

Abstract
Dyskeratosis congenita is a highly pleotropic genetic disorder. This heterogeneity can lead to difficulties in making an accurate diagnosis and delays in appropriate management. The aim of this study was to determine the underlying genetic basis in patients presenting with features of dyskeratosis congenita and who were negative for mutations in the classical dyskeratosis congenita genes. By whole exome and targeted sequencing, we identified biallelic variants in genes that are not associated with dyskeratosis congenita in 17 individuals from 12 families. Specifically, these were homozygous variants in USB1 (8 families), homozygous missense variants in GRHL2 (2 families) and identical compound heterozygous variants in LIG4 (2 families). All patients had multiple somatic features of dyskeratosis congenita but not the characteristic short telomeres. Our case series shows that biallelic variants in USB1, LIG4 and GRHL2, the genes mutated in poikiloderma with neutropenia, LIG4/Dubowitz syndrome and the recently recognized ectodermal dysplasia/short stature syndrome, respectively, cause features that overlap with dyskeratosis congenita. Strikingly, these genes also overlap in their biological function with the known dyskeratosis congenita genes that are implicated in telomere maintenance and DNA repair pathways. Collectively, these observations demonstrate the marked overlap of dyskeratosis congenita with four other genetic syndromes, confounding accurate diagnosis and subsequent management. This has important implications for establishing a genetic diagnosis when a new patient presents in the clinic. Patients with clinical features of dyskeratosis congenita need to have genetic analysis of USB1, LIG4 and GRHL2 in addition to the classical dyskeratosis congenita genes and telomere length measurements.
Introduction
Dyskeratosis congenita (DC) is a highly heterogeneous genetic and clinical syndrome. The classical presentation of DC is characterized by the mucocutaneous triad of abnormal skin pigmentation (hyper/hypopigmentation with atrophy and telangiectasia, termed poikiloderma), oral leukoplakia and nail dystrophy. Individuals with DC frequently develop bone marrow (BM) failure, are at a high risk of developing cancer and can develop disease features in virtually every system in the body. Pathologically, DC is characterized by selective exhaustion of highly proliferative cells1 that have critically short telomeres and exhibit an abnormal DNA damage response.2
The Dyskeratosis Congenita Registry (DCR, London, UK) is a collection of patients that have a clinical diagnosis of DC or an overlapping phenotype as defined by Dokal et al.3 The diagnostic inclusion criteria are:
All three mucocutaneous features (abnormal skin pigmentation, nail dystrophy and oral leukoplakia).
One mucocutaneous feature plus BM failure and two other somatic features of DC.
Aplastic anaemia (AA), myelodysplastic syndrome (MDS) or idiopathic pulmonary fibrosis (IPF) associated with a pathogenic telomerase variant.
Hoyeraal-Hreidarsson syndrome (HHS, growth retardation, developmental delay, microcephaly, cerebellar hypoplasia, BM failure and immunodeficiency).
Two or more features seen in DC plus very short telomeres (<1st centile).
Based on these criteria the phenotype of DC is highly variable. Furthermore, not all physical malformations are present at the point of diagnosis, with many additional features developing with age or, in the case of severe disease, death may occur before more typical clinical presentations become apparent.
To date, pathogenic mutations have been identified in 11 genes that cause DC according to the criteria above: dyskeratosis congenita 1, dyskerin (DKC1), telomerase RNA component (TERC), telomerase reverse transcriptase (TERT), NOP10 ribonucleoprotein (NOP10), NHP2 ribonucleoprotein (NHP2), TERF1 (TRF1)-interacting nuclear factor 2 (TINF2), WD repeat containing antisense to TP53 (WRAP53, also known as TCAB1), CST telomere maintenance complex component 1 (CTC1), regulator of telomere elongation helicase 1 (RTEL1), adrenocortical dysplasia homolog (mouse) (ACD), and poly(A)-specific ribonuclease (PARN). DKC1, TERC, TERT, NOP10 and NHP2 all encode components of the telomerase complex which is involved in telomere elongation. TINF2 and ACD encode components of the shelterin complex which is involved in telomere protection. CTC1 encodes a member of the CST complex which facilitates recruitment and docking of telomerase on to the telomere. WRAP53 is involved in telomerase trafficking and RTEL1 is involved in telomere replication, and DNA replication and repair.3–7 The final gene, PARN, is not exclusively involved in telomere biology but is mutated in a small number of cases of DC, and is an exoribonuclease involved in the control of mRNA stability and the maturation of TERC snoRNA.8–10 As the majority of DC genes are involved in telomere biology and DC patients usually have short telomeres, this has led to DC being classed as a telomeropathy.11 However these mutations do not explain all the cases that are present in the DCR (Data from the DCR, London, UK, April 2016). The advent of next-generation sequencing (NGS), particularly whole-exome sequencing (WES), has become an invaluable tool for determining the causal variant(s) underlying a specific disease. By sequencing all the coding regions and comparing variants in many individuals with an overlapping phenotype, it is possible to elucidate potential disease genes in a way that would not have been possible five years ago.12 This can give the clinical diagnosis a genetic basis. The main problem in obtaining a definitive clinical diagnosis is the degree of phenotypic overlap that exists between many different diseases. The diagnosis, in many cases, depends on the interpretation of the initial clinician and this is often dependent on their specialty. Although subsequent opinions may be sought, the preliminary diagnosis tends to remain with the patient until proven otherwise. An accurate genetic diagnosis can aid clinical diagnosis and subsequent patient management. This has the potential to suggest alternative treatment avenues that may not have been considered, and also aid in the selection of healthy sibling donors for hematopoietic stem cell transplantation, should this become necessary.
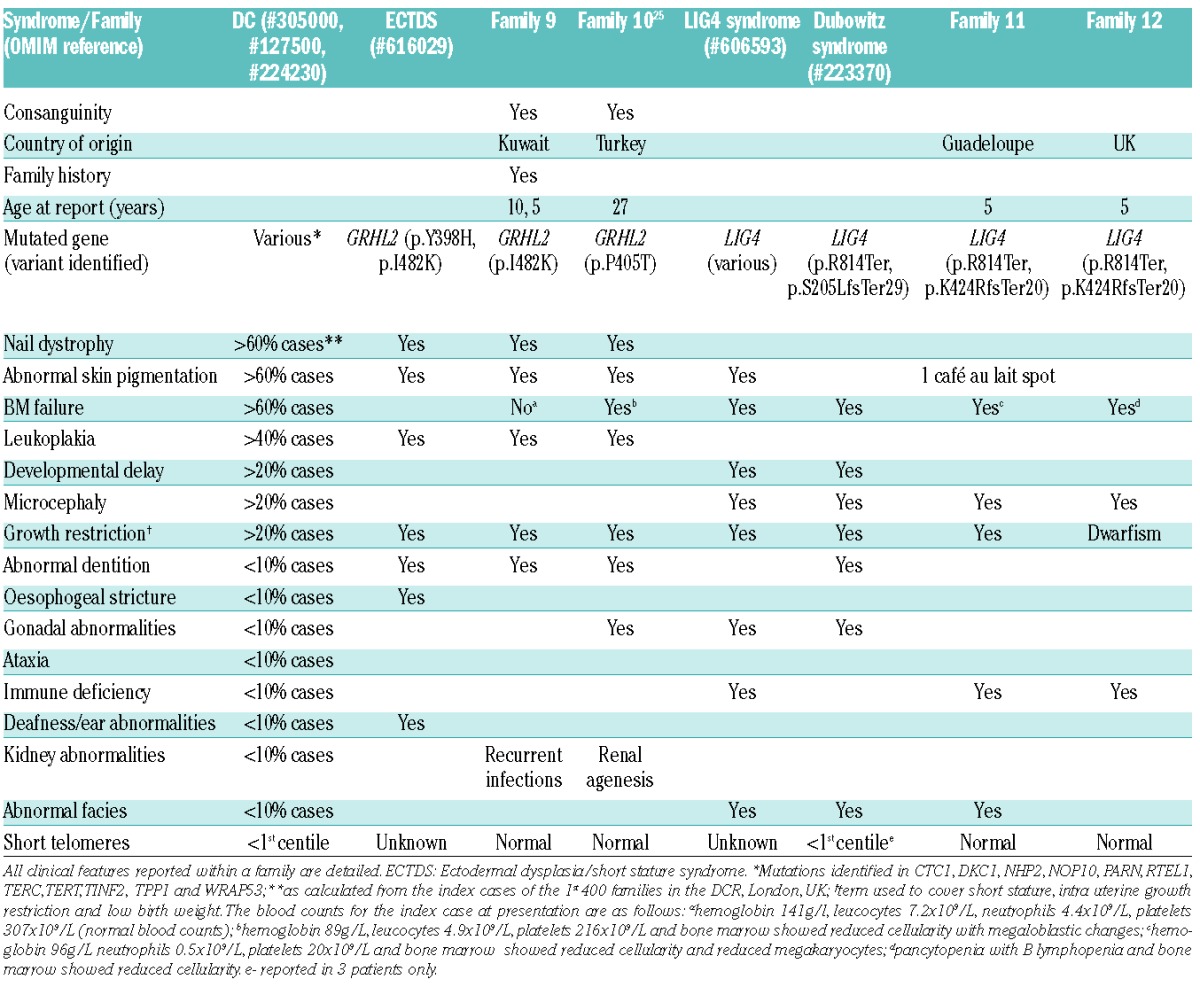
Here we report on 12 families with an initial diagnosis of DC who were found to have biallelic variants in one of three genes linked to a disease distinct from DC. The genes identified were USB1 (U6 snRNA biogenesis 1) associated with poikiloderma with neutropenia (PN), GRHL2 (grainyhead-like transcription factor 2) which is associated with ectodermal dysplasia/short stature syndrome (ECTDS), and LIG4 (Ligase IV, DNA, ATP-dependent) which is predominantly associated with LIG4 syndrome and occasionally Dubowitz syndrome. The key presenting features of PN are poikiloderma with non-cycling neutropenia, recurrent infections, short stature and nail abnormalities.13 The typical clinical features associated with ECTDS are short stature, nail dystrophy, abnormal oral pigmentation, and keratoderma and hyperkeratosis of the hands and feet.14 LIG4 syndrome is characterized by immune deficiency and developmental and growth delay.15 Patients can also display unusual facial features, microcephaly, pancytopenia and various skin abnormalities. Dubowitz syndrome is characterized by growth failure/short stature, characteristic facial features, microcephaly, mild mental retardation and eczema.16 All of these disorders have a high degree of phenotypic overlap with DC, thus leading to difficulties in making an accurate clinical diagnosis. The identification of biallelic USB1, GRHL2 and LIG4 mutations in 12 different families, initially diagnosed to have DC, therefore demonstrate the marked phenotypic overlap of ‘classical DC’ with four other genetic syndromes.
Methods
Patient samples
Exome capture or targeted gene screening was performed on a series of genetically uncharacterized index cases in the Dyskeratosis Congenita Registry (held at Barts and The London Hospital, London, UK). Presenting features included some/all of the classic mucocutaneous abnormalities (abnormal skin pigmentation, nail dystrophy, leukoplakia), with or without bone marrow failure. Peripheral blood samples were obtained with written consent under the approval of our local research ethics committee (London – City and East). Genomic DNA was extracted from these peripheral blood samples for use in all the subsequent analyses (Puregene, Qiagen).
Telomere length measurement
Telomere lengths were measured using the monochrome multiplex quantitative PCR (MMqPCR) method (modified from Cawthon),17 on a LightCycler 480 real-time thermocycler (Roche). Briefly, in each well, amplification of telomeric DNA (T) and a single copy gene (S) were quantified against standard curves obtained from the dilution of a reference DNA sample. The T/S ratio, obtained in triplicate for each sample, is proportional to the telomere length. This ratio was normalized to the T/S ratio of a second reference sample that was run on every plate to give a relative T/S ratio.
Whole exome sequencing (WES)
50ng genomic DNA was subjected to library preparation and exome capture using the Nextera Rapid Capture Exome kit (Illumina). Sequencing was performed using the Illumina HiSeq 2000 system and 100bp paired-end reads were generated and the data processed through the Illumina pipeline. Variants were called as described previously.8 All relevant variants were confirmed by Sanger sequencing.
Targeted gene screening
We designed an NGS assay covering the coding regions and some 5′UTR’s from 31 genes associated with genetic bone marrow failure syndromes (Online Supplementary Table S1). We used the Illumina TruSeq custom amplicon kit for library preparation and capture according to the manufacturer’s instructions. The resultant targeted fragments were indexed by dual barcodes and then sequenced on the Illumina MiSeq platform. Read alignment was performed automatically using the online MSR: TruSeq amplicon software. Annotation of the variants was performed using ANNOVAR.
Variant filtering
Exome data from our patients were jointly called with 2500 WES internal control samples (UCL-EX consortium) with unrelated conditions to minimize artificial batch effects. Where there was a family history, we assumed an autosomal recessive mode of inheritance as all parents were reportedly asymptomatic. Filtering was performed using the following criteria: all variants had to pass the Illumina filter, have a read depth of >10×, be present in the general population at a frequency of <0.0002 as reported on The Exome Aggregation Consortium database, (ExAC), 1000 genome project, and from the UCL-EX consortium. Variants that were predicted to be tolerated and benign by the Sorting Intolerant from Tolerant algorithm (SIFT) and Polymorphism Phenotyping v2 (PolyPhen-2) were then removed from further analysis. All relevant variants were confirmed by Sanger sequencing.
Results
Telomere length analysis
It has been well established that in the majority of patients with “classical” DC who have mutations in components of the telomere maintenance pathway have short telomeres, usually below the 1st centile.18,19 In fact, this measure is often used as a diagnostic screening tool to perform a differential diagnosis of DC from other bone marrow failure syndromes such as Fanconi anemia.20 Telomere lengths were therefore measured in our patients by MMqPCR, and this was used as an indicator of whether we could be looking for a mutation in a known telomere biology related gene or not. As none of the patients reported in this study had short telomeres when compared with controls (Figure 1), this suggested that we were looking for mutations outside the spectrum of those usually associated with DC, but they could still be associated with other bone marrow failure diseases or else have a previously unidentified association to another pathology.
Figure 1.
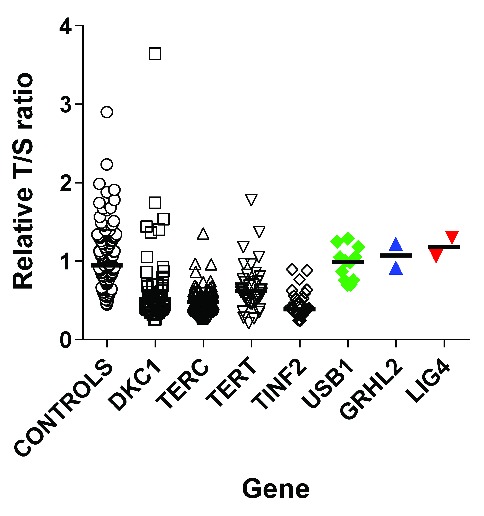
Telomere lengths in patients with GRHL2, LIG4 and USB1 mutations are not short. Relative T/S ratios are within the normal range. Open circles: controls (n=130); green diamonds: patients with biallelic USB1 variants (n=12), blue triangles: patients with homozygous GRHL2 variants (n=2); inverted red triangles – patients with compound heterozygous LIG4 variants (n=2). For comparison short telomeres are seen in patients with mutations in the known telomere associated genes DKC1 (n=50, open squares), TERC (n=66, open triangles), TERT (n=36, open inverted triangles) and TINF2 (n=33, open diamonds). The black line represents the median for each data set.
Identification of causal genetic variants
Analysis of our data has identified significant biallelic variants in three genes as detailed below. Five families underwent WES (Families 4, 9–12), and the remaining underwent targeted sequencing either using a 31 bone marrow failure disease gene panel (Online Supplementary Table S1) or direct sequencing.
USB1 (U6 snRNA biogenesis 1)
Biallelic USB1 mutations have been previously described in patients with DC and the overlapping disease poikiloderma with neutropenia (PN).21,22 Since our previous publication,21 we have identified homozygous USB1 variants in an additional 8 families with features of DC. In 7 of the 8 families, the variant was identified by targeted sequencing and the remaining family (2 cases) underwent WES. The only variant that was homozygous and shared by both affected siblings was in USB1 so this was deemed to be causal. Five out of the 8 index cases had the previously documented recurring homozygous variant c.531delA, p.His179MetfsTer8613 (Figure 2A) which is not described on ExAC. The 6th index case was found to have the homozygous variant c.673C>T, p.Gln225Ter (Figure 2B), which has been reported previously as part of a compound heterozygote.23 The remaining 2 families (Family 7 and Family 8) have the same homozygous missense mutation c.623A>G, p.His208Arg (Figure 2C). This is the first report of a homozygous missense being associated with disease in USB1, as to date all 19 disease-causing variants reported in USB1 are predicted to be loss of function (splicing, nonsense or frame shift).13 Segregation analysis confirmed an autosomal recessive mode of inheritance, and homozygous variants were confirmed in an additional 4 affected siblings within these families (Figure 2A). In silico analysis of mutations on USB1 crystal structure (PDB id: 4W7H, Figure 2D) revealed that both p.His179MetfsTer86 and p.Gln225Ter truncates several α-helices and β-sheets in USB1 (Figure 2E,F). Furthermore the homozygous missense change p.His208Arg identified in families 7 and 8 affects the highly conserved histidine residue His208, (Online Supplementary Figure S1 and Figure 2G,H). This change is predicted to be probably damaging by PolyPhen-2; (HumVar score 1.0) and disease causing by Mutation Taster. This His208 residue is implicated in the catalytic mechanism of USB1, where it facilitates the displacement of uridine nucleoside on RNA substrates to stabilize U6 small nuclear RNA, which in turn plays a critical role in RNA splicing.24
Figure 2.
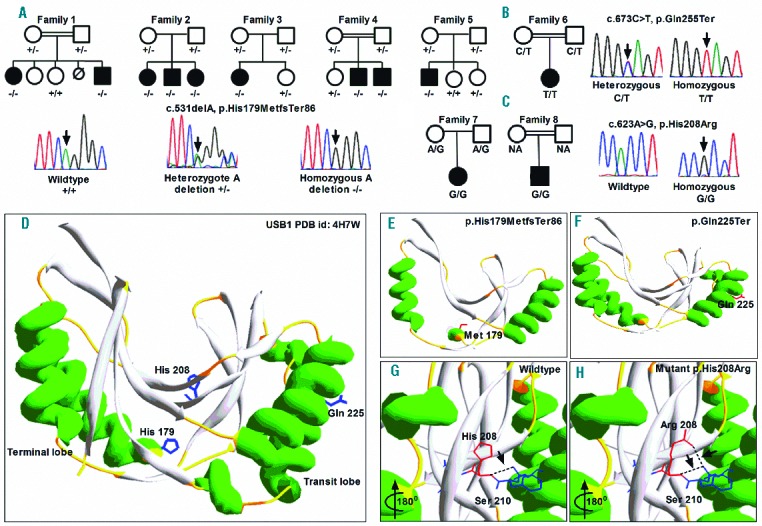
Identification and segregation of disease-causing variants in USB1. (A) The c.531delA variant is identified in 5 families with a DC phenotype. Representative sequencing traces are shown for the wild-type (+/+), heterozygous (+/−) and homozygous (−/−) forms of the mutation. (B) Family tree and sequence traces of c.673C>T variant seen in Family 6. A representative heterozygous trace and the homozygous trace are shown. (C) Family tree and sequence traces of the novel c.623A>G homozygous variant (G/G) as seen in Family 7 and 8. NA: sample not available. D) Protein crystal structure of human USB1 (PDB id: 4W7H) depicted in ribbon form indicating both the transit and terminal lobes indicated in green α helices. The position of identified residues mutated in USB1 deficiency patients is indicated as a stick model in blue. (E and F) In silico modeling of truncation mutations reveals the loss of several α helices and β sheets in both the transit and terminal lobes of the USB1 secondary structure. (G and H) The invariant H-x-S motif in the active site is indicated as a stick model. The missense change histidine to arginine at position 208 (red) introduces an extra hydrogen bond (dotted black line indicated by arrows), with Ser210 (blue), which in turn might cause aberrant oligoadenylation of the RNA substrate.
Table 1 summarizes the clinical presentation of these families, and the similarity between the clinical presentation of DC and PN is further highlighted in Figure 3. The degree of overlap between the reticular pigmentation observed on the legs (Figure 3A–C), and nail dystrophy of the toes (Figure 3D–F) is marked. Similar pigmentation is seen on the trunk (Figure 3G,H) and nail dystrophy and blistering is shown on the hands (Figure 3I). In addition to highlighting the similarity between the cutaneous presentation of DC and PN, panels B, C, E, F, H and I also emphasize the difference in presentation that can be caused by mutations within the same gene. The blood results detailed in Table 1 show that although there is neutropenia in all the patients, the overall blood pathology is more global rather than being restricted to just one lineage. This investigation has expanded the repertoire of USB1 variants identified to date, by adding a recurrent novel homozygous missense variant.
Table 1.
Overlap between DC, PN and patients with homozygous variants in USB1.
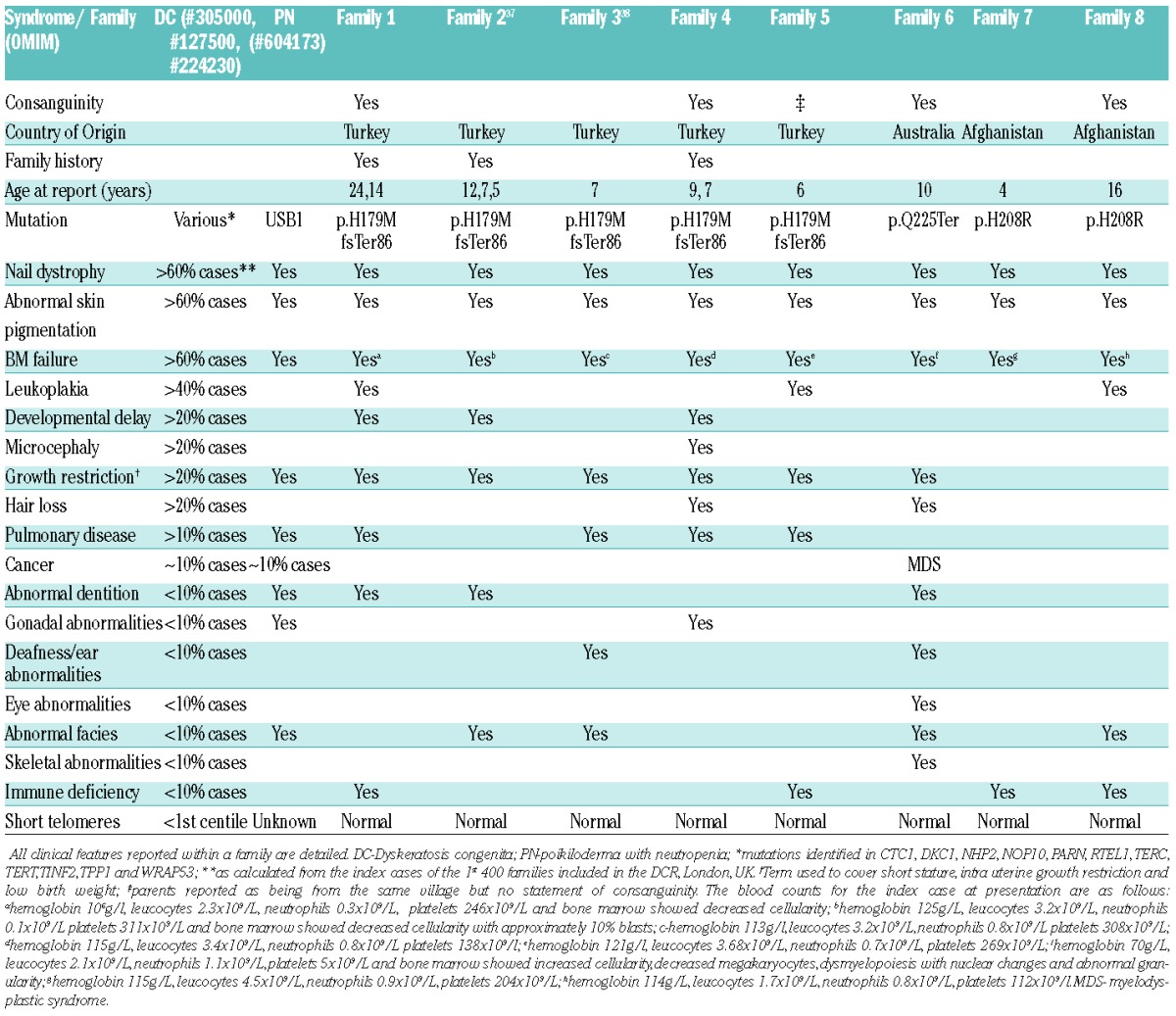
Figure 3.
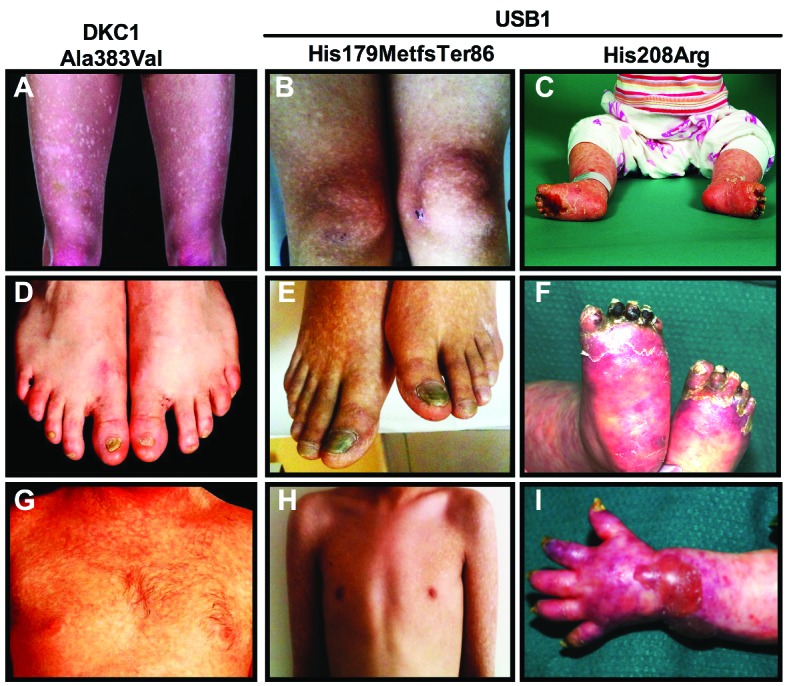
Similar clinical presentation is observed between DC and PN highlighting the marked overlap between these two syndromes. Panels A, D, G are from a male patient with p.Ala353Val DKC1 mutation, panels B, E and H are from a male patient with the homozygous USB1 p.His127MetfsTer86 mutation and panels C, F and I are from a child with the homozygous USB1 p.His208Arg mutation. Panels A–C show similar patterns of reticular pigmentation on the legs. Panels D–F show nail dystrophy on the toes. Panels G and H show reticular pigmentation on the trunk and panel I shows pigmentation on the arm along with nail dystrophy on the fingers. It is notable that the mucocutaneous features of the patient with USB1 p.His179MetfsTer86 mutation appear more like those of the patient with DKC1 p.Ala353Val mutation than the one with the USB1 p.His208Arg mutation.
GRHL2 (grainyhead-like transcription factor 2)
The index case from both families 9 and 1025 underwent WES, and as both families were consanguineous, we assumed an autosomal recessive mode of inheritance. Using the filtering approach described, it was noted that both families had homozygous non synonymous variants in the gene GRHL2; c.1445T>A p.Ile482Lys in Family 9 and c.1213C>A p.Pro405Thr in Family 10 (other homozygous calls that fulfilled the filtering criteria are detailed in the Online Supplementary Table S2). Segregation analysis confirmed that both variants were inherited in an autosomal recessive manner (Figure 4A). Both of these single base substitutions affect highly conserved nucleotides (Online Supplementary Figure S2). As neither variant is reported on ExAC and both variants are predicted by PolyPhen-2 to be probably damaging (HumVar scores p.P405T - 0.995 and p.I482K - 0.917) and disease causing by Mutation Taster, GRHL2 is the disease causing gene in these two families. GRHL2 is a member of a highly conserved family of transcription factors that are essential for epithelial development. At the molecular level, the GRHL transcription factors regulate the expression of proteins involved in cell proliferation, differentiation, adhesion and polarity. These factors adopt a DNA-binding immunoglobulin fold homologous to the DNA-binding domain of key tumor suppressor p53.26 Recently, Petrof and colleagues described homozygous mutations in GRHL2 in two families with ectodermal dysplasia/short stature syndrome (ECTDS).14 One of these was p.Ile482Lys, which is the same variant as identified in Family 9. All the variants described by us and Petrof et al., affect the DNA binding domain of the protein (Figure 4A). Phenotypically there are many shared features between our two families (Families 9 and 10) and the ECTDS families reported in the literature (Table 2). Another recent study27 examined the role of GRHL2 in the developing kidney, and clarified the involvement of GRHL2 in a network that controls the development of lumen expansion and barrier formation in renal epithelia, which may explain the previously reported renal agenesis seen in Family 10.25 This suggests that homozygous variants in the gene GRHL2, previously linked to ECTDS, are disease-causing in these 2 families.
Figure 4.
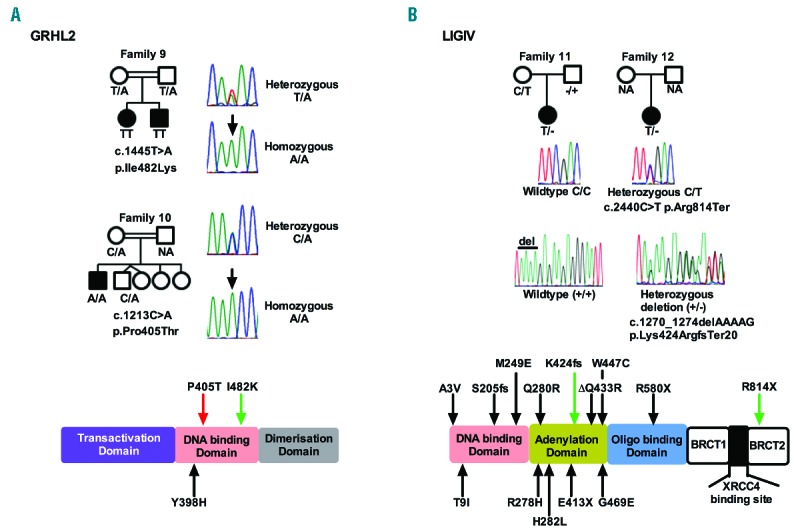
Identification of disease-causing variants in two different genes in patients with clinical features of DC. (A) Segregation and sequence traces for the homozygous GRHL2 variants identified in Families 9 and 10. Representative heterozygous and homozygous traces are shown for both variants. Schematic representation of the protein shows the identified domains and the location of the variants. The red arrow highlights the variant identified in this study. The black arrow highlights a variant identified in a previous study. The green arrow highlights the variant observed in both this and a previous study. NA-sample not available. (B) Segregation and sequence traces for the compound heterozygous LIG4 variants identified in Families 11 and 12. A representative wild-type and heterozygous trace is shown for each. The nucleotides deleted are indicated by a line above the corresponding bases on the wild-type (+/+) trace. Schematic representation of the protein showing the precise location of variants in different domains. Black arrows highlight variants identified in previous studies. Green arrows highlight variants observed in both this and previous studies.
Table 2.
Clinical overlap between the different genetic syndromes and the families with biallelic variants identified in this study.
LIG4 (Ligase IV, DNA, ATP-dependent)
Analysis of exome data from the index cases of Families 11 and 12 failed to reveal any homozygous variants that passed the filtering strategy. However, a biallelic analysis showed that they both shared the same compound heterozygous variants in LIG4, namely c.2440C>T, p.Arg814Ter and c.1270_1274 delAAAAG, p.Lys424ArgfsTer20 (Figure 4B, other biallelic calls are listed in the Online Supplementary Table S2). Although both variants observed in Families 11 and 12 are reported on ExAC at a very low frequency (0.000082 and 0.00014, respectively) in the heterozygous state, the likelihood of them occurring together (as is the case here) by chance is 2.4×10−8. LIG4 is involved in DNA non-homologous end joining and V(D)J recombination.28 Biallelic mutations in LIG4 are rare, with only 28 cases being reported to date in the literature,29 and are usually associated with LIG4 syndrome, and more recently in Dubowitz syndrome (DS).16 Both LIG4 syndrome and DS belong to a group of disorders that are associated with impaired DNA damage response mechanisms.28 LIG4 syndrome is a hereditary disorder associated with impaired DNA double-strand break repair mechanisms. It is characterized by growth restriction, developmental delay, microcephaly, facial dysmorphism, pancytopenia, variable immune deficiency and an increased predisposition to leukemia. DS is a rare multiple congenital syndrome characterized by cognitive delay, growth failure, microcephaly, distinctive facial dysmorphism, immune defects, pancytopenia, hematological malignancy and neuroblastoma. The degree of clinical overlap between LIG4 syndrome, DS and our 2 patients is marked (Table 2). Furthermore, the fact that both of the variants in LIG4 described here have been previously documented to be pathogenic and have been shown to occur in the same combination,30 suggests that these are causal genetic defects in these two unrelated individuals.
Discussion
Historically the diagnosis of disease is defined by the presenting clinical features, but it is becoming more apparent that this can give conflicting diagnoses as shown in Tables 1 and 2. Recent advances in whole exome and targeted sequencing have enabled a genetic diagnosis to be obtained more readily than ever before. This now enables the clinician to consider the possibility of diseases with overlapping phenotype based on a genetic result, rather than solely relying on the clinical presentation. In this report we have elucidated the underlying genetic causal variant(s) in 12 families (comprising 17 affected individuals) who were given a clinical diagnosis of dyskeratosis congenita. Initially we undertook whole exome sequencing on uncharacterized patients who presented with sufficient clinical features to be diagnosed as DC. As targeted sequencing methodologies improved, we then chose to use this approach as a screening tool to identify patients who had mutations in the known DC and bone marrow failure genes (Online Supplementary Table S1). The data reported herein represent a subset of all the cases analyzed, where we believe the disease-causing gene has been identified and where the identified gene has been previously associated with another genetic syndrome. By using a combination of these two strategies we identified disease-causing variants in the genes USB1 (8 families, 12 cases), GRHL2 (2 families, 3 cases) and LIG4 (2 families, 2 cases). Clinically, all patients show a high degree of overlap with the phenotypic profile for DC as described by Dokal et al.3 and the other diseases with which the disease-causing variants have been associated. This presents a problem in defining the disease: is it best to use phenotype or genotype¿
Significantly short telomeres are often used as a biomarker for DC. A telomere length below the 1st centile is considered diagnostic for DC, but this measurement is not routinely reported in patients with other bone marrow failure diseases. Figure 1 shows how telomeres in patients with variants in the known telomere genes (DKC1, TERC, TERT and TINF2) are significantly short when compared with a control population. Telomere lengths in all affected cases reported herein were not significantly short when compared with controls, regardless of the underlying genetic variant (Figure 1). This measure should perhaps be used as a screening tool where a diagnosis of classical DC is suspected, in order to decide the best course of action to determine a genetic diagnosis. Reduced telomere length has also been associated with an increased risk of cancer, and DC is regarded as a cancer prone syndrome. Patients with USB1 mutations tend to have an earlier presentation of myelodysplastic syndrome (MDS) than the general population, as most reports describe the disease in children. In this study and in our previous study21 we report 6 patients with MDS and 2 with acute myeloid leukemia, suggesting there is an increased cancer risk despite the patients not having short telomeres. Based on the small number of cases in our series, there does not appear to be an increased cancer risk for patients with either GRHL2 or LIG4 mutations. It is notable, however, that in the literature LIG4 syndrome is described as a disorder with a predisposition to leukemia. Although an increased cancer risk is not observed in our families, this may be due to the small numbers of patients described herein.
Due to the extensive overlap in clinical features between DC and PN as described in Table 1, telomere length seems to be one way of separating these two diagnostic definitions. Given that these cases do not have short telomeres but do possess DC features (Table 1), present us with a perplexing situation in accurately defining these patients. We propose it is timely to combine features of these two entities as a new syndrome, “USB1 deficiency syndrome”, characterized by bone marrow failure, abnormal skin pigmentation (poikiloderma), nail dystrophy, growth restriction, cancer predisposition and normal length telomeres.
It is also notable that the genes mutated in these overlapping syndromes have similarities with the biological functions of the known DC genes (Figure 5). USB1 has a role in snRNA processing, as is the case for dyskerin, NHP2 and NOP10.31 It also functions as an exoribonuclease reminiscent to the function of PARN that has been recently found to be mutated in both DC and pulmonary fibrosis.32 Both LIG4 and RTEL1 are involved in the maintenance of genomic stability, as they are both involved in pathways that repair double-strand breaks. LIG4 is an ATP-dependent DNA ligase that joins double-strand breaks during non-homologous end joining, and RTEL1 (a protein that is mutated in a subset of DC) is an ATP-dependent DNA helicase, which has an important role in telomere-length regulation. Both function via interactions with another shelterin component TRF2 (Telomere repeat binding-factor 2).33,34 Phenotypically, many of the features seen in LIG4/Dubowitz syndrome are also seen in DC patients with biallelic RTEL1 mutations (Table 2).7 This supports the idea that mutations in LIG4 can give rise to a phenotype that is reminiscent of DC as caused by a gene that has a known association with telomere biology. GRHL2 affects TERT expression by acting on its promoter35,36 thereby providing an important biological link to classical DC. While these proteins do have different functions or interactions, we speculate that it is this similarity, as suggested in Figure 5, that gives rise to the overlapping phenotypes. It is also notable that many of the DC genes have functions other than telomere maintenance. It is therefore possible that those overlapping shared functions may also be contributing to the similar clinical features observed herein. In the future it will be interesting to determine whether therapeutic agents that work in one disease have a beneficial effect in any of the overlapping syndromes.
Figure 5.
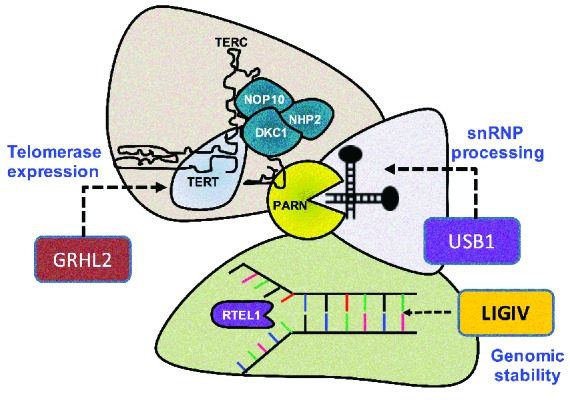
Biological overlap of classical DC proteins (TERT, DKC1, NOP10, NHP2, PARN and RTEL1) and those (GRHL2, USB1 and LIG4) mutated in patients in this study. Proteins with similar functions are grouped together. The schematic shows that GRHL2 can be grouped broadly with the molecules which have a role in telomerase expression, USB1 with small nuclear ribonucleoprotein (snRNP) processing and LIG4 with genomic stability. Previous studies have shown that biallelic mutations in GRHL2, USB1 and LIG4 are associated with ectodermal dysplasia, poikiloderma with neutropenia (or USB1 deficiency syndrome) and Lig4/Dubowitz syndrome, respectively. Mutations in DKC1, NHP2, NOP10, PARN and RTEL1 have been linked to dyskeratosis congenita.
The findings of this study raise a potential issue as to what constitutes DC. Taking into account the clinical features, germline genetic mutation (s) and telomere length we can recognize three categories of patients. “Category 1”: patients with sufficient clinical features (such as abnormal skin pigmentation, nail dystrophy, leukoplakia and bone marrow failure) to be labelled as DC, who have a germline mutation (s) in a telomere biology gene (such as TERT, TERC, DKC1, TINF2, RTEL1 and so forth) and who have short telomeres (described as being less than 1st centile). “Category 2”: patients with some clinical features of DC (but not sufficient to be classified as DC) who are found to have a germline mutation (s) in one of the telomere biology genes and who have short telomeres. “Category 3”: patients with sufficient clinical features of DC but who harbor a germline mutation (s) in a non-telomere biology gene (such as USB1, LIG4 and GRHL2) and have normal length telomeres. Category 1 patients can be considered to represent “classical or pure DC”. Category 1 and Category 2 patients can both be considered to represent disorders of telomeres – “telomeropathies”. Category 3 could be considered as “DC-like” or “DC-overlap”.
In summary, in 9 out of the 12 families highlighted in this study, the biallelic variants identified in USB1, GRHL2 and LIG4 have been shown to be pathogenic in previous studies.13,14,20,23,24 In the remaining families we have provided strong genetic and in silico evidence that the identified mutations are pathogenic. This study has demonstrated that the pleotropic clinical phenotype of DC markedly overlaps with the recognized disease entities LIG4 syndrome, Dubowitz syndrome and PN as well as the recently recognized ECTDS. In doing so it has substantiated the last category as a significant disease entity. The marked overlap of features of DC with PN, ECTDS, LIG4 and Dubowitz syndromes has important implications for establishing genetic diagnosis when a new patient presents in the clinic, specifically in patients with a clinical diagnosis of DC the genetic analysis needs to include GRHL2, LIG4 and USB1 in addition to the classical DC genes.
Acknowledgments
The authors would like to thank all the clinicians and patients who have helped us over the years, particularly Drs Babik, Balci, Ghoulam and Leblanc. We would also like to thank the staff at Barts and The London Genome Centre for Sanger sequencing analysis.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/10/1180
Funding
Financial support is provided by The Medical Research Council-MR/K000292/1, Children with Cancer- 2013/144 and Blood Wise-14032 (AJW, LC, SC, AE, TV, HT and ID). KMG is supported by the National Institute for Health Research through the NIHR Southampton Biomedical Research Centre.
References
- 1.Pereboeva L, Westin E, Patel T, et al. DNA damage responses and oxidative stress in dyskeratosis congenita. PLoS One. 2013;8(10):e76473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kirwan M, Beswick R, Walne AJ, et al. Dyskeratosis congenita and the DNA damage response. Br J Haematol. 2011;153(5):634–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dokal I, Vulliamy T, Mason P, Bessler M. Clinical utility gene card for: Dyskeratosis congenita - update 2015. Eur J Hum Genet. 2015;23(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gramatges MM, Bertuch AA. Short telomeres: from dyskeratosis congenita to sporadic aplastic anemia and malignancy. Transl Res. 2013;162(6):353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Savage SA. Human telomeres and telomere biology disorders. Prog Mol Biol Transl Sci. 2014;125:41–66. [DOI] [PubMed] [Google Scholar]
- 6.Kocak H, Ballew BJ, Bisht K, et al. Hoyeraal-Hreidarsson syndrome caused by a germline mutation in the TEL patch of the telomere protein TPP1. Genes Dev. 2014;28(19):2090–2102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Walne AJ, Vulliamy T, Kirwan M, Plagnol V, Dokal I. Constitutional mutations in RTEL1 cause severe dyskeratosis congenita. Am J Hum Genet. 2013;92(3):448–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tummala H, Walne A, Collopy L, et al. Poly(A)-specific ribonuclease deficiency impacts telomere biology and causes dyskeratosis congenita. J Clin Invest. 2015;125(5):2151–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moon DH, Segal M, Boyraz B, et al. Poly(A)-specific ribonuclease (PARN) mediates 3′-end maturation of the telomerase RNA component. Nat Genet. 2015;47(12):1482–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dhanraj S, Gunja SM, Deveau AP, et al. Bone marrow failure and developmental delay caused by mutations in poly(A)-specific ribonuclease (PARN). J Med Genet. 2015;52(11):738–748. [DOI] [PubMed] [Google Scholar]
- 11.Townsley DM, Dumitriu B, Young NS. Bone marrow failure and the telomeropathies. Blood. 2014;124(18):2775–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ng SB, Buckingham KJ, Lee C, et al. Exome sequencing identifies the cause of a mendelian disorder. Nat Genet. 2010;42(1):30–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koparir A, Gezdirici A, Koparir E, et al. Poikiloderma with neutropenia: genotypeethnic origin correlation, expanding phenotype and literature review. Am J Med Genet A. 2014;164A(10):2535–2540. [DOI] [PubMed] [Google Scholar]
- 14.Petrof G, Nanda A, Howden J, et al. Mutations in GRHL2 result in an autosomal-recessive ectodermal Dysplasia syndrome. Am J Hum Genet. 2014;95(3):308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chistiakov DA. Ligase IV syndrome. Adv Exp Med Biol. 2010;685:175–185. [DOI] [PubMed] [Google Scholar]
- 16.Stewart DR, Pemov A, Johnston JJ, et al. Dubowitz syndrome is a complex comprised of multiple, genetically distinct and phenotypically overlapping disorders. PLoS One. 2014;9(6):e98686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cawthon RM. Telomere length measurement by a novel monochrome multiplex quantitative PCR method. Nuc Acids Res. 2009;37(3):e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Alter BP, Baerlocher GM, Savage SA, et al. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood. 2007;110(5):1439–1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vulliamy TJ, Kirwan MJ, Beswick R, et al. Differences in disease severity but similar telomere lengths in genetic subgroups of patients with telomerase and shelterin mutations. PLoS One. 2011;6(9):e24383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alter BP, Giri N, Savage SA, Rosenberg PS. Telomere length in inherited bone marrow failure syndromes. Haematologica. 2015; 100(1):49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. Mutations in C16orf57 and normal-length telomeres unify a subset of patients with dyskeratosis congenita, poikiloderma with neutropenia and Rothmund-Thomson syndrome. Hum Mol Genet. 2010;19(22):4453–4461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arnold AW, Itin PH, Pigors M, et al. Poikiloderma with neutropenia: a novel C16orf57 mutation and clinical diagnostic criteria. Br J Dermatol. 2010;163(4):866–869. [DOI] [PubMed] [Google Scholar]
- 23.Clericuzio C, Harutyunyan K, Jin W, et al. Identification of a novel C16orf57 mutation in Athabaskan patients with Poikiloderma with Neutropenia. Am J Med Genet A. 2011;155A(2):337–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hilcenko C, Simpson PJ, Finch AJ, et al. Aberrant 3′ oligoadenylation of spliceosomal U6 small nuclear RNA in poikiloderma with neutropenia. Blood. 2013;121(6):1028–1038. [DOI] [PubMed] [Google Scholar]
- 25.Balci S, Engiz O, Erekul A, Gozdasoglu S, Vulliamy T. An atypical form of dyskeratosis congenita with renal agenesis and no mutation in DKC1, TERC and TERT genes. J Eur Acad Dermatol Venereol. 2009; 23(5):607–608. [DOI] [PubMed] [Google Scholar]
- 26.Mlacki M, Kikulska A, Krzywinska E, Pawlak M, Wilanowski T. Recent discoveries concerning the involvement of transcription factors from the Grainyhead-like family in cancer. Exp Biol Med (Maywood). 2015;240(11):1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aue A, Hinze C, Walentin K, et al. A Grainyhead-Like 2/Ovo-Like 2 Pathway Regulates Renal Epithelial Barrier Function and Lumen Expansion. J Am Soc Nephrol. 2015;26(11):2704–2715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chistiakov DA, Voronova NV, Chistiakov AP. Ligase IV syndrome. Eur J Med Genet. 2009;52(6):373–378. [DOI] [PubMed] [Google Scholar]
- 29.Tamura S, Higuchi K, Tamaki M, et al. Novel compound heterozygous DNA ligase IV mutations in an adolescent with a slowly-progressing radiosensitive-severe combined immunodeficiency. Clin Immunol. 2015;160(2):255–260. [DOI] [PubMed] [Google Scholar]
- 30.Murray JE, Bicknell LS, Yigit G, et al. Extreme growth failure is a common presentation of ligase IV deficiency. Hum Mutat. 2014;35(1):76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meier UT. The many facets of H/ACA ribonucleoproteins. Chromosoma. 2005; 114(1):1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berndt H, Harnisch C, Rammelt C, et al. Maturation of mammalian H/ACA box snoRNAs: PAPD5-dependent adenylation and PARN-dependent trimming. RNA. 2012;18(5):958–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell. 2012;47(4):497–510. [DOI] [PubMed] [Google Scholar]
- 34.Arnoult N, Karlseder J. Complex interactions between the DNA-damage response and mammalian telomeres. Nat Struct Mol Biol. 2015;22(11):859–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen W, Dong Q, Shin KH, et al. Grainyhead-like 2 enhances the human telomerase reverse transcriptase gene expression by inhibiting DNA methylation at the 5′-CpG island in normal human keratinocytes. J Biol Chem. 2010; 285(52):40852–40863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang X, Chen W, Kim RH, Kang MK, Park NH. Regulation of the hTERT promoter activity by MSH2, the hnRNPs K and D, and GRHL2 in human oral squamous cell carcinoma cells. Oncogene. 2009;28(4):565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Patiroglu T, Akar HH. Clericuzio-type Poikiloderma with Neutropenia Syndrome in a Turkish Family: a Three Report of Siblings with Mutation in the C16orf57 gene. Iran J Allergy Asthma Immunol. 2015;14(3):331–337. [PubMed] [Google Scholar]
- 38.Kilic SS, Cekic S. Juvenile Idiopathic Inflammatory Myopathy in a Patient With Dyskeratosis Congenita Due to C16orf57 Mutation. J Pediatr Hematol Oncol. 2015; 38(2):e75–77. [DOI] [PubMed] [Google Scholar]