

ABSTRACT
Inflammatory bowel diseases (IBD) are chronic intestinal inflammatory disorders characterized by a complex disruption of the physiologic interaction between the host immune system and intestinal microbes precipitated by environmental factors. Numerous observations indicate the altered composition and function of the intestinal microbiome of patients with ulcerative colitis (UC), a subtype of IBD. The accuracy of these results may be limited by confounding factors, such as concurrent medication use. To address these limitations, we examined the colonic mucosal microbiome of pediatric patients with UC prior to initiating treatment. Based on bacterial 16S rRNA gene sequencing, we identified a significant decrease in the phylum Verrucomicrobia in patients with UC. At the genus level, we observed a significant decrease in the short chain fatty acid producer Roseburia. Despite these compositional changes, we did not identify inferred gene content differences between the UC and control groups. To determine if microbial taxa may be associated with clinical outcomes, we retrospectively assessed the clinical course of the UC patients. Despite similar metrics of OTU richness and diversity, multiple OTU differences were observed between patients who responded to therapy and those who did not. Our observations regarding the mucosal microbiome and the associations with differential clinical outcomes support the contributions of gut microbes to disease onset and modulation.
KEYWORDS: Inflammatory bowel disease, microbiome, microbiota, metagenome, ulcerative colitis
Introduction
Inflammatory bowel diseases (IBD) are gastrointestinal disorders with a key characteristic of chronic and incurable intestinal inflammation. IBD is classically subdivided into ulcerative colitis (UC) and Crohn's disease (CD). IBD pathogenesis involves a dysregulated interaction between the host immune system and intestinal microbes, in a genetically susceptible host.1 From patients with IBD, genome wide association studies identified numerous susceptibility polymorphisms, which are related to microbial sensing and tolerance.2 These polymorphisms are linked to alterations in the composition of the intestinal microbiome3 (i.e. dysbiosis) and multiple studies, in IBD patients, have demonstrated dysbiosis.4-9
The dysbiosis found in patients with UC can be described in terms of richness and diversity. Richness delineates the unique number of bacteria present in a microbial community and diversity extends this to account for their relative abundance. In patients with UC, richness and diversity have been reported as similar4,8,10 or reduced5,11,12 compared to controls. These changes may have prognostic value, since declines in both metrics were associated with an increased risk of disease relapse.13
Similarly, specific bacterial taxa have been associated with therapeutic outcomes in IBD. Mouse models revealed that Faecalibacterium prausnitzii has anti-inflammatory properties and, in patients with CD, reductions in ileal F. prausnitzii abundance were associated with disease recurrence.14 In patients with UC, studies have reported either no change4,10,15 or lower4,9,16-18 abundance of F. prausnitzii. However, select patients with UC may carry increased amounts of this bacterium in the colonic mucosa.19 From stool samples, reductions in F. prausnitzii abundance were associated with an increased number of disease flares, a shorter time in remission, and a greater disease extent (pancolitis versus proctitis) in adult UC patients.9
The significance of prior studies, however, may be limited by other confounding factors. The majority of prior work in UC included patients on treatment,4,5,11 which can independently influence microbiome composition.20 Additionally, age may independently alter the gut microbiome in patients with IBD, so studies of adult populations may not generalize to children.21 Finally, microbial communities differ between stool and mucosa,7,15,22,23 and studies of mucosal samples may provide a more accurate model of microbe-host interaction. Therefore, the aim of our study was to characterize the mucosal microbiome of pediatric UC patients prior to beginning therapies and identify bacteria associated with specific clinical outcomes.
Results
Patient and sample data
We analyzed colonic biopsy samples from 10 patients with UC and 13 non-IBD controls. Baseline demographics are shown in Table 1. The groups were similar in terms of age, sex and race. The mean age of patients with UC was 12.9 (range 5–17) and for controls was 13.9 (range 11-16). More males were included in the UC group (40%) compared with controls (31%). The majority of subjects were Caucasian in both groups (60%, patients with UC; 77%, controls).
Table 1.
Baseline characteristics. There were no significant differences in terms of age, sex or race.
| UC (n = 10) | Control (n = 13) | |
|---|---|---|
| Age, mean (SD) | 12.9 (3.7) | 13.9 (1.8) |
| Sex, n (%) | ||
| Male | 4 (40) | 4 (30.8) |
| Race, n (%) | ||
| Caucasian | 6 (60) | 10 (76.9) |
| Black | 1 (10) | 0 |
| Hispanic | 2 (20) | 3 (23.1) |
| Other | 1 (10) | 0 |
Initially, a total of 2,468,305 16S rRNA gene sequences were produced. After quality filtering, 2,030,690 sequences remained. An average of 21,048 (SD 17,583) sequences per sample were retained following open-reference operational taxonomic unit (OTU)-picking (568,309 reads in total), and, through this process, 4 samples were removed from further analysis due to a low number of quality sequences. The remaining samples were rarified to 2350 sequences per sample before further analysis.
Richness and diversity were similar between patients with UC and controls
The average richness for patients with UC was lower than controls (186 [standard deviation; SD 30] vs. 207 [SD 24] OTUs per sample), but this difference was not significantly different (p = 0.11). Similarly, Shannon diversity was comparable (p = 0.64) between patients with UC (4.76 SD 0.72) and controls (4.89 SD 0.40). The OTU level β diversity was visualized in a principal coordinates analysis (PCoA) plot using unweighted and weighted UniFrac distances (Fig. 1). Nonparametric testing was used to compare the distance metrics of cases and controls. Comparison of unweighted distances revealed a significant difference (p = 0.01) between the groups, but comparison of weighted distances failed to reach statistical significance (p = 0.08).
Figure 1.

No significant differences were seen between richness (A) and diversity (B) between patients with UC and controls. Solids bars represent the mean and the error bars the standard deviation. Principal coordinate analysis of unweighted (C) Unifrac distances showing a significant separation (p = 0.01) and weighted (D) UniFrac distances without a significant separation (p = 0.08) for patients with UC and controls. Red dots represent patients with UC and green dots represent controls. Values in the parenthesis describe the amount of community variation explained along each respective axes.
Patients with UC had taxonomically distinct mucosal microbiomes
We tested for significant differences with respect to bacterial relative abundances between patients with UC and controls from the phylum to the genus levels. At the phylum level, the groups were similar, with the exception of Verrucomicrobia being reduced in patients with UC (Table 2). When we examined the genus level differences, the relative abundance of the short chain fatty acid (SCFA) producing organism Roseburia was significantly reduced in patients with UC while the relative abundance of the genus Haemophilus was increased in patients with UC.
Table 2.
Bacterial clades differentially present in patients with UC and controls. Bacterial clades were compared using the White's non-parametric t test with Benjamini Hochberg FDR correction. Clades with a corrected p value <0.05 were considered statistically significant. Data is presented at mean with a standard deviation.
| Mean Abundance, % (SD) |
|||
|---|---|---|---|
| Ulcerative Colitis (n = 10) | Controls (n = 13) | Corrected p value | |
| Phylum | |||
| Verrucomicrobia | 0.01 (0.02) | 0.84 (1.79) | 0.02 |
| Class | |||
| Verrucomicrobiae | 0.01 (0.02) | 0.84 (1.79) | 0.05 |
| Order | |||
| Pasteurellales | 0.54 (0.70) | 0.01 (0.02) | 0.03 |
| RF32 | 0.23 (0.49) | 0 | 0.02 |
| Verrucomicrobiales | 0.01 (0.02) | 0.02 (1.79) | 0.04 |
| Family | |||
| Pasteurellaceae | 0.54 (0.70) | 0.01 (0.02) | 0.02 |
| Genus | |||
| Phenylobacterium | 0.11 (0.34) | 0 | 0.05 |
| Haemophilus | 0.47 (0.60) | 0.01 (0.02) | 0.02 |
| Roseburia | 0.04 (0.04) | 0.34 (0.33) | 0.03 |
Multiple OTUs were differentially abundant between the groups. Specifically, OTUs for the mucolytic bacteria Akkermansia and Roseburia were decreased in UC patients. Multiple OTUs identified as F. prausnitzii and a single OTU for Haemophilus were present at increased relative abundances in patients with UC compared to controls (Figs. 2 and S1). Consistent with this observation, a trend toward an increased abundance of F. prausnitzii in patients with UC (12% versus 10%) was observed (corrected p = 0.85).
Figure 2.
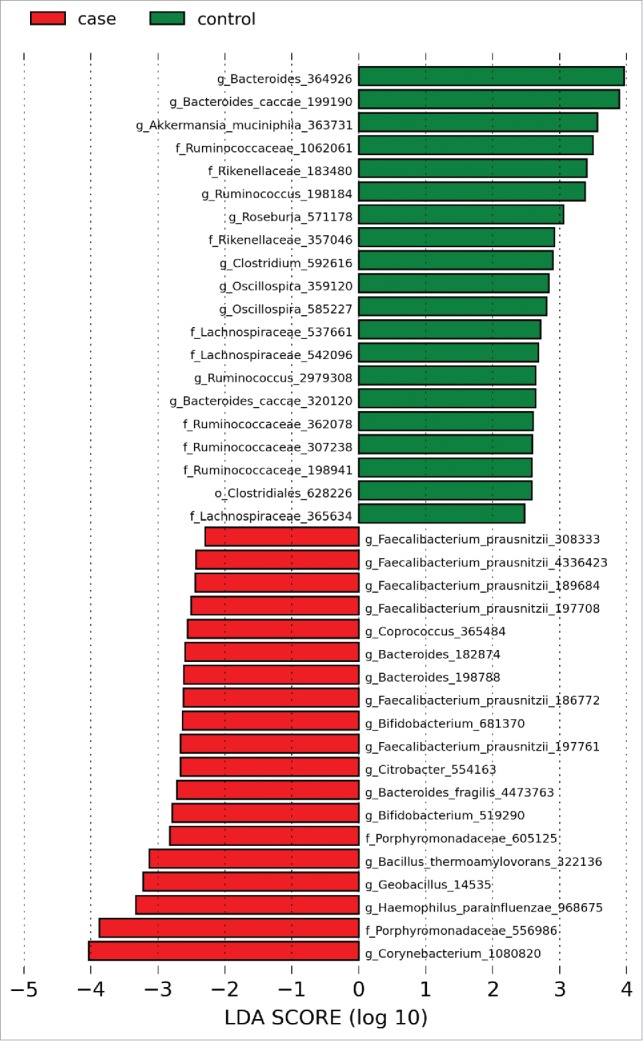
Histogram of LDA effect sizes differentially present between patients with UC and controls with a threshold LDA score of 2. Green bars indicate taxa enriched in controls relative to patients with UC and red bars indicate taxa enriched in patients with UC relative to controls. Numerical identifiers indicate individual OTUs with taxonomic characterization performed against the Greengenes database to the furthest extent.
Inferred metagenomic analysis revealed no significant functional differences between groups
The 16S rRNA gene data was used to generate inferred functional metagenomic profiles of patients with UC and non-IBD controls (see methods for details). The average nearest sequenced taxon index (NSTI), a metric to account for divergence of closed reference taxonomic reads and their corresponding inferred metagenomes, was 0.06 (range 0.03-0.08). This indicates greater than 90% of our closed reference reads matched to fully sequenced genomes, which allowed for accurate metagenomic assignment. When KEGG (Kyoto Encyclopedia of Genes and Genomes) orthologs and their associated KEGG pathways were compared between the groups, no significant differences were found in their relative abundances. Specifically, no differences were detected among pathways involved in carbohydrate, lipid or amino acid metabolism, in contrast to prior reports in pediatric patients with CD.22
UC patients with clinical improvement have a discrete mucosal microbiome
Among the patients with UC, we examined compositional and functional features to identify determinants of clinical response. There were no significant differences in baseline demographics for the UC patients with and without response to initial therapy (Table 3). Richness, Shannon diversity and the distribution of communities across PCoA plots were similar between the groups (Fig. 3). Comparisons of the response groups from the phylum to genus levels failed to identify differentially abundant taxa, but OTU level analysis from LEfSe identified multiple differences between those who responded to initial therapy and those who did not (Fig. 4). OTUs from F. prausnitzii were differentially abundant between responders and non-responders, while OTUs from the genera Clostridium and Bacteroides were specific to patients without a clinical response. PICRUSt analysis did not identify discrete differences between the groups.
Table 3.
Baseline characteristics for the patients with UC. The groups were not significantly different and were initiated on similar therapies. The responders consisted of patients with less extensive disease, but similar numbers of patients with pancolitis as the non-responder group.
| Responders (n = 6) | Non-responders (n = 4) | |
|---|---|---|
| Age, mean | 14.3 | 10.8 |
| Sex | ||
| Male, n (%) | 3 (50) | 1 (25) |
| Race, n (%) | ||
| Caucasian | 3 (50) | 3 (75) |
| Black | 1 (16.7) | – |
| Hispanic | 1 (16.7) | 1 (25) |
| Other | 1 (16.7) | – |
| Montreal classification, n (%) | ||
| E1 | 1 (16.7) | – |
| E2 | 1 (16.7) | – |
| E3 | 4 (66.7) | 4 (100) |
| Initial therapy, n (%) | ||
| 5-aminosalicylates | 4 (66.7) | 2 (50) |
| Steroids | 2 (33.3) | 2 (50) |
Figure 3.

Richness (A) and diversity (B) were similar between patients with and without a clinical response. Solids bars represent the mean and the error bars the standard deviation. No distinct separation of the groups was appreciated with visualization of either unweighted (C) or weighted (D) UniFrac distances (p = 0.87 and p = 0.92, respectively). Yellow dots represent patients with clinical improvement and blue dots those without clinical improvement. Values in the parenthesis describe the amount of community variation explained along each respective axes.
Figure 4.
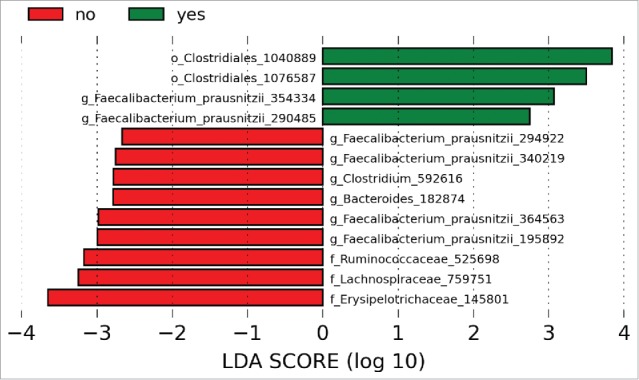
LDA scores demonstrated OTUs for F. prausnitzii associated with both responders and non-responders. OTUs for the genera Clostridium and Bacteroides were increased in patients with UC compared to controls. Green bars indicate taxa enriched in controls relative to patients with UC and red bars indicate taxa enriched in patients with UC relative to controls. Numerical identifiers represent unique OTUs and taxonomic characterization was performed against the Greengenes database to the furthest extent.
Discussion
Our results replicate the lack of community level differences (richness and diversity) in the mucosal microbiome of pediatric patients with UC relative to non-IBD controls.8,10 Despite those similarities, we found significant compositional variation at several taxonomic (phylum to genus) levels. In contrast, we were not able to detect differences among inferred metagenomic profiles. Multiple OTUs associated differentially with patients depending on clinical improvement, but these associations did not remain when examined at higher taxonomic levels.
Reductions in richness and diversity were previously linked to an increased risk of adverse outcomes in IBD.9,11,13 Since age,22 IBD phenotype,6 sample type,22 geographic location,24 and medications20 can independently alter microbiome composition, a limited number of studies have included similar patients to facilitate comparisons.8,10,21 These studies, from adult and pediatric populations, also used mucosal samples taken prior to treatment and consistently showed no differences in α diversity.8,10 These prior studies, and our findings herein reinforce the concept of similar overall gut mucosal microbiome community structures between UC patients and non-IBD controls.
Compositionally, the only phylum level difference to demonstrate statistical significance was a decrease in Verrucomicrobia (p = 0.02) for patients with UC. This decrease was characterized by a loss of the mucolytic bacteria Akkermansia, which was demonstrated at the OTU level. Akkermansia is an anaerobic mucolytic bacterium25,26 that can be detected early in life27 and is found at its highest concentrations in the cecum.28 The loss of Akkermansia is consistent with prior observations in UC patients regardless of treatment or disease status.29-32 Since mucin is the primary energy source for Akkermansia, the low abundance of Akkermansia may be related to the relative lack of mucins (MUC2, MUC3, MUC4) in patients with UC.33 Investigators have suspected that this bacterium may provide intestinal epithelial access to other pathogenic organisms by degrading mucus. This concept was demonstrated by experiments co-colonizing Akkermansia and Salmonella in gnotobiotic mice.34 Mice co-colonized had significantly more inflammation compared to mice colonized with either organism separately. In contrast, other groups showed that Akkermansia derived vesicles reduced DSS induced colitis, which suggested an anti-inflammatory phenotype for this bacterial genus.35
We found a significant decrease in the relative abundance of the genus Roseburia in the UC patients, which is consistent with previously reported studies.15,20,36 The genus Roseburia includes known butyrate producing organisms and butyrate can interact with the intestinal epithelium to produce an anti-inflammatory environment37 and modulate intestinal barrier function.38 In patients with UC, a decrease in SCFA production is correlated with reductions in Roseburia abundance.17,39 Therefore, the loss of Roseburia may be a contributor to the uncontrolled colonic inflammation of UC. Consistent with this hypothesis, we previously reported a decreased abundance of Roseburia to be associated with increased susceptibility to acute colitis, in murine models, indicating a role for this genus in the pathology of mammalian large bowel inflammation.40
The only genus in patients with UC that had a statistically significant increase in relative abundance was Haemophilus. It is a gram negative bacterium belonging to the family Pasteurellaceae. Our observed increase in mucosal relative abundance is consistent with previous studies examining stool samples from treatment-naive UC patients.21 Contrary to these fecal findings, sequencing of the oral microbiota from patients with IBD showed a reduction in the relative abundance of Haemophilus, which correlated with a loss of the innate immunity molecule, lysozyme.41 Additionally, serologic studies from patients with IBD revealed a trend toward higher concentrations of antibodies directed at Haemophilus influenzae.42 Together these findings illustrate the complex relationship between the host immune system and gut microbes, as well as the topographic distribution of intestinal microbes. Targeted studies of immunologic responses against individual microbes may provide new insight into the pathogenesis of IBD.
Prior studies are conflicting regarding the abundance of F. prausnitzii in patients with UC.10,17,19 Although we did not detect differences at the genus level, the relative abundances of multiple F. prausnitzii OTUs were increased in patients with UC. A case report from a young patient with UC demonstrated similar increases in F. prausnitzii from mucosal biopsies,19 but this finding was not replicated in larger cohorts.10 Additionally, when we compared patients who clinically improved to those who did not, different F. prausnitzii OTUs associated with each outcome. Prior work with surgical specimens from patients with CD showed that reductions in F. prausnitzii were associated with increased risk of disease recurrence14 and similar findings were later shown in UC with fecal samples.9 Though no conclusions can be drawn, our results, in conjunction with prior observations, support pursuing studies into defining the physiologic differences between F. prausnitzii strains and their relevance in respect to IBD pathogenesis and disease modulation.
This study is the first to examine the predicted metagenome of the mucosal microbiome in treatment-naïve pediatric UC patients. We found no significant differences compared to controls. Previously, when subgroups (UC or CD) stratified by the presence of inflammation within biopsies were compared, differences were detected in gene content related to carbohydrate, amino acid and lipid metabolism.43 Similarly from stool samples, patients with IBD had alterations in inferred gene content related to carbohydrate and amino acid metabolism, which was confirmed with whole genome sequencing.20 These results suggest that microbiome functional differences are present in IBD, but become more apparent in stool and inflamed mucosa. Overall, the inferred metagenome approach has shown up to 80% agreement with shotgun metagenomic approaches (i.e., whole genome sequencing, WGS),44 but this approach may miss significant differences depending on the bacterial species and strains under consideration. Inferred metagenome methodology is limited by the extent of reliable reference genome databases. Therefore, our negative results may partially be explained by inherent limitations of our methodology. Future studies incorporating WGS techniques and methods are needed to clarify UC specific mucosal metagenome alterations.
Specific bacteria have been associated with IBD related clinical outcomes,9,14,45 which we observed at the OTU level. Previous studies have linked clinical response in patients with UC to the composition of the microbiome. Adults, in particular, experienced declines in richness and diversity prior to disease relapse, but no OTU level features were associated with relapse.13 Similarly, in pediatric patients with severe UC, reductions in richness were associated with a non-response to steroids.11 Although richness and diversity were similar between responders and non-responders, we identified multiple OTU level differences. OTUs from F. prausnitzii were differentially abundant between the groups, but OTUs from Clostridium and Bacteroides were more abundant in patients without a clinical response. The differential abundance of F. prausnitzii OTUs emphasizes the importance of studying strain level physiology, especially since studies have demonstrated bacterial strains within the same species can initiate different physiologic responses.46 Although we identified OTU level differences between responders and non-responders, these differences must by confirmed using larger patient populations, since our study was limited by the small number of patients with UC. Future optimally powered cohort studies incorporating longitudinal sampling will be needed to identify potential microbes or microbial products associated with clinical outcomes in UC.
As with most translational work with respect to the human microbiome, our study also carries significant limitations. We examined a small number of patients, which limited our ability to detect potential significant differences. As with most studies using mucosal samples, our control subjects had symptoms prompting a colonoscopy, so comparisons to completely health subjects may identify other alterations. With the use of mucosal samples, we observed a relatively increased proportion of human DNA content, which may have further limited our ability to detect meaningful differences. Regarding generalizability, our study was conducted in a pediatric population and the results may not apply to adults. Despite removing the confounding effects of medications, diet and host genetics were not accounted for, which could independently alter the microbiome.3,47 In spite of these limitations, our findings of a similar overall microbiome structure (i.e. α diversity) and alterations in select genera (e.g. Roseburia, Akkermansia) are consistent with prior reports. Our observations and the associations with differential clinical outcomes in pediatric patients with UC advance the concept that intestinal microbiota may contribute to disease onset, and the intestinal microbiome may be a potential target to alter the course of inflammatory bowel disease.
Materials and methods
Population and design
All patients were initially seen at the Texas Children's Hospital Gastroenterology Clinic and then referred for colonoscopy to evaluate symptoms consistent with IBD (e.g., abdominal pain, rectal bleeding, weight loss or diarrhea). They had a standard colonic preparation prior to colonoscopy and mucosal biopsies were taken throughout the colon. For this study, only colonic samples taken from the descending or sigmoid colon were included for analysis. Mucosal biopsy tissue samples were flash frozen in liquid nitrogen or dry ice prior to transfer to the Baylor College of Medicine Digestive Diseases Center Tissue Bank. The Bank is IRB approved (H-17654, P30 DK56338) and maintains a secure database of tissues with associated identifiers.
The initial cohort consisted of 12 patients with UC and 15 non-IBD controls. Patients with UC had to have a clinical history, endoscopic appearance and histologic findings consistent with UC. Controls were defined by the lack of endoscopic or histologic evidence of IBD. All controls had a complete chart review following their colonoscopy to confirm no subsequent diagnosis of IBD or other colonic diseases. We collected basic demographic information including age at diagnosis, sex (male or female) and race (Caucasian, Black, Hispanic, other). Clinical information included clinical status defined by the Physicians Global Assessment (PGA) scale and disease extent categorized by the Montreal classification system (E1 proctitis, E2 left sided colitis, E3 pancolitis). Initial UC therapies were categorized into 5-aminosalicylates, anti-tumor necrosis factors (anti-TNFs) or steroids.
Clinical outcomes were defined using the change in PGA scale over time. The PGA scale categorized patients as inactive (no pain, diarrhea or bleeding), mild (mild pain, diarrhea without bleeding or nocturnal symptoms), moderate (moderate pain, diarrhea with nocturnal symptoms or bleeding, but non-toxic) or severe (severe pain, diarrhea with nocturnal symptoms and bleeding, toxic appearance). Clinical status was compared between each patient's first visit, which occurred before their colonoscopy, and their first visit at least 1 month after, but within 12 months of the initial colonoscopy. Improvement (i.e., responder) was considered as a binary variable (i.e., yes or no) with yes occurring if the PGA classification improved between these time periods. If the PGA remained the same or worsened, then improvement was classified as no (i.e. non-responder).
DNA extraction
Tissue samples were thawed to room temperature. ATL buffer (540 ul, Qiagen, Valencia, CA) was added to the sample tissue (10 mg) in an Eppendorf tube (1.5 ml) and homogenized completely using an IKA Ultra-Turrax T8 (IKA Works, USA). Proteinase K (60 µl, Qiagen, Valencia, CA) was added and the samples were vortexed before an overnight incubation on a shaking platform at 50°C. Each tissue sample digest was then placed into a primed heavy phase-lock gel separation tube and 600 µl phenol/chloroform/isoamyl alcohol (25:24:1; PCI) was added to each. Sample tubes were shaken vigorously for 15 seconds and then centrifuged at 14000 rcf for 1 minute at 4°C. The top phase of each was transferred to a new primed Phase-Lock tube and PCI extraction was repeated followed by a chloroform only extraction. The top phase was then used for sodium acetate ethanol precipitation. Precipitated DNA was dissolved into TE buffer (EMD Millipore, Darmstadt, Germany) and its concentration measured by Nanodrop 1000 Spectrophotometer (Thermo Scientific, Wilmington, DE). Samples were diluted to 20 ng/µl concentration and stored at 4°C before transport to the sequencing provider.
16S rRNA V4-V6 amplicon library preparation
Targeted locus 16S rRNA gene sequencing of the V4-V6 variable regions was performed by the MR DNA laboratory (Shallowater, TX, USA) with an Illumina MiSeq instrument. Genomic DNA preparations were amplified with 530F 5′- GTGCCAGCMGCNGCGG −3′ and 926R 5′- CCGTCAATTYYTTTRAGTTT −3′ primers with each sample barcoded on both the forward and reverse ends (Supplemental Table 1) using HotStarTaq Plus Master Mix Kit (Qiagen, Valencia CA). The following PCR amplification conditions were used: 1 cycle 94°C for 3 minutes; 28 cycles 94°C for 30 seconds, 53°C for 40 seconds, and 72°C for 1 minute; finally, 1 cycle 72°C for 5 minutes. Amplification products were checked and relative intensity was examined by UV illumination in 2% agarose gel after electrophoresis. PCR samples were then pooled in equal proportions and purified using calibrated Ampure XP beads (Beckman Coulter, Brea CA). The pooled and purified product was used in a TruSeq DNA sample prep kit (Illumina, San Diego CA) following the supplied protocol. Then sequencing was performed following the manufacturer's guidelines. These sequence data have been submitted to the NCBI SRA database under BioProject ID number PRJNA276700.
Sequence data analysis
QIIME (version 1.8) was used for post sequence processing and analysis.48 After sequencing, the forward read was converted from the fastq format to the corresponding fasta and quality files. Due to poor joining of the corresponding reads and previous literature showing the forward read is acceptable for taxonomic assignment49 we preceded with the forward reads. Sequence data were separated by barcode and quality filtered in QIIME using the default parameters (minimum sequence length 200, maximum sequence length 1000, minimum average quality score 25, maximum ambiguous bases 6, maximum homopolymers in sequence 6, and maximum primer mismatch 0). Operational taxonomic units (OTUs) were picked using an open reference strategy against the Greengenes database (Version 13_8), at 97% identity.50,51 ChimeraSlayer52 was used to identify chimeric sequences and the resulting chimera filtered OTU Table was rarefied to 2350 sequences per sample, which was chosen to retain the majority of samples and remove samples with very low quality sequence levels (2 patients with UC and 2 controls; range 327-1610 sequences). Alpha diversity was evaluated by the Shannon metric. Beta diversity was compared using non-parametric methods (PERMANOVA) and was visualized using EMPeror with unweighted and weighted UniFrac distances.53-55
To perform inferred metagenome analysis, de novo picked OTUs were removed from the previously created OTU table. The resulting closed reference OTU Table was then analyzed using the Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt)44 pipeline available through the Huttenhower lab Galaxy56-58 online module. First, the OTU Table was normalized for 16S rRNA gene copy number followed by metagenome predications against the KEGG59,60 reference (v3.5 of IMG). The resulting KEGG orthologs were then compiled into pathways for downstream analysis. Features with a low abundance (< 1%) in all samples were filtered from further analysis.
Statistical analysis
Both community compositional and functional pathway data were analyzed using Statistical analysis of taxonomic and functional profiles (STAMP)61 and linear discriminant analysis (LDA) and effect size (LEfSe).62 Data analysis was performed in STAMP to identify differences between patients with UC and controls from the phylum to genus levels. Differences were compared using White's non-parametric t-test63 with Benjamini Hochberg FDR correction for multiple comparisons. Features with a corrected p value less than 0.05 were considered statistically significant. To analyze differences in OTUs between groups, the Galaxy web-based implementation of LEfSe was utilized for statistical analysis and plotting of effect size. Clinical and microbiome characteristics with a normal distribution are presented as a mean with a standard deviation.
Supplementary Material
{kind=link}
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors would like to thank the Gutsy Kids Fund, a philanthropic donation from the Karen and Brock Wagner family and other generous families, and the Houston Men of Distinction for their support.
Funding
This project was supported in part by PHS grant DK56338 awarded to the Texas Medical Center Digestive Diseases Center.
References
- [1].Li J, Butcher J, Mack D, Stintzi A. Functional impacts of the intestinal microbiome in the pathogenesis of inflammatory bowel disease. Inflamm Bowel Dis 2015; 21:139-53; PMID:25248007; http://dx.doi.org/ 10.1097/MIB.0000000000000215 [DOI] [PubMed] [Google Scholar]
- [2].Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY, Lee JC, Schumm LP, Sharma Y, Anderson CA, et al.. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491:119-24; PMID:23128233; http://dx.doi.org/ 10.1038/nature11582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Frank DN, Robertson CE, Hamm CM, Kpadeh Z, Zhang T, Chen H, Zhu W, Sartor RB, Boedeker EC, Harpaz N, et al.. Disease phenotype and genotype are associated with shifts in intestinal-associated microbiota in inflammatory bowel diseases. Inflamm Bowel Dis 2011; 17:179-84; PMID:20839241; http://dx.doi.org/ 10.1002/ibd.21339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Willing BP, Dicksved J, Halfvarson J, Andersson AF, Lucio M, Zheng Z, Jarnerot G, Tysk C, Jansson JK, Engstrand L. A pyrosequencing study in twins shows that gastrointestinal microbial profiles vary with inflammatory bowel disease phenotypes. Gastroenterology 2010; 139:1844-54 e1; PMID:20816835; http://dx.doi.org/ 10.1053/j.gastro.2010.08.049 [DOI] [PubMed] [Google Scholar]
- [5].Ott SJ, Musfeldt M, Wenderoth DF, Hampe J, Brant O, Folsch UR, Timmis KN, Schreiber S. Reduction in diversity of the colonic mucosa associated bacterial microflora in patients with active inflammatory bowel disease. Gut 2004; 53:685-93; PMID:15082587; http://dx.doi.org/ 10.1136/gut.2003.025403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Frank DN, St Amand AL, Feldman RA, Boedeker EC, Harpaz N, Pace NR. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 2007; 104:13780-5; PMID:17699621; http://dx.doi.org/ 10.1073/pnas.0706625104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lepage P, Seksik P, Sutren M, de la Cochetière M-F, Jian R, Marteau P, Doré J. Biodiversity of the mucosa-associated microbiota is stable along the distal digestive tract in healthy individuals and patients with Ibd. Inflamm Bowel Dis 2005; 11:473-80; PMID:15867587; http://dx.doi.org/ 10.1097/01.MIB.0000159662.62651.06 [DOI] [PubMed] [Google Scholar]
- [8].Bibiloni R, Mangold M, Madsen KL, Fedorak RN, Tannock GW. The bacteriology of biopsies differs between newly diagnosed, untreated, Crohn's disease and ulcerative colitis patients. J Med Microbiol 2006; 55:1141-9; PMID:16849736; http://dx.doi.org/ 10.1099/jmm.0.46498-0 [DOI] [PubMed] [Google Scholar]
- [9].Varela E, Manichanh C, Gallart M, Torrejon A, Borruel N, Casellas F, Guarner F, Antolin M. Colonisation by Faecalibacterium prausnitzii and maintenance of clinical remission in patients with ulcerative colitis. Aliment Pharmacol Ther 2013; 38:151-61; PMID:23725320; http://dx.doi.org/ 10.1111/apt.12365 [DOI] [PubMed] [Google Scholar]
- [10].Hansen R, Russell RK, Reiff C, Louis P, McIntosh F, Berry SH, Mukhopadhya I, Bisset WM, Barclay AR, Bishop J, et al.. Microbiota of de-novo pediatric IBD: increased Faecalibacterium prausnitzii and reduced bacterial diversity in Crohn's but not in ulcerative colitis. Am J Gastroenterol 2012; 107:1913-22; PMID:23044767; http://dx.doi.org/ 10.1038/ajg.2012.335 [DOI] [PubMed] [Google Scholar]
- [11].Michail S, Durbin M, Turner D, Griffiths AM, Mack DR, Hyams J, Leleiko N, Kenche H, Stolfi A, Wine E. Alterations in the gut microbiome of children with severe ulcerative colitis. Inflamm Bowel Dis 2012; 18:1799-808; PMID:22170749; http://dx.doi.org/ 10.1002/ibd.22860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lepage P, Hasler R, Spehlmann ME, Rehman A, Zvirbliene A, Begun A, Ott S, Kupcinskas L, Dore J, Raedler A, et al.. Twin study indicates loss of interaction between microbiota and mucosa of patients with ulcerative colitis. Gastroenterology 2011; 141:227-36; PMID:21621540; http://dx.doi.org/ 10.1053/j.gastro.2011.04.011 [DOI] [PubMed] [Google Scholar]
- [13].Ott SJ, Plamondon S, Hart A, Begun A, Rehman A, Kamm MA, Schreiber S. Dynamics of the mucosa-associated flora in ulcerative colitis patients during remission and clinical relapse. J Clin Microbiol 2008; 46:3510-3; PMID:18701655; http://dx.doi.org/ 10.1128/JCM.01512-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al.. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci U S A 2008; 105:16731-6; PMID:18936492; http://dx.doi.org/ 10.1073/pnas.0804812105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Chen L, Wang W, Zhou R, Ng SC, Li J, Huang M, Zhou F, Wang X, Shen B, M AK, et al.. Characteristics of fecal and mucosa-associated microbiota in chinese patients with inflammatory bowel disease. Medicine 2014; 93:e51; PMID:25121355; http://dx.doi.org/ 10.1097/MD.0000000000000051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Fite A, Macfarlane S, Furrie E, Bahrami B, Cummings JH, Steinke DT, Macfarlane GT. Longitudinal analyses of gut mucosal microbiotas in ulcerative colitis in relation to patient age and disease severity and duration. J Clin Microbiol 2013; 51:849-56; PMID:23269735; http://dx.doi.org/ 10.1128/JCM.02574-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Machiels K, Joossens M, Sabino J, De Preter V, Arijs I, Eeckhaut V, Ballet V, Claes K, Van Immerseel F, Verbeke K, et al.. A decrease of the butyrate-producing species Roseburia hominis and Faecalibacterium prausnitzii defines dysbiosis in patients with ulcerative colitis. Gut 2014; 63:1275-83; PMID:24021287; http://dx.doi.org/ 10.1136/gutjnl-2013-304833 [DOI] [PubMed] [Google Scholar]
- [18].Lopez-Siles M, Martinez-Medina M, Busquets D, Sabat-Mir M, Duncan SH, Flint HJ, Aldeguer X, Garcia-Gil LJ. Mucosa-associated Faecalibacterium prausnitzii and Escherichia coli co-abundance can distinguish Irritable Bowel Syndrome and Inflammatory Bowel Disease phenotypes. Int J Med Microbiol 2014; 304:464-75; PMID:24713205; http://dx.doi.org/ 10.1016/j.ijmm.2014.02.009 [DOI] [PubMed] [Google Scholar]
- [19].Wang M, Molin G, Ahrne S, Adawi D, Jeppsson B. High proportions of proinflammatory bacteria on the colonic mucosa in a young patient with ulcerative colitis as revealed by cloning and sequencing of 16S rRNA genes. Dig Dis Sci 2007; 52:620-7; PMID:17265126; http://dx.doi.org/ 10.1007/s10620-006-9461-1 [DOI] [PubMed] [Google Scholar]
- [20].Morgan XC, Tickle TL, Sokol H, Gevers D, Devaney KL, Ward DV, Reyes JA, Shah SA, LeLeiko N, Snapper SB, et al.. Dysfunction of the intestinal microbiome in inflammatory bowel disease and treatment. Genome Biol 2012; 13:R79; PMID:23013615; http://dx.doi.org/ 10.1186/gb-2012-13-9-r79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Nwosu FC, Thorkildsen LT, Avershina E, Ricanek P, Perminow G, Brackmann S, Vatn MH, Rudi K. Age-dependent fecal bacterial correlation to inflammatory bowel disease for newly diagnosed untreated children. Gastroenterol Res Pract 2013; 2013:302398; PMID:23690761; http://dx.doi.org/ 10.1155/2013/302398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Gevers D, Kugathasan S, Denson LA, Vazquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, et al.. The treatment-naive microbiome in new-onset Crohn's disease. Cell Host Microbe 2014; 15:382-92; PMID:24629344; http://dx.doi.org/ 10.1016/j.chom.2014.02.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Zoetendal EG, von Wright A, Vilpponen-Salmela T, Ben-Amor K, Akkermans AD, de Vos WM. Mucosa-associated bacteria in the human gastrointestinal tract are uniformly distributed along the colon and differ from the community recovered from feces. Appl Environ Microbiol 2002; 68:3401-7; PMID:12089021; http://dx.doi.org/ 10.1128/AEM.68.7.3401-3407.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kellermayer R, Mir SA, Nagy-Szakal D, Cox SB, Dowd SE, Kaplan JL, Sun Y, Reddy S, Bronsky J, Winter HS. Microbiota separation and C-reactive protein elevation in treatment-naive pediatric granulomatous Crohn disease. J Pediatr Gastroenterol Nutr 2012; 55:243-50; PMID:22699834; http://dx.doi.org/ 10.1097/MPG.0b013e3182617c16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Derrien M, Vaughan EE, Plugge CM, de Vos WM. Akkermansia muciniphila gen. nov., sp. nov., a human intestinal mucin-degrading bacterium. Int J Syst Evol Microbiol 2004; 54:1469-76; PMID:15388697; http://dx.doi.org/ 10.1099/ijs.0.02873-0 [DOI] [PubMed] [Google Scholar]
- [26].van Passel MW, Kant R, Zoetendal EG, Plugge CM, Derrien M, Malfatti SA, Chain PS, Woyke T, Palva A, de Vos WM, et al.. The genome of Akkermansia muciniphila, a dedicated intestinal mucin degrader, and its use in exploring intestinal metagenomes. PloS One 2011; 6:e16876; PMID:21390229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Collado MC, Derrien M, Isolauri E, de Vos WM, Salminen S. Intestinal integrity and Akkermansia muciniphila, a mucin-degrading member of the intestinal microbiota present in infants, adults, and the elderly. Appl Environ Microbiol 2007; 73:7767-70; PMID:17933936; http://dx.doi.org/ 10.1128/AEM.01477-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Derrien M, Van Baarlen P, Hooiveld G, Norin E, Muller M, de Vos WM. Modulation of Mucosal Immune Response, Tolerance, and Proliferation in Mice Colonized by the Mucin-Degrader Akkermansia muciniphila. Front Microbiol 2011; 2:166; PMID:21904534; http://dx.doi.org/ 10.3389/fmicb.2011.00166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Png CW, Linden SK, Gilshenan KS, Zoetendal EG, McSweeney CS, Sly LI, McGuckin MA, Florin TH. Mucolytic bacteria with increased prevalence in IBD mucosa augment in vitro utilization of mucin by other bacteria. Am J Gastroenterol 2010; 105:2420-8; PMID:20648002; http://dx.doi.org/ 10.1038/ajg.2010.281 [DOI] [PubMed] [Google Scholar]
- [30].Rajilic-Stojanovic M, Shanahan F, Guarner F, de Vos WM. Phylogenetic analysis of dysbiosis in ulcerative colitis during remission. Inflamm Bowel Dis 2013; 19:481-8; PMID:23385241; http://dx.doi.org/ 10.1097/MIB.0b013e31827fec6d [DOI] [PubMed] [Google Scholar]
- [31].James SL, Christophersen CT, Bird AR, Conlon MA, Rosella O, Gibson PR, Muir JG. Abnormal fibre usage in UC in remission. Gut 2014; 64:562-70; PMID:25037189; http://dx.doi.org/ 10.1136/gutjnl-2014-307198 [DOI] [PubMed] [Google Scholar]
- [32].Vigsnaes LK, Brynskov J, Steenholdt C, Wilcks A, Licht TR. Gram-negative bacteria account for main differences between faecal microbiota from patients with ulcerative colitis and healthy controls. Benef Microbes 2012; 3:287-97; PMID:22968374; http://dx.doi.org/ 10.3920/BM2012.0018 [DOI] [PubMed] [Google Scholar]
- [33].Dorofeyev AE, Vasilenko IV, Rassokhina OA, Kondratiuk RB. Mucosal barrier in ulcerative colitis and Crohn's disease. Gastroenterol Res Pract 2013; 2013:431231; PMID:23737764; http://dx.doi.org/ 10.1155/2013/431231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Ganesh BP, Klopfleisch R, Loh G, Blaut M. Commensal Akkermansia muciniphila exacerbates gut inflammation in Salmonella Typhimurium-infected gnotobiotic mice. PloS One 2013; 8:e74963; PMID:24040367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kang CS, Ban M, Choi EJ, Moon HG, Jeon JS, Kim DK, Park SK, Jeon SG, Roh TY, Myung SJ, et al.. Extracellular vesicles derived from gut microbiota, especially Akkermansia muciniphila, protect the progression of dextran sulfate sodium-induced colitis. PloS One 2013; 8:e76520; PMID:24204633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rehman A, Rausch P, Wang J, Skieceviciene J, Kiudelis G, Bhagalia K, Amarapurkar D, Kupcinskas L, Schreiber S, Rosenstiel P, et al.. Geographical patterns of the standing and active human gut microbiome in health and IBD. Gut 2015; 65:238-48; PMID:25567118 [DOI] [PubMed] [Google Scholar]
- [37].Louis P, Hold GL, Flint HJ. The gut microbiota, bacterial metabolites and colorectal cancer. Nat Rev Microbiol 2014; 12:661-72; PMID:25198138; http://dx.doi.org/ 10.1038/nrmicro3344 [DOI] [PubMed] [Google Scholar]
- [38].Kelly CJ, Zheng L, Campbell EL, Saeedi B, Scholz CC, Bayless AJ, Wilson KE, Glover LE, Kominsky DJ, Magnuson A, et al.. Crosstalk between Microbiota-Derived Short-Chain Fatty Acids and Intestinal Epithelial HIF Augments Tissue Barrier Function. Cell Host Microbe 2015; PMID:25865369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kumari R, Ahuja V, Paul J. Fluctuations in butyrate-producing bacteria in ulcerative colitis patients of North India. World J Gastroenterol 2013; 19:3404-14; PMID:23801832; http://dx.doi.org/ 10.3748/wjg.v19.i22.3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Schaible TD, Harris RA, Dowd SE, Smith CW, Kellermayer R. Maternal methyl-donor supplementation induces prolonged murine offspring colitis susceptibility in association with mucosal epigenetic and microbiomic changes. Hum Mol Genet 2011; 20:1687-96; PMID:21296867; http://dx.doi.org/ 10.1093/hmg/ddr044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Said HS, Suda W, Nakagome S, Chinen H, Oshima K, Kim S, Kimura R, Iraha A, Ishida H, Fujita J, et al.. Dysbiosis of salivary microbiota in inflammatory bowel disease and its association with oral immunological biomarkers. DNA Res 2014; 21:15-25; PMID:24013298; http://dx.doi.org/ 10.1093/dnares/dst037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Song CH, Vadheim CM, Snape WJ, Heiner DC. Antibodies in patients with inflammatory bowel disease and the apparent influence of medications. J Clin Lab Immunol 1995; 46:143-54; PMID:8733027 [PubMed] [Google Scholar]
- [43].Davenport M, Poles J, Leung JM, Wolff MJ, Abidi WM, Ullman T, Mayer L, Cho I, Loke P. Metabolic alterations to the mucosal microbiota in inflammatory bowel disease. Inflamm Bowel Dis 2014; 20:723-31; PMID:24583479; http://dx.doi.org/ 10.1097/MIB.0000000000000011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA, Clemente JC, Burkepile DE, Vega Thurber RL, Knight R, et al.. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol 2013; 31:814-21; PMID:23975157; http://dx.doi.org/ 10.1038/nbt.2676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Rajca S, Grondin V, Louis E, Vernier-Massouille G, Grimaud JC, Bouhnik Y, Laharie D, Dupas JL, Pillant H, Picon L, et al.. Alterations in the intestinal microbiome (dysbiosis) as a predictor of relapse after infliximab withdrawal in Crohn's disease. Inflamm Bowel Dis 2014; 20:978-86; PMID:24788220 [DOI] [PubMed] [Google Scholar]
- [46].Preidis GA, Saulnier DM, Blutt SE, Mistretta TA, Riehle KP, Major AM, Venable SF, Finegold MJ, Petrosino JF, Conner ME, et al.. Probiotics stimulate enterocyte migration and microbial diversity in the neonatal mouse intestine. FASEB J 2012; 26:1960-9; PMID:22267340; http://dx.doi.org/ 10.1096/fj.10-177980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Carmody RN, Gerber GK, Luevano JM Jr., Gatti DM, Somes L, Svenson KL, Turnbaugh PJ. Diet Dominates Host Genotype in Shaping the Murine Gut Microbiota. Cell Host Microbe 2014; PMID:25532804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Pena AG, Goodrich JK, Gordon JI, et al.. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010; 7:335-6; PMID:20383131; http://dx.doi.org/ 10.1038/nmeth.f.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Goodrich JK, Di Rienzi SC, Poole AC, Koren O, Walters WA, Caporaso JG, Knight R, Ley RE. Conducting a microbiome study. Cell 2014; 158:250-62; PMID:25036628; http://dx.doi.org/ 10.1016/j.cell.2014.06.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].DeSantis TZ, Hugenholtz P, Larsen N, Rojas M, Brodie EL, Keller K, Huber T, Dalevi D, Hu P, Andersen GL. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006; 72:5069-72; PMID:16820507; http://dx.doi.org/ 10.1128/AEM.03006-05 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J 2012; 6:610-8; PMID:22134646; http://dx.doi.org/ 10.1038/ismej.2011.139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla D, Tabbaa D, Highlander SK, Sodergren E, et al.. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res 2011; 21:494-504; PMID:21212162; http://dx.doi.org/ 10.1101/gr.112730.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lozupone C, Knight R. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 2005; 71:8228-35; PMID:16332807; http://dx.doi.org/ 10.1128/AEM.71.12.8228-8235.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Vazquez-Baeza Y, Pirrung M, Gonzalez A, Knight R. EMPeror: a tool for visualizing high-throughput microbial community data. GigaScience 2013; 2:16; PMID:24280061; http://dx.doi.org/ 10.1186/2047-217X-2-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Caporaso JG, Knight R, Kelley ST. Host-associated and free-living phage communities differ profoundly in phylogenetic composition. PloS One 2011; 6:e16900; PMID:21383980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Goecks J, Nekrutenko A, Taylor J, Galaxy T. Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 2010; 11:R86; PMID:20738864; http://dx.doi.org/ 10.1186/gb-2010-11-8-r86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J. Galaxy: a web-based genome analysis tool for experimentalists Current protocols in molecular biology / edited by Ausubel Frederick M [et al]. 2010; Chapter 19:Unit 19 0 1-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Doerks T, Copley RR, Schultz J, Ponting CP, Bork P. Systematic identification of novel protein domain families associated with nuclear functions. Genome Res 2002; 12:47-56; PMID:11779830; http://dx.doi.org/ 10.1101/gr.203201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 2000; 28:27-30; PMID:10592173; http://dx.doi.org/ 10.1093/nar/28.1.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kanehisa M, Goto S, Sato Y, Kawashima M, Furumichi M, Tanabe M. Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 2014; 42:D199-205; PMID:24214961; http://dx.doi.org/ 10.1093/nar/gkt1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Parks DH, Tyson GW, Hugenholtz P, Beiko RG. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 2014; 30:3123-4; PMID:25061070; http://dx.doi.org/ 10.1093/bioinformatics/btu494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol 2011; 12:R60; PMID:21702898; http://dx.doi.org/ 10.1186/gb-2011-12-6-r60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].White JR, Nagarajan N, Pop M. Statistical methods for detecting differentially abundant features in clinical metagenomic samples. PLoS Comput Biol 2009; 5:e1000352; PMID:19360128; http://dx.doi.org/ 10.1371/journal.pcbi.1000352 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.