

Abstract
Introduction
The under-reporting of adverse drug events (ADEs) is an international health concern. A number of studies have assessed the root causes but, to our knowledge, little information exists relating under-reporting to practices and systems used for the recording and tracking of drug‐related adverse event observations in ambulatory settings, institutional settings, and retail pharmacies.
Objectives
Our objective was to explore the process for reporting ADEs in US hospitals, ambulatory settings, and retail pharmacies; to explore gaps and inconsistencies in the reporting process; and to identify the causes of under-reporting ADEs in these settings.
Methods
The Tufts Center for the Study of Drug Development (Tufts CSDD) interviewed 11 thought leaders and conducted a survey between May and August 2014 among US-based healthcare providers (HCPs) in diverse settings to assess their experiences with, and processes for, reporting ADEs.
Results
A total of 123 individuals completed the survey (42 % were pharmacists; 27 % were nurses; 15 % were physicians; and 16 % were classified as ‘other’). HCPs indicated that the main reasons for under-reporting were difficulty in determining the cause of the ADE, given that most patients receive multiple therapies simultaneously (66 % of respondents); that HCPs lack sufficient time to report ADEs (63 % of respondents); poor integration of ADE-reporting systems (53 % of respondents); and uncertainty about reporting procedures (52 % of respondents).
Discussion
The results of this pilot study identify that key factors contributing to the under-reporting of ADEs relate to a lack of standardized process, a lack of training and education, and a lack of integrated health information technologies.
Electronic supplementary material
The online version of this article (doi:10.1007/s40264-016-0455-4) contains supplementary material, which is available to authorized users.
Key Points
| This pilot study evaluated adverse drug event (ADE) reporting processes in US hospitals, private practices, and retail pharmacies. |
| The results highlight gaps in the reporting process that fall into three categories: technology, education, and the overall process. |
| Recommendations include integrating health information systems to streamline the reporting process, training and educating both healthcare providers and patients on ADE reporting, and creating a standardized ADE-reporting process. |
Introduction
Adverse drug events (ADEs)—defined by the World Health Organization (WHO) as “a response to a medicine which is noxious and unintended, and which occurs at doses normally used in man” [1]—are extremely costly to society. For example, half of all ADEs in the USA are the result of preventable medication errors; they affect more than 7 million patients, cause 7000 deaths, and cost more than $US20 billion across all care settings each year [2–4].
When a drug is approved, its efficacy and safety is accepted with the expectation that its benefit–risk profile will be further expanded by exposure to more patients in real-world healthcare settings, some of whom may have been excluded from registrational study enrollment. In the USA, this passive post-approval monitoring system relies on voluntary and accurate reporting that identifies a product and its manufacturer [5–7]. The majority of ADE reports are sent directly to drug manufacturers; some are sent directly to the US FDA via its MedWatch program. The FDA’s Sentinel Initiative is another source of administrative and claims-based data that enables the study of real-world effects of a medication [8, 9].
Following the launch of a drug, the FDA requires biopharmaceutical companies to report any and all spontaneous ADEs once an event has been observed and the patient, observer, and suspect product identified [10]. Other regulatory agencies have similar programs: the European Medicines Agency’s EudraVigilance is the European equivalent.
Standardized reporting systems with ongoing institution-based surveillance have assisted healthcare providers (HCPs) in voluntarily reporting ADE observations and complying with regulatory guidance. ADE reports have led to drugs being withdrawn from the market because of risks outweighing benefits [6]. However, a number of studies indicate that the under-reporting of ADEs is a pervasive and widespread problem [6, 11–18]. For example, two studies noted that the FDA receives reports for less than 1 % of suspected serious ADEs [8, 19].
In 2015, the results of a study by Ma et al. [20] suggested that some pharmaceutical organizations and manufacturers add to the under-reporting issue by delaying reports of serious ADEs. Their study indicated that roughly 10 % of all serious ADEs were not reported to the FDA within the 15-day required time period. The study concluded that one possible explanation was that pharmaceutical companies could be taking longer to investigate the reports; however, they concluded, “this discretion is outside the scope of the current regulatory regime.” [20].
In the USA, ADE reporting to the FDA or manufacturer is voluntary. However, policies around ADEs are created and decided upon at the state level. For example, some states require pharmacists to report vaccine ADEs. As of January 2008, 26 states had created regulations related to hospital adverse event (AE) reporting, and one had recently decided to create policies [21]. Although roughly half of the states had systems in place, under-reporting was still considered a large problem in 2010 [11, 22]. It remained a considerable issue in 2012; the Office of the Inspector General had seen no improvements in reporting, noting that hospitals were still recording only 1 % of ADEs and that, although 60 % of ADEs occurred in hospitals with infrastructure in place for reporting, only 12 % of ADEs were reported by hospitals with such infrastructure [23].
Although some hospitals have implemented technology systems to capture ADEs, the quality of the data are questionable as misclassification of an ADE for a given drug in a multisource market is prevalent. Krahn et al. [17] discussed a European project, SALUS (Scalable, Standard based Interoperability Framework for Sustainable Proactive Post Market Safety Studies), and tools and algorithms that assess a patient’s electronic health record (EHR)/electronic medical record (EMR) and notify physicians of any possible ADEs. They noted that the algorithms were only as strong as the data entered by individual HCPs, which was a limitation of the project [17]. Elliott et al. [16] evaluated an electronic clinical safety reporting system implemented in Newfoundland and Labrador, Canada, and noted an increase in the number of ADEs reported with the use of this tool by hospital staff (i.e., not physicians or nurses). However, they also noted that challenges with customizing the tool increased the time it took to report an event [16]. Klein et al. [24] looked at the traceability of biological medicines in the Netherlands and concluded that deficiencies in the systematic capture of biological drug brand names and lot (batch) numbers for ADE reporting could be due to “shortcomings in the recording of information in clinical practice.” Studies conducted by the Tufts Center for the Study of Drug Development (Tufts CSDD) [25] and by Vermeer and colleagues [26, 27] also demonstrated that the quality and completeness of voluntary ADE reporting results in misclassification and an inability to trace observations to the source or suspect product.
Several studies have assessed the root causes of under-reporting. Varallo et al. [15] conducted a systematic review and concluded that the main causes for under-reporting included William Howard Wallace Inman’s description, “ignorance, insecurity, and indifference.” Goldman et al. [28] investigated pediatric ADEs and observed that inclusion of a drug safety service resulted in a fourfold increase in reporting. Hirose et al. [29] found that signal detection time, i.e., lag time, was longer for physicians reporting ADEs than for nurses. However, Pagotto et al. [14] noted that primary educational interventions in Europe increased both the number and the quality of ADE reports. However, in the USA, while studies indicate that hospitals are still under-reporting, few have focused on why that under-reporting occurs [14, 15] and, specifically, the process for reporting ADEs and the gaps in that process.
In 2014, under an unrestricted grant from Amgen Inc., we conducted a two-part study to explore the process for reporting ADEs in US hospitals, ambulatory settings, and retail pharmacies; to explore the gaps and inconsistencies in the reporting process; and to further identify reasons for under-reporting of ADEs in these settings. We hope the results of this study will inform policy makers on best practices for ADE reporting.
Methodology
In January 2014, Tufts CSDD first conducted a literature review to map the ADE-reporting process and to identify any known gaps within that process. From February to May 2014, Tufts CSDD interviewed 11 thought leaders to gain insight into the systems and technologies supporting each step in the process. Individuals interviewed were claims data experts, pharmacists, and physicians and from the FDA and generics manufacturing companies.
Based on the interview findings, Tufts CSDD created a data-collection instrument (i.e., survey) assessing the ADE-reporting process in hospitals (i.e., institutional), ambulatory (i.e., private practice), and retail pharmacy settings. Survey respondents were asked about the following:
their experience in healthcare and reporting ADEs;
the process for reporting AEs at their primary treatment setting;
thoughts on the reasons for ADEs not being reported;
health information systems used for ADE reporting.
To assess the ADE-reporting process, respondents were provided 10–18 possible steps in the ADE-reporting process, depending on professional setting. Respondents selected and ordered the steps to create a reporting process map. Using a Likert scale, respondents then reported whether the selected steps were consistently completed at their institution. Respondents employed by hospitals were provided 18 possible steps to select and order, respondents in private practice were provided 13 possible steps, and respondents employed by retail pharmacies were provided 14. The complete survey questionnaire may be found in the Electronic Supplementary Material.
Data were analyzed using descriptive statistics. Group comparisons were conducted using the Chi-squared test for independence (alpha level 0.05). Qualitative responses were coded and categorized into main themes.
The survey was sent to individuals in internal and external databases via e-mail, social media, and HCP and pharmacist associations from three states: New Jersey, New York, and Washington. The number of individuals who received an invitation to participate is unknown, as the HCP and pharmacist associations did not disclose the total number of individuals in their associations. The survey was conducted from 16 May 2014 to 31 August 2014.
Results
Phase I
Tufts CSDD conducted 11 interviews with thought leaders in pharmacovigilance. One individual had worked in the Office of Surveillance and Epidemiology at the FDA, one individual worked for a generics manufacturer, three were professors of epidemiology with expertise in claims data; the remaining six were pharmacists or physicians in an ambulatory setting, a retail pharmacy setting, or an institutional setting.
Interviewees indicated that many health information technology (HIT) systems are typically used. Table 1 lists the HIT systems found in a typical US hospital and a definition of each.
Table 1.
Health information technology systems within a typical hospital practice. Phase IV post-marketing clinical trials are also used to track and report adverse drug events if the hospital is taking part in an established post-marketing study
| Acronym | Electronic system | Definition of electronic system | Occupation accessing data | Used for ADE tracking |
|---|---|---|---|---|
| EHR/EMR | Electronic health records/electronic medical records | Database containing standard medical and clinical data collected by HCP [30] | Accessed by billing department to determine diagnoses | Yes |
| CPOE | Computerized physician order entry system | Database containing medication orders as prescribed and directly entered by a licensed HCP [30] | Orders entered by physicians. Entries communicated to pharmacy, laboratory, or radiology | No |
| eMAR | Electronic medication administration record | Database used to track medication from order to administration [31] | Entered by nurses when administrating medicine to patients | No |
| BCMA | Barcode-enabled medication administration | Database that combines eMAR with bar coding (item-specific identification) [32] | Barcodes scanned by nurses; contains information on providers, patients, and drugs to facilitate entering of information into eMAR | No |
| BCMP | Barcode medication preparation technologies | Database that contains barcoding for inventory of items used in medicine preparation [33] | Used by pharmacy technicians when preparing medicines such as those for intravenous administration. Entries allow pharmacists to check technician’s work | No |
| IR | Electronic incident reporting system | Database that captures instances of patient harm [34] | Information entered by nurses, physician’s assistants, or pharmacists. Reviewed by Quality and Safety or Risk Management teams. Results in interventions deployed within the institution and reporting to the state | Yes |
| EBS | Electronic billing system | Database that captures ICD code as defined by the WHO. Data used for reimbursement | Information from EHRs is retrieved and converted to codes (ICD-9) that represent events or diagnoses such as heart attack. Coded information is forwarded to payers | Yes |
ADE adverse drug event, HCP healthcare provider, ICD-9 International Classification of diseases and related health problems, ninth revision, WHO World Health Organization
The systems listed in Table 1 were also in place in ambulatory settings. Employees in such a setting also relied on the pharmacy’s Patient Medication Record (PMR) system, which forwards patient diagnoses to health insurance companies. A PMR captures patient information, medication prescription information (dose, quantity, name), dispenser of the medication, and prescriber information [35]. Interviewees noted that the incident-reporting systems were not integrated with any of the other electronic systems (e.g. computerized physician order entry [CPOE], EHR/EMR), and that the incident reporting systems did not have a feature that could push ADE data directly to the FDA or drug manufacturer.
Phase II
Respondent Demographics and Adverse Drug Event (ADE) Reporting History
Of the 284 respondents who participated, 123 completed the survey (47 worked in hospitals, 31 in ambulatory settings, 14 in retail pharmacies, and 31 in other, e.g., medical professionals employed by correction facilities, senior centers, the military, school systems, etc.). The survey outreach design described in the methodology meant the majority of respondents who completed the survey worked in New Jersey, New York, and Washington State. Table 2 contains their demographic information.
Table 2.
Survey demographics
| Demographic | Pharmacists | Nurses | Physicians | Other HCP | Total |
|---|---|---|---|---|---|
| Setting | |||||
| Hospital/institutional | 32 (26) | 15 (12) | 0 (0) | 0 (0) | 47 (38) |
| Ambulatory/private practice | 0 (0) | 4 (3) | 18 (15) | 9 (7) | 31 (25) |
| Retail pharmacy | 11 (9) | 0 (0) | 0 (0) | 3 (2) | 14 (11) |
| Other | 9 (7) | 14 (11) | 1 (1) | 7 (6) | 31 (25) |
| Total | 52 (42) | 33 (27) | 19 (15) | 19 (15) | 123a |
| State | |||||
| NJ | 0 (0) | 29 (24) | 0 (0) | 10 (8) | 39 (32) |
| NY | 24 (20) | 0 (0) | 13 (11) | 4 (3) | 41 (33) |
| WA | 21 (17) | 0 (0) | 0 (0) | 0 (0) | 21 (17) |
| Other | 7 (6) | 4 (3) | 6 (5) | 5 (4) | 22 (18) |
| Total | 52 (42) | 33 (27) | 19 (15) | 19 (15) | 123a |
| From NJ | From NY | From WA | From other | Total | |
|---|---|---|---|---|---|
| Setting | |||||
| Hospital/institutional | 12 (10) | 20 (16) | 7 (6) | 8 (7) | 47 (38) |
| Ambulatory/private practice | 10 (8) | 13 (11) | 0 (0) | 8 (7) | 31 (25) |
| Retail pharmacy | 0 (0) | 4 (3) | 8 (7) | 2 (2) | 14 (11) |
| Other | 17 (14) | 4 (3) | 6 (5) | 4 (3) | 31 (25) |
| Total | 39 (32) | 41 (33) | 21 (17) | 22 (18) | 123a |
Data are presented as N (%) unless otherwise indicated
HCP healthcare provider, NJ New Jersey, NY New York, WA Washington
aReflects only respondents who completed the survey. Individuals in the ‘other’ category are employed by institutions such as correction facilities, senior centers, academic/school systems, and the military
The majority (51 %) of the 123 respondents who completed the survey had not reported any ADEs to the FDA or drug manufacturer in the last 5 years. There was a statistically significant difference in the number of times an ADE had been reported by occupation; pharmacists had reported one or more ADEs more often than nurses or physicians (p = 0.005; Chi-squared test for independence). Table 3 shows the percentage breakdown by occupation and number of times an ADE was reported in the last 5 years.
Table 3.
Frequency of adverse drug events reported to the FDA or the drug manufacturer in the last 5 years by occupation
| Occupation | ADEs reported to the FDA/drug manufacturer | p value (Chi-squared) | ||
|---|---|---|---|---|
| None | 1–4 times | ≥5 times | ||
| Physician | 63 | 32 | 5 | 0.005 |
| Nurse | 68 | 27 | 5 | |
| Pharmacist | 33 | 48 | 19 | |
| Overall | 51 | 38 | 11 | |
Data are presented as % unless otherwise stated
Chi-squared test for independence, alpha level 0.05; sample size is 123 respondents (those who completed the survey)
ADE adverse drug event
Of respondents who reported an ADE in the last 5 years, 92 % had reported to two or more organizations. The main organizations reported to were as follows: internal reporting, FDA MedWatch, and the drug manufacturer. Pharmacists reported ADEs internally and to FDA MedWatch more often than either physicians or nurses (p < 0.0001 and p = 0.002, respectively, Chi-squared test for independence). Table 4 provides a percentage breakdown of where ADEs were reported by occupation.
Table 4.
Organization to which respondent has reported an adverse drug event, by occupation
| Pharmacist | Nurse | Physician | Total | p value | |
|---|---|---|---|---|---|
| Sample size (n) | 52 | 44 | 19 | 115 | |
| Agency reported to | |||||
| Internal Reporting | 75 | 45 | 21 | 55 | <0.0001 |
| FDA MedWatch program | 58 | 25 | 26 | 40 | 0.002 |
| Drug manufacturer | 42 | 20 | 26 | 31 | 0.06 |
| State Department of Health | 6 | 14 | 11 | 10 | 0.42 |
| State Board of Pharmacy | 8 | 7 | 0 | 6 | 0.47 |
| Never reported an ADE | 8 | 34 | 42 | 23 | 0.001 |
Data are presented as % unless otherwise indicated
Chi-squared test for independence, alpha level 0.05; sample size is 115 respondents (the total number of respondents answering the question, e.g., a total of 44 nurses answered the question, whereas only 33 completed the entire survey)
ADE adverse drug event
By setting, of the respondents who selected internal reporting, 61 % worked in a hospital, 18 % in an ambulatory setting, 6 % in a retail pharmacy, and 15 % elsewhere (p = 0.0001, Chi-squared test for independence).
ADE Reporting Process Flow
Among all respondents, no single ADE-reporting process dominated. However, although not in the same order, six steps were identified as being a part of the ADE-reporting process by all parties across treatment settings. Figure 1 depicts the common elements in the process for reporting an ADE across all settings, along with the main gaps identified for four of the six steps in the ADE-reporting process.
Fig. 1.
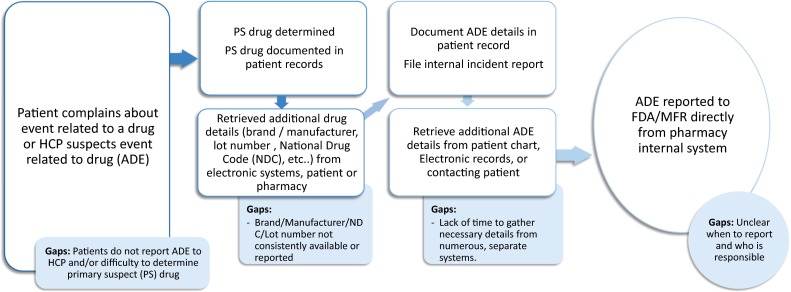
Process flow and main gaps identified for adverse drug event reporting across all settings. ADE adverse drug event, HCP healthcare provider, MFR manufacturer, NDC National Drug Code, PS primary suspect
After determining the ADE-reporting process within their organization, respondents determined how consistently each selected step was performed. “Reporting the ADE” was often one of the least consistent steps in the process across all treatment settings. Table 5 highlights the most and least consistent steps in the process by setting.
Table 5.
Most and least consistent steps in the adverse drug event reporting process, stratified by setting
| Setting | Step most consistently completed when reporting ADEs | Respondents indicating step is ‘very/somewhat’ consistently completed (%) | Step least consistently completed when reporting ADEs | Respondents indicating step is ‘not very/not at all’ consistently completed (%) |
|---|---|---|---|---|
| Hospital (n = 57) | Reporter retrieves drug details from the EHR/EMR | 90 | Drug and ADE details documented in internal incident report | 34 |
| Reporter retrieves ADE details from the EHR/EMR | 83 | Reporter reports the ADE | 29 | |
| Ambulatory (n = 29) | Clinician determines the primary suspect | 96 | Reporter reports the ADE | 73 |
| ADE details documented in patient record | 95 | Dispensing pharmacy contacted for drug details | 33 | |
| Retail pharmacy (n = 14) | Reporter retrieves details about the drug dispensed from the patient profile | 88 | Reporter reports the ADE by accessing MedWatch website/calling manufacturer | 75 |
| Pharmacist determines the primary suspect drug | 83 | Reporter contacts the patient for additional information about the ADE | 33 |
Sample size is 90 respondents (the total number of respondents answering the question, e.g., 57 physicians answered the question, whereas only 47 completed the entire survey)
ADE adverse drug event, EHR electronic health record, EMR electronic medical record
Within the hospital setting, 44 % of respondents (n = 75) were unsure whether a formal procedure for reviewing reports submitted to incident reporting existed, 22 % indicated no formal procedure existed, and 34 % indicated a formal procedure was in place.
Parties Accountable for ADE Reporting
When respondents were asked who was accountable for reporting ADEs, 33 % of those from a hospital or institutional setting, 20 % from an ambulatory or private practice, and 36 % from a retail pharmacy were uncertain.
Of those from a hospital or institutional setting, 36 % retail pharmacy; 33 % unsure, 11 % risk-mitigation department, 20 % indicated ‘other’. When stratifying by occupation, there was less clarity on who was responsible for reporting ADEs: 45 % of pharmacists stated the pharmacist was responsible for reporting, but only 21 % of nurses and 38 % of physicians agreed with this (n = 75; all respondents, not only those who completed the entire survey).
Of those from an ambulatory or private practice setting, 20 % were unsure, 27 % indicated the physician, and 20 % indicated the nurse was responsible for reporting ADEs. By occupation, there was less clarity on who was responsible for reporting ADEs in this setting: 47 % of physicians stated the physician was responsible for reporting, but only 6 % (n = 30) of nurses agreed with this.
Of those from a retail pharmacy setting, five of 14 indicated the pharmacy manager, four indicated the staff pharmacist, and five answered ‘other’.
Potential Reasons for the Under-Reporting of ADEs
Figure 1 shows the main gaps associated with each main step in the process:
The patient did not report the ADE to the HCP or pharmacist.
HIT did not consistently capture drug manufacturer, National Drug Code (NDC) number, and lot number of the drug.
HCPs did not have enough time to gather all the required information.
HCPs were unclear about when to report and who was responsible for reporting.
Figure 2 contains respondents’ perceptions on why HCPs may not have reported ADEs to the FDA or drug manufacturer. The top three reasons were that the patient was receiving more than one therapy so it was difficult to establish which drug caused the ADE (66 % of respondents selected ‘often’ or ‘very often’); that [the HCP] did not have enough time to devote to reporting given the priority placed on the provision of care (63 % of respondents selected ‘often’ or ‘very often’); and that integration between the disparate electronic systems and the reporting form is lacking (53 % of respondents selected ‘often’ or ‘very often’).
Fig. 2.
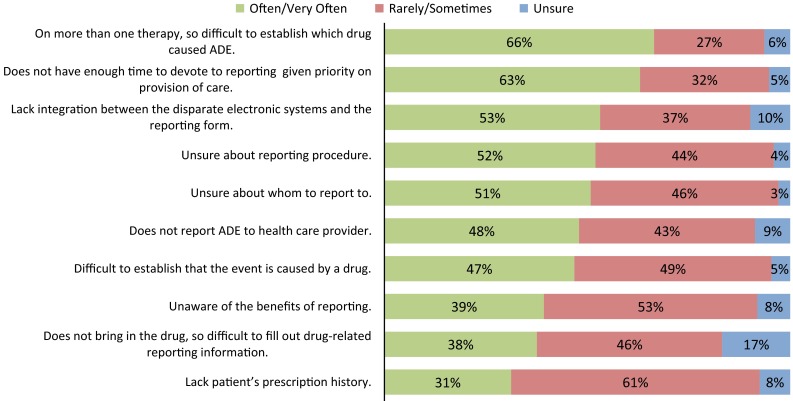
Respondent perceptions on reasons preventing health care providers from reporting adverse drug events to the FDA or to the drug manufacturer. Sample size is 173, all respondents answering the question. ADE adverse drug event
Overall, respondent perceptions were consistent across the three different settings; however, the following exceptions were noted:
76 % of respondents employed in hospitals selected ‘often/very often’ for not having enough time, compared with 47 % of respondents in an ambulatory setting and 60 % of respondents in a retail pharmacy (p = 0.005; Chi-squared test for independence).
93 % of respondents employed in a retail pharmacy selected ‘often/very often’ for [patient] does not report ADE to healthcare provider, compared with 40 % of respondents employed in a hospital and 37 % of respondents employed in an ambulatory setting (p = 0.005; Chi-squared test for independence).
72 % of respondents employed in an ambulatory setting selected ‘rarely/sometimes’ for being unaware of the benefits of reporting, compared with 43 % of respondents employed in a hospital and 60 % of respondents employed in a retail pharmacy (p = 0.03; Chi-squared test for independence).
Gaps in Health Information Technology Solutions
When respondents employed in hospital or institutional settings were asked about what HIT could be accessed to retrieve details about the suspect drug to aid in reporting the ADE, respondents selected an average of 3.2 sources (range 1–7); 46 % gathered data from four or more sources. Respondents indicated an average of 2.6 sources from which to retrieve details about the ADE, with 51 % of respondents selecting three or more sources for ADE details.
However, not all electronic systems contained all relevant information. Hospital electronic systems did not routinely capture manufacturer information, NDC, drug expiry date and lot number. Table 6 lists the percentage of respondents from a hospital setting that stated key variables useful for identifying specific products and batches (e.g., for complex and sensitive products such as biologics) were available within a specific hospital electronic system. No hospital electronic system routinely captured manufacturer, expiry date, lot number, and NDC, which are used as US product identifiers in the International Organization for Standardization’s Identification of Medicinal Products (ISO IDMP).
Table 6.
Availability of variables required for adverse drug reporting by the main hospital electronic system
| Variables to identify suspect products | EHR/EMR | CPOE | eMAR | BCMA | BCMP | IR |
|---|---|---|---|---|---|---|
| Sample size (n) | 57 | 57 | 59 | 38 | 23 | 33 |
| Dose | 95 | 95 | 98 | 95 | 91 | 85 |
| Administration route | 96 | 93 | 97 | 89 | 78 | 88 |
| Label strength | 86 | 82 | 86 | 82 | 87 | 79 |
| Brand name | 82 | 81 | 81 | 76 | 74 | 76 |
| Active ingredient | 53 | 60 | 54 | 50 | 43 | 55 |
| Concomitant medications | 67 | 63 | 61 | 39 | 35 | 33 |
| Manufacturer | 23 | 14 | 15 | 34 | 70 | 30 |
| NDC | 28 | 18 | 15 | 37 | 52 | 27 |
| Expiry date | 19 | 14 | 15 | 29 | 57 | 27 |
| Lot number | 14 | 9 | 10 | 16 | 39 | 27 |
Data are presented as % unless otherwise indicated
Sample size is 59 respondents employed in a hospital setting
ADE adverse drug event, BCMA Barcode-enabled Medication Administration, BCMP Bar-Code Medication Preparation Technologies, CPOE Computerized Physician Order Entry system, EHR electronic health record, eMAR Electronic Medication Administration Record, EMR electronic medical record, IR electronic incident reporting system, NDC National Drug Code
Respondents relying on paper-based systems exhibited a similar trend when asked about data capture. NDC and lot numbers were never (11 of 25 respondents) or rarely (13 of 25) captured, whereas active ingredient and brand was often (14 of 25) or always (12 of 25) captured.
Of the 30 respondents from an ambulatory or private practice setting, 27 indicated at least one electronic system was available at their private practice. When asked about electronic systems available to track and report ADEs, only five of 23 respondents with an EHR system indicated it could be used to track ADEs. None of the respondents had one-click forward ADE to the drug manufacturer capability. When asked about services allowing access to information about a patient’s medication history, 17 % of respondents from this setting were aware of or subscribed to such a service; 63 % were not aware of such a service.
Seven of 14 respondents from retail pharmacies indicated no tracking system was in place for recording ADEs. Respondents also indicated that electronic systems lack the ability to record the active ingredient and lot number: three and four of 14 respondents, respectively, indicated that the active ingredient and lot number were available in the patient’s profile.
Discussion
This Tufts CSDD study examined the process for reporting ADEs in US hospitals, private practice, and retail pharmacies and identified factors contributing to ADE under-reporting. Survey results indicate that pharmacists, predominantly those in a hospital setting, have the most experience reporting ADEs compared with physicians and nurses. Across all settings, factors for not reporting ADEs could be classified into three categories:
Gaps in technology integration was lacking between disparate electronic systems and the reporting process form (53 % of respondents selected this ‘often/very often’ prevented HCPs from reporting ADEs to the FDA or drug manufacturer). Additionally, the patient often did not bring drug packaging, making it difficult to complete drug-related reporting information (38 % of respondents). Lastly, patients’ prescription history was lacking (31 % of respondents).
Gaps in education there were reported gaps in education around ADEs and the ADE-reporting process. Examples of gaps in education around ADEs include how to establish which drug caused the ADE when the patient is receiving more than one therapy (66 %) and how to establish that the event was caused by a drug (47 %). Examples of gaps in education around the ADE-reporting process include that respondents were unsure about reporting procedures (52 %) and to whom the report should be made (51 %) and that respondents were unaware of the benefits of reporting (39 %).
Gaps in process respondents indicated they did not have time to devote to reporting given the priority placed on provision of care (63 %) and that patients did not report the ADE to the HCP (48 %).
HCPs indicated the main reason for not reporting ADEs is that the patient typically receives more than one therapy, making it difficult to establish which drug caused the ADE, followed by not having enough time to report given the priority placed on provision of care. Respondents also indicate a lack of integration between electronic systems within the hospital and that they are not sure about the reporting process or to whom the report should be made.
To some extent, difficulty in identifying a primary suspect drug is an intrinsic limitation of the ADE-reporting system, but the other major obstacles identified in the survey involve extrinsic factors that may be open to improvement. Suggestions for improving ADE reporting involve improving integration between electronic systems, simplifying the reporting process, and increasing awareness of the benefits of reporting.
Although respondents indicated the inability to determine which drug caused the ADE and lack of integration in HIT are the main gaps, there is no set process for hospitals and private practices to follow. This lack of standardized process could explain why 52 % of respondents are unsure about the reporting process and 51 % are unsure about to whom they should report. Moreover, it may explain why 33 % of respondents in a hospital setting and 20 % of those in private practice indicated they were uncertain about who is responsible for ADE reporting. Those who were certain about who is responsible could provide no consensus as to who was accountable: physician, nurse, or pharmacist. This could be partly because the FDA and professional associations such as the American Medical Association and American Pharmacists Association have not offered guidance on best practices for voluntary ADE reporting. While 27 states have regulations covering hospital ADE reporting, each state’s regulations may vary, which will make the creation of a standard operating procedure more challenging. For example, Levinson [21] noted that key differences existed in the definitions of a reportable event, the reporting procedure for the ADE, and other requirements for submission.
This study corroborates the findings of Varallo et al. [15] that the lack of education within both hospital and ambulatory settings is noteworthy. In addition to not being familiar with the overall process for reporting ADEs and the benefits of reporting, respondents were unclear as how to determine which therapy caused the ADE or whether the event was even established by a drug. These concepts can be addressed in training and continuing education. However, there are two targetable intervention groups: the reporter (physician, nurse, pharmacists) and the patient. Pagotto et al. [14] demonstrated that training sessions and education increased ADE reporting in European hospitals, and—through separate patient and healthcare provider surveys—Yi et al. [36] noted that patients would prefer education and training on ADEs from their physician, followed by their pharmacist. However, in the HCP survey, physicians indicated they were not likely to educate patients on ADEs. Thus, a lack of education may play a large role in ADE under-reporting.
Not having enough time to devote to reporting, given priorities placed on the provision of care, was the second most common reason selected by HCPs for not reporting ADEs. The third reason was lack of integration between different HITs. Key details are spread among multiple data sources: 46 % of respondents (N = 74) indicated that drug details were spread across four or more sources and 51 % (N = 69) indicated that ADE details are spread across three or more sources. Moreover, Table 6 indicates that details important for ADE reporting—manufacturer, NDC, expiry date, and lot number—are not commonly found in EHR, CPOE, eMAR (Electronic Medication Administration Record), or BCMA (Barcode-enabled Medication Administration). These drug details may be especially important for complex or sensitive medications, such as biologics, for which it may be important to link AEs to specific manufacturers and lot numbers [25, 27]. Thus, integration of and improvements in access to information are needed to streamline the reporting process. One suggestion would be to integrate product-specific data from barcode-based systems into other HIT systems to improve the availability of drug–data elements currently missing in other systems. Table 7 contains recommendations for improving ADE reporting.
Table 7.
Recommendations for improving adverse drug event reporting
| Integrate health information systems to streamline the reporting process |
| Train and educate both healthcare providers and patients on adverse drug event reporting |
| Create a standardized adverse drug event-reporting process |
The current Tufts CSDD study has some limitations and biases. This study had a small sample size of 123 HCPs completing the survey and so may not be generalizable. The majority of hospital-based survey responses originated from three US states (New York, New Jersey, Washington). These three states have regulations related to mandatory hospital reporting of AEs [21, 22]. The findings of this study may not be generalizable to states that do not have regulations in place. Additionally, 23 % (n = 52) of hospital respondents had never reported an ADE (internally or externally) and 48 % had never reported an ADE to the FDA or a manufacturer. Individuals who have not reported an ADE may be less aware of the process than those who have, thereby skewing some of the results. Additionally, while physicians in a hospital setting started the study, none completed it, suggesting that physician perceptions were not captured for questions later in the survey. Additionally, this study was cross-sectional and used a survey tool; therefore, no causal inferences can be made. Lastly, the survey did not assess reporting requirements for near misses; i.e., an event that could have resulted in injury due to medication error and not due to an ADE, and what should be done to report near misses to the FDA. Future directions and steps will attempt to address these limitations by expanding the study to more geographic regions as well as furthering our understanding of the root causes of ADE under-reporting and assessing the types of training and education systems within hospitals and private practices.
In 1976, William Howard Wallace Inman identified seven “deadly sins” for ADE under-reporting: complacency; fear of litigation; guilt from incorrect prescribing; ambition to publish a case study; ignorance of the process; insecurity about reporting suspicions (diffidence); and indifference in role of sharing medical knowledge [15, 37]. Now, 40 years later, all sins still apply but two particularly stand out: ignorance of process and insecurity about accurately identifying the drug causing the ADE. As such, not only should improvements be made in HIT to streamline the reporting process but stakeholders such as regulatory agencies and associations should also provide stronger guidance and continuing training for HCPs to increase awareness of both the importance of reporting events and the processes that should be followed in each setting.
Conclusion
This Tufts CSDD pilot study evaluated the process for ADE reporting in hospitals (i.e., institutional settings), private practice (i.e., ambulatory settings), and retail pharmacies. Tufts CSDD administered a survey to physicians, nurses, and pharmacists to determine the overall process and gaps in ADE reporting. These gaps fell into three areas: technology, education, and the overall process. Recommendations include integrating HIT systems to streamline the reporting process, training and education for both HCPs and patients on ADE reporting, and creation of a standardized ADE reporting process.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
The authors would like to thank Paulami Naik and Jesse Moskowitz for their contributions to this study.
Compliance with Ethical Standards
Declaration of conflicting interests
Gustavo Grampp and Thomas Felix are employed by Amgen Inc., own stock, and/or hold stock options in Amgen, Inc., and have provided expert testimony on behalf of Amgen Inc. in support of legislation in US states that allows for automatic substitution of FDA-approved interchangeable biologic products with provisions to communicate and record biologic product dispensed. Gustavo Grampp and Thomas Felix have no conflicts of interest that are directly relevant to the content of the study. Carrie A. Brown, Kenneth A. Getz, and Stella Stergiopoulos have no conflicts of interest that are directly relevant to the content of this study. Carrie A. Brown was employed at Tufts University at the time the study was conducted and is now affiliated with Boston University.
Funding
This study was funded by a non-restricted grant from Amgen.
References
- 1.Couper M. Safety of medicines: a guide to detecting and reporting adverse drug reactions. Geneva: World Health Organisation. 2002. http://apps.who.int/iris/bitstream/10665/67378/1/WHO_EDM_QSM_2002.2.pdf. Accessed 13 July 2016.
- 2.Lahue BJ, Pyenson B, Iwasaki K, Blumen HE, Forray S, Rothschild JM. National burden of preventable adverse drug events associated with inpatient injectable medications: healthcare and medical professional liability costs. Am Health Drug Benefits. 2012;5(7):1–10. [PMC free article] [PubMed] [Google Scholar]
- 3.Preventing medication errors: a $21 billion opportunity. Cambridge, MA: New England Health Institute. 2012. http://www.nehi.net/bendthecurve/sup/documents/Medication_Errors_%20Brief.pdf. Accessed 18 July 2016.
- 4.Errors in Health Care: A leading Cause of Death and Injury. In: Kohn LT, Corrigan JM, Donaldson MS (Committee on Quality of Health Care in America, Institute of Medicine), editors. To err is human: building a safer health system. Washington, DC: National Academies; 2000. http://www.nap.edu/catalog/9728/to-err-is-human-building-a-safer-health-system. [PubMed]
- 5.Kohn LT, Corrigan JM, Donaldson MS. To err is human: building a safer health system. Washington, DC: Institute of Medicine; 1999. [PubMed] [Google Scholar]
- 6.Wysowski DK, Swartz L. Adverse drug event surveillance and drug withdrawals in the United States, 1969–2002: the importance of reporting suspected reactions. Arch Intern Med. 2005;165(12):1363–1369. doi: 10.1001/archinte.165.12.1363. [DOI] [PubMed] [Google Scholar]
- 7.Woo EJ. Postmarketing safety of biologics and biological devices. Spine J. 2014;14(3):560–565. doi: 10.1016/j.spinee.2013.09.056. [DOI] [PubMed] [Google Scholar]
- 8.Ahmad SR, Goetsch RA, Marks NS. Spontaneous reporting in the United States. In: Strom BL, editor. Pharmacoepidemiology. 4. Chichester: Wiley; 2006. pp. 131–159. [Google Scholar]
- 9.McClure DL, Raebel MA, Yih WK, Shoaibi A, Mullersman JE, Anderson-Smits C, et al. Mini-Sentinel methods: framework for assessment of positive results from signal refinement. Pharmacoepidemiol Drug Saf. 2014;23(1):3–8. doi: 10.1002/pds.3547. [DOI] [PubMed] [Google Scholar]
- 10.US Department of Health and Human services; Food and Drug Administration; Center for Drug Evaluation and Research (CDER); Center for Biologics Evaluation and Research (CBER). Guidance for Industry: Postmarketing Safety Reporting for Human Drug and Biological Products Including Vaccines. Draft guidance. Rockville: US FDA; 2001.
- 11.No authors listed OIG ‘state of the union’: hospitals still underreporting adverse events. Hosp Peer Rev. 2010;35(6):61–64. [PubMed] [Google Scholar]
- 12.Figueiras A, Tato F, Fontainas J, Gestal-Otero JJ. Influence of physicians’ attitudes on reporting adverse drug events: a case-control study. Med Care. 1999;37(8):809–814. doi: 10.1097/00005650-199908000-00010. [DOI] [PubMed] [Google Scholar]
- 13.Howe CL. A review of the Office of Inspector General’s reports on adverse event identification and reporting. J Healthc Risk Manag. 2011;30(4):48–54. doi: 10.1002/jhrm.20068. [DOI] [PubMed] [Google Scholar]
- 14.Pagotto C, Varallo F, Mastroianni P. Impact of educational interventions on adverse drug events reporting. Int J Technol Assess Health Care. 2013;29(4):410–417. doi: 10.1017/S0266462313000457. [DOI] [PubMed] [Google Scholar]
- 15.Varallo FR. Guimaraes Sde O, Abjaude SA, Mastroianni Pde C. Causes for the underreporting of adverse drug events by health professionals: a systematic review. Rev Esc Enferm USP. 2014;48(4):739–747. doi: 10.1590/S0080-623420140000400023. [DOI] [PubMed] [Google Scholar]
- 16.Elliott P, Martin D, Neville D. Electronic clinical safety reporting system: a benefits evaluation. JMIR Med Inform. 2014;2(1):e12. doi: 10.2196/medinform.3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Krahn T, Eichelberg M, Muller F, Gonul S, Laleci Erturkmen GB, Sinaci AA, et al. Adverse drug event notification on a semantic interoperability framework. Stud Health Technol Inform. 2014;205:111–115. [PubMed] [Google Scholar]
- 18.National Action Plan for Adverse Drug Event Prevention. Washington, DC: US Department of Health and Human Services, Office of Disease Prevention and Health Promotion; 2013.
- 19.Scott H, Rosenbaum S, Waters W, Colt A, Andrews L, Juergens J. Rhode Island physicians’ recognition and reporting of adverse drug reactions. RI Med J. 1987;70:311–316. [PubMed] [Google Scholar]
- 20.Ma P, Marinovic I, Karaca-Mandic P. Drug manufacturers’ delayed disclosure of serious and unexpected adverse events to the US food and drug administration. JAMA Int Med. 2015;175(9):1565–1566. doi: 10.1001/jamainternmed.2015.3565. [DOI] [PubMed] [Google Scholar]
- 21.Levinson D. Adverse events in hospitals: state reporting systems OEI-06-07-00471. Washington, DC: US Department of Health and Human Services, Office of Inspector General; 2008. [Google Scholar]
- 22.Hanlon C, Sheedy K, Kniffin T, Rosenthal J. 2014 guide to state adverse event reporting systems. Washington, DC: National Academy for State Health Policy; 2015. [Google Scholar]
- 23.Wright S. Memorandum report: few adverse events in hospitals were reported to state adverse event reporting systems, OEI-06-09-00092. Washington, DC: US Department of Health and Human Services, Office of Inspector General; 2012.
- 24.Klein K, Scholl JH, Vermeer NS, Broekmans AW, Van Puijenbroek EP, De Bruin ML, et al. Traceability of biologics in The Netherlands: an analysis of information-recording systems in clinical practice and spontaneous ADR reports. Drug Saf. 2016;39(2):185–192. doi: 10.1007/s40264-015-0383-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stergiopoulos S, Brown CA, Grampp G, Felix T, Getz KA. Identifying and quantifying the accuracy of product name attribution of US-sourced adverse event reports in medwatch of somatropins and insulins. Ther Innov Regul Sci. 2015;49(5):706–716. doi: 10.1177/2168479015578156. [DOI] [PubMed] [Google Scholar]
- 26.Vermeer NS, Spierings I, Mantel-Teeuwisse AK, Straus SM, Giezen TJ, Leufkens HG, et al. Traceability of biologicals: present challenges in pharmacovigilance. Exp Opin Drug Safety. 2015;14(1):63–72. doi: 10.1517/14740338.2015.972362. [DOI] [PubMed] [Google Scholar]
- 27.Vermeer NS, Straus SM, Mantel-Teeuwisse AK, Domergue F, Egberts TC, Leufkens HG, et al. Traceability of biopharmaceuticals in spontaneous reporting systems: a cross-sectional study in the FDA Adverse Event Reporting System (FAERS) and EudraVigilance databases. Drug Saf. 2013;36(8):617–625. doi: 10.1007/s40264-013-0073-3. [DOI] [PubMed] [Google Scholar]
- 28.Goldman JL, Sullins A, Sandritter T, Leeder JS, Lowry J. Pediatric pharmacovigilance: enhancing adverse drug reaction reporting in a tertiary Care Children’s Hospital. Ther Innov Regul Sci. 2013;47(5):566–571. doi: 10.1177/2168479013499153. [DOI] [PubMed] [Google Scholar]
- 29.Hirose M, Regenbogen SE, Lipsitz S, Imanaka Y, Ishizaki T, Sekimoto M, et al. Lag time in an incident reporting system at a university hospital in Japan. Qual Saf Health Care. 2007;16(2):101–104. doi: 10.1136/qshc.2006.019851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.What is an Electronic Medical Record. Washington, DC: Office of the National Coordinator for Health Information Technology; US Department of Health and Human Services. 2014 [updated 29 August 2014]. https://www.healthit.gov/providers-professionals/electronic-medical-records-emr. Accessed 15 July 2016.
- 31.Stage 2: Eligibile Hospital and Critical Access Hospital Meaningful Use Core Measures. Measure 16 of 16. Washington, DC: Centers for Medicare & Medicaid Services; 2012 [updated October 2012]. https://www.cms.gov/Regulations-and-Guidance/Legislation/EHRIncentivePrograms/downloads/Stage2_HospitalCore_16_ElectronicMedicationAdminRec_eMAR.pdf. Accessed 31 Aug 2016.
- 32.Bar-coded Medication Administration. Rockville, MD: US Department of Health and Human Services, Agency for Healthcare Research and Quality. https://healthit.ahrq.gov/ahrq-funded-projects/emerging-lessons/bar-coded-medication-administration. Accessed 15 July 2016.
- 33.American Society of Health-System Pharmacists ASHP statement on barcode verification during inventory, preparation, and dispensing of medications. Am J Health-Syst Pharm. 2011;68:442–445. doi: 10.2146/sp100012. [DOI] [PubMed] [Google Scholar]
- 34.Levinson D. Hospital incident reporting systems do not capture most patient harm. Washington, DC: US Department of Health and Human Services; 2012. [Google Scholar]
- 35.Pharmacy–Patient Medication Record Systems. Washington State; 1992. http://apps.leg.wa.gov/wac/default.aspx?cite=246-875&full=true. Accessed 15 July 2016.
- 36.Yi ZM, Zhi XJ, Yang L, Sun SS, Zhang Z, Sun ZM, et al. Identify practice gaps in medication education through surveys to patients and physicians. Patient Prefer Adherence. 2015;9:1423–1430. doi: 10.2147/PPA.S93219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Inman WHW. Assessment of drug safety problems. In: Gent M, Shigematsu I, editors. Epidemiological issues in reported drug-induced illnesses - SMON and other examples. Hamilton, ON: McMaster University Press; 1978. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.