

Abstract
Background
Oxidative stress is implicated in increased vascular permeability associated with metabolic disorders, but the underlying redox mechanism is poorly defined. S-glutathionylation, a stable adduct of glutathione with protein sulfhydryl, is a reversible oxidative modification of protein and is emerging as an important redox signaling paradigm in cardiovascular physiopathology. The present study determines the role of protein S-glutathionylation in metabolic stress-induced endothelial cell permeability.
Methods and results
In endothelial cells isolated from patients with type-2 diabetes mellitus, protein S-glutathionylation level was increased. This change was also observed in aortic endothelium in ApoE deficient (ApoE-/-) mice fed on Western diet. Metabolic stress-induced protein S-glutathionylation in human aortic endothelial cells (HAEC) was positively correlated with elevated endothelial cell permeability, as reflected by disassembly of cell-cell adherens junctions and cortical actin structures. These impairments were reversed by adenoviral overexpression of a specific de-glutathionylation enzyme, glutaredoxin-1 in cultured HAECs. Consistently, transgenic overexpression of human Glrx-1 in ApoE-/- mice fed the Western diet attenuated endothelial protein S-glutathionylation, actin cytoskeletal disorganization, and vascular permeability in the aorta. Mechanistically, glutathionylation and inactivation of Rac1, a small RhoGPase, were associated with endothelial hyperpermeability caused by metabolic stress. Glutathionylation of Rac1 on cysteine 81 and 157 located adjacent to guanine nucleotide binding site was required for the metabolic stress to inhibit Rac1 activity and promote endothelial hyperpermeability.
Conclusions
Glutathionylation and inactivation of Rac1 in endothelial cells represent a novel redox mechanism of vascular barrier dysfunction associated with metabolic disorders.
Keywords: Protein S-glutathionylation, Glutaredoxin-1, Actin cytoskeleton, Small Rho GTPase Rac1, Endothelial barrier function, ApoE-deficient mice
Graphical abstract
Highlights
-
•
In metabolically stressed endothelial cells, protein S-glutathionylation is elevated.
-
•
glutaredoxin-1 diminishes protein S-glutathionylation and preserves aortic barrier function.
-
•
Pharmacological inhibition of Rac1 abrogates Glrx1-mediated barrier protection.
-
•
Glutathionylation of Rac1 is associated with a defect in Rac1 activation status.
1. INTRODUCTION
Metabolic disorders such as obesity, diabetes mellitus, and hyperlipidemia promote cardiovascular diseases, which are responsible for most of the morbidity, hospitalizations, and mortality in patients [1]. A common element in the complex pathogenesis of vascular complications is oxidative stress, which arises as results of sustained overproduction of reactive oxygen species (ROS) and impaired intrinsic antioxidant system, such as glutathione (GSH) [2], [3]. Protein S-glutathionylation is a reversible oxidative modification of protein cysteineyl residues by the addition of GSH, and represents an important mode of redox signal transduction [4]. As a redox signaling switch, glutathionylation can be efficiently and specifically removed by glutaredoxin-1 (Glrx-1) via a thiol-disulfide exchange reaction in the presence of glutathione, NADPH and glutathione reductase [5]. Glrx-1 has served as an important tool to elucidate glutathionylation-mediated redox signaling which appears to be an important regulator of various cellular processes [4], [6], [7]. Glutathionylation has been detected in proteins involved in cellular response to diabetic conditions, including protein kinase C [8]; NF-κB [9], protein tyrosine phosphatase 1B [10], and cAMP-dependent protein kinase [11]. However, little is known regarding how glutathionylation participates in metabolic stress-induced endothelial cell dysfunction.
Increased vascular permeability has been established as an early characteristic of vascular dysfunction associated with metabolic diseases [12], [13], [14]. It is thought to be one of initiate steps of atherosclerosis by increasing lipoprotein entry and oxidation within the sub-endothelial space, propagating endothelial cell inflammation and favoring leukocyte transmigration [15], [16]. Although overproduction of oxidants and decrease in the ratio of reduced to oxidized GSH (GSH/GSSG) have been linked to impaired endothelial cell barrier integrity [17], [18], the redox-dependent mechanisms remain elusive. Endothelial cell barrier function is largely determined by the integrity of adherens junctions (AJs) and actomyosin-based cell contractility, both of which are tightly controlled by the dynamics of actin cytoskeletal architecture [19]. These dynamic processes are coordinately governed by Rho GTPases including RhoA, Rac1 and Cdc42, whose activity is very carefully controlled by their upstream regulatory proteins such as guanine nucleotide exchange factors (GEFs), as well as by post-transcriptional modifications including cysteinyl thiol oxidation under physiological conditions [20], [21], [22]. Recently, accumulating evidence showed that a defect in Rac1 activity and its mediated cortical actin reorganization is mechanistically linked to peripheral insulin resistance and neuronal complications associated with obesity and diabetes. It is thus intriguing to investigate how glutathionylation of Rho GTPases regulates endothelial barrier function and atherosclerosis under conditions of diabetes and hyperlipidemia.
The present study is prompted by the observation of elevated protein S-glutathionylation in endothelial cells isolated from patients with type 2 diabetes mellitus and hypercholesterolemia mice. Our studies demonstrated that glutathionylation and inhibition of Rac1 may represent an important redox mechanism for endothelial cell barrier failure caused by metabolic abnormalities. Overexpression of Glrx-1 attenuates Western diet-induced protein S-glutathionylation and preserves aortic endothelial barrier integrity. Collectively, these results suggest a causative role of protein S-glutathionylation in metabolic stress-induced endothelial barrier dysfunction.
2. Materials and methods
2.1. Animals
All animal experimental procedures were approved by the Institutional Animal Care and Use Committee (IACUC) at Boston University Medical Campus. Glrx-1 transgenic mice with human Glrx-1 overexpression driven by the mouse β-actin promotor (a kind gift from Dr. Y.S. Ho, Wayne State University, Detroit, MI) were fully backcrossed onto a C57BL/6 background and crossed with ApoE-deficient mice (B.6 129P2) purchased from Jackson Laboratory (Bar Harbor, ME). Global Glrx-1 transgenic /ApoE-/- mice and WT littermates were fed a control (normal chow, ND: 4.5% fat, 0.02% cholesterol by weight), or a Western diet (WD: 21% fat, 0.21% cholesterol by weight and 35% sucrose) purchased from Research Diets Inc. (D12079B, New Brunswick, NJ) for two weeks starting at 8 weeks of age. We use only male mice for experiments.
2.2. Immunofluorescence histochemistry and atherosclerotic lesion quantification
To analyze glutathionylated proteins in aorta with immunohistochemistry, the arterial tree of mice was perfused with ice-cold PBS containing NEM (10 mM) to alkylate the free protein thiols [23]. The cross sections of aorta were incubated with NEM (10 mM) in PBS on ice for 15 min and fixed with acetone for 15 min at −20 °C, followed by immunofluorescence staining procedure described in online-only data supplement.
To analyze glutathionylated proteins and actin cytoskeleton structures in aortic endothelium in vivo, aortas from ApoE-/-and Glrx-1 TG on ApoE-/-mice fed WD for 2 weeks were perfused with NEM buffer and 2% paraformaldehyde, sequentially. The dissected ascending aorta and proximal arch segments were opened longitudinally and subjected to immunofluorescence staining for actin and glutathionylated proteins.
2.3. Human venous endothelial cell collection, immunostaining and quantification
We enrolled adults with type 2 diabetes mellitus defined as fasting serum glucose ≥126 mg/dL or ongoing treatment for type 2 diabetes mellitus at Boston Medical Center and control individuals without diabetes mellitus defined as fasting glucose ≤100 mg/dL. All subjects were studied in the fasting state, and a blood sample was taken for measurement of lipid levels and glucose levels in the Boston Medical Center Clinical Laboratory. Peripheral venous endothelial cell biopsy was performed as previously described [24]. The isolated ECs were co-immunostained for glutathionylated proteins and Von Willebrand Factor (vWF) to aid in endothelial cell identification. Fluorescence intensity was quantified in 20–30 cells from each subject. The detailed procedure is described in online-only data supplement.
2.4. Cell culture and treatments
Human aortic endothelial cells (HAECs), purchased from Lonza (Walkersville, MD), were treated for indicated period of time with vehicle (5 mM glucose, 20 mM mannitol and fatty acid free BSA at indicated concentrations in starvation medium supplemented with 0.1% FBS) or high palmitate and high glucose at indicated concentrations (HPHG). HAECs were incubated with an inhibitor of Rac1, 100 μM NSC-23766 (Santa Cruz Biotech., Dallas, TX) for 30 mins, followed by HPHG treatments.
Transfection of siRNA was performed using siRNA transfection reagent and medium according to the manufacture’s protocol from Santa Cruz Biotechnology. HAECs were infected with adenoviruses encoding LacZ (control) or human Glrx-1 (a generous gift from Dr. Young J. Lee, the University of Pittsburgh) [6] for 24 h before further treatments. Rac1-deficent COS-7 cells were transiently transfected with plasmids encoding Rac1 WT and mutants using Lipofectamine 3000 according to the manufacturer’s instruction. The detailed procedure is described in online-only data supplement.
2.5. Generation of cysteine to serine Rac1 mutants
Single amino acid substitutions (Cys18S, Cys81S, and Cys157S) of Rac1 were created using pGFP-Rac1WT as a template and the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). Primers were designed using the QuickChange Primer Design program and are available upon request. All mutations were confirmed by DNA sequencing (Genewiz, Inc., South Plainfield, NJ).
2.6. Rac1 knockdown by CRISPR/Cas-9 system
pSpCas9/sgRNA(BB)−2A-Puro (PX459) V2.0 plasmid was used to generate Cas9 endonuclease targeted to Rac1 gene in green monkey kidney fibroblast cells (COS-7) following the procedure described in [25]. COS-7 cells transfected with sequence-verified empty or Rac1 knockout CRISPR/Cas9 plasmids were harvested for Western blotting analysis of endogenous Rac1 expression. The detailed procedure is described in online-only data supplement.
2.7. Rho GTPases activation assays
Activation of RhoA, Rac1 and Cdc42 GTPases was determined in HAECs lysates using pull-down assay kits purchased from Cytokeleton (Denver, CO).
2.8. Endothelial permeability measurement in vitro and in vivo
HAEC monolayers cultured onto Transwell culture inserts were transfected with adenoviral LacZ (control) or Glrx-1, or with non-targeting siRNA or Glrx-1 siRNA. Cells then were exposed to HPHG at indicated concentrations followed by assay of FITC-dextran influx across the Transwell membrane described in online-only data supplement.
To measure aortic permeability in vivo, 0.5% Evans Blue Dye (EBD) in PBS containing 4% BSA at the dosage of 30 mg/Kg was intravenously administrated to mice and allowed to circulate for 60 min before euthanasia. The extravasation of EBD-BSA into aortic wall was assessed as described in online-only data supplement.
2.9. Biotin-switch assay for labeling of reversible cysteine oxidation in proteins extracted from HAECs and whole aortae
Total reversible cysteinyl thiols of proteins extracted from HAECs were assessed as previously described in [26], [27]. The detailed procedure is described in online-only data supplement.
2.10. Analysis of glutathionylated Rac1 in COS-7 cells using biotinylated GSH ethyl ester
Glutathionylation of Rac1 was accessed using biotinylation method as previously described [28]. COS-7 cells loaded with 500 μM BioGEE were exposed to vehicle or HPHG as indicated. Glutathionylated proteins in cleared cell lysates were enriched by streptavidin magnetic beads, followed by Western blotting analysis of Rac1 and β-actin. The detailed procedure is described in online-only data supplement.
2.11. Quantitative real-time PCR
Total RNA isolated from the whole aorta using TRIzol™ reagent (Invitrogen) was converted to cDNA serving as a template for PCR. Quantitative PCR was conducted using inventory mouse (Mm) gene-specific TaqMan™ Primers with StepOne™ real-time PCR software (Applied Biosystems).
2.12. Statistical analysis
Statistical analysis was performed using Prism 6.0 (GraphPad Software). Means were compared between two groups by the Mann-Whitney U test. Multiple comparisons were conducted with 1-way ANOVA followed by Dunnett test. A value of p<0.05 was considered statistically significant. Analysis of fluorescence intensity of ECs from human subjects was conducted using a mixed model. The average of intensity in arbitrary units obtained from 20 to 30 measurements per human subject was used as the dependent variable, and sample types (control vs. DM) were used as the independent variable adjusting for different batches of patient cells, with a random effect for each to account for within-person correlation.
3. RESULTS
3.1. Protein S-glutathionylation is increased in endothelial cells under conditions of diabetes and hypercholesterolemia
To gain insight into the clinical relevance of protein S-glutathionylation (PrS-SG) to vascular endothelial cell dysfunction associated with metabolic disorders, endothelial cells isolated from patients with Type 2 diabetes mellitus and non-diabetic control subjects were obtained and co-immunostained for PrS-SG and Von Willebrand factor (vWF, endothelial cell marker). As shown in Fig. 1A and B, the level of PrS-SG was significantly increased in diabetic endothelial cells. As Type 2 diabetes is usually associated with hyperlipidemia, a major risk factor for cardiovascular diseases [30], we next test whether in hypercholesteremic mice the level of PrS-SG in the aorta is altered through comparing PrS-SG staining density in cross sections of aortae from ApoE-/- mice fed normal chow (ND) and Western diet (WD) for two weeks. As shown in Fig. 1D and E, two-week WD feeding dramatically increased the plasma levels of cholesterol, but not triglycerides. In WD-fed ApoE-/- mice, the staining intensity of PrS-SG was dramatically higher in the aortic vessel wall than that in ND-fed ApoE-/- mice. In a more detailed analysis (Fig. 1C), the intense staining for PrS-SG was highly colocalized with CD31 positive endothelial cells, but not in smooth muscle cells stained with α-actin. The specificity of PrS-SG staining was verified by demonstrating that immunofluorescence signals in aortic specimens were abolished by treatment with thiol reducing agent, dithiothreitol (DTT) for cleaving protein disulfide bonds (Supplemental Fig. 1). Also, we demonstrated in Fig. 1F and G that PrS-SG was induced in cultured human aortic EC cells (HAECs) exposed to high levels of palmitate and glucose, which mimics high circulating levels of saturated free fatty acids (FFA) and glucose in vivo associated with metabolic disorders. In vitro treatment of endothelial cells with palmitate and/or high level of glucose have been well documented to induce endothelial dysfunctions including oxidative stress, inflammation, apoptosis, impaired eNOS signaling [31], [32], [33], [34], [35]. These results obtained from human samples and experimental models of metabolic disorders both in vivo and in vitro together clearly indicate that PrS-SG is induced in endothelial cells under the conditions of metabolic stress, suggesting a role of glutathionylation in the regulation of endothelial cell responses to metabolic cues.
Fig. 1.

Protein S-glutathionylation in endothelial cells is increased under conditions of diabetes and hypercholesteremia. Glutathionylated proteins (PrS-SG) is elevated in diabetic endothelial cells (ECs). A: Representative images of immunofluorescence staining of PrS-SG in ECs isolated from diabetic and non-diabetic control subjects. B: The intensity of PrS-SG staining in ECs from diabetic subjects (n=5) and control subjects (n=5) was quantified. From each subject sample, 20–30cells were immunostained for PrS-SG, and the fluorescence density was measured and averaged. Data represent mean±SEM. *p≤0.05. C: PrS-SG is elevated in aortic ECs in hypercholesterolemic mice. Representative images showing the localization and levels of PrS-SG in the cross sections of aortae from ApoE-/- mice fed a normal chow (ND) and a Western diet (WD) for two weeks. Freshly prepared aortic tissue cross sections were incubated with 10 mM NEM to alkylate free thiols, fixed with ice-cold acetone, and immunostained for PrS-SG (red channel), CD31(EC marker, green channel) and α-actin (smooth muscle cell marker, green channel). The first and the third row shows high magnification views of yellow-boxed areas. Scale bars are 50 µm. D: Two-week WD feeding dramatically increased the plasma levels of cholesterol, but not triglycerides in AopE-/- mice. Data represent mean±SEM (ND: n=7; WD: n=12). *p<0.05, compared with ND group. E: PrS-SG is elevated in cultured HAECs exposed to diabetic conditions. HAECs were treated with vehicle (50 μM BSA, 5 mM D-glucose, 20 mM mannitol) or high levels of palmitate-BSA conjugate and high glucose (HPHG) at indicated concentrations for 24 h. Cells were then incubated with 10 mM NEM for 15 min to alkylate free protein thiols, and the cleared cell lysates underwent Western blotting analysis with the anti-GSH antibody under nonreducing condition. The right panel is the image with short exposure. The β-actin blot serves as a loading control. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.2. Overexpression of Glrx-1 reduces protein S-glutathionylation and improves aortic endothelial barrier function in response to metabolic stress in vitro
To test the hypothesis of that glutathionylation plays a role in the redox regulation of endothelial barrier function, which has long been recognized as an early phenotypic change associated with metabolic disorders [12]. Genetic manipulation of Glrx-1 expression in endothelial cells was employed to control glutathionylation level. As shown in Fig. 2A and B, adenoviral overexpression of Glrx-1 efficiently inhibited PrS-SG induction in HAECs challenged by HPHG. We next examined the impact of HPHG treatment on HAEC permeability using a well-established two-compartment chamber in vitro model as described. In considering the apoptotic effect of chronic exposure to HPHG on endothelial cells [31], [32], we chose to challenge HAECs with HPHG for two hours after ensuring this condition could not stimulate robust apoptotic signals (supplemental Fig. 2). HPHG treatment increased the permeability of HAEC monolayer to fluorescein-labeled dextran in a dose-dependent manner (Fig. 2C). More importantly, The HPHG-induced endothelial hyperpermeability was protected by overexpression of Glrx-1 (Fig. 2D), and aggravated by siRNA-mediated downregulation of Glrx-1 (Fig. 2E), supporting a critical role of PrS-SG in metabolic stress-induced EC barrier regulation. We next directly visualized and accessed the EC barrier integrity and actin cytoskeletal structure through immunostaining of VE-cadherin (a molecular marker of adhesion junctions) and F-actin in HAECs under control and metabolic stress conditions. Consistently, HPHG treatment induced disappearance of VE-Cadherin from contact cell borders associated with intercellular gap formation, which was prevented by overexpression of Glrx-1 (Fig. 2F and G). As shown in Fig. 2H and I, under basal condition, overexpression of Glrx-1 stimulated F-actin polymerization. HPHG challenge significantly increased the formation of stress fibers in HAECs infected with AdLacZ, but not in the cells overexpressing Glrx-1. These results together suggest a protective role of Glrx-1 in metabolic stress-induced barrier dysfunction.
Fig. 2.

Adenoviral overexpression of Glrx-1 attenuates metabolic stress-induced protein S-glutathionylation and endothelial cell permeability. A–B: Overexpression of Glrx-1 abrogates HPHG-induced PrS-SG in ECs. HAECs were infected with an adenovirus expressing human Glrx-1 or a control adenovirus (AdlacZ) for 24 h. Cells were then treated with vehicle (25 μM BSA, 5 mM glucose, 20 mannitol) and HPHG (100 μM palmitate-BSA conjugate, 25 mM glucose) in a serum-free medium for another 24 h, followed by immunostaining for PrS-SG (red channel) and Glrx-1 (green channel) with antibodies against GSH and Glrx-1, respectively. The first row shows high magnification views of yellow boxed areas. Green outlined cells overexpress Glrx-1. Fifteen randomly selected image fields from three independent experiments were used for quantification (n=15). *p≤0.05 between indicated groups. C: HPHG treatment increases endothelial cell permeability in a dose-dependent manner. HAEC monolayers cultured onto Transwell inserts were exposed to HPHG at indicated concentrations for 2 h, followed by FITC-dextran influx assay. Results are the normalized percentages of total FITC-dextran passing across monolayers relative to the control group and represented as mean ± SD of three independent experiments, each performed in triplicates. *p<0.05, compared with vehicle group. D-E: Adenoviral overexpression of Glrx-1 (D) protected against, siRNA-mediated downregulation of Glrx-1(E) exacerbated HPHG-induced permeability. The expression of Glrx-1 in HAECs infected with adenovirus or siRNAs were assessed by immunoblotting shown in the lower panels of D and E, respectively. *p<0.05 compared between indicated groups. F–I: Overexpression of Glrx-1 in HAECs protected cell-cell adherens junctions and actin cytoskeletal structures against HPHG challenge. Adenovirus-infected HAECs were exposed to vehicle (50 μM BSA, 5 mM glucose, 20 mM mannitol) or HPHG (200 μM palmitate-BSA conjugate, 25 mM glucose) for 2 h, followed by immunostaining for VE-Cadherin, a marker for cell-cell junctions (F, red channel), and Glrx-1 (F and H, green channel). F-actin was stained with red fluorescence-labeled phalloidin (H). The second and last column shows high magnified views of yellow boxed areas. Junctional index (G) as a measure of barrier integrity (VE-Cadherin staining density/total area of image field)×100/cell number), and F-actin staining intensity (F) were measured in 15 randomly selected image fields from three independent experiments. Intercellular gaps were indicated by white arrows. Results represent mean±SEM (n=15). *p<0.05, compared between indicated groups. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.3. Vascular permeability is attenuated in the aorta of transgenic overexpression of Glrx-1 in ApoE-/- mice fed the Western diet-
To gain further insights into the role of protein S-glutathionylation in vascular barrier dysfunction associated metabolic disorders in vivo, human Glrx-1 transgenic mice were bred onto hypercholesteremic ApoE deficient background (Glrx-1-TG/ApoE-/-) after verifying the expression of human Glrx-1 and increased enzymatic activity of Glrx-1 in endothelial cells from Glrx-1 TG mice (Fig. 3A). Glrx-1-TG/ApoE-/- and their littermates (WT/ApoE-/-) were fed ND or WD for two weeks when no significant aortic atherosclerotic plaques were observed. Consistently, two-week WD diet increased the level of PrS-SG in cross-sections of the aorta from WT mice, but to a much less extent in Glrx-1 TG mice (Fig. 3B). En face co-immunostaining for PrS-SG and F-actin of aortic endothelial cells at lesion-prone sites on the lesser curvature of the aortic arch showed that overexpression of Glrx-1 attenuated the formation of PrS-SG and preserved F-actin structure in aortic endothelium in WD-fed ApoE-/- mice (Fig. 3C). Furthermore, Aortic permeability was assayed by measuring permeation of BSA-conjugated Evans blue dye (EBD) into the vessel wall, a well-established approach to access vascular permeability in vivo [36]. Compared with WD-fed WT/ApoE-/- mice, BSA-EBD leakage in the aorta of Glrx-1 TG was significantly decreased (Fig. 3D and E). Of note, overexpression of Glrx-1 in ApoE-/- mice does not affect plasma lipid profiles and fasting glucose level (Supplemental Table 1). As the increased vascular permeability may result from EC inflammatory responses to leukocyte transendothelial migration and activation, we examined the expression of adhesion molecules, VCAM-1 and ICAM-1, as well as the leukocyte chemoattractant, MCP-1, in aortae of Glrx-1-TG/ApoE-/- and WT/Apo E -/- mice on ND and WD. As shown in Fig. 3F, gene expression of VCAM-1 and MCP-1 was elevated in aortae from WD-fed mice, but there were no statistically significant differences between Glrx-1-TG/ApoE-/- and their littermates. Consistent with a previous report, aortic mRNA level of ICAM-1 was not increased in response to WD [37]. Taken together, these data demonstrated an important role of protein S-glutathionylation in regulating vascular barrier dysfunction associated with metabolic abnormalities in vivo.
Fig. 3.

Transgenic overexpression of human Glrx-1 in ApoE-/- mice attenuates hypercholesterolemia-induced protein S-glutathionylation and aortic barrier dysfunction in vivo. A. ECs isolated from the lung of human Glrx-1 transgenic (hGlrx-1 TG) mice express human Glrx-1. B-C: hypercholesterolemia-induced PrS-SG is diminished in aortic endothelium of hGlrx-1 TG mice, accompanied by preserved EC actin cytoskeletal structure. WT and hGlrx-1 TG mice on ApoE-/- background were fed ND or WD for 2 weeks. B: Representative immunofluorescence images show PrS-SG staining in aortic cross-sections from WT and hGlrx-1 TG. Inserts are high magnification views of yellow boxed areas. Scale bar is 50 µm. C: Representative en face confocal fluorescence images of PrS-SG (red channel) and cytoskeletal organization (F-actin, green channel) in aortic endothelium at the lesser curvature of aortic arch segments from WT and hGlrx-1 TG mice. The second and last column show high magnification view of yellow box areas. Scale bars are 20 µm. D–E: Hypercholesterolemia-induced aortic permeability is attenuated in hGlrx-1 TG mice. Aortic permeability in WT and hGlrx-1 TG mice on ND and WD was assessed by BSA-Evans blue dye (BSA-EBD) conjugate permeation method. F: Representative photographs of BSA-EBD leakage in aortic arch segments of mice. Scale bar is 200 µm. G: The permeated EBD in aortae was extracted, measured, and presented as means±SEM (WT: n=5–9; hGlrx-1 TG: 5–10). *p<0.05 between indicated groups. H: Hypercholesterolemia-induced inflammatory genes including MCP1, VCAM1, and ICAM1, in aorta is not affected by overexpression of hGlrx-1. Bar graphs show normalized mRNA level of inflammatory genes relative to ND group. Data are represented as mean±SEM. WT on ND (n=5) and WD (n=5). hGlrx-1 TG on ND (n=5) and WD (n=7). *p<0.05 vs. WT mice on ND group, † p<0.05 vs. hGlrx-1 TG mice on ND group. (For interpretation of the references to color in this figure legend, the reader is referred to the web version of this article.)
3.4. Overexpression of Glrx-1 preserves the activity of small Rho GTPase Rac1 in endothelial cells exposed to metabolic stress
Small Rho GTPases including RhoA and Rac1 are putative master regulators of dynamic actin cytoskeleton arrangement and are well recognized redox-sensitive signaling molecules as well [20], [22], [38]. We thus tested their role in redox regulation of actin cytoskeletal reorganization and barrier function.
To assess the effect of metabolic stress on the activities of Rho GTPases in endothelial cells, we performed affinity pull-down assays to detect the activity of RhoA, Rac1 and Cdc42 in HAECs exposed to increasing concentrations of HPHG for two hours. The data shown in Fig. 4A and B indicate that HPHG treatment differentially regulated their activities. Specifically, HPHG challenge activated RhoA but inhibited Rac1 activity dose-dependently. The activity of Cdc42 determined from the same pull-down samples used for the Rac1 activity assay was not changed significantly. To specifically investigate the impact of protein S-glutathionylation induction on the activation status of RhoA and Rac1, oxidized GSH (GSSG) was utilized to induce protein S-glutathionylation via thiol-disulfide bond exchange. The activity of RhoA and Rac1 was assessed in, i) HAEC lysate incubated with GSSG; ii) HAECs exposed to diamide, which is a well-established thiol-oxidizing agent and shown to increase intracellular GSSG. As shown in Fig. 4C and D, both GSSG and diamide treatments inhibited Rac1 activity, demonstrating a negative regulatory effect of protein S-glutathionylation on Rac1 activity in endothelial cells. In contrast with HPHG treatment, RhoA activity was not increased by either GSSG or diamide, suggesting a negligible effect of glutathionylation on RhoA activity in endothelial cells. Focusing therefore on Rac1, we found that overexpression of Glrx-1 in HAECs prevented inactivation of Rac1 caused by HPHG challenge (Fig. 4E and F).
Fig. 4.

Overexpression of Glrx-1 preserves Rac1 activity in endothelial cells under conditions of metabolic stress. A–B: Effect of HPHG treatment on the activity of RhoA, Rac1, and Cdc42 in HAECs. HAECs were exposed to HPHG at the indicated concentrations for 2 h, followed by activity assays for RhoA, Rac1, and Cdc42. A: Representative Western blotting result for RhoA, Rac1, and Cdc42 in the GTP-bound form and 10% of total input. B: Bar graph shows the corresponding densitometric analysis of active RhoGTPases (ratio of GTP-bound form over total GTPases) from three independent experiments. *p<0.05 vs. vehicle group. C–F: Effect of PrS-SG induction on the activity of Rac1 and RhoA in HAECs. HAECs exposed to 50 μM diamide (C and E, left panel), or 20 μg cell lysates incubated with a mixture of 1 mM GSSG/3 mM GSH on ice for 30 mins (C and E, right panel), were subject to RhoA and Rac1 pull-down activity assays. D and F: The corresponding Densitometric analysis of blot data for the activity of Rac1 and RhoA, respectively. *p<0.05 vs. vehicle group. G–H: Effect of Glrx-1 overexpression on Rac1 activity in HAECs exposed to HPHG. HAECs were infected with an adenovirus expressing hGlrx-1 or a control adenovirus (AdLacZ) for 24 h, followed by HPHG treatment (200 μM palmitate-BSA conjugate, 25 mM glucose) for 2 h. Cells were then subject to Rac1 activity assay. *p<0.05 between indicated group.
3.5. Pharmacological inhibition of Rac1 abrogates the ability of Glrx-1 to protect against endothelial barrier dysfunction in response to metabolic stress in vitro
To examine whether Rac1 is involved in the effect of Glrx-1 on EC barrier function, a specific inhibitor of Rac1 activation (NSC23766), which interferes with the interaction between Rac1 and the Rac1-specific guanine nucleotide exchange factors (GEFs: Trio and Tiam1) was employed. Pretreatment of HAEC with 100 μM NSC23766 increased the formation of stress fibers in both vehicle and HPHG-treated cells (Fig. 5A). Inhibition of Rac1 activity also abrogated Glrx-1-mediated preservation of cell-cell junctions (Fig. 5B). In concert with the effects on cytoskeletal organization and cell-cell junctional integrity, Rac1 inactivation mitigated Glrx-1-mediated protection of endothelial permeability against HPHG (Fig. 5C). This set of data reaffirms the critical role of Rac1 in redox-regulation of EC permeability. In support of this notion, HAECs expressing green fluorescence protein (GFP) tagged constitutively active forms of Rac1 (Rac1CA), but not wild-type (Rac1WT), exhibited an increase in cortical actin rim structure and resisted against HPHG-induced cytoskeletal reorganization (supplemental Fig. 3).
Fig. 5.

Pharmacological inhibition of Rac1 abolishes Glrx-1-mediated protection of EC barrier integrity against metabolic stress. HAECs were pre-incubated with or without NSC-23766, a Rac1 specific inhibitor, at 100 μM for 30 mins. Cells were then exposed to vehicle (50 μM BSA, 5 mM glucose, 20 mM mannitol) or HPHG (200 μM palmitate-BSA conjugate, 25 mM glucose) for additional 2 h. A–D: The effect of Glrx-1 on HPHG-induced formation of F-actin and disassembly of cell-cell adherens junctions is abrogated by NSC-23766. Pre-incubation of HAECs with NSC-23766 induced the formation of F-actin (A and B) and delocalization of VE-Cadherin away from cell adherens junctions (C and D) under both basal and HPHG challenge conditions. Scale bar=25 µm. White arrows indicate intercellular gaps. Data are represented as mean±SEM. *p<0.05, between indicated groups. E: The effect of Glrx-1 on HPHG-induced permeability is abolished by NSC-23766. HAEC monolayers cultured onto Transwell inserts were pre-incubated with NSC-23766 at 100 μM for 30 mins. Cells were then exposed to vehicle (50 μM BSA, 5 mM glucose, 20 mM mannitol) or HPHG (200 μM palmitate-BSA conjugate, 25 mM glucose) for additional 2 h, followed by FITC-dextran influx assay. Values are the normalized percentages of total FITC-dextran passing across monolayers about the control group. Results represent Mean±SD of three independent experiments, each performed in triplicates. *p<0.05, between indicated groups.
3.6. Glutathionylation of Rac1 on Cysteine 81 and 157 is responsible for Rac1 inactivation caused by metabolic stress
Data from the biotin switch assay of HAECs demonstrated that HPHG treatment promoted the reversible oxidations of Rac1 and β-actin (serving as a positive control), which were effectively reversed by up-regulation of Glrx-1, suggesting that Rac1 is glutathionylated in metabolically stressed endothelial cells (Fig. 6A and B). To further investigate the mechanism of glutathionylation-induced Rac1 inactivation, we sought to determine the specific cysteine residues modified. As Cys18, Cys81, and Cys157 of Rac1 are located adjacent to the guanine nucleotide binding site, and their thiol oxidations are reported to be associated with the alteration of GTPase activity status [39], [40], [41], we generated GFP-tagged Rac1 mutants in which Cys18, Cys81, or Cy157 was mutated to oxidation resistant serine, and expressed them into a COS-7 cell line deficient in endogenous Rac1 using CRISPR-Cas9 system (Supplemental Fig. 4), allowing to assess the importance of these residues on the redox regulation of Rac1. We determined the level of glutathionylated Rac1 WT and cysteine oxidation resistant mutants in Rac1 knockout COS-7 cells, which were loaded with cell-permeable BioGEE and then treated with HPHG. In comparison with Rac1WT, HPHG-induced glutathionylation of Rac1C18S, Rac1C81S, or Rac1C157S was significantly decreased (Fig. 6C and D), suggesting that all of these cysteine residues can undergo glutathionylation. We further examined the impact of these modified cysteine residues on the Rac1 activity using PAK pull-down assay. As shown in Fig. 6E and F, the activity of Rac1C18S was inhibited by HPHG treatment to the extent similar as that of Rac1WT, whereas Rac1 C81S and Rac1C157S resisted against the decrease in activity. Collectively, these results suggest a causative role of glutathionylation of Rac1, especially on Cys81 and Cys157, in metabolic stress-induced Rac1 inactivation and consequent impairment of barrier function.
Fig. 6.

Cysteine mutants (C81S and C157S) of Rac1 resist S-glutathionylation and inactivation caused by metabolic stress. A–B: HPHG-induced oxidation of cysteine thiols of Rac1 is inhibited by overexpression of Glrx-1. HAECs overexpressing LacZ (control) or hGlrx-1 were exposed to vehicle (50 μM BSA, 5 mM glucose, and 20 mM mannitol) or HPHG (200 μM palmitate-BSA conjugate and 25 mM glucose) for 2 h. Cells were then lysed with RIPA buffer containing 10 mM NEM and subject to Biotin-switch assay. A: Immunoblotting of reversibly oxidized Rac1 and β-actin from biotinylated proteins pulled down by streptavidin magnetic beads (upper panel), and Rac1 and β-actin from 10% of total cell lysates, serving as loading controls (lower panel). B: Bar graph showing the fold changes in oxidized Rac1 (ratio of Biotinylated over total Rac1) relative to the vehicle group from three independent experiments. * p<0.05, compared between indicated groups. C–D: Mutation of cysteine residue 18, 81, or 157 to serine renders Rac1 resistant to HPHG-induced glutathionylation. Rac1 knock-out COS-7 cells generated by the CRISPR-Cas9 system were transiently transfected with GFP-Rac1 WT and indicated cysteine mutants for 24 h. Cells were then loaded with 500 μM BioGEE for 1 h before challenges with vehicle (100 μM BSA, 5 mM Glucose, 20 mM mannitol) or HPHG (400 μM palmitate-BSA conjugate and 25 mM glucose) for another 2 h. BioGEE labeled proteins pulled down with streptavidin magnetic beads were stained with MemCode protein staining kit (A, upper panel), and underwent immunoblotting for Rac1 and β-actin (as a positive control). B: Bar graph showing the fold changes in glutathionylated Rac1 (ratio of biotinylated over total input Rac1) relative to the vehicle group from three independent experiments.*p<0.05 compared between indicated groups. E and F: cysteine mutants (C18S and C157S) of Rac1 were resistant to HPHG-induced inactivation of Rac1. Rac1 knock-out COS-7 cells expressing GFP-Rac1 WT and indicated mutants were subject to Rac1 pull-down activation assay. Values were the fold changes in the active form of Rac1 (ratio of GTP-bound form over total input Rac1) about to the vehicle group from four independent experiments. *p<0.05 compared between indicated groups.
4. DISCUSSION
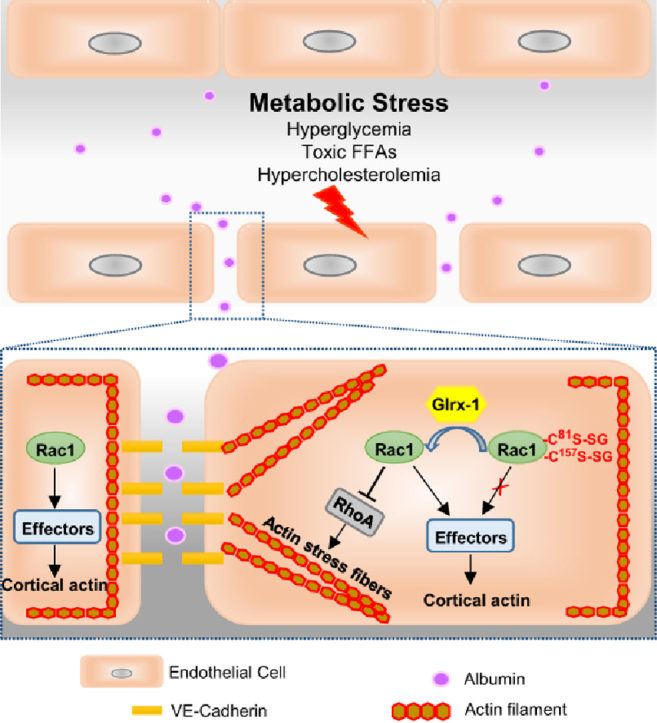
The novel findings of the present study include the following: 1) in endothelial cells under conditions of diabetes and hypercholesterolemia, the levels of protein S-glutathionylation are elevated; 2) removing protein S-glutathionylation by overexpressing Glrx-1 protects against vascular barrier dysfunction in hypercholesterolemic mice; 3) glutathionylation of Rac1 on cysteine residue 81 and 157 is associated with the defect in Rac1 activation status, representing an important redox mechanism for metabolic stress-induced endothelial cell barrier failure (Fig. 7).
Fig. 7.

The proposed redox mechanism for vascular barrier dysfunction associated with metabolic disorders. Metabolic abnormalities including hyperglycemia, toxic free fatty acids, and hypercholesterolemia, impair endothelial cell barrier function, an early characteristic of vascular dysfunction associated with metabolic diseases. In endothelial cells undergoing metabolic stress, the level of glutathionylated proteins is elevated. Glutathionylation of Rac1 on Cys81 and Cys157 reduces the activity of Rac1, resulting in loss of cortical actin structure, increased stress fibers, and disassembly of cell-cell adherens junctions. Inactivation of Rac1 increases actin stress fibers likely through release of its suppression on RhoA activation, a key driver of stress fiber formation and actomyosin-based cell contractility. Overexpression of Glrx-1 removes GSH adducts from Rac1 and restores its activity, which in turn improves endothelial barrier dysfunction under conditions of metabolic stress.
Protein S-glutathionylation is generally associated with increased oxidative or nitrosative stress during such pathological conditions as ischemia-reperfusion and hypertension [42], [43], regulating cardiovascular responses to stress and injury. Sustained overproduction of ROS has emerged as a common element in a variety of mechanisms controlling vascular complications of metabolic disorders [44], but how protein S-glutathionylation in endothelial cells is involved in these pathogenic processes has not been investigated. In the present study, we show that glutathionylated proteins are markedly increased in endothelial cells under conditions of diabetes and hyperlipidemia (Fig. 1). This elevated thiol oxidation plays a causative role in metabolic stress-induced aortic barrier dysfunction through altering cytoskeletal organization and cell-cell adhesion integrity (Fig. 2, Fig. 3). In particular, at the atherosclerotic lesion-prone aortic arch segment, vascular permeability is increased and can be attenuated by overexpressing Glrx-1 in hypercholesteremic mice. As endothelial hyperpermeability is an important initiating factor in atherogenesis by increasing lipoprotein entry and favoring leukocyte transmigration into the arterial wall [14], [15], [16], [45], [46]. we speculate that protein S-glutathionylation in endothelial cells may participate in atherosclerosis development.
Endothelial barrier integrity is a key manifestation of endothelial function and tightly associated with cellular redox status [47], but the underlying mechanism is not completely defined. In particular, little is known about the role of protein S-glutathionylation in redox regulation of endothelial permeability, although it has been reported that the ratio of reduced to oxidized GSH is essential for maintenance of endothelial cell barrier function [18]. The novel finding in the present study is the role of protein S-glutathionylation in cytoskeletal reorganization in response to metabolic stress, resulting in increased permeability (Fig. 2, Fig. 3, Fig. 4, Fig. 5). β-actin is one of the earliest identified cytoskeletal protein susceptible to glutathionylation. Glrx-1-mediated removal of GSH from β-actin leads to a 6-fold increase in the rate of polymerization [48], [49]. Consistently, our results showed that in endothelial cells, Glrx-1 reversed glutathionylation of β-actin with a concomitant increase in F-actin content under basal condition. Metabolic stress imposed by high levels of palmitate and glucose increased glutathionylation of β-actin and stimulated stress fiber formation, which is very unlikely mediated by glutathionylation of β-actin itself. Overexpression of Glrx-1 resisted against metabolic stress-induced stress fiber formation (Fig. 2, Fig. 4). We further showed that glutathionylation of Rac1 on Cys81 and Cys157, but not RhoA, is involved in redox-regulated cytoskeleton reorganization and permeability (Fig. 5, Fig. 6). Consistently, a defect in Rac1-mediated cortical actin structure, but not in RhoA/ROCK signaling, was recently shown to drive endothelial barrier failure in response to hypoxia/reoxygenation [50], which is known to increase protein S-glutathionylation [26], [51]. The impaired Rac1/cytoskeleton signaling was also linked to insulin and glucose intolerance in obesity and diabetes [52], [53], [54]. The present study provides a novel redox mechanism linking Rac1 inactivation to vascular barrier impairment caused by metabolic stress. Herein, Rac1 activity is accessed by measuring the level of association with PAK1, which is a major downstream effector conveying the role of Rac1 in actin cytoskeleton reorganization and endothelial barrier integrity enhancement [55], our data thus do not provide information about the impact of glutathionylation on the interactions of Rac1 with other downstream effectors such as p67, a component of NADPH oxidase [56], neither on the guanine nucleotide exchange and GTP hydrolysis [22]. A recent study reported that glutathionylation of Rac1 on Cys18 contributes to oxidant-induced activation by promoting guanine nucleotide exchange [39]. In line with this finding, our results suggest that glutathionylation of Cys18 is not required for metabolic stress-induced Rac1 inactivation, and may counteract the inhibitory effect on Rac1 activity exerted by modification of Cys81 and Cys157. It is also appreciated that Rac1, as a convergence point for multiple signaling pathways involving endothelial barrier enhancement, and can be activated by such signaling cascades as PKA/cAMP and PI3K/Akt [57], [58]. Both PKA and Akt can be modulated by S-glutathionylation [59], [60]. It is also possible that the observed change in Rac1 activity is a net effect of multiple glutathionylation-mediated signaling events. The detailed mechanisms warrant further investigations. Because Rac1 also supports NADPH-oxidase-mediated ROS generation via translocating to membrane and facilitating the assembly Nox subunits, it is likely to contribute to oxidative stress-induced vascular pathology and proposed as a therapeutic target for cardiovascular diseases [38]. This present study together with other reports demonstrates an essential role of Rac1 in vascular and metabolic hemostasis. It necessitates a better understanding of stimulus-specific regulation of Rac1 activation status and precise mechanisms how Rac1 is involved in cardiovascular pathologies, which may lead to highly selective therapeutic targeting.
Here, we chose to induce metabolic stress to endothelial cells in vitro using combined treatment with high glucose and high palmitate, which mimic elevated circulating glucose and free fatty acids (FFA) in patients with type 2 diabetes mellitus [61]. It has been well documented that this in vitro treatment induces endothelial cell dysfunction, including impaired eNOS signaling, apoptosis, inflammation and autophagy [32], [33], [34], [35], [62]. Of note, plasma levels of FFA and glucose in patients are positively correlated with the severity of diabetes mellitus, ranging from 200 to 800 μM for FFA, from 130 to 400 mg/dL for glucose [63]. In ApoE-/- mice, a mouse model of hypercholesterolemia used in the present study, the FFA concentration is elevated after two weeks on the Western diet (200 μM). These mice display increased fasting glucose levels on a normal diet (146 mg/dL) and the Western diet (155 mg/dL), which is consistent with a recent report [64] (Supplemental Table 1). Based on this information and experimental literature [32], [33], [34], [35], [62], we treated endothelial cells with high glucose (450 mg/dL) together with palmitate at concentrations ranging from 50 to 300 μM. We are aware of that this in vitro model does not completely recapitulate metabolic stress imposed on vascular endothelium under diabetic conditions in vivo, although our studies focus on the role of protein glutathionylation in redox regulation of endothelial barrier function, and the combined treatment successfully induces glutathionylation of endothelial proteins, in particular Rac1, and barrier integrity failure, allowing us to investigate their causal relationship. Oxidative stress appears to be a major mechanism mediating metabolic stress-induced endothelial dysfunction [31]. High levels of glucose or palmitate alone are reported to stimulate intracellular ROS generation and may contribute to the increased protein S-glutathionylation in endothelial cells. We tested this possibility (Supplemental Fig. 5) and found that high palmitate, but not high glucose, stimulated protein S-glutathionylation. Interestingly, the combined treatment exerted a synergic effect, possibly due to an additive increase in ROS generation, particularly derived from mitochondria, whose function is severely impaired by this combination treatment [65].
Our in vivo findings on Glrx-1 TG mice do not distinguish possible effects of downregulation of protein S-glutathionylation in cell types other than ECs that may have influenced aortic permeability. However, our in vitro experiments with overexpression of Glrx-1 in aortic endothelial cells showed strong parallels with the in vivo findings that include stabilized actin cytoskeletal structures and preserved barrier integrity.
In summary, we have demonstrated that S-glutathionylation, a reversible oxidative modification of proteins, is elevated in endothelial cells under conditions of diabetes and hyperlipidemia, contributing to metabolic stress-induced endothelial barrier failure. Glutathionylation of Rac1 on cysteine residue 81 and 157 appears as an important redox mechanism controlling endothelial cell barrier function in response to metabolic abnormalities.
Sources of funding
This study is supported by AHA SDG20140036 (J.H.), 1UL1TR001430 (BU CTSI), T32 HL07224, and R37 HL104017 (R.A.C.).
Disclosures
No.
Footnotes
Supplementary data associated with this article can be found in the online version at http://dx.doi.org/10.1016/j.redox.2016.09.003.
Appendix A. Supplementary material
Supplementary material
.
References
- 1.Faxon D.P., Creager M.A., Smith S.C., Jr, Pasternak R.C., Olin J.W., Bettmann M.A., Criqui M.H., Milani R.V., Loscalzo J., Kaufman J.A., Jones D.W., Pearce W.H. American Heart Association. Atherosclerotic vascular disease conference: executive summary: atherosclerotic vascular disease conference proceeding for healthcare professionals from a special writing group of the american heart association. Circulation. 2004;109:2595–2604. doi: 10.1161/01.CIR.0000128517.52533.DB. [DOI] [PubMed] [Google Scholar]
- 2.Kondo T., Hirose M., Kageyama K. Roles of oxidative stress and redox regulation in atherosclerosis. J. Atheroscler. Thromb. 2009;16:532–538. doi: 10.5551/jat.1255. [DOI] [PubMed] [Google Scholar]
- 3.Brown D.I., Griendling K.K. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ. Res. 2015;116:531–549. doi: 10.1161/CIRCRESAHA.116.303584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mieyal J.J., Chock P.B. Posttranslational modification of cysteine in redox signaling and oxidative stress: focus on s-glutathionylation. Antioxid. Redox Signal. 2012;16:471–475. doi: 10.1089/ars.2011.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lillig C.H., Berndt C. Glutaredoxins in thiol/disulfide exchange. Antioxid. Redox Signal. 2013;18:1654–1665. doi: 10.1089/ars.2012.5007. [DOI] [PubMed] [Google Scholar]
- 6.Adachi T., Weisbrod R.M., Pimentel D.R., Ying J., Sharov V.S., Schoneich C., Cohen R.A. S-glutathiolation by peroxynitrite activates SERCA during arterial relaxation by nitric oxide. Nat. Med. 2004;10:1200–1207. doi: 10.1038/nm1119. [DOI] [PubMed] [Google Scholar]
- 7.Chen F.C., Ogut O. Decline of contractility during ischemia-reperfusion injury: actin glutathionylation and its effect on allosteric interaction with tropomyosin. Am. J. Physiol. Cell Physiol. 2006;290:C719–C727. doi: 10.1152/ajpcell.00419.2005. [DOI] [PubMed] [Google Scholar]
- 8.Ward N.E., Stewart J.R., Ioannides C.G., O'Brian C.A. Oxidant-induced S-glutathiolation inactivates protein kinase C-alpha (PKC-alpha): a potential mechanism of PKC isozyme regulation. Biochemistry. 2000;39:10319–10329. doi: 10.1021/bi000781g. [DOI] [PubMed] [Google Scholar]
- 9.Shelton M.D., Mieyal J.J. Regulation by reversible S-glutathionylation: molecular targets implicated in inflammatory diseases. Mol. Cells. 2008;25:332–346. [PMC free article] [PubMed] [Google Scholar]
- 10.Barrett W.C., DeGnore J.P., Konig S., Fales H.M., Keng Y.F., Zhang Z.Y., Yim M.B., Chock P.B. Regulation of PTP1B via glutathionylation of the active site cysteine 215. Biochemistry. 1999;38:6699–6705. doi: 10.1021/bi990240v. [DOI] [PubMed] [Google Scholar]
- 11.Humphries K.M., Juliano C., Taylor S.S. Regulation of cAMP-dependent protein kinase activity by glutathionylation. J. Biol. Chem. 2002;277:43505–43511. doi: 10.1074/jbc.M207088200. [DOI] [PubMed] [Google Scholar]
- 12.Kilzer P., Chang K., Marvel J., Rowold E., Jaudes P., Ullensvang S., Kilo C., Williamson J.R. Albumin permeation of new vessels is increased in diabetic rats. Diabetes. 1985;34:333–336. doi: 10.2337/diab.34.4.333. [DOI] [PubMed] [Google Scholar]
- 13.Williamson J.R., Chang K., Tilton R.G., Prater C., Jeffrey J.R., Weigel C., Sherman W.R., Eades D.M., Kilo C. Increased vascular permeability in spontaneously diabetic BB/W rats and in rats with mild versus severe streptozocin-induced diabetes. Prevention by aldose reductase inhibitors and castration. Diabetes. 1987;36:813–821. doi: 10.2337/diab.36.7.813. [DOI] [PubMed] [Google Scholar]
- 14.Davies P.F., Civelek M., Fang Y., Fleming I. The atherosusceptible endothelium: Endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc. Res. 2013;99:315–327. doi: 10.1093/cvr/cvt101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Miyazaki T., Taketomi Y., Takimoto M., Lei X.F., Arita S., Kim-Kaneyama J.R., Arata S., Ohata H., Ota H., Murakami M., Miyazaki A. m-calpain induction in vascular endothelial cells on human and mouse atheromas and its roles in VE-cadherin disorganization and atherosclerosis. Circulation. 2011;124:2522–2532. doi: 10.1161/CIRCULATIONAHA.111.021675. [DOI] [PubMed] [Google Scholar]
- 16.Wu C., Huang R.T., Kuo C.H., Kumar S., Kim C.W., Lin Y.C., Chen Y.J., Birukova A., Birukov K.G., Dulin N.O., Civelek M., Lusis A.J., Loyer X., Tedgui A., Dai G., Jo H., Fang Y. Mechanosensitive PPAP2B regulates endothelial responses to atherorelevant hemodynamic forces. Circ. Res. 2015;117:e41–e53. doi: 10.1161/CIRCRESAHA.117.306457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boueiz A., Hassoun P.M. Regulation of endothelial barrier function by reactive oxygen and nitrogen species. Microvasc. Res. 2009;77:26–34. doi: 10.1016/j.mvr.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 18.Edelhauser H.F., Van Horn D.L., Miller P., Pederson H.J. Effect of thiol-oxidation of glutathione with diamide on corneal endothelial function, junctional complexes, and microfilaments. J. Cell Biol. 1976;68:567–578. doi: 10.1083/jcb.68.3.567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vandenbroucke E., Mehta D., Minshall R., Malik A.B. Regulation of endothelial junctional permeability. Ann. NY Acad. Sci. 2008;1123:134–145. doi: 10.1196/annals.1420.016. [DOI] [PubMed] [Google Scholar]
- 20.Guilluy C., Garcia-Mata R., Burridge K. Rho protein crosstalk: another social network? Trends Cell Biol. 2011;21:718–726. doi: 10.1016/j.tcb.2011.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tzima E. Role of small GTPases in endothelial cytoskeletal dynamics and the shear stress response. Circ. Res. 2006;98:176–185. doi: 10.1161/01.RES.0000200162.94463.d7. [DOI] [PubMed] [Google Scholar]
- 22.Mitchell L., Hobbs G.A., Aghajanian A., Campbell S.L. Redox regulation of ras and rho GTPases: mechanism and function. Antioxid. Redox Signal. 2013;18:250–258. doi: 10.1089/ars.2012.4687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Paulech J., Solis N., Cordwell S.J. Characterization of reaction conditions providing rapid and specific cysteine alkylation for peptide-based mass spectrometry. Biochim. Biophys. Acta. 2013;1834:372–379. doi: 10.1016/j.bbapap.2012.08.002. [DOI] [PubMed] [Google Scholar]
- 24.Tabit C.E., Shenouda S.M., Holbrook M., Fetterman J.L., Kiani S., Frame A.A., Kluge M.A., Held A., Dohadwala M.M., Gokce N., Farb M.G., Rosenzweig J., Ruderman N., Vita J.A., Hamburg N.M. Protein kinase C-beta contributes to impaired endothelial insulin signaling in humans with diabetes mellitus. Circulation. 2013;127:86–95. doi: 10.1161/CIRCULATIONAHA.112.127514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murdoch C.E., Shuler M., Haeussler D.J., Kikuchi R., Bearelly P., Han J., Watanabe Y., Fuster J.J., Walsh K., Ho Y.S., Bachschmid M.M., Cohen R.A., Matsui R. Glutaredoxin-1 up-regulation induces soluble vascular endothelial growth factor receptor 1, attenuating post-ischemia limb revascularization. J. Biol. Chem. 2014;289:8633–8644. doi: 10.1074/jbc.M113.517219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shao D., Fry J.L., Han J., Hou X., Pimentel D.R., Matsui R., Cohen R.A., Bachschmid M.M. A redox-resistant sirtuin-1 mutant protects against hepatic metabolic and oxidant stress. J. Biol. Chem. 2014;289:7293–7306. doi: 10.1074/jbc.M113.520403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Clavreul N., Bachschmid M.M., Hou X., Shi C., Idrizovic A., Ido Y., Pimentel D., Cohen R.A. S-glutathiolation of p21ras by peroxynitrite mediates endothelial insulin resistance caused by oxidized low-density lipoprotein. Arterioscler. Thromb. Vasc. Biol. 2006;26:2454–2461. doi: 10.1161/01.ATV.0000242791.28953.4c. [DOI] [PubMed] [Google Scholar]
- 30.Beckman J.A., Creager M.A. Vascular complications of diabetes. Circ. Res. 2016;118:1771–1785. doi: 10.1161/CIRCRESAHA.115.306884. [DOI] [PubMed] [Google Scholar]
- 31.Burgoyne J.R., Haeussler D.J., Kumar V., Ji Y., Pimental D.R., Zee R.S., Costello C.E., Lin C., McComb M.E., Cohen R.A., Bachschmid M.M. Oxidation of HRas cysteine thiols by metabolic stress prevents palmitoylation in vivo and contributes to endothelial cell apoptosis. FASEB J. 2012;26:832–841. doi: 10.1096/fj.11-189415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Inoguchi T., Li P., Umeda F., Yu H.Y., Kakimoto M., Imamura M., Aoki T., Etoh T., Hashimoto T., Naruse M., Sano H., Utsumi H., Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C--dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes. 2000;49:1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 33.Sathanoori R., Sward K., Olde B., Erlinge D. Correction: the ATP receptors P2X7 and P2X4 modulate high glucose and palmitate-induced inflammatory responses in endothelial cells. PLoS One. 2015;10:e0133346. doi: 10.1371/journal.pone.0125111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singh H., Brindle N.P., Zammit V.A. High glucose and elevated fatty acids suppress signaling by the endothelium protective ligand angiopoietin-1. Microvasc. Res. 2010;79:121–127. doi: 10.1016/j.mvr.2010.01.005. [DOI] [PubMed] [Google Scholar]
- 35.Symons J.D., McMillin S.L., Riehle C., Tanner J., Palionyte M., Hillas E., Jones D., Cooksey R.C., Birnbaum M.J., McClain D.A., Zhang Q.J., Gale D., Wilson L.J., Abel E.D. Contribution of insulin and Akt1 signaling to endothelial nitric oxide synthase in the regulation of endothelial function and blood pressure. Circ. Res. 2009;104:1085–1094. doi: 10.1161/CIRCRESAHA.108.189316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huynh J., Nishimura N., Rana K., Peloquin J.M., Califano J.P., Montague C.R., King M.R., Schaffer C.B., Reinhart-King C.A. Age-related intimal stiffening enhances endothelial permeability and leukocyte transmigration. Sci. Transl. Med. 2011;3 doi: 10.1126/scitranslmed.3002761. 112ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakashima Y., Raines E.W., Plump A.S., Breslow J.L., Ross R. Upregulation of VCAM-1 and ICAM-1 at atherosclerosis-prone sites on the endothelium in the ApoE-deficient mouse. Arterioscler. Thromb. Vasc. Biol. 1998;18:842–851. doi: 10.1161/01.atv.18.5.842. [DOI] [PubMed] [Google Scholar]
- 38.Sawada N., Li Y., Liao J.K. Novel aspects of the roles of Rac1 GTPase in the cardiovascular system. Curr. Opin. Pharm. 2010;10:116–121. doi: 10.1016/j.coph.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hobbs G.A., Mitchell L.E., Arrington M.E., Gunawardena H.P., DeCristo M.J., Loeser R.F., Chen X., Cox A.D., Campbell S.L. Redox regulation of Rac1 by thiol oxidation. Free Radic. Biol. Med. 2015;79:237–250. doi: 10.1016/j.freeradbiomed.2014.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kil I.S., Shin S.W., Park J.W. S-glutathionylation regulates GTP-binding of Rac2. Biochem. Biophys. Res. Commun. 2012;425:892–896. doi: 10.1016/j.bbrc.2012.07.169. [DOI] [PubMed] [Google Scholar]
- 41.Osborn-Heaford H.L., Ryan A.J., Murthy S., Racila A.M., He C., Sieren J.C., Spitz D.R., Carter A.B. Mitochondrial Rac1 GTPase import and electron transfer from cytochrome c are required for pulmonary fibrosis. J. Biol. Chem. 2012;287:3301–3312. doi: 10.1074/jbc.M111.308387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Popov D. Protein S-glutathionylation: from current basics to targeted modifications. Arch. Physiol. Biochem. 2014;120:123–130. doi: 10.3109/13813455.2014.944544. [DOI] [PubMed] [Google Scholar]
- 43.Hill B.G., Bhatnagar A. Protein S-glutathiolation: redox-sensitive regulation of protein function. J. Mol. Cell Cardiol. 2012;52:559–567. doi: 10.1016/j.yjmcc.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shah M.S., Brownlee M. Molecular and cellular mechanisms of cardiovascular disorders in diabetes. Circ. Res. 2016;118:1808–1829. doi: 10.1161/CIRCRESAHA.116.306923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hirase T., Node K. Endothelial dysfunction as a cellular mechanism for vascular failure. Am. J. Physiol. Heart Circ. Physiol. 2012;302:H499–H505. doi: 10.1152/ajpheart.00325.2011. [DOI] [PubMed] [Google Scholar]
- 46.Bartels E.D., Christoffersen C., Lindholm M.W., Nielsen L.B. Altered metabolism of LDL in the arterial wall precedes atherosclerosis regression. Circ. Res. 2015 doi: 10.1161/CIRCRESAHA.115.307182. [DOI] [PubMed] [Google Scholar]
- 47.Usatyuk P.V., Vepa S., Watkins T., He D., Parinandi N.L., Natarajan V. Redox regulation of reactive oxygen species-induced p38 MAP kinase activation and barrier dysfunction in lung microvascular endothelial cells. Antioxid. Redox Signal. 2003;5:723–730. doi: 10.1089/152308603770380025. [DOI] [PubMed] [Google Scholar]
- 48.Dalle-Donne I., Giustarini D., Rossi R., Colombo R., Milzani A. Reversible S-glutathionylation of cys 374 regulates actin filament formation by inducing structural changes in the actin molecule. Free Radic. Biol. Med. 2003;34:23–32. doi: 10.1016/s0891-5849(02)01182-6. [DOI] [PubMed] [Google Scholar]
- 49.Wang J., Sun L., Si Y.F., Li B.M. Overexpression of actin-depolymerizing factor blocks oxidized low-density lipoprotein-induced mouse brain microvascular endothelial cell barrier dysfunction. Mol. Cell Biochem. 2012;371:1–8. doi: 10.1007/s11010-012-1415-7. [DOI] [PubMed] [Google Scholar]
- 50.Aslam M., Schluter K.D., Rohrbach S., Rafiq A., Nazli S., Piper H.M., Noll T., Schulz R., Gunduz D. Hypoxia-reoxygenation-induced endothelial barrier failure: Role of RhoA, Rac1 and myosin light chain kinase. J. Physiol. 2013;591:461–473. doi: 10.1113/jphysiol.2012.237834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.De Pascali F., Hemann C., Samons K., Chen C.A., Zweier J.L. Hypoxia and reoxygenation induce endothelial nitric oxide synthase uncoupling in endothelial cells through tetrahydrobiopterin depletion and S-glutathionylation. Biochemistry. 2014;53:3679–3688. doi: 10.1021/bi500076r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.JeBailey L., Wanono O., Niu W., Roessler J., Rudich A., Klip A. Ceramide- and oxidant-induced insulin resistance involve loss of insulin-dependent rac-activation and actin remodeling in muscle cells. Diabetes. 2007;56:394–403. doi: 10.2337/db06-0823. [DOI] [PubMed] [Google Scholar]
- 53.Sylow L., Kleinert M., Pehmoller C., Prats C., Chiu T.T., Klip A., Richter E.A., Jensen T.E. Akt and Rac1 signaling are jointly required for insulin-stimulated glucose uptake in skeletal muscle and downregulated in insulin resistance. Cell Signal. 2014;26:323–331. doi: 10.1016/j.cellsig.2013.11.007. [DOI] [PubMed] [Google Scholar]
- 54.Guleria R.S., Pan J., Dipette D., Singh U.S. Hyperglycemia inhibits retinoic acid-induced activation of Rac1, prevents differentiation of cortical neurons, and causes oxidative stress in a rat model of diabetic pregnancy. Diabetes. 2006;55:3326–3334. doi: 10.2337/db06-0169. [DOI] [PubMed] [Google Scholar]
- 55.Ke Y., Lei M., Wang X., Solaro R.J. Unique catalytic activities and scaffolding of p21 activated kinase-1 in cardiovascular signaling. Front Pharmacol. 2013;4:116. doi: 10.3389/fphar.2013.00116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Miyano K., Sumimoto H. Assessment of the role for rho family GTPases in NADPH oxidase activation. Methods Mol. Biol. 2012;827:195–212. doi: 10.1007/978-1-61779-442-1_14. [DOI] [PubMed] [Google Scholar]
- 57.Gunduz D., Thom J., Hussain I., Lopez D., Hartel F.V., Erdogan A., Grebe M., Sedding D., Piper H.M., Tillmanns H., Noll T., Aslam M. Insulin stabilizes microvascular endothelial barrier function via phosphatidylinositol 3-kinase/Akt-mediated Rac1 activation. Arterioscler. Thromb. Vasc. Biol. 2010;30:1237–1245. doi: 10.1161/ATVBAHA.110.203901. [DOI] [PubMed] [Google Scholar]
- 58.Birukova A.A., Burdette D., Moldobaeva N., Xing J., Fu P., Birukov K.G. Rac GTPase is a hub for protein kinase A and epac signaling in endothelial barrier protection by cAMP. Microvasc. Res. 2010;79:128–138. doi: 10.1016/j.mvr.2009.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Humphries K.M., Juliano C., Taylor S.S. Regulation of cAMP-dependent protein kinase activity by glutathionylation. J. Biol. Chem. 2002;277:43505–43511. doi: 10.1074/jbc.M207088200. [DOI] [PubMed] [Google Scholar]
- 60.Murata H., Ihara Y., Nakamura H., Yodoi J., Sumikawa K., Kondo T. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of akt. J. Biol. Chem. 2003;278:50226–50233. doi: 10.1074/jbc.M310171200. [DOI] [PubMed] [Google Scholar]
- 61.Bergman R.N., Ader M. Free fatty acids and pathogenesis of type 2 diabetes mellitus. Trends Endocrinol. Metab. 2000;11:351–356. doi: 10.1016/s1043-2760(00)00323-4. [DOI] [PubMed] [Google Scholar]
- 62.Weikel K.A., Cacicedo J.M., Ruderman N.B., Ido Y. Glucose and palmitate uncouple AMPK from autophagy in human aortic endothelial cells. Am. J. Physiol. Cell Physiol. 2015;308:C249–C263. doi: 10.1152/ajpcell.00265.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Reaven G.M., Hollenbeck C., Jeng C.Y., Wu M.S., Chen Y.D. Measurement of plasma glucose, free fatty acid, lactate, and insulin for 24 h in patients with NIDDM. Diabetes. 1988;37:1020–1024. doi: 10.2337/diab.37.8.1020. [DOI] [PubMed] [Google Scholar]
- 64.Li J., Wang Q., Chai W., Chen M.H., Liu Z., Shi W. Hyperglycemia in apolipoprotein E-deficient mouse strains with different atherosclerosis susceptibility. Cardiovasc. Diabetol. 2011;10 doi: 10.1186/1475-2840-10-117. 117-2840-10-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gao C.L., Zhu C., Zhao Y.P., Chen X.H., Ji C.B., Zhang C.M., Zhu J.G., Xia Z.K., Tong M.L., Guo X.R. Mitochondrial dysfunction is induced by high levels of glucose and free fatty acids in 3T3-L1 adipocytes. Mol. Cell Endocrinol. 2010;320:25–33. doi: 10.1016/j.mce.2010.01.039. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material