

Summary
Tertiary lymphoid follicles (TLFs) can develop in the respiratory tract in response to infections or chronic inflammation. However, their functional relevance remains unclear because they are implicated in both protective and pathological responses. In contrast to homeostatic conditions, external antigens and damage to the lung tissue may drive TLF formation in inflamed lungs, and once established, the presence of pulmonary TLFs may signal the progression of chronic lung disease. This novel concept will be discussed in light of recent work in chronic obstructive pulmonary disease and how changes in the pulmonary microbiota may drive and direct TLF formation and function. We will also discuss the cellularity of TLFs at the pulmonary mucosa, with emphasis on the potential roles of lymphoid tissue inducer cells, and B‐ and T‐cell aggregates, and will examine the function of key chemokines and cytokines including CXCL13 and interleukin‐17, in the formation and maintenance of pulmonary TLFs.
Keywords: B cells, inflammation, lung, lymphoid follicles
Abbreviations
- COPD
chronic obstructive pulmonary disease
- CPs
cryptopatches
- CS
cigarette smoke
- DCs
dendritic cells
- IL‐17
interleukin‐17
- ILFs
isolated lymphoid follicles
- LTi
lymphoid tissue inducer cells
- LTo
lymphoid tissue organizer cells
- NOD‐2
nucleotide‐binding oligomerization domain‐containing protein‐2
- SLOs
secondary lymphoid organs
- TLFs
tertiary lymphoid follicles
Introduction
Lymphoid organs are vital components of the mammalian adaptive immune system. Based on their function, they can be categorized as either primary or secondary. Primary lymphoid organs are found in most vertebrates and are sites for the development and maturation of lymphocytes. Secondary lymphoid organs (SLOs), such as lymph nodes, evolved in higher vertebrates as structures that enable the initiation of adaptive immunity.1 They are formed during embryogenesis or in the early postnatal period at predetermined anatomical locations.1, 2 They contribute to immune surveillance by providing an optimal site for the interaction between antigen‐presenting cells from diverse tissues and rare populations of circulating antigen‐specific T and B lymphocytes. Upon recognizing antigens specific to their cognate receptors, T and B cells clonally expand within the SLOs and are subsequently recruited to the inflamed tissue where they exert their specified effector function. Hence SLOs provide a controlled environment in the host to elicit an effective and appropriate response against antigens.
In addition to SLOs, tertiary lymphoid follicles (TLFs) can serve as sites of immune induction and are often associated with mucosal tissues, such as the lungs and intestines. In contrast to SLOs, TLFs form postnatally in response to chronic antigenic stimulation, inflammation or persistent infection.3 However, there are similarities in the structural organization of both TLFs and SLOs including their cellular composition and compartmentalized organization. Furthermore, the molecular and cellular mechanisms that regulate the development of SLOs can impact the formation of TLFs.4
Several questions remain regarding TLF origin and function. It is unclear whether TLFs represent a functional extension of SLOs. It is also uncertain whether TLFs primarily bolster protective immunity or amplify inflammation and contribute to localized immunopathology. In this review we first discuss the factors that can contribute to the formation and maintenance of TLFs at the pulmonary mucosa, followed by their functional role in chronic lung diseases, specifically chronic obstructive pulmonary disease (COPD).
What constitutes TLFs at the pulmonary mucosa
Tertiary lymphoid follicles formed at the pulmonary mucosa exhibit considerable heterogeneity in their localization and structural organization. TLFs can be found in association with a bronchus, a pulmonary vessel or be placed without a clear association in the interstitium.5 The TLFs found associated with an airway or in the perivascular space are most frequently reported in the lungs of patients with COPD.5
Structurally, TLFs can range from less complex, loose aggregates of B and T cells, to highly organized structures closely resembling SLOs.3, 6 The latter are referred to as tertiary lymphoid organs and have distinct B‐cell and T‐cell areas, germinal centres, high endothelial venules and follicular dendritic cell networks but unlike SLOs, TLFs are not encapsulated.7 Overall the consensus on the minimal requirements to qualify as a TLF is the presence of a densely packed B‐cell follicle associated with follicular dendritic cells.3, 8 Within the B‐cell follicles of TLFs, germinal centres can be identified using peanut agglutinin or the antibody GL7.9 In addition, plasma cells also appear at the periphery of the TLFs.10
Tertiary lymphoid follicles can also exhibit structural adaptations that can assist in the acquisition and presentation of antigen locally at the pulmonary mucosa. For instance it has been reported that the epithelium overlying the bronchus‐associated lymphoid tissue can be non‐ciliated and specialized to acquire and transport antigens directly from the airway lumen.11, 12 Furthermore, dendritic cell (DC) networks reported at the alveolar interface of TLFs may also facilitate antigen uptake.5 The development of high endothelial venules and lymphatic vessels around the TLFs illustrates yet another path for the trafficking of cells transporting antigens in and out of the TLFs.13, 14 Overall, TLFs at the pulmonary mucosa are structurally adapted to facilitate immune induction locally. But it remains unclear how the varying degrees of organization influence their functionality.
Developmental pathways involved in TLF formation
Tertiary lymphoid follicles are a normal component of the healthy lung tissue of certain species like rabbits, but they are not a constitutive feature of the healthy human or mouse lungs.15, 16 In humans, comparable structures develop only upon microbial stimulation or inflammation, a process termed lymphoid neogenesis.17
There is some overlap between the factors that govern the development of pre‐programmed SLOs and the de novo formation of TLFs.18 Development of SLOs is critically dependent on the interaction between specialized cells called lymphoid tissue inducer cells (LTi) with stromal lymphoid tissue organizer cells (LTo).19 The LTi cells signal through the lymphotoxin β‐receptor on LTo cells to drive the expression of chemokines CCL19, CCL21 and CXCL13, which recruit and organize B and T lymphocytes.20, 21 Certainly these chemokines play a role in the development of TLFs but some studies suggest that LTi cells may be dispensable for this process. Notably, following influenza infection and endotoxin‐induced inflammation TLFs developed normally in RORγt and Id2‐deficient mice, which lack LTi cells.9, 22
Other immune cells such as DCs or T cells can promote TLF formation in the absence of LTi cells. During the development of TLFs, DCs can provide instructional cues by directly interacting with stromal cells.10, 23 In line with this, intratracheal instillation of DCs was sufficient to induce the formation of TLFs in the lungs. Conversely, depleting DCs disintegrated pre‐existing TLFs.10, 23 Overall, there is clear evidence that DCs are important in the formation and maintenance of TLFs although the underlying mechanism is unclear. Additionally, DCs could impact TLF formation indirectly by activating and polarizing CD4+ T‐cell responses.
The cytokine IL‐17 may also contribute to TLF formation. Rangel‐Moreno et al.22 demonstrated that endotoxin‐induced TLFs in neonatal lungs were dependent on interleukin‐17 (IL‐17) and independent of LTi cells. The IL‐17‐producing T cells were sufficient for their formation through the induction of lymphotoxin‐independent expression of CXCL13 and CCL19. However, TLFs also develop in inflammatory conditions not clearly associated with IL‐17. Studies from the Forster laboratory demonstrated that following infection with modified vaccinia virus Ankara both TLF formation and CXCL13 expression were independent of IL‐17.24, 25 TLFs can also be found in models of allergic asthma, where the inflammatory milieu is biased towards T helper type 2 responses.26, 27 In line with these data, overexpression of IL‐5 is sufficient to induce TLF formation in the lungs.28 Hence multiple pathways and cells have been attributed functional roles in TLF formation and maintenance. Yet the relative importance of one over the other appears to be linked to the initial inflammatory trigger.
Tertiary lymphoid follicles in COPD
Chronic obstructive pulmonary disease is a complex, inflammatory disease, characterized by chronic bronchitis, formation of TLFs and emphysema (parenchymal destruction) that lead to irreversible airflow limitation (Fig. 1). Patients with COPD show considerable heterogeneity with respect to the involvement of small airway disease versus emphysema as well as the degree of airflow limitation. Exposure to cigarette smoke (CS) is the most widely associated environmental risk factor for disease development. Though smoking cessation lead to an improvement in lung function, beyond a certain age smokers did not restore lung function.29 In addition, Willemse et al.30 found that smoking cessation did not impact disease progression in a subset of patients. This suggests that in susceptible individuals, disease can progress through a self‐perpetuated inflammatory loop.29 The pathogenesis of COPD is multidimensional comprising aberrant innate and adaptive immune responses and has been extensively reviewed.31, 32 Here, we will focus specifically on the role of TLFs.
Figure 1.
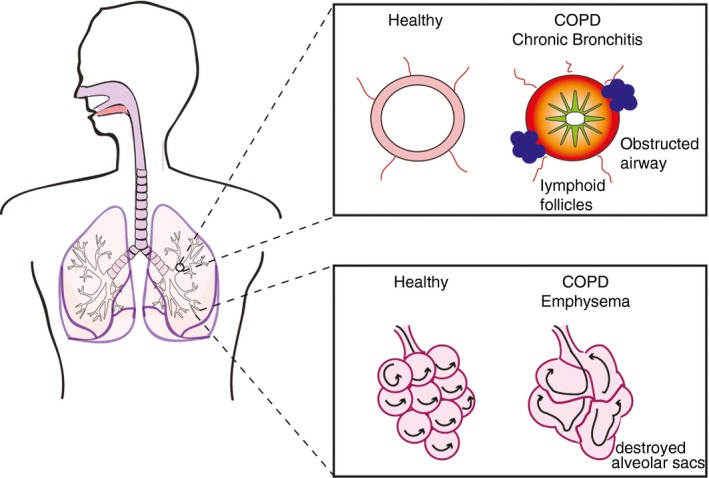
Pathological changes in chronic obstructive pulmonary disease (COPD).
Tertiary lymphoid follicles may present as such a candidate in marking this point of no return in disease progression. There is increasing evidence for a role of TLFs in COPD.33, 34 Structures similar to TLFs were described as early as 1992,35 but Hogg and colleagues provided the first compelling evidence for a correlation between disease severity and the incidence of TLFs.36 Since then, several others have shown that TLFs form in both human disease and preclinical models.5, 37, 38, 39 If TLFs mediate the self‐sustained inflammatory processes in disease, disrupting their formation or function may arrest the disease progression. But before targeting TLFs in disease, it is vital to understand what triggers their formation and carefully evaluate their beneficial or deleterious impact on disease.
Antigenic triggers and mechanisms that induce TLFs in COPD
In humans, there is an evident connection between the incidence of TLFs at the pulmonary mucosa and the degree of antigenic stimulation.6, 15, 16 However, the characteristics of the antigens that promote the formation of TLFs in COPD are poorly understood. While discussing antigenic triggers for TLF formation, it is also necessary to distinguish between the triggers that incite COPD versus those that incite TLF formation. TLFs are not as frequently encountered in inflamed lungs of patients with mild to moderate disease.36 This supports the view that there may not always be an overlap between factors that affect lung function and antigenic triggers for TLF formation.
Bringing together insight from different studies, including our recent work,39, 40, 41, 42, 43 we propose that the antigenic stimuli for TLF formation in COPD can be provided by both self, as well as non‐self components. Cigarette smoke‐induced irritation to bronchial epithelium may contribute to the release of neo‐ or self‐antigens44 and induce inflammatory cytokines that initiate TLF formation. Accumulation of self‐antigens in COPD lungs could be further amplified by defective clearance of damaged tissue and apoptotic cells by resident immune cells,45 and by increased enzymatic degradation of tissue by activated macrophages and neutrophils. Thaunat et al.46 have proposed that defective lymphatic drainage of inflamed tissue can trigger lymphoid neogenesis and provide some evidence from studies in chronic allograft rejection. Whether a similar mechanism is at work in COPD is not known.
Non‐self antigens’ triggers for TLF formation include components of CS or microbial antigens derived from the commensal and non‐commensal microbial colonizers. CS is a complex mixture of an estimated 5000 chemicals47 and can also contain a number of bacteria‐ or fungi‐derived products including endotoxin,48 which can directly induce the formation of TLFs.22 Whether non‐microbially derived components of CS can directly induce TLF formation has not been investigated.
CS components may also impact TLF formation indirectly by compromising the innate immune mechanisms and barrier function of the epithelium49 leading to the dysbiosis of the pulmonary microbiome and an increased susceptibility to infection. Although the field of pulmonary microbiome research is still in its nascent stages, breakthroughs in non‐culture‐based detection methods have provided some evidence for the presence of a commensal microbial community in the respiratory tract during homeostasis, which is altered in disease.50 A compromised barrier function in COPD51, 52, 53 may cause a dissemination of microbial products to distal locations in the respiratory tract. Alternatively, chronic inflammation and tissue damage may promote the outgrowth of specific species within commensal microbial communities that alter the cytokine milieu to ultimately facilitate the formation of TLFs. In a similar vein the structural and chemical changes in the inflamed lungs may also predispose patients with COPD to colonization by non‐commensal microbes.54 Recently, we demonstrated that microbial signals amplify IL‐17 production. This is in line with research that implicates IL‐17 in lymphoid neogenesis following endotoxin exposure as well as in systemic autoimmune conditions.22, 55, 56 Whether or not specific bacteria can polarize cytokine responses and trigger TLF formation similar to what is observed in the gastrointestinal tract (Box 1) is not known.
Box 1. Triggers for lymphoid neogenesis: Lessons from the intestinal tract.
The gut‐associated lymphoid tissue also contains small organized lymphoid tissues termed cryptopatches (CPs), and isolated lymphoid follicles (ILFs) that function as immune inductive sites.63, 64, 65 CPs/ILFs develop postnatally around day 14 in mice, coinciding with a critical window of opportunity for microbial expansion in the gastrointestinal tract. Despite this correlation, germ‐free mice still develop CPs/ILFs in the intestine in the absence of intestinal colonization; albeit the total numbers of CPs/ILFs are dramatically reduced compared with colonized mice.63, 64, 65 To support this concept, it has been shown that mice lacking microbial signalling through innate receptors MyD88 (differentiation primary response gene 88), NOD‐2 (nucleotide‐binding oligomerization domain‐containing protein‐2) and/or NOD1 have a reduction or lack of intestinal ILFs, indicating the involvement of innate microbial signalling molecules for normal development of intestinal ILFs.66 Furthermore Donaldson et al.67 recently found that a subset of colonic regulatory T cells may potentially regulate the transition of CPs into ILFs based on cytokine cues induced from the microbial environment (namely IL‐25 and IL‐23 expression). Collectively, these data suggest that the intestinal microbiota plays a critical role in providing feedback for CP/ILF development, and the maturation of CPs and ILFs in the gut‐associated lymphoid tissue is largely influenced by the presence of microbial stimulus and burden. In comparison, the respiratory tract harbours a much lower microbial load and perhaps consequently it does not require TLFs constitutively in humans and mice.
A role for IL‐17 in driving TLF formation in COPD is further supported by the observation that similar to the incidence of TLFs, the number of IL‐17‐producing cells correlates with airflow limitation.57, 58 Recently Roos et al.59 showed that the lymphoid neogenesis in end‐stage COPD is linked to IL‐17 levels. Mechanistically they found that this effect was mediated by IL‐17‐dependent expression of CXCL12, a chemokine that promotes TLF formation in the absence of differentiated follicular DCs.25
In some studies, IL‐17 promotes TLF formation through CXCL13 production.22 Interestingly, although lymphotoxin can increase CXCL13 expression in a CS exposure model of COPD,37 TLFs develop to a similar extent in lymphotoxin α‐deficient mice. However, treating CS‐exposed animals with CXCL13 neutralizing antibody completely abrogated the formation of lymphoid follicles as well as reducing alveolar wall destruction.38
Taken together these data suggest that in COPD, lymphotoxin‐independent mechanisms associated with inflammation such as IL‐17 may promote the formation of TLFs. However, the complexity of the antigen assault in COPD argues against the requirement for the persistence of a single antigen for TLF formation or maintenance. The thresholds set for TLF formation are probably determined by antigenic load (self or non‐self), tissue damage and remodelling and warrant further investigation.
Further insight into the role of microbial dysbiosis in lymphoid neogenesis in the respiratory tract may come from studies in bronchiectasis. Bronchiectasis is a chronic inflammatory lung disease characterized by the irreversible and abnormal enlargement of the bronchi and bronchioles.60 Though the underlying causes of the disease are variable, in many patients the condition precipitates from persistent microbial infection and a defective host immune response that leads to a self‐sustained cycle of inflammation.60 Damage induced by microbial infections may introduce changes in the lung environment that potentiate pathogenicity of certain species within the commensal communities. Similar to COPD, follicle‐like structures have also been described in lung biopsies of patients with bronchiectasis.61 Certainly there appears to be an underappreciated overlap in the pathogenic changes across bronchiectasis and COPD. In addition, bronchiectasis can also occur in patients in COPD and is associated with an increased risk of mortality and morbidity in moderate to severe COPD.62 Studies investigating patient subsets with bronchiectasis and COPD could reveal the link between specific microbial signatures and lymphoid neogenesis in the respiratory tract.
Functional relevance of TLFs in disease
The capacity to form TLFs de novo in the presence of specialized, preprogrammed SLOs suggests a protective function for TLFs in pulmonary immunity. Certainly, the benefits of TLFs have been demonstrated in the context of antimicrobial immunity.7, 68
Tertiary lymphoid follicless in COPD may consequently arise to counteract the defect in innate immune control of microbial colonization and to protect more distal parts of the lower respiratory tract that are not adapted to the chronic presence of microbes. Studies on the pulmonary microbiome suggest that the most marked changes in the microbiome, occur in severe disease, the time when TLFs are also most frequent.69, 70, 71 We speculate that microbial dysbiosis triggers TLF formation. Hence, disrupting TLFs may enhance the infection‐induced episodes of exacerbation in the disease. This notion is supported by studies that have implicated pulmonary TLFs in protective immunity to respiratory viral infections.7, 9, 68 In addition, Wiley et al.68 found that the induction of TLFs by protein cage nanoparticles enhanced anti‐viral responses to diverse viruses and limited collateral damage to lung tissue. Studies in mice lacking SLOs suggest that responses generated in pulmonary TLFs following high‐dose influenza infection are protective but less pathological than responses generated systemically.7, 9
Insight into the pathogenic role of TLFs has come from studies trying to target TLF formation directly in preclinical models.33, 38 Recently Seys and colleagues showed that antagonizing B‐cell‐activating factor (BAFF) in a CS‐induced model of COPD reduced TLF formation, pulmonary inflammation and alveolar wall destruction.33 In addition, this also impacted the phenotype of lung‐resident macrophages illustrating that TLFs also influence innate immunity in the lung environment.
The pathological role of TLFs in a number of autoimmune conditions has been attributed to the production of autoreactive antibodies.72 Though sequence analysis of B‐cell clones from TLFs from COPD lungs has confirmed the presence of an oligoclonal, antigen‐specific humoral response39 there are conflicting reports on autoreactive antibodies in disease.41, 73, 74, 75 Packard et al.43 showed that higher titres of self‐reactive antibodies are found in patients with worse emphysema. This suggests that perhaps a subset of patients may have autoimmune emphysema. The pathogenic potential of autoreactive antibodies in patients with COPD was further demonstrated by Feghali‐Bostwick et al.41 when they found autoantibodies directed against the pulmonary epithelium, which could mediate antibody‐dependent cell‐mediated cytotoxicity. Morissette et al.76 showed that anti‐nuclear antibodies could only be detected in animals that had been exposed to CS and had TLFs in the lungs. Moreover, these antibodies persisted in animals that had TLFs, even upon cessation of CS exposure. In addition to autoreactive antibodies, autoreactive T cells are also implicated in COPD.73, 77, 78 Taken together these studies suggest a possible role for TLFs and autoreactivity in perpetuating an inflammatory loop in COPD.
However, these studies have several limitations. Several of these studies investigate the changes in the systemic antibody levels and this may not be necessarily representative of the local immune responses in the lungs. Indeed studies in preclinical models elaborate the discrepancies between the antibody responses within the local compartment; i.e. the broncho‐alveolar lavage versus systemic circulating antibody levels.38, 42 Consistent with these findings, immune complexes and complement can be present in human lungs of patients with COPD.41 Another limitation of these studies is the lack of a specific autoantigen or a specific pathogenic role for antibodies derived from TLFs in disease. Mild or severe COPD is defined by the degree of decline in lung function without consideration of the heterogeneity in the underlying pathological mechanisms.79 The release of self‐antigens or their altered immunogenicity may vary significantly in patients depending on differences in CS exposure,44 tissue degradation by the innate immune cells and other factors. This is further illustrated by conflicting reports regarding the presence of anti‐elastin antibodies in COPD.73, 75, 80 It is also important to bear in mind that not all autoimmune conditions depend exclusively on autoantibodies and the presence of autoantibodies may not be indicative of disease. Rather it would be more informative to demonstrate a functional role for autoantibodies in disease.
Conclusion
The available data suggest that TLFs may be key players in the pathophysiology of COPD by mediating the self‐sustained inflammatory processes in disease. It seems likely that TLFs arise in response to antigenic stimuli, from microbial dysbiosis, and from tissue damage during chronic inflammation (Fig. 2). Although TLFs may arise to counteract environmental insults, they may also facilitate the generation of autoimmune pathogenic reactions. Moreover, it remains unclear whether TLFs persist even in the absence of inflammatory stimulus. Specifically in COPD, could smoking cessation or antibiotic therapy lead to the disintegration of the TLFs? There is some preliminary evidence that TLFs may persist and contribute to autoreactive antibodies even upon cessation of CS exposure.76 Further insight into the functional role of TLFs could be gained by investigating whether cells primed within TLFs display a distinct effector function compared to cells primed in SLOs. Hence, multiple questions remain regarding the origins, prognostic significance and mechanisms by which TLFs contribute to disease. This is further complicated by the disease heterogeneity observed in patients with COPD. It may be possible to arrest the inflammatory progression of COPD by disrupting the formation or function of TLFs. However, first it is vital to understand and carefully assess their beneficial or deleterious impact on disease.
Figure 2.
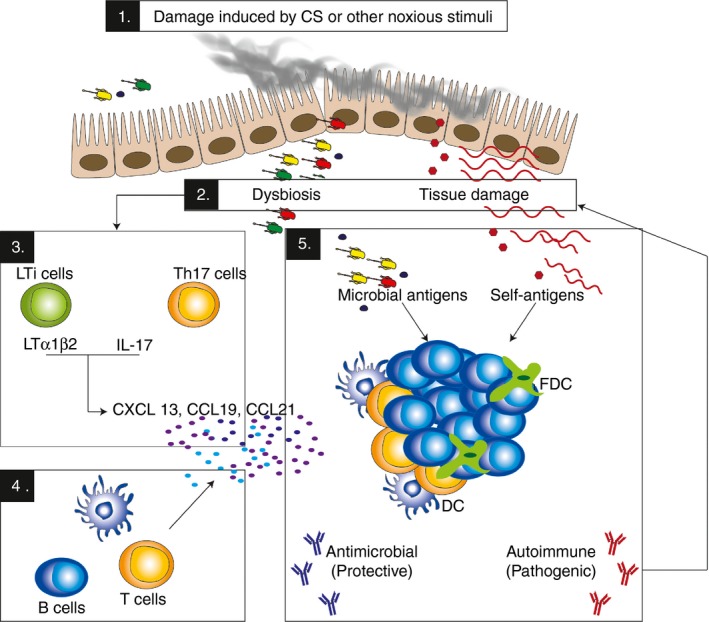
New insights into tertiary lymphoid follicles (TLFs) formation and function in chronic obstructive pulmonary disease. Exposure to noxious stimuli such as cigarette smoke (CS) [1] can lead to tissue damage and consequently the release of self‐antigens and dysbiosis of the pulmonary microbiome [2]. Antigenic stimuli provided by these, can activate lymphoid tissue inducer cells or interleukin‐17 (IL‐17) ‐producing cells which may in turn stimulate the production of chemokines such as CXCL13, CCL21 and CCL19 [3], which recruit T and B cells into the lungs and initiate lymphoid neogenesis [4]. Within the TLFs that are thus formed, microbial and self‐antigens may be presented leading concomitant generation of local responses that may be both protective and detrimental simultaneously [5].
Disclosure
The authors declare no conflict of interest.
Acknowledgements
Koshika Yadava is supported by the Swiss National Science Foundation early postdoctoral mobility grant and Child Health Research Institute and the Stanford NIH‐NCATS‐CTSA (grant no. UL1 TR001085). Melissa Lawson is supported by Horizon 2020 (H2020‐MSCA‐IF‐2014; grant # 661594).
References
- 1. Boehm T, Hess I, Swann JB. Evolution of lymphoid tissues. Trends Immunol Elsevier; 2012; 33:315–21. [DOI] [PubMed] [Google Scholar]
- 2. Boehm T. Form follows function, function follows form: how lymphoid tissues enable and constrain immune reactions. Immunol Rev 2016; 271:4–9. [DOI] [PubMed] [Google Scholar]
- 3. Carragher DM, Rangel‐Moreno J, Randall TD. Ectopic lymphoid tissues and local immunity. Semin Immunol 2008; 20:26–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Rangel‐Moreno J, Carragher D, Randall TD. Role of lymphotoxin and homeostatic chemokines in the development and function of local lymphoid tissues in the respiratory tract. Inmunologia 2007; 26:13–28. [PMC free article] [PubMed] [Google Scholar]
- 5. Mori M, Andersson CK, Svedberg KA, Glader P, Bergqvist A, Shikhagaie M, et al Appearance of remodelled and dendritic cell‐rich alveolar–lymphoid interfaces provides a structural basis for increased alveolar antigen uptake in chronic obstructive pulmonary disease. Thorax 2013; 68:521–31. [DOI] [PubMed] [Google Scholar]
- 6. Richmond I, Pritchard GE, Ashcroft T, Avery A, Corris PA, Walters EH. Bronchus associated lymphoid tissue (BALT) in human lung: its distribution in smokers and non‐smokers. Thorax 1993; 48:1130–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moyron‐Quiroz J, Rangel‐Moreno J, Carragher DM, Randall TD. The function of local lymphoid tissues in pulmonary immune responses. Adv Exp Med Biol 2007; 590:55–68. [DOI] [PubMed] [Google Scholar]
- 8. Pabst R. Plasticity and heterogeneity of lymphoid organs. What are the criteria to call a lymphoid organ primary, secondary or tertiary? Immunol Lett 2007; 112:1–8. [DOI] [PubMed] [Google Scholar]
- 9. Moyron‐Quiroz JE, Rangel‐Moreno J, Kusser K, Hartson L, Sprague F, Goodrich S, et al Role of inducible bronchus associated lymphoid tissue (iBALT) in respiratory immunity. Nat Med 2004; 10:927–34. [DOI] [PubMed] [Google Scholar]
- 10. GeurtsvanKessel CH, Willart MAM, Bergen IM, van Rijt LS, Muskens F, Elewaut D, et al Dendritic cells are crucial for maintenance of tertiary lymphoid structures in the lung of influenza virus‐infected mice. J Exp Med 2009; 206:2339–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gould SJ, Isaacson PG. Bronchus‐associated lymphoid tissue (BALT) in human fetal and infant lung. J Pathol 1993; 169:229–34. [DOI] [PubMed] [Google Scholar]
- 12. Tango M, Suzuki E, Gejyo F, Ushiki T. The presence of specialized epithelial cells on the bronchus‐associated lymphoid tissue (BALT) in the mouse. Arch Histol Cytol 2000; 63:81–9. [DOI] [PubMed] [Google Scholar]
- 13. Baluk P, Adams A, Phillips K, Feng J, Hong Y‐K, Brown MB, et al Preferential lymphatic growth in bronchus‐associated lymphoid tissue in sustained lung inflammation. Am J Pathol 2014; 184:1577–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ruddle NH.Lymphatic vessels and tertiary lymphoid organs. J Clin Invest American Society for Clinical Investigation; 2014; 124:953–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Pabst R, Gehrke I. Is the bronchus‐associated lymphoid tissue (BALT) an integral structure of the lung in normal mammals, including humans? Am J Respir Cell Mol Biol 1990; 3:131–5. [DOI] [PubMed] [Google Scholar]
- 16. Tschernig T, Pabst R. Bronchus‐associated lymphoid tissue (BALT) is not present in the normal adult lung but in different diseases. Pathobiology 2000; 68:1–8. [DOI] [PubMed] [Google Scholar]
- 17. Bienenstock J, Johnston N, Perey DY. Bronchial lymphoid tissue. I. Morphologic characteristics. Lab Invest 1973; 28:686–92. [PubMed] [Google Scholar]
- 18. Jones GW, Jones SA. Ectopic lymphoid follicles: inducible centres for generating antigen‐specific immune responses within tissues. Immunology 2015; 147:141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mebius RE, Rennert P, Weissman IL. Developing lymph nodes collect CD4+ CD3− LTβ + cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 1997; 7:493–504. [DOI] [PubMed] [Google Scholar]
- 20. De Togni P, Goellner J, Ruddle NH, Streeter PR, Fick A, Mariathasan S, et al Abnormal development of peripheral lymphoid organs in mice deficient in lymphotoxin. Science 1994; 264:703–7. [DOI] [PubMed] [Google Scholar]
- 21. van de Pavert SA, Olivier BJ, Goverse G, Vondenhoff MF, Greuter M, Beke P, et al Chemokine CXCL13 is essential for lymph node initiation and is induced by retinoic acid and neuronal stimulation. Nat Immunol 2009; 10:1193–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rangel‐Moreno J, Carragher DM, de la Luz Garcia‐Hernandez M, Hwang JY, Kusser K, Hartson L, et al The development of inducible bronchus‐associated lymphoid tissue depends on IL‐17. Nat Immunol 2011; 12:639–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Halle S, Dujardin HC, Bakocevic N, Fleige H, Danzer H, Willenzon S, et al Induced bronchus‐associated lymphoid tissue serves as a general priming site for T cells and is maintained by dendritic cells. J Exp Med 2009; 206:2593–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Fleige H, Haas JD, Stahl FR, Willenzon S, Prinz I, Förster R. Induction of BALT in the absence of IL‐17. Nat Immunol Nature Publishing Group, a division of Macmillan Publishers Limited. All Rights Reserved; 2012; 13:1; author reply 2. [DOI] [PubMed] [Google Scholar]
- 25. Fleige H, Ravens S, Moschovakis GL, Bölter J, Willenzon S, Sutter G, et al IL‐17‐induced CXCL12 recruits B cells and induces follicle formation in BALT in the absence of differentiated FDCs. J Exp Med 2014; 211:643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chvatchko Y. Germinal center formation and local immunoglobulin E (IgE) production in the lung after an airway antigenic challenge. J Exp Med 1996; 184:2353–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Elliot JG, Jensen CM, Mutavdzic S, Lamb JP, Carroll NG, James AL. Aggregations of lymphoid cells in the airways of nonsmokers, smokers, and subjects with asthma. Am J Respir Crit Care Med 2004; 169:712–8. [DOI] [PubMed] [Google Scholar]
- 28. Lee JJ, McGarry MP, Farmer SC, Denzler KL, Larson KA, Carrigan PE, et al Interleukin‐5 expression in the lung epithelium of transgenic mice leads to pulmonary changes pathognomonic of asthma. J Exp Med 1997; 185:2143–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Sethi S, Mallia P, Johnston SL. New paradigms in the pathogenesis of chronic obstructive pulmonary disease II. Proc Am Thorac Soc American Thoracic Society 2009; 6:532–4 [DOI] [PubMed] [Google Scholar]
- 30. Willemse BWM, Postma DS, Timens W, ten Hacken NHT. The impact of smoking cessation on respiratory symptoms, lung function, airway hyperresponsiveness and inflammation. Eur Respir J 2004; 23:464–76. [DOI] [PubMed] [Google Scholar]
- 31. Barnes PJ. New concepts in chronic obstructive pulmonary disease. Annu Rev Med Annual Reviews 4139 El Camino Way, P.O. Box 10139, Palo Alto, CA 94303‐0139, USA; 2003; 54:113–29. [DOI] [PubMed] [Google Scholar]
- 32. Tuder RM, Petrache I. Pathogenesis of chronic obstructive pulmonary disease. J Clin Invest American Society for Clinical Investigation; 2012; 122:2749–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Seys LJM, Verhamme FM, Schinwald A, Hammad H, Cunoosamy DM, Bantsimba‐Malanda C, et al Role of B cell‐activating factor in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2015; 192:706–18. [DOI] [PubMed] [Google Scholar]
- 34. Brusselle GG, Demoor T, Bracke KR, Brandsma C‐A, Timens W. Lymphoid follicles in (very) severe COPD: beneficial or harmful? Eur Respir J 2009; 34:219–30. [DOI] [PubMed] [Google Scholar]
- 35. Bosken CH, Hards J, Gatter K, Hogg JC. Characterization of the inflammatory reaction in the peripheral airways of cigarette smokers using immunocytochemistry. Am Rev Respir Dis American Lung Association; 1992; 145(4 Pt 1):911–7. [DOI] [PubMed] [Google Scholar]
- 36. Hogg JC, Chu F, Utokaparch S, Woods R, Elliott WM, Buzatu L, et al The nature of small‐airway obstruction in chronic obstructive pulmonary disease. N Engl J Med 2004; 350:2645–53. [DOI] [PubMed] [Google Scholar]
- 37. Demoor T, Bracke KR, Maes T, Vandooren B, Elewaut D, Pilette C, et al Role of lymphotoxin‐α in cigarette smoke‐induced inflammation and lymphoid neogenesis. Eur Respir J 2009; 34:405–16. [DOI] [PubMed] [Google Scholar]
- 38. Bracke KR, Verhamme FM, Seys LJM, Bantsimba‐Malanda C, Cunoosamy DM, Herbst R, et al Role of CXCL13 in cigarette smoke‐induced lymphoid follicle formation and chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2013; 188:343–55. [DOI] [PubMed] [Google Scholar]
- 39. van der Strate BWA, Postma DS, Brandsma C‐A, Melgert BN, Luinge MA, Geerlings M, et al Cigarette smoke‐induced emphysema: a role for the B cell? Am J Respir Crit Care Med 2006; 173:751–8. [DOI] [PubMed] [Google Scholar]
- 40. Gadgil A, Duncan SR. Role of T‐lymphocytes and pro‐inflammatory mediators in the pathogenesis of chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2008; 3:531–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Feghali‐Bostwick CA, Gadgil AS, Otterbein LE, Pilewski JM, Stoner MW, Csizmadia E, et al Autoantibodies in patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2008; 177:156–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yadava K, Pattaroni C, Sichelstiel AK, Trompette A, Gollwitzer ES, Salami O, et al Microbiota promotes chronic pulmonary inflammation by enhancing IL‐17A and autoantibodies. Am J Respir Crit Care Med 2016; 193:975–87. [DOI] [PubMed] [Google Scholar]
- 43. Packard TA, Li QZ, Cosgrove GP, Bowler RP, Cambier JC. COPD is associated with production of autoantibodies to a broad spectrum of self‐antigens, correlative with disease phenotype. Immunol Res 2013; 55:48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kirkham PA, Caramori G, Casolari P, Papi AA, Edwards M, Shamji B, et al Oxidative stress‐induced antibodies to carbonyl‐modified protein correlate with severity of chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2011; 184:796–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Henson PM, Vandivier RW, Douglas IS. Cell death, remodeling, and repair in chronic obstructive pulmonary disease? Proc Am Thorac Soc 2006; 3:713–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Thaunat O, Kerjaschki D, Nicoletti A. Is defective lymphatic drainage a trigger for lymphoid neogenesis? Trends Immunol 2006; 27:441–5. [DOI] [PubMed] [Google Scholar]
- 47. Talhout R, Schulz T, Florek E, van Benthem J, Wester P, Opperhuizen A. Hazardous compounds in tobacco smoke. Int J Environ Res Public Health 2011; 8:613–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Larsson L, Szponar B, Ridha B, Pehrson C, Dutkiewicz J, Krysińska‐Traczyk E, et al Identification of bacterial and fungal components in tobacco and tobacco smoke. Tob Induc Dis 2008; 4:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Thorley AJ, Tetley TD. Pulmonary epithelium, cigarette smoke, and chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 2007; 2:409–28. [PMC free article] [PubMed] [Google Scholar]
- 50. Marsland BJ, Yadava K, Nicod LP. The airway microbiome and disease. Chest 2013; 144:632–7. [DOI] [PubMed] [Google Scholar]
- 51. Heijink IH, Noordhoek JA, Timens W, vanOosterhout AJM , Postma DS. Abnormalities in airway epithelial junction formation in chronic obstructive pulmonary disease. Am J Respir Crit Care Med American Thoracic Society; 2014; 189:1439–42. [DOI] [PubMed] [Google Scholar]
- 52. Shaykhiev R, Otaki F, Bonsu P, Dang DT, Teater M, Strulovici‐Barel Y, et al Cigarette smoking reprograms apical junctional complex molecular architecture in the human airway epithelium in vivo . Cell Mol Life Sci 2011; 68:877–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. van den Berge M, Steiling K, Timens W, Hiemstra PS, Sterk PJ, Heijink IH, et al Airway gene expression in COPD is dynamic with inhaled corticosteroid treatment and reflects biological pathways associated with disease activity. Thorax 2014; 69:14–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sethi S, Maloney J, Grove L, Wrona C, Berenson CS. Airway inflammation and bronchial bacterial colonization in chronic obstructive pulmonary disease. Am J Respir Crit Care Med American Thoracic Society; 2006; 173:991–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Peters A, Pitcher LA, Sullivan JM, Mitsdoerffer M, Acton SE, Franz B, et al Th17 cells induce ectopic lymphoid follicles in central nervous system tissue inflammation. Immunity 2011; 35:986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hsu H‐C, Yang P, Wang J, Wu Q, Myers R, Chen J, et al Interleukin 17‐producing T helper cells and interleukin 17 orchestrate autoreactive germinal center development in autoimmune BXD2 mice. Nat Immunol Nature Publishing Group2008; 9:166–75. [DOI] [PubMed] [Google Scholar]
- 57. Zhang J, Chu S, Zhong X, Lao Q, He Z, Liang Y. Increased expression of CD4+ IL‐17+ cells in the lung tissue of patients with stable chronic obstructive pulmonary disease (COPD) and smokers. Int Immunopharmacol 2013; 15:58–66. [DOI] [PubMed] [Google Scholar]
- 58. Di Stefano A, Caramori G, Gnemmi I, Contoli M, Vicari C, Capelli A, et al T helper type 17‐related cytokine expression is increased in the bronchial mucosa of stable chronic obstructive pulmonary disease patients. Clin Exp Immunol 2009; 157:316–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Roos AB, Sandén C, Mori M, Bjermer L, Stampfli MR, Erjefält JS. IL‐17A is elevated in end‐stage chronic obstructive pulmonary disease and contributes to cigarette smoke‐induced lymphoid neogenesis. Am J Respir Crit Care Med American Thoracic Society; 2015; 191:1232–41 [DOI] [PubMed] [Google Scholar]
- 60. Boyton RJ, Altmann DM. Bronchiectasis: current concepts in pathogenesis, immunology, and microbiology. Annu Rev Pathol 2016; 23:523–54. [DOI] [PubMed] [Google Scholar]
- 61. Whitwell F. A study of the pathology and pathogenesis of bronchiectasis. Thorax BMJ Group1952; 7:213–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Suarez‐Cuartin G, Chalmers JD, Sibila O. Diagnostic challenges of bronchiectasis. Respir Med 2016; 116:70–7. [DOI] [PubMed] [Google Scholar]
- 63. Lochner M, Ohnmacht C, Presley L, Bruhns P, Si‐Tahar M, Sawa S, et al Microbiota‐induced tertiary lymphoid tissues aggravate inflammatory disease in the absence of RORγt and LTi cells. J Exp Med 2011; 208:125–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pabst O, Herbrand H, Friedrichsen M, Velaga S, Dorsch M, Berhardt G, et al Adaptation of solitary intestinal lymphoid tissue in response to microbiota and chemokine receptor CCR7 signaling. J Immunol 2006; 177:6824–32. [DOI] [PubMed] [Google Scholar]
- 65. Eberl G. Inducible lymphoid tissues in the adult gut: recapitulation of a fetal developmental pathway? Nat Rev Immunol 2005; 5:413–20. [DOI] [PubMed] [Google Scholar]
- 66. Bouskra D, Brézillon C, Bérard M, Werts C, Varona R, Boneca IG, et al Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 2008; 456:507–10. [DOI] [PubMed] [Google Scholar]
- 67. Donaldson DS, Bradford BM, Artis D, Mabbott NA. Reciprocal regulation of lymphoid tissue development in the large intestine by IL‐25 and IL‐23. Mucosal Immunol 2015; 8:582–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wiley JA, Richert LE, Swain SD, Harmsen A, Barnard DL, Randall TD, et al Inducible bronchus‐associated lymphoid tissue elicited by a protein cage nanoparticle enhances protection in mice against diverse respiratory viruses. PLoS One 2009; 4:e7142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Erb‐Downward JR, Huffnagle GB, Martinez FJ. The microbiota in respiratory disease. Am J Respir Crit Care Med American Thoracic Society; 2012;185:1037–8. [DOI] [PubMed] [Google Scholar]
- 70. Sze MA, Dimitriu PA, Suzuki M, McDonough JE, Campbell JD, Brothers JF, et al Host response to the lung microbiome in chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2015; 192:438–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sze MA, Hogg JC, Sin DD. Bacterial microbiome of lungs in COPD. Int J Chron Obstruct Pulmon Dis 2014; 9:229–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Aloisi F, Pujol‐Borrell R. Lymphoid neogenesis in chronic inflammatory diseases. Nat Rev Immunol 2006; 6:205–17. [DOI] [PubMed] [Google Scholar]
- 73. Lee S‐H, Goswami S, Grudo A, Song L‐Z, Bandi V, Goodnight‐White S, et al Antielastin autoimmunity in tobacco smoking‐induced emphysema. Nat Med Nature Publishing Group; 2007; 13:567–9. [DOI] [PubMed] [Google Scholar]
- 74. Rinaldi M, Lehouck A, Heulens N, Lavend'homme R, Carlier V, Saint‐Remy J‐M, et al Antielastin B‐cell and T‐cell immunity in patients with chronic obstructive pulmonary disease. Thorax 2012; 67:694–700. [DOI] [PubMed] [Google Scholar]
- 75. Cottin V, Fabien N, Khouatra C, Moreira A, Cordier J‐F. Anti‐elastin autoantibodies are not present in combined pulmonary fibrosis and emphysema. Eur Respir J 2009; 33:219–21. [DOI] [PubMed] [Google Scholar]
- 76. Morissette MC, Jobse BN, Thayaparan D, Nikota JK, Shen P, Labiris NR, et al Persistence of pulmonary tertiary lymphoid tissues and anti‐nuclear antibodies following cessation of cigarette smoke exposure. Respir Res BioMed Central; 2014; 15:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kheradmand F, Shan M, Xu C, Corry DB. Autoimmunity in chronic obstructive pulmonary disease: clinical and experimental evidence. Expert Rev Clin Immunol 2012; 8:285–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Shan M, Cheng H‐F, Song L‐Z, Roberts L, Green L, Hacken‐Bitar J, et al Lung myeloid dendritic cells coordinately induce TH1 and TH17 responses in human emphysema. Sci Transl Med 2009; 1:4ra10. [DOI] [PubMed] [Google Scholar]
- 79. Viegi G, Pistelli F, Sherrill DL, Maio S, Baldacci S, Carrozzi L. Definition, epidemiology and natural history of COPD. Eur Respir J 2007; 30:993–1013. [DOI] [PubMed] [Google Scholar]
- 80. Brandsma CA, Kerstjens HAM, Geerlings M, Kerkhof M, Hylkema MN, Postma DS, et al The search for autoantibodies against elastin, collagen and decorin in COPD. Eur Respir J 2011;37:1289–92 [DOI] [PubMed] [Google Scholar]