

Abstract
RGS14 is a multifunctional scaffolding protein possessing two distinct G protein interaction sites including a regulator of G protein signaling (RGS) domain that acts as a GTPase activating protein (GAP) to deactivate Gαi/o‐GTP proteins, and a G protein regulatory (GPR) motif that binds inactive Gαi1/3‐GDP proteins independent of Gβγ. GPR interactions with Gαi recruit RGS14 to the plasma membrane to interact with Gαi‐linked GPCRs and regulate Gαi signaling. While RGS14 actions on Gα proteins are well characterized, consequent effects on Gβγ signaling remain unknown. Conventional RGS proteins act as dedicated GAPs to deactivate Gα and Gβγ signaling following receptor activation. RGS14 may do the same or, alternatively, may coordinate its actions to deactivate Gα‐GTP with the RGS domain and then capture the same Gα‐GDP via its GPR motif to prevent heterotrimer reassociation and prolong Gβγ signaling. To test this idea, we compared the regulation of G protein activation and deactivation kinetics by a conventional RGS protein, RGS4, and RGS14 in response to GPCR agonist/antagonist treatment utilizing bioluminescence resonance energy transfer (BRET). Co‐expression of either RGS4 or RGS14 inhibited the release of free Gβγ after agonist stimulation and increased the deactivation rate of Gα, consistent with their roles as GTPase activating proteins (GAPs). Overexpression of inactive Gαi1 to recruit RGS14 to the plasma membrane did not alter RGS14′s capacity to act as a GAP for a second Gαo protein. These results demonstrate the role of RGS14 as a dedicated GAP and suggest that the G protein regulatory (GPR) motif functions independently of the RGS domain and is silent in regulating GAP activity in a cellular context.
Keywords: G protein, GPCR, Gαi1, Gβγ, RGS protein, RGS14.
Abbreviations
- BRET
bioluminescence resonance energy transfer
- GAP
GTPase‐activating protein
- GPCR
G protein‐coupled receptor
- GPR
G protein regulatory
- Luc
luciferase
- RBD
Ras‐binding domain
- RGS
regulator of G protein signaling
- SE
standard error
- Ven
venus
- α2‐AR
α2 adrenergic receptor
Introduction
Canonical G protein‐coupled receptor (GPCR) signaling begins with binding of an extracellular ligand to the receptor. Upon conformational rearrangement of the GPCR, the receptor is able to stimulate the exchange of GDP for GTP in the Gα subunit of the heterotrimeric G protein (Gαβγ) (Bourne 1997). Binding of GTP by Gα results in rearrangement and sometimes dissociation of the Gαβγ heterotrimer, allowing both Gα and Gβγ to signal to downstream effector molecules (Gilman 1987; Hepler and Gilman 1992; Hamm 1998). Regulators of G protein signaling (RGS) proteins negatively regulate G protein signaling by serving as GTPase activating proteins (GAPs) that stimulate the intrinsic GTPase of the Gα subunit. Upon hydrolysis of GTP to GDP, Gα rebinds Gβγ thereby terminating the G protein signaling event (De Vries et al. 2000; Ross and Wilkie 2000; Hollinger and Hepler 2002).
Many RGS proteins have a relatively simple structure, lacking domains outside the canonical RGS domain. However, other RGS proteins have a more complicated structure. One such protein, RGS14, is a multifunctional scaffold that is highly expressed in the brain (Traver et al. 2000; Lee et al. 2010; Evans et al. 2014), and has been identified as a natural suppressor of synaptic plasticity in CA2 hippocampal neurons and of hippocampal‐based learning and memory (Lee et al. 2010). As a member of the R12 family of RGS proteins, RGS14 possesses an N‐terminal RGS domain that engages Gαi/o family members (Cho et al. 2000; Traver et al. 2000; Hollinger et al. 2001). In addition, RGS14 also has a C‐terminal G protein regulatory (GPR) motif (Hollinger et al. 2001; Kimple et al. 2001) and two tandem Ras/Rap‐binding domains (RBDs) (Traver et al. 2000). The GPR motif binds inactive Gαi1/3 proteins while the first RBD binds activated Ras/Rap proteins (Traver et al. 2000; Mittal and Linder 2004; Willard et al. 2009; Shu et al. 2010; Vellano et al. 2013).
Given its unique molecular architecture, RGS14 is primed to intercept incoming Gα signals. Previously we showed that activation of Gαi/o proteins can recruit cytosolic RGS14 to the plasma membrane through the RGS domain (Brown et al. 2015b). Moreover, co‐expression of inactive Gαi1 targets RGS14 to the plasma membrane through the GPR motif (Shu et al. 2007; Brown et al. 2015b) where RGS14 can form a Gα‐dependent complex with GPCRs (Vellano et al. 2013). Considering these two distinct Gα‐interacting sites, we have proposed a model of RGS14 function (Brown et al. 2015b) in which the RGS domain “senses” G protein activation, thereby recruiting cytosolic RGS14 to the plasma membrane. At the membrane, RGS14 accelerates the GTPase of the Gα to hydrolyze GTP to GDP. At this time, RGS14 would then be optimally positioned to bind the newly formed Gα‐GDP through its GPR motif and form a stable complex at the plasma membrane. Alternatively, a subset of RGS14 may exist in a preformed complex with Gαi‐GDP at the plasma membrane, independent of Gβγ.
Previous studies have indicated that the binding of Gα by Gβγ and the GPR motif of RGS14 are mutually exclusive (Mittal and Linder 2006; Shu et al. 2007). In support of this idea, structural characterization demonstrated that the binding site of the RGS14 GPR motif on Gα overlaps with that of Gβγ (Kimple et al. 2002). While biochemical studies have suggested the RGS14 GPR motif cannot disrupt preformed Gαβγ heterotrimers (Mittal and Linder 2006), other studies have suggested the GPR motif may prevent heterotrimer reassembly after GPCR stimulation (Webb et al. 2005). As such, RGS14 interference with the reassociation of Gα with Gβγ may prolong Gβγ signaling. To test the idea that RGS14 might prolong Gβγ signaling more than conventional RGS proteins, we utilized a bioluminescence resonance energy transfer (BRET) based biosensor for Gβγ release to monitor the activation and deactivation of heterotrimeric G proteins that interact with RGS14 (Gαo and Gαi1). Using this biosensor, we compared RGS4, a conventional RGS protein, with the unconventional RGS14 to understand the regulation of G protein heterotrimers. We examined whether RGS14 interrupts formation of Gαβγ heterotrimers by examining basal BRET ratios prior to agonist addition. Additionally, we examined whether BRET signals returned to baseline after antagonist addition to assess whether RGS14 disrupts heterotrimer reassembly after a signaling event.
Here, we show that co‐expression of RGS4 or RGS14 each limits the release of free Gβγ as well as stimulates the deactivation rate of G proteins in live cells. RGS14 does not appear to interfere with formation of Gαβγ heterotrimers either before or after receptor stimulation. Co‐expression of inactive Gαi1 with RGS14 did not alter the GAP effect on Gαo proteins. Based on these findings, we propose that the RGS domain and the GPR motif of RGS14 function independently in live cells.
Materials and Methods
Cell culture and transfection
HEK 293 cells were maintained in 1× Dulbecco's modified Eagle's medium without phenol red indicator supplemented with 10% fetal bovine serum (5% after transfection), 2 mmol/L l‐glutamine, 100 units/mL penicillin, and 100 mg/mL streptomycin. Cells were kept in a 37°C incubator supplied with 5% CO2. Transfections were carried out using polyethyleneimine (PEI) as described previously (Brown et al. 2015a).
Constructs and reagents
The hemagglutinin (HA) epitope‐tagged α2a‐adrenergic receptor (HA‐α2a‐AR) used in this study was kindly provided by Dr. Joe Blumer (Medical University of South Carolina). Hemagglutinin epitope‐tagged RGS4 (HA‐RGS4) and FLAG‐tagged RGS14 (FLAG‐RGS14) were generated as described previously (Bernstein et al. 2004; Shu et al. 2007). The pertussis‐resistant mutants (C351G) of Gαo and Gαi1 were purchased from the cDNA Resource Center (cDNA.org, Bloomsberg, PA). Mas‐GRK3ct‐Luc and Ven‐Gβγ were described previously (Hollins et al. 2009). UK 14,304 was obtained from Sigma‐Aldrich (St. Louis, MO) while rauwolscine was purchased from Tocris Bioscience (Bristol, United Kingdom). Pertussis toxin was purchased from List Biological Laboratories, Inc (Campbell, CA).
Kinetic BRET assay
For kinetic BRET experiments, HEK 293 cells seeded in six‐well plates were transfected with 25 ng mas‐GRK3ct‐Luc, 200 ng HA‐α2A‐AR, 200 ng Ven‐Gβ1, 200 ng Ven‐Gγ2, and 400 ng of either Gαo‐C351G or Gαi1‐C351G. HA‐RGS4 or FLAG‐RGS14 was also transfected in 3, 10, 30, and 100 ng amounts. For each experiment, a control using pertussis‐sensitive Gαo or Gαi1 was included to record any noise within the system. Pertussis toxin (100 ng/mL) was added at the time of transfection to limit activation of endogenous G proteins. Cells were transfected for 24 h before resuspension in Tyrode's solution (140 mmol/L NaCl, 5 mmol/L KCl, 1 mmol/L MgCl2, 1 mmol/L CaCl2, 0.37 mmol/L NaH2PO4, 24 mmol/L NaHCO3, 10 mmol/L HEPES, and 0.1% glucose, pH 7.4) and plated on white 96‐well Optiplates (Perkin Elmer Life Sciences, Waltham, MA). Fluorescence measurements to confirm acceptor expression were made using the TriStar LB 941 plate reader (Berthold Technologies, Bad Wildbad, Germany) with 485‐nm excitation and 530‐nm emission filters. After a 5 min application of 5 μmol/L coelenterazine H (Nanolight Technologies, Pinetop, AZ), kinetic BRET was monitored using sequential measurements through 485‐ and 530‐nm emission filters. BRET was recorded for 30 sec with no stimulation to establish basal BRET. After basal BRET measurements, agonist was applied for 60 sec followed by 90 sec of antagonist application. The change in BRET (ΔBRET) was calculated by dividing the mas‐GRK3ct‐Luc signal (530 nm) by the Ven‐Gβγ signal (485 nm) and subtracting the average BRET signal observed from the first 30 sec of observation (basal BRET). With each experiment, a kinetic BRET control was performed utilizing pertussis‐sensitive Gα to ensure the effectiveness of the pertussis toxin. Any signal recorded in these controls was regarded as noise and subtracted from experimental kinetic BRET recordings. Data were collected using the MikroWin 2000 software (Mikrotek Laborsysteme GmbH, Overath, Germany) and analyzed using Microsoft Excel and GraphPad Prism 5. Deactivation curves were fitted to a single‐phase decay exponential function. Statistical data analysis was performed using a one‐way analysis of variance (ANOVA) with Tukey's or Dunnett's post hoc test where indicated.
Immunoblotting
Representative immunoblots were performed using primary antibodies for α2A‐AR, Gαo, and Gαi1 from Santa Cruz Biotechnology. Primary GFP antibodies (MBL International) were used to detect Venus‐Gβ expression while mas‐GRK3ct‐Luc expression was assessed with the anti‐Luciferase antibody from Millipore. HRP‐conjugated FLAG antibodies (Sigma) were used to detect FLAG‐tagged RGS14 constructs. Proteins were then detected with enhanced chemiluminescence.
Results
Activation of α2a‐AR releases free Gβγ from Gαo and Gαi1 proteins
The goal of these studies was to compare the regulation of G protein activation and deactivation and Gβγ signaling in live cells by a conventional RGS protein (RGS4) and an unconventional RGS protein containing a second G protein‐binding GPR motif (RGS14). Ideally, this would involve measuring the real‐time kinetics of Gα and Gβγ dissociation and reassociation directly in live cells. Our initial studies attempted this using a live cell BRET assay with luciferase‐tagged Gαi (Gαi‐Luc) and a Venus‐tagged Gβγ (Gβγ‐Ven), activated with an appropriate Gαi/o‐coupled GPCR. However, we were unable to detect significant heterotrimer dissociation following receptor activation using this assay (data not shown), consistent with previous reports of G protein heterotrimer rearrangement in cells (Bunemann et al. 2003; Hepler 2014).
As an alternative approach, we took advantage of a previously reported live cell biosensor assay for Gβγ activation that involves transfecting into cells a GPCR, Gα protein, Gβγ, and a biosensor for Gβγ activation (Fig. 1A). The biosensor for Gβγ activation, designated as mas‐GRK3ct‐Luc (Hollins et al. 2009), features the C‐terminal Gβγ‐binding region of G protein‐coupled receptor kinase 3 (GRK3). A myristate attachment sequence (mas) was attached to the N‐terminus of the GRK3 Gβγ‐binding domain while Renilla Luciferase (RLuc8) was attached to the C‐terminus. The mas sequence allows the Gβγ biosensor to be targeted to the plasma membrane while the Luciferase tag serves as a BRET donor. A bimolecular fluorescence complementation technique was employed to generate a Venus‐Gβ1γ2 BRET acceptor. Residues 156–239 of Venus were fused to the N‐terminus of Gβ1 while residues 1–155 of Venus were fused to the N‐terminus of Gγ2. Upon heterodimerization, the Venus‐tagged Gβ1γ2 constructs form a functional Gβγ subunit (Hollins et al. 2009; Hynes et al. 2011). We chose this Gβγ biosensing system because the mas‐GRK3ct‐Luc biosensor provides a clear indication of heterotrimer activation (Hollins et al. 2009; Masuho et al. 2015a,b). Additionally, we chose the α2A‐adrenergic receptor as we have studied it previously and demonstrated that RGS14 can associate with the receptor in a Gαi1‐dependent manner (Vellano et al. 2011, 2013).
Figure 1.
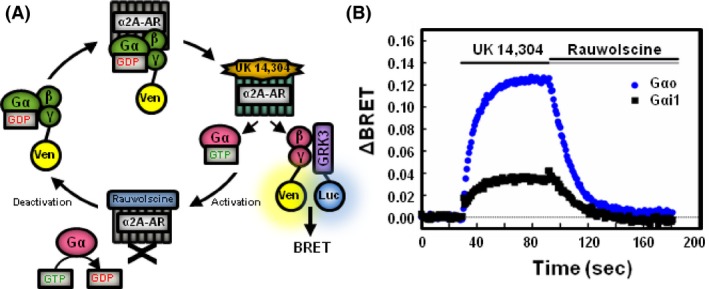
Gαo releases more free Gβγ than Gαi1 following receptor activation. (A) Schematic representation of kinetic BRET experiment. Venus‐tagged Gβ1 and Gγ2 form a dimer that binds inactive Gα‐GDP. Addition of α2A‐adrenergic receptor agonist UK 14,304 stimulates Gα to bind GTP and releases Venus‐Gβγ. During the activation phase, free Venus‐Gβγ binds the mas‐GRK3ct‐Luc to produce a rise in BRET. Addition of rauwolscine halts the activation of Gα proteins. During the deactivation phase, Gα hydrolyzes GTP to GDP and reforms a heterotrimer with Gβγ, quenching the BRET signal. (B) HEK 293 cells were transfected with 200 ng of α2A‐adrenergic receptor, 200 ng of Venus‐Gβ1, 200 ng of Venus‐Gγ2, 25 ng of mas‐GRK3ct‐Luc, and 400 ng of either Gαo or Gαi1. Baseline BRET was measured for 30 sec prior to addition of agonist. Alpha2A‐AR agonist (1 μmol/L UK 14,304) was added for 60 sec followed by a 90 sec application of antagonist (100 μmol/L rauwolscine). Data are expressed as average whole traces of BRET in cells expressing Gαo (blue, n = 3) and Gαi1 (black, n = 4).
HEK 293 cells were transfected with the α2A‐adrenergic receptor (α2A‐AR), mas‐GRK3ct‐Luc, Venus‐Gβγ and either Gαo or Gαi1, each containing a mutation conferring pertussis toxin‐resistance (C351G) and then treated with pertussis toxin to eliminate signaling contributions from endogenous G proteins. Addition of α2a‐AR agonist UK 14,304 (1 μmol/L) activates the G protein heterotrimer and produces dissociation of the Gα and Gβγ subunits. Free Gβγ binds the Gβγ activation biosensor as indicated by the rise in ΔBRET (Fig. 1B). After 60 sec of agonist application, addition of α2a‐AR antagonist rauwolscine (100 μmol/L) rapidly halts activation of G proteins and allows Gα proteins to hydrolyze GTP to GDP. Gβγ dissociates from the Gβγ biosensor to reform a heterotrimer with Gα as indicated by a decrease in ΔBRET.
Agonist‐induced activation of Gαo proteins generated a larger ΔBRET than Gαi1. Cells expressing Gαo produced a maximum ΔBRET of 0.129 (±0.020 SE) while cells expressing Gαi1 produced a maximum ΔBRET of 0.040 (±0.002 SE) (Fig. 1B). These results are consistent with other reports suggesting Gαi1 proteins do not release as much free Gβγ as Gαo proteins (Digby et al. 2008; Masuho et al. 2015b).
Expression of RGS4 reduces the release of free Gβγ and accelerates the deactivation rate of G proteins
To determine the effect of an RGS protein on free Gβγ release, we co‐expressed increasing amounts of RGS4 (Figs. 2A, 3A). We chose RGS4 because it is a well‐studied RGS protein that features a relatively simple protein structure, lacking domains outside the RGS domain. In cells expressing Gαo, expression of 100 ng of RGS4 significantly reduced the maximum ΔBRET from 0.112 (±0.010 SE) to 0.078 (±0.003 SE, P < 0.01) (Fig. 2B). In cells expressing Gαi1, co‐expression of RGS4 did not result in a significant decrease in maximum ΔBRET though a trend appears to be present (Fig. 3B).
Figure 2.
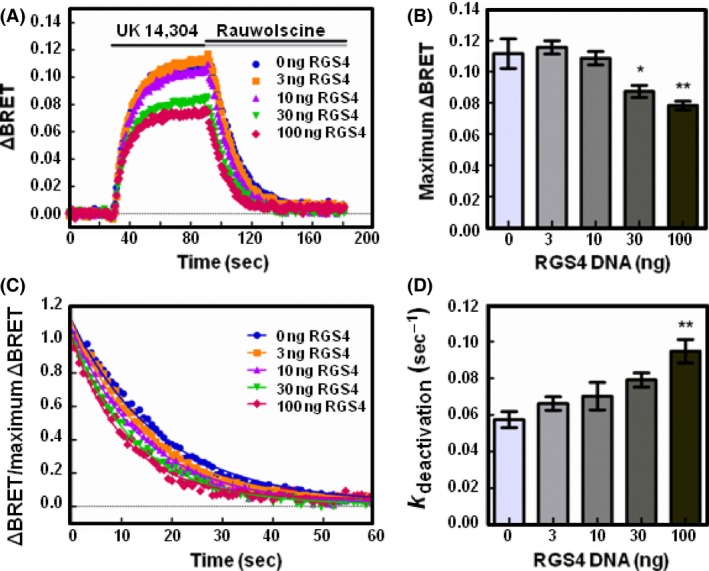
RGS4 reduces the release of free Gβγ and accelerates the deactivation of Gαo proteins following receptor activation. (A) HEK 293 cells were transfected with 200 ng of α2A‐adrenergic receptor, 200 ng of Venus‐Gβ1, 200 ng of Venus‐Gγ2, 25 ng of mas‐GRK3ct‐Luc, and 400 ng of Gαo and monitored for kinetic BRET as in Figure 1. Average whole traces of BRET signal over time are shown from cells expressing Gαo alone and 3, 10, 30, or 100 ng of HA‐RGS4 (n = 3). (B) Maximum ΔBRET observed from data presented in (A). (C) Deactivation curves normalized to maximum BRET and fit using a single exponential decay function. (D) Deactivation rates (k deactivation) determined from curves fit in (C). Error bars represent ± S.E. Statistical analysis was performed using a one‐way analysis of variance (ANOVA) with Tukey's post hoc test (*P < 0.05, **P < 0.01).
Figure 3.
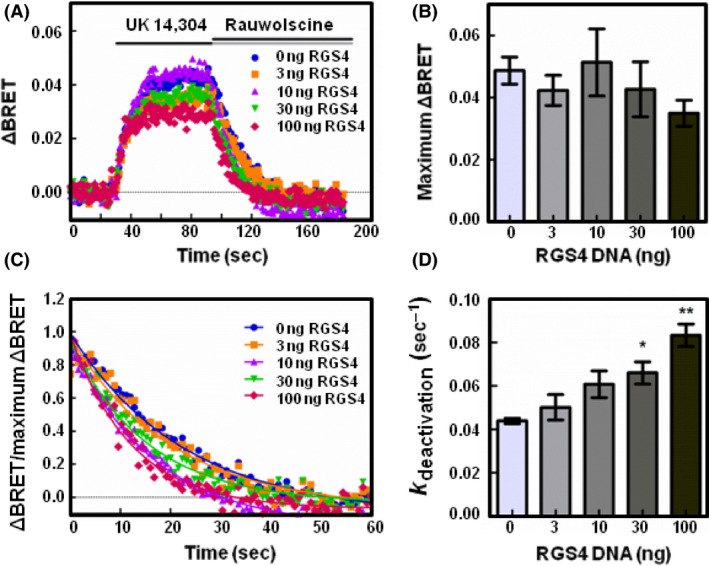
RGS4 accelerates the deactivation of Gαi1 proteins following receptor activation. (A) HEK 293 cells were transfected with 200 ng of α2A‐adrenergic receptor, 200 ng of Venus‐Gβ1, 200 ng of Venus‐Gγ2, 25 ng of mas‐GRK3ct‐Luc, and 400 ng of Gαi1 and monitored for kinetic BRET as in Figure 1. Average whole traces of BRET signal over time are shown from cells expressing Gαi1 alone and 3, 10, 30, or 100 ng of HA‐RGS4 (n = 3). (B) Maximum ΔBRET observed from data presented in (A). (C) Deactivation curves normalized to Maximum BRET and fit using a single exponential decay function. (D) Deactivation rates (k deactivation) determined from curves fit in (C). Error bars represent ± S.E. Statistical analysis was performed using a one‐way analysis of variance (ANOVA) with Tukey's post hoc test (*P < 0.05, **P < 0.01).
We then examined the effect of an RGS on G protein deactivation. The association of Gβγ with the biosensor is governed by the activation state of Gα. Upon heterotrimer activation and formation of Gα‐GTP, Gβγ is released and binds the Gβγ biosensor. Upon hydrolysis of GTP to GDP, Gα‐GDP regains affinity for Gβγ and Gβγ dissociates from the biosensor to reassociate with Gα‐GDP. As Gβγ association with the biosensor is directly tied to the activation state of Gα, the deactivation rate serves as an indirect measure of the Gα GTPase rate (Lambert et al. 2010). To determine the deactivation rate, we fit the BRET curve dissociation phase with a single‐phase exponential decay function (Figs. 2C, 3C).
In cells expressing Gαo alone, the deactivation rate was determined to be 0.057 sec−1 (±0.004 SE) (Fig. 2D). In cells expressing 100 ng RGS4, the deactivation rate increased significantly to 0.095 sec−1 (±0.006 SE, P < 0.01). In cells expressing Gαi1, 100 ng of RGS4 expression significantly increased the deactivation rate from 0.044 sec−1 (±0.001 SE) to 0.083 sec−1 (±0.005 SE, P < 0.01) (Fig. 3D). These results correspond with the decreased maximum ΔBRET observed in Figures 2B, 3B and highlight the utility of the Gβγ biosensor in detecting free Gβγ release.
Expression of RGS14 reduces the release of free Gβγ and accelerates the deactivation rate of G proteins
We then sought to compare the effects of RGS14 with RGS4 on the release of Gβγ and deactivation rate. Unlike most RGS proteins including RGS4, RGS14 contains tandem RBDs and a GPR motif in addition to the RGS domain that could regulate the RGS14 effects on G protein heterotrimer activity. Similar to RGS4 (Figs. 2, 3), increasing expression of RGS14 reduced the maximum ΔBRET observed in both Gαo‐ and Gαi1‐expressing cells (Figs. 4A, 5A). In cells expressing Gαo, co‐expression of 100 ng of RGS14 decreased the maximum ΔBRET from 0.129 (±0.020 SE) to 0.023 (±0.009 SE, P < 0.01) (Fig. 4B). Similar results were obtained in cells expressing Gαi1 where the maximum ΔBRET recorded for Gαi1 decreased from 0.040 (±0.002 SE) to 0.014 (±0.001 SE P < 0.05) in cells co‐expressing 100 ng RGS14 (Fig. 5B). These results indicate that RGS14 limits the release of free Gβγ from both Gαo and Gαi1 proteins.
Figure 4.
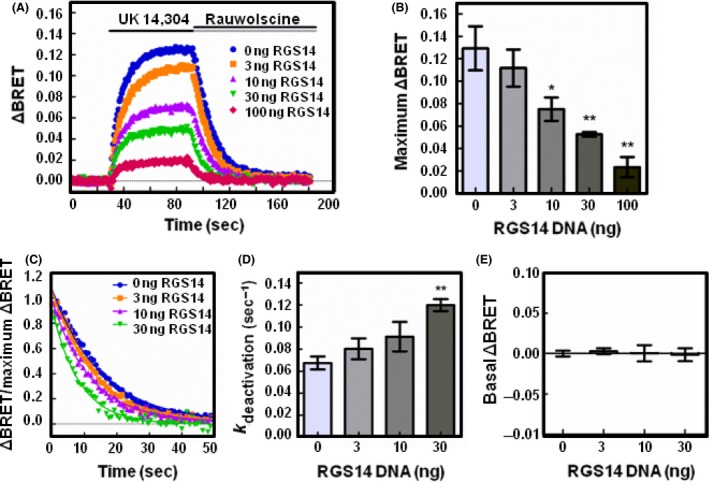
RGS14 expression reduces the release of free Gβγ and accelerates the deactivation of Gαo proteins following receptor activation. (A) HEK 293 cells were transfected with 200 ng of α2A‐adrenergic receptor, 200 ng of Venus‐Gβ1, 200 ng of Venus‐Gγ2, 25 ng of mas‐GRK3ct‐Luc, and 400 ng of Gαo and monitored for kinetic BRET as in Figure 1. Average whole traces of BRET signal over time are shown from cells expressing Gαo alone and 3, 10, 30, or 100 ng of FLAG‐RGS14 (n = 3). (B) Maximum ΔBRET observed from data presented in (A). (C) Deactivation curves normalized to maximum ΔBRET and fit using a single exponential decay function. (D) Deactivation rates (k deactivation) determined from curves fit in (C). (E) Average BRET measured for 30 sec at baseline, prior to agonist stimulation. Error bars represent ± S.E. Statistical analysis was performed using a one‐way analysis of variance (ANOVA) with Tukey's post hoc test (*P < 0.05, **P < 0.01).
Figure 5.
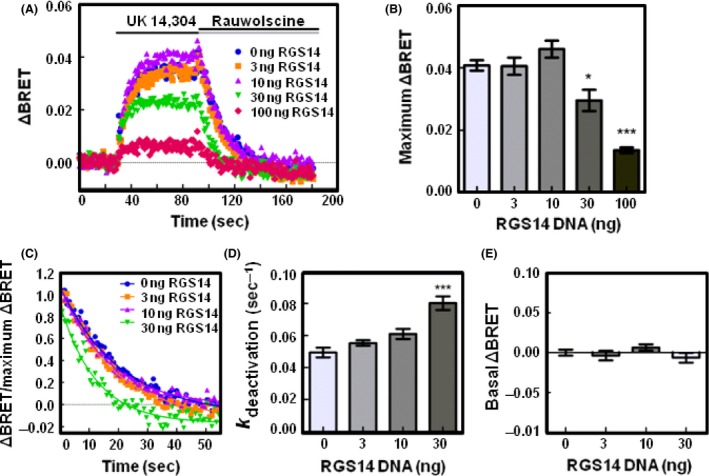
RGS14 reduces the release of free Gβγ and accelerates the deactivation of Gαi1 proteins following receptor activation. (A) HEK 293 cells were transfected with 200 ng of α2A‐adrenergic receptor, 200 ng of Venus‐Gβ1, 200 ng of Venus‐Gγ2, 25 ng of mas‐GRK3ct‐Luc, and 400 ng of Gαi1 and monitored for kinetic BRET as in Figure 1. Average whole traces of BRET signal over time are shown from cells expressing Gαi1 alone and 3, 10, 30, or 100 ng of FLAG‐RGS14 (n = 4). (B) Maximum ΔBRET observed from data presented in (A). (C) Deactivation curves normalized to maximum BRET and fit using a single exponential decay function. (D) Deactivation rates (k deactivation) determined from curves fit in (C). (E) Average BRET measured for 30 sec at baseline, prior to agonist stimulation. Error bars represent ± S.E. Statistical analysis was performed using a one‐way analysis of variance (ANOVA) with Tukey's post hoc test (*P < 0.05, ***P < 0.001).
Next, we determined the deactivation kinetics of G proteins in response to RGS14 expression (Figs. 4C, 5C). In conditions transfected with 100 ng of RGS14, the ΔBRET was not large enough to reliably fit the deactivation curve, thus we limited this analysis to conditions transfected with 0, 3, 10, and 30 ng of RGS14. In cells expressing Gαo, the deactivation rate was 0.067 sec−1 (±0.006 SE) (Fig. 4D). Co‐expression of 30 ng of RGS14 significantly increased the deactivation rate to 0.120 sec−1 (±0.006 SE, P < 0.05), indicative of RGS14 GAP activity. In cells expressing Gαi1, the deactivation rate was 0.049 sec−1 (±0.003 SE) which increased to 0.080 sec−1 (±0.004 SE) upon co‐expression of 30 ng of RGS14 (Fig. 5D).
RGS14 does not interrupt formation of Gαβγ heterotrimers
To assess whether RGS14 alters the formation of Gαβγ heterotrimers prior to agonist stimulation, we then examined the basal BRET. As the GPR motif only interacts with Gαi1/3 proteins, we did not expect RGS14 to alter basal BRET of Gαo proteins. Prior to activation with receptor agonist, we recorded a basal ΔBRET value of 0.00 (±0.003 SE). Upon expression of RGS14, no significant difference in basal ΔBRET values was observed across a range of increasing RGS14 amounts (Fig. 4E). For Gαi1, we observed a basal ΔBRET value of 0.00 (±0.004 SE) and no significant difference was observed upon expression of increasing amounts of RGS14 (Fig. 5E). These results suggest co‐expression of RGS14 does not alter the basal formation of Gαβγ heterotrimers.
Additionally, based on previous observations (Webb et al. 2005; Mittal and Linder 2006), we predicted that RGS14 may be able to interfere with the capacity of the Gαi1 and Gβγ subunits to reform a heterotrimer. If RGS14 binding to Gαi1 through the GPR motif prevented Gβγ from rebinding Gαi1, we would not expect the ΔBRET to return to baseline after addition of antagonist. Upon examination of deactivation curves, we did not observe any significant difference in the return to baseline after addition of antagonist. These results suggest that RGS14 does not interfere with heterotrimer reformation after receptor stimulation.
RGS‐null mutant of RGS14 cannot accelerate GTPase while the GPR‐null RGS14 mutant retains GAP activity
Next, to elucidate the roles of the RGS domain and GPR motif on heterotrimer kinetics, we employed RGS‐null and GPR‐null mutants of RGS14 (Fig. 6). To examine whether the accelerated deactivation rates were due to the RGS domain, we employed an RGS‐null mutation of RGS14 (E92A/N93A). In cells expressing Gαo, co‐expression of 30 ng RGS‐null RGS14 resulted in a deactivation rate of 0.065 sec−1 (±0.009 SE), which did not differ from Gαo alone. Moreover, expression of a GPR‐null mutant of RGS14 (Q515A/R516A) increased the deactivation rate to 0.132 sec−1 (±0.012 SE), which did not differ from RGS14‐WT. Similar results were obtained in cells co‐expressing RGS14 mutants with Gαi1 proteins (Fig. 7). Expression of 30 ng of RGS‐null RGS14 resulted in a deactivation rate of 0.052 sec−1 (±0.004 SE), which did not differ from Gαi1 alone. Expression of 30 ng of GPR‐null RGS14 resulted in a deactivation rate of 0.093 sec−1 (±0.009 SE), which did not differ from RGS14‐WT.
Figure 6.
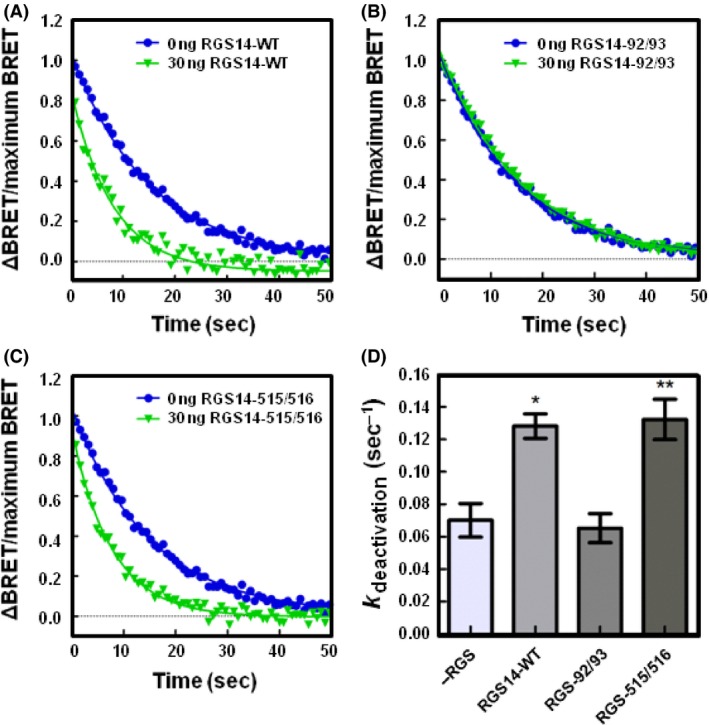
RGS14 accelerates the deactivation of Gαo proteins through its RGS domain following receptor activation. (A) HEK 293 cells were transfected with 200 ng of α2A‐adrenergic receptor, 200 ng of Venus‐Gβ1, 200 ng of Venus‐Gγ2, 25 ng of mas‐GRK3ct‐Luc, and 400 ng of Gαo and monitored for kinetic BRET as in Figure 1. Deactivation curves were normalized to Maximum BRET and fit using a single exponential decay function in cells expressing Gαo and either 0 ng or 30 ng FLAG‐RGS14‐WT (n = 3). (B) Deactivation curves as in (A) in cells expressing Gαo and either 0 ng or 30 ng FLAG‐RGS14‐E92A/N93A (n = 3). (C) Deactivation curves as in (A) in cells expressing Gαo and either 0 ng or 30 ng FLAG‐RGS14‐Q515A/R516A (n = 3). (D) Deactivation rates (k deactivation) determined from curves fit in (A, B, and C). Error bars represent ± S.E. Statistical analysis was performed using a one‐way analysis of variance (ANOVA) with Dunnett's post hoc test (*P < 0.05, **P < 0.01).
Figure 7.
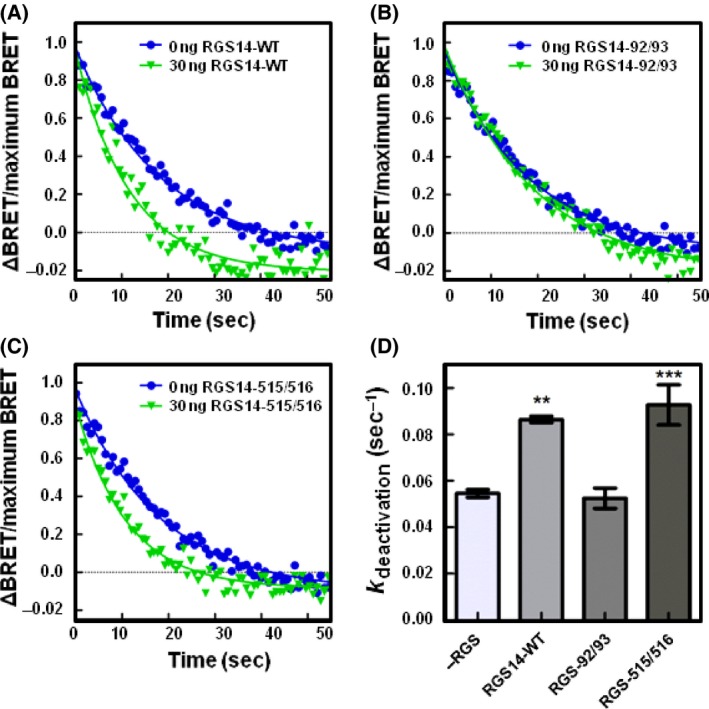
RGS14 accelerates the deactivation of Gαi1 proteins through its RGS domain following receptor activation. (A) HEK 293 cells were transfected with 200 ng of α2A‐adrenergic receptor, 200 ng of Venus‐Gβ1, 200 ng of Venus‐Gγ2, 25 ng of mas‐GRK3ct‐Luc, and 400 ng of Gαi1 and monitored for kinetic BRET as in Figure 1. Deactivation curves were normalized to maximum BRET and fit using a single exponential decay function in cells expressing Gαi1 and either 0 ng or 30 ng FLAG‐RGS14‐WT (n = 3). (B) Deactivation curves as in (A) in cells expressing Gαi1 and either 0 ng or 30 ng FLAG‐RGS14‐E92A/N93A (n = 3). (C) Deactivation curves as in (A) in cells expressing Gαi1 and either 0 ng or 30 ng FLAG‐RGS14‐Q515A/R516A (n = 3). (D) Deactivation rates (k deactivation) determined from curves fit in (A, B, and C). Error bars represent ± S.E. Statistical analysis was performed using a one‐way analysis of variance (ANOVA) with Dunnett's post hoc test (**P < 0.01, ***P < 0.001). RGS, Regulators of G protein signaling.
RGS14 GAP function on Gαo is unaltered by GPR binding of Gαi1
Our previous investigations of RGS14 revealed that, when bound to an inactive Gα at the GPR motif, RGS14 undergoes conformational rearrangements within the RGS domain (Brown et al. 2015b). Upon measurement of RGS domain function utilizing in vitro GTPase assays, the preformed RGS14:Gαi1‐GDP complex showed no difference in its ability to catalyze GTPase activity in Gαo when compared to apo‐RGS14 (Brown et al. 2015b). Thus, despite binding an inactive G protein to the GPR motif, RGS14 retained RGS function to stimulate the GTPase of a second Gα at the RGS domain (Brown et al. 2015b). Moreover, we were able to demonstrate the capacity of RGS14 to form a ternary complex with an inactive Gα at the GPR motif and an active Gα at the RGS domain (Brown et al. 2015b).
To further demonstrate the role of the GPR motif in regulating RGS function in live cells, we examined whether co‐expression of Gαi1 altered RGS14 effects on Gαo heterotrimers (Fig. 8A). For these experiments, we utilized pertussis‐resistant Gαo and pertussis‐sensitive Gαi1 proteins to ensure that the recorded signal was due to Gαo activation. As a control, we examined the interaction with RGS14 and Gαi1 in the presence and absence of pertussis toxin. While treatment with pertussis toxin uncouples G proteins from GPCRs, pertussis toxin treatment did not alter the interaction between Gαi1 and the GPR motif of RGS14 (Fig. 8B).
Figure 8.
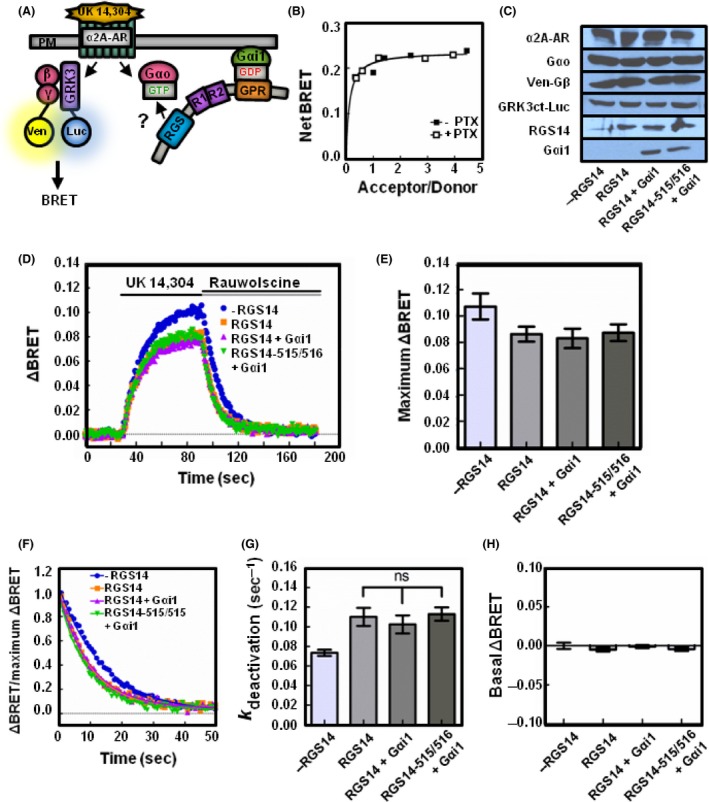
RGS14 GAP function on Gαo is unaltered by GPR binding of Gαi1. (A) Schematic representation of BRET experiment. Expression of inactive Gαi1 recruits RGS14 to the plasma membrane through the GPR motif. Agonist binding of the α2A‐adrenergic receptor activates the heterotrimeric G protein. Venus‐Gβ1γ2 binds mas‐GRK3ct to produce a rise in the BRET signal. RGS14 placement at the plasma membrane may accelerate GTP hydrolysis by Gαo. (B) HEK 293 cells were transfected with 10 ng RGS14‐Luc and 0, 50, 100, 250, 500, or 750 ng of Gαi1‐YFP in the presence or absence of 100 ng/mL of pertussis toxin. BRET ratios were recorded, and net BRET was calculated by subtracting the BRET signal from the luciferase alone (n = 3). (C) Representative immunoblots for HEK 293 cells transfected with 200 ng of α2A‐adrenergic receptor, 200 ng of Venus‐Gβ1, 200 ng of Venus‐Gγ2, 25 ng of mas‐GRK3ct‐Luc, 400 ng of pertussis‐resistant Gαo as well as 10 ng of FLAG‐RGS14/FLAG‐RGS14‐515/516 and 300 ng of pertussis‐sensitive Gαi1 where indicated. (D) Kinetic BRET was monitored for as in Figure 1 (n = 4). (E) Maximum ΔBRET observed from data presented in (D). (F) Deactivation curves were normalized to Maximum BRET and fit using a single exponential decay function (n = 4). (G) Deactivation rates (k deactivation) were determined from curves fit in (D). (H) Average BRET measured for 30 sec at baseline, prior to agonist stimulation. Error bars represent ± S.E. Statistical analysis was performed using a one‐way analysis of variance (ANOVA) with Tukey's post hoc test. GAP, GTPase activating protein; GPR, G protein regulatory.
In cells expressing Gαo, co‐expression of 10 ng of RGS14 (Fig. 8C) resulted in a maximum ΔBRET of 0.0862 (±0.006 SE) (Fig. 8D and E). Upon co‐expression of RGS14 and 300 ng of pertussis‐sensitive Gαi1, the observed maximum ΔBRET was 0.0830 (±0.007 SE) (Fig. 8D and E), demonstrating no difference from RGS14 expressed alone. Co‐expression of a GPR‐null (RGS14‐515/516) confirmed the observed effect on ΔBRET was due to an intact RGS domain (Fig. 8E). Additionally, the enhanced deactivation rate observed with expression of RGS14 was unchanged with co‐expression of Gαi1. In cells expressing RGS14 alone, the observed deactivation rate was 0.110 sec−1 (±0.009 SE) while cells expressing RGS14 and 300 ng of pertussis‐sensitive Gαi1 demonstrated an observed deactivation rate of 0.1023 sec−1 (±0.009 SE) (Fig. 8F and G). Co‐expression of either RGS14 or RGS14 and Gαi1 did not alter the basal BRET (Fig. 8H). These results suggest that Gα interactions at the GPR motif do not alter RGS domain function on a separate G protein.
Discussion
The primary goal of this work was to compare and determine the effects of a conventional RGS protein (RGS4) with those of an unconventional RGS protein with two G protein‐binding domains (RGS14) on G protein activation/deactivation kinetics and Gβγ signaling. Many conventional RGS proteins interact directly with the receptor/G protein complex by one or more mechanisms to deactivate both Gα and Gβγ signaling (Neitzel and Hepler 2006). RGS14, by contrast, with both a GPR motif and an RGS domain, exerts unique regulation of G protein signaling that has yet to be fully elucidated. Our previous work revealed that RGS14 can bind two Gα subunits simultaneously, one active Gα‐GTP at the RGS domain and one inactive Gα‐GDP at the GPR motif (Brown et al. 2015b). In vitro GTPase assays showed that binding of inactive Gα‐GDP to the GPR motif does not alter the GAP function of the RGS domain (Brown et al. 2015b). While this biochemical assessment provided key insights into the mechanics of RGS14 function, how this information would translate to a cellular environment remained unclear. Moreover, these previous findings did not clarify the effects RGS14 regulation of Gα would have on Gβγ signaling. Two possible models exist: (1) the RGS domain of RGS14 could operate independently of the GPR motif and serve as a dedicated GAP that deactivates both Gα and Gβγ signaling, similar to other conventional RGS proteins, or (2) the RGS domain and the GPR motif could work cooperatively to deactivate and capture a single inactive Gαi‐GDP, thereby preventing G protein heterotrimer reassociation and prolonging Gβγ signaling. Our findings in this study are most consistent with the former model, and suggest that the GPR motif is functionally silent in regulating RGS14 GAP activity in a cellular context. This model suggests that RGS14 may exist as a preformed dimeric complex with Gαi‐GDP at membranes, positioned to act as a GAP on a separate nearby GPCR/G protein signaling event without prolonging Gβγ signaling.
Gαo releases more free Gβγ than Gαi1
Upon examining ΔBRET in our reconstituted GPCR/G protein signaling system, an apparent difference in free Gβγ released by Gαo and Gαi1 emerged. Activation of Gαo‐containing heterotrimers resulted in an approximately threefold greater increase in Gβγ‐GRK BRET activity (indicative of release of free Gβγ) than that observed for activation of Gαi1‐containing heterotrimers. Similar studies examining Gβγ release have also demonstrated larger maximum ΔBRET signals from Gαo than Gαi1 transfected with similar Gα:Gβγ ratios as we present here (Masuho et al. 2015b). Reasons for this difference in free Gβγ release from the two different Gα are unclear, but could be due to differential delivery of Gαo heterotrimers to the plasma membrane than Gαi1. Alternatively, differential coupling efficiency of heterotrimers with the GPCR or differential dissociation of heterotrimers after activation could underlie these differences. These possibilities are not mutually exclusive and both may contribute to the phenomena observed. In a study examining the release of free Gβγ dimers from Gαo and Gαi1, Gβγ was more readily released from Gαo activation suggesting Gαi1 may have a higher affinity for Gβγ than Gαo (Digby et al. 2008). In vitro analysis of G protein affinities revealed Gαi1 has a higher affinity for Gβγ than Gαo (Sarvazyan et al. 2002). Thus, the differences we observe in free Gβγ release may be due to the higher affinity of Gαi1 for Gβγ than Gαo.
RGS14 and RGS4 both limit the release of free Gβγ and accelerate the deactivation rates of Gαo‐ and Gαi1‐containing G proteins
We examined the effects of a conventional RGS protein, RGS4, and an unconventional RGS protein, RGS14, on the kinetics of G protein heterotrimer dissociation and reassociation. Somewhat surprisingly, RGS14 exhibited similar effects as RGS4 on G protein subunit kinetics, despite having a second distinct G protein‐binding domain in addition to the RGS domain. Increased expression of either RGS protein significantly inhibited the release of free Gβγ (ΔBRET). Moreover, increased RGS expression resulted in increased rates of deactivation for both Gαo and Gαi1 proteins.
The BRET signal amplitude is governed by the balance of activation (release of Gβγ from Gα), and deactivation of G protein heterotrimers, measured indirectly as release of Gβγ from the GRK BRET sensor. As RGS proteins stimulate G protein GTPase activity, RGS proteins accelerate the deactivation rate and lower the ΔBRET signal as fewer G proteins are active upon addition of agonist. This was observed in the present work since increased RGS expression resulted in decreased ΔBRET. The increase in deactivation rate due to RGS14 was relatively small (approximately twofold), especially when compared to the profound (more than 100‐fold) increase in the rate of Gα GTPase stimulated by RGS proteins in biochemical assays using pure proteins (Ross and Wilkie 2000). Several factors likely contribute to this discrepancy. At a technical level, our assays were limited by the amount of RGS14 that could be transfected (30 ng) because additional RGS14 greatly reduced the ΔBRET so that deactivation curves could not be reliably fit, thereby limiting our interpretation. Other cellular factors also must be considered. First, the GTPase acceleration by RGS defined biochemically compares a Gα in the absence of RGS to one in the presence of RGS. However, our cell‐based studies compare a G protein with endogenous RGS to a G protein with endogenous plus additional exogenous RGS. Therefore, the basal Gα GTPase state in HEK cells is not RGS‐null, and the basal deactivation rate of Gα in cells is faster due to the presence of endogenous RGS (Lambert et al. 2010). Thus, the relatively small increase in deactivation rate (twofold) we observe in HEK cells with addition of recombinant RGS14 or RGS4 is very similar to the modest effects previously reported. Second, the Gβγ‐GRK BRET assay does not directly measure Gα GTPase activity and heterotrimer reassociation, but rather it directly measures Gβγ association with and dissociation from the GRK BRET sensor. Thus, one possible contribution to the slow Gβγ deactivation rate may be that the rate of release of Gβγ from GRK is slower than the rate of RGS‐directed formation of Gα‐GDP. Consistent with this idea, the rate of deactivation of GIRK channels is increased only four‐ to fivefold by RGS4 (Doupnik et al. 2004), in this case also reflecting the rate of dissociation of Gβγ from GIRK, and is much slower than the accelerated rate of Gα‐GTP hydrolysis stimulated by RGS4 (Ross and Wilkie 2000). Lastly, the BRET tag on the Gβγ may impede heterotrimer reassociation and/or the presence of endogenous Gβγ may compete with recombinant protein.
Given these caveats, we also determined whether the increase in rate of G protein deactivation by RGS14 was due solely to the RGS domain or had contributions from the GPR motif. Expression of an RGS‐null mutant of RGS14 (E92A/N93A) abolished the increase in deactivation rate observed with RGS14‐WT, whereas expression of a GPR‐null mutant of RGS14 (Q515A/R516A) did not alter the deactivation rate observed with RGS14‐WT. These results highlight the fact that the deactivation of G protein signaling by RGS14 is due solely to the RGS domain, and that the GPR motif does not slow reassociation of heterotrimers. We postulate that the RGS14 association with Gαi1‐GDP via the GPR motif likely serves a primary role of stably anchoring RGS14 to membranes (Shu et al. 2007), but plays no role in the regulation of RGS14 GAP activity (Brown et al. 2015b).
RGS14 does not alter the formation of Gαi1 heterotrimers with Gβγ
Binding of Gαi1 by the RGS14 GPR motif and Gβγ are mutually exclusive (Kimple et al. 2002; Mittal and Linder 2004; Shu et al. 2007). Previous in vitro studies have suggested that the RGS14 GPR motif cannot displace Gβγ from a preformed Gαβγ heterotrimer (Mittal and Linder 2006). However, other studies have suggested that an isolated RGS14 GPR motif peptide can prevent heterotrimer assembly in vitro, and alter Gβγ signaling to GIRK channels in response to D2S dopamine receptor activation (Webb et al. 2005). To examine whether co‐expression of RGS14 alters the basal formation of Gαβγ heterotrimers in cells, we examined basal BRET prior to agonist stimulation. Since the RGS14 GPR motif is selective for Gαi1 (Hollinger et al. 2001), we expected co‐expression of RGS14 to disrupt basal BRET in Gαi1‐expressing cells, but not Gαo‐expressing cells. However, in cells expressing either Gαi1 or Gαo, we saw no difference in basal BRET upon co‐expression of RGS14. These results suggest that RGS14 does not alter the assembly of Gαβγ heterotrimers in live cells, consistent with previous observations with purified proteins (Mittal and Linder 2006).
Although RGS14 does not appear to disrupt the basal formation of Gαβγ heterotrimers, it may be able to bind Gα‐GDP following a GPCR‐mediated signaling event. As the GPR motif does not bind Gαo (Hollinger et al. 2001), we did not see a difference in return to baseline BRET in cells expressing Gαo (Fig. 4). Surprisingly, we also did not see a difference in return to baseline in cells expressing Gαi1 (Fig. 5). These results suggest that RGS14 does not interfere with heterotrimer assembly prior to or after agonist stimulation, and does not prolong Gβγ signaling, suggesting that RGS14 may be prebound to the plasma membrane by a distinct Gαi that is bound to the GPR motif and uninvolved with the receptor signaling event.
Previous experiments demonstrating the RGS14 GPR motif could disrupt heterotrimer formation were performed with isolated GPR motif peptides (Webb et al. 2005; Mittal and Linder 2006), while the present experiments were performed with the full‐length RGS14 protein. It is possible that an isolated GPR motif behaves differently than a GPR motif in a full‐length RGS14 protein. The effect of an isolated RGS14 GPR motif on Gβγ release was not investigated in this study because a mammalian truncation expression construct containing only the GPR motif does not express well in HEK 293 cells (unpubl. obs.). Moreover, our investigation focused on the full‐length RGS14 as we attempt to understand the interdomain regulation of G proteins by RGS14.
Interactions of the GPR motif with Gαi1 does not alter RGS14 GAP activity of the RGS domain toward Gαo
The presence of a second G protein‐binding motif (GPR) in RGS14 presents a unique mechanism for further regulation of G protein signaling in addition to the RGS domain. Previously we have demonstrated that upon co‐expression of inactive Gαi1, RGS14 is targeted to the plasma membrane (Shu et al. 2007; Brown et al. 2015b) where it can associate with GPCRs via Gα (Vellano et al. 2011). We previously proposed that GPR associations with membrane‐bound G proteins could facilitate RGS14 RGS GAP activity by physically bringing RGS14 to the site of G protein signaling (Brown et al. 2015b). Recent evidence suggests that membrane association of R7 RGS family members, specifically RGS7 and RGS9‐2, potentiated the GAP effect of the RGS domain (Muntean and Martemyanov 2016). Results obtained in this study suggest that RGS14 association with the plasma membrane via the GPR motif does not enhance the deactivation kinetics of RGS14‐accelerated G protein deactivation (Fig. 8). Taken together, these results suggest that the RGS domain and GPR motif function independently from one another, perhaps on distinct Gα – one involved with receptor signaling and a second that anchors RGS14 to the plasma membrane.
While we did not observe a change in Gαo kinetics upon co‐expression of Gαi1, our studies do not address the possibility that our treatment of cells with pertussis toxin may have altered the ability of Gαi1 to recruit RGS14 to the site of G protein activation. Previously, we and others have shown that GPR‐containing proteins can associate with GPCRs (Vellano et al. 2011; Robichaux et al. 2015), an association that is dependent on Gα. Pertussis toxin‐mediated uncoupling of receptor/Gαi1 may not optimally position RGS14:Gαi1 complexes to favor interaction with GPCRs. However, given that these studies were completed with expressed recombinant proteins, the density of G proteins at the plasma membrane likely favored RGS14:Gαi1 complex placement near GPCRs.
Concluding remarks
Here, we compared the effects of distinct RGS proteins on Gαi/o heterotrimeric signaling. Our results indicate that the conventional RGS protein RGS4 and the unconventional RGS14 each similarly limit the release of free Gβγ and accelerate the deactivation rate of Gα. The GAP effect of RGS14 is solely due to the RGS domain since binding of an inactive G protein to the GPR motif does not alter the GAP effect of the RGS domain. The results presented here suggest the RGS domain and GPR motif do not coordinate to regulate G protein signaling. We propose that despite RGS14 containing two Gα interaction sites, the GPR motif serves to anchor RGS14 in complex with Gαi‐GDP at membranes independently of Gβγ and is functionally silent with regard to RGS domain‐mediated GAP activity.
Author Contributions
Participated in research design: Brown, Lambert, Hepler. Conducted experiments: Brown. Contributed new reagents or analytic tools: Lambert. Performed data analysis: Brown, Hepler. Wrote or contributed to the writing of the manuscript: Brown, Lambert, Hepler.
Dislosures
None declared.
Acknowledgements
We acknowledge Suneela Ramineni for technical advice in support of this project. We also acknowledge Kyle Gerber and Katherine Squires for insightful discussions on RGS14 function.
Brown N. E., Lambert N. A., Hepler J. R..RGS14 regulates the lifetime of Gα‐GTP signaling but does not prolong Gβγ signaling following receptor activation in live cells, Pharma Res Per, 4(5), 2016, e00249, doi: 10.1002/prp2.249
References
- Bernstein LS, Ramineni S, Hague C, Cladman W, Chidiac P, Levey AI, et al. (2004). RGS2 binds directly and selectively to the M1 muscarinic acetylcholine receptor third intracellular loop to modulate Gq/11alpha signaling. J Biol Chem 279: 21248–21256. [DOI] [PubMed] [Google Scholar]
- Bourne HR (1997). How receptors talk to trimeric G proteins. Curr Opin Cell Biol 9: 134–142. [DOI] [PubMed] [Google Scholar]
- Brown NE, Blumer JB, Hepler JR (2015a). Bioluminescence resonance energy transfer to detect protein‐protein interactions in live cells. Methods Mol Biol 1278: 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown NE, Goswami D, Branch MR, Ramineni S, Ortlund EA, Griffin PR, et al. (2015b). Integration of G protein alpha (Galpha) signaling by the regulator of G protein signaling 14 (RGS14). J Biol Chem 290: 9037–9049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunemann M, Frank M, Lohse MJ (2003). Gi protein activation in intact cells involves subunit rearrangement rather than dissociation. Proc Natl Acad Sci USA 100: 16077–16082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho H, Kozasa T, Takekoshi K, De Gunzburg J, Kehrl JH (2000). RGS14, a GTPase‐activating protein for Gialpha, attenuates Gialpha‐ and G13alpha‐mediated signaling pathways. Mol Pharmacol 58: 569–576. [DOI] [PubMed] [Google Scholar]
- De Vries L, Zheng B, Fischer T, Elenko E, Farquhar MG (2000). The regulator of G protein signaling family. Annu Rev Pharmacol Toxicol 40: 235–271. [DOI] [PubMed] [Google Scholar]
- Digby GJ, Sethi PR, Lambert NA (2008). Differential dissociation of G protein heterotrimers. J Physiol 586: 3325–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doupnik CA, Jaen C, Zhang Q (2004). Measuring the modulatory effects of RGS proteins on GIRK channels. Methods Enzymol 389: 131–154. [DOI] [PubMed] [Google Scholar]
- Evans PR, Lee SE, Smith Y, Hepler JR (2014). Postnatal developmental expression of regulator of G protein signaling 14 (RGS14) in the mouse brain. J Comp Neurol 522: 186–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman AG (1987). G proteins: transducers of receptor‐generated signals. Annu Rev Biochem 56: 615–649. [DOI] [PubMed] [Google Scholar]
- Hamm HE (1998). The many faces of G protein signaling. J Biol Chem 273: 669–672. [DOI] [PubMed] [Google Scholar]
- Hepler JR (2014). G protein coupled receptor signaling complexes in live cells. Cellular Logist 4: e29392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepler JR, Gilman AG (1992). G proteins. Trends Biochem 17: 383–387. [DOI] [PubMed] [Google Scholar]
- Hollinger S, Hepler JR (2002). Cellular regulation of RGS proteins: modulators and integrators of G protein signaling. Pharmacol Rev 54: 527–559. [DOI] [PubMed] [Google Scholar]
- Hollinger S, Taylor JB, Goldman EH, Hepler JR (2001). RGS14 is a bifunctional regulator of Galphai/o activity that exists in multiple populations in brain. J Neurochem 79: 941–949. [DOI] [PubMed] [Google Scholar]
- Hollins B, Kuravi S, Digby GJ, Lambert NA (2009). The c‐terminus of GRK3 indicates rapid dissociation of G protein heterotrimers. Cell Signal 21: 1015–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hynes TR, Yost EA, Yost SM, Berlot CH (2011). Multicolor BiFC analysis of G protein betagamma complex formation and localization. Methods Mol Biol 756: 229–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimple RJ, De Vries L, Tronchere H, Behe CI, Morris RA, Gist Farquhar M, et al. (2001). RGS12 and RGS14 GoLoco motifs are G alpha(i) interaction sites with guanine nucleotide dissociation inhibitor activity. J Biol Chem 276: 29275–29281. [DOI] [PubMed] [Google Scholar]
- Kimple RJ, Kimple ME, Betts L, Sondek J, Siderovski DP (2002). Structural determinants for GoLoco‐induced inhibition of nucleotide release by Galpha subunits. Nature 416: 878–881. [DOI] [PubMed] [Google Scholar]
- Lambert NA, Johnston CA, Cappell SD, Kuravi S, Kimple AJ, Willard FS, et al. (2010). Regulators of G‐protein signaling accelerate GPCR signaling kinetics and govern sensitivity solely by accelerating GTPase activity. Proc Natl Acad Sci USA 107: 7066–7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SE, Simons SB, Heldt SA, Zhao M, Schroeder JP, Vellano CP, et al. (2010). RGS14 is a natural suppressor of both synaptic plasticity in CA2 neurons and hippocampal‐based learning and memory. Proc Natl Acad Sci USA 107: 16994–16998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuho I, Martemyanov KA, Lambert NA (2015a). Monitoring G protein activation in cells with BRET. Methods Mol Biol 1335: 107–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuho I, Ostrovskaya O, Kramer GM, Jones CD, Xie K, Martemyanov KA (2015b). Distinct profiles of functional discrimination among G proteins determine the actions of G protein‐coupled receptors. Sci Signal 8: ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittal V, Linder ME (2004). The RGS14 GoLoco domain discriminates among Galphai isoforms. J Biol Chem 279: 46772–46778. [DOI] [PubMed] [Google Scholar]
- Mittal V, Linder ME (2006). Biochemical characterization of RGS14: RGS14 activity towards G‐protein alpha subunits is independent of its binding to Rap2A. Biochem J 394(Pt 1): 309–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntean BS, Martemyanov KA (2016). Association with the plasma membrane is sufficient for potentiating catalytic activity of regulators of g protein signaling (RGS) proteins of the R7 subfamily. J Biol Chem 291: 7195–7204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neitzel KL, Hepler JR (2006). Cellular mechanisms that determine selective RGS protein regulation of G protein‐coupled receptor signaling. Semin Cell Dev Biol 17: 383–389. [DOI] [PubMed] [Google Scholar]
- Robichaux WG 3rd, Oner SS, Lanier SM, Blumer JB (2015). Direct coupling of a seven‐transmembrane‐span receptor to a galphai G‐protein regulatory motif complex. Mol Pharmacol 88: 231–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross EM, Wilkie TM (2000). GTPase‐activating proteins for heterotrimeric G proteins: regulators of G protein signaling (RGS) and RGS‐like proteins. Annu Rev Biochem 69: 795–827. [DOI] [PubMed] [Google Scholar]
- Sarvazyan NA, Lim WK, Neubig RR (2002). Fluorescence analysis of receptor‐G protein interactions in cell membranes. Biochemistry 41: 12858–12867. [DOI] [PubMed] [Google Scholar]
- Shu FJ, Ramineni S, Amyot W, Hepler JR (2007). Selective interactions between Gi alpha1 and Gi alpha3 and the GoLoco/GPR domain of RGS14 influence its dynamic subcellular localization. Cell Signal 19: 163–176. [DOI] [PubMed] [Google Scholar]
- Shu FJ, Ramineni S, Hepler JR (2010). RGS14 is a multifunctional scaffold that integrates G protein and Ras/Raf MAPkinase signalling pathways. Cell Signal 22: 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traver S, Bidot C, Spassky N, Baltauss T, De Tand MF, Thomas JL, et al. (2000). RGS14 is a novel Rap effector that preferentially regulates the GTPase activity of galphao. Biochem J 350(Pt 1): 19–29. [PMC free article] [PubMed] [Google Scholar]
- Vellano CP, Maher EM, Hepler JR, Blumer JB (2011). G protein‐coupled receptors and resistance to inhibitors of cholinesterase‐8A (Ric‐8A) both regulate the regulator of g protein signaling 14 RGS14.Galphai1 complex in live cells. J Biol Chem 286: 38659–38669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vellano CP, Brown NE, Blumer JB, Hepler JR (2013). Assembly and function of the regulator of G protein signaling 14 (RGS14):H‐Ras signaling complex in live cells are regulated by galphai1 and galphai‐linked GPCRs. J Biol Chem 288: 3620–3631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb CK, McCudden CR, Willard FS, Kimple RJ, Siderovski DP, Oxford GS (2005). D2 dopamine receptor activation of potassium channels is selectively decoupled by Galpha‐specific GoLoco motif peptides. J Neurochem 92: 1408–1418. [DOI] [PubMed] [Google Scholar]
- Willard FS, Willard MD, Kimple AJ, Soundararajan M, Oestreich EA, Li X, et al. (2009). Regulator of G‐protein signaling 14 (RGS14) is a selective H‐Ras effector. PLoS ONE 4: e4884. [DOI] [PMC free article] [PubMed] [Google Scholar]