Trayanova-R01-HL103428, R01-HL105216 and N.A. Trayanova and J. Gorelik-RO1-HL126802), NIH Director’s Pioneer Award DP1 HL123271 (N.A. Trayanova).
Disclosures
None.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- HF
- heart failure
- LTCC
- L-type Ca2+ channel
- RyRs
- ryanodine receptors
- CaMKII
- calcium–calmodulin kinase II
- TT
- T-tubule
- EADs
- early afterdepolarizations
- AP
- action potential
- Po
- open probability
In July 2016, the average time from submission to first decision for all original research papers submitted to Circulation Research was 13.27 days.
These authors contributed equally to this article.
The online-only Data Supplement is available with this article at http://circres.ahajournals.org/lookup/suppl/doi:10.1161/CIRCRESAHA.116.308698/-/DC1.
Novelty and Significance
What Is Known?
Heart failure (HF) alters cardiac function and the functionality of ion channels.
Cells from HF models show a progressive loss of membrane structures and organization.
L-Type calcium channels (LTCCs) are considered an important component of alterations seen in HF cells.
What New Information Does This Article Contribute?
Loss of structure in HF cardiac myocytes leads to the redistribution of LTCCs.
Relocated LTCCs have different characteristics and may be a source of arrhythmias.
Translational modeling studies suggest that these specific LTCCs could be a potential drug target.
HF is a complex and multifactorial disease. At the single-cell level, HF causes hypertrophy, disorganization of the T-tubular structures, and alterations of ion channels profiles, including the LTCCs. To assess the impact of these alterations on ion channels, we studied isolated myocytes from the failing human heart. We found that the loss of T-tubules leads to the redistribution of a portion of LTCCs on the cell surface. This redistribution leads to a dramatic increase in the activity of the LTCCs. They become a possible source of arrhythmias (because of the manifestation of early afterdepolarizations) and are partly controlled by the increase in the Ca2+ calmodulin-dependent protein kinase II activity seen in HF. We extrapolated these findings observed at the single-cell level to whole heart using mathematical modeling to deduce the consequence of the redistribution of LTCCs on the propagation of arrhythmias. Our results suggest that nonphysiological ion channels in specific microdomains could be novel therapeutic targets in HF.
References
- 1.Al Chekakie MO. Traditional heart failure medications and sudden cardiac death prevention: a review. J Cardiovasc Pharmacol Ther. 2013;18:412–426. doi: 10.1177/1074248413491496. doi: 10.1177/1074248413491496. [DOI] [PubMed] [Google Scholar]
- 2.Anderson ME. Calmodulin kinase and L-type calcium channels; a recipe for arrhythmias? Trends Cardiovasc Med. 2004;14:152–161. doi: 10.1016/j.tcm.2004.02.005. doi: 10.1016/j.tcm.2004.02.005. [DOI] [PubMed] [Google Scholar]
- 3.Aimond F, Alvarez JL, Rauzier JM, Lorente P, Vassort G. Ionic basis of ventricular arrhythmias in remodeled rat heart during long-term myocardial infarction. Cardiovasc Res. 1999;42:402–415. doi: 10.1016/s0008-6363(99)00070-x. [DOI] [PubMed] [Google Scholar]
- 4.Tomaselli GF, Zipes DP. What causes sudden death in heart failure? Circ Res. 2004;95:754–763. doi: 10.1161/01.RES.0000145047.14691.db. doi: 10.1161/01.RES.0000145047.14691.db. [DOI] [PubMed] [Google Scholar]
- 5.Tomaselli GF, Marbán E. Electrophysiological remodeling in hypertrophy and heart failure. Cardiovasc Res. 1999;42:270–283. doi: 10.1016/s0008-6363(99)00017-6. [DOI] [PubMed] [Google Scholar]
- 6.Lyon AR, MacLeod KT, Zhang Y, Garcia E, Kanda GK, Lab MJ, Korchev YE, Harding SE, Gorelik J. Loss of T-tubules and other changes to surface topography in ventricular myocytes from failing human and rat heart. Proc Natl Acad Sci U S A. 2009;106:6854–6859. doi: 10.1073/pnas.0809777106. doi: 10.1073/pnas.0809777106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Glukhov AV, Fedorov VV, Lou Q, Ravikumar VK, Kalish PW, Schuessler RB, Moazami N, Efimov IR. Transmural dispersion of repolarization in failing and nonfailing human ventricle. Circ Res. 2010;106:981–991. doi: 10.1161/CIRCRESAHA.109.204891. doi: 10.1161/CIRCRESAHA.109.204891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nivala M, Song Z, Weiss JN, Qu Z. T-tubule disruption promotes calcium alternans in failing ventricular myocytes: mechanistic insights from computational modeling. J Mol Cell Cardiol. 2015;79:32–41. doi: 10.1016/j.yjmcc.2014.10.018. doi: 10.1016/j.yjmcc.2014.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bénitah JP, Kerfant BG, Vassort G, Richard S, Gómez AM. Altered communication between L-type calcium channels and ryanodine receptors in heart failure. Front Biosci. 2002;7:e263–e275. doi: 10.2741/benitah. [DOI] [PubMed] [Google Scholar]
- 10.Bito V, Heinzel FR, Biesmans L, Antoons G, Sipido KR. Crosstalk between L-type Ca2+ channels and the sarcoplasmic reticulum: alterations during cardiac remodelling. Cardiovasc Res. 2008;77:315–324. doi: 10.1093/cvr/cvm063. doi: 10.1093/cvr/cvm063. [DOI] [PubMed] [Google Scholar]
- 11.Zhang XQ, Moore RL, Tillotson DL, Cheung JY. Calcium currents in postinfarction rat cardiac myocytes. Am J Physiol. 1995;269(6)(pt 1):C1464–C1473. doi: 10.1152/ajpcell.1995.269.6.C1464. [DOI] [PubMed] [Google Scholar]
- 12.Bhargava A, Lin X, Novak P, Mehta K, Korchev Y, Delmar M, Gorelik J. Super-resolution scanning patch clamp reveals clustering of functional ion channels in adult ventricular myocyte. Circ Res. 2013;112:1112–1120. doi: 10.1161/CIRCRESAHA.111.300445. doi: 10.1161/CIRCRESAHA.111.300445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Novak P, Gorelik J, Vivekananda U, Shevchuk AI, Ermolyuk YS, Bailey RJ, Bushby AJ, Moss GW, Rusakov DA, Klenerman D, Kullmann DM, Volynski KE, Korchev YE. Nanoscale-targeted patch-clamp recordings of functional presynaptic ion channels. Neuron. 2013;79:1067–1077. doi: 10.1016/j.neuron.2013.07.012. doi: 10.1016/j.neuron.2013.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Glukhov AV, Balycheva M, Sanchez-Alonso JL, Ilkan Z, Alvarez-Laviada A, Bhogal N, Diakonov I, Schobesberger S, Sikkel MB, Bhargava A, Faggian G, Punjabi PP, Houser SR, Gorelik J. Direct evidence for microdomain-specific localization and remodeling of functional L-type calcium channels in rat and human atrial myocytes. Circulation. 2015;132:2372–2384. doi: 10.1161/CIRCULATIONAHA.115.018131. doi: 10.1161/CIRCULATIONAHA.115.018131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Hara T, Virág L, Varró A, Rudy Y. Simulation of the undiseased human cardiac ventricular action potential: model formulation and experimental validation. PLoS Comput Biol. 2011;7:e1002061. doi: 10.1371/journal.pcbi.1002061. doi: 10.1371/journal.pcbi.1002061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Grandi E, Pasqualini FS, Bers DM. A novel computational model of the human ventricular action potential and Ca transient. J Mol Cell Cardiol. 2010;48:112–121. doi: 10.1016/j.yjmcc.2009.09.019. doi: 10.1016/j.yjmcc.2009.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J. 2004;87:3351–3371. doi: 10.1529/biophysj.104.047449. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moreno JD, Zhu ZI, Yang PC, Bankston JR, Jeng MT, Kang C, Wang L, Bayer JD, Christini DJ, Trayanova NA, Ripplinger CM, Kass RS, Clancy CE. A computational model to predict the effects of class I anti-arrhythmic drugs on ventricular rhythms. Sci Transl Med. 2011;3:98ra83. doi: 10.1126/scitranslmed.3002588. doi: 10.1126/scitranslmed.3002588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bayer JD, Blake RC, Plank G, Trayanova NA. A novel rule-based algorithm for assigning myocardial fiber orientation to computational heart models. Ann Biomed Eng. 2012;40:2243–2254. doi: 10.1007/s10439-012-0593-5. doi: 10.1007/s10439-012-0593-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prassl AJ, Kickinger F, Ahammer H, Grau V, Schneider JE, Hofer E, Vigmond EJ, Trayanova NA, Plank G. Automatically generated, anatomically accurate meshes for cardiac electrophysiology problems. IEEE Trans Biomed Eng. 2009;56:1318–1330. doi: 10.1109/TBME.2009.2014243. doi: 10.1109/TBME.2009.2014243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vigmond EJ, Aguel F, Trayanova NA. Computational techniques for solving the bidomain equations in three dimensions. IEEE Trans Biomed Eng. 2002;49:1260–1269. doi: 10.1109/TBME.2002.804597. doi: 10.1109/TBME.2002.804597. [DOI] [PubMed] [Google Scholar]
- 22.Vigmond EJ, Weber dos Santos R, Prassl AJ, Deo M, Plank G. Solvers for the cardiac bidomain equations. Prog Biophys Mol Biol. 2008;96:3–18. doi: 10.1016/j.pbiomolbio.2007.07.012. doi: 10.1016/j.pbiomolbio.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Novak P, Li C, Shevchuk AI, Stepanyan R, Caldwell M, Hughes S, Smart TG, Gorelik J, Ostanin VP, Lab MJ, Moss GW, Frolenkov GI, Klenerman D, Korchev YE. Nanoscale live-cell imaging using hopping probe ion conductance microscopy. Nat Methods. 2009;6:279–281. doi: 10.1038/nmeth.1306. doi: 10.1038/nmeth.1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorelik J, Yang LQ, Zhang Y, Lab M, Korchev Y, Harding SE. A novel Z-groove index characterizing myocardial surface structure. Cardiovasc Res. 2006;72:422–429. doi: 10.1016/j.cardiores.2006.09.009. doi: 10.1016/j.cardiores.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 25.Schröder F, Handrock R, Beuckelmann DJ, Hirt S, Hullin R, Priebe L, Schwinger RH, Weil J, Herzig S. Increased availability and open probability of single L-type calcium channels from failing compared with nonfailing human ventricle. Circulation. 1998;98:969–976. doi: 10.1161/01.cir.98.10.969. [DOI] [PubMed] [Google Scholar]
- 26.Kirchhefer U, Schmitz W, Scholz H, Neumann J. Activity of cAMP-dependent protein kinase and Ca2+/calmodulin-dependent protein kinase in failing and nonfailing human hearts. Cardiovasc Res. 1999;42:254–261. doi: 10.1016/s0008-6363(98)00296-x. [DOI] [PubMed] [Google Scholar]
- 27.Lee TS, Karl R, Moosmang S, Lenhardt P, Klugbauer N, Hofmann F, Kleppisch T, Welling A. Calmodulin kinase II is involved in voltage-dependent facilitation of the L-type Cav1.2 calcium channel: Identification of the phosphorylation sites. J Biol Chem. 2006;281:25560–25567. doi: 10.1074/jbc.M508661200. doi: 10.1074/jbc.M508661200. [DOI] [PubMed] [Google Scholar]
- 28.Grueter CE, Abiria SA, Wu Y, Anderson ME, Colbran RJ. Differential regulated interactions of calcium/calmodulin-dependent protein kinase II with isoforms of voltage-gated calcium channel beta subunits. Biochemistry. 2008;47:1760–1767. doi: 10.1021/bi701755q. doi: 10.1021/bi701755q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao L, Blair LA, Marshall J. CaMKII-independent effects of KN93 and its inactive analog KN92: reversible inhibition of L-type calcium channels. Biochem Biophys Res Commun. 2006;345:1606–1610. doi: 10.1016/j.bbrc.2006.05.066. doi: 10.1016/j.bbrc.2006.05.066. [DOI] [PubMed] [Google Scholar]
- 30.Wickenden AD, Kaprielian R, Kassiri Z, Tsoporis JN, Tsushima R, Fishman GI, Backx PH. The role of action potential prolongation and altered intracellular calcium handling in the pathogenesis of heart failure. Cardiovasc Res. 1998;37:312–323. doi: 10.1016/s0008-6363(97)00256-3. [DOI] [PubMed] [Google Scholar]
- 31.Qin D, Zhang ZH, Caref EB, Boutjdir M, Jain P, el-Sherif N. Cellular and ionic basis of arrhythmias in postinfarction remodeled ventricular myocardium. Circ Res. 1996;79:461–473. doi: 10.1161/01.res.79.3.461. [DOI] [PubMed] [Google Scholar]
- 32.Fozzard HA. Afterdepolarizations and triggered activity. Basic Res Cardiol. 1992;87(suppl 2):105–113. doi: 10.1007/978-3-642-72477-0_10. [DOI] [PubMed] [Google Scholar]
- 33.Xie Y, Sato D, Garfinkel A, Qu Z, Weiss JN. So little source, so much sink: requirements for afterdepolarizations to propagate in tissue. Biophys J. 2010;99:1408–1415. doi: 10.1016/j.bpj.2010.06.042. doi: 10.1016/j.bpj.2010.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beuckelmann DJ, Erdmann E. Ca(2+)-currents and intracellular [Ca2+]i-transients in single ventricular myocytes isolated from terminally failing human myocardium. Basic Res Cardiol. 1992;87(suppl 1):235–243. doi: 10.1007/978-3-642-72474-9_19. [DOI] [PubMed] [Google Scholar]
- 35.Mewes T, Ravens U. L-type calcium currents of human myocytes from ventricle of non-failing and failing hearts and from atrium. J Mol Cell Cardiol. 1994;26:1307–1320. doi: 10.1006/jmcc.1994.1149. doi: 10.1006/jmcc.1994.1149. [DOI] [PubMed] [Google Scholar]
- 36.Piacentino V, III, Weber CR, Chen X, Weisser-Thomas J, Margulies KB, Bers DM, Houser SR. Cellular basis of abnormal calcium transients of failing human ventricular myocytes. Circ Res. 2003;92:651–658. doi: 10.1161/01.RES.0000062469.83985.9B. doi: 10.1161/01.RES.0000062469.83985.9B. [DOI] [PubMed] [Google Scholar]
- 37.Chen X, Zhang X, Harris DM, Piacentino V, III, Berretta RM, Margulies KB, Houser SR. Reduced effects of BAY K 8644 on L-type Ca2+ current in failing human cardiac myocytes are related to abnormal adrenergic regulation. Am J Physiol Heart Circ Physiol. 2008;294:H2257–H2267. doi: 10.1152/ajpheart.01335.2007. doi: 10.1152/ajpheart.01335.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bryant SM, Kong CH, Watson J, Cannell MB, James AF, Orchard CH. Altered distribution of ICa impairs Ca release at the t-tubules of ventricular myocytes from failing hearts. J Mol Cell Cardiol. 2015;86:23–31. doi: 10.1016/j.yjmcc.2015.06.012. doi: 10.1016/j.yjmcc.2015.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hashambhoy YL, Greenstein JL, Winslow RL. Role of CaMKII in RyR leak, EC coupling and action potential duration: a computational model. J Mol Cell Cardiol. 2010;49:617–624. doi: 10.1016/j.yjmcc.2010.07.011. doi: 10.1016/j.yjmcc.2010.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ibrahim M, Navaratnarajah M, Siedlecka U, Rao C, Dias P, Moshkov AV, Gorelik J, Yacoub MH, Terracciano CM. Mechanical unloading reverses transverse tubule remodelling and normalizes local Ca(2+)-induced Ca(2+)release in a rodent model of heart failure. Eur J Heart Fail. 2012;14:571–580. doi: 10.1093/eurjhf/hfs038. doi: 10.1093/eurjhf/hfs038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Louch WE, Bito V, Heinzel FR, Macianskiene R, Vanhaecke J, Flameng W, Mubagwa K, Sipido KR. Reduced synchrony of Ca2+ release with loss of T-tubules-a comparison to Ca2+ release in human failing cardiomyocytes. Cardiovasc Res. 2004;62:63–73. doi: 10.1016/j.cardiores.2003.12.031. doi: 10.1016/j.cardiores.2003.12.031. [DOI] [PubMed] [Google Scholar]
- 42.He J, Conklin MW, Foell JD, Wolff MR, Haworth RA, Coronado R, Kamp TJ. Reduction in density of transverse tubules and L-type Ca(2+) channels in canine tachycardia-induced heart failure. Cardiovasc Res. 2001;49:298–307. doi: 10.1016/s0008-6363(00)00256-x. [DOI] [PubMed] [Google Scholar]
- 43.Hong TT, Smyth JW, Chu KY, Vogan JM, Fong TS, Jensen BC, Fang K, Halushka MK, Russell SD, Colecraft H, Hoopes CW, Ocorr K, Chi NC, Shaw RM. BIN1 is reduced and Cav1.2 trafficking is impaired in human failing cardiomyocytes. Heart Rhythm. 2012;9:812–820. doi: 10.1016/j.hrthm.2011.11.055. doi: 10.1016/j.hrthm.2011.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Balycheva M, Faggian G, Glukhov AV, Gorelik J. Microdomain–specific localization of functional ion channels in cardiac myocytes: an emerging concept of local regulation and remodelling. Biophys Rev. 2015;7:43–62. doi: 10.1007/s12551-014-0159-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Makarewich CA, Correll RN, Gao H, Zhang H, Yang B, Berretta RM, Rizzo V, Molkentin JD, Houser SR. A caveolae-targeted L-type Ca2+ channel antagonist inhibits hypertrophic signaling without reducing cardiac contractility. Circ Res. 2012;110:669–674. doi: 10.1161/CIRCRESAHA.111.264028. doi: 10.1161/CIRCRESAHA.111.264028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sossalla S, Fluschnik N, Schotola H, Ort KR, Neef S, Schulte T, Wittköpper K, Renner A, Schmitto JD, Gummert J, El-Armouche A, Hasenfuss G, Maier LS. Inhibition of elevated Ca2+/calmodulin-dependent protein kinase II improves contractility in human failing myocardium. Circ Res. 2010;107:1150–1161. doi: 10.1161/CIRCRESAHA.110.220418. doi: 10.1161/CIRCRESAHA.110.220418. [DOI] [PubMed] [Google Scholar]
- 47.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE, Jr, Thiel W, Guatimosim S, Song LS, Madu EC, Shah AN, Vishnivetskaya TA, Atkinson JB, Gurevich VV, Salama G, Lederer WJ, Colbran RJ, Anderson ME. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–417. doi: 10.1038/nm1215. doi: 10.1038/nm1215. [DOI] [PubMed] [Google Scholar]
- 48.Bers DM. CaMKII inhibition in heart failure makes jump to human. Circ Res. 2010;107:1044–1046. doi: 10.1161/CIRCRESAHA.110.231902. doi: 10.1161/CIRCRESAHA.110.231902. [DOI] [PubMed] [Google Scholar]
- 49.Koval OM, Guan X, Wu Y, Joiner M, Gao Z, Chen B, Grumbach IM, Luczak ED, Colbran RJ, Song LS, Hunh TJ, Mohler PJ, Anderson ME. Cav1.2 β-subunit coordinates CaMKII-triggered cardiac myocyte death and afterdepolarizations. Proc Natl Acad Sci USA. 2010;107:4996–5000. doi: 10.1073/pnas.0913760107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hullin R, Matthes J, von Vietinghoff S, Bodi I, Rubio M, D’Souza K, Friedrich Khan I, Rottländer D, Hoppe UC, Mohacsi P, Schmitteckert E, Gilsbach R, Bünemann M, Hein L, Schwartz A, Herzig S. Increased expression of the auxiliary beta(2)-subunit of ventricular L-type Ca(2)+ channels leads to single-channel activity characteristic of heart failure. PLoS One. 2007;2:e292. doi: 10.1371/journal.pone.0000292. doi: 10.1371/journal.pone.0000292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hashambhoy YL, Winslow RL, Greenstein JL. CaMKII-induced shift in modal gating explains L-type Ca(2+) current facilitation: a modeling study. Biophys J. 2009;96:1770–1785. doi: 10.1016/j.bpj.2008.11.055. doi: 10.1016/j.bpj.2008.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Morotti S, Grandi E, Summa A, Ginsburg KS, Bers DM. Theoretical study of L-type Ca(2+) current inactivation kinetics during action potential repolarization and early afterdepolarizations. J Physiol. 2012;590:4465–4481. doi: 10.1113/jphysiol.2012.231886. doi: 10.1113/jphysiol.2012.231886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elshrif MM, Shi P, Cherry EM. Representing variability and transmural differences in a model of human heart failure. IEEE J Biomed Health Inform. 2015;19:1308–1320. doi: 10.1109/JBHI.2015.2442833. doi: 10.1109/JBHI.2015.2442833. [DOI] [PubMed] [Google Scholar]
- 54.Swaminathan PD, Purohit A, Hund TJ, Anderson ME. Calmodulin-dependent protein kinase II: linking heart failure and arrhythmias. Circ Res. 2012;110:1661–1677. doi: 10.1161/CIRCRESAHA.111.243956. doi: 10.1161/CIRCRESAHA.111.243956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, Gao Z, He BJ, Luczak ED, Joiner ML, Kutschke W, Yang J, Donahue JK, Weiss RM, Grumbach IM, Ogawa M, Chen PS, Efimov I, Dobrev D, Mohler PJ, Hund TJ, Anderson ME. Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J Clin Invest. 2011;121:3277–3288. doi: 10.1172/JCI57833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Glynn P, Musa H, Wu X, Unudurthi SD, Little S, Qian L, Wright PJ, Radwanski PB, Gyorke S, Mohler PJ, Hund TJ. Voltage-gated sodium channel phosphorylation at Ser571 regulates late current, arrhythmia, and cardiac function in vivo. Circulation. 2015;132:567–577. doi: 10.1161/CIRCULATIONAHA.114.015218. doi: 10.1161/CIRCULATIONAHA.114.015218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Undrovinas AI, Maltsev VA, Sabbah HN. Repolarization abnormalities in cardiomyocytes of dogs with chronic heart failure: role of sustained inward current. Cell Mol Life Sci. 1999;55:494–505. doi: 10.1007/s000180050306. doi: 10.1007/s000180050306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zeng J, Rudy Y. Early afterdepolarizations in cardiac myocytes: mechanism and rate dependence. Biophys J. 1995;68:949–964. doi: 10.1016/S0006-3495(95)80271-7. doi: 10.1016/S0006-3495(95)80271-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chang MG, Sato D, de Lange E, Lee JH, Karagueuzian HS, Garfinkel A, Weiss JN, Qu Z. Bi-stable wave propagation and early afterdepolarization-mediated cardiac arrhythmias. Heart Rhythm. 2012;9:115–122. doi: 10.1016/j.hrthm.2011.08.014. doi: 10.1016/j.hrthm.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.January CT, Riddle JM. Early afterdepolarizations: mechanism of induction and block. A role for L-type Ca2+ current. Circ Res. 1989;64:977–990. doi: 10.1161/01.res.64.5.977. [DOI] [PubMed] [Google Scholar]
- 61.Madhvani RV, Angelini M, Xie Y, Pantazis A, Suriany S, Borgstrom NP, Garfinkel A, Qu Z, Weiss JN, Olcese R. Targeting the late component of the cardiac L-type Ca2+ current to suppress early afterdepolarizations. J Gen Physiol. 2015;145:395–404. doi: 10.1085/jgp.201411288. doi: 10.1085/jgp.201411288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yamada M, Ohta K, Niwa A, Tsujino N, Nakada T, Hirose M. Contribution of L-type Ca2+ channels to early afterdepolarizations induced by I Kr and I Ks channel suppression in guinea pig ventricular myocytes. J Membr Biol. 2008;222:151–166. doi: 10.1007/s00232-008-9113-9. doi: 10.1007/s00232-008-9113-9. [DOI] [PubMed] [Google Scholar]
- 63.Maltsev VA, Silverman N, Sabbah HN, Undrovinas AI. Chronic heart failure slows late sodium current in human and canine ventricular myocytes: implications for repolarization variability. Eur J Heart Fail. 2007;9:219–227. doi: 10.1016/j.ejheart.2006.08.007. doi: 10.1016/j.ejheart.2006.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]