

Abstract
Current range expansions of large terrestrial carnivores are occurring following human-induced range contraction. Contractions are often incomplete, leaving small remnant groups in refugia throughout the former range. Little is known about the underlying ecological and evolutionary processes that influence how remnant groups are affected during range expansion. We used data from a spatially explicit, long-term genetic sampling effort of grizzly bears (Ursus arctos) in the Northern Continental Divide Ecosystem (NCDE), USA, to identify the demographic processes underlying spatial and temporal patterns of genetic diversity. We conducted parentage analysis to evaluate how reproductive success and dispersal contribute to spatio-temporal patterns of genetic diversity in remnant groups of grizzly bears existing in the southwestern (SW), southeastern (SE) and east-central (EC) regions of the NCDE. A few reproductively dominant individuals and local inbreeding caused low genetic diversity in peripheral regions that may have persisted for multiple generations before eroding rapidly (approx. one generation) during population expansion. Our results highlight that individual-level genetic and reproductive dynamics play critical roles during genetic assimilation, and show that spatial patterns of genetic diversity on the leading edge of an expansion may result from historical demographic patterns that are highly ephemeral.
Keywords: genetic diversity, parentage, reproduction, migration, range expansion, dispersal
1. Introduction
Worldwide, many large terrestrial carnivores suffered population declines and extirpation from much of their range during the nineteenth and early twentieth centuries [1–4]. Though some populations continue to decline or remain imperilled, many populations are now stable or rapidly recovering [5]. Given the crisis nature of conservation biology [6], most research focuses on determining the mechanisms underpinning population decline, range contraction, or extirpation. On the other hand, understanding of the recovery process and subsequent range expansion following contraction is based largely on theoretical simulations [7–13] and historical reconstructions [14–17], and thus relies heavily on idealized scenarios. This is particularly true for large carnivores, as detailed data collected at appropriate spatial and temporal scales necessary for tracking expansion processes in long-lived and highly mobile organisms are extraordinarily rare.
Population genetic theory generally predicts greater differentiation and lower genetic diversity on the leading edge of range expansions [7–11,13]. This pattern arises from the long-distance dispersal of small numbers of founding individuals at the edges of the expansion zone. Typically, gene flow (dispersal) between populations increases as the contiguous population continues to expand, leading to greater similarity between populations at the range core and on the periphery [7,12,18]. However, when remnant groups exist in the direct path of the core expansion, the manner and timescale of assimilation is currently unknown, and probably depends on reproductive success, dispersal rate, and habitat carrying capacity [7,10,12]. Initially, remnant groups tend to have low genetic diversity due to generations of relative isolation, low effective population sizes, and subsequent genetic drift. Theoretical simulations suggest that during population expansion an infusion of dispersers from the neighbouring core and subsequent interbreeding between populations function to increase genetic diversity within peripheral populations [7,8,13].
Numerous populations of carnivores persist as remnants [19–29], largely due to habitat fragmentation and persecution, with climate change expected to exacerbate the problem in additional populations [30]. Nevertheless, few studies have examined fine-scale interactions between expanding core populations and existing remnant groups because the temporal and spatial data necessary to do so are rarely available. Empirical studies at the ecosystem scale at which large carnivores function can reveal patterns in spatial genetic diversity and help identify the underlying ecological and evolutionary processes responsible for observed patterns. Demographic dynamics (e.g. dispersal, reproduction, and carrying capacity), in particular, probably have a very strong impact on contemporary evolutionary processes responsible for spatio-temporal patterns of genetic diversity observed during ongoing range expansions.
Logistically, investigating reproductive success and dispersal in large carnivores is exceedingly difficult given the challenges posed by the life history and ecology of most species. Many large carnivores are solitary and most are wide-ranging, making observational assessments of maternity difficult and paternity nearly impossible to ascertain. Migration and dispersal studies traditionally use mark–recapture or tracking technologies to follow individuals over time, but often suffer from insufficient sample sizes and focus on subsets of ecosystems due to the monetary expenses and time involved. Inferred methods (e.g. parentage/kinship analysis) have therefore been the preferred method of reconstructing kin relationships [31–41]. However, large carnivores are often long-lived and have overlapping generations, making it difficult to obtain the spatial and temporal genetic data needed to construct accurate pedigrees. Given these challenges, no study has evaluated how demographic phenomena occurring in remnant, low-density groups and an expanding core influence subsequent patterns of intraspecific genetic diversity in large carnivores.
Grizzly bears (Ursus arctos) in the contiguous USA survive in four geographically separated populations [42], a fraction of their former range. Only two of these, the Northern Continental Divide Ecosystem (NCDE) and the Greater Yellowstone Ecosystem (GYE) support more than approximately 80 individuals. Dispersal between populations is rare, especially for females [43], but does occur in limited cases [44], and the NCDE population remains connected to Canadian populations. The NCDE population was listed as threatened in 1975 [45], but has been showing signs of recovery, both in terms of occupied range [46] (Mace R, Roberts L. 2012 Northern Continental Divide Ecosystem Grizzly Bear Monitoring Team Annual Report, 2012. Montana Fish, Wildlife & Parks, 490 N. Meridian Road, Kalispell, MT 59901, 2012, unpublished data) and population abundance, growing from approximately 765 individuals in 2004 [46] to nearly 1 000 individuals in 2009 [47]. Historically, Glacier National Park (GNP), in the most northern NCDE, was a stronghold for the population with higher protection afforded to individuals within park boundaries resulting in fewer human-caused mortalities [45]. Partly as a result, GNP and adjacent lands, henceforth referred to as the core, had high densities of grizzly bears, while lands further south supported low densities of bears that were probably semi-isolated when populations were smaller, as evidenced by historical genetic structure [46].
The combination of an incomplete range contraction and subsequent expansion provides a valuable natural experiment using grizzly bears as a model carnivore species. Grizzly bears can reach nearly 30 years of age [47], have overlapping generations with a generation length of approximately 10 years [48], and exhibit many of the same traits that make studying other large carnivores difficult. Here, we use extremely rare, highly rigorous spatial and temporal grizzly bear genetic data collected at an ecosystem scale to (i) evaluate the spatial and temporal dynamics of genetic diversity during grizzly bear population growth and range expansion in the NCDE and (ii) identify the underlying demographic processes responsible for spatial and temporal fluctuations in genetic diversity.
2. Material and methods
(a). Study area
The 32 000 km2 NCDE is one of the largest and most intact ecosystems in the USA. The vast majority of land is protected mountainous terrain, encompassing GNP, all or portions of five national forests (Flathead, Lolo, Lewis & Clark, Lincoln, and Kootenai), five wilderness areas (Bob Marshall, Scapegoat, Great Bear, Mission Mountain, and Rattlesnake), three wilderness study areas (Mount Hefty Tuchuck, Thompson-Seton, and Ten Lakes), parts of the Blackfeet Nation and Confederated Salish and Kootenai Reservations, along with private land holdings located mostly on the eastern, western, and southern periphery. Waterton Lakes National Park in Alberta, Canada, is adjacent to the northern boundary of the NCDE.
(b). Genetic samples
Two independent sampling methods were used to collect genetic samples (bear hair) from 1998–2012: (i) hair traps—corrals of barbed wire with lure in the centre systematically distributed using an 8 × 8 km (1998, 2000) or 7 × 7 km (2004) grid and (ii) bear rubs—naturally occurring trees or other objects that bears rub on fitted with barbed wire (1998–2000, 2004, and 2009–2012). From 1998 to 2000, sampling occurred on 8 000 km2 in the northern extent of the NCDE (north of Highway 2 (figure 1)), whereas systematic and consistent, ecosystem-wide sampling occurred in 2004 and 2009–2012. In total, there were 6 160 confirmed grizzly bear detections, leading to the identification of 1 115 unique individual genotypes (520 male, 595 female). All samples were originally genotyped at seven variable microsatellite loci to identify unique individuals (G10 J, G1A, G10B, G1D, G10H, G10M, and G10P). We then attempted to extend genotypes of a subset of samples to 16 (n = 142) (previous seven loci in addition to G10C, CXX110, CXX20, G10L, MU50, MU59, G10U, MU23, and G10X) and 24 markers (n = 637) (previous 16 loci in addition to REN145.P07, MSUT2, CPH9, MU51, REN144.A06, MU26, D123, and D1A) as resources and sample material allowed (electronic supplementary material, tables S1 and S2). Individuals were also genotyped at amelogenin to identify gender [49]. We sampled 71% (95% CI = 66–76%) of the population in 2004 [46] suggesting that most individuals first identified in 2009–2012 are younger bears. Further details on data collection, laboratory protocol, molecular analysis, and data quality control are available in Kendall et al. [46]. Samples collected from 2009 to 2012 were processed in the same laboratory with the same procedures as Kendall et al. [46]. Assuming a 2–3% annual rate of increase in the NCDE population [47], approximately 30–35% of the population was sampled annually from 2009 to 2012.
Figure 1.
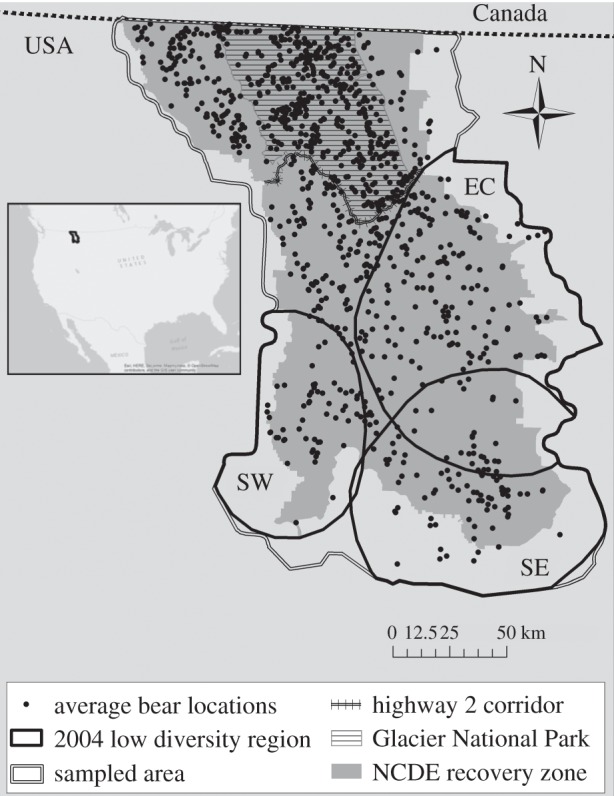
Average locations of 1 115 individual grizzly bears in the Northern Continental Divide Ecosystem (NCDE), USA, between 1998 and 2012. Black polygons encompass buffered regions of interest due to low heterozygosity (HO) in 2004, clipped by the sampled study area (double lines).
(c). Spatial genetic diversity
To describe spatial patterns of genetic diversity (Objective 1), we split samples into three time periods: samples collected in 2004 (545 individuals), 2009–2010 (449 individuals), and 2011–2012 (494 individuals). Within each time period, we averaged locations of individuals that were detected more than once. We performed calculations with the seven loci that were genotyped consistently (electronic supplementary material, table S2) across study years and individuals. We found positive spatial autocorrelation of genotypes out to 66 km using Genalex 6.5 [50] (electronic supplementary material, figure S1), and subsequently used this to define the extent of the neighbourhood used for calculating spatial patterns in genetic diversity. We calculated six metrics of local genetic diversity using a radius of 33 km in a moving-window analysis with the spatial Genetic Diversity package (sGD) [51] for the R statistical environment [52]. We calculated metrics when more than or equal to 10 individuals were contained within the 33 km radius of each focal individual.
(d). Parentage analysis
We used the full-pedigree likelihood approach implemented in COLONY 2.0 [52–54] to identify familial relationships among grizzly bears in the NCDE. COLONY iteratively substitutes individuals into clusters of family groups based on genotypes to determine the most parsimonious family tree configuration [53–55] and has been shown to be quite accurate (80–100% correct assignments) with more than or equal to 10 loci genotyped when run on a simulated population with similar attributes to the NCDE grizzly bear population [56]. This method also infers genotypes of non-sampled parents. All 1 115 genotypes were included in the analysis. Missing alleles are dropped from analysis so that individuals genotyped at fewer loci are compared only at available loci. We report parentage results from a single long run in COLONY (approx. 300 million iterations) using all data where we allowed polygamy in both sexes, and estimated that the proportion of sampled parents was 0.4 for each sex and genotyping error was set at 0.001 for each locus. We used COLONY output to quantify the number of offspring and partners per individual, without considering inferred parents.
(e). Reproductive success
To quantify individual reproductive success, we used samples collected in 2004 as our baseline adult generation because sampling in this year was systematic across the entire ecosystem, the number of loci genotyped was consistent (electronic supplementary material, table S1, more than 93% individuals had 24 markers genotyped), and because we had high probabilities of detecting their offspring in later years of sampling (2009–2012) based on age of primiparity. Female grizzly bears in the NCDE reach primiparity at a mean of 5.4 years old and average 2 cubs per litter, with each litter separated by approximately 3 years [47]. Male first age of reproduction can be as early as 4 years old, but can be later when older, more dominant males are present. Reproductive success refers only to the sampling period and incorporates individuals varying in age at the end of the study, and thus differs from lifetime reproductive success. We averaged the proportion of reproductively successful adult bears within each genetic neighbourhood based on a 33 km radius in a moving-window analysis (analogous to the above approach (sGD) used to calculate genetic diversity). We then compared results to weighted neighbourhood estimates of relative median density of male and female bears with original 10 km resolution [57].
(f). Low-diversity persistence and assimilation
We used spatial patterns in observed heterozygosity (HO) from 2004 to identify peripheral regions in the NCDE where grizzly bears had low genetic diversity. Peripheral regions were bounded by selecting individuals with HO values less than 0.70, creating minimum convex polygons of individuals in each of the regions, and then buffering the polygons by 33 km to encompass individuals factoring into the low sGD estimates. To assess temporal changes in genetic diversity within the regions with low HO, we used the same boundaries and tracked changes in regional AR, HO, HE between time periods using the diveRsity package [58] for the R statistical environment [52]. We assessed significance of the difference across time in genetic metrics through bootstrap sampling of individuals within each region with 1 000 replicates. Additionally, mean FIS was calculated using 1 000 bootstrap replicates. We then used patterns in individual reproductive success and dispersal to identify the demographic mechanisms potentially responsible for spatio-temporal patterns in genetic diversity (Objective 2).
Individuals within the buffered regions of low HO were classified as contemporary residents or immigrants (individuals dispersing from outside of the region). We used the mean location of mothers assigned in the parentage analysis as individual origins (i.e. locations of natal home ranges, as in [37,39,59]). When mothers were inferred and individuals were assigned fathers only, we used the mean location of the father as the origin. If no parents were assigned, we had no information on origin location, and classified the individual as unknown. Individuals within each region that originated from outside of the region were classified as immigrants and individuals that had originated within the region were classified as residents unless they had descended from one of the identified immigrants, in which case they were classified as immigrant offspring or grandoffspring. Immigrants that originated from more than 66 km away from the region were additionally classified as long-distance migrants.
3. Results
(a). Spatial and temporal patterns in genetic diversity
Observed HO was relatively low in three regions of the NCDE in 2004; the southwest (SW), east-central (EC), and southeast (SE) (figure 2). In the SE, SW, and EC regions, we sampled 45, 28, and 98 different bears in 2004, 64, 32, and 104 bears in 2009–2010, and 72, 32, and 120 bears in 2011–2012. Five other measures of diversity: (i) the average number of alleles per locus (A), (ii) proportion of alleles in a genetic neighbourhood (AP), (iii) allelic richness (AR), (iv) Nei's genetic diversity (HS), and (v) the inbreeding coefficient (FIS) exhibit similar patterns of diversity as HO (electronic supplementary material, figures S2–S6). Varying the minimum number of neighbours and neighbourhood size had little effect on results.
Figure 2.

Observed heterozygosity (HO) in the NCDE. Small, light-coloured circles represent low HO, while large, dark-coloured circles represent higher HO. Black polygons encompass regions of interest due to low HO in 2004. Small black dots indicate areas where individuals were detected but there were insufficient sample sizes (n < 10) to accurately calculate summary statistics.
(b). Reproductive success
The parentage analysis yielded 1 287 assignments, including 435 triads (offspring with two assigned parents), 621 mother–offspring (M–O) dyads, and 666 father–offspring (F–O) dyads. Nearly all assignments contained zero mismatching loci, with eight assignments containing one mismatched loci, and one assignment containing two mismatched loci. Respectively, 205 and 139 unique mothers and fathers were identified from the full dataset.
Individual reproductive success varied greatly for both sexes (electronic supplementary material, figure S7a). Female reproductive success ranged from 0 offspring for nearly 50% of females up to 10 offspring for one individual in the SE, with a mean of 1.55 (median = 1). Male reproductive success ranged from 0 to 18 offspring with a mean of 2.06 (median = 0). Variability of success was substantial; over half of the males (59%) produced 0 detected offspring, while the top four reproducers (located in the central and southern NCDE) fathered between 16 and 18 detected offspring each. A higher proportion of females (0.50) had reproductive success than did males (0.41) (p = 0.03, two sample test for equality of proportions).
There was a strong inverse relationship between bear density and the number of offspring per adult and the proportion of successful adults of both sexes. In the north part of the NCDE, bear density was highest and reproductive success was lowest (less than 40% of males with more than one offspring; electronic supplementary material, figure S8). In the lowest density areas, males produced an average of nearly twice as many detected offspring compared with males in high density areas (approx. 50% of males with more than or equal to one offspring), and females produced approximately 40% more offspring.
(c). Demographic mechanisms explaining patterns of genetic diversity
Several individuals with extremely high reproductive success had a large number of descendants in two of the three regions with relatively low genetic diversity (SE and SW), probably compounding the effects of contemporary and historical regional inbreeding associated with historical population decline and fragmentation. The relatively low genetic diversity in the SE region can be partially explained by the reproductive success of one individual. All of the SE residents (13 of 13) and nearly half of the individuals with unknown origins (8 of 18) detected in 2004 descend from one inferred male, and nearly all residents (11 of 13) also descended from one inferred female. The male fathered a highly productive male (8 offspring) and the two most productive females (10 and 8 offspring, respectively) in the entire ecosystem with two inferred females. The productive half-siblings (the male and females had the same father, but different mothers) also subsequently mated with each other, producing six offspring together. We sampled 101 descendants of the inferred male during the entire study period, primarily in the SE region (electronic supplementary material, figure S9b). The proportion of immigrants in the SE increased between 2004 and 2009–2010 samples (31–41%). This was largely due to an increase in the number of immigrant offspring between 2004, when three immigrant offspring were detected, and 2009–2010, when 10 immigrant offspring were detected (table 1). This increase in immigrant reproductive success, as well as an increase in the number of bears detected, coincides with a significant increase in HO (table 2) that continued through 2012 (2004 to 2009–2010 mean differences in HO = 0.06 (lower 95% CI 0.00), 2004 to 2011–2012 mean differences in HO = 0.06 (lower 95% CI 0.00)) and the erosion of the low diversity signature visible in 2004 in less than one generation (figure 2a–c).
Table 1.
Number of immigrants into each region through time. Note that the same individual may be labelled as a migrant in multiple years. Long-distance migrants (LDM) are a subsample of immigrants in the region, not additional individuals.
| 2004 | 2009–2010 | 2011–2012 | |
|---|---|---|---|
| southeast (SE) immigration | |||
| class | total (LDM) | total (LDM) | total (LDM) |
| immigrants | 11 (6) | 16 (7) | 22 (10) |
| immigrant offspring | 3 (3) | 10 (7) | 4 (3) |
| immigrant grandoffspring | 0 (0) | 0 (0) | 0 (0) |
| total immigrants in region | 14 (9) | 26 (14) | 26 (13) |
| total bears sampled in region | 45 | 64 | 72 |
| percentage of region of immigrant origin | 31.11% (20.00%) | 40.63% (21.88%) | 36.11% (18.06%) |
| southwest (SW) immigration | |||
| immigrants | 5 (1) | 8 (3) | 7 (2) |
| immigrant offspring | 0 (0) | 4 (1) | 6 (0) |
| immigrant grandoffspring | 0 (0) | 0 (0) | 0 (0) |
| total immigrants in region | 5 (1) | 12 (4) | 13 (2) |
| total bears sampled in region | 28 | 32 | 32 |
| percentage of region of immigrant origin | 17.86% (3.57%) | 37.50% (12.50%) | 40.63% (6.25%) |
| east-central (EC) immigration | |||
| immigrants | 27 (6) | 30 (11) | 41 (11) |
| immigrant offspring | 7 (5) | 14 (5) | 14 (5) |
| immigrant grandoffspring | 7 (3) | 4 (3) | 8 (4) |
| total immigrants in region | 41 (14) | 48 (19) | 63 (20) |
| total bears sampled in region | 98 | 104 | 120 |
| percentage of region of immigrant origin | 41.84% (14.29%) | 46.15% (18.27%) | 52.50% (16.67%) |
Table 2.
Bootstrapped differences of genetic metrics through time within regions. Lower 95% confidence intervals not encompassing zero were considered significant differences between time periods.
| region | allelic richness (AR) |
observed heterozygosity (HO) |
expected heterozygosity (HE) |
|||
|---|---|---|---|---|---|---|
| mean difference | lower 95% CI | mean difference | lower 95% CI | mean difference | lower 95% CI | |
| EC 2004 to 2009–2010 | 0.05 | −0.32 | 0.05 | 0.01 | 0.02 | 0.00 |
| EC 2004 to 2011–2012 | 0.12 | −0.25 | 0.07 | 0.03 | 0.03 | 0.01 |
| EC 2009–2010 to 2011–2012 | 0.06 | −0.25 | 0.02 | −0.01 | 0.01 | −0.01 |
| SE 2004 to 2009–2010 | 0.25 | −0.23 | 0.06 | 0.00 | 0.06 | 0.03 |
| SE 2004 to 2011–2012 | 0.24 | −0.16 | 0.06 | 0.00 | 0.06 | 0.02 |
| SE 2009–2010 to 2011–2012 | −0.01 | −0.39 | 0.00 | −0.04 | −0.01 | −0.03 |
| SW 2004 to 2009–2010 | 0.19 | −0.31 | 0.01 | −0.06 | 0.00 | −0.04 |
| SW 2004 to 2011–2012 | 0.20 | −0.27 | 0.00 | −0.09 | −0.01 | −0.05 |
| SW 2009–2010 to 2011–2012 | 0.01 | −0.47 | −0.02 | −0.10 | −0.01 | −0.05 |
| core (GNP) 2004 to 2009–2010 | 0.05 | −0.19 | 0.01 | −0.02 | 0.00 | −0.01 |
| core (GNP) 2004 to 2011–2012 | 0.15 | −0.13 | 0.01 | −0.02 | 0.00 | −0.01 |
| core (GNP) 2009–2010 to 2011–2012 | 0.10 | −0.18 | 0.00 | −0.04 | 0.00 | −0.02 |
In the SW region, one male dominated reproduction, siring 17 offspring, most of which dispersed only short distances (electronic supplementary material, figure S9a). Of those offspring, 14 descended from four females, three of which were three generations of a single family (‘grandmother’–‘daughter’–‘granddaughter’). In 2004, the majority (10 of 16) of all residents sampled in the region were the offspring of this male. We detected 61 descendants of this male throughout the study. Dispersal into the region was substantial, and increased over time from a low of 5 in 2004 to 8 and 7 in 2009–2010 and 2011–2012, respectively, and included several immigrant offspring in the later sampling periods although not in 2004 (table 1). However, we identified only one long-distance disperser in each sampling period that did not come from one of the other two low diversity regions, and most immigrants were concentrated in the eastern side of the region. Despite increasing dispersal into the region, and a very slight increase in the number of bears detected, no significant difference in genetic diversity was observed over time (table 2).
The demographic processes underlying low initial diversity and subsequent increase in diversity in the EC region were more complicated than in the SE or SW. Despite low levels of HO in 2004 (figure 2a), no sampled or inferred individuals contributed disproportionately to the local gene pool. In 2004, the individual with the most resident descendants in the region is the same inferred male that contributed disproportionately to the SE region (10 residents), while 10 immigrants in the region could be traced back to an individual detected far outside of the region which fathered two male immigrants, one of which was quite reproductively successful in the region (eight descendants). In 2004, we identified 27 immigrants, 7 immigrant offspring, and 7 immigrant grandoffspring (table 1). The presence of these immigrants and especially immigrant grandoffspring in 2004, which were not detected in either of the other two regions, suggests that dispersal into the EC began at least one or two generations prior to the other two regions. In spite of the large numbers of detected immigrants, genetic diversity was relatively low in 2004, suggesting that genetic diversity in this region may have been even lower in the past. HO increased between 2004 and each of the latter two time periods, rising 0.05 (lower 95% CI 0.01) between 2004 and 2009–2010 and 0.07 (lower 95% CI 0.03) between 2004 and 2011–2012 (table 2). This also coincides with an increasing proportion of immigrants in the region, from 42% in 2004 to 46% in 2009–2010 and 53% in 2011–2012 (table 1). Levels of inbreeding (FIS) in the EC were higher than expected under random mating (i.e. there was a deficit of heterozygotes) in 2004 (FIS = 0.04 (95% CI 0.00–0.09)), but had decreased by 2009–2010 (FIS = 0.00 (95% CI −0.04–0.04)), and by 2011–2012, were lower still (FIS = −0.02 (95% CI −0.06–0.02)); (electronic supplementary material, figure S5a–c and table S3).
4. Discussion
We used data from an extensive and rigorous ecosystem-scale genetic monitoring programme to demonstrate that genetic diversity in a large carnivore can be ephemeral and dynamic on the periphery of its range. Shifts in distribution, range expansions, and recolonizations primarily driven by climate change [60–62], species invasions [63], and more recently population growth of recovering species [64] are becoming increasingly ubiquitous for all biota, and increasingly complicated by the effects of habitat degradation and fragmentation [65–68]. Despite rapidly changing species distributions worldwide, our understanding of temporal patterns of genetic diversity in expanding populations is limited, and the underlying demographic mechanisms driving evolutionary changes at range margins have largely been ignored [69]. We found that idiosyncratic demographic processes consistently led to rapid local changes in genetic diversity in a recovering carnivore found across northern latitudes.
Range contraction and habitat fragmentation of NCDE grizzly bears occurred for approximately a century, with the population reaching a low point in the 1920s or 1930s [45]. The protracted reduction in the population probably caused the reduced genetic diversity found in peripheral remnant groups at the beginning of this study, as expected when lengthy range contractions occur [70]. In this system, the effects of over 10 generations of semi-isolation and reproductive bottlenecks, common to small populations of carnivores globally [27,71–73], were erased via an influx of immigrants as the core population underwent range expansion. Our study supports the importance of dispersal and connectivity in facilitating range expansions and restoring genetic diversity [74–78] by empirically demonstrating that high landscape permeability coupled with the intrinsic dispersal capacity of large carnivores [79] can lead to rapid restoration of genetic diversity in species of conservation concern.
The small size and isolation of animals living on the range periphery often predispose remnant groups to lower genetic diversity due to reduced gene flow [80] and smaller effective population sizes [81]. In early years of the study, we found relatively few immigrants entering the peripheral regions and highly skewed reproductive success among individuals, with several individuals dominating reproduction. Over less than a single generation, two of the three remnant groups showed significant increases in HO, a pattern consistent with expectations of extreme temporal variation on the range periphery [81]. The increases of HO in the SE and EC periphery were six and seven times the magnitude of genetic change in the core over the same period of time, respectively. The increase of genetic diversity occurred in conjunction with an influx of immigrants and a surge of successful breeders. Despite the significant increase in genetic diversity, nearly all (37/45–82%) of the southernmost females detected in the SE periphery descended from three females, illustrating a trend that is likely to continue at the expansion front due to male-biased dispersal of grizzly bears [39,59,82]. A similar pattern is likely for many mammals owing to their tendency for male-biased dispersal [83], where persistently female relatedness may exist despite increases in overall genetic diversity within populations.
The evolutionary dynamics on the expansion front of a species' range has been a major focus of both theoretical and empirical studies. Theoretical studies have focused primarily on the patterns of colonization during range expansion in the absence of existing remnant groups (e.g. [7,11]), and the influence dispersal has on those patterns [8]. Similarly, empirical studies have focused on genetic drift and adaptive dynamics along the expansion axis (e.g. [84,85]). Edge populations may be particularly important sources of genetic diversity that may enable species persistence particularly in changing environments [80,81]. However, the transient nature of genetic diversity at range margins [81] greatly complicates conservation efforts that focus on protecting genetic diversity or understanding patterns of connectivity (i.e. landscape genetics; [86]) based on data with restricted spatial or temporal replication. Ultimately, the data presented here highlight that a broad spatial and temporal perspective is necessary for understanding evolutionary dynamics at the edge of species distributions where adaptive and stochastic dynamics may be at their strongest.
The patterns in genetic diversity caused by the assimilation of existing remnant groups by a core population range expansion (reduced genetic diversity in peripheral groups, founder effects, etc.) closely resemble the outcome of a pure range expansion (e.g. [87]). However, they are in fact the product of both a prolonged contraction and a subsequent recolonization. Interestingly, populations resulting from the two distinct processes may be adaptively and demographically very different. Considering the prevalence of remnant groups in carnivore populations around the world [19–29], the ongoing recovery of many populations [64], and the increasing probability of range shifts, contractions and expansions related to climate change, we expect that neutral and adaptive evolutionary changes caused by recolonization and assimilation will be increasingly common.
Patterns of genetic diversity in populations are dynamic in space and time, particularly for species undergoing changes in population dynamics and geographical distributions. Extreme changes can occur in less than a single generation and are surprisingly idiosyncratic and stochastic. Individual-level, local demography, an oft-neglected driver of contemporary evolutionary dynamics plays a critical role in shaping patterns of genetic diversity, although the overall trajectory is influenced by historical events (e.g. inbreeding, isolation, and random genetic drift) and also contemporary gene flow (e.g. dispersal). As such, we suggest caution when making landscape-scale conservation and management decisions based on a single sampling snapshot in time. Long-term, ecosystem-scale monitoring of focal populations illuminate processes that govern spatial and temporal patterns in ecological and evolutionary dynamics, providing key insights that might otherwise be missed.
Supplementary Material
Acknowledgements
We thank the agencies that provided substantial logistical, funding, and in-kind support: Blackfeet Nation; Confederated Salish and Kootenai Tribes, Montana Department of Fish, Wildlife, and Parks, Montana Department of Natural Resources and Conservation; National Park Service, Northwest Connections, US Bureau of Land Management, US Fish and Wildlife Service, and the US Forest Service. Robin Waples and Cecily Costello provided helpful comments on an earlier version of the manuscript. We also thank all of the agency personnel supporting this work and the hundreds of employees and volunteers who made this possible. Any use of trade, firm, or product names is for descriptive purposes only and does not imply endorsement by the US Government.
Data accessibility
The genetic profiles have been deposited in ScienceBase and are freely available.
Authors' contributions
N.M., T.A.G., and R.K. designed the study and analysis. N.M. performed the analyses. K.C.K., A.C.M., T.A.G., and many others painstakingly collected the data. All authors contributed to the writing of the manuscript.
Competing interests
We have no competing interests.
Funding
In addition to primary funding from USGS and USFS, NSF DEB grant no. 0919239, the David H. Smith Postdoctoral fellowship, and especially support from the Glacier National Park Conservancy enabled this analysis.
References
- 1.Ceballos G, Ehrlich PR. 2002. Mammal population losses and the extinction crisis. Science 296, 904–907. ( 10.1126/science.1069349) [DOI] [PubMed] [Google Scholar]
- 2.Laliberte AS, Ripple WJ. 2004. Range contractions of North American carnivores and ungulates. Bioscience 54, 123–138. ( 10.1641/0006-3568(2004)054%5B0123:rconac%5D2.0.co;2) [DOI] [Google Scholar]
- 3.Morrison JC, Sechrest W, Dinerstein E, Wilcove DS, Lamoreux JF. 2007. Persistence of large mammal faunas as indicators of global human impacts. J. Mamm. 88, 1363–1380. ( 10.1644/06-mamm-a-124r2.1) [DOI] [Google Scholar]
- 4.Proctor MF, McLellan BN, Strobeck C, Barclay RMR. 2005. Genetic analysis reveals demographic fragmentation of grizzly bears yielding vulnerably small populations. Proc. R. Soc. B 272, 2409–2416. ( 10.1098/rspb.2005.3246) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ripple WJ, et al. 2014. Status and ecological effects of the world's largest carnivores. Science 343, 1241484 ( 10.1126/science.1241484) [DOI] [PubMed] [Google Scholar]
- 6.Soule ME. 1985. What is conservation biology. Bioscience 35, 727–734. ( 10.2307/1310054) [DOI] [Google Scholar]
- 7.Austerlitz F, JungMuller B, Godelle B, Gouyon PH. 1997. Evolution of coalescence times, genetic diversity and structure during colonization. Theor. Popul. Biol. 51, 148–164. ( 10.1006/tpbi.1997.1302) [DOI] [Google Scholar]
- 8.Excoffier L, Foll M, Petit RJ. 2009. Genetic consequences of range expansions. Annu. Rev. Ecol. Evol. Syst. 40, 481–501. ( 10.1146/annurev.ecolsys.39.110707.173414) [DOI] [Google Scholar]
- 9.Excoffier L, Ray N. 2008. Surfing during population expansions promotes genetic revolutions and structuration. Trends Ecol. Evol. 23, 347–351. ( 10.1016/j.tree.2008.04.004) [DOI] [PubMed] [Google Scholar]
- 10.Ibrahim KM, Nichols RA, Hewitt GM. 1996. Spatial patterns of genetic variation generated by different forms of dispersal during range expansion. Heredity 77, 282–291. ( 10.1038/hdy.1996.142ER) [DOI] [Google Scholar]
- 11.Le Corre V, Kremer A. 1998. Cumulative effects of founding events during colonisation on genetic diversity and differentiation in an island and stepping-stone model. J. Evol. Biol. 11, 495–512. ( 10.1007/s000360050102) [DOI] [Google Scholar]
- 12.Peischl S, Dupanloup I, Kirkpatrick M, Excoffier L. 2013. On the accumulation of deleterious mutations during range expansions. Mol. Ecol. 22, 5972–5982. ( 10.1111/mec.12524) [DOI] [PubMed] [Google Scholar]
- 13.Slatkin M, Excoffier L. 2012. Serial founder effects during range expansion: a spatial analog of genetic drift. Genetics 191, 171–181. ( 10.1534/genetics.112.139022) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arenas M, Francois O, Currat M, Ray N, Excoffier L. 2013. Influence of admixture and paleolithic range contractions on current european diversity gradients. Mol. Biol. Evol. 30, 57–61. ( 10.1093/molbev/mss203) [DOI] [PubMed] [Google Scholar]
- 15.Hewitt G. 2000. The genetic legacy of the Quaternary ice ages. Nature 405, 907–913. ( 10.1038/35016000) [DOI] [PubMed] [Google Scholar]
- 16.Hewitt GM. 1996. Some genetic consequences of ice ages, and their role in divergence and speciation. Biol. J. Linn. Soc. 58, 247–276. ( 10.1111/j.1095-8312.1996.tb01434.x) [DOI] [Google Scholar]
- 17.Hirata D, et al. 2013. Molecular phylogeography of the brown bear (Ursus arctos) in Northeastern Asia based on analyses of complete mitochondrial DNA sequences. Mol. Biol. Evol. 30, 1644–1652. ( 10.1093/molbev/mst077) [DOI] [PubMed] [Google Scholar]
- 18.Ramakrishnan AP, Musial T, Cruzan MB. 2010. Shifting dispersal modes at an expanding species’ range margin. Mol. Ecol. 19, 1134–1146. ( 10.1111/j.1365-294X.2010.04543.x) [DOI] [PubMed] [Google Scholar]
- 19.Cegelski CC, Waits LP, Anderson NJ. 2003. Assessing population structure and gene flow in Montana wolverines (Gulo gulo) using assignment-based approaches. Mol. Ecol. 12, 2907–2918. ( 10.1046/j.1365-294X.2003.01969.x) [DOI] [PubMed] [Google Scholar]
- 20.Ciucci P, Gervasi V, Boitani L, Boulanger J, Paetkau D, Prive R, Tosoni E. 2015. Estimating abundance of the remnant Apennine brown bear population using multiple noninvasive genetic data sources. J. Mamm. 96, 206–220. ( 10.1093/jmammal/gyu029) [DOI] [Google Scholar]
- 21.Clevenger AP, Purroy FJ, Campos MA. 1997. Habitat assessment of a relict brown bear Ursus arctos population in northern Spain. Biol. Conserv. 80, 17–22. ( 10.1016/s0006-3207(96)00081-x) [DOI] [Google Scholar]
- 22.Darimont CT, Paquet PC. 2002. The gray wolves, Canis lupus, of British Columbia's central and north coast: distribution and conservation assessment. Can. Field Nat. 116, 416–422. [Google Scholar]
- 23.Doko T, Fukui H, Kooiman A, Toxopeus AG, Ichinose T, Chen W, Skidmore AK. 2011. Identifying habitat patches and potential ecological corridors for remnant Asiatic black bear (Ursus thibetanus japonicus) populations in Japan. Ecol. Modell. 222, 748–761. ( 10.1016/j.ecolmodel.2010.11.005) [DOI] [Google Scholar]
- 24.Gil-Sanchez JM, Ballesteros-Duperon E, Bueno-Segura JF. 2006. Feeding ecology of the Iberian lynx Lynx pardinus in eastern Sierra Morena (Southern spain). Acta Theriologica 51, 85–90. ( 10.1007/bf03192659) [DOI] [Google Scholar]
- 25.Kyle CJ, Strobeck C. 2001. Genetic structure of North American wolverine (Gulo gulo) populations. Mol. Ecol. 10, 337–347. ( 10.1046/j.1365-294x.2001.01222.x) [DOI] [PubMed] [Google Scholar]
- 26.Miotto RA, Cervini M, Figueiredo MG, Begotti RA, Galetti PM. 2011. Genetic diversity and population structure of pumas (Puma concolor) in southeastern Brazil: implications for conservation in a human-dominated landscape. Conserv. Genet. 12, 1447–1455. ( 10.1007/s10592-011-0243-8) [DOI] [Google Scholar]
- 27.Straka M, Paule L, Ionescu O, Stofik J, Adamec M. 2012. Microsatellite diversity and structure of Carpathian brown bears (Ursus arctos): consequences of human caused fragmentation. Conserv. Genet. 13, 153–164. ( 10.1007/s10592-011-0271-4) [DOI] [Google Scholar]
- 28.van Gils H, Westinga E, Carafa M, Antonucci A, Ciaschetti G. 2014. Where the bears roam in Majella National Park, Italy. J. Nat. Conserv. 22, 23–34. ( 10.1016/j.jnc.2013.08.001) [DOI] [Google Scholar]
- 29.Weckworth BV, Talbot SL, Cook JA. 2010. Phylogeography of wolves (Canis lupus) in the Pacific Northwest. J. Mamm. 91, 363–375. ( 10.1644/09-mamm-a-036.1) [DOI] [Google Scholar]
- 30.Stirling I, Derocher AE. 2012. Effects of climate warming on polar bears: a review of the evidence. Glob. Change Biol. 18, 2694–2706. ( 10.1111/j.1365-2486.2012.02753.x) [DOI] [PubMed] [Google Scholar]
- 31.Castilho CS, Marins-Sa LG, Benedet RC, Freitas TO. 2011. Landscape genetics of mountain lions (Puma concolor) in southern Brazil. Mamm. Biol. 76, 476–483. ( 10.1016/j.mambio.2010.08.002) [DOI] [Google Scholar]
- 32.Costello CM, Creel SR, Kalinowski ST, Vu NV, Quigley HB. 2009. Determinants of male reproductive success in American black bears. Behav. Ecol. Sociobiol. 64, 125–134. ( 10.1007/s00265-009-0828-0) [DOI] [Google Scholar]
- 33.De Barba M, Waits LP, Garton EO, Genovesi P, Randi E, Mustoni A, Groff C. 2010. The power of genetic monitoring for studying demography, ecology and genetics of a reintroduced brown bear population. Mol. Ecol. 19, 3938–3951. ( 10.1111/j.1365-294X.2010.04791.x) [DOI] [PubMed] [Google Scholar]
- 34.Frosch C, Dutsov A, Zlatanova D, Valchev K, Reiners TE, Steyer K, Pfenninger M, Nowak C. 2014. Noninvasive genetic assessment of brown bear population structure in Bulgarian mountain regions. Mamm. Biol. 79, 268–276. ( 10.1016/j.mambio.2014.04.001) [DOI] [Google Scholar]
- 35.Hedmark E, Ellegren H. 2007. DNA-based monitoring of two newly founded Scandinavian wolverine populations. Conserv. Genet. 8, 843–852. ( 10.1007/s10592-006-9231-9) [DOI] [Google Scholar]
- 36.Itoh T, Sato Y, Kobayashi K, Mano T, Iwata R. 2012. Effective dispersal of brown bears (Ursus arctos) in eastern Hokkaido, inferred from analyses of mitochondrial DNA and microsatellites. Mamm. Study 37, 29–41. ( 10.3106/041.037.0104) [DOI] [Google Scholar]
- 37.Moore JA, Draheim HM, Etter D, Winterstein S, Scribner KT. 2014. Application of large-scale parentage analysis for investigating natal dispersal in highly vagile vertebrates: a case study of American black bears (Ursus americanus). PLoS ONE 9, e91168 ( 10.1371/journal.pone.0091168) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Moore JA, Xu R, Frank K, Draheim H, Scribner KT. 2015. Social network analysis of mating patterns in American black bears (Ursus americanus). Mol. Ecol. 24, 4010–4022. ( 10.1111/mec.13290) [DOI] [PubMed] [Google Scholar]
- 39.Proctor MF, McLellan BN, Strobeck C, Barclay RMR. 2004. Gender-specific dispersal distances of grizzly bears estimated by genetic analysis. Can. J. Zool. 82, 1108–1118. ( 10.1139/z04-077) [DOI] [Google Scholar]
- 40.Sawaya MA, Kalinowski ST, Clevenger AP. 2014. Genetic connectivity for two bear species at wildlife crossing structures in Banff National Park. Proc. R. Soc. B 281, 20131705 ( 10.1098/rspb.2013.1705) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Horn RC, Engh AL, Scribner KT, Funk SM, Holekamp KE. 2004. Behavioural structuring of relatedness in the spotted hyena (Crocuta crocuta) suggests direct fitness benefits of clan-level cooperation. Mol. Ecol. 13, 449–458. ( 10.1046/j.1365-294X.2003.02071.x) [DOI] [PubMed] [Google Scholar]
- 42.U.S. Fish and Wildlife Service [USFWS]. 2011. Grizzly bear (Ursus arctos horribilis) 5-year review: summary and evaluation, 205 Missoula, MT: U.S. Fish and Wildlife Service, Grizzly Bear Recovery Office. [Google Scholar]
- 43.Proctor MF, et al. 2012. Population fragmentation and inter-ecosystem movements of grizzly bears in western Canada and the northern United States. Wildl. Monogr. 180, 1–46. ( 10.1002/wmon.6) [DOI] [Google Scholar]
- 44.Kendall KC, et al. 2015. Density, distribution, and genetic structure of grizzly bears in the Cabinet-Yaak Ecosystem. J. Wildl. Manage. 80, 314–331. ( 10.1002/jwmg.1019) [DOI] [Google Scholar]
- 45.U.S. Fish and Wildlife Service [USFWS]. 1993. Grizzly bear recovery plan. Missoula, MT: U.S. Fish Wildlife Service. [Google Scholar]
- 46.Kendall KC, Stetz JB, Boulanger J, Macleod AC, Paetkau D, White GC. 2009. Demography and genetic structure of a recovering grizzly bear population. J. Wildl. Manage. 73, 3–17. ( 10.2193/2008-330) [DOI] [Google Scholar]
- 47.Mace RD, et al. 2012. Grizzly bear population vital rates and trend in the Northern Continental Divide Ecosystem, Montana. J. Wildl. Manage. 76, 119–128. ( 10.1002/jwmg.250) [DOI] [Google Scholar]
- 48.Kamath PL, Haroldson MA, Luikart G, Paetkau D, Whitman C, Van Manen FT. 2015. Multiple estimates of effective population size for monitoring a long-lived vertebrate: an application to Yellowstone grizzly bears. Mol. Ecol. 24, 5507–5521. ( 10.1111/mec.13398) [DOI] [PubMed] [Google Scholar]
- 49.Ennis S, Gallagher TF. 1994. A PCR-based sex-determination assay in cattle based on the bovine amelogenin locus. Anim. Genet. 25, 425–427. ( 10.1111/j.1365-2052.1994.tb00533.x) [DOI] [PubMed] [Google Scholar]
- 50.Peakall R, Smouse PE. 2012. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28, 2537–2539. ( 10.1093/bioinformatics/bts460) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Shirk AJ.2014. sGD: spatially explicit estimation of genetic diversity indices and Wright's neighborhood size (NS). R package version 2.0. 2008;73, 158–70. (doi:10.3389/fevo.2014.00062)
- 52.R Core Team. 2015. R: a language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; See http://www.R-project.org/. [Google Scholar]
- 53.Jones OR, Wang JL. 2010. COLONY: a program for parentage and sibship inference from multilocus genotype data. Mol. Ecol. Resources 10, 551–555. ( 10.1111/j.1755-0998.2009.02787.x) [DOI] [PubMed] [Google Scholar]
- 54.Wang JL. 2004. Sibship reconstruction from genetic data with typing errors. Genetics 166, 1963–1979. ( 10.1534/genetics.166.4.1963) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang JL, Santure AW. 2009. Parentage and sibship inference from multilocus genotype data under polygamy. Genetics 181, 1579–1594. ( 10.1534/genetics.108.100214) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Harrison HB, Saenz-Agudelo P, Planes S, Jones GP, Berumen ML. 2013. Relative accuracy of three common methods of parentage analysis in natural populations. Mol. Ecol. 22, 1158–1170. ( 10.1111/mec.12138) [DOI] [PubMed] [Google Scholar]
- 57.Graves TA, Royle JA, Kendall KC, Beier P, Stetz JB, Macleod AC. 2012. Balancing Precision and Risk: Should Multiple Detection Methods Be Analyzed Separately in N-Mixture Models? PLoS ONE 7(12). ( 10.1371/journal.pone.0049410) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Keenan K, McGinnity P, Cross TF, Crozier WW, Prodöhl PA. 2013. diveRsity: an R package for the estimation of population genetics parameters and their associated errors. Methods Ecol. Evol. 4, 782–788. ( 10.1111/2041-210X.12067) [DOI] [Google Scholar]
- 59.Graves T, Chandler RB, Royle JA, Beier P, Kendall KC. 2014. Estimating landscape resistance to dispersal. Landscape Ecol. 29, 1201–1211. ( 10.1007/s10980-014-0056-5) [DOI] [Google Scholar]
- 60.Chen IC, Hill JK, Ohlemueller R, Roy DB, Thomas CD. 2011. Rapid range shifts of species associated with high levels of climate warming. Science 333, 1024–1026. ( 10.1126/science.1206432) [DOI] [PubMed] [Google Scholar]
- 61.Parmesan C, Yohe G. 2003. A globally coherent fingerprint of climate change impacts across natural systems. Nature 421, 37–42. ( 10.1038/nature01286) [DOI] [PubMed] [Google Scholar]
- 62.Root TL, Price JT, Hall KR, Schneider SH, Rosenzweig C, Pounds JA. 2003. Fingerprints of global warming on wild animals and plants. Nature 421, 57–60. ( 10.1038/nature01333) [DOI] [PubMed] [Google Scholar]
- 63.Simberloff D, et al. 2013. Impacts of biological invasions: what's what and the way forward. Trends Ecol. Evol. 28, 58–66. ( 10.1016/j.tree.2012.07.013) [DOI] [PubMed] [Google Scholar]
- 64.Crooks KR, Burdett CL, Theobald DM, Rondinini C, Boitani L. 2011. Global patterns of fragmentation and connectivity of mammalian carnivore habitat. Phil. Trans. R. Soc. B 366, 2642–2651. ( 10.1098/rstb.2011.0120) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hodgson JA, Thomas CD, Dytham C, Travis JMJ, Cornell SJ. 2012. The speed of range shifts in fragmented landscapes. PLoS ONE 7, e47141 ( 10.1371/journal.pone.0047141) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Honnay O, Verheyen K, Butaye J, Jacquemyn H, Bossuyt B, Hermy M. 2002. Possible effects of habitat fragmentation and climate change on the range of forest plant species. Ecol. Lett. 5, 525–530. ( 10.1046/j.1461-0248.2002.00346.x) [DOI] [Google Scholar]
- 67.Opdam P, Wascher D. 2004. Climate change meets habitat fragmentation: linking landscape and biogeographical scale levels in research and conservation. Biol. Conserv. 117, 285–297. ( 10.1016/j.biocon.2003.12.008) [DOI] [Google Scholar]
- 68.Eckert CG, Samis KE, Lougheed SC. 2008. Genetic variation across species’ geographical ranges: the central-marginal hypothesis and beyond. Mol. Ecol. 17, 1170–1188. ( 10.1111/j.1365-294X.2007.03659.x) [DOI] [PubMed] [Google Scholar]
- 69.Arenas M, Ray N, Currat M, Excoffier L. 2012. Consequences of range contractions and range shifts on molecular diversity. Mol. Biol. Evol. 29, 207–218. ( 10.1093/molbev/msr187) [DOI] [PubMed] [Google Scholar]
- 70.Aspi J, Roininen E, Ruokonen M, Kojola I, Vila C. 2006. Genetic diversity, population structure, effective population size and demographic history of the Finnish wolf population. Mol. Ecol. 15, 1561–1576. ( 10.1111/j.1365-294X.2006.02877.x) [DOI] [PubMed] [Google Scholar]
- 71.Jones ME, Paetkau D, Geffen E, Moritz C. 2004. Genetic diversity and population structure of Tasmanian devils, the largest marsupial carnivore. Mol. Ecol. 13, 2197–2209. ( 10.1111/j.1365-294X.2004.02239.x) [DOI] [PubMed] [Google Scholar]
- 72.Wisely SM, Buskirk SW, Fleming MA, McDonald DB, Ostrander EA. 2002. Genetic diversity and fitness in black-footed ferrets before and during a bottleneck. J. Heredity 93, 231–237. ( 10.1093/jhered/93.4.231) [DOI] [PubMed] [Google Scholar]
- 73.Keller LF, Waller DM. 2002. Inbreeding effects in wild populations. Trends Ecol. Evol. 17, 230–241. ( 10.1016/s0169-5347(02)02489-8) [DOI] [Google Scholar]
- 74.McEachern MB, Van Vuren DH, Floyd CH, May B, Eadie JM. 2011. Bottlenecks and rescue effects in a fluctuating population of golden-mantled ground squirrels (Spermophilus lateralis). Conserv. Genet. 12, 285–296. ( 10.1007/s10592-010-0139-z) [DOI] [Google Scholar]
- 75.Beier P, Brost B. 2010. Use of land facets to plan for climate change: conserving the arenas, not the actors. Conserv. Biol. 24, 701–710. ( 10.1111/j.1523-1739.2009.01422.x) [DOI] [PubMed] [Google Scholar]
- 76.Vila C, et al. 2003. Rescue of a severely bottlenecked wolf (Canis lupus) population by a single immigrant. Proc. R. Soc. Lond. B 270, 91–97. ( 10.1098/rspb.2002.2184) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zgurski JM, Hik DS. 2014. Gene flow and the restoration of genetic diversity in a fluctuating collared pika (Ochotona collaris) population. Conserv. Genet. 15, 37–48. ( 10.1007/s10592-013-0519-2) [DOI] [Google Scholar]
- 78.Schloss CA, Nunez TA, Lawler JJ. 2012. Dispersal will limit ability of mammals to track climate change in the Western Hemisphere. Proc. Natl Acad. Sci. USA 109, 8606–8611. ( 10.1073/pnas.1116791109) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lesica P, Allendorf FW. 1995. When are peripheral-populations valuable for conservation? Conserv. Biol. 9, 753–760. ( 10.1046/j.1523-1739.1995.09040753.x) [DOI] [Google Scholar]
- 80.Vucetich JA, Waite TA. 2003. Spatial patterns of demography and genetic processes across the species’ range: null hypotheses for landscape conservation genetics. Conserv. Genet. 4, 639–645. ( 10.1023/a:1025671831349) [DOI] [Google Scholar]
- 81.McLellan BN, Hovey FW. 2001. Natal dispersal of grizzly bears. Can. J. Zool. 79, 838–844. ( 10.1139/cjz-79-5-838) [DOI] [Google Scholar]
- 82.Dobson FS. 1982. Competition for mates and predominant juvenile male dispersal in mammals. Anim. Behav. 30, 1183–1192. ( 10.1016/S0003-3472(82)80209-1) [DOI] [Google Scholar]
- 83.Buckley J, Butlin RK, Bridle JR. 2012. Evidence for evolutionary change associated with the recent range expansion of the British butterfly, Aricia agestis, in response to climate change. Mol. Ecol. 21, 267–280. ( 10.1111/j.1365-294X.2011.05388.x) [DOI] [PubMed] [Google Scholar]
- 84.White TA, Perkins SE, Heckel G, Searle JB. 2013. Adaptive evolution during an ongoing range expansion: the invasive bank vole (Myodes glareolus) in Ireland. Mol. Ecol. 22, 2971–2985. ( 10.1111/mec.12343) [DOI] [PubMed] [Google Scholar]
- 85.Manel S, Schwartz MK, Luikart G, Taberlet P. 2003. Landscape genetics: combining landscape ecology and population genetics. Trends Ecol. Evol. 18, 189–197. ( 10.1016/s0169-5347(03)00008-9) [DOI] [Google Scholar]
- 86.Chapron G, et al. 2014. Recovery of large carnivores in Europe's modern human-dominated landscapes. Science 346, 1517–1519. ( 10.1126/science.1257553) [DOI] [PubMed] [Google Scholar]
- 87.Hagen SB, Kopatz A, Aspi J, Kojola I, Eiken HG. 2015. Evidence of rapid change in genetic structure and diversity during range expansion in a recovering large terrestrial carnivore. Proc. R. Soc. B 282, 20150092 ( 10.1098/rspb.2015.0092) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The genetic profiles have been deposited in ScienceBase and are freely available.