

Abstract
Juvenile myelomonocytic leukemia is a rare myeloproliferative neoplasm characterized by hyperactive RAS signaling. Neurofibromin1 (encoded by the NF1 gene) is a negative regulator of RAS activation. Patients with neurofibromatosis type 1 harbor loss-of-function mutations in NF1 and have a 200- to 500-fold increased risk of juvenile myelomonocytic leukemia. Leukemia cells from patients with juvenile myelomonocytic leukemia display hypersensitivity to certain cytokines, such as granulocyte-macrophage colony-stimulating factor. The granulocyte-macrophage colony-stimulating factor receptor utilizes pre-associated JAK2 to initiate signals after ligand binding. JAK2 subsequently activates STAT5, among other downstream effectors. Although STAT5 is gaining recognition as an important mediator of growth factor signaling in myeloid leukemias, the contribution of STAT5 to the development of hyperactive RAS-initiated myeloproliferative disease has not been well described. In this study, we investigated the consequence of STAT5 attenuation via genetic and pharmacological approaches in Nf1-deficient murine models of juvenile myelomonocytic leukemia. We found that homozygous Stat5 deficiency extended the lifespan of Nf1-deficient mice and eliminated the development of myeloproliferative neoplasm associated with Nf1 gene loss. Likewise, we found that JAK inhibition with ruxolitinib attenuated myeloproliferative neoplasm in Nf1-deficient mice. Finally, we found that primary cells from a patient with KRAS-mutant juvenile myelomonocytic leukemia displayed reduced colony formation in response to JAK2 inhibition. Our findings establish a central role for STAT5 activation in the pathogenesis of juvenile myelomonocytic leukemia and suggest that targeting this pathway may be of clinical utility in these patients.
Introduction
Juvenile myelomonocytic leukemia (JMML) is a rare myeloproliferative neoplasm (MPN) with no effective chemotherapy or targeted therapy options. Hematopoietic stem cell transplantation, with its considerable morbidity and morality burden, remains the only modality that can improve survival in patients with this condition.1,2 Nearly all patients (80–90%) harbor somatic or germline mutations that lead to hyperactive RAS signaling.2–4 Recent deep sequencing efforts have discovered that some patients harbor two, co-occurring RAS-pathway activating mutations and that these compound mutations are associated with more aggressive disease,3,4 underscoring the importance of hyperactive RAS in JMML. Neurofibromin, encoded by NF1, negatively regulates RAS activity.5 Patients with inherited mutations of NF1 have a 200- to 500-fold increased risk of developing JMML.1 Mice harboring activated Ras genes or Nf1 deficiency develop MPN that resembles human JMML.6–14 Likewise, mice that harbor compound activating mutations that activate the RAS pathway also display a more aggressive JMML phenotype.15 Notably, transplantation of Nf1-null fetal liver cells or somatic deletion of Nf1 in the hematopoietic compartment results in progressive myeloid expansion.9,10,16,17 Furthermore, induced pluripotent stem cells, generated from two patients with JMML, differentiated into myeloid cells with high proliferative capacity and enhanced basal ERK (a well-known mediator of RAS activation) and STAT5 activation.18 Malignant cells from JMML patients and JMML mouse models display hypersensitivity to certain cytokines, in particular granulocyte-macrophage colony-stimulating factor (GM-CSF).5,9,14,19 The absence of GM-CSF receptor signaling prevents the development of MPN in recipient mice receiving hematopoietic stem cells doubly deficient for Nf1 and the GM-CSF receptor common β chain.16 Similarly, in an NrasG12D/+ model of MPN, β common chain deficiency did not prevent initiation of disease, but reduced splenomegaly and spontaneous colony formation and prolonged survival.20 GM-CSF receptor signaling promotes proliferation and differentiation by activating a variety of signal transduction pathways including Janus kinase 2 - signal transducer and activator of transcription 5 (Jak2-Stat5) and Ras.21,22
Mek inhibitors to modulate RAS activation have had variable therapeutic efficacy in JMML models. Myeloid cells, derived from the induced pluripotent stem cells described above, displayed reduced GM-CSF independence in response to Mek inhibition. In an activated Kras model of MPN, Mek inhibition abrogated the disease.23 In mouse models of Nf1-deficient or Kras-mutant MPN, Mek inhibition enhanced erythropoiesis and reduced spleen size, but failed to eradicate Nf1-deficient or Kras-mutant cells.23,24 These studies support a central role of aberrant Raf/MEK/ERK signaling in the abnormal growth of JMML cells.
The importance of STAT5a/b activation in JAK2-mutant MPN has been well described. STAT5 is an important contributor to hematopoiesis and cancer.25–28 Hyperphosphorylation of STAT5 in response to minimal concentrations of GM-CSF is a hallmark of JMML.29 JAK2 mutations are common in other MPN, including 95% of cases of polycythemia vera and 50–60% of cases of primary myelofibrosis and essential thrombocythemia.30 Treatment with the JAK2 inhibitor ruxolitinib improves the clinical parameters and symptoms associated with these disorders31–34 and leads to a reduction of STAT5 activation in the cells of treated patients.35 JAK2 inhibition reduces the viability of primary cells from patients with chronic myelomonocytic leukemia displaying hypersensitivity to GM-CSF signaling.36 Likewise, Stat5 deficiency abrogates disease in mouse models of JAK2V617F MPN.37,38 These findings highlight the critical role of STAT5 signaling in JAK2-mutant and other MPN featuring hyperactive GM-CSF signaling.
The possible contribution of the JAK2/STAT5 pathway to MPN with hyperactive RAS signaling, such as JMML, has not been well described. JMML cells derived from NF1-deficient patients display differential STAT5 activation,29 implicating this pathway in diseases with hyperactive RAS signaling. In a mouse model of NrasG12D CMML, a subset of cells developed hyperactive Erk and Stat5 activation in response to GM-CSF signaling.39 Mek inhibition prolonged the life of 40% of CMML mice harboring NrasG12D/G12D, while combined Mek inhibition with Jak2 inhibition abolished the disease in these mice.40 These findings implicate STAT5 as a potential contributor to the pathogenesis of MPN with activated RAS. Since the therapeutic options in JMML are severely limited, identifying effective drug targets in this devastating disease of infancy is an important clinical priority.
To elucidate the contribution of the Jak2-Stat5a/b signaling pathway to MPN derived from loss of Nf1, we attenuated Stat5 signaling in Nf1-deficient mice using either a genetic Stat5a/b hypomorphic knockout41,42 (which harbors a loss of both Stat5a and Stat5b genes) or pharmacological Jak2 inhibition with ruxolitinib.
Methods
Mice
Animals were treated in accordance with protocols approved by the Institutional Animal Care and Use Committee at the University of Minnesota
A complex breeding scheme was established to generate animals of the appropriate genotypes (Figure 1A). The Stat5a and Stat5b alleles used in this study produce low amounts of an N-terminally deleted, partially functional form of their respective proteins.42,43 Henceforth, Stat5 refers to both Stat5a and Stat5b loci on mouse chromosome 11, with the status of both alleles indicated simultaneously as either + for both wild-type alleles or ΔN for the hypomorphic double knockout. The murine Stat5 loci map approximately 15 cM away from the Nf1 locus on chromosome 11. Therefore, two separate recombinant chromosomes were generated, one chromosome with the Stat544,45 combined with the Nf1Fcr (null) allele46 and the other with Stat5 combined with the Nf1flox allele.47 Breeding was complicated because Stat5ΔN/ΔN females are infertile and Stat5ΔN/ΔN offspring often fail to thrive. The low ratio of useful animals per litter necessitated transplantation of donor bone marrow into histocompatible recipient animals.
Figure 1.

Stat5/Nf1-deficient bone marrow engrafts recipient animals. (A) Diagram depicting the breeding scheme to generate the five genetic backgrounds used in these studies. Stat5ΔN/+/Nf1Fcr/+ were generated and crossed with Stat5ΔN/+/Nf1Fcr/+/Mx1-Cre animals to generate the required genotypes. (B) Bone marrow was harvested from mice in each group and transplanted into syngeneic recipients. Four weeks after transplant and 2 weeks after induction of Cre recombinase, peripheral blood of recipient animals was immune-stained to measure the level of engraftment by Ly5.2+/Ly5.1− donor cells. Recipient mice showed greater than 70% engraftment by donor cells. Typical results are shown. (C) Eight weeks after transplantation, DNA was extracted from peripheral blood nucleated cells of recipient animals. Polymerase chain reaction analysis was performed on genomic DNA from each animal to determine the degree of deletion of the floxed Nf1 allele. A band indicating deletion was detected in all animals from which adequate DNA was obtained. Typical results are shown for three animals. W: water; Δ: recombined flox allele.
Mx1-Cre transgenic animals (C57BL/6) were crossed with Nf1flox mice (C57BL/6) to generate Nf1flox/+/Mx1-Cre animals. Separately, Stat5ΔN mice on a C57BL/6 × 129/Sv background were crossed with Nf1Fcr mice (C57BL/6) to generate Nf1Fcr/+/Stat5ΔN//+ animals and with Nf1flox/+/Mx1-Cre animals to generate Nf1flox/+/Stat5ΔN//+/Mx1-Cre animals. These animals were crossed to provide donor animals of the following genotypes: Nf1flox/Fcr/Stat5+/+/Mx1-Cre, Nf1flox/+/Stat5ΔN//+/Mx1-Cre, Nf1flox/Fcr/Stat5ΔN//+/Mx1-Cre, Nf1flox/Fcr/Stat5ΔN/ΔN/Mx1-Cre and Nf1flox/+/Stat5ΔN/ΔN/Mx1-Cre animals (Figure 1A).
Stat5ΔN/ΔN/Nf1 heterozygous mice, whether with the Nf1Fcr or Nf1flox allele, had particularly poor health and frequently died by 6 to 8 weeks of age. Transplants involving these genotypes were, therefore, done with single donors, rather than donor cells pooled from multiple mice. Multiple transplants were performed to achieve adequate numbers of experimental transplant recipients. For these and all of the other genotypes, all donor animals also carried the Mx1-Cre transgene.
Donor animals were F1 offspring from a cross of two strain backgrounds, C57BL/6 and 129/Sv, both of which express Ly5.2 (Ptpcra) on the surface of hematopoietic cells. Recipient animals were generated as F1 offspring from a cross between 129/Sv (Ly5.2, Ptpcra) and C57BL/6J (Ly5.1, Ptpcrb) animals. The recipient offspring were therefore congenic at the Ly5.1 locus, providing a mechanism by which to distinguish recipient Ly5.1+Ly5.2+ cells from donor Ly5.1− Ly5.2+ cells by immune staining for surface expression of Ly5.1 and Ly5.2.
Colony forming assays
Methylcellulose cultures were performed as previously described.5 Briefly, peripheral blood mononuclear cells were isolated using Lymphoprep™ (Stemcell Technologies, Vancouver, BC, Canada) according to the manufacturer’s instructions and plated at 5×103 cells/mL with MethoCult (Stemcell Technologies, Vancouver, BC, Canada), 100 U/mL penicillin G, 10 mg/mL streptomycin, and inhibitor. The inhibitors used were ruxolitinib at 0.4 μM, PD325901 at 13 μM (both from Selleckchem, Houston, TX, USA), or dimethylsulfoxide vehicle. Cultures were incubated at 37°C in 5% CO2 and scored 7 days later. Experiments in each condition were performed in triplicate.
Results
Generation of Stat5, Nf1 double-knockout mice
Mice were bred to generate five experimental groups: wild-type at the Stat5 loci and homozygous deficient at the Nf1 locus (Stat5+/+/Nf1flox/Fcr), heterozygous at the Stat5 loci and either heterozygous or homozygous deficient at the Nf1 locus (Stat5ΔN/+/Nf1+/Fcr or Stat5ΔN/+/Nf1flox/Fcr), and homozygous deficient at the Stat5 loci and either heterozygous or homozygous deficient at the Nf1 locus (Stat5ΔN/ΔN/Nf1+/Fcr or Stat5ΔN/ΔN/Nf1flox/Fcr) (Figure 1A). All donor animals also carried the Mx1-Cre transgene. Bone marrow from mice from each of these groups was transplanted into histocompatible recipients. One week after the transplant, recipient animals were injected with polyinosinic-polycytidylic acid (pIpC) to induce expression of the interferon responsive Mx1-Cre transgene; this led to deletion of the Nf1flox allele (Nf1Δ). Recipient animals heterozygous at the Ptpcr locus, Ptpcra/b, expressed both Ly5.1 and Ly5.2, and donor animals homozygous at the Ptpcr locus, Ptpcra/a, expressed only Ly5.2. This difference allowed us to identify cell origin by cell surface immune-staining. Recipient animals for all genotypes tested showed 70–90% engraftment 4 weeks after transplantation by flow cytometric analysis of circulating white blood cells (Figure 1B). Eight weeks after transplantation, peripheral blood was analyzed for deletion of the floxed Nf1 allele (Figure 1C). In all animals from which adequate DNA was obtained, Cre-mediated recombination was detected in peripheral blood mononuclear cell DNA (data not shown).
Stat5 deficiency attenuates Nf1-deficient myeloproliferative neoplasm
To determine the potential contribution of Stat5 to the development of Nf1-deficient MPN, cells derived from Nf1flox/floxmice induced with pIpC to cause biallelic Nf1 deletion (Nf1Δ/Δ) were used. Baseline levels of STAT5 phosphorylation were comparable between Nf1Δ/Δ and Nf1+/+ controls. GM-CSF-stimulation of cKit+ lineage−/Low populations (enriched for stem cells and progenitor cells) from these mice led to increased levels of STAT5 phosphorylation, a measure of STAT5 activation, in both populations. Notably, Nf1Δ/Δ cells achieved maximal levels of STAT5 phosphorylation within 10 min of stimulation, while Nf1+/+ populations took 60 min to achieve comparable levels of STAT5 activation (Figure 2A). A more rapid response to cytokines in Nf1Δ/Δ cells tightly correlates with the hypersensitivity to GM-CSF observed in the hematopoietic compartment of these Nf1-mutant mice10 and recapitulates the cytokine hyperresponsiveness described in other studies of MPN.5,9,14,48–50 In contrast, these cells displayed a trend toward elevated levels of phosphorylated ERK (pERK) under basal conditions, without significant differences in induction of pERK. Phosphorylated STAT3 levels were similar between the two genotypes; GM-CSF stimulation did not significantly increase STAT3 phosphorylation in either group. Transplantation of Stat5+/+/Nf1Δ/Fcr bone marrow in this study resulted in MPN similar to that found in previous studies as assessed by elevated white blood cell counts and spleen weights9,51 (Figure 2B,C and Online Supplementary Tables S1–S5). Hemoglobin concentration did not vary significantly by genotype and remained within the normal range (Online Supplementary Figure S1A). The platelet count remained in the low-normal range for all genotypes for the first year after transplantation (Online Supplementary Figure S1B), likely reflecting low-level radiation-induced bone marrow toxicity. After 1 year after transplantation, Stat5ΔN/ΔN/Nf1Δ/+ animals developed platelet counts that were significantly higher than those of the animals with other genotypes, but well within the normal range. Since these animals had an intact Nf1 allele, this phenotype likely reflects the effect of isolated Stat5 deficiency on the platelet count. Stat5+/+/Nf1Δ/Fcr animals succumbed to MPN at a median of 55 weeks after transplantation (Figure 2D and Online Supplementary Figure S1C). In contrast, animals with a single, intact Nf1 allele (Nf1Δ/+) did not develop MPN, as assessed by white blood cell counts and spleen weights (Figure 2B,C). Animals that received bone marrow harboring a single, intact Stat5 allele and homozygous Nf1 deficiency (Stat5Δ/+/Nf1Δ/Fcr) also developed MPN. These Stat5Δ/+/Nf1Δ/Fcr recipients had a comparable median survival to that of Stat5+/+/Nf1Δ/Fcr recipients (58 versus 55 weeks) and comparable spleen sizes (average spleen 1.03 g versus 0.75 g, P=0.25) but did display a delay in the development of MPN as measured by peripheral white blood cell counts (Figure 2B). In contrast, recipients of Nf1Δ/Fcr bone marrow lacking both copies of wild-type Stat5 (Stat5Δ/Δ/Nf1Δ/Fcr) did not develop MPN (as determined by white blood cell counts and spleen weights) and had a prolonged median survival of 79 weeks. The experiment was terminated 80 weeks after the transplants. Three Stat5Δ/Δ/Nf1Δ/Fcr animals died prior to this time point with no obvious cause of death but their premature death may be attributed to the complications of radiation exposure. The survival of animals of all genotypes along with a comprehensive table of their clinical status is shown in the Online Supplementary Material (Online Supplementary Figure S1C and Online Supplementary Table S1). Chimerism was measured via Ly5.1/5.2 mismatching for all animals at the termination of the experiment and necropsy. All surviving Stat5ΔN/ΔN animals were sufficiently reconstituted with donor hematopoietic cells (average % donor cells ± SE in bone marrow = 83.3 ± 13.4 and 73.1 ± 16.2 for Stat5ΔN/ΔNNf1Δ/Fcr and Stat5ΔN/ΔNNf1Δ/+mice, respectively), yet did not develop disease. Only one animal showed chimerism with less than 60% donor cells (49.7%). Thus, the failure to observe MPN in Stat5ΔN/ΔN recipients could not be attributed to engraftment failure. These data show that the absence of any wild-type STAT5 abrogates the development of NF1-deficient MPN and demonstrate that STAT5 activity is a critical contributor to NF1-deficient MPN.
Figure 2.
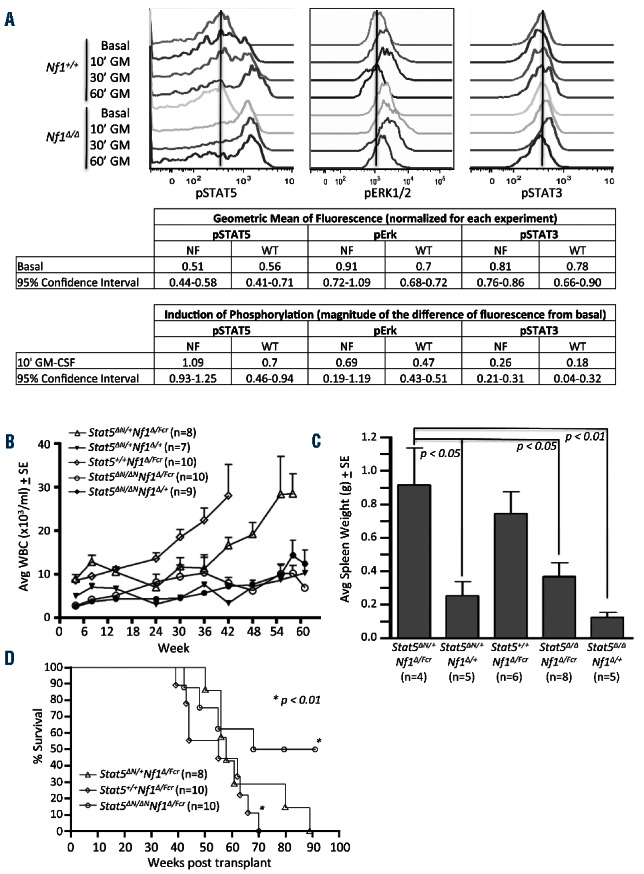
Stat5 insufficiency alleviates MPN in Nf1-deficient mice. Bone marrow was harvested from donor mice, transplanted into syngeneic recipients, and allowed to engraft (as described in Figure 1). (A) Bone marrow was harvested from recipient animals [Nf1∆/∆ (n=4) and Nf1+/+ (n=3)], serum- and cytokine-starved, then stimulated with GM-CSF (10 ng/mL) for 10, 30 and 60 min. Levels of phosphorylated STAT5 (pSTAT5), ERK1/2 (pErk1/2), and STAT3 (pSTAT3) were measured using phospho-specific, intracellular flow cytometry of c-Kit+/lineage− cells. A representative histogram is shown. Geometric mean of fluorescence is normalized for each experiment by dividing the geometric mean of the fluorescence of each sample by the average of the geometric mean of all the samples in each experiment. Induction of phosphorylation is reported as the fraction of basal levels. Induction of phosphorylation is calculated by subtracting the basal geometric mean of fluorescence from the geometric mean of fluorescence from each GM-CSF-stimulated sample; this difference is normalized for each sample by dividing by the basal geometric mean of fluorescence. (B) Peripheral blood from transplant recipients was collected every 6 weeks for the duration of experiments and with increased frequency in diseased animals. Total white blood cells counts (WBC) and peripheral blood smears (data not shown) were used to monitor the development of myeloproliferative disease in recipient mice. P<0.01, one way ANOVA followed by the Bartlett test for equal variance and the Tukey multiple comparison were performed for the comparison between Stat5+/+/Nf1∆N/Fcr bone marrow and Stat5∆N/+/Nf1∆N/+, Stat5∆N/∆N/Nf1∆N/Fcr, and Stat5∆N/∆N/Nf1∆N/+ bone marrow. (C) Moribund animals were sacrificed and spleen weights assessed. ANOVA followed by the Tukey test for significant differences were performed. (D) Kaplan-Meier survival plot comparing overall survival of recipients with the indicated genetic background. n ≥ 7 recipients per group; Log-rank followed by chi square testing was performed. In all figure panels, error bars represent standard errors of the mean.
Stat5 deficiency reverses myeloid precursor accumulation characteristic of myeloproliferative neoplasm
MPN in Stat5ΔN/+/Nf1Δ/Fcr animals was typical of MPN associated with Stat5+/+/Nf1−/− animals and did not differ significantly from that reported by other investigators10 in terms of absolute leukocytosis, splenomegaly, and morphology (Figure 2 and Online Supplementary Figure S1). Furthermore, as has been described in other models of Nf1-deficient MPN,10 homozygous Nf1-deficient animals displayed an expansion of immature myeloid precursors (Mac1+Gr1Low double-positive cells) in the bone marrow (Figure 3A,B), spleen (Figure 3A,C), and peripheral blood (data not shown). Homozygous Stat5 deficiency reversed this expansion (Figure 3). Interestingly, mice with Stat5 heterozygous, Nf1-deficient MPN displayed an expansion of the Mac1+Gr1Low (immature precursors) compartment that was intermediate between the expansion of this compartment in Stat5ΔN/ΔN and Stat5+/+ animals. This intermediate phenotype suggests that haploinsufficiency at the Stat5 locus may abrogate MPN as well.
Figure 3.

STAT5 insufficiency reverses immature myeloid expansion in Nf1-deficient mice. Surface immunophenotyping was performed on bone marrow and spleen mononuclear cells. (A) Representative immunophenotyping flow cytometry plot of Nf1-deficient bone marrow. (B) The proportion of cells from each immunophenotypic compartment in the bone marrow is indicated for each genotype. The inset shows the ratio of Mac1+Gr1High to Mac1+Gr1Low cells for each genotype. Two-way ANOVA followed by the Bonferroni post-test for significance was used for the main panel, and one way ANOVA followed by the Tukey multiple comparison test was used for the inset Mac1+Gr1High/Low ratio. (C) The proportion of Mac1+Gr1Low cells in the spleen is indicated for each genotype. ANOVA followed by the Tukey test was used to determine significant differences. In all figure panels, n = 10 for Stat5+/+/Nf1∆N/Fcr, n = 4 for Stat5∆N/+/Nf1∆N/Fcr, and n = 6 for Stat5∆N/∆N/Nf1∆N/Fcr; error bars represent standard errors of the mean.
The Jak2 inhibitor, ruxolitinib, diminishes myeloproliferative neoplasm
Next, we investigated whether pharmacological inhibition of the Jak/Stat pathway abrogates MPN initiated by Nf1 deficiency. We used ruxolitinib because Jak inhibition with ruxolitinib has been shown to attenuate Stat5 activation.52 We analyzed the bone marrow and spleen compartments of Mx1-Cre, Nf1flox/flox animals.10 Treatment with pIpC homozygously ablates the Nf1 locus (Nf1Δ/Δ) in the hematopoietic compartment of these mice leading to JMML-like MPN.10 These mice were treated with pIpC at 2 months of age and aged for an additional 6 months to allow MPN to develop before treatment with ruxolitinib. The mice were treated twice daily with ruxolitinib for 6 weeks. Complete blood counts were obtained weekly throughout the treatment period. At the completion of treatment, mice were sacrificed and bone marrow and spleens were harvested. Nf1-deficient animals developed MPN characterized by splenomegaly, leukocytosis, and anemia (Figure 4A, Online Supplementary Figure S2A,B, Online Supplementary Tables S6–S9). Platelet counts remained within the normal range but were higher in Nf1-deficient, vehicle-treated animals than in animals retaining a wild-type copy of the Nf1 allele (Online Supplementary Figure S2C). Ruxolitinib treatment attenuated MPN in mice, as evidenced by reduced spleen size (Figure 4A). White blood cell counts varied significantly among the mice with MPN (Online Supplementary Figure S2A). Ruxolitinib therapy reduced the white blood cell count by 50% in these MPN animals (Figure 4B). Ruxolitinib treatment was also associated with worsening anemia in animals with MPN (as has been described in clinical trials with this agent) but did not reduce the hemoglobin concentration of animals without MPN (Online Supplementary Figure S2B). Additionally, ruxolitinib was associated with a reduction in platelet count that was more pronounced in animals with MPN (Online Supplementary Figure S2C). All of the ruxolitinib-treated Nf1Δ/Δ mice survived until completion of the experiment. In contrast, 5/13 (38%) of vehicle-treated Nf1Δ/Δ mice succumbed to MPN during the treatment course (Figure 4C). Although this experiment was not designed to detect the effect of ruxolitinib on survival, the difference that we observed is statistically significant (P<0.05). Since the mice were sacrificed at completion of therapy, the difference in survival between the two treatment groups likely under-estimates the effect of ruxolitinib on survival. In accordance with our findings in STAT5Δ/Δ mice, Jak/Stat inhibition with ruxolitinib tended to reduce the percentage of Mac1+Gr1Low cells in the bone marrow of mice with Nf1Δ/Δ MPN, although this trend was not statistically significant (Figure 4D). These data demonstrate that a targeted inhibitor of Jak/Stat signaling is efficacious in attenuating the clinical features of Nf1-deficient MPN in mice.
Figure 4.

The Jak/Stat inhibitor, ruxolitinib significantly reduces disease burden in mice with MPN. Mice were treated daily with ruxolitinib for 6 weeks and sacrificed at the completion of treatment or when moribund. (A) Spleens were harvested and weighed at the time of sacrifice. Average spleen weights are indicated for each genotype. (B) For each mouse with MPN, reduction of disease burden was assessed by dividing the white blood cell count (WBC) at the time of sacrifice to maximum WBC measured during the animal’s lifetime. The average of these ratios was compared between mice with MPN treated with vehicle and ruxolitinib. (C) Kaplan-Meier survival curves are shown for vehicle- or ruxolitinib-treated Nf1∆N/∆N animals with MPN. (D) The average percentage of Mac1+Gr1Low cells in the bone marrows of MPN mice is shown for mice treated with vehicle and ruxolitinib. The number of mice (n) per group is indicated. In all figure panels, error bars represent standard errors of the mean.
Stat5 activity is implicated in maintaining signaling through the Ras/Nf1 pathway
Bone marrow cells of Nf1-deficient (Mx1-Cre, Nf1Δ/Δ) mice with MPN displayed hyperactive Erk signaling in comparison to bone marrow from Mx1-Cre, Nf1Δ/+ controls with no MPN (Figure 5A). Ruxolitinib treatment, which inhibits the Jak/Stat pathway, reduced levels of phosphorylated Erk in Nf1-deficient bone marrow (Figure 5A). This finding, along with our data from Stat5ΔN/ΔN/Nf1Δ/Fcr mice (Figures 2 and 3), suggests that sustained Stat5 signaling may be required to maintain hyperactive Mek/Erk signaling conferred by Nf1 deficiency. Indeed, mononuclear cells of mice treated with ruxolitinib showed a trend to reduced pSTAT5 induction in response to in vitro GM-CSF stimulation, although this trend was not statistically significant (Online Supplementary Figure S2D). To investigate whether the STAT5 and RAS pathways are similarly inter-connected in human MPN, we studied a patient with JMML harboring a KRAS mutation (KRASG13D). As in our murine model, ruxolitinib treatment led to reduced levels of pErk in bone marrow mononuclear cells from this patient (Figure 5B). Treatment with a MEK inhibitor (PD325901) or a JAK inhibitor (ruxolitinib) led to a decrease in colony formation in methylcellulose by peripheral blood mononuclear cells from this patient (Figure 5C). Notably, JAK inhibition had a more profound effect on colony formation than had MEK inhibition. Simultaneous treatment with both inhibitors gave results similar to those with inhibition of JAK alone. These data suggest that active JAK/STAT is required for the proliferative phenotype of MPN with hyperactive RAS.
Figure 5.
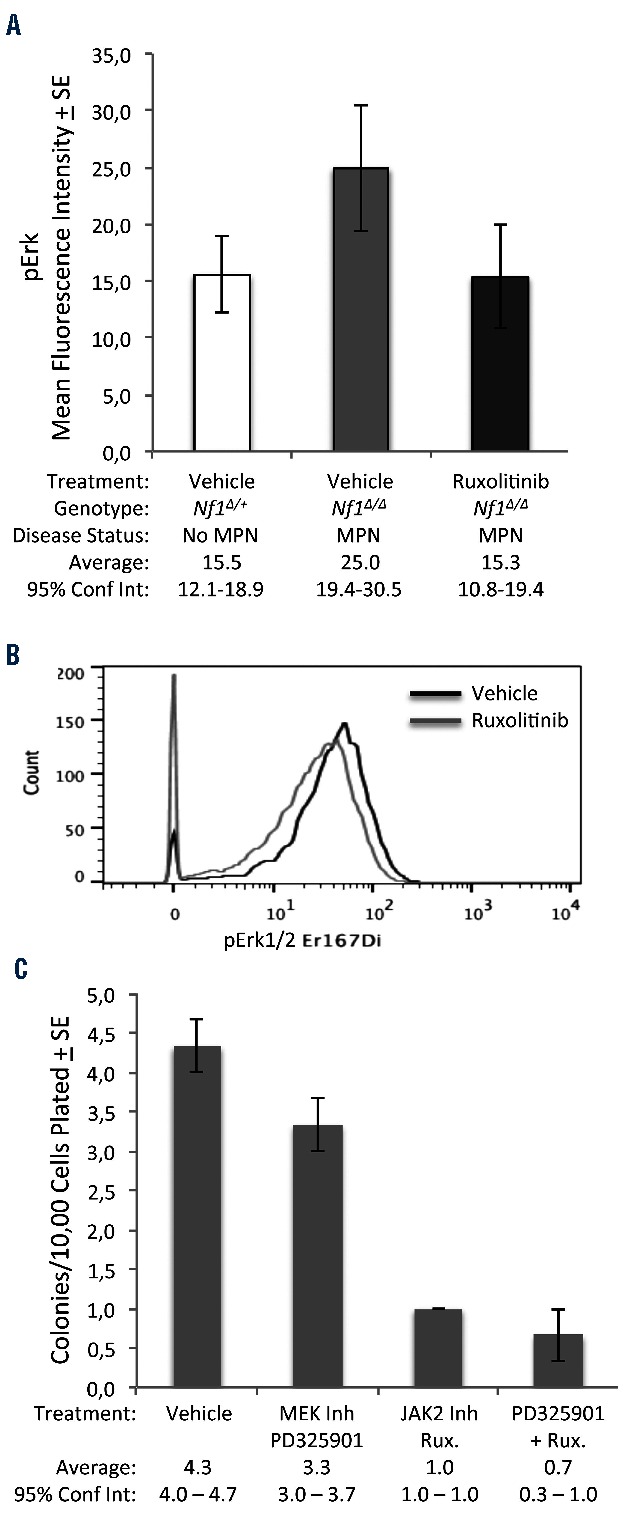
Jak/Stat inhibition inhibits ERK phosphorylation and colony formation in RAS-activated MPN. (A) Intracellular, phospho-specific flow cytometry was performed on bone marrow cells from vehicle-treated Nf1∆/+ control mice (n=12), vehicle-treated Nf1∆/∆ MPN mice (n=3), and ruxolitinib-treated Nf1∆/∆ MPN mice (n=7). Average mean fluorescence intensity representing levels of phospho-Erk are shown for each cohort. (B) Primary bone marrow mononuclear cells from a patient with KRAS-mutant (KRASG13D) JMML were incubated with vehicle or ruxolitinib (4 μm) for 30 min before fixation and permeabilization. Levels of phospho-Erk were assessed by mass cytometry. (C) Peripheral blood mononuclear cells from a patient with KRAS-mutant JMML were plated in methylcellulose containing MEK inhibitor, JAK inhibitor, both inhibitors, or vehicle. Each condition was plated in three replicates. Colony formation was scored after 7 days. In all figure panels, error bars represent standard errors of the mean. 95% confidence intervals are indicated.
Discussion
In this study, we used genetic and pharmacological approaches to demonstrate the importance of Stat5 in the pathogenesis of MPN initiated by Nf1 inactivation. We showed that MPN in Nf1-deficient, Stat5 hypomorphic mice is significantly diminished, leading to prolonged survival, improvement in blood count indices, and reduced spleen size in comparison to Nf1-deficient mice with intact Stat5 genes. Similarly, Nf1-deficient mice treated with ruxolitinib, an inhibitor of Jak/Stat signaling, had attenuated MPN with reduced white blood cell counts and smaller spleens. Both approaches tended to reverse the Mac1+GrLow immature myeloid cell accumulation seen in Nf1-deficient MPN bone marrow. We showed that ruxolitinib treatment diminished Erk signaling in these mice and in the bone marrow of a KRAS-mutated JMML patient. Ruxolitinib treatment also inhibited colony formation of primary cells from this JMML patient. Our ruxolitinib data implicate the Jak/Stat pathway in the pathogenesis of MPN but do not rule out effects of other STAT in the phenotype we observed. However, the ruxolitinib data, together with the data from our genetic Stat5-deficient model, suggest that STAT5 can modulate RAS-activated MAPK pathway activity.
The Stat5 alleles utilized in these experiments express an N-terminally deleted form of Stat5 that retains partial Stat5 function.42,43 Nevertheless, we showed that attenuation of Stat5 with retention of residual Stat5 function was sufficient to alleviate MPN in our genetic model. In a mouse model of MPN mediated by Mpl mutation, which leads to tonic activation of the Jak2/Stat5 pathway, conditional genetic ablation of Jak2 (via floxed alleles) was sufficient to induce complete remission of MPN, while ruxolitinib treatment of these Mpl mutant mice could only attenuate the disease.53 This study indicates that ruxolitinib does not completely inhibit Jak2 signaling, a finding that is consistent with clinical trials that show that ruxolitinib improves the clinical parameters of MPN but does not cure the disease.32–34,54 Likewise, incomplete inactivation of Stat5 with ruxolitinib alleviated many features of MPN in our model. Our data indicate a reduction of STAT5 activity, as is clinically attainable with ruxolitinib, may be sufficient to alleviate disease.
Activation of JAK-STAT and RAS signaling are both common features of myeloid leukemias.30,55,56 Previous work has demonstrated that MEK inhibition can attenuate myeloid neoplasia but is insufficient to cure this disease in either mouse models or human patients.23,56–58 KrasG12D myeloid cells remain hypersensitive to cytokines (according to colony-forming assays) despite MEK inhibition.23 Jak inhibition abrogates GM-CSF-dependent ERK phosphorylation in KrasG12D myeloid cells.59 These results indicate that other pathways contribute to disease in leukemias with hyperactive RAS signaling. Our work suggests activation of the STAT5 pathway may provide these critical signals in leukemia. Two recent reports from independent groups show that activated NRAS directs self-renewal in hematopoietic60 and leukemia stem cells.61 Gene expression analyses by both of these groups revealed that oncogenic NRAS led to activation of Stat5-mediated gene transcription and confirmed a relationship between RAS and STAT5 activity.
Early T-precursor acute lymphoblastic leukemia (ETP ALL) is a treatment-resistant, fatal leukemia with a mutational and gene expression signature comparable to that of poor-risk acute myeloid leukemia.62–65 Like acute myeloid leukemia, most ETP ALL harbor mutations that activate RAS and RAS pathway components.63,65,66 JAK1 and JAK3 are also commonly mutated in ETP ALL.65,66 Recently, activated STAT5 (phospho-STAT5) levels were found to be elevated in all ETP-ALL cases tested. This elevation was not related to JAK mutational status but to surface levels of interleukin-7 receptor (a receptor known to activate RAS, JAK/STAT, and PI3K signaling67,68). Intriguingly, ruxolitinib treatment of ETP-ALL xenografts led to profound reduction in disease, independently of JAK mutational status.67 Analogous to our work, this study also demonstrated the efficacy of STAT5 inhibition in leukemia with hyperactive RAS signaling.
Despite the considerable strides in the development of targeted therapies for treating myeloid neoplasms, there are no chemotherapy or targeted treatment options that have been shown to improve outcomes in JMML. As >90% of JMML patients exhibit activated RAS signaling, our data suggest that combination therapy with RAS-pathway inhibitors and ruxolitinib may be an effective, rational therapeutic strategy in this disease. MEK inhibition in mice with Nf1-deficient or Ras-activated MPN led to improvements in disease parameters but failed to eradicate leukemia cells.24,40 Likewise, MEK inhibition in early phase clinical trials in acute myelogenous leukemia has yielded largely disappointing results.57 In contrast, combined MEK and JAK/STAT inhibition in an NRASG12D/G12D model of MPN significantly improved survival of these mice.40 Our JMML patient harbors a KRAS mutation, yet ruxolitinib was more effective than MEK inhibition at controlling colony formation of the patient’s cells. These data provide a rationale for clinical trials combining ruxolitinib with RAS-pathway inhibitors to control activated RAS myeloid neoplasia.
Acknowledgments
The authors would like to thank and acknowledge the Comparative Pathology, Flow Cytometry and Biostatistics and Informatics Shared Resources of the Masonic Cancer Center and the Mass Cytometry Shared Resource (which is supported by the Office of the Vice President for Research) at the University of Minnesota and Michael Franklin, a scientific writing editor supported by the Division of Hematology, Oncology, and Transplantation, Department of Medicine, University of Minnesota, for his editorial assistance.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/10/1190
Funding
This work was supported in part by gifts made to the Minnesota Medical Foundation for the Matthew Kyle Nordos Pecha Memorial Fund and Dylan’s Wish Memorial Fund. This work was partially funded by a Leukemia and Lymphoma Society of America Specialized Center of Research Grant (LLS 7019-04) and National Cancer Institute (U01 CA84221) grant to DAL. This was supported in part by an NIH Heart, Lung, and Blood Institute training grant (T32HL007062) (ZS), an NIH/NCATS training grant (ULI RR033183 & KL2 RR0333182) (ZS), and funds from the Division of Hematology, Oncology, and Transplantation, Department of Medicine, University of Minnesota (ZS).
References
- 1.Dvorak CC, Loh ML. Juvenile myelomonocytic leukemia: molecular pathogenesis informs current approaches to therapy and hematopoietic cell transplantation. Front Pediatr. 2014;2(25):2296–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Locatelli F, Niemeyer CM. How I treat juvenile myelomonocytic leukemia (JMML). Blood. 2015;125(7):1083–1090. [DOI] [PubMed] [Google Scholar]
- 3.Stieglitz E, Taylor-Weiner AN, Chang TY, et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet. 2015;47(11):1326–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Caye A, Strullu M, Guidez F, et al. Juvenile myelomonocytic leukemia displays mutations in components of the RAS pathway and the PRC2 network. Nat Genet. 2015;47(11):1334–1340. [DOI] [PubMed] [Google Scholar]
- 5.Bollag G, Clapp DW, Shih S, et al. Loss of NF1 results in activation of the Ras signaling pathway and leads to aberrant growth in haematopoietic cells. Nat Genet. 1996;12(2):144–148. [DOI] [PubMed] [Google Scholar]
- 6.Amir el AD, Davis KL, Tadmor MD, et al. viSNE enables visualization of high dimensional single-cell data and reveals phenotypic heterogeneity of leukemia. Nat Biotechnol. 2013;31(6):545–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Braun BS, Tuveson DA, Kong N, et al. Somatic activation of oncogenic Kras in hematopoietic cells initiates a rapidly fatal myeloproliferative disorder. Proc Natl Acad Sci USA. 2004;101(2):597–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chan IT, Gilliland DG. Oncogenic K-ras in mouse models of myeloproliferative disease and acute myeloid leukemia. Cell Cycle. 2004;3(5):536–537. [DOI] [PubMed] [Google Scholar]
- 9.Largaespada DA, Brannan CI, Jenkins NA, Copeland NG. Nf1 deficiency causes Ras-mediated granulocyte/macrophage colony stimulating factor hypersensitivity and chronic myeloid leukaemia. Nat Genet. 1996;12(2):137–143. [DOI] [PubMed] [Google Scholar]
- 10.Le DT, Kong N, Zhu Y, et al. Somatic inactivation of Nf1 in hematopoietic cells results in a progressive myeloproliferative disorder. Blood. 2004;103(11):4243–4250. [DOI] [PubMed] [Google Scholar]
- 11.Li Q, Haigis KM, McDaniel A, et al. Hematopoiesis and leukemogenesis in mice expressing oncogenic NrasG12D from the endogenous locus. Blood. 2011;117(6):2022–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang J, Liu Y, Li Z, et al. Endogenous oncogenic Nras mutation initiates hematopoietic malignancies in a dose- and cell type-dependent manner. Blood. 2011;118(2):368–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang J, Liu Y, Tan LX, et al. Distinct requirements of hematopoietic stem cell activity and Nras G12D signaling in different cell types during leukemogenesis. Cell Cycle. 2011;10(17):2836–2839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang YY, Vik TA, Ryder JW, et al. Nf1 regulates hematopoietic progenitor cell growth and ras signaling in response to multiple cytokines. J Exp Med. 1998;187(11):1893–1902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu YL, Yan Y, Webster C, et al. Timing of the loss of Pten protein determines disease severity in a mouse model of myeloid malignancy. Blood. 2016;127(15):1912–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim A, Morgan K, Hasz DE, et al. Beta common receptor inactivation attenuates myeloproliferative disease in Nf1 mutant mice. Blood. 2007;109(4):1687–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahgoub N, Taylor BR, Gratiot M, et al. In vitro and in vivo effects of a farnesyltransferase inhibitor on Nf1-deficient hematopoietic cells. Blood. 1999;94(7): 2469–2476. [PubMed] [Google Scholar]
- 18.Gandre-Babbe S, Paluru P, Aribeana C, et al. Patient-derived induced pluripotent stem cells recapitulate hematopoietic abnormalities of juvenile myelomonocytic leukemia. Blood. 2013;121(24):4925–4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emanuel PD, Bates LJ, Castleberry RP, Gualtieri RJ, Zuckerman KS. Selective hypersensitivity to granulocyte-macrophage colony-stimulating factor by juvenile chronic myeloid leukemia hematopoietic progenitors. Blood. 1991;77(5):925–929. [PubMed] [Google Scholar]
- 20.Zhang J, Ranheim EA, Du J, et al. Deficiency of beta common receptor moderately attenuates the progression of myeloproliferative neoplasm in NrasG12D/+ mice. J Biol Chem. 2015;290(31):19093–19103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Guthridge MA, Stomski FC, Thomas D, et al. Mechanism of activation of the GM-CSF, IL-3, and IL-5 family of receptors. Stem Cells. 1998;16(5):301–313. [DOI] [PubMed] [Google Scholar]
- 22.Okuda K, Foster R, Griffin JD. Signaling domains of the beta c chain of the GM-CSF/IL-3/IL-5 receptor. Ann N Y Acad Sci. 1999;872:305–312; discussion 312–313. [DOI] [PubMed] [Google Scholar]
- 23.Lyubynska N, Gorman MF, Lauchle JO, et al. A MEK inhibitor abrogates myeloproliferative disease in Kras mutant mice. Sci Transl Med. 2011;3(76):76ra27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chang T, Krisman K, Theobald EH, et al. Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J Clin Invest. 2013;123(1):335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grimley PM, Fang H, Rui H, et al. Prolonged STAT1 activation related to the growth arrest of malignant lymphoma cells by interferon-alpha. Blood. 1998;91(8): 3017–3027. [PubMed] [Google Scholar]
- 26.Hennighausen L, Robinson GW. Interpretation of cytokine signaling through the transcription factors STAT5A and STAT5B. Genes Dev. 2008;22(6):711–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Paukku K, Silvennoinen O. STATs as critical mediators of signal transduction and transcription: lessons learned from STAT5. Cytokine Growth Factor Rev. 2004;15(6): 435–455. [DOI] [PubMed] [Google Scholar]
- 28.Sternberg DW, Gilliland DG. The role of signal transducer and activator of transcription factors in leukemogenesis. J Clin Oncol. 2004;22(2):361–371. [DOI] [PubMed] [Google Scholar]
- 29.Kotecha N, Flores NJ, Irish JM, et al. Single-cell profiling identifies aberrant STAT5 activation in myeloid malignancies with specific clinical and biologic correlates. Cancer Cell. 2008;14(4):335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Muxi PJ, Oliver AC. Jak-2 positive myeloproliferative neoplasms. Curr Treat Options Oncol. 2014;15(2):147–156. [DOI] [PubMed] [Google Scholar]
- 31.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. [DOI] [PubMed] [Google Scholar]
- 32.Harrison C, Vannucchi AM. Ruxolitinib: a potent and selective Janus kinase 1 and 2 inhibitor in patients with myelofibrosis. An update for clinicians. Ther Adv Hematol. 2012;3(6):341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vannucchi AM, Kiladjian JJ, Griesshammer M, et al. Ruxolitinib versus standard therapy for the treatment of polycythemia vera. N Engl J Med. 2015;372(5):426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Quintas-Cardama A, Verstovsek S. Molecular pathways: Jak/STAT pathway: mutations, inhibitors, and resistance. Clin Cancer Res. 2013;19(8):1933–1940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Padron E, Painter JS, Kunigal S, et al. GM-CSF-dependent pSTAT5 sensitivity is a feature with therapeutic potential in chronic myelomonocytic leukemia. Blood. 2013;121(25):5068–5077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walz C, Ahmed W, Lazarides K, et al. Essential role for Stat5a/b in myeloproliferative neoplasms induced by BCR-ABL1 and JAK2(V617F) in mice. Blood. 2012;119(15): 3550–3560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan D, Hutchison RE, Mohi G. Critical requirement for Stat5 in a mouse model of polycythemia vera. Blood. 2012;119(15): 3539–3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Liu Y, Li Z, et al. Endogenous oncogenic Nras mutation promotes aberrant GM-CSF signaling in granulocytic/monocytic precursors in a murine model of chronic myelomonocytic leukemia. Blood. 2010;116(26):5991–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kong G, Wunderlich M, Yang D, et al. Combined MEK and JAK inhibition abrogates murine myeloproliferative neoplasm. J Clin Invest. 2014;124(6):2762–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cain JA, Xiang Z, O’Neal J, et al. Myeloproliferative disease induced by TEL-PDGFRB displays dynamic range sensitivity to Stat5 gene dosage. Blood. 2007;109(9):3906–3914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li G, Wang Z, Zhang Y, et al. STAT5 requires the N-domain to maintain hematopoietic stem cell repopulating function and appropriate lymphoid-myeloid lineage output. Exp Hematol. 2007;35(11):1684–1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cui Y, Riedlinger G, Miyoshi K, et al. Inactivation of Stat5 in mouse mammary epithelium during pregnancy reveals distinct functions in cell proliferation, survival, and differentiation. Mol Cell Biol. 2004;24(18): 8037–8047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Socolovsky M, Nam H, Fleming MD, Haase VH, Brugnara C, Lodish HF. Ineffective erythropoiesis in Stat5a(−/−)5b(−/−) mice due to decreased survival of early erythroblasts. Blood. 2001;98(12):3261–3273. [DOI] [PubMed] [Google Scholar]
- 45.Teglund S, McKay C, Schuetz E, et al. Stat5a and Stat5b proteins have essential and nonessential, or redundant, roles in cytokine responses. Cell. 1998;93(5):841–850. [DOI] [PubMed] [Google Scholar]
- 46.Brannan CI, Perkins AS, Vogel KS, et al. Targeted disruption of the neurofibromatosis type-1 gene leads to developmental abnormalities in heart and various neural crest-derived tissues. Genes Dev. 1994;8(9): 1019–1029. [DOI] [PubMed] [Google Scholar]
- 47.Zhu Y, Romero MI, Ghosh P, et al. Ablation of NF1 function in neurons induces abnormal development of cerebral cortex and reactive gliosis in the brain. Genes Dev. 2001;15(7):859–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Irish JM, Hovland R, Krutzik PO, et al. Single cell profiling of potentiated phospho-protein networks in cancer cells. Cell. 2004;118(2): 217–228. [DOI] [PubMed] [Google Scholar]
- 49.Krutzik PO, Irish JM, Nolan GP, Perez OD. Analysis of protein phosphorylation and cellular signaling events by flow cytometry: techniques and clinical applications. Clin Immunol. 2004;110(3):206–221. [DOI] [PubMed] [Google Scholar]
- 50.Van Meter ME, Diaz-Flores E, Archard JA, et al. K-RasG12D expression induces hyperproliferation and aberrant signaling in primary hematopoietic stem/progenitor cells. Blood. 2007;109(9):3945–3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kogan SC, Ward JM, Anver MR, et al. Bethesda proposals for classification of non-lymphoid hematopoietic neoplasms in mice. Blood. 2002;100(1):238–245. [DOI] [PubMed] [Google Scholar]
- 52.Quintas-Cardama A, Vaddi K, Liu P, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115(15): 3109–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bhagwat N, Koppikar P, Keller M, et al. Improved targeting of JAK2 leads to increased therapeutic efficacy in myeloproliferative neoplasms. Blood. 2014;123(13): 2075–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verstovsek S, Passamonti F, Rambaldi A, et al. A phase 2 study of ruxolitinib, an oral JAK1 and JAK2 inhibitor, in patients with advanced polycythemia vera who are refractory or intolerant to hydroxyurea. Cancer. 2014;120(4):513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Spiekermann K, Biethahn S, Wilde S, Hiddemann W, Alves F. Constitutive activation of STAT transcription factors in acute myelogenous leukemia. Eur J Haematol. 2001;67(2):63–71. [PubMed] [Google Scholar]
- 56.Ward AF, Braun BS, Shannon KM. Targeting oncogenic Ras signaling in hematologic malignancies. Blood. 2012;120(17):3397–3406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jain N, Curran E, Iyengar NM, et al. Phase II study of the oral MEK inhibitor selumetinib in advanced acute myelogenous leukemia: a University of Chicago phase II consortium trial. Clin Cancer Res. 2014;20(2):490–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lauchle JO, Kim D, Le DT, et al. Response and resistance to MEK inhibition in leukaemias initiated by hyperactive Ras. Nature. 2009;461(7262):411–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Diaz-Flores E, Goldschmidt H, Depeille P, et al. PLC-gamma and PI3K link cytokines to ERK activation in hematopoietic cells with normal and oncogenic Kras. Sci Signal. 2013;6(304):ra105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Q, Bohin N, Wen T, et al. Oncogenic Nras has bimodal effects on stem cells that sustainably increase competitiveness. Nature. 2013;504(7478):143–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sachs Z, LaRue RS, Nguyen HT, et al. NRASG12V oncogene facilitates self-renewal in a murine model of acute myelogenous leukemia. Blood. 2014;124(22):3274–3283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Coustan-Smith E, Mullighan CG, Onciu M, et al. Early T-cell precursor leukaemia: a subtype of very high-risk acute lymphoblastic leukaemia. Lancet Oncol. 2009;10(2):147–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neumann M, Coskun E, Fransecky L, et al. FLT3 mutations in early T-cell precursor ALL characterize a stem cell like leukemia and imply the clinical use of tyrosine kinase inhibitors. PloS One. 2013;8(1):e53190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Neumann M, Heesch S, Gokbuget N, et al. Clinical and molecular characterization of early T-cell precursor leukemia: a high-risk subgroup in adult T-ALL with a high frequency of FLT3 mutations. Blood Cancer J. 2012;2(1):e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang J, Ding L, Holmfeldt L, et al. The genetic basis of early T-cell precursor acute lymphoblastic leukaemia. Nature. 2012;481(7380):157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Neumann M, Heesch S, Schlee C, et al. Whole-exome sequencing in adult ETP-ALL reveals a high rate of DNMT3A mutations. Blood. 2013;121(23):4749–4752. [DOI] [PubMed] [Google Scholar]
- 67.Maude SL, Dolai S, Delgado-Martin C, et al. Efficacy of JAK/STAT pathway inhibition in murine xenograft models of early T-cell precursor (ETP) acute lymphoblastic leukemia. Blood. 2015;125(11):1759–1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reth M, Nielsen P. Signaling circuits in early B-cell development. Adv Immunol. 2014; 122:129–175. [DOI] [PubMed] [Google Scholar]