

Abstract
Currently, the analyses of most genome-wide association studies (GWAS) have been performed on a single phenotype. There is increasing evidence showing that pleiotropy is a widespread phenomenon in complex diseases. Therefore, using only one single phenotype may lose statistical power to identify the underlying genetic mechanism. There is an increasing need to develop and apply powerful statistical tests to detect association between multiple phenotypes and a genetic variant. In this paper, we develop an Adaptive Fisher’s Combination (AFC) method for joint analysis of multiple phenotypes in association studies. The AFC method combines p-values obtained in standard univariate GWAS by using the optimal number of p-values which is determined by the data. We perform extensive simulations to evaluate the performance of the AFC method and compare the power of our method with the powers of TATES, Tippett’s method, Fisher’s combination test, MANOVA, MultiPhen, and SUMSCORE. Our simulation studies show that the proposed method has correct type I error rates and is either the most powerful test or comparable with the most powerful test. Finally, we illustrate our proposed methodology by analyzing whole-genome genotyping data from a lung function study.
To date, genome-wide association studies (GWAS) have become a tool of choice for the identification of genetic variants associated with complex human diseases. Currently, the analyses of most GWAS have been performed on a single phenotype. There is increasing evidence showing that pleiotropy, the effect of one variant on multiple phenotypes, is a widespread phenomenon in complex diseases1,2. Therefore, using only one single phenotype may lose statistical power to identify the underlying genetic mechanism. By taking into account the correlated structure of multiple phenotypes, we can not only discover genetic variants influencing multiple phenotypes that may lead to better understanding of etiology of complex human diseases3,4, but also can improve the statistical power by aggregating multiple weak effects and provide new biological insights by revealing pleiotropic variants5,6,7. Consequently, there is an increasing need to develop powerful statistical methods to detect association between multiple phenotypes and genetic variants.
Recently, several statistical methods for detecting association using multivariate phenotypes have been developed8,9,10,11,12,13. These methods can be divided into three groups: regression models, variable reduction method, and combining test statistics from univariate analysis14. Regression models, such as linear mixed effects models, generalized mixed effects models, and generalized estimating equations, can be used to test the association between a genetic variant and multiple phenotypes. By using random effects to account for correlation among individuals, linear and generalized mixed effect models can model the covariance structure not only caused by correlated phenotypes, but also caused by population structure9,15,16,17,18. Generalized estimating equations collapse random effects and random residual errors in the model19. Existing variable reduction methods can be roughly divided into three categories, principal components analysis of phenotypes (PCP)20, canonical correlation analysis (CCA)10 and principal component of heritability (PCH)11,21. The PCP approach applies a dimension reduction technique and tests for associations between genetic variants and the principle components of the phenotypes rather than the individual phenotypes. CCA provides a convenient statistical framework to simultaneously test the association between any number of quantitative phenotypes and any number of genetic variants genotyped across a gene or region of interest for unrelated individuals. For each genetic variant, the PCH approach reduces the phenotypes to a linear combination of phenotypes that has the highest heritability among all linear combinations of the phenotypes. Based on PCH, several advanced methods have been proposed such as penalized PCH applicable to high-dimensional data22,23 and principle components of heritability with coefficients maximizing the quantitative phenotype locus heritability (PCQH)11,24,25. The third group, combining test statistics from univariate tests, is to conduct univariate analysis on each phenotype, then combine the univariate test statistics26. The Trait-based Association Test that uses Extended Simes procedure (TATES)12 belongs to this group. TATES combines p-values obtained in standard univariate GWAS while correcting for the correlation between p-values.
Motivated by TATES, in this article, we propose an Adaptive Fisher’s Combination (AFC) method for joint analysis of multiple phenotypes in genetic association studies. We first test the association between each of the phenotypes and a genetic variant of interest using standard GWAS software. Then, AFC uses the optimal number of p-values which is determined by the data to test the association. Using extensive simulation studies, we evaluate the performance of the proposed method and compare the power of the proposed method with the powers of TATES, Tippett’s method27, Fisher’s Combination test (FC)28, Multivariate Analysis of Variance (MANOVA)29, joint model of Multiple Phenotypes (MultiPhen)8, and Sum Score method (SUMSCORE)12. Our simulation studies show that the proposed method has correct type I error rates and is either the most powerful test or comparable with the most powerful tests. Finally, we illustrate our proposed methodology by analyzing whole-genome genotyping data from a lung function study.
Method
Consider a sample of n unrelated individuals. Each individual has K phenotypes. Denote Yk = (y1k, …, ynk)T as the kth phenotype of n individuals. Denote X = (x1, …, xn)T as the genotypic score of n individuals at a genetic variant of interest, where xi ∈ {0, 1, 2} is the number of minor alleles that the ith individual carries at the genetic variant. We propose a new method to test the null hypothesis H0: none of the K phenotypes are associated with the genetic variant.
We test the association between each phenotypic vector Yk (k = 1, 2, …, K) and the genotypic score X using a standard GWAS software (e.g. PLINK, Gen/ProbABEL, MaCH, SNPTEST, and FaST-LMM)30,31,32,33,34,35,36. Let p1, p2, …, pK denote the p-values obtained by the standard univariate GWAS. Based on these p-values, we propose an Adaptive Fisher’s Combination (AFC) method to test the association between multiple phenotypes and the genetic variant. Let p(k) denote the kth smallest p-value, 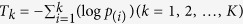 , and
, and 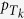 denote the p-value of Tk. The statistic of AFC to test the association between the K phenotypes and the genetic variant is given by
denote the p-value of Tk. The statistic of AFC to test the association between the K phenotypes and the genetic variant is given by 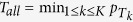 . We use the following permutation procedure to evaluate the p-values of Tk and Tall.
. We use the following permutation procedure to evaluate the p-values of Tk and Tall.
1. In each permutation, we randomly shuffle the genotypes and recalculate p(1), …, p(K) and T1, …, TK. Suppose that we perform B. times of permutations. Let 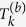 (b = 0, 1, …, B) denote the value of Tk based on the bth. permuted data, where b = 0 represents the original data.
(b = 0, 1, …, B) denote the value of Tk based on the bth. permuted data, where b = 0 represents the original data.
2. We transfer 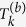 to
to 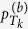 by
by
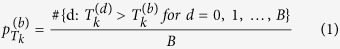 |
3. Let 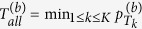 . Then, the p-vae of Tall is given by
. Then, the p-vae of Tall is given by
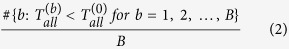 |
As shown in Appendix, the null distributions of p1, p2, …, pK and thus of Tall do not depend on the genetic variant being tested. Thus, the permutation procedure described above to generate an empirical null distribution of Tall needs to be done only once for a GWAS.
The R code of AFC is available at Shuanglin Zhang’s homepage http://www.math.mtu.edu/~shuzhang/software.html.
Comparison of Methods
We compare the performance of our method with those of TATES12, Tippett’s method27, Fisher’s Combination test (FC)28, Multivariate Analysis of Variance (MANOVA)29, joint model of Multiple Phenotypes (MultiPhen)8, and Sum Score method (SUMSCORE)12. Here we briefly introduce each of those methods using the notations in the method section.
TATES
Combine the K phenotype-specific p-values obtained in standard univariate GWAS to acquire one overall p-value, 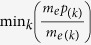 , where me denotes the effective number of independent p-values of all K phenotypes, and me(k) denotes the effective number of p-values among the top k p-values.
, where me denotes the effective number of independent p-values of all K phenotypes, and me(k) denotes the effective number of p-values among the top k p-values.
MANOVA
Consider a multivariate multiple linear regression model: Y = XβT + ε, where Y denotes the n × K matrix of phenotypes, βT = (β1, …, βK) is a vector of coefficients corresponding to the K phenotypes, and ε. is the n × K matrix of random errors with each row of ε to be i.i.d. MVN (0, Σ), where Σ is the covariance matrix of ε. To test H0 : β = 0, the likelihood ratio test is equivalent to the Wilk’s Lambda test statistic of MANOVA37, that is, 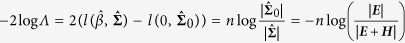 . Here Λ denote the ratio of the likelihood function under H0 to the likelihood function under H1, l(β, Σ) is the log-likelihood function,
. Here Λ denote the ratio of the likelihood function under H0 to the likelihood function under H1, l(β, Σ) is the log-likelihood function, 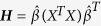 and
and 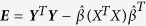 , where
, where 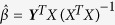 is the maximum likelihood estimator (MLE) of β, and |·| denotes the determinant of a matrix. Then the test statistic has an asymptotic
is the maximum likelihood estimator (MLE) of β, and |·| denotes the determinant of a matrix. Then the test statistic has an asymptotic 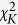 distribution38.
distribution38.
MultiPhen
By performing ordinaregression, MultiPhen develops a reversed analysis for joint analysis of multiple phenotypes by considering a genetic variant of interest X = (x1, …, xn)T as a response variable, and the correlated phenotypes Yk = (y1k, …, ynk)T as predictors.
SUMSCORE
Let 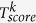 denote the score test statistic to test the association between the kth phenotype and the genetic variant. The test statistic of SUMSCORE is given by
denote the score test statistic to test the association between the kth phenotype and the genetic variant. The test statistic of SUMSCORE is given by 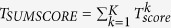 . The p-value of TSUMSCORE is estimated using a permutation procedure.
. The p-value of TSUMSCORE is estimated using a permutation procedure.
Tippett
The test statistic of Tippett is given by 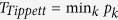 . The p-value of TTippett is estimated using a permutation procedure.
. The p-value of TTippett is estimated using a permutation procedure.
FC
The Fisher’s combination test statistic is defined as 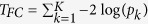 . Yang et al.28 adopted three different approaches to calculate the p-value for correlated phenotypes. In this article, we calculate the p-value using a permutation procedure.
. Yang et al.28 adopted three different approaches to calculate the p-value for correlated phenotypes. In this article, we calculate the p-value using a permutation procedure.
AFC, FC, and Tippett are closely related. Intuitively, when only one phenotype or very few phenotypes are associated with the variant, Tippett is more powerful than FC because in this case FC contains a lot of noises. When all phenotypes or a large proportion of the phenotypes are associated with the variant, FC is more powerful than Tippett because in this case Tippett only uses the minimum p-value and loses information. AFC can be adaptive to the number of phenotypes associated with the variant.
Simulation
We generate genotype data at a genetic variant according to a minor allele frequency (MAF) under Hardy-Weinberg equilibrium. Phenotypes are generated similarly to that of van der Sluis et al.12. The phenotypic correlation structures mimic that of UK10K39, that is, the phenotypes are divided into several groups (factors) and the within-group correlation is larger than the between-group correlation. Denote Yk = (y1k, …, ynk)T as the kth phenotype of n individuals and X = (x1, …, xn)T as the genotypic score of the n individuals at the genetic variant.
Scenario 1
considering one factor model with genetic variant effect on the factor. We first generate a common factor, f = βX, where f is the n by 1 common factor and β is the effect size. Then we simulate K phenotypes by
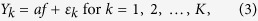 |
where a is a factor loading, εk = (ε1k, …, εnk)T ~ MVN(0, In), and In is the identity matrix.
Scenario 2
considering 4 factor model with the genetic variant effect on the fourth factor, each factor has (K)/4 (K is a multiple of 4) phenotypes. We generate 4 correlated factors using (f1, f2, f3, f4)T ~ MVN(0, Σ), where Σ = (1−ρfa)I + ρfa A, A is a matrix with elements of 1, I is the identity matrix, and ρfa is the correlation between any two factors. Then, we transform the fourth factor f4 to 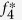 by
by 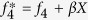 and simulate K phenotypes using
and simulate K phenotypes using
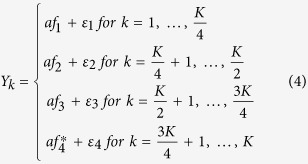 |
where a is a factor loading, εj = (ε1j, …, εnj)T ~ MVN(0, In) for j = 1, …, 4, and β is the effect size.
Scenario 3
considering two factor model with the genetic variant effect on the second factor, each factor has (K)/(2) (K is a multiple of 2) phenotypes. We generate two correlated factors using (f1, f2)T ~ MVN(0, Σ), where Σ = (1−ρfa)I + ρfaA, A is a matrix with elements of 1, I is the identity matrix, and ρfa is the correlation between any two factors. Then, we transform the second factor f2 to 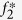 by
by 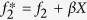 and simulate K phenotypes using
and simulate K phenotypes using
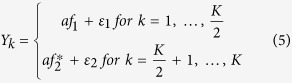 |
where a is a factor loading, εj = (ε1j, …, εnj)T ~ MVN(0, In) for j = 1, 2, and β is the effect size.
Scenario 4
considering 4 factor model with genetic variant effect specific to the Kth phenotype, each factor has (K)/4 (K is a multiple of 4) phenotypes. By using the original factors f1, f2, f3, f4 in scenario 2, we simulate K phenotypes using
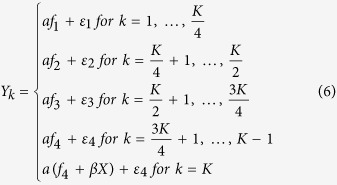 |
where a is a factor loading, εj = (ε1j, …, εnj)T ~ MVN(0, In) for j = 1, …, 4, and β is the effect size.
Scenario 5
considering one factor model with the genetic variant effect specific to the Kth phenotype. We simulate K phenotypes by
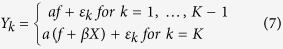 |
where f~MVN(0, In), a is a factor loading, εk = (ε1k, …, εnk)T ~ MVN(0, In), and β is the effect size.
Scenario 6
considering a network model, where the K phenotypes are correlated and the correlation structure is not due to one or multiple underlying common factors. We generate K phenotypes independent of genotypes for each individual by using 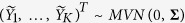 , where Σ = (1 − ρphe)I + ρpheA, A is a matrix with elements of 1, I is the identity matrix, and ρphe is the correlation between any two phenotypes. After generating
, where Σ = (1 − ρphe)I + ρpheA, A is a matrix with elements of 1, I is the identity matrix, and ρphe is the correlation between any two phenotypes. After generating , let
, let 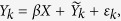 where εk = (ε1k, …, εnk)T ~ MVN(0, In).
where εk = (ε1k, …, εnk)T ~ MVN(0, In).
In scenarios 2–5, the within-factor correlation is a2 and between-factor correlaiton is a2ρfa. To evaluate type I error of the proposed method, we generate phenotypic values independent of genotypes by assigning β = 0. To evaluate power, we generate phenotypic values according to the six scenarios described above.
Simulation results
We use two sets of simulations to evaluate the type I error rates of the proposed method. The first set of simulations is normal simulation studies and includes 10,000 replicated samples for each sample size under each scenario. The p-values are estimated using 10,000 permutations. For 10,000 replicated samples, the 95% confidence intervals (CIs) for type I error rates at the nominal levels 0.01 and 0.001 are (0.008, 0.012) and (0.0004, 0.0016), respectively. The estimated type I error rates of the proposed test (AFC) are summarized in Table 1. From Table 1, we can see that most of the estimated type I error rates are within the 95% CIs and those type I error rates not within the 95% CIs are very close to the bound of the corresponding 95% CI, which indicates that the proposed method is valid.
Table 1. The estimated type I error rates of the proposed method for MAF equals 0.3.
| α | Sample size | Scenario | |||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | ||
| 0.01 | 1000 | 0.0088 | 0.0110 | 0.0105 | 0.0083 | 0.0083 | 0.0108 |
| 2000 | 0.0095 | 0.0107 | 0.0094 | 0.0083 | 0.0098 | 0.0110 | |
| 0.001 | 1000 | 0.0008 | 0.0015 | 0.0012 | 0.0008 | 0.0007 | 0.0012 |
| 2000 | 0.0015 | 0.0014 | 0.0007 | 0.0009 | 0.0011 | 0.0014 | |
α is the significance level. 10,000 replicates are used in the simulations.
The second set of simulations mimics GWAS. To be comparable with the real data analysis, we generate 6,000 unrelated individuals with 8 phenotypes at 106 variant sites under each scenario. The phenotypes are independent of genotypes. The MAF at each variant is a random number between 0.05 and 0.5. The null distributions of T1, …, TK and Tall are generated by 106 permutations using the genotypes at the first variant. We consider genotypes at 106 variants as 106 replicated samples. For 106 replicated samples, the 95% confidence intervals (CIs) for the type I error rates at the nominal levels 10−3, 10−4, and 10−5 are (0.94×10−3, 1.06×10−3), (0.80 × 10−4, 1.20 × 10−4) and (0.38 × 10−5, 1.62 × 10−5), respectively. The estimated type I error rates of the proposed test (AFC) are summarized in Table 2. From Table 2, we can see that all of the estimated type I error rates are within the 95% CIs, which indicates that the proposed method is valid.
Table 2. The estimated type I error rates of the proposed method that mimic GWAS.
| α | Scenario | |||||
|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | |
| 1.00 × 10−3 | 1.02 × 10−3 | 1.06 × 10−3 | 0.94 × 10−3 | 1.03 × 10−3 | 1.00 × 10−3 | 1.05 × 10−3 |
| 1.00 × 10−4 | 1.03 × 10−4 | 1.20 × 10−4 | 0.80 × 10−4 | 0.97 × 10−4 | 1.20 × 10−4 | 0.82 × 10−4 |
| 1.00 × 10−5 | 1.30 × 10−5 | 1.10 × 10−5 | 1.50 × 10−5 | 1.40 × 10−5 | 1.00 × 10−5 | 0.50 × 10−5 |
α is the significance level.
For power comparisons, we consider (1) power as a function of the effect size under all six scenarios, and (2) power as a function of factorial correlation (ρfa) under scenarios 2–4 and power as a function of phenotypic correlation (ρphe) under scenario 6 because scenarios 1 and 5 are one factor model and thus have no ρfa and ρphe involved. For Figs 1 and 2, the p-values of AFC, FC, Tippett, and SUMSCORE are estimated using 10,000 permutations and the p-values of TATES, MultiPhen, and MANOVA are estimated using asymptotic distributions. The powers of all tests are evaluated using 1,000 replicated samples at 0.1% significance level. For Fig. 3, the p-values of AFC, FC, Tippett, and SUMSCORE are estimated using 107 permutations. The powers of all tests are evaluated using 1,000 replicated samples at 10−6 significance level.
Figure 1. Power comparisons of the 7 tests for power as a function of effect size (β) under the 6 scenarios.

The total number of phenotypes is 20. The sample size is 1,000. MAF is 0.3. The factor loadings are 0.75. In scenarios 2, 3 and 4, the factorial correlation (ρfa) is 0.1. In scenario 6, the phenotypic correlation (ρphe) is 0.1. The powers are evaluated at 0.1% significance level.
Figure 2. Power comparisons of the 7 tests for power as a function of factorial correlation (ρfa) under scenarios 2 to 4, and as a function of phenotypic correlation (ρphe) under scenario 6.

The total number of phenotypes is 20. The sample size is 1,000. MAF is 0.3. The factor loadings are 0.75. In scenarios 2 and 3, the effect size (β) is 0.2. In scenario 4, the effect size (β) is 0.3. In scenario 6, the effect size (β) is 0.1. The powers are evaluated at 0.1% significance level.
Figure 3. Power comparisons of the 7 tests for power as a function of effect size (β) under scenario 2.
The total number of phenotypes is 20. The sample size is 1,000. MAF is 0.3. The factor loadings are 0.75. The factorial correlation (ρfa) is 0.1. The powers are evaluated at 10-6 significance level while p-values of AFC, FC, Tippet, and SUMSCORE are evaluated by 107 permutations.
Figure 1 gives the power comparisons of the 7 tests (AFC, TATES, Tippett, FC, MANOVA, MultiPhen, and SUMSCORE) for the power as a function of the effect size based on the six scenarios for 20 phenotypes. This figure shows that (1) AFC is either the most powerful test (genotypes directly impact on a portion of the phenotypes: scenarios 2–3) or comparable to the most powerful test (genotypes directly impact on all phenotypes or a single phenotype: scenarios 1, 4, 5, and 6); (2) TATES and Tippett have similar power and are much less powerful than other methods when genotypes directly impact on all phenotypes (scenarios 1 and 6); (3) MANOVA and MultiPhen have similar power and are much less powerful than other methods when genotypes directly impact on a portion of the phenotypes (scenarios 2–3); and (4) SUMSCORE and FC have similar power and are much less powerful than other methods when genotypes directly impact on a single phenotype (scenarios 4–5).
Power comparisons of the 7 tests for the power as a function of the factorial correlation (ρfa) under scenarios 2–4 and as a function of the phenotypic correlation (ρphe) under scenario 6 are given by Fig. 2. This figure shows that under scenario 4, the powers of all tests do not change with the factorial correlation because only one phenotype is associated with the variant and thus the factorial correlation does not change the information of association between the variant and phenotypes. Under scenarios 2, 3 and 6, (1) the powers of SUMSCORE and FC decrease with the increasing of the factorial or phenotypic correlation because SUMSCORE and FC involve all phenotypes and thus information contained by all phenotypes will decrease with the increasing of the factorial or phenotypic correlation; (2) the powers of TATES and Tippett do not change with the increasing of the factorial or phenotypic correlation because TATES and Tippett essentially only depend on the phenotype that has the strongest association with the variant; (3) under scenario 6, the power of AFC decreases with the increasing of the phenotypic correlation; under scenarios 2–3, the power of AFC does not change much with the factorial correlation; and (4) under scenario 6, the powers of MANOVA and MultiPhen decrease with the increasing of the phenotypic correlation; under scenarios 2–3, the powers of MANOVA and MultiPhen increase with the increasing of the factorial correlation, which is consistent with the results of Ray et al.38. We also give power comparisons of the 7 tests using a significance level of 10−6 with 107 permutations and 1,000 replicates for the power as a function of effect size (β) under scenario 2 (Fig. 3). Figure 3 shows that the patterns of the power comparisons using significance level 10−6 are similar to that using a significance level of 0.1% in Fig. 1 (scenario 2).
In summary, the proposed method has correct type I error rates and is either the most powerful test or comparable with the most powerful tests. No other methods have consistently good performance under the six simulation scenarios.
Application to the COPDGene
Chronic obstructive pulmonary disease (COPD) is one of the most common lung diseases characterized by long term poor airflow and is a major public health problem40. The COPDGene Study41 (http://www.ncbi.nlm.nih.gov/projects/gap/cgi-bin/study.cgi?study_id=phs000179.v1.p1) is a multi-center genetic and epidemiologic investigation to study COPD. This study is sufficiently large and appropriately designed for genome-wide association analysis of COPD. In this study, a total of more than 10,000 subjects have been recruited including non-Hispanic Whites (NHW) and African-Americans (AA). The participants in this study have completed a detailed protocol, including questionnaires, pre- and post-bronchodilator spirometry, high-resolution CT scanning of the chest, exercise capacity (assessed by six-minute walk distance), and blood samples for genotyping. The participants were genotyped using the Illumina OmniExpress platform. The genotype data have gone through standard quality-control procedures for genome-wide association analysis detailed at http://www.copdgene.org/sites/default/files/GWAS_QC_Methodology_20121115.pdf. Variants with MAF <1% were excluded in the data set.
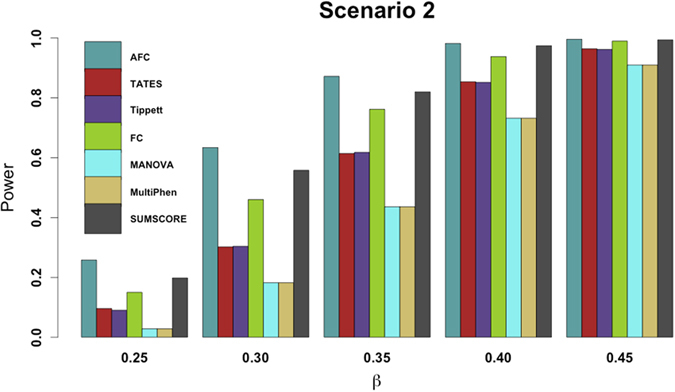
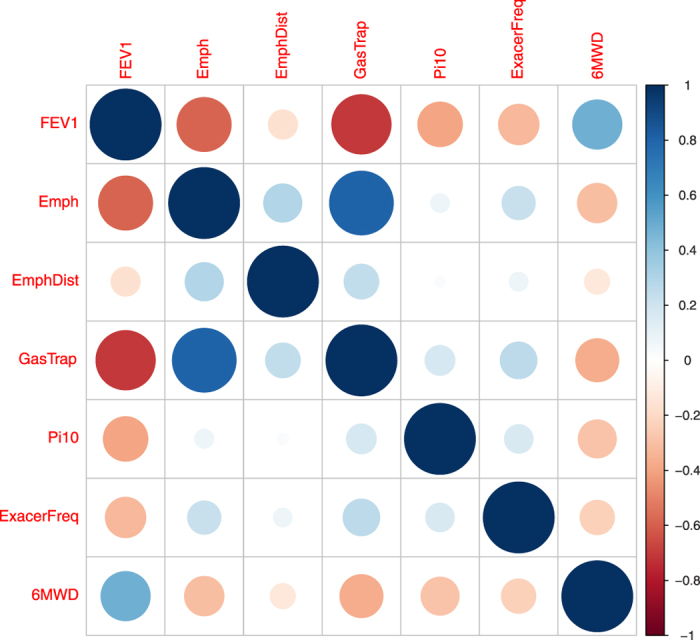
To evaluate the performance of our proposed method on a real data set, we applied all of the 7 methods to the COPDGene of NHW population to carry out GWAS of COPD-related phenotypes. Based on the literature studies of COPD42,43, we selected 7 key quantitative COPD-related phenotypes, including FEV1 (% predicted FEV1), Emphysema (Emph), Emphysema Distribution (EmphDist), Gas Trapping (GasTrap), Airway Wall Area (Pi10), Exacerbation frequency (ExacerFreq), Six-minute walk distance (6MWD), and 4 covariates, including BMI, Age, Pack-Years (PackYear) and Sex. EmphDist is the ratio of emphysema at −950 HU in the upper 1/3 of lung fields compared to the lower 1/3 of lung fields. Followed by Chu et al.42, we did a log transformation on EmphDist in the following analysis. The correlation structure of the 7 COPD-related phenotypes is given in Fig. 4. In the analysis, participants with missing data in any of the 11 variables were excluded. Therefore, a complete set of 5,430 individuals across 630,860 SNPs were used in the following analyses. In the analysis, we first adjusted each of the 7 phenotypes for the 4 covariates using linear models. Then, we performed the analysis based on the adjusted phenotypes.
Figure 4. The correlation matrix plot of the 7 COPD-related phenotypes.
To identify SNPs associated with the phenotypes, we adopted the commonly used genome-wide significance level 5 × 10−8. The results were summarized in Table 3. There were total 14 SNPs in Table 3. All of the 14 SNPs had previously been reported to be in association with COPD by eligible studies44,45,46,47,48,49,50,51,52,53,54,55,56,57. From Table 3, we can see that MultiPhen identified 14 SNPs; MANOVA identified 13 SNPs; AFC identified 12 SNPs, FC and SUMSCORE identified 10 SNPs; and TATES and Tippett identified 9 SNPs. Among the five methods based on combining test statistics from univariate analysis (AFC, TATES, Tippett, FC, and SUMSCORE), AFC identified 2 or 3 more genome-wide significant SNPs than the other 4 methods.
Table 3. Significant SNPs and the corresponding p-values in the analysis of COPDGene.
| Chr | Position | Variant identifier | AFC | TATES | Tippett | FC | MANOVA | MultiPhen | SUM-SCORE |
|---|---|---|---|---|---|---|---|---|---|
| 4 | 145431497 | rs1512282 | 1.10 × 10−8 | 5.77 × 10−9 | 8.00 × 10−9 | 6.00 × 10−9 | 1.69 × 10−9 | 1.03 × 10−9 | 2.00 × 10−8 |
| 4 | 145434744 | rs1032297 | 0 | 6.22 × 10−13 | 0 | 0 | 6.52 × 10−14 | 7.69 × 10−14 | 0 |
| 4 | 145474473 | rs1489759 | 0 | 2.52 × 10−16 | 0 | 0 | 1.11 × 10−16 | 1.22 × 10−16 | 0 |
| 4 | 145485738 | rs1980057 | 0 | 9.35 × 10−17 | 0 | 0 | 6.68 × 10−17 | 8.14 × 10−17 | 0 |
| 4 | 145485915 | rs7655625 | 0 | 1.64 × 10−16 | 0 | 0 | 7.12 × 10−17 | 9.13 × 10−17 | 0 |
| 15 | 78882925 | rs16969968 | 0 | 2.98 × 10−8 | 4.90 × 10−8 | 1.00 × 10−8 | 1.32 × 10−11 | 7.84 × 10−12 | 3.33 × 10−8 |
| 15 | 78894339 | rs1051730 | 0 | 2.63 × 10−8 | 4.20 × 10−8 | 9.00 × 10−9 | 1.41 × 10−11 | 8.16 × 10−12 | 1.00 × 10−8 |
| 15 | 78898723 | rs12914385 | 0 | 5.14 × 10−10 | 0 | 0 | 1.76 × 10−12 | 1.48 × 10−12 | 0 |
| 15 | 78911181 | rs8040868 | 0 | 2.40 × 10−9 | 5.00 × 10−9 | 0 | 2.74 × 10−12 | 2.59 × 10−12 | 0 |
| 15 | 78878541 | rs951266 | 0 | 5.17 × 10−8 | 8.10 × 10−8 | 1.50 × 10−8 | 1.77 × 10−11 | 1.02 × 10−11 | 2.15 × 10−8 |
| 15 | 78806023 | rs8034191 | 1.40 × 10−8 | 1.02 × 10−7 | 1.70 × 10−7 | 9.50 × 10−8 | 2.14 × 10−10 | 7.74 × 10−11 | 8.43 × 10−8 |
| 15 | 78851615 | rs2036527 | 2.90 × 10−8 | 1.56 × 10−7 | 2.41 × 10−7 | 1.12 × 10−7 | 3.99 × 10−10 | 1.77 × 10−10 | 2.01 × 10−7 |
| 15 | 78826180 | rs931794 | 6.30 × 10−8 | 1.18 × 10−7 | 1.94 × 10−7 | 2.67 × 10−7 | 2.35 × 10−10 | 9.09 × 10−11 | 3.32 × 10−7 |
| 15 | 78740964 | rs2568494 | 5.00 × 10−6 | 2.88 × 10−5 | 3.42 × 10−5 | 1.34 × 10−5 | 1.05 × 10−7 | 4.23 × 10−8 | 2.11 × 10−5 |
The p-values of AFC, Tippett, FC, and SUMSCORE are evaluated using 109 permutations. The p-values of TATES, MANOVA, and MultiPhen are evaluated using asymptotic distributions. The bold p-values indicate the p-values > 5 × 10−8.
Discussion
GWAS have identified many variants with each variant affecting multiple phenotypes, which suggests that pleiotropic effects on human complex phenotypes may be widespread. Therefore, statistical methods that can jointly analyze multiple phenotypes in GWAS may have advantages over analyzing each phenotype individually. In this article, we developed a new method AFC to jointly analyze multivariate phenotypes in genetic association studies. We used extensive simulation studies as well as real data application to compare the performance of AFC with TATES, Tippett, FC, MANOVA, MultiPhen, and SUMSCORE. Our simulation results showed that AFC has correct type I error rates. With respect to power, AFC is either the most powerful test or has similar power with the most powerful test under a variety of simulation scenarios. Additionally, the real data analysis results demonstrated that the proposed method has great potential in GWAS on complex diseases with multiple phenotypes such as COPD.
AFC has several important advantages. First, it allows researchers to test genetic associations using standard GWAS software. Second, phenotypes of different types (e.g., dichotomous, ordinal, continuous) can be easily analyzed simultaneously. Third, since AFC is based on p-values obtained from standard univariate GWAS, it can not only test the association between multiple phenotypes and one genetic variant of interest, but also can test the association between multiple phenotypes and multiple genetic variants. For common variants, multiple-variant AFC can be applied based on p-values obtained in standard univariate GWAS for each variant and each phenotype. For rare variants, we can first combine genotypes of rare variants by giving different weights, hoping that we give big weights to the variants having strong associations with the phenotypes. Then, we can apply AFC to test the association between the combined genotypes and multiple phenotypes. In conclusion, we showed that our proposed method provides a useful framework for joint analysis of multiple phenotypes in association studies.
It is well known that the effect sizes of identified variants are often small and that a large sample size is necessary to ensure sufficient power to detect such variants. A common strategy to increase sample size is to perform a meta-analysis by combining summary statistics from a series of studies. The proposed AFC method can be applied to meta-analysis. Suppose that there are L independent studies containing the variant of interest and each study has K phenotypes. Let T1l, …, TKl denote the summary statistics from the lth study. Suppose that Tl = (T1l, …, TKl)T~N(0,Σl) under the null hypothesis, where Σl can be estimated from the values of Tl for all independent SNPs in the GWAS from the lth study58. Then, 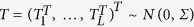 , where Σ = diag (Σ1, …, ΣL). From T, we can calculate the corresponding p-values
, where Σ = diag (Σ1, …, ΣL). From T, we can calculate the corresponding p-values 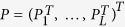 , where Pl = (p1l, …, pKl)T. The AFC test statistic is based on the p-values P. In the permutation procedure, we can generate T according to the distribution N(0,Σ) and then we can calculate the p-values P in each permutation.
, where Pl = (p1l, …, pKl)T. The AFC test statistic is based on the p-values P. In the permutation procedure, we can generate T according to the distribution N(0,Σ) and then we can calculate the p-values P in each permutation.
Additional Information
How to cite this article: Liang, X. et al. An Adaptive Fisher’s Combination Method for Joint Analysis of Multiple Phenotypes in Association Studies. Sci. Rep. 6, 34323; doi: 10.1038/srep34323 (2016).
Supplementary Material
Acknowledgments
Research reported in this publication was supported by the National Human Genome Research Institute of the National Institutes of Health under Award Number R15HG008209. This research used data generated by the COPDGene study, which was supported by National Institutes of Health (NIH) grants U01HL089856 and U01HL089897. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health. The COPDGene project is also supported by the COPD Foundation through contributions made by an Industry Advisory Board comprised of Pfizer, AstraZeneca, Boehringer Ingelheim, Novartis, and Sunovion.
Footnotes
Author Contributions Q.S. and S.Z. designed research, X.L. and S.Z. performed statistical analysis, Z.W. performed real data analysis, and X.L., Q.S. and S.Z. wrote the manuscript.
References
- Sivakumaran S. et al. Abundant pleiotropy in human complex diseases and traits. Am. J. Hum. Genet. 89(5), 607–618 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Wang X., Sha Q. & Zhang S. Joint analysis of multiple traits in rare variant association studies. Ann. Hum. Genet. 80(3), 162–171 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Q., Avery C. L. & Lin D. Y. A general framework for association tests with multivariate traits in large-scale genomics studies. Genet. Epidemiol. 37(8), 759–767 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K. Testing genetic association by regressing genotype over multiple phenotypes. PLoS One 9(9), e106918 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos C. I. & Laing A. E. A comparison of univariate and multivariate tests for genetic linkage. Genet. Epidemiol. 10(6), 671–676 (1993). [DOI] [PubMed] [Google Scholar]
- Jiang C. & Zeng Z. B. Multiple trait analysis of genetic mapping for quantitative trait loci. Genetics 140(3), 1111–1127 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schifano E. D., Li L., Christiani D. C. & Lin X. Genome-wide association analysis for multiple continuous secondary phenotypes. Am. J. Hum. Genet. 92(5), 744–759 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly P. F. et al. MultiPhen: joint model of multiple phenotypes can increase discovery in GWAS. PLoS One 7(5), e34861 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan T., Li Q., Li Y., Li Z. & Zheng G. Genetic association with multiple traits in the presence of population stratification. Genet. Epidemiol. 37(6), 571–580 (2013). [DOI] [PubMed] [Google Scholar]
- Tang C. S. & Ferreira M. A. A gene-based test of association using canonical correlation analysis. Bioinformatics 28(6), 845–850 (2012). [DOI] [PubMed] [Google Scholar]
- Klei L., Luca D., Devlin B. & Roeder K. Pleiotropy and principal components of heritability combine to increase power for association analysis. Genet. Epidemiol. 32(1), 9–19 (2008). [DOI] [PubMed] [Google Scholar]
- van der Sluis S., Posthuma D. & Dolan C. V. TATES: Efficient multivariate genotype-phenotype analysis for genome-wide association studies. PLoS Genet. 9(1), e1003235 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. X., Gui H. S., Kwan J. S. & Sham P. C. GATES: a rapid and powerful gene-based association test using extended Simes procedure. Am. J. Hum. Genet. 88(3), 283–293 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q. & Wang Y. Methods for analyzing multivariate phenotypes in genetic association studies. J. Probab. Stat. 2012, 652569 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird N. M. & Ware J. H. Random-effects models for longitudinal data. Biometrics 38, 963–974 (1982). [PubMed] [Google Scholar]
- Fitzmaurice G. M. & Laird N. M. A likelihood-based method for analysing longitudinal binary responses. Biometrika 80(1), 141–151 (1993). [Google Scholar]
- Breslow N. E. & Clayton D. G. Approximate inference in generalized linear mixed models. JASA 88(421), 9–25 (1993). [Google Scholar]
- Bates D. M. & DebRoy S. Linear mixed models and penalized least squares. J. Multivar. Anal. 91(1), 1–17 (2004). [Google Scholar]
- Liang K. Y. & Zeger S. L. Longitudinal data analysis using generalized linear models. Biometrika 73(1), 13–22 (1986). [Google Scholar]
- Aschard H. et al. Maximizing the power of principal-component analysis of correlated phenotypes in genome-wide association studies. Am. J. Hum. Genet. 94(5), 662–676 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott J. U. R. & Rabinowitz D. A principal-components approach based on heritability for combining phenotype information. Hum. Hered. 49(2), 106–111 (1999). [DOI] [PubMed] [Google Scholar]
- Wang Y., Fang Y. & Jin M. A ridge penalized principal-components approach based on heritability for high-dimensional data. Hum. Hered. 64(3), 182–191 (2007). [DOI] [PubMed] [Google Scholar]
- Wang Y., Fang Y. & Wang S. Clustering and principal-components approach based on heritability for mapping multiple gene expressions. BMC Proc 1 (Suppl 1), S121 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira M. A. & Purcell S. M. A multivariate test of association. Bioinformatics 25(1), 132–133 (2009). [DOI] [PubMed] [Google Scholar]
- Lange C., DeMeo D. L. & Laird N. M. Power and design considerations for a general class of family-based association tests: quantitative traits. Am. J. Hum. Genet. 71(6), 1330–1341 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q., Wu H., Guo C. Y. & Fox C. S. Analyze multivariate phenotypes in genetic association studies by combining univariate association tests. Genet. Epidemiol. 34(5), 444–454 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesarin F. & Salmaso L. Permutation tests for complex data: theory, applications and software 128–134 (John Wiley & Sons, 2010). [Google Scholar]
- Yang J. J., Li J., Williams L. K. & Buu A. An efficient genome-wide association test for multivariate phenotypes based on the Fisher combination function. BMC Bioinform. 17(1), 1 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole D. A., Maxwell S. E., Avrey R. & Salas E. How the power of MANOVA can both increase and decrease as a funcion of the intercorrelations among the dependent variables. Psychol. Bull. 115(3), 465 (1994). [Google Scholar]
- Purcell S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81(3), 559–575 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulchenko Y. S., Ripke S., Isaacs A. & Van Duijn C. M. GenABEL: an R library for genome-wide association analysis. Bioinformatics 23(10), 1294–1296 (2007). [DOI] [PubMed] [Google Scholar]
- Aulchenko Y. S., Struchalin M. V. & van Duijn C. M. ProbABEL package for genome-wide association analysis of imputed data. BMC bioinform. 11(1), 1 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Willer C., Sanna S. & Abecasis G. Genotype imputation. Annu Rev Genomics Hum Genet 10, 387 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Willer C. J., Ding J., Scheet P. & Abecasis G. R. MaCH: using sequence and genotype data to estimate haplotypes and unobserved genotypes. Genet. Epidemiol. 34(8), 816–834 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchini J., Howie B., Myers S., McVean G. & Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 39(7), 906–913 (2007). [DOI] [PubMed] [Google Scholar]
- Lippert C. et al. FaST linear mixed models for genome-wide association studies. Nat. Methods 8(10), 833–835 (2011). [DOI] [PubMed] [Google Scholar]
- Rencher A. C. Methods of multivariate analysis. 161–164 (John Wiley & Sons, 2003). [Google Scholar]
- Ray D., Pankow J. S. & Basu S. USAT: A Unified Score-Based Association Test for Multiple Phenotype-Genotype Analysis. Genet. Epidemiol. 40(1), 20–34 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- UK10K Consortium. The UK10K project identifies rare variants in health and disease. Nature 526(7571), 82–90 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nazir S. A. & Erbland M. L. Chronic Obstructive Pulmonary Disease. Drug & Aging 26(10), 813–831 (2009). [DOI] [PubMed] [Google Scholar]
- Regan E. A. et al. Genetic epidemiology of COPD (COPDGene) study design. COPD 7(1), 32–43 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J. H. et al. Analyzing networks of phenotypes in complex diseases: methodology and applications in COPD. BMC Syst. Biol. 8(1), 1 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han M. K. et al. Chronic obstructive pulmonary disease exacerbations in the COPDGene study: associated radiologic phenotypes. Radiology 261(1), 274–282 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutz S. M. et al. A genome-wide association study identifies risk loci for spirometric measures among smokers of European and African ancestry. BMC Genet. 16(1), 1 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X. et al. Importance of hedgehog interacting protein and other lung function genes in asthma. J. Allergy Clin. Immunol. 127(6), 1457–1465 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho M. H. et al. Variants in FAM13A are associated with chronic obstructive pulmonary disease. Nat. Genet. 42(3), 200–202 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young R. P. et al. Chromosome 4q31 locus in COPD is also associated with lung cancer. Eur. Respir. J. 36(6), 1375–1382 (2010). [DOI] [PubMed] [Google Scholar]
- Hancock D. B. et al. Meta-analyses of genome-wide association studies identify multiple loci associated with pulmonary function. Nat. Genet. 42(1), 45–52 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk J. B. et al. A genome-wide association study of pulmonary function measures in the Framingham Heart Study. PLoS Genet. 5(3), e1000429 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilk J. B. et al. Genome-wide association studies identify CHRNA5/3 and HTR4 in the development of airflow obstruction. Am. J. Respir. Crit. Care Med. 186(7), 622–632 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Summah H., Zhu Y. G. & Qu J. M. Nicotinic acetylcholine receptor variants associated with susceptibility to chronic obstructive pulmonary disease: a meta-analysis. Respir. Res. 12(1), 1 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillai S. G. et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): identification of two major susceptibility loci. PLoS Genet. 5(3), e1000421 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm J. M. et al. Identification of FGF7 as a novel susceptibility locus for chronic obstructive pulmonary disease. Thorax 66(12), 1085–1090 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho M. H. et al. Risk loci for chronic obstructive pulmonary disease: a genome-wide association study and meta-analysis. Lancet Respir. Med. 2(3), 214–225 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui K., Ge X. & Ma H. Four SNPs in the CHRNA3/5 alpha-neuronal nicotinic acetylcholine receptor subunit locus are associated with COPD risk based on meta-analyses. PloS One 9(7), e102324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu A. Z. et al. Association of CHRNA5-A3-B4 SNP rs2036527 with smoking cessation therapy response in African-American smokers. Clin. Pharmacol. Ther. 96(2), 256–265 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y., Xue Y. & Xiao W. Association of IREB2 gene rs2568494 polymorphism with risk of chronic obstructive pulmonary disease: a meta-analysis. Med. Sci. Monit. 22, 177 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X. et al. Meta-analysis of correlated traits via summary statistics from GWASs with an application in hypertension. Am. J. Hum. Genet. 96(1), 21–36 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.