

The nuclear factor of kappa light polypeptide gene enhancer in B-cells 1 (NF-κB1) is a master regulator of immune and inflammatory responses.1,2 NF-κB1 belongs to the NF-κB/Rel family of transcription factors that consists of five members in humans: NF-κB1 (p105/p50), NF-κB2 (p100/p52), RelA, c-Rel, and RelB. The p105 and p100 precursors are proteolytically processed by the proteasome to generate the shorter p50 and p52 isoforms. Homo- and heterodimers are formed by p50, p52 and the Rel proteins. Unstimulated, these dimeric complexes are sequestered in the cytoplasm by inhibitory IκB proteins in an inactive state. Upon stimulation, the phosphorylation and subsequent degradation of IκB proteins is rapidly triggered, releasing the NF-κB/Rel complexes to enter the nucleus where they bind to DNA at κB sites and activate or repress the expression of their target genes.3
Recently, heterozygous mutations affecting the NFKB1 gene were identified in three human families. Haploinsufficiency of NF-κB1 was causative for combined variable immunodeficiency (CVID) characterized by recurrent infections due to immunoglobulin deficiency (pan-IgG, IgA and/or IgM).4
We report here on two pediatric patients from unrelated families with two novel NFKB1 gene mutations identified by whole-exome sequencing (Figure 1A,B). Both patients had early onset of disease during their teenage years and presented in addition to hypogammaglobulinemia or selective IgA deficiency with a striking lack of specific antibodies (clinical characteristics are summarized in Table 1 and Online Supplementary Table S1).
Figure 1.
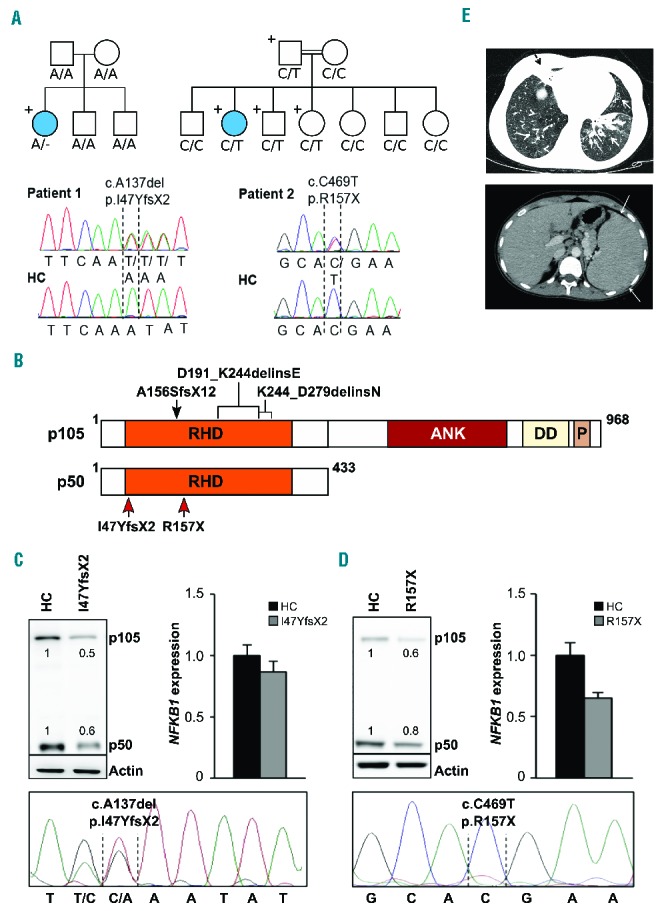
Two novel heterozygous NFKB1 mutations detected in two families decrease the protein levels of p105 and p50. (A) Upper left: Whole exome sequencing identified a heterozygous NFKB1 mutation (A/−) in patient 1. The patient is the only carrier of the mutation in the family pedigree (indicated by “+”) and the only diseased family member (indicated by a filled circle). Lower left: Capillary sequencing using genomic DNA confirmed an NFKB1 frameshift mutation (c.A137del, p.I47YfsX2) in patient 1. Representative chromatograms of patient 1 and a healthy control (HC) are shown. Upper right: Patient 2 descended from consanguineous parents and harbors an inherited heterozygous NFKB1 mutation (C/T). The patient is the only diseased family member. The father and two siblings carry the same mutation but are not affected. Sanger sequencing of NFKB1 confirmed the heterozygous missense mutation (c.C469T, p.R157X) in patient 2. Representative chromatograms are shown. (B) Schematic drawing of the proteins p105 and p50 and their domains which are both encoded by the NFKB1 gene. The mutations in the Rel homology domain (RHD) identified in the two patients (red arrows) lead to early truncation of both proteins. Previously reported heterozygous germline mutations associated with CVID are indicated on top (black arrow and brackets). ANK, ankyrin repeats; DD, death domain; P, PEST domain enriched for proline (P), glutamic acid (E), serine (S), and threonine (T) residues. (C, D) Expression of p105 and p50 proteins is decreased in the affected patients. (C) Primary T cells of patient 1 and healthy controls were activated by phytohemagglutinin in the presence of interleukin-2. Protein and RNA extracts were prepared. Western blot analysis was carried out employing a specific p105/p50 antibody using β-actin as a loading control (left panel). NFKB1 mRNA expression was measured by real-time polymerase chain reaction (right panel). The fold-change in cells of the patient compared to a representative healthy control is shown, GAPDH and β-actin expression were used as internal standards. Mean values of representative experiments performed in triplicates and corresponding SDs are shown. Sanger sequencing using reverse transcribed mRNA of the patient demonstrates the presence of mutated NFKB1 transcripts (lower panel). (D) Epstein-Barr virus-transformed B cells of patient 2 were used for protein and RNA extraction. Analysis of NFKB1 protein and RNA expression was carried out as described in (C). Capillary sequencing of cDNA from patient 2 failed to detect the NFKB1 mutation indicating that the mutation leads to mRNA instability (lower panel). (E) Upper panel: axial high resolution chest computer tomography image of patient 2 at the level of lung bases demonstrating multiple areas of bronchiectasis (white arrows) and consolidation with an atelectatic component surrounding bronchiectases in the right middle lobe (black dashed arrow). Mosaic pattern of perfusion of the lung parenchyma is noted, with multiple areas of low attenuation in the right low lobe (arrowheads). Lower panel: axial computer tomography image of patient 2 at the level of the upper abdomen demonstrating the enlarged spleen.
Table 1.
Clinical and laboratory characteristics of two patients with novel NFKB1 mutations.
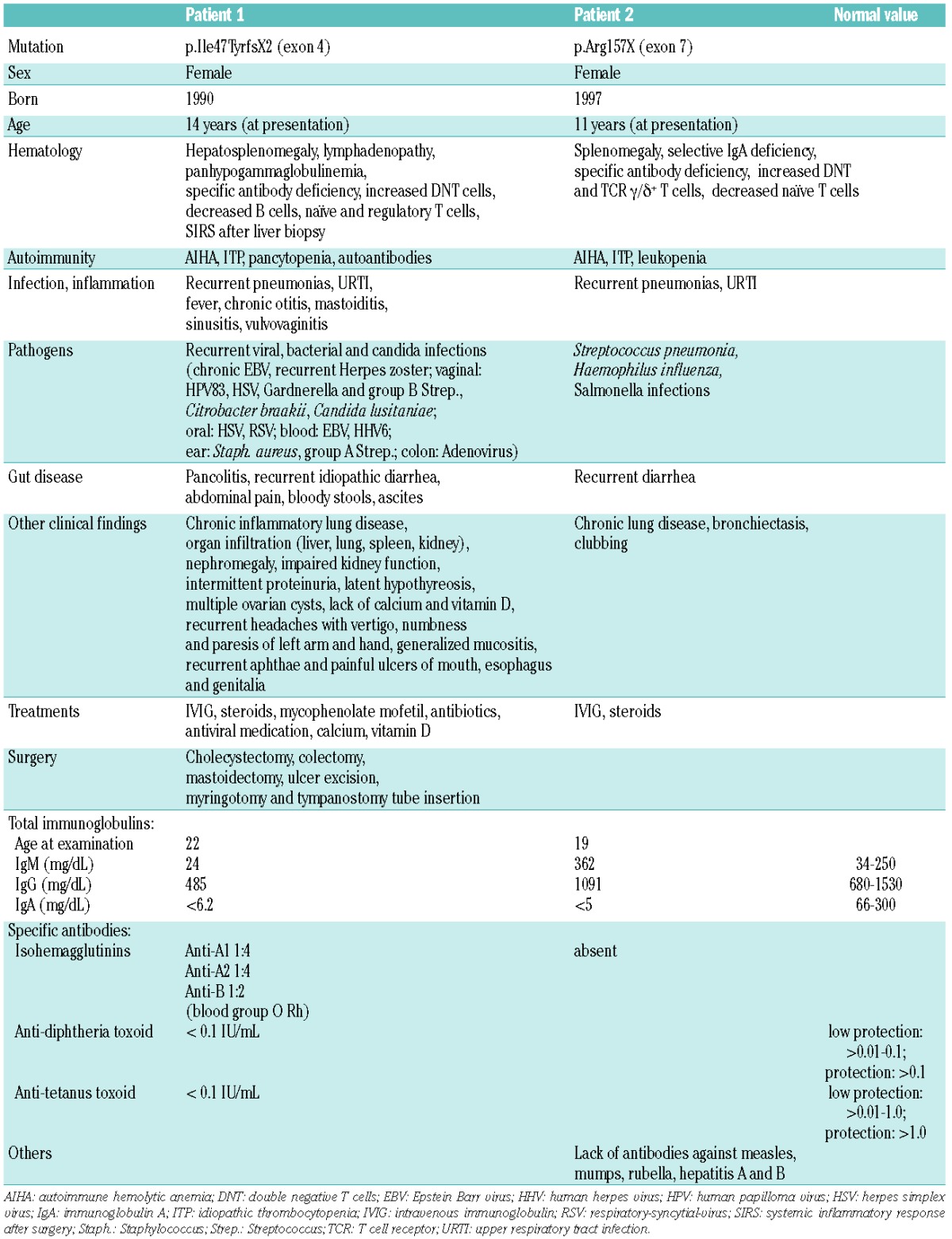
Patient 1 is a now 26-year old female who first presented with recurrent autoimmune hemolytic anemia at the age of 14. Hypogammaglobulinemia (IgG2 subclass deficiency), deficient production of specific antibodies, decreased class-switched and memory B cells, naïve CD4-positive and regulatory T cells, increased activated and double-negative T cells (DNT cells, CD4−CD8− TCRα/β+), autoimmune phenomena (hemolytic anemia, thrombocytopenia and leukopenia), lymphadenopathy, and hepatosplenomegaly were observed. She developed chronic lung disease with bronchiectasis, frequent respiratory tract infections and pneumonia. Infections with viral, bacterial and fungal pathogens were frequent. She suffered from intractable abdominal pain and bloody diarrhea without evidence of infection. After a liver biopsy she developed pancolitis with subsequent sepsis and multi-organ failure and was successfully resuscitated. The patient is being treated with intravenous immunoglobulin. Steroids were intermittently given to reduce pulmonary infiltrates with partial response. Mycophenolate mofetil stabilized blood counts, but pulmonary symptoms and infections remained.
To identify the genetic cause of disease whole exome sequencing was performed for the patient and her family (Online Supplementary Methods, Online Supplementary Table S2). A heterozygous de novo NFKB1 frameshift mutation was detected (Figure 1A). The NFKB1 gene encodes two proteins: p50 and its precursor p105. The mutation led to early protein truncation (I47YfsX2) and decreased expression of both protein isoforms (Figure 1B,C). RNA expression was slightly reduced.
Patient 2 is a 19-year old female from a consanguineous family (Figure 1A), who presented at the age of 11 with recurrent respiratory infections leading to chronic lung disease with bronchiectasis. She suffered from lymphadenopathy, splenomegaly, recurrent autoimmune hemolytic anemia and diarrhea. Whole exome sequencing revealed an inherited heterozygous nonsense NFKB1 mutation (R157X) leading to early protein truncation and decreased p50/p105 expression (Figure 1B,D). NFKB1 mRNA levels were low suggesting that the mutation may lead to mRNA instability and nonsense-mediated decay. Sequencing of reverse transcribed mRNA (Figure 1C,D, lower panels) indicated the absence of mutated mRNA in cells of patient 2, but not patient 1. Patient 2 had a selective IgA deficiency; her IgG and IgM levels were within the normal ranges. Similar to patient 1, she lacked specific antibodies including isohemagglutinins. Immunophenotyping demonstrated normal absolute numbers of T, B and natural killer cells, but decreased numbers of naïve T cells and increased levels of activated, DNT and TCRγ/δ+ T cells. Her splenomegaly and bronchiectasis are shown in Figure 1E. The father and two siblings carry the same mutation, so far without clinical signs except low IgM, IgG1 and IgA levels (Online Supplementary Table S3). Elevated levels of IgG2, IgG3 or IgG4 may compensate for lack of IgG1 in these mutation carriers.
To analyze the impact of the mutations on NF-κB mediated signaling, we tested the response to stimuli known to trigger the canonical NF-κB pathway (Figure 2A). We treated Epstein Barr virus-transformed B cells of patient 2 and healthy controls with lipopolysaccharide (LPS), tumor necrosis factor α or interleukin 1β and analyzed expression of NFKB1 (itself a target of NF-κB-mediated regulation). NFKB1 expression was upregulated in control, but not in R157X-mutant cells. The lack of a measurable NFKB1 transcriptional response in the heterozygous cells suggested a more profound effect of the mutation than mere p50/p105 dose reduction would explain. Upregulation of other target genes, such as CFLAR, upon activation was deficient in R157X-mutant cells (Figure 2B). To analyze the alteration of NF-κB-regulated gene expression in more detail we used predesigned real-time polymerase chain reaction assays that include 84 NF-κB targets (Online Supplementary Figure S1). The basal expression pattern of target genes was largely different in wild-type compared to R157X-mutant cells. LPS stimulation demonstrated stark differences in inducible NF-κB target gene expression. Of the NF-κB genes, RELA and RELB were similarly expressed in mutant and control cells (at baseline and stimulated), whereas NFKB1, NFKB2 and cREL were expressed less in mutant cells and only cREL showed inducible expression upon LPS treatment (Online Supplementary Figure S2). Only 12 of 79 evaluable genes showed a comparable expression pattern in wild-type and NFKB1 mutant cells indicating compensatory regulation or independence of p50/p105 in the presence of other NF-κB proteins. When we compared primary T cells of patient 1 to those of two healthy controls (Figure 2C) baseline expression of all NF-κB genes was reduced and 40 more known targets were differentially regulated. Both patients demonstrated disturbed expression and regulation of the death receptor FAS. Instead of downregulating FAS expression during infection to allow for effective immune response, LPS induced FAS upregulation (Online Supplementary Figure S1). Accordingly, in vitro stimulation of primary T cells with recombinant Fas ligand resulted in significantly decreased cell death in the presence of LPS or tumor necrosis factor α in control cells, but not in cells derived from both patients (Figure 2D). Both patients presented with clinical features similar to those of the Fas signaling pathway associated autoimmune lymphoproliferative syndrome (increased DNT cells, hepatosplenomegaly, lymphadenopathy, autoimmune symptoms, low regulatory T cells), but Fas ligand-induced apoptosis was not deficient, autoimmune lymphoproliferative syndrome-associated gene defects were absent and infections were too frequent.
Figure 2.
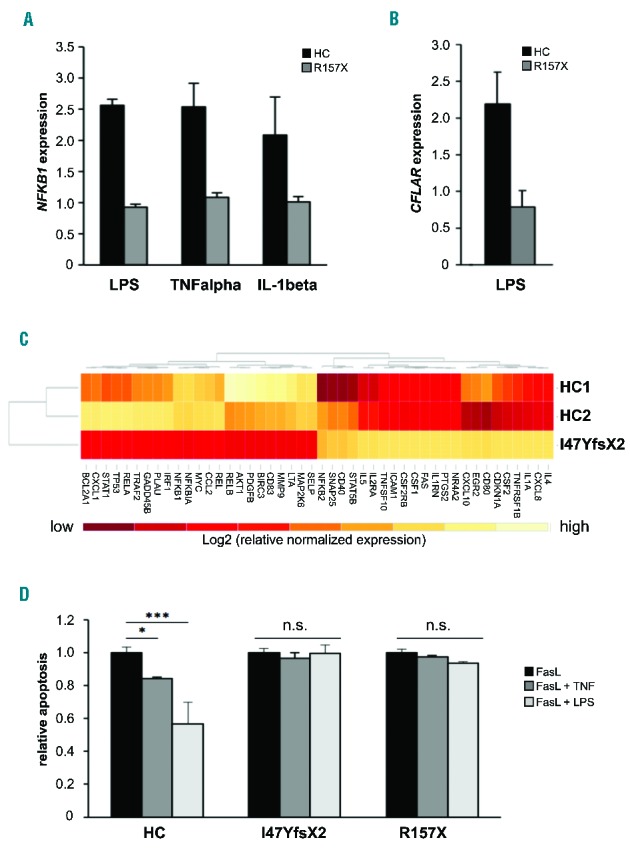
NF-κB-mediated signaling is affected in the patients. (A) Upregulation of NFKB1 mRNA expression in response to NF-κB activating stimuli is deficient in NFKB1-mutated cells. Epstein-Barr virus (EBV)-immortalized B cells of patient 2 and of a representative healthy control (HC) were treated for 16 h with LPS (5 μg/mL), tumor necrosis factor α (TNFα) (50 ng/mL) or IL-1β (10 ng/mL). Fold change of NFKB1 mRNA expression in untreated compared to treated samples was determined by real-time polymerase chain reaction (PCR). The relative expression was normalized to the respective untreated controls (=1). GAPDH and β-actin expression were used as internal standards. Mean values of representative experiments performed in triplicate and corresponding standard deviations are shown. (B) Expression of the NF-κB target gene CFLAR (synonymous for cFLIP) is not induced by LPS in the NFKB1-mutated patients’ cells. EBV-immortalized B cells of patient 2 and healthy control cells were treated for 16 h with LPS (5 μg/mL). Fold change of mRNA expression of CFLAR was determined by real-time PCR as described in Figure 1C. (C) Differential expression of NF-κB target genes in NFKB1-mutated primary T cells of patient 1 and two healthy wild-type controls. Baseline expression of a panel of NF-κB target genes was analyzed by real-time PCR using predesigned arrays (NF-κB signaling targets RT2 Profiler PCR arrays, Qiagen, Hilden, Germany). Mean values of two independent assays are shown. High gene expression is indicated in light color, low gene expression in dark red. The analysis was supervised and results are shown scaled. (D) LPS does not protect NFKB1 mutant primary T cells from apoptosis. Primary T cells of patients 1 and 2 and healthy controls were activated by phytohemagglutinin and IL2. Cells were treated with 100 ng/mL recombinant Fas ligand to induce apoptosis in the presence or absence of 100 ng/mL LPS or 10 ng/mL TNFα. Apoptosis was determined by flow cytometric measurement of phosphatidylserine exposure indicated by binding of annexin V-FITC. Dead cells were detected with propidium iodide. Significance was tested using two-way ANoVA (*P<0,05; ***P<0,001).
The gastrointestinal symptoms of patient 1 were reminiscent of ulcerative colitis, which is acknowledged to be a systemic autoimmune disease of unknown genetic cause. A candidate gene is NFKB1.5 Susceptibility to inflammatory bowel disease can be caused by mutant NOD2, which fails to activate NF-κB in response to bacterial infection.6 An NFKB1 promoter polymorphism (-94delATTG) leading to lower LPS-induced gene expression was described in inflammatory bowel disease.7
A characteristic of both presented patients was inadequate development of specific antibodies. The genetic cause of specific antibody deficiency is not known, but heterozygous deletion of IkBα led to functional NF-κB haploinsufficiency and lack of specific antibody responses after immunization.8 Reduced p50 expression may specifically play a role. Mice with targeted p50 disruption were deficient in producing specific antibodies in response to vaccination,9 whereas mice lacking only the C terminal half of the p105 precursor, but expressing the N terminal p50 protein (p105−/−), were not.10
Immunoglobulin class switching is known to be regulated by NF-κB. Mutations in the NF-κB essential modulator (NEMO) cause a class switch defective hyper-IgM syndrome in humans.12 Decreases in IgG, IgA, IgE, but not IgM were observed in p50 knockout mice.9–11 NF-κB regulates the expression of activation-induced cytidine deaminase (AID encoded by the AICDA gene), an enzyme required for DNA cleavage during class switch recombination. AICDA-knockout mice and patients with AICDA mutations are both class switch deficient. Thus, low levels of NF-κB may impair AID-mediated class switch recombination.13
Loss of p105 in the patients may play a role in the pathogenesis of infections and inflammatory disease, lymphadenopathy and splenomegaly that were also observed in the mouse model.10 p105 knockout led to constitutive activation of p50 homodimer repressors/activators indicating that low expression of NFKB1 may result in a severe dysregulation of the NF-κB network.
Similar to other gene defects with an impact on immune system control, NFKB1 mutations lack complete penetrance.14 In three pedigrees with 20 individuals harboring mutant NFKB1 alleles, the age at onset of disease varied from 2–64 years. Two gene carriers were completely healthy, while others had mild phenotypes with transient hypogammaglobulinemia or mild infections.4 Gender may be a bias, because most clinically affected individuals were females (17 of 22 cases). In our patients, we did not detect mutations in possible modifier genes on the X chromosome (e.g. FOXP3, TLR7). Taking into account that our and most of the reported cases (18 of 20) were affected during or after puberty we could speculate that X-linked hormonal differences may contribute to the higher penetrance of the disease in females. In this line, one of our patients showed reduced numbers of regulatory T cells. The number of these cells can also be affected by hormonal fluctuations.15
Our results broaden the phenotypic spectrum of NFKB1-associated disease and have novel implications for the diagnosis and treatment of patients harboring NFKB1 mutations and the therapeutic application of NF-κB directed drugs.
Acknowledgments
The authors would like to thank Katayoun Alemazkour, Bianca Killing and Monika Schmidt for excellent technical assistance.
Footnotes
Funding: this work was funded by an intramural grant (05/2014) of the research commission of the Medical Faculty of the Heinrich-Heine-University Düsseldorf.
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Baeuerle PA, Henkel T. Function and activation of NF-kappa B in the immune system. Ann Rev Immunol. 1994;12:141–179. [DOI] [PubMed] [Google Scholar]
- 2.Perkins ND. Integrating cell-signalling pathways with NF-kappaB and IKK function. Nat Rev Mol Cell Biol. 2007;8(1):49–62. [DOI] [PubMed] [Google Scholar]
- 3.Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18(49):6853–6866. [DOI] [PubMed] [Google Scholar]
- 4.Fliegauf M, Bryant VL, Frede N, et al. Haploinsufficiency of the NF-kappaB1 subunit p50 in common variable immunodeficiency. Am J Hum Gen. 2015. 3;97(3):389–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Baldwin AS., Jr Series introduction: the transcription factor NF-kappaB and human disease. J Clin Invest. 2001;107(1):3–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inohara N, Ogura Y, Fontalba A, et al. Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J Biol Chem. 2003;278(8):5509–5512. [DOI] [PubMed] [Google Scholar]
- 7.Karban AS, Okazaki T, Panhuysen CI, et al. Functional annotation of a novel NFKB1 promoter polymorphism that increases risk for ulcerative colitis. Hum Mol Genet. 2004;13(1):35–45. [DOI] [PubMed] [Google Scholar]
- 8.McDonald DR, Mooster JL, Reddy M, Bawle E, Secord E, Geha RS. Heterozygous N-terminal deletion of IkappaBalpha results in functional nuclear factor kappaB haploinsufficiency, ectodermal dysplasia, and immune deficiency. J Allergy Clin Immunol. 2007;120(4):900–907. [DOI] [PubMed] [Google Scholar]
- 9.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80(2):321–330. [DOI] [PubMed] [Google Scholar]
- 10.Ishikawa H, Claudio E, Dambach D, Raventos-Suarez C, Ryan C, Bravo R. Chronic inflammation and susceptibility to bacterial infections in mice lacking the polypeptide (p)105 precursor (NF-kappaB1) but expressing p50. J Exp Med. 1998;187(7):985–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snapper CM, Zelazowski P, Rosas FR, et al. B cells from p50/NF-kappa B knockout mice have selective defects in proliferation, differentiation, germ-line CH transcription, and Ig class switching. J Immunol. 1996;156(1):183–191. [PubMed] [Google Scholar]
- 12.Jain A, Ma CA, Liu S, Brown M, Cohen J, Strober W. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol. 2001;2(3):223–228. [DOI] [PubMed] [Google Scholar]
- 13.Cerutti A. The regulation of IgA class switching. Nat Rev Immunol. 2008;8(6):421–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rieux-Laucat F, Casanova JL. Immunology. Autoimmunity by haploinsufficiency. Science. 2014; 345(6204):1560–1561. [DOI] [PubMed] [Google Scholar]
- 15.Nie J, Li YY, Zheng SG, Tsun A, Li B. FOXP3+ Treg cells and gender bias in autoimmune diseases. Front Immunol. 2015;6:493. [DOI] [PMC free article] [PubMed] [Google Scholar]