

Calpain-1, a calcium-activated cysteine protease, is ubiquitously expressed in hematopoietic cells, and is known to play a functional role in a myriad of cellular processes by regulating limited cleavage of multiple substrates.1 Using a calpain-1 null model (CKO) previously generated in our laboratory, recent studies revealed a functional role of calpain-1 in IgE-dependent mast cell activation.2 Interestingly, in the Berkeley sickle mice, mast cell activation contributes to neurogenic inflammation, chronic pain, and hypoxia/reoxygenation (H/R)-evoked hyperalgesia, which were ameliorated upon treatment with mast cell inhibitor imatinib, and cannabinoids as well as nociception receptor ligand AT200.3–5 Therefore, we examined if calpain-1 contributes to chronic and/or hypoxia/reoxygenation (H/R)-evoked acute pain in sickle mice. We generated calpain-1 knockout Townes sickle (SSCKO) mice by cross-breeding HbSS-Townes sickle mice with calpain-1 KO mice. Systemic deletion of calpain-1 in Townes sickle mice6 ameliorated chronic pain behaviors including mechanical, heat, cold, and deep tissue/musculoskeletal hyperalgesia.
Calpains in mammals are encoded by 14 genes; however, two conventional calpains termed calpain-1 (μ-calpain) and calpain-2 (m-calpain), with 61% amino acid identity, are highly expressed.1 Calpain-1 is activated at micromolar calcium, whereas active calpain-2 is detected at millimolar calcium concentration in vitro.1 Calpastatin, an endogenous inhibitor of both calpains, provides a regulatory mechanism for suppressing calpain activity under steady state conditions.1 While both calpains are ubiquitously expressed, calpain-1 generally dominates in hematopoietic cells such as RBCs and platelets. In contrast, calpain-2 is more prominent in the nervous system. Sickle RBCs are known to exhibit high levels of intracellular calcium.7 Our previous study demonstrated that dense/dehydrated RBCs from patients with sickle cell disease show enhanced calpain-1 activity as measured by the calpastatin levels, and pharmacological inhibition of calpain-1 in the transgenic SAD mouse model of mild sickle cell disease reduced sickle RBC density/dehydration.8
Due to high sequence similarity between the two calpains, gene targeting and not the use of pharmacological inhibitors is preferable to elucidate the unique function of each calpain isoform in vivo. While the calpain-1 knockout mice are viable and fertile,9 calpain-2 gene inactivation causes early embryonic lethality in mice. Following up on our recent investigation of the role of calpain-1 in the SAD mouse model of mild sickle cell disease (SCD),8 in the present study we utilized genetic inactivation of calpain-1 in the homozygous, HbSS-Townes mice with severe SCD to define the role of calpain-1 in pain sensitivity. A comprehensive breeding strategy was designed to generate viable calpain-1 knockout Townes sickle (SSCKO) mice (Figure 1A and B), which express human but not mouse α- and β-globin genes. After 15 generations of breeding, the SSCKO mice were generated by back-crossing HbSS-Townes with calpain-1 KO mice (Figure 1C). The absence of mouse globins, and presence of human alpha and beta sickle and calpain-1 deletion were confirmed by PCR genotyping (Figure 1B). Additionally, phenotyping for homozygous (SS), heterozygous (AS), and normal human hemoglobin (HbA) was performed by cellulose acetate differential hemoglobin electrophoresis (Figures 1D). Only homozygous Townes mice with and without calpain-1 deletion and control HbAA-Townes mice expressing normal human hemoglobin A were used. The lack of casein cleavage confirmed the absence of calpain-1 enzymatic activity in the RBCs and reticulocytes of SSCKO mice as compared to HbAA-Townes and HbSS-Townes sickle mice (Figure 1E). Complete blood count analysis indicated no deleterious effects of calpain-1 deletion on the hematologic profile of SSCKO mice as compared to their HbSS counterparts, which displayed expected severe anemia (Online Supplementary Table S1). Together, we report here the first successful generation of Townes sickle mice with systemic deletion of calpain-1 gene. To our knowledge, calpain-1 is the second gene to be successfully deleted globally in the Townes sickle mice. The first gene BCL11A was deleted in both Townes and Berkeley mice.10
Figure 1.
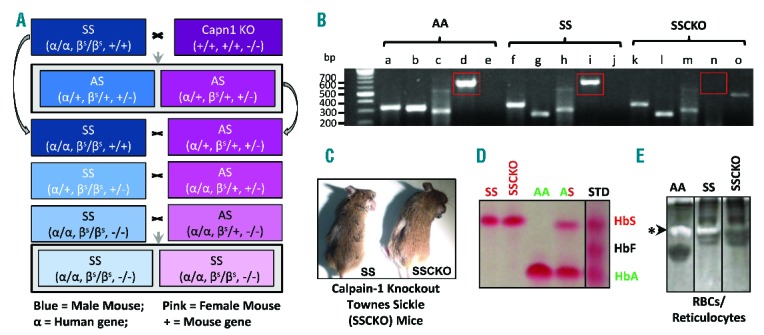
Generation of humanized calpain-1 knockout Townes sickle mice. (A) Schematic representation of the breeding strategy used to develop calpain-1 knockout Townes sickle (SSCKO) mice. (B) PCR/Gel electrophoresis confirming the genotypes of AA, SS, and SSCKO mice (a: human α-globin (330bp), b: human β-globin (320bp), c: absence of mouse β-globin (432bp), d: calpain-1 (650bp), e: absence of PGK-Neo cassette inserted to knockout calpain-1 (415bp), f: human α-globin (330bp), g: human sickle βs-globin (250bp), h: absence of mouse β-globin (432bp), i: calpain-1 (650bp), j: absence of PGK-Neo cassette inserted to knockout calpain-1 (415bp), k: human α-globin (330bp), l: human sickle βs-globin (250bp), m: absence of mouse β-globin (432bp), n: absence of calpain-1 (650bp), o: presence of PGK-Neo cassette inserted to knockout calpain-1 (415bp). For each mouse, PCR genotyping was conducted on 3 loci: α-globin, β-globin, and calpain-1, and the results are represented as genotypes in parentheses. For example, the CKO mouse has two alleles for the mouse alpha globin gene (+/+), two alleles for the mouse beta globin gene (+/+), and genetic inactivation of calpain-1 (−/−). The humanized Townes SS mouse has two alleles for the human alpha globin gene (α/α), two alleles for the human sickle hemoglobin gene (βS/βS), and intact calpain-1 gene (+/+). Blue: male; Pink: female. Calpain-1 is only the second gene to be systematically deleted successfully in the Townes sickle mice. (C) SS and SSCKO mice after 15 generations. (D) Phenotypic analysis of hemoglobins in different groups of Townes mice. Differential hemoglobin electrophoresis on cellulose acetate membrane was performed on each mouse used in this study. The Ponceau S stained bands show the presence of human sickle hemoglobin only in homozygous sickle mice (labeled SS and SSCKO), normal human hemoglobin A only in control mice (labeled AA) and both human sickle and normal human hemoglobin A (labeled AS) in heterozygous mice. Representative images from several mice phenotyped are shown. (E) Casein zymography confirming the absence of calpain-1 enzymatic activity in the RBCs/reticulocytes of SSCKO mice compared to their AA and SS counterparts.
The ultimate goal of any sickle cell therapy is the reduction of chronic pain and episodes of acute pain crises in patients with sickle cell disease. In rat models of spinal nerve injury, inhibition of calpain-1 reduced neuropathic pain.11 Thus, we evaluated whether SSCKO would show a reduction in pain behaviors that have been previously characterized in the Berkeley and Townes sickle mice.3,12 Townes SS mice showed a significant increase in mechanical (P<0.0001), heat (P<0.0001), cold (P<0.001), and deep tissue/musculoskeletal (P<0.01) hyperalgesia, as compared to control AA mice (Figure 2A–D). In comparison to HbSS-Townes, SSCKO mice exhibited a significant decrease in four measures of hyperalgesia: i) mechanical (P<0.0001); ii) thermal sensitivity to heat (P<0.01); iii) thermal sensitivity to cold (P<0.01); and iv) deep tissue/musculoskeletal hyperalgesia (P<0.0001). Notably, there were no significant differences in the thermal and deep tissue hyperalgesia between SSCKO and AA mice (Figure 2B–D) but mechanical hyperalgesia was significantly increased in SSCKO as compared to AA mice (P<0.05) (Figure 2A). These results show that chronic thermal and deep tissue hyperalgesia are completely ameliorated by the deletion of calpain-1 gene in SS mice. Thus, calpain-1 may contribute to the regulation of chronic hyperalgesia in SCD.
Figure 2.
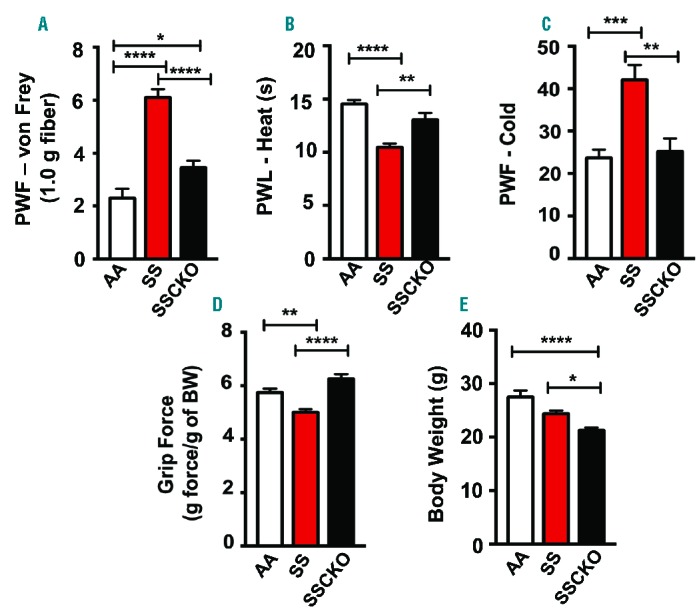
Global knockout of calpain-1 ameliorates chronic pain in Townes sickle mice. (A) Mechanical hyperalgesia, (B) sensitivity to heat, (C) sensitivity to cold, (D) deep tissue hyperalgesia, and (E) body weight are shown. Mean age of each group of female mice in months ± SEM are AA: 5.58 ± 0.21 (n = 10); SS: 5.51 ± 0.35 (n = 10); SSCKO: 6.23 ± 0.1 (n = 10). Values on the graphs are mean ± SEM. Differences between groups were analyzed by one-way ANOVA with Bonferroni multiple comparison. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001. PWF: paw withdrawal frequency; PWL: paw withdrawal latency; BW: body weight; AA: Townes control mice; SS: Townes sickle mice; SSCKO: calpain-1 knockout Townes sickle mice.
In contrast, calpain-1 deletion did not reduce H/R-evoked hyperalgesia (Figure 3A–D). Following the first H/R treatment, the SS mice showed an increase in all 4 measures of hyperalgesia that were sustained following the second H/R treatment. Although no further H/R treatment was administered, SS mice continued to show significant hyperalgesia for five days for cold (P<0.05), and seven days for heat and deep tissue hyperalgesia (P<0.05 for both), as compared to baseline pain levels. In contrast, mechanical hyperalgesia was not sustained beyond the second treatment in SS mice (Figure 3A). Similarly, SSCKO mice displayed an increase in H/R-evoked hyperalgesia, which was sustained until day 7 for mechanical, heat, and deep tissue, and until day 5 for cold hyperalgesia. Together, these results indicate that calpain-1 did not influence H/R-evoked hyperalgesia or recovery from H/R-evoked pain response. Thus, calpain-1 may not contribute to acute pain due to vaso-occlusive crisis in sickle cell disease.
Figure 3.
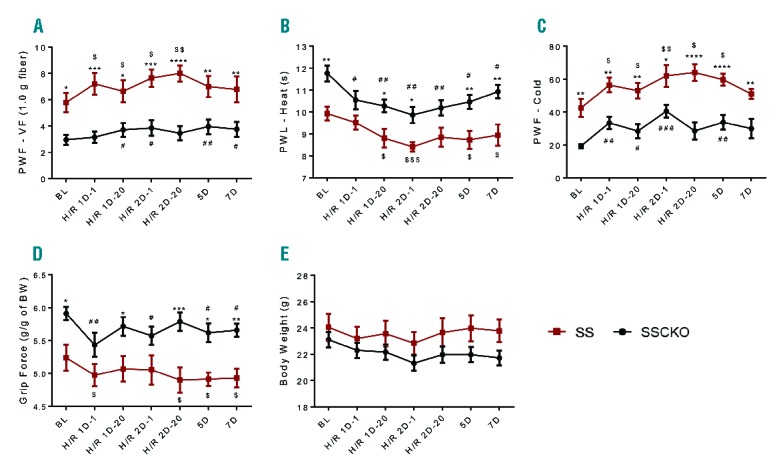
Effect of calpain-1 deletion on hypoxia/reoxygenation-evoked hyperalgesia in sickle mice. (A) Mechanical hyperalgesia, (B) sensitivity to heat, (C), sensitivity to cold, (D) deep tissue hyperalgesia, and (E) body weight were assessed at baseline (BL) before H/R treatment. Mice were then exposed to 8% O2/92% N2 hypoxia (H) for 3 h followed by reoxygenation (R) at room air. Pain behaviors were analyzed after 1 h (1D-1) and 20 h (1D-20). Twenty-four h later, the hypoxia treatment was repeated and pain behaviors were analyzed at 1 h (H/R 2D-1) and 20 h (2D-20). No further hypoxia treatment was performed, but pain behaviors were analyzed up to the 5th and 7th days from baseline measures. Mean age of each group of mice in months ± SEM are SS: 6.56 ± 0.38 (n=7); SSCKO: 7.86 ± 0.11 (n=10). Values on the graphs are mean ± SEM. *P<0.05; **P<0.01; ***P<0.001; ****P<0.0001 for SSCKO vs. SS (two-way ANOVA, Bonferroni). #P<0.05; ##P<0.01 vs. BL in SSCKO group (one-way ANOVA, paired t-test); $P<0.05; $$P<0.01, $$$P<0.001 vs. BL in SS group (one-way ANOVA, paired t-test). PWF-VF: paw withdrawal frequency-Von Frey; PWL: paw withdrawal latency; BW: body weight; BL: baseline; AA: Townes control mice; SS: Townes sickle mice; SSCKO: calpain-1 knockout Townes sickle mice.
Pain characteristics are well defined in the Berkeley sickle mice, which show tonic mechanical, deep tissue, and thermal hyperalgesia.12 However, the pain characteristics in Townes sickle mice are not yet well characterized. Our group (Gupta) observed that tonic hyperalgesia in Townes sickle mice is significantly reduced as compared to age- and gender-matched Berkeley SS mice.13 In rat models, calpain inhibition reduced cancer-induced bone pain,14 inflammatory pain, and neuropathic pain11 via inhibition of osteoclastogenesis, COX-2, and NMDA receptors, respectively. Inhibition of CaMKIIα, a calpain substrate1 and an emerging player in sickle pain, also reversed neuropathic pain in mice.15 Thus, it appears likely that calpain-1 activation under elevated calcium concentrations exacerbates sickle pain possibly by inhibiting mast cell activation and/or other mechanisms. Peripheral activation of mast cells in the skin, glial activation in the spinal cord, and central sensitization of the spinal dorsal horn neurons are known to contribute to chronic hyperalgesia in the Berkeley sickle mice.3,16 Our data demonstrating that calpain-1 deletion reduces all four measures of chronic hyperalgesia in HbSS-Townes mice are novel and supportive of the hypothesis that mast cell activation contributes to chronic hyperalgesia in sickle mice. On the contrary, acute pain incited by H/R injury to simulate pain due to vaso-occlusive crisis remained unaffected with calpain-1 deletion in Townes sickle mice.
Mechanisms underlying pain in sickle cell disease are emerging but their specificity with respect to chronic and acute pain is not yet known. Younger HbSS-Berk sickle mice show significantly lower pain than older mice, suggesting that sickle pathobiology influences the activation of nociceptive system leading to chronic pain. Acute pain on the other hand, may result from spontaneous activation of the nociceptive system evoked by VOC. Since calpain-2 dominates in the nervous system in contrast to calpain-1 that is active in hematopoietic cells,1 calpain-2 might be up-regulated as a compensatory mechanism in SSCKO mice. This important feature of calpain biology will require pharmacological inhibition of calpain-2 in both SS and SSCKO mice to examine the specific contribution of each calpain isoform in chronic and acute sickle pain.
In conclusion, we demonstrate that genetic ablation of calpain-1 ameliorates the characteristic chronic pain behaviors of sickle mice: mechanical, deep tissue, heat, and cold hyperalgesia. These findings are the first to establish a functional role of calpain-1 in the pathobiology of sickle pain. Future studies would throw light on the role of calpain-1 in other blood cells involved in whole blood platelet aggregation, including leukocytes and reticulocytes, as well as possible compensatory roles of calpain-2 in H/R-induced platelet hyperactivity and acute pain. We posit that calpain-1 is a potentially treatable novel target for SCD.
Acknowledgments
The authors thank Drs. Betty S. Pace, Levi H.C. Makala, Tim M. Townes, Solomon F. Ofori-Acquah, and Carlo Brugnara for guidance in the breeding, genotyping, and phenotyping of the calpain-1 knockout Townes sickle mice; Drs. John Castellot, David J. Greenblatt, Henry H. Wortis, William F. Dietrich, and Richard J. Labotka for guiding JON in the completion of her doctoral dissertation research. JON performed these studies in the laboratory of Dr. Athar Chishti.
Footnotes
The online version of this letter has a Supplementary Appendix.
Funding: funding provided by the Mentors in Medicine Program at the Washington University School of Medicine in Saint Louis.
Funding: this work was supported by the Howard Hughes Medical Institute International Student Research Fellowship (JON), National Institutes of Health grants HL089517 and HL095050, American Society of Hematology Bridge Grant, and American Heart Association Grant-in-Aid (AHC), and National Institutes of Health grants R01HL103773 and U01 HL117664, and grants from the Institute for Engineering in Medicine (KG).
Information on authorship, contributions, and financial & other disclosures was provided by the authors and is available with the online version of this article at www.haematologica.org.
References
- 1.Goll DE, Thompson VF, Li H, Wei W, Cong J. The calpain system. Physiol Rev. 2003;83(3):731–801. [DOI] [PubMed] [Google Scholar]
- 2.Wu Z, Chen X, Liu F, et al. Calpain-1 contributes to IgE-mediated mast cell activation. J Immunol. 2014;192(11):5130–5139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vincent L, Vang D, Nguyen J, et al. Mast cell activation contributes to sickle cell pathobiology and pain in mice. Blood. 2013;122(11):1853–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vincent L, Vang D, Nguyen J, Benson B, Lei J, Gupta K. Cannabinoid receptor-specific mechanisms to ameliorate pain in sickle cell anemia via inhibition of mast cell activation and neurogenic inflammation. Haematologica. 2016;101(5):566–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vang D, Paul JA, Nguyen J, et al. Small-molecule nociceptin receptor agonist ameliorates mast cell activation and pain in sickle mice. Haematologica. 2015;100(12):1517–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ryan TM, Ciavatta DJ, Townes TM. Knockout-transgenic mouse model of sickle cell disease. Science. 1997;278(5339):873–876. [DOI] [PubMed] [Google Scholar]
- 7.Eaton JW, Skelton TD, Swofford HS, Kolpin CE, Jacob HS. Elevated erythrocyte calcium in sickle cell disease. Nature. 1973;246(5428):105–106. [DOI] [PubMed] [Google Scholar]
- 8.De Franceschi L, Franco RS, Bertoldi M, et al. Pharmacological inhibition of calpain-1 prevents red cell dehydration and reduces Gardos channel activity in a mouse model of sickle cell disease. FASEB J. 2013;27(2):750–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Azam M, Andrabi SS, Sahr KE, Kamath L, Kuliopulos A, Chishti AH. Disruption of the mouse mu-calpain gene reveals an essential role in platelet function. Mol Cell Biol. 2001;21(6):2213–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xu J, Peng C, Sankaran VG, et al. Correction of sickle cell disease in adult mice by interference with fetal hemoglobin silencing. Science. 2011;334(6058):993–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou HY, Chen SR, Byun HS, et al. N-methyl-D-aspartate receptor-and calpain-mediated proteolytic cleavage of K+-Cl− cotransporter-2 impairs spinal chloride homeostasis in neuropathic pain. J Biol Chem. 2012;287(40):33853–33864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kohli DR, Li Y, Khasabov SG, et al. Pain-related behaviors and neurochemical alterations in mice expressing sickle hemoglobin: modulation by cannabinoids. Blood. 2010;116(3):456–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lei J, Benson B, Tran H, et al. Comparative Analysis of Pain Behaviours in Humanized Mouse Models of Sickle Cell Anemia. PlosOne. 5 August 2016. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xu JY, Jiang Y, Liu W, Huang YG. Calpain inhibitor reduces cancer-induced bone pain possibly through inhibition of osteoclastogenesis in rat cancer-induced bone pain model. Chin Med J (Engl). 2015;128(8):1102–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Y, Luo F, Yang C, Kirkmire CM, Wang ZJ. Acute inhibition of Ca2+/calmodulin-dependent protein kinase II reverses experimental neuropathic pain in mice. J Pharmacol Exp Ther. 2009;330(2):650–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Valverde Y, Benson B, Gupta M, Gupta K. Spinal glial activation and oxidative stress are alleviated by treatment with curcumin or coenzyme Q in sickle mice. Haematologica. 2016;101(2):e44–47. [DOI] [PMC free article] [PubMed] [Google Scholar]