

Abstract
The PML/RARA fusion protein occurs as a result of the t(15;17) translocation in the acute promyelocytic leukemia subtype of human acute myeloid leukemia. Gain of chromosome 8 is the most common chromosomal gain in human acute myeloid leukemia, including acute promyelocytic leukemia. We previously demonstrated that gain of chromosome 8-containing MYC is of central importance in trisomy 8, but the role of the nearby TRIB1 gene has not been experimentally addressed in this context. We have now tested the hypothesis that both MYC and TRIB1 have functional roles underlying leukemogenesis of trisomy 8 by using retroviral vectors to express MYC and TRIB1 in wild-type bone marrow and in marrow that expressed a PML/RARA transgene. Interestingly, although MYC and TRIB1 readily co-operated in leukemogenesis for wild-type bone marrow, TRIB1 provided no selective advantage to cells expressing PML/RARA. We hypothesized that this lack of co-operation between PML/RARA and TRIB1 reflected a common pathway for their effect: both proteins targeting the myeloid transcription factor C/EBPα. In support of this idea, TRIB1 expression abrogated the all-trans retinoic acid response of acute promyelocytic leukemia cells in vitro and in vivo. Our data delineate the common and redundant inhibitory effects of TRIB1 and PML/RARA on C/EBPα providing a potential explanation for the lack of selection of TRIB1 in human acute promyelocytic leukemia, and highlighting the key role of C/EBPs in acute promyelocytic leukemia pathogenesis and therapeutic response. In addition, the co-operativity we observed between MYC and TRIB1 in the absence of PML/RARA show that, outside of acute promyelocytic leukemia, gain of both genes may drive selection for trisomy 8.
Introduction
Acute myeloid leukemia (AML) is the most common leukemia subtype in adults representing 80% of cases for a worldwide incidence of 3.8/100,000 cases per year.1 The relapse risk for AML remains unacceptably high and relapse is the most common cause of death. Multiple courses of chemotherapy including combinations of anthracycline and cytarabine remain the mainstay of treatment but a ceiling of benefit has been reached and toxicity is significant. One particular exception is patients with acute promyelocytic leukemia (APL), a distinctive subtype representing 5%–15% of AML cases.2 APL patients typically express the PML/RARA fusion protein as a result of a t(15;17)(q22;q12) translocation, which renders cells exquisitely sensitive to all-trans retinoic acid (ATRA) leading to remission in the majority of cases.3 APL is characterized by clonal proliferation of myeloid blasts that have lost their differentiation capacity, and ATRA is thought to bypass this maturation block by relieving the repressive properties of PML/RARA on myeloid differentiation genes.4
Alteration of transcription factors often seen in AMLs such as RUNX1, GATA or CEBPA are commonly associated with mutations in tyrosine kinase receptors or other important signaling molecules, such as RAS or FLT3. Recent studies and the development of whole-genome sequencing technologies have revealed a complex process of leukemic transformation.5,6 These studies highlight the importance of transcriptional regulators controlling gene expression and differentiation, mutations affecting cell signaling and, most recently, mutations affecting epigenetic modifiers as co-operating factors in leukemic disease.
It therefore appears that the path to cellular transformation can be a complex and multistep-process. In APL, secondary karyotypic lesions are often seen, with trisomy 8 being the most common. Trisomy 8 is also the most common unbalanced gain in AML in general.7,8 Interestingly, the segment that is often gained carries the well-known MYC proto-oncogene.9 MYC encodes a transcription factor controlling expression of downstream targets such as cyclins, thereby promoting proliferation, but is also able to limit cellular differentiation, including via deregulation of the master regulator of myeloid differentiation C/EBPα.10 Importantly, MYC and C/EBPα expression require tight regulation to maintain myeloid and stem cell homeostasis.11 Previous analyses in our laboratory have shown that cells characterized by a gain of MYC through trisomy 8 display approximately 45% higher MYC RNA levels. Using a PML/RARA transgenic model, we also showed that MYC overexpression both accelerated the development of leukemia and impaired myeloid cell maturation, and that gain of MYC underlines the recurrent trisomy of this gene commonly seen in APL.9
Interestingly, TRIB1 is located contiguously to MYC on chromosome 8, and is thus expected to be duplicated in the chromosomal gain containing the MYC fragment. Not surprisingly, overexpression of TRIB1 has been found in several AML patients12 and TRIB proteins have been implicated in AML pathogenesis.13 Initially the Tribbles gene was identified in drosophila (dTribbles) and mammalian TRIB genes are comprised of three human homologs: TRIB1, TRIB2 and TRIB3.14 Supporting a role for TRIB1 in disease initiation, murine recipients of hematopoietic stem cells transduced with TRIB1 or TRIB2, but not TRIB3 developed AML and mediated COP1 ubiquitin ligase-dependent C/EBPα degradation.15–17 This TRIB-mediated degradation of C/EBPα was critical for TRIB-induced AML, and possibly necessary for the maturation block seen in AMLs.
In APL, the PML/RARA fusion protein retains the DNA binding domain of the endogenous RARA, therefore acting as a chimeric transcription factor.18 By dimerizing and associating with an altered set of co-factors the DNA binding specificity and repressive ability of the fusion protein is expanded,19 a characteristic that is believed to play a key role in the leukemogenic process. PML/RARA has been shown to interact with key transcription factors of the granulocytic differentiation program, such as PU.120 and C/EBPα downregulation has been observed in PML/RARA expressing cells,21,22 both events possibly participating in the transformation process. Interestingly, TRIB1 has been shown to co-localize with RARA/RXR leading to negative regulation of the transcriptional activity of the complex,23 although the precise mechanism mediating this inhibition is still unclear.
Given previous studies and clinical data implicating PML/RARA, MYC and TRIB1 in AML and APL leukemogenesis, we aimed to investigate the co-operation of these oncogenes in the development of leukemia. Using a retroviral bone marrow (BM) transduction and BM transplantation (BMT) approach to over-express MYC, TRIB1, or both in wild type (Wt) or PML/RARA BM from transgenic animals, we characterized the resultant neoplasms arising in the different groups. We found that MYC and TRIB1 cooperated in AML, but that MYC alone was able to drive APL development in the presence of PML/RARA. These data indicate that PML/RARA and TRIB1 could share redundant functions. We showed that TRIB1 (and TRIB2, but not a mutant of TRIB1) prevents differentiation of PML/RARA-expressing cells in the presence of ATRA, which normally derepresses the fusion protein to allow terminal maturation. We also showed that TRIB1 overexpression leads to sustained decreased C/EBPα protein levels in these cells, providing an explanation for the inability of these cells to respond to ATRA. Indeed, leukemias generated in secondary recipient animals transplanted with PML/RARA+MYC+TRIB1+ leukemia were unresponsive to ATRA, in contrast to the ATRA responsiveness of PML/RARA+MYC+ leukemia.
Methods
Expression constructs and retrovirus production
The HA-tagged human MYC cDNA24 was subcloned into MSCV-IRES-mCherry vector. Mouse Trib1 cDNA25 was subcloned into MSCV-IRES-GFP (MigR1) vector. Mutant Trib1 (ΔC4) expressed in MigR1 was previously published.26 Mouse Trib2 cDNA was subcloned into MSCV-IRES-GFP (MigR1) and MSCV-IRES-NGFR (NGFR).15,17 For mouse BM transduction experiments, BOSC23 packaging cell line was transfected with pCL-Eco and retroviral expression plasmids, as previously described.9 For NB4 transduction experiments, HEK293T packaging cell line was transfected with retroviral (pCGP, VSV-G) packaging vectors and retroviral expression plasmids. Viral supernatants (sups) were harvested at 24–48 h post transfection.
Cell culture and transduction
NB4 cells were cultured in RPMI supplemented with 10% fetal bovine serum (FBS). Cells were transduced by spinoculation with virus and 4 μg/mL Polybrene at 1290g for 90 mins at room temperature (RT). Transduced cells were sorted by flow cytometry 48 h post transduction for GFP expression (MigR1, TRIB1, ΔC4) or using anti-biotin beads (NGFR, TRIB2). Sorted cells were plated in presence of 1 uM ATRA at a density of 0.05×106 cells/mL.
Bone marrow harvest, retroviral transduction and transplantation
Donor animals (6–12 weeks old) were injected intraperitoneally with 5-Fluorouracil (5 FU, 150 mg/kg animal) to enrich for hemopoietic stem and progenitor cells and push them into cycle for the facilitation of retrovirus transduction. Leg and pelvic bones were harvested and 1×106 white blood cells/mL plated into pre-stimulation media (Myelocult M5300 Stemcell Technologies, 15% FBS, 10% of IL-3 and IL-6 conditioned medium, 0.4 mM of L-Glutamine and 10 ng/mL of murine recombinant SCF). Two spinoculations were performed (at 4 h and 24 h after harvest), and cells were injected retro-orbitally into lethally-irradiated 6–16-week old recipients (3×105−1×106 cells/mouse).
Cytomorphology
Cytospins were prepared by harvesting 25,000 cells and slides were stained with the Kwik-Diff staining kit (Thermo Scientific) as per the manufacturer’s instructions. Chromatin condensation and granularity was used to define differentiation on a Leica DM2000 and photographs taken on an Olympus DP70. A sample of slides were blinded and reviewed by Dr Mike Leech, Consultant Hematologist with expertise in diagnostic morphology and there was 100% correlation with our findings. Paraffin embedded sections were stained with hematoxylin & eosin (H&E). Photographs were taken on a Nikon Eclipse 80i microscope with a Nikon Digital Sight camera using NIS-Elements F2.30 or F4.30 software at a resolution of 2560 Å~ 1920.
Flow cytometry
A total of 10,000–50,000 cells were assessed by staining with CD45.1, CD45.2, Gr1 (Ly6G), and c-Kit on a LSRFortessa (BD Biosciences) or with CD15 (MMA) and CD11c (3.9) (eBioscience) on a FACSCanto II (BD Biosciences). Dead cells were excluded by DAPI staining (Sigma). Analysis was performed on FlowJo (Treestar).
RNA extraction and real time q-PCR
Total RNA was extracted using RNeasy Mini Kit (QIAGEN) and reverse transcribed with the High Capacity cDNA Reverse Transcription Kit (Life Technologies). QPCR was performed using Fast SYBR® Green Master Mix (Life Technologies) on a 7900HT Fast Real-Time PCR System (Life Technologies). hENOX2, hABL and hβ-2 microglobulin were used as internal controls and averaged; relative mRNA levels were calculated using the 2-δδCT method, and absolute mRNA levels calculated using the 2-δCT method. Experiments were performed on technical triplicates and biological duplicate samples. Details of primers are available in the Online Supplementary Appendix.
Immunoblotting
Lysates from cultured NB4 cells (50,000–150,000) +/− ATRA were prepared by direct lysis in 2X SDS sample buffer. Antibodies used were: anti-C/EBPα (Santa Cruz sc-61), anti-Actin (Sigma Aldrich A5441).
Animals
Mice were bred and maintained at University of California San Francisco (UCSF) (USA) and Cardiff University (UK) and were cared for in accordance with Institutional Animal Care and Use Committee guidelines. The recipients FVB/n CD45.1/45.2 mice were generated by crossing FVB/n (CD45.1) to FVB/n CD45.2 congenic animals. hMRP8-PML/RARA mice have been previously described.27 Fisher’s exact test was used to assess whether Wt and PML/RARA leukemias were equivalent in their likelihood to express TRIB1.
In vivo ATRA treatment of recipients transplanted with PML/RARA leukemic cells
After one passage into recipient animals, resurrected cryopreserved leukemic cells (2×106) of the PML/RARA−MYC+ and PML/RARA−MYC+TRIB1+ phenotype were intravenously injected into unirradiated or sublethally irradiated (500 cGy) FVB/n mice (n=4–5 per group in 2 independent experiments). Ten days post injection, 10 mg 21-day release ATRA or placebo pellets (Innovative Research of America) were implanted into the dorsal neck scruff. Mice were sacrificed when moribund or upon veterinary advice. Statistical significance was calculated using a log rank test.
Bioinformatics analysis
GSE6891 and GSE12662 datasets reflect results on the Affymetrix Human Genome U133 Plus 2.0 Array. Three TRIB1 probes are present: 202241_at, 239818_x_at, 235641_at. Signal strength was more than 10-fold higher for 202241_at as compared to the other probes and the results presented reflect this probe. P values were obtained using Microsoft EXCEL t-test (two-tailed, unequal variance). The MYC gene is represented in GSE6891 by probe 202431_s_at. Correlations of MYC and TRIB1 expression were assessed using Microsoft EXCEL, including calculation of R2 value.
Results and Discussion
In order to investigate if MYC and TRIB1 function cooperatively in the context of myeloid leukemia, we utilized a retroviral transduction system to concurrently over-express MYC and Trib1 in 5-FU-treated murine BM from Wt or PML/RARA transgenic animals. The PML/RARA mouse model expresses a human PML/RARA cDNA from the hMRP8 promoter cassette, which drives transgene expression in myeloid cells.27 Three groups of donor cells (PML/RARA, Wt, or a combination of Wt + PML/RARA mixed in a 1:1 ratio) were transduced and transplanted into lethally-irradiated recipients (n=5 PML/RARA donor group, n=4 Wt donor group, n=15 combined donor group) (Figure 1A). In the combined donor experiments, injected cells were comprised of all possible combinations, including cells that lacked oncogene integration. With this approach, we anticipated that the cells expressing all three oncogenes (PML/RARA, MYC and TRIB1) would be most able to initiate leukemia. At disease manifestation (or on day 260–272 at experiment termination), BM from these animals was harvested and analyzed by flow cytometry to look at chimerism and characterization of the arising neoplasm. Every leukemia which arose from the Wt donor BM expressed both MYC and TRIB1, confirming that these oncogenes co-operate and co-express to drive AML transformation in this transplant model (Table 1, red color-coded in column 2). In contrast, leukemias originating from PML/RARA-expressing donor BM displayed a different phenotype, with 5 leukemias expressing MYC only (Table 1, green color-coded in column 2), and only 2 leukemias expressing both MYC and TRIB1 (Table 1, blue color-coded in column 2). With regard to the non-leukemic mice in our study, at the time of experiment termination, 4 of 24 recipient mice had failed to develop disease and 3 others were identified as bearing recipient-derived malignancies likely due to irradiation. In the presence of PML/RARA, the dominant leukemic phenotype was MYC co-expression alone, as opposed to the Wt setting where MYC and TRIB1 were always co-expressed to drive leukemia. It is highly unlikely that this result was due to chance. If the selective pressure to express TRIB1 had been equivalent in Wt and PML/RARA-expressing BM, then we would have expected equal proportions of Wt and PML/RARA leukemias to express TRIB1. In fact, the observed result of TRIB1 expression in 10 of 10 Wt leukemias but only 2 of 7 PML/RARA leukemias indicates that selection for TRIB1 was indeed abrogated by the presence of PML/RARA (P=0.003). Of note, this was not a result of different TRIB1 levels in the Wt and PML/RARA leukemias as similar transduction levels were obtained in the groups (data not shown). To our surprise, transduced PML/RARA marrows did not outcompete transduced wild-type marrows in the combined donor experiments, further supporting the idea that PML/RARA and TRIB1 did not co-operate in leukemia initiation. When looking at all the Wt or PML/RARA leukemias combined (Figure 1B), we do not observe statistically significant differences in terms of numbers or time to disease, although a wider range of latencies was observed for the Wt leukemias. The median latency to disease observed in Wt and PML/RARA leukemias was 140 and 153 days, respectively, suggesting that additional events are likely necessary to complete the transformation process, which could further explain why some recipients did not develop disease within the time-frame of the experiment. Morphologically, Wt and PML/RARA leukemias resembled each other and were characterized by the expansion of immature myeloid cells. Flow cytometry analysis and histology staining from a representative Wt−MYC+TRIB1+, PML/RARA−MYC+ and a PML/RARA−MYC+TRIB1+ leukemia are shown in Figure 1D, showing similar leukemic cell organ infiltration and morphology of the indicated leukemias. Flow cytometric immunophenotyping confirmed an immature myeloid character, with some variation in levels of expression of KIT and Gr1. Thus, MYC and TRIB1 co-expression were able to drive myeloid leukemia, similar to leukemias driven by PML/RARA and MYC co-expression.
Figure 1.
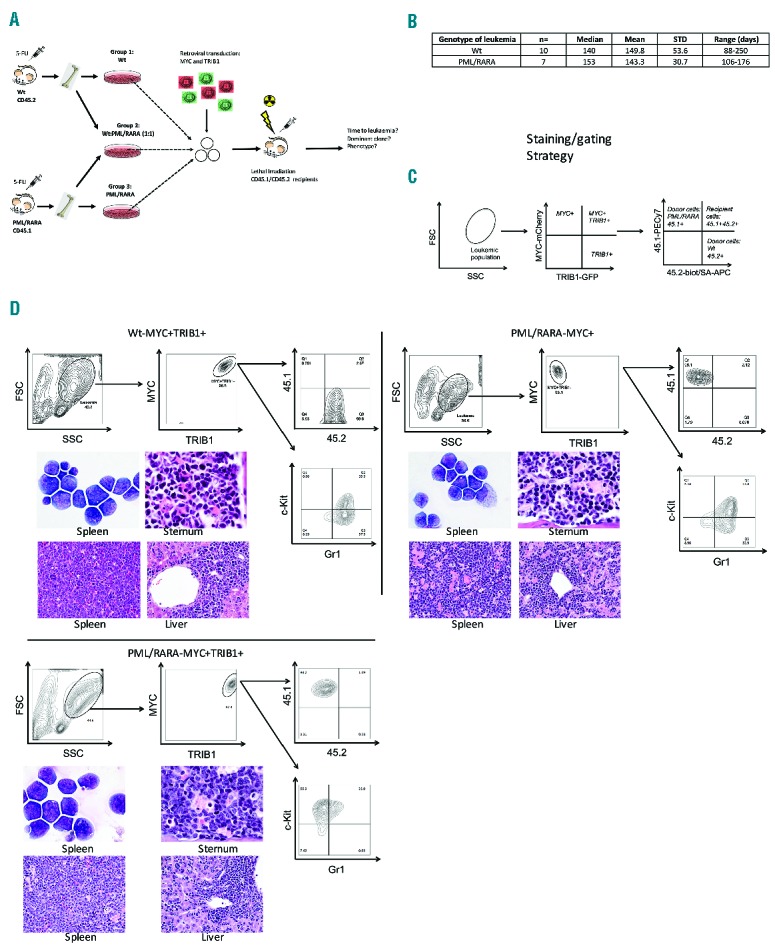
MYC and TRIB1 co-operate to initiate acute myeloid leukemia (AML), whereas MYC alone combined with PML/RARA is able to drive acute promyelocytic leukemia (APL). (A) Schematic representation of the in vivo experimental strategy used to assess the selective advantage of MYC and/or TRIB1 in transforming wild-type (Wt) or PML/RARA bone marrow (BM). Lethally-irradiated recipients were reconstituted with transduced bone marrow, animals were monitored for disease development and the arising neoplasm characterized. (B) Summary table combining all Wt leukemias and all PML/RARA leukemias. As seen, no differences were observed in the mean or median time to disease in AML or APL initiation. (C) Panels show the flow cytometry staining and gating strategy used. (D) Phenotypic analysis of resultant leukemia in representative examples of a Wt−MYC+TRIB1+, PML/RARA−MYC+ and PML/RARA−MYC+TRIB1+ leukemias. Panels show representative Wt−MYC+TRIB1+ (top left), PML/RARA−MYC+ (top right), and PML/RARA−MYC+TRIB1+ (bottom left) leukemias staining for CD45.1, CD45.2, Gr-1 and c-Kit through the indicated gate of parental population; cytomorphology of spleen, sternum, and liver from representative Wt−MYC+TRIB1+ (top left), PML/RARA−MYC+ (top right) and PML/RARA−MYC+TRIB1+ (bottom left) leukemias showing leukemic cell organ infiltration.
Table 1.
Summary table of the outcome of the in vivo leukemogenesis experiment, specifying the phenotype of the leukemia or cause of death in the first column, disease latency (time to death column), mean time to disease, and input (donor) bone marrow (BM) specified in the last column.
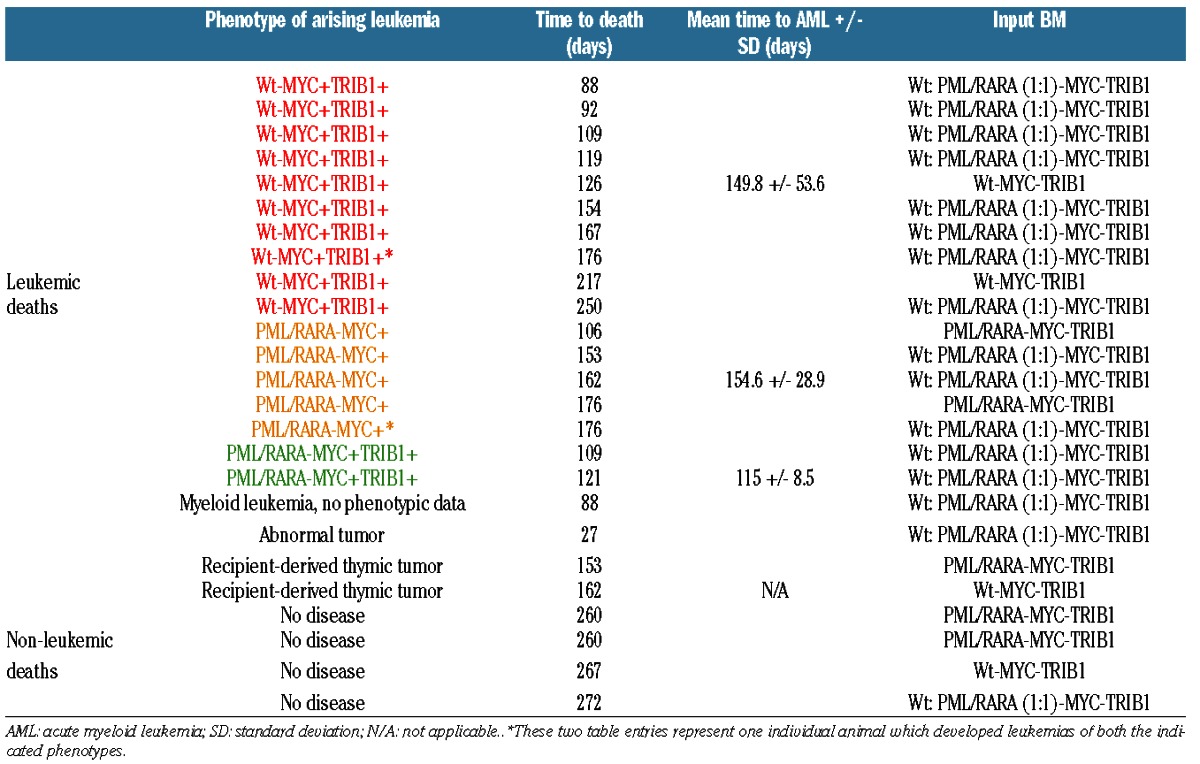
We hypothesized that if TRIB1 levels play a pathogenic role in some AMLs, but not in APL, that APL might show lower TRIB1 expression than other AMLs. Using a published dataset (GSE689128) we compared TRIB1 transcript levels in 21 APLs to those seen in 440 non-APL AMLs. On average, APLs expressed less than 50% the level of TRIB1 present in non-APL AMLs (P<0.001) (Figure 2A). In order to assess if this simply reflected the normal expression levels in promyelocytes as compared to less mature myeloid blasts, we examined TRIB1 levels in a published dataset (GSE1266229) that includes normal CD34 cells, normal promyelocytes and APLs. TRIB1 increases 5-fold with maturation from normal CD34 cells to normal promyelocytes, indicating that the low levels in APL as compared to other non-APL AMLs is not simply a reflection of cell maturation. Furthermore, in this dataset, APLs expressed much lower levels of TRIB1 than do normal promyelocytes (>4 fold decrease; P<0.003) (Figure 2B). While recognizing that mRNA levels may not reflect protein levels, these results are compatible with the idea that TRIB1 does not contribute to the pathogenesis of APL. We also examined the published dataset of 461 AMLs (including 21 APLs) to evaluate whether there is a correlation between increased TRIB1 transcripts and increased MYC transcripts, which would suggest co-selection for expression. Although our data in mice show that MYC and TRIB1 can co-operate in AML, in the human dataset there was no significant correlation between TRIB1 levels and MYC levels in AML as a whole, as well as no significant correlation in the normal karyotype (n=187), +8 (n=20), and APL (n=21) subsets (R2 <0.5 for these four comparisons) (Figure 2C).
Figure 2.
TRIB1 expression is decreased in human acute promyelocytic leukemia (APL) and does not correlate with MYC expression. (A) TRIB1 transcript levels in 21 cases of human APL are compared to levels in 440 cases of human non-APL acute myeloid leukemia (AML). Midlines represent median values, boxes represent 25th–75th percentiles, whiskers represent range of all values. TRIB1 levels are lower in APL than in non-APL AML (P<0.001). (Dataset reported values in log2 scale of GSE6891 have been transformed to linear values for clarity.) (B) TRIB1 values rise with maturation from human normal CD34 (n=5) to normal promyelocytes (n=5), but are low in human APL (n=14). Individual values are shown. [Intensity signal on arrays were scaled differently in the GSE6891 and GSE12662 datasets resulting in the different relative expression scales in (A) and (B)]. (C) Correlation plots of TRIB1 expression and MYC expression in GSE6891 for 461 AMLs, including APL, as well as for AMLs with isolated trisomy 8 (+8, n=20), AMLs with normal karyotype (NK, n=187), and APL alone (n=21). Trendlines are shown but R2 <0.5 for each of the comparisons. [Dataset log2 scale GSE6891 values were graphed for these comparisons; in contrast to (A), the values have not been linear transformed.]
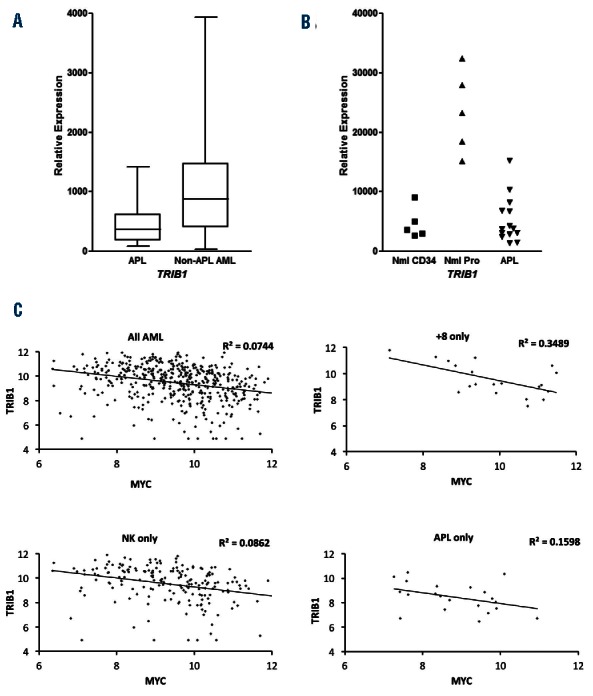
Our in vivo leukemogenesis experiments showed that TRIB1 overexpression does not co-operate with PML/RARA for disease initiation but rather appears to phenocopy the PML/RARA-induced disease in the context of MYC co-expression. These results raise the possibility that PML/RARA and TRIB1 share overlapping functions or have redundant functions in leukemic transformation, thereby explaining the lack of selective advantage for cells to express PML/RARA, MYC and TRIB1 concomitantly. C/EBPα is a key myeloid transcription factor shown to be a target of both PML/RARA and TRIB family members, potentially representing the common target of the two oncogenes in our model. To test this hypothesis, we used an established in vitro system in which PML/RARA-expressing APL cells are induced to terminally differentiate by exposure to all-trans retinoic acid (ATRA). As a master regulator of granulocyte maturation, activation of C/EBPα, C/EBPβ30 and its downstream targets is mandatory to bypass the maturation block in these cells, which ATRA can circumvent by directly targeting PML/RARA. Therefore, we hypothesized that over-expressing TRIB proteins in PML/RARA-expressing cells might abrogate the differentiating effect of ATRA due to C/EBPα inhibition or degradation. NB4 cells transduced with control, TRIB1 or TRIB1 mutant (ΔC4) unable to bind and degrade C/EBPα26 expressing retroviruses were cultured in presence of ATRA for six days to induce myeloid differentiation. NB4 cells differentiated after six days in ATRA, as seen by loss of nucleoli, maturation/condensation of the chromatin and granularity, and CD15 and CD11c surface marker expression, indicating that the maturation block had been bypassed following targeting of PML/RARA by ATRA (Figure 3A–C; MigR1 controls). TRIB1 overexpression was able to block ATRA-induced differentiation and this was lost upon overexpression of mutant TRIB1 (ΔC4) (Figure 3A–C). These data indicate that TRIB1 abrogates ATRA-induced terminal differentiation and suggest the mechanism is via C/EBP degradation.
Figure 3.
TRIB1 overexpression blocks the ATRA-mediated differentiation of NB4 cells. (A) NB4 cells were transduced with control MigR1, MigR1-TRIB1, and MigR1-ΔC4 (Trib1 mutant unable to degrade C/EBPα), sorted and treated with 1 μM ATRA for six days and cell morphology assessed by manual differential counts. (B) Representative cell morphology at day 0 and day 6. (C) Flow cytometry analysis of CD15 and CD11c expression in vehicle control (VC top) and ATRA treated transduced NB4 cells at day 6. (D) QPCR analysis of: exogenous Trib1 (top) and endogenous TRIB1 (bottom) in MigR1 and MigR1-TRIB1 transduced NB4 cells treated with ATRA at the indicated time points. Top graph depicts absolute gene expression relative to internal controls. Bottom graphs depict relative gene expression normalized to MigR1-control at day 0. (E) QPCR analysis of G-CSFR in MigR1, MigR1-TRIB1, and MigR1-ΔC4 transduced NB4 cells treated with ATRA at the indicated time points. Graphs depict relative gene expression normalized to MigR1-control at day 0. Significance determined by one-way ANOVA and Bonferroni post test. *P<0.05; **P<0.01. (F) Western blot analysis for C/EBPα in direct lysis samples prepared from NB4 cells transduced with control MigR1, MigR1-TRIB1, and MigR1-ΔC4 and treated with 1 μM ATRA for indicated time points. Actin was used as a loading control. h: hours.
Using primers specific for endogenous human TRIB1, we observed that ATRA treatment (which degrades PML/RARA) up-regulated TRIB1 in NB4 cells (Figure 3D). In combination with the finding (see above) that TRIB1 transcript levels are lower in human APL than they are in normal human promyelocytes, it appears possible PML/RARA may suppress TRIB1 expression. Although we have not investigated the mechanism for low endogenous TRIB1 levels in APL cells, PML/RARA suppression of TRIB1 may be permissive for increased C/EBP activity in response to ATRA and therefore permissive for maturation beyond the promyelocyte stage. NB4 cell transduction with TRIB1 expressing vectors resulted in significant expression of exogenous TRIB1 (detected using mouse TRIB1 specific primers) and, of note, there was no increase in endogenous TRIB1 in response to ATRA when exogenous TRIB1 was present (Figure 3D).
To further investigate TRIB-mediated block of ATRA-induced differentiation via C/EBPα, we assessed the gene expression of a C/EBPα-dependent target important for granulocytic maturation, G-CSF receptor (G-CSFR).31 G-CSFR gene expression levels increased over time in the control groups, as expected, which was blocked upon expression of TRIB1 and lost upon overexpression of mutant TRIB1 (ΔC4) (Figure 3E), demonstrating that expression of TRIB1 protein inhibits a C/EBPα-dependent target gene important for driving the myeloid maturation program. To confirm that the inhibitory phenotypic and transcriptional activities of TRIB proteins are mediated via C/EBP protein inhibition, we assessed C/EBPα protein levels. Expression of TRIB1 attenuated ATRA-induced C/EBPα protein expression and this was lost upon overexpression of mutant TRIB1 (ΔC4) (Figure 3F). We also assessed C/EBPβ protein levels and found that TRIB1 attenuated ATRA-induced C/EBPβ protein levels but not with the same impact as on C/EBPα protein expression (Online Supplementary Figure S1A). Similarly, the overexpression of TRIB2 in NB4 cells blocked ATRA-induced differentiation assessed by morphology and G-CSFR gene expression, and abrogated the ATRA-mediated induction of C/EBPα, C/EBPβ and PU.1 transcription factors (Online Supplementary Figure S1B–D). These data provide strong evidence that TRIB1 or TRIB2 overexpression prevents ATRA-induced differentiation of APL cells via the inhibition of C/EBPα.
To further study the clinical relevance of TRIB1 expression in APL, we investigated ATRA (or placebo) response in vivo in recipient animals inoculated with PML/RARA−MYC+ compared to PML/RARA−MYC+TRIB1+ leukemia. Leukemias generated in our initial leukemogenesis experiment (Figure 1) were tested (see Figure 4A for details of the experimental strategy) in two independent experiments. Results of these experiments were concordant and the combined results are shown (Figure 4B). Significantly, the response of the leukemias to ATRA treatment was markedly different. ATRA treatment significantly delays disease progression and extends median survival from 31 to 53 days (P<0.0001) in recipients of PML/RARA−MYC+ leukemias compared to placebo treated controls, as expected. However, ATRA did not extend survival in mice injected with PML/RAR−MYC+TRIB1+ leukemia. These results show that TRIB1 expression abrogates ATRA response in vivo confirming our in vitro findings. Our data indicate that TRIB1 expression provides critical oncogenic functions preventing APL cells from responding to ATRA therapy, presumably via repression of C/EBPα.
Figure 4.
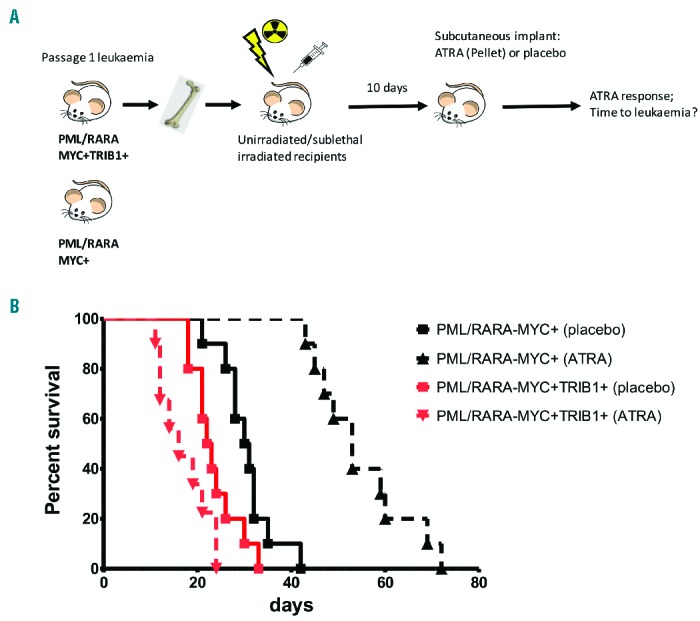
TRIB1 overexpression abrogates ATRA response in vivo. (A) Schematic representation of the in vivo experimental strategy to assess ATRA response in PML/RARA+MYC+ versus PML/RARA+MYC+TRIB1+ leukemias (n=10 each group). Recipient animals were either unirradiated or sublethally-irradiated and transplanted with PML/RARA−MYC+ or PML/RARA−MYC+TRIB1+ leukemias. (B) Kaplan-Meier survival curves of transplanted animals treated with placebo or ATRA pellet implantation. ATRA is able to extend survival of PML/RARA+MYC+ but not PML/RARA+MYC+TRIB1+ secondary transplanted mice. Using the log rank (Mantel-Cox) test for each group, PML/RARA−MYC+: placebo versus ATRA P<0.0001 (longer survival with ATRA; PML/RARA−MYC+TRIB1+: placebo versus ATRA; P<0.05 (shorter survival with ATRA).
To conclude, in this study we aimed to investigate the co-operation of MYC and TRIB1 in the pathogenesis of AML and APL, two genes located on a contiguous fragment of chromosome 8 in humans. Given the common gain of the MYC/TRIB1-containing segment in AMLs, we hypothesized that TRIB1 could also play an important role in leukemic transformation, including for APL. Using a retroviral transduction model of in vivo leukemogenesis, we found that both TRIB1 and MYC oncogenes co-operate to initiate AML, but that in most cases, MYC is sufficient to accelerate APL leukemogenesis. C/EBPα downregulation is an overlapping function of PML/RARA and TRIB1, and this common feature explains the lack of selective leukemic outgrowth for clones expressing both these oncogenes. The responsiveness to ATRA treatment was severely impaired by the expression of TRIB1, a phenotype observed both in vitro and in vivo. Overall, our results provide critical information about the role of TRIB1 in leukemogenesis and responsiveness to ATRA treatment. Our data support a role for PML/RARA in altering C/EBPα and C/EBPβ in APL leukemogenesis and provide strong genetic evidence for a key role of C/EBP family members in myeloid leukemias beyond those with mutations in or methylation of the CEBPA gene.
Acknowledgments
We thank Dr. Huimin Geng for assistance with statistical analysis and Dr Mike Leech for morphology assessment.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/10/1228
Funding
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under Award Number R01CA095274 (to SCK), by the Howat Foundation, Children with Cancer UK and Bloodwise (LLR 13011) (to KK) and Bloodwise (to NO).
References
- 1.Estey E, Döhner H. Acute myeloid leukaemia. Lancet. 2006;368(9550):1894–1907. [DOI] [PubMed] [Google Scholar]
- 2.Douer D. The epidemiology of acute promyelocytic leukaemia. Best Pract Res Clin Haematol. 2003;16(3):357–367. [DOI] [PubMed] [Google Scholar]
- 3.Coombs CC, Tavakkoli M, Tallman MS. Acute promyelocytic leukemia: where did we start, where are we now, and the future. Blood Cancer J. 2015;5:e304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Arteaga MF, Mikesch J-H, Fung T-K, So CWE. Epigenetics in acute promyelocytic leukaemia pathogenesis and treatment response: a TRAnsition to targeted therapies. Br J Cancer. 2015;112(3):413–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cauchy P, James SR, Zacarias-Cabeza J, et al. Chronic FLT3-ITD Signaling in Acute Myeloid Leukemia Is Connected to a Specific Chromatin Signature. Cell Rep. 2015;12(5):821–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Welch JS, Ley TJ, Link DC, et al. The origin and evolution of mutations in acute myeloid leukemia. Cell. 2012; 150(2):264–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grimwade D, Walker H, Oliver F, et al. The importance of diagnostic cytogenetics on outcome in AML: analysis of 1,612 patients entered into the MRC AML 10 trial. The Medical Research Council Adult and Children’s Leukaemia Working Parties. Blood. 1998;92(7):2322–2333. [PubMed] [Google Scholar]
- 8.Grimwade D, Hills RK, Moorman AV, et al. Refinement of cytogenetic classification in acute myeloid leukemia: determination of prognostic significance of rare recurring chromosomal abnormalities among 5876 younger adult patients treated in the United Kingdom Medical Research Council trials. Blood. 2010;116(3):354–365. [DOI] [PubMed] [Google Scholar]
- 9.Jones L, Wei G, Sevcikova S, et al. Gain of MYC underlies recurrent trisomy of the MYC chromosome in acute promyelocytic leukemia. J Exp Med. 2010;207(12):2581–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoffman B, Amanullah A, Shafarenko M, Liebermann DA. The proto-oncogene c-myc in hematopoietic development and leukemogenesis. Oncogene. 2002; 21(21): 3414–3421. [DOI] [PubMed] [Google Scholar]
- 11.Ye M, Zhang H, Amabile G, et al. C/EBPa controls acquisition and maintenance of adult haematopoietic stem cell quiescence. Nat Cell Biol. 2013;15(4):385–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Röthlisberger B, Heizmann M, Bargetzi MJ, Huber AR. TRIB1 overexpression in acute myeloid leukemia. Cancer Genet Cytogenet. 2007;176(1):58–60. [DOI] [PubMed] [Google Scholar]
- 13.Liang KL, Rishi L, Keeshan K. Tribbles in acute leukemia. Blood. 2013;121(21):4265–4270 [DOI] [PubMed] [Google Scholar]
- 14.Lohan F, Keeshan K. The functionally diverse roles of tribbles. Biochem Soc Trans. 2013;41(4):1096–1100. [DOI] [PubMed] [Google Scholar]
- 15.Keeshan K, He Y, Wouters BJ, et al. Tribbles homolog 2 inactivates C/EBPalpha and causes acute myelogenous leukemia. Cancer Cell. 2006;10(5):401–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dedhia PH, Keeshan K, Uljon S, et al. Differential ability of Tribbles family members to promote degradation of C/EBPalpha and induce acute myelogenous leukemia. Blood. 2010;116(8):1321–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Keeshan K, Bailis W, Dedhia PH, et al. Transformation by Tribbles homolog 2 (Trib2) requires both the Trib2 kinase domain and COP1 binding. Blood. 2010; 116(23):4948–4957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Melnick A, Licht JD. Deconstructing a disease: RARalpha, its fusion partners, and their roles in the pathogenesis of acute promyelocytic leukemia. Blood. 1999; 93(10):3167–3215. [PubMed] [Google Scholar]
- 19.Saeed S, Logie C, Stunnenberg HG, Martens JHA. Genome-wide functions of PML-RARα in acute promyelocytic leukaemia. Br J Cancer. 2011;104(4):554–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang K, Wang P, Shi J, et al. PML/RARalpha targets promoter regions containing PU.1 consensus and RARE half sites in acute promyelocytic leukemia. Cancer Cell. 2010;17(2):186–197. [DOI] [PubMed] [Google Scholar]
- 21.Guibal FC, Alberich-Jorda M, Hirai H, et al. Identification of a myeloid committed progenitor as the cancer-initiating cell in acute promyelocytic leukemia. Blood. 2009; 114(27):5415–5425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gaillard C, Tokuyasu TA, Rosen G, et al. Transcription and methylation analyses of preleukemic promyelocytes indicate a dual role for PML/RARA in leukemia initiation. Haematologica. 2015;100(8):1064–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Imajo M, Nishida E. Human Tribbles homolog 1 functions as a negative regulator of retinoic acid receptor. Genes Cells. 2010; 15(10):1089–1097. [DOI] [PubMed] [Google Scholar]
- 24.Hemann MT, Bric A, Teruya-Feldstein J, et al. Evasion of the p53 tumour surveillance network by tumour-derived MYC mutants. Nature. 2005;436(7052):807–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jin G, Yamazaki Y, Takuwa M, et al. Trib1 and Evi1 cooperate with Hoxa and Meis1 in myeloid leukemogenesis. Blood. 2007; 109(9):3998–4005. [DOI] [PubMed] [Google Scholar]
- 26.Yokoyama T, Kanno Y, Yamazaki Y, Takahara T, Miyata S, Nakamura T. Trib1 links the MEK1/ERK pathway in myeloid leukemogenesis. Blood. 2010; 116(15):2768–2775. [DOI] [PubMed] [Google Scholar]
- 27.Brown D, Kogan S, Lagasse E, et al. A PMLRARalpha transgene initiates murine acute promyelocytic leukemia. Proc Natl Acad Sci USA. 1997;94(6):2551–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Verhaak RGW, Wouters BJ, Erpelinck CAJ, et al. Prediction of molecular subtypes in acute myeloid leukemia based on gene expression profiling. Haematologica. 2009; 94(1):131–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Payton JE, Grieselhuber NR, Chang L-W, et al. High throughput digital quantification of mRNA abundance in primary human acute myeloid leukemia samples. J Clin Invest. 2009;119(6):1714–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Duprez E, Wagner K, Koch H, Tenen DG. C/EBPbeta: a major PML-RARA-responsive gene in retinoic acid-induced differentiation of APL cells. EMBO J. 2003; 22(21):5806–5816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith LT, Hohaus S, Gonzalez DA, Dziennis SE, Tenen DG. PU.1 (Spi-1) and C/EBP alpha regulate the granulocyte colony-stimulating factor receptor promoter in myeloid cells. Blood. 1996;88(4):1234–1247. [PubMed] [Google Scholar]