

Abstract
We use dispersion-corrected density functional theory molecular dynamics simulations to predict the ionic, electronic and vibrational properties of the SrTiO3/H2O solid–liquid interface. Approximately 50% of surface oxygens on the planar SrO termination are hydroxylated at all studied levels of water coverage, the corresponding number being 15% for the planar TiO2 termination and 5% on the stepped TiO2-terminated surface. The lateral ordering of the hydration structure is largely controlled by covalent-like surface cation to H2O bonding and surface corrugation. We find a featureless electronic density of states in and around the band gap energy region at the solid–liquid interface. The vibrational spectrum indicates redshifting of the O–H stretching band due to surface-to-liquid hydrogen bonding and blueshifting due to high-frequency stretching vibrations of OH fragments within the liquid, as well as strong suppression of the OH stretching band on the stepped surface. We find highly varying rates of proton transfer above different SrTiO3 surfaces, owing to differences in hydrogen bond strength and the degree of dissociation of incident water. Trends in proton dynamics and the mode of H2O adsorption among studied surfaces can be explained by the differential ionicity of the Ti–O and Sr–O bonds in the SrTiO3 crystal.
Keywords: strontium titanate, density functional theory, molecular dynamics, hydration structure
1. Introduction
Understanding the interaction of metal-oxide surfaces and water is a topic of broad interest. Research of these systems is inspired by applications in such fields as geochemistry, catalysis and chemical sensing [1,2]. A particular focus of interest in recent decades has been water-splitting using a metal-oxide anode such as SrTiO3 or TiO2 in an electrochemical cell as a photocatalyst, enabling the conversion of solar radiation into chemical potential energy in the form of H2 [3]. Making this environmentally appealing process efficient and economically viable requires a thorough understanding of the relevant oxide–water interface.
While the photocatalytic capability of SrTiO3 was demonstrated experimentally over 40 years ago [4], a good understanding of the SrTiO3/H2O solid–liquid interface has remained elusive. The general view emerging from temperature-programmed desorption spectroscopy (TPD) [5–7], friction-force microscopy [8], X-ray photoemission spectroscopy [9] and surface X-ray diffraction [10] is that H2O tends to adsorb strongly and through dissociation on step edges, vacancy sites and onto the SrO-terminated (001) surface, but molecularly and weakly onto the stoichiometric TiO2-terminated surface. This view is broadly corroborated by static DFT [7,11,12] and hybrid functional [13] calculations as well as molecular dynamics based on empirical interaction potentials [14], all up to one monolayer of H2O coverage on the planar SrO- and TiO2-terminated surfaces. In spite of this large body of work, a detailed understanding of the hydration structure of SrTiO3, the electronic structure of the SrTiO3/H2O interface, as well as the vibrational properties at the interface, in particular at high levels of H2O coverage and non-planar surfaces, is missing.
In this work, we use dispersion-corrected density functional theory molecular dynamics (DFT MD) to attack this problem. We compute the hydration structure of SrTiO3 at varying levels of H2O coverage for both planar and non-planar SrTiO3 surfaces. A strongly mixed mode of molecular and dissociative adsorption is found for planar (001) surfaces, with a crossover to more molecular adsorption on a TiO2-terminated stepped surface and the ( or ‘RT13’ surface reconstruction [15]. Bonding of covalent character plays an important role alongside surface corrugation, hydrogen bonding and the dispersion interaction in the ordering of the hydration structure. While the electronic structure around the band gap region of the solid–liquid interface displays no features of interest, the vibrational spectrum of adsorbed H2O is significantly altered at the O–H stretching band frequencies. Proton transfer rates and the degree of surface hydroxylation are highest on the SrO-terminated surface, an effect we trace to the high ionicity of the Sr–O bond with respect to the Ti–O bond in the crystal.
2. Methodology
To gain a thorough understanding of the solid–liquid interface of SrTiO3 and H2O, we studied four different surfaces for the oxide: bulk-like SrO-terminated (001), bulk-like TiO2-terminated (001), the TiO2-dominated and strongly corrugated RT13 surface reconstruction [15] and a stepped TiO2-terminated (001) surface. The first two of these are commonly observed in experiment [16], whereas the RT13 surface is believed to be among the most stable SrTiO3 surfaces near ambient conditions and particularly suitable for homoepitaxial growth of SrTiO3 and heteroepitaxial growth of other perovskite oxides [17]. We chose the TiO2 termination for our stepped (001) surface, as experiments suggest that this termination dominates over the SrO termination in step-and-terrace systems [18,19].
The four different SrTiO3 surfaces were each modelled with a slab geometry (figure 1). The supercell was periodic in all three dimensions, and a vacuum gap of approximately 20 Å was created in the [001] direction between periodic slab images. In order to achieve a vanishing surface dipole for the overall neutral slabs, all slabs were made mirror-symmetric with respect to the central plane. The SrO and TiO2-terminated slabs were four and a half unit cells in thickness and 4×4 unit cells in the lateral dimension, whereas the RT13 slab consisted of four unit cells of bulk SrTiO3 beneath the surface reconstruction and one single surface unit cell in the lateral plane. The size of the stepped surface was 6×3 unit cells in the lateral dimension and six unit cells in the vertical dimension. One to three central atomic planes of each slab were fixed, depending on thickness of the slab (figure 1), with the lattice constant for fixed layers set to its equilibrium bulk value for the exchange-correlation functional in question.
Figure 1.

Visualizations [20] of the relaxed SrO-, TiO2- and RT13-terminated slabs and the TiO2-terminated stepped surface in vacuum. Definitions for the surface and subsurfacelayer used in this work are indicated by solid and dashed lines, and the shaded areas indicate fixed atomic layers. O ions are red, Sr is yellow and Ti is pink. Only the top Ti–O layer is shown in the top view of the RT13 surface. (Online version in colour.)
For all static structure optimizations and molecular dynamics runs, we used the CP2K code [21], which hosts a hybrid LCAO and planewave basis set. This set-up is efficient for computations involving surfaces and large vacuum regions, as is the case in this work. We sample the Brillouin zone at the Γ-point, which is justified by the large size of the supercell. Goedecker-Teter-Hutter pseudopotentials [22–24] are used to avoid explicit calculation of the core electrons. We use the DZVP localized basis set [25] as a good compromise between precision and speed.
The standard semi-local formulations of the exchange–correlation functional in DFT exclude, by definition, the long-range dispersion interaction between atoms and molecules [26]. As this electron correlation effect can be important for surface stability, surface–molecule interaction and molecule–molecule interaction [27], dispersion effects should be considered when modelling the present systems of interest. To enable us to include dispersion effects in our model and to still perform large-scale DFT MD simulations, we use the Grimme D3 dispersion correction [28]. To guide us in the choice of exchange–correlation functional, we initially employed a range of functionals (LDA [29], PBE [30], PBEsol [31], PBE-D3, BLYP [32,33], BLYP-D3) to compute the lattice constant, bulk modulus and band gap of SrTiO3 in the bulk cubic perovskite phase (4×4×4 unit cells), and the ionic displacements upon relaxation of the (001) SrO- and TiO2-terminated surfaces as well as the surface energy of the bulk-like (001)-terminated crystal. Additionally, using the subset of functionals PBE, PBE-D3 and PBEsol, we performed a set of static structural optimizations of single-molecule adsorption of H2O onto the SrO- and TiO2-terminated planar slabs, where the H2O was initially placed over the most important high-symmetry points on each surface termination. As shown in §3a, this set of static calculations ultimately showed PBE-D3 to offer the overall best results. Therefore, all subsequent MD calculations were done using PBE-D3.
We performed all MD runs at 300 K in the NV T ensemble, employing a time-step of 0.5 fs and the Nosé–Hoover chain thermostat (to all except the fixed atoms in the system) with three links and a time constant of 20 fs, i.e. a frequency of 5×1013 Hz, which is in the range of the dominant vibrational frequencies expected in the system (1013 Hz for SrTiO3 and 2–10×1013 Hz for H2O [34]). The total duration of the MD was 10 ps for the SrO-, TiO2- and RT13-terminated surfaces (448-734 atoms, 3072-3944 electrons per system, 60–150 s ionic−1 step on a Cray XC40 over 384 cores), and 5 ps for the stepped surface (1332 atoms, 6816 electrons, 170 s ionic−1 step over 864 cores) due to the very large size of this system. A drift of no more than −1 meV atom−1 ps−1 was observed in any of the MD runs after an initial equilibration period of 5 and 3 ps, respectively, which indicates that the systems were reasonably close to dynamic equilibrium. We considered three levels of H2O coverage: 0.5 monolayers (ML, defined here as one H2O per surface unit cell or cation), 1.0 ML, and bulk H2O. For the 0.5 ML and 1.0 ML coverages, the H2O molecules were initially positioned in the middle of the vacuum layer in two or four sheets of 4×4 molecules each, respectively, the molecules being spaced laterally in units of the crystal lattice constant and each molecule being uniformly randomly rotated about the O ion. For bulk water coverage, the gap between two periodic slab surfaces and the number of water molecules were adjusted to give the experimental density of 1 g cm−3 for the total density of H2O between the surfaces. To gain an understanding of how the level of water coverage affects the hydration structure of SrTiO3, all three levels of coverage were simulated for the SrO- and TiO2-terminated planar surfaces. On the RT13 and the stepped surface, we considered only bulk water coverage, as this is most relevant for real-world applications in photocatalysis using SrTiO3.
After an equilibration period of 5 ps (3 ps for the stepped surface), all MD runs were analysed for the vertical density profile of H2O and the histogram of lateral positioning of H2O in each hydration layer for the remaining duration of each simulation trajectory (over all time steps). These quantities yield a detailed description of the hydration structure of the SrTiO3 surface. In addition, to analyse the vibrational properties of H2O on the photocatalyst surface, we computed the vibrational density of states (VDOS) of H2O by performing a Fourier transform of the velocity auto-correlation function [35]. The fraction of surface hydroxylation, the fraction of dissociated H2O in the liquid, and rates of proton transfer within the system were assessed by studying the O–H coordination statistics of O in the slab surface and in the liquid region. By computing the O–H radial distribution function, we found a radial cutoff of 1.25 Å to define the first O–H coordination shell for oxygens in both the liquid and in the slab surface.
Finally, to compute the electronic density of states (EDOS) more precisely than allowed for by the Γ-point sampling of CP2K, we employed the VASP [36–39] code to compute the mean EDOS over five uncorrelated snapshots off each MD trajectory. For the planar surfaces and RT13, a 2×2×1 Γ-centred Monkhorst-Pack [40] grid was used to sample the Brillouin zone, and the energy cutoff for the planewave basis was set to 750 eV. For the stepped surface, a 1×3×1 grid was used, and the planewave cutoff energy was set to 500 eV to fulfil memory constraints on the computation. Only a single representative density of states (DOS) computation is presented for this system, due to computational constraints. The projector-augmented wave method was used in all VASP calculations to avoid explicit treatment of the core electrons.
3. Results
In this section, we first present results from our static calculations, including single-molecule adsorption of H2O onto SrTiO3. Then, we present results from our MD simulations at finite temperature and higher levels of water coverage.
(a). Static calculations
(i). Bulk SrTiO3
Results for bulk SrTiO3 are presented in table 1. For each functional, we find the equilibrium lattice constant (λ) and bulk modulus at zero pressure (B0) by fitting a third-order Birch–Murnaghan equation of state [44] to a set of (V,E) data points from a set of static NV T calculations on the crystal. Whereas of the GGA-type functionals PBE and BLYP slightly overestimate λ with respect to experiment, PBEsol gives excellent results. LDA in turn slightly underestimates λ. Correspondingly, the GGA functionals give a softer crystal than LDA does, the former being closer to experiment. Overall, these results on the mechanical properties of SrTiO3 reflect the usual underbinding exhibited by GGA and overbinding given by LDA. For all functionals, the band gap is about 1.5 eV lower than in experiment. Our findings are in agreement with earlier DFT results on SrTiO3 [45].
Table 1.
Computed equilibrium lattice constant (λ), bulk modulus at zero pressure (B0) and minimum band gap (Eg) for cubic SrTiO3 perovskite at static conditions, along with experimental results at 300 K.
(ii). Clean surface in vacuum
As cleaving the SrTiO3 crystal reveals both the SrO and TiO2 terminations, we define the surface energy of the bulk-like (001) termination to be [46]
| 3.1 |
where
| 3.2 |
here ESrO being the total energy of the SrO-terminated slab, NSrTiO3 being the number of bulk unit cells of SrTiO3 in the slab (), being the total energy per unit cell of bulk SrTiO3, A being the area of each exposed surface of the slab and correspondingly for . Before relaxing the structure, the symmetry of the surface was broken by introducing small random displacements for the outermost layer of ions on each side of the slab. Then, to find ESrO and ETiO2, we ran conjugate-gradient relaxation on the ionic structure until the total force acting on any ion was below 0.05 eV Å −1.
The results for the surface energy are given in table 2. Except for BLYP, our results agree broadly with previous DFT calculations on the quantity [13,46,47]. A slight exception is our result for γ obtained using PBE, which is somewhat lower than previously published values. It is possible that by breaking the symmetry of the surface prior to relaxation and allowing displacements of the ions in all spatial dimensions, we have found a slightly more favourable arrangement for the ionic structure.
Table 2.
Computed surface energy of SrTiO3 in units of eV/surface (1×1) unit cell and J m−2 (see text) and the minimum band gap of the slab system with either the SrO or TiO2 termination at static conditions.
| functional | γ (eV/1×1) | γ (J m−2) | ESrOg (eV) | ETiO2g (eV) |
|---|---|---|---|---|
| LDA | 1.28 | 1.33 | 1.69 | 0.87 |
| PBE | 0.99 | 1.03 | 1.68 | 0.99 |
| PBE-D3 | 1.23 | 1.28 | 1.72 | 0.94 |
| PBEsol | 1.13 | 1.18 | 1.64 | 0.95 |
| BLYP | 2.11 | 2.20 | 1.81 | 0.99 |
| BLYP-D3 | 2.53 | 2.64 | 1.82 | 0.91 |
We report the mean atomic displacements in the [001] direction (perpendicular to the slab surface) upon relaxation from the bulk-like structure in table 3. Our results are again in good qualitative agreement with previous DFT calculations [13,46–48]. Experimental results are available for the surface rumpling (S), i.e. the relative displacement of O with respect to the metal cation in the outermost plane of the slab (table 4), and the change in the cation–cation distance between neighbouring planes i and j upon relaxation (Dij, table 5). While for S our results agree with results from low-energy electron diffraction (LEED) [49] and reflection high-energy electron diffraction (RHEED) [50] experiments, overall our calculated values for the quantity Dij cannot be reconciled with these measurements. We note, however, that the experimental results on Dij for the first two planes on the SrO termination and the second and third planes on the TiO2 termination conflict each other on the sign of the displacement Dij, a discrepancy which has been noted before and the reason for which is not clear [46]. Our values for these structural parameters are again in good agreement with previous calculations [13,46–48].
Table 3.
Mean displacements of ions (in Å) in the [001] direction upon relaxation of the slab from the bulk-like structure.
| layer | ion type | LDA | PBE | PBE-D3 | PBEsol |
|---|---|---|---|---|---|
| SrO termination | |||||
| 1 | Sr | −0.19 | −0.17 | −0.17 | −0.22 |
| O | 0.016 | 0.048 | 0.052 | 0.0059 | |
| 2 | Ti | 0.059 | 0.093 | 0.091 | 0.047 |
| O | 0.016 | 0.045 | 0.044 | −0.0048 | |
| 3 | Sr | −0.048 | −0.036 | −0.039 | −0.062 |
| O | −0.0026 | 0.017 | 0.016 | −0.014 | |
| TiO2 termination | |||||
| 1 | Ti | −0.092 | −0.096 | −0.092 | −0.11 |
| O | −0.016 | 0.0083 | 0.0082 | −0.029 | |
| 2 | Sr | 0.12 | 0.13 | 0.12 | 0.11 |
| O | 0.012 | 0.0054 | 0.0028 | −0.0064 | |
| 3 | Ti | −0.015 | −0.030 | −0.030 | −0.030 |
| O | −0.0033 | −0.027 | −0.028 | −0.026 | |
Table 4.
Surface rumpling (S), i.e. the relative displacement of O in the [001] direction with respect to the metal cation in the topmost plane of the slab (in Å).
Table 5.
Change in cation–cation distance (in Å) between neighbouring planes i and j (Dij) in the [001] direction upon relaxing the slab.
(iii). Single-molecule adsorption of H2O onto SrTiO3
We present the energy of adsorption of a single molecule of H2O onto SrTiO3 from our static conjugate-gradient relaxations (equivalent to a 1/32 ML water coverage of the surface) along with the adsorption energy from previous computational studies (with varying levels of water coverage) in table 6. For each exchange–correlation functional and surface termination, we report the highest, i.e. most favourable adsorption energy in both the molecular and dissociated configurations of the molecule. In the former, water adsorbs as an intact molecule onto the surface, whereas in the latter, the molecule breaks up into an OH fragment and H, and a covalent bond is formed between the H and a surface O ion. We have applied the counterpoise correction to the basis set superposition error in our results for the adsorption energy, the correction being within the range of −0.001 to −0.065 eV molecule−1 throughout all configurations.
Table 6.
The most favourable adsorption energy of H2O in the molecular (Emolads) and dissociated (Edissads) mode for each bulk-like surface termination of (001) SrTiO3. Results from previous studies are presented for comparison. The numbers in parentheses for PBE-D3 signify the contribution of dispersion energy in the total adsorption energy given.
| Emolads (eV) | Edissads (eV) | coverage (ML) | |
|---|---|---|---|
| SrO termination | |||
| PBE | 0.64 | 1.0 | 1/32 |
| PBE-D3 | 0.82 (0.46) | 1.1 (0.43) | 1/32 |
| PBEsol | 1.0 | 1.3 | 1/32 |
| PBE [11] | — | 1.4 | 1/18 |
| PBE [12] | 0.90 | 1.3 | 1/8 |
| B3LYP [13] | 0.91 | 0.92 | 1/2 |
| TiO2 termination | |||
| PBE | 0.65 | 0.52 | 1/32 |
| PBE-D3 | 0.86 (0.21) | 0.94 (0.20) | 1/32 |
| PBEsol | 0.94 | 0.99 | 1/32 |
| PBE [11] | 0.72 | 0.99 | 1/18 |
| PBE [12] | 0.89 | 1.1 | 1/8 |
| PBEsol [12] | 2.67 | 2.83 | 1/8 |
| B3LYP [13] | 0.84 | 0.74 | 1/2 |
Our values for these adsorption energies are slightly lower than the previous literature DFT values, a finding we attribute to the much lower effective H2O coverage in our calculations. Overall the agreement is reasonable and largely within the spread of previous DFT results. A striking difference can be seen, however, between our PBEsol results and those of Hinojosa et al. [12]. The extremely high adsorption energy of 2.67 eV in the latter work is clearly at odds with the present results as well as those of previous studies. The value of 0.92 eV for molecular adsorption onto the TiO2-termination as deduced [12] from TPD experiments [6] compares well with our corresponding PBE-D3 value of 0.91 eV. In terms of thermodynamics, these static results imply that the SrO termination clearly favours dissociation, while the difference in preference between the molecular and dissociative modes of adsorption is smaller on the TiO2 termination. We find the dispersion interaction to make up a significant fraction of the total adsorption energy, contributing approximately 0.4 eV molecule−1 on the SrO termination and 0.1–0.2 eV molecule−1 on the TiO2 termination, thus making up some 20–50% of the total adsorption energy.
The most favourable adsorption geometries for H2O on each of the two planar surface terminations as given by PBE-D3 are presented in figure 2, along with the electronic charge density in a two-dimensional plane through the molecule. In the molecular adsorption configuration on the SrO termination, we find the O ion of the H2O molecule to form an ionic bond with an underlying surface cation, with additional attraction to the surface provided by hydrogen bonds donated from the molecule to the surface. In the dissociated configuration, the OH fragment above the surface is bonded to the surface H through a bond of strong covalent character. In the molecular adsorption configuration on the TiO2 termination, a bond of covalent character is formed between the O in H2O and a surface Ti cation, in addition to a hydrogen bond between the molecule and the surface. In the dissociated configuration, the O in the OH fragment above the surface is bonded covalently to the surface cation, similar to the molecular case, with the surface proton additionally donating a hydrogen bond to the OH fragment.
Figure 2.
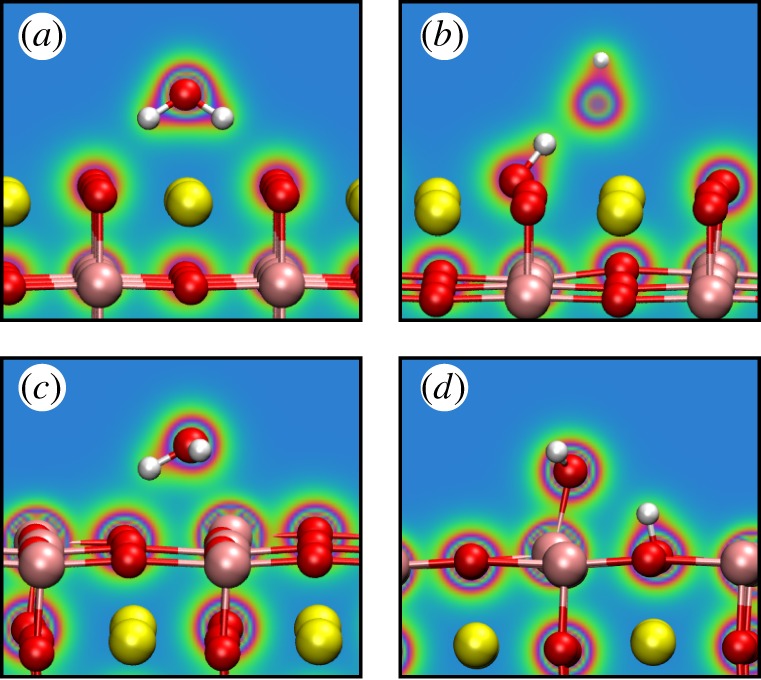
The most favourable adsorption geometries of H2O onto SrTiO3, as calculated using PBE-D3, corresponding to the energies in table 6. Colours in the plane indicate electronic charge density, with red being high and blue low. (a) Molecular adsorption onto SrOtermination, (b) dissociative adsorption onto the same, (c) molecular adsorption onto the TiO2 termination, (d) dissociative adsorption onto the same. The upper oxygen ion in (b) is behind the plane in which the electronic density is plotted. (Online version in colour.)
Based on the presented static results on the SrTiO3 bulk crystal, the surface in vacuum and H2O adsorption onto SrTiO3, we deemed PBE-D3 the overall best functional to use for modelling the SrTiO3/H2O solid–liquid interface. Therefore, all simulations to follow were performed using PBE-D3.
(b). Molecular dynamics
(i). Hydration structure
We present the ionic structure of the SrTiO3/H2O interface at 0.5 ML, 1.0 ML and bulk H2O coverage in figures 3, 4 and 5, respectively. Regardless of the level of water coverage and surface termination, the vertical density profiles show that water is strongly structured on the oxide surface, up to at least 7 Å at bulk water coverage. Clear lateral patterns are also evident in the ordering of the water on the surface, as revealed by the histograms of lateral positions. An analysis of the hydration structure of the two surfaces of each slab revealed qualitatively very similar results for the ordering of water and practically identical results for the degree of surface hydroxylation. Therefore, for simplicity, the results presented below are for one surface of each crystal slab. The similarity of results on the two faces of each slab implies that our results for the hydration structure are repeatable and robust.
Figure 3.

Results from MD simulations with 0.5 ML of H2O coverage on the planar SrO termination (a) and TiO2 termination (b). For each surface, we present a snapshot of the equilibrated structure, the vertical density profile, and the histogram of lateral positions for selected peaks in the hydration structure. In the vertical density profiles, the dashed verticallines denote the first hydration layer. In the lateral position histograms, the surface cation positions are schematically marked by yellow circles and the surface anion positions by red ones. The dashed squares denote simulation cell boundaries. (Online version in colour.)
Figure 4.

Results from MD simulations with 1.0 ML of H2O coverage on the planar SrO termination (a) and TiO2 termination (b). For each surface, we present a snapshot of the equilibrated structure, the vertical density profile and the histogram of lateral positions for selected peaks in the hydration structure. In the vertical density profiles, the dashed verticallines denote the first hydration layer. In the lateral position histograms, the surface cation positions are schematically marked by yellow circles and the surface anion positions by red ones. The dashed squares denote simulation cell boundaries. (Online version in colour.)
Figure 5.

Results from MD simulations of bulk H2O coverage on the four studied SrTiO3 terminations (from top down): planar SrO termination, planar TiO2 termination, RT13 and stepped TiO2-terminated (001) surface. For each surface, we present a snapshot of the equilibrated structure, the vertical density profile and the histogram of lateral positions for selected peaks in the hydration structure. In the vertical density profiles, the dashed vertical lines denote the first hydration layer. In the lateral position histograms, the surface cation positions areschematically marked by yellow circles and the surface anion positions by red ones. For the stepped surface, we also show position histograms in the xz-plane. The dashed squares denote simulation cell boundaries. (Online version in colour.)
Adsorption of H2O onto the SrO-terminated surface (figures 3–5) is characterized by a strong tendency for the water to dissociate. Some 50–60% of surface O ions are hydroxylated into OH groups at all water coverages on this termination (figure 6a), the observed large degree of hydroxylation being in qualitative agreement with experiment [8,10]. Also, our static DFT results above as well as previous DFT [11,12] and hybrid functional calculations [13] imply a tendency for water to dissociate, and a slightly lower affinity for molecular adsorption, overall in agreement with the observed strongly mixed mode of adsorption on the SrO termination. The double peak structure of O density seen at low coverages in the first hydration layer (figures 3 and 4) is due to the OH fragments generally assuming a smaller distance from the surface than molecular H2O. The first hydration layer consists of OH fragments alongside molecular H2O, the OH dominating this region at 0.5 ML but with a crossover to the molecular component dominating at higher coverages (figures 5 and 6b). The majority of the formed OH fragments are localized in the first hydration layer, the fraction of all OH fragments found in this region of the liquid being 100, 85 and 70% for coverages of 0.5 ML, 1.0 ML and bulk H2O, respectively. At the lowest coverage of 0.5 ML, we witness some hopping of the OH fragments within the first hydration layer between neighbouring Sr–Sr bridge sites, as observed recently experimentally for dissociated water on the SrO-terminated surface of the perovskite Sr3Ru2O7 [51].
Figure 6.

(a) The fraction of surface O ions which are hydroxylated into OH groups as a function of simulation time for the planar SrO and TiO2 terminations and the stepped surface (red, blue and magenta, respectively) at all studied water coverages. (b) The fraction of O ions in the first hydration layer of the planar SrO termination which are part of OH fragments as a function of simulation time (after the 5 psequilibration period), the remaining O being part of molecular H2O. (c) The same for the planar TiO2 termination. (Online version in colour.)
We find a much lower degree of hydroxylation on the TiO2-termination (figures 3–5) than on the SrO-termination, with only 10–15% of surface O hydroxylated at all coverages (figure 6a), again in qualitative agreement with experiment [6,8,10]. Our static DFT results, previous DFT [11,12] and hybrid functional [13] calculations indicate an increased tendency for molecular adsorption on the TiO2 termination with respect to the SrO termination, consistent with the observed shift in the degree of hydroxylation in our MD results. For this surface termination, the first hydration layer is represented at all coverages by a sharp peak in the O density profile (figures 3–5), the peak consisting of both dissociated and molecular H2O (figure 6c) bound to underlying Ti cations via Ti–O bonds of significant covalent character. The 2 Å distance of this peak from the surface is consistent with results from surface X-ray diffraction experiments of the TiO2-terminated (001) surface in air, which indicate an overlayer of O (most likely OH) on top of the TiO2 termination due to adsorbed water vapour [52]. This overlayer was proposed to completely cover the lateral fractional sites (0,0), (0,1/2), (1/2,0) and (1/2, 1/2) of the surface unit cell. At 1.0 ML of water coverage, we find the OH and H2O to be positioned on most of the cation and some of the bridging O sites (figure 4), and at bulk coverage, O ions of the first hydration layer are on top of the cations (figure 5). Our result for the hydration structure of the TiO2-terminated surface therefore resembles the surface X-ray diffraction result, but differs crucially in that the overlayer we find does not uniformly cover all surface sites.
Interestingly, although the degree of surface hydroxylation is much lower for the TiO2-terminated surface than for the SrO termination, the composition of the first hydration layer at bulk water coverage is very similar for the two terminations (some 30% of oxygens belonging to OH fragments, 70% to molecular H2O, figure 6b,c). This seemingly contradictory result is possible because on the TiO2 termination, 100% of the OH fragments remain in the first hydration layer, with the total O density of the hydration layer simultaneously being lower than on the SrO termination. All in all, the mode of adsorption is a mixed case of molecular and dissociated H2O for both terminations at all coverages.
On the RT13 surface (figure 5), water adsorbs almost entirely in the molecular mode, in the first hydration layer filling the cavities between the crests formed by TiO5 polyhedra on the surface reconstruction (figure 1). This layer of H2O binds to the surface structure mainly through hydrogen bonding. In the second hydration layer, the adsorbed H2O forms O–Ti bonds of partly covalent character with the topmost surface cations and also occupies volume above the central cavity of the surface unit cell, the cavity already being occupied by a single water molecule in the first hydration layer. In this system, at the central cavity of the surface reconstruction, we observe a single dissociation event of the form , where no stable surface OH group is formed. We discuss the mechanism behind this event in §4.
The stepped TiO2-terminated surface (figure 5) also induces a low degree of dissociation of the incident H2O, again in agreement with experiment [10]. Some 5% of all surface O ions are hydroxylated (figure 6a), these being observed at both terrace and edge sites in the system. Dissociation at step sites has been suggested from experiment decades ago [9], and indeed step edge anion sites are more likely than terrace sites to be hydroxylated in our simulations (1/6 of all step sites being hydroxylated versus 1/30 of terrace sites). The vertical density profiles at the terraces reveal a broader first hydration layer than on the planar TiO2 termination, with H2O positioned both on top of the Ti sites through partly covalent bonds and on top of the underlying Sr sites, bonded by hydrogen bonding and dispersion to the crystal surface. This configuration is closer to the hydration structure of TiO2-terminated SrTiO3 suggested by Vonk et al. [52] than what we find for the planar TiO2 surface. We observe strong covalent Ti–H2O bonding at the step edges alongside the hydroxylated edge anions. Finally, H2O molecules are strongly hydrogen-bonded to the crystal at the lower corner where the upper and lower terraces meet.
(ii). Electronic structure
In figure 7a, we present the EDOS for bulk SrTiO3, the SrTiO3 surface in vacuum and the surface under H2O coverage. In all simulated systems, the valence band is dominated by O 2p orbitals and the conduction band by Ti 3d states. The EDOS projected onto either the planar SrO-terminated or RT13 surface is very similar to that of bulk SrTiO3, including the magnitude of the band gap. The planar and stepped TiO2-terminated surfaces, however, reveal strongly localized sets of shallow O gap states of px and py character at the bottom of the band gap. We attribute this difference between the SrO and TiO2 terminations to the fact that on the SrO termination, all Ti–O6 octahedra remain intact, while on the TiO2 termination, the undercoordinated Ti–O5 polyhedra exposed on the planar surface lead to the formation of the observed ‘dangling bonds’. While such dangling states have been predicted also in earlier DFT studies [48,53–55], they have not been observed in experiment to date. We note that this feature in the EDOS is completely removed already at 0.5 ML of H2O coverage, and we find even a 1/32 coverage at static conditions to substantially subdue the peak at the bottom of the band gap. Furthermore, these shallow gap states are fully due to the outermost SrTiO3 layer of the slab, with no contribution from lower lying atomic planes. For these reasons, the present feature may have escaped experimental observation. Interestingly, on the RT13 surface, we see no such dangling states, despite an excess of Ti–O5 polyhedra on the surface. Instead, the EDOS on the RT13 surface hints that the surface layer is actually more stable than the subsurface layer. It is clearly the particular geometry of the reconstruction which allows for the stability of the surface states and the low surface energy we find for RT13 (0.815 J m−2) with respect to the SrO and TiO2 terminations (1.28 J m−2, table 2).
Figure 7.

(a) The electronic density of states (EDOS) of the SrTiO3 surface in vacuum and the surface at different levels of H2O coverage. The EDOS of bulk SrTiO3 and the surface in vacuum are from static geometric relaxations, results for the systems with H2O are from MD. All EDOS involving the surface arealigned to the valence and conduction bands of bulk SrTiO3 (also shown). The Fermi level is indicated by the dashed orange line. (b) Visualization of the charge density for the surface systems in (a) within an energy window down to 0.5 eV below the Fermi level (HOMO) and for the indicated set of states due to the first hydration layer (denoted by P in (a)). We plot the isosurface of the charge density (in blue) at 10% of the maximum density. For definitions of the surface and subsurface layers, see figure 1. (Online version in colour.)
The electronic states due to the first hydration layer overlap significantly in energy with the valence band energy range for all surfaces and coverages. In addition to these broad bands of Op character, states due to the first hydration layer appear at the bottom of the valence band and also below the valence band minimum upon adsorption of H2O (denoted by P in figure 7a). On all surfaces, these sharp features in the EDOS are due to O–H bonds in surface OH groups, in OH fragments within the liquid, in H2O within the liquid, as well as the lone pair charge density in both OH fragments and molecular H2O (figure 7b).
The band gap of the surface-projected EDOS generally changes by some 0.1 to 0.2 eV upon introduction of the water, decreasing for the stepped TiO2-surface but increasing for the SrO-terminated and RT13 surfaces. An exception to this is the planar TiO2-terminated surface, where the band gap increases by approximately 0.5 eV upon saturation of the dangling bond states. These results are, with the exception of the TiO2-terminated crystal, similar to previous results on rutile TiO2 (110) [56]. For rutile TiO2 (110), it was found that the conduction band edge shifts upwards in energy by 1.6 eV upon the adsorption of a monolayer of water [56]. Similarly, we find an upward shift of 0.7 eV for the conduction band minimum of our TiO2-terminated crystal upon introduction of a monolayer of water, assessed by aligning the computed Kohn–Sham states to the vacuum level between periodic slab surfaces. For the SrO-terminated crystal, in contrast, we find the conduction band minimum to shift downwards by 1.3 eV when a monolayer of water is adsorbed onto the surface. It is clearly the different character of Ti–O and Sr–O bonding which is responsible for the observed difference.
The orbital structure of the top of the valence band (highest occupied molecular orbitals, HOMO) remains fairly intact on the SrO termination upon adding H2O. By contrast, the dangling surface HOMO states of the TiO2-terminated slab are stabilized upon introducing the H2O, as the surface Ti–O5 polyhedra are effectively made into coordination-saturated Ti–O6 octahedra. The HOMO then resembles that of the SrO-termination, as can be expected. On the RT13 surface, the interaction between the H2O and the surface stabilizes the highest energy orbitals of the very top layer of the reconstruction to energies below those of the subsurface layer, leaving the latter to form the HOMO. The H2O molecules sitting in the cavities of the surface evidently induce a very favourable crystal-to-liquid interaction, which is thus seen in the EDOS.
(iii). Vibrational properties
We present the VDOS of H2O on each of the four surfaces at all considered water coverages in figure 8. In addition, for reference, we plot the computed VDOS of pure H2O and the frequencies of the vibrational modes of liquid H2O as found in experiment [34]. Focusing first on the planar and RT13 surfaces, it is immediately apparent that introducing the SrTiO3 crystal and forming the solid–liquid interface induces significant changes into the vibrational mode as well as the O–H stretching mode of H2O with respect to pure bulk H2O. These changes in the VDOS are the largest at the lowest levels of H2O coverage, which is to be expected, as the strength of the surface-to-molecule interaction per molecule is highest then.
Figure 8.

The vibrational density of states (VDOS) of H2O on SrTiO3 for all studied surfaces and all levels of water coverage, along with the VDOS for a pure, periodic supercell of liquid H2O. The dashed vertical lines are results for the vibrational modes of liquid H2O from experiment [34]. (Online version in colour.)
Redshifting, i.e. broadening of the low-frequency shoulder of the O–H stretching band is observed for the SrO, TiO2 and RT13 surfaces at all levels of water coverage. The fundamental reason behind this effect is the weakening of O–H bonds in H2O due to the molecules donating hydrogen bonds to surface O ions [57–59]. As is well known from previous work on, e.g. TiO2 [57,58], the stronger the water-to-water or water-to-surface hydrogen bonding, the weaker the covalent O–H bonding within H2O, and hence the lower the O–H vibrational frequencies. Therefore, as expected, the redshifting effect is strongest for monolayer and sub-monolayer H2O coverages, where more acceptors of hydrogen bonds are available on the surface per molecule of H2O than in the case of bulk water coverage. High-resolution electron energy loss spectroscopy experiments find the O–H stretching peak at 3662 cm−1 to become broader and move towards lower energies as water coverage is increased [59]. These observations, reported with respect to the sharp stretching peak of an isolated molecule of H2O, are consistent with our VDOS results.
Interestingly, we also find a slight blueshifting of the high-frequency shoulder of the O–H stretching band beyond approximately 3700 cm−1 for the case of the SrO-terminated surface. An analysis of the individual velocity autocorrelation functions of H ions reveals that this feature in the VDOS is due to the strong covalent O–H bond of the OH fragments within the liquid. As shown in figure 6, a larger fraction of H2O dissociates on the SrO than on the TiO2 termination, which then leads to the blueshifting effect being seen more prominently on the former surface termination.
A conspicuous effect is seen for the vibrational spectrum of H2O on the stepped TiO2-terminated surface. The VDOS is altered so that the O–H stretching band is strongly subdued, with the libration band instead being strengthened significantly. While exceptionally strong hydrogen bonding between the H2O and the surface and ensuing weakening of the O–H bonds within the liquid might explain such a curious effect, we explore other options in §4.
(iv). Proton dynamics
We find various different kinds of proton transfer events taking place in our dynamical simulations. The first type of event is observed in the initial phase of the simulation, at the onset of the dissociation process, when a fraction of the water molecules incident on the slab surface each donate a proton to a surface oxygen. Concurrent to this process, we see resulting OH fragments and neighbouring H2O molecules exchanging protons in the liquid region of the system. After reaching equilibrium, this latter process continues. Furthermore, in equilibrium, we observe a process where a surface proton is captured back up into the liquid by a nearby OH fragment and then donated back down to the surface again.
To quantify the rate of these proton transfer events in our simulations, we track the O–H coordination in each system as a function of time. For a given oxygen ion i in the liquid, a change from 2 to 1 in the O–H coordination number ZiOH is considered a proton donation event, a change from 1 to 2 conversely being considered a proton acceptance event. Correspondingly, for surface oxygens, a change of ZiOH from 1 to 0 implies a proton donation event and 0 to 1 means acceptance. We find all other changes of ZOH to have a negligible presence in our simulations.
As an example, in figure 9a,b we plot ZiOH for all oxygen ions in the liquid and in the slab surface, respectively, for the SrO-terminated SrTiO3 surface at bulk H2O coverage. From these data, we compute the mean rate of proton transfer events (treating all donation and acceptance events as separate transfer events), the results being presented for all the relevant simulations in table 7. To enable additional insight, the results are separated into the rate of proton jump events involving oxygens in the liquid region and those involving oxygens in the slab surface, along with the total rate which is the sum of these two. Also, the rates are presented in units scaled by the number of dissociated H2O molecules in each simulation as well as in units scaled by surface area. Two inferences are immediately apparent. First, in equilibrium, proton transfer takes place mainly among oxygens within the liquid region, the transfer of protons between surface and liquid oxygens being much less efficient for water on all considered surfaces. Second, proton transfer is much more efficient on the SrO termination than either on the planar or stepped TiO2 termination. We also note that no proton transfer is observed to take place directly between hydroxylated surface oxygens.
Figure 9.

The O–H coordination of oxygen ions as a function of time (after the 5 ps equilibration period) for the SrO-terminated SrTiO3 slab with bulk H2O coverage. Results are shown for O ions (a) between the two periodic slab surfaces, i.e. in the liquid region and (b) in the outermost surface layer of the slab. (Online version in colour.)
Table 7.
Mean rate of proton transfer events in all simulated systems, in units of ‘jumps per ps per OH fragment’ and ‘jumps per ps per (1×1) surface unit cell’. Proton transfer events involving changes in O–H coordination for O in the liquid and within the surface are separated.
| jumps/ps/OH |
jumps/ps/(1×1) |
|||||
|---|---|---|---|---|---|---|
| 0.5 ML | 1.0 ML | Bulk | 0.5 ML | 1.0 ML | Bulk | |
| oxygen in liquid | ||||||
| SrO termination | 5.1 | 13 | 7.4 | 2.4 | 6.3 | 4.3 |
| TiO2 termination | 0.82 | 1.3 | 2.2 | 0.17 | 0.33 | 0.69 |
| stepped surface | — | — | 2.2 | — | — | 0.25 |
| RT13 | — | — | 24 | — | — | 0.91 |
| oxygen at slab surface | ||||||
| SrO termination | 0.58 | 1.4 | 0.96 | 0.28 | 0.65 | 0.56 |
| TiO2 termination | 0.092 | 0.050 | 0.00 | 0.019 | 0.013 | 0.00 |
| stepped surface | — | — | 0.75 | — | — | 0.083 |
| RT13 | — | — | 2.8 | — | — | 0.11 |
| total | ||||||
| SrO termination | 5.7 | 14 | 8.3 | 2.7 | 6.9 | 4.8 |
| TiO2 termination | 0.91 | 1.4 | 2.2 | 0.19 | 0.34 | 0.69 |
| stepped surface | — | — | 3.0 | — | — | 0.33 |
| RT13 | — | — | 26 | — | — | 1.0 |
4. Discussion
Our DFT MD results show that although the degrees of surface hydroxylation on the planar SrO- and TiO2-terminated (001) surfaces of SrTiO3 are very different, the hydration layer in closest proximity to the surface has a similar composition in terms of H2O and OH in the two cases. However, while the lateral ordering of the liquid on the SrO termination appears close to amorphous, the TiO2 termination leads to a lattice-like configuration of OH and H2O covalently bonded to underlying surface cations. These two archetypal surfaces of SrTiO3 therefore present both striking similarities and differences in hydration structure. Introducing a stepped geometry to the surface leads to covalent bonding between the step edge and incident H2O and a large density of hydrogen-bond acceptors at the bottom of the step. A qualitatively different situation is found on the RT13 surface, where the lowest lying hydration structure appears completely dominated by the surface corrugation due to the ordering of Ti–O5 octahedra on the surface, and H2O remains molecular. The second hydration layer of RT13 already appears dictated by Ti–O bonding, as in the case of the planar TiO2 termination.
The degree of surface hydroxylation varies in descending order from the planar SrO termination to the TiO2 termination and finally to the stepped TiO2-terminated surface. We explain this trend in the basicity of the three surfaces by differences in the ionicities of the bonds between surface cations and anions among the surfaces. In bulk SrTiO3, the Sr–O bond is known to be much more ionic in character than the Ti–O bond [41,45], a finding we confirm in our own calculations of the bulk crystal. On the surface in vacuum, our computed Bader charges show that this inherent difference in the character of Sr–O and Ti–O bonding leads to the O ion of the planar SrO termination assuming a higher mean accumulation of charge (−1.28 e) than O on either the planar (−1.15 e) or stepped TiO2 termination (−1.13 e). The more negative the O charge, the stronger the hydrogen-bonding between the average surface anion and the liquid, and therefore the higher the degree of dissociation of H2O and the basicity of the surface. In table 8, we tabulate the degree of dissociation in units of dissociated molecules per surface anion (as above) alongside the number of dissociated molecules per surface bulk-like (1×1) unit cell.
Table 8.
The equilibrium number of dissociated H2O molecules per (1×1) surface unit cell and per surface anion in each simulation.
| molecules/(1×1) |
molecules/surface O |
|||||
|---|---|---|---|---|---|---|
| 0.5 ML | 1.0 ML | bulk | 0.5 ML | 1.0 ML | bulk | |
| SrO termination | 0.47 | 0.48 | 0.58 | 0.47 | 0.48 | 0.58 |
| TiO2 termination | 0.21 | 0.25 | 0.31 | 0.11 | 0.13 | 0.16 |
| stepped surface | — | — | 0.11 | — | — | 0.042 |
| RT13 | — | — | 0.002 | — | — | 0.0005 |
Sprik and co-workers have analysed the degree of dissociation of water at solid–liquid interfaces according to the acidity of available hydroxide groups on the crystal surface [56,60]. Specifically, they have shown [56] that the free energy of water dissociation on rutile TiO2 (110) can be expressed as
| 4.1 |
where pKa1 and pKa2 are the acidities of the two types of sites capable of binding protons under normal pH conditions, these sites being the hydroxylated fivefold coordinated Ti site (Ti–OH−) and the bridging oxygen sites. Considering the planar SrO- and TiO2-terminated surfaces of SrTiO3, our simulations clearly indicate a favourable (i.e. negative) free energy of water dissociation on both surfaces. If we identify two active sites for each surface, a surface-bound OH fragment (bound to the Ti cation on the TiO2-terminated surface and more loosely bound at bridging Sr–Sr sites on the SrO-terminated surface) and the oxygen anion of the crystal surface, the favourable free energy of dissociation implies that on both surfaces, it is the surface anion which is more basic of the two active sites. Correspondingly, molecularly bound H2O can be considered more acidic than a surface-anion-to-H group on both terminations of SrTiO3.
Proton transfer rates were found to be much higher on the SrO-terminated surface than on the TiO2-terminated planar and stepped surfaces, both between the surface and liquid and within the liquid region. We identify two effects to this end. First, a larger fraction of incident H2O dissociates on the SrO termination, which simply implies more available hopping sites for protons jumping from H2O to OH or from surface OH to liquid OH to form molecular H2O. The second effect is more subtle. It is known that oxygen ions which donate hydrogen bonds accumulate electronic charge at the site of the oxygen ion [61], making strong hydrogen bond donors also strong hydrogen bond acceptors. As noted above, the SrO-terminated surface is the strongest acceptor of hydrogen bonds from the liquid among these three surfaces, which implies that this charge accumulation on the liquid O ions will be strongest on the SrO surface. This strengthens hydrogen-bonding within the liquid region [61], which we see as a smaller mean hydrogen bond length on the planar SrO termination than on the TiO2 one (1.60 Åversus 1.65 Åat 0.5 ML of water coverage, with the same trend visible for higher coverages and the stepped surface displaying the weakest hydrogen bonds). The decreased hydrogen-to-hydrogen-bond-acceptor distance implies a lower barrier for proton transfer and hence an increased rate. We note that both of the effects contributing to the high proton transfer rate on the planar SrO-terminated surface (higher fraction of dissociation, decreased hydrogen bond length) are thus ultimately attributable to the higher ionicity of the Sr–O bond as compared with the Ti–O bond in the surface of the crystal. In figure 10, we illustrate the clear trend in proton transfer rate and the affinity for water dissociation with respect to the surface anion charge.
Figure 10.

The number of dissociated H2O molecules per surface (1×1) unit cell and the mean proton transfer rate at bulk H2O coverage as a function of surface anion charge for the surface in vacuum. The RT13 surface does not follow the same trends in degree of dissociation and proton transfer rate with anion charge as the other surfaces do (see text). (Online version in colour.)
The RT13 surface fails to obey this simple analysis, displaying the lowest surface anion charge (−1.11 e) and correspondingly the lowest degree of dissociation (0.002 molecules/(1×1)), yet having the smallest hydrogen-bond length in the liquid, and correspondingly higher proton transfer rates (1.0 jumps/ps/(1×1)) than the TiO2-terminated surfaces, despite only a single dissociation event occurring in the system during the simulation. This discrepancy with respect to the other surfaces is likely due to the unusual corrugation of RT13 and the qualitatively different process of dissociation ( instead of ) observed on this surface. We suggest that the dissociation process we witness on the RT13 surface, whereby a chain of three H2O molecules, the first one of which sits inside the central cavity of the surface (figures 1 and 5), breaks up into H3O in the cavity and H2O and OH above the cavity, can be explained from the above mechanism of strong hydrogen bond donors becoming strong acceptors. In this view, the H2O initially inside the cavity is ideally positioned to form strong hydrogen bonds to the O ions at the edge of the central cavity. The strength of these hydrogen bonds then propagates through the chain of three molecules, the last one which ends up donating a proton down the chain to the H2O in the cavity.
As shown in figure 9a,b, the analysis method for proton transfer rates in our simulations, based on a hard cutoff for O–H coordination, produces some noise into ZiOH, presumably when a proton is in transition between two O ions. Therefore, the absolute rates of proton jump events presented here are probably overestimates of the actual rates implied by our simulations. In addition, our DFT MD method is by definition a framework of classical nuclear dynamics, whereas taking into account the quantum nature of the proton may be important for the kinds of transfer processes described here [62]. Regardless of these shortcomings, we expect trends in the presented rates between the different regions in the systems (water, surface) and between different surface terminations to be robust. Proton transfer rates of 1.6 jumps/ps/(1×1) or 2.7 jumps/ps/OH found for the rutile TiO2 (110) surface from DFT MD [61] are within the range of rates that we find here on SrTiO3.
The drastic effect seen in the VDOS of H2O on the stepped TiO2-terminated surface (figure 8) does not seem explainable by the mean strength of surface-to-water hydrogen bonding, because as noted above, hydrogen bonding within this system is weaker than on the planar surfaces, where changes in the VDOS of H2O upon forming the solid–liquid interface are much smaller. Clearly, the stepped geometry of the surface is an important factor in the observed difference between the two TiO2-terminated systems. The hydration structure on both surfaces shows a grid-like ordering of O ions on top of the Ti sites (figure 5), but on the stepped surface, this lattice-like Ti-O ordering is complemented by O sitting on the bridging O sites within the same layer, whereas on the planar surface these two components to the vertical density profile are clearly separated. While such an adsorbed grid-like arrangement of O and OH on the stepped surface might lead to the observed anomalous vibrational properties of H2O in the system, the actual mechanism for this is unclear. We have recently observed a similar effect for H2O adsorbed onto the (210) surface of brookite TiO2, and further work is needed to fully understand this feature in the vibrational spectrum of the adsorbed H2O.
Finally, we examine the significance of our results for the efficiency of photocatalytic water-splitting using SrTiO3. While electron/hole transfer is generally considered the rate-limiting step for water-splitting using metal-oxide photocatalysts [63], it is interesting to analyse the reaction in terms of proton transfer. We focus on the reduction of H+ into H via the electron of the electron-hole pair photogenerated in the SrTiO3 anode [3]. To the extent that rate of this reaction, , depends on diffusivity of the protons within the solid–liquid interface, higher rates of proton hopping imply higher rates of 2H+ reduction. Comparing the proton hopping rates among the different surfaces at bulk water coverage (in units of jumps/ps/(1×1), table 7), it would seem that the SrO-terminated crystal should provide highest efficiency for the reduction process. However, the high reactivity of the surface to H2O seen as the high degree of surface hydroxylation (table 8) implies that the SrO-terminated surface is not stable with respect to water exposure. RT13, on the other hand, provides a higher rate of proton transfer than the TiO2-terminated surfaces do, as well as an extremely stable surface, which makes it the most promising candidate for efficient photocatalysis of the surfaces considered here.
5. Conclusion
We used dispersion-corrected DFT MD to predict the structural, electronic and vibrational properties of the SrTiO3/H2O solid–liquid interface. A strongly mixed mode of molecular and dissociative adsorption is found on the planar SrO- and TiO2-terminated (001) surfaces, with a low and negligible degree of dissociation on the stepped TiO2-terminated and RT13 surfaces, respectively. The ordering of adsorbed water appears dictated by covalent-like surface-cation-to-H2O bonding and surface corrugation on the various studied SrTiO3 surfaces. The electronic structure of the solid–liquid interface is featureless in the band gap region, and the vibrational spectrum exhibits features traced back to liquid-to-surface hydrogen bonding and the vibrational frequency of OH fragments in the liquid. We find the inherent difference in the ionicity of the Ti–O and Sr–O bonds of the SrTiO3 crystal to qualitatively explain observed trends in proton transfer rates and degrees of dissociation of water among the studied surfaces. Based on surface reactivity and proton mobility at the solid–liquid interface, RT13 is suggested to be more suitable than the TiO2- or SrO-terminated SrTiO3 surfaces for photocatalytic water-splitting.
Acknowledgements
We acknowledge financial support by the Academy of Finland through the Centres of Excellence Program (Project No. 251748). We wish to acknowledge CSC-IT Center for Science, Finland, for generous grants of computational resources. We thank S. Kawasaki, M. Lippmaa, H. Onishi, V. Vonk and T. Ghosh for insightful discussions.
Data accessibility
All relevant information and data for reproducing and verifying the results of this work are contained in the manuscript.
Authors' contributions
E.H. designed and performed the simulations and analysis. P.S. and A.S.F. supported the simulations and analysis. A.S.F. conceived the study. All authors contributed to writing the article and gave final approval for publication.
Competing interests
We have no competing interests.
Funding
We were funded through the Academy of Finland through the Centres of Excellence Program (Project No. 251748).
References
- 1.Brown GE. et al. 1999. Metal oxide surfaces and their interactions with aqueous solutions and microbial organisms. Chem. Rev. 99, 77–174. (doi:10.1021/cr980011z) [DOI] [PubMed] [Google Scholar]
- 2.Henderson MA. 2002. The interaction of water with solid surfaces: fundamental aspects revisited. Surf. Sci. Rep. 46, 1–308. (doi:10.1016/S0167-5729(01)00020-6) [Google Scholar]
- 3.Catlow CRA, Guo ZX, Miskufova M, Shevlin SA, Smith AGH, Sokol AA, Walsh A, Wilson DJ, Woodley SM. 2010. Advances in computational studies of energy materials. Phil. Trans. R. Soc. A 368, 3379–3456. (doi:10.1098/rsta.2010.0111) [DOI] [PubMed] [Google Scholar]
- 4.Wrighton MS, Ellis AB, Wolczanski PT, Morse DL, Abrahamson HB, Ginley DS. 1975. Strontium titanate photoelectrodes: efficient photoassisted electrolysis of water at zero applied potential. J. Am. Chem. Soc. 98, 2774–2779. (doi:10.1021/ja00426a017) [Google Scholar]
- 5.Lopez A, Heller T, Bitzer T, Chen Q, Richardson NV. 2001. The influence of sodium on the adsorption of water on SrTiO3(100)-1×1 surfaces. Surf. Sci. Lett. 494, L811–L814. (doi:10.1016/S0039-6028(01)01494-7) [Google Scholar]
- 6.Wang L-Q, Ferris KF, Herman GS. 2002. Interactions of H2O with SrTiO3(100) surfaces. J. Vac. Sci. Tech. A 20, 239–244. (doi:10.1116/1.1430246) [Google Scholar]
- 7.Baniecki JD, Ishii M, Kurihara K, Yamanaka K, Yano T, Shinozaki K, Imada T, Kobayashi Y. 2009. Chemisorption of water and carbon dioxide on nanostructured BaTiO3-SrTiO3(001). J. Appl. Phys. 106, 054109 (doi:10.1063/1.3169654) [Google Scholar]
- 8.Iwahori K, Watanabe S, Kawai M. 2003. Effect of water adsorption on microscopic friction force on SrTiO3 (001). J. Appl. Phys. 93, 3223–3227. (doi:10.1063/1.1540223) [Google Scholar]
- 9.Brookes NB, Quinn FM, Thornton G. 1987. The involvement of step and terrace sites in H2O adsorption on SrTiO3(100). Phys. Scr. 36, 711–714. (doi:10.1088/0031-8949/36/4/016) [Google Scholar]
- 10.Hussain H, Torrelles X, Rajput P, Nicotra M, Thornton G, Zebenhagen J. 2014. A quantitative structural investigation of the 0.1 wt % Nb-SrTiO3(001)/H2O interface. J. Phys. Chem. C 118, 10 980–10 988. (doi:10.1021/jp5034118) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guhl H, Miller W, Reuter K. 2010. Water adsorption and dissociation on SrTiO3(001) revisited: a density functional theory study. Phys. Rev. B 81, 155455 (doi:10.1103/PhysRevB.81.155455) [Google Scholar]
- 12.Hinojosa BB, Cleve TV, Asthagiri A. 2010. A first-principles study of H2O adsorption and dissociation on the SrTiO3(100) surface. Mol. Sim. 36, 604–617. (doi:10.1080/08927021003762746) [Google Scholar]
- 13.Evarestov RA, Bandura AV, Alexandrov VE. 2007. Adsorption of water on (001) surface of SrTiO3 and SrZrO3 cubic perovskites: hybrid HF-DFT LCAO calculations. Surf. Sci. 601, 1844–1856. (doi:10.1016/j.susc.2007.02.010) [Google Scholar]
- 14.Lukyanov SI, Bandura AV, Evarestov RA. 2013. Quantum mechanics based classical molecular dynamics study of water adsorption on (001) SrMO3 surfaces (M=Ti, Zr). Surf. Sci. 611, 10–24. (doi:10.1016/j.susc.2013.01.002) [Google Scholar]
- 15.Kienzle DM, Becerra-Toledo AE, Marks LD. 2011. Vacant-site octahedral tilings on SrTiO3 (001), the ()r33.7° surface, and related structures. Phys. Rev. Lett. 106, 176102 (doi:10.1103/PhysRevLett.106.176102) [DOI] [PubMed] [Google Scholar]
- 16.Guisinger NP, Santos TS, Guest JR, Chien T-Y, Bhattacharya A, Freeland JW, Bode M. 2009. Nanometer-scale striped surface terminations on fractured SrTiO3 surfaces. ACS Nano 3, 4132–4136. (doi:10.1021/nn901086x) [DOI] [PubMed] [Google Scholar]
- 17.Shimizu R, Iwaya K, Ohsawa T, Shiraki S, Hasegawa T, Hashizume T, Hitosugi T. 2011. Atomic-scale visualization of initial growth of homoepitaxial SrTiO3 thin film on an atomically ordered substrate. ACS Nano 5, 7967–7971. (doi:10.1021/nn202477n) [DOI] [PubMed] [Google Scholar]
- 18.Iwahori K, Watanabe S, Komeda T, Kawai M, Saito A, Kuwahara Y, Aono M. 1999. Force microscopy study of SrTiO3(001) surfaces with single atomic-layer steps. Jpn. J. Appl. Phys. 38, 3949–3948. (doi:10.1143/JJAP.38.3946) [Google Scholar]
- 19.Biswas A, Rossen PB, Yang C-H, Siemons W, Jung M-H, Yang IK, Ramesh R, Jeong YH. 2011. Universal ti-rich termination of atomically flat SrTiO3(001), (110), and (111) surfaces. Appl. Phys. Lett. 98, 051904 (doi:10.1063/1.3549860) [Google Scholar]
- 20.Humphrey W, Dalke A, Schulten K. 1996. Vmd - visual molecular dynamics. J. Mol. Graph. 14, 33–38. (doi:10.1016/0263-7855(96)00018-5) [DOI] [PubMed] [Google Scholar]
- 21.CP2K software package. See http://www.cp2k.org.
- 22.Goedecker S, Teter M, Hutter J. 1996. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 54, 1703–1710. (doi:10.1103/PhysRevB.54.1703) [DOI] [PubMed] [Google Scholar]
- 23.Hartwigsen C, Goedecker S, Hutter J. 1998. Relativistic separable dual-space Gaussian pseudopotentials from H to Rn. Phys. Rev. B 58, 3641–3662. (doi:10.1103/PhysRevB.58.3641) [DOI] [PubMed] [Google Scholar]
- 24.Krack M. 2005. Pseudopotentials for H to Kr optimized for gradient-corrected exchange-correlation functionals. Theor. Chem. Acc. 114, 145–152. (doi:10.1007/s00214-005-0655-y) [Google Scholar]
- 25.VandeVondele J, Hutter J. 2007. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 127, 114105 (doi:10.1063/1.2770708) [DOI] [PubMed] [Google Scholar]
- 26.Allen MJ, Tozer DJ. 2002. Helium dimer dispersion forces and correlation potentials in density functional theory. J. Chem. Phys. 117, 11113 (doi:10.1063/1.1522715) [Google Scholar]
- 27.Israelachvili JN. 2011. Intermolecular and surface forces. Burlington, VT: Academic Press. [Google Scholar]
- 28.Grimme S, Antony J, Ehrlich S, Krieg H. 2010. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 132, 154104 (doi:10.1063/1.3382344) [DOI] [PubMed] [Google Scholar]
- 29.Martin RM. 2008. Electronic structure: basic theory and practical methods. Cambridge, UK: Cambridge University Press. [Google Scholar]
- 30.Perdew JP, Burke K, Ernzerhof M. 1996. Generalized gradient approximation made simple. Phys. Rev. Lett. 77, 3865–3868. (doi:10.1103/PhysRevLett.77.3865) [DOI] [PubMed] [Google Scholar]
- 31.Perdew JP, Ruzsinsky A, Csonka GI, Vydrov OA, Scuseria GE, Constantin LA, Zhou X, Burke K. 2008. Restoring the density-gradient expansion for exchange in solids and surfaces. Phys. Rev. Lett. 100, 136406 (doi:10.1103/PhysRevLett.100.136406) [DOI] [PubMed] [Google Scholar]
- 32.Becke ADJ. 1988. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 38, 3098–3100. (doi:10.1103/PhysRevA.38.3098) [DOI] [PubMed] [Google Scholar]
- 33.Lee C, Yang W, Parr RG. 1988. Development of the Colle–Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 37, 785–789. (doi:10.1103/PhysRevB.37.785) [DOI] [PubMed] [Google Scholar]
- 34.Chen S-H, Toukan K, Loong C-K, Price DL, Teixeira J. 1984. Hydrogen-bond spectroscopy of water by neutron scattering. Phys. Rev. Lett. 53, 1360–1363. (doi:10.1103/PhysRevLett.53.1360) [Google Scholar]
- 35.Dickey JM, Paskin A. 1969. Computer simulation of the lattice dynamics of solids. Phys. Rev. 188, 1407–1418. (doi:10.1103/PhysRev.188.1407) [Google Scholar]
- 36.Kresse G, Hafner J. 1993. Ab initio molecular dynamics for liquid metals. Phys. Rev. B 47, 558–561. (doi:10.1103/PhysRevB.47.558) [DOI] [PubMed] [Google Scholar]
- 37.Kresse G, Hafner J. 1994. Ab initio molecular-dynamics simulation of the liquid-metal–amorphous-semiconductor transition in germanium. Phys. Rev. B 49, 14251–14269. (doi:10.1103/PhysRevB.49.14251) [DOI] [PubMed] [Google Scholar]
- 38.Kresse G, Hafner J. 1996. Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput. Mat. Sci. 6, 15–50. (doi:10.1016/0927-0256(96)00008-0) [Google Scholar]
- 39.Kresse G, Hafner J. 1996. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11 169–11 186. (doi:10.1103/PhysRevB.54.11169) [DOI] [PubMed] [Google Scholar]
- 40.Monkhorst HJ, Pack JD. 1976. Special points for Brillouin-zone integrations. Phys. Rev. B 13, 5188–5192. (doi:10.1103/PhysRevB.13.5188) [Google Scholar]
- 41.Abramov YA, Tsirelson vG, Zavodnik VE, Ivanov SA, Brown ID. 1995. The chemical bond and atomic displacements in SrTiO3 from X-ray diffraction analysis. Acta Cryst. B51, 942–951. (doi:10.1107/S0108768195003752) [Google Scholar]
- 42.Beattie AG, Samara GA. 1971. Pressure dependence of the elastic constants of SrTiO3. J. Appl. Phys. 42, 2376 (doi:10.1063/1.1660551) [Google Scholar]
- 43.van Benthem K, Elsässer C, French RH. 2001. Bulk electronic structure of SrTiO3: experiment and theory. J. Appl. Phys. 90, 6156 (doi:10.1063/1.1415766) [Google Scholar]
- 44.Birch F. 1986. Equation of state and thermodynamic parameters of nacl to 300 kbar in the high-temperature domain. J. Geophys. Res. 91, 4949–4954. (doi:10.1029/JB091iB05p04949) [Google Scholar]
- 45.Piskunov S, Heifets E, Eglitis RI, Borstel G. 2004. Bulk properties and electronic structure of SrTiO3, BaTiO3, PbTiO3 perovskites: an ab initio HF/DFT study. Comput. Mater. Sci. 29, 165–178. (doi:10.1016/j.commatsci.2003.08.036) [Google Scholar]
- 46.Heifets E, Eglitis RI, Kotomin EA, Maier J, Borstel G. 2001. Ab initio modeling of surface structure for SrTiO3 perovskite crystals. Phys. Rev. B 64, 235417 (doi:10.1103/PhysRevB.64.235417) [Google Scholar]
- 47.Cheng C, Kunc K, Lee MH. 2000. Structural relaxation and longitudal dipole moment of SrTiO3(001)(1×1) surfaces. Phys. Rev. B 62, 10 409–10 418. (doi:10.1103/PhysRevB.62.10409) [Google Scholar]
- 48.Padilla J, Vanderbilt D. 1998. Ab initio study of SrTiO3 surfaces. Surf. Sci. 418, 64–70. (doi:10.1016/S0039-6028(98)00670-0) [Google Scholar]
- 49.Bickel N, Schmidt G, Heinz K, Müller K. 1989. Ferroelectric relaxation of the SrTiO3(100) surface. Phys. Rev. Lett. 62, 2009–2011. (doi:10.1103/PhysRevLett.62.2009) [DOI] [PubMed] [Google Scholar]
- 50.Hikita T, Hanada T, Kudo M. 1993. Structure and electronic state of the TiO2 and SrO terminated SrTiO3(100) surfaces. Surf. Sci. 287/288, 377–381. (doi:10.1016/0039-6028(93)90806-U) [Google Scholar]
- 51.Halwidl D. et al. 2016. Adsorption of water at the SrO surface of ruthenates. Nat. Mater. 15, 450–456. (doi:10.1038/nmat4512) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vonk V, Konings S, van Hummel GJ, Harkema S, Graafsma H. 2005. The atomic surface structure of SrTiO3(001) in air studied with synchrotron X-rays. Surf. Sci. 595, 183–193. (doi:10.1016/j.susc.2005.08.010) [Google Scholar]
- 53.Bandura AV, Evarestov RA, Zhukovskii YF. 2015. Energetic stability and photocatalytic activity of SrTiO3 nanowires: ab initio simulations. RSC Adv. 5, 24 115–24 125. (doi:10.1039/C5RA00306G) [Google Scholar]
- 54.Guo Y, Qiu X, Dong H, Zhou X. 2015. Trends in non-metal doping of the SrTiO3 surface: a hybrid density functional study. Phys. Chem. Chem. Phys. 17, 21 611–21 621. (doi:10.1039/C5CP03005F) [DOI] [PubMed] [Google Scholar]
- 55.Raghavan S, Carvalho A, Formal FL, Setter N, Öberg S, Briddon PR. 2011. Adsorbate-localized states at water-covered (100) SrTiO3 surfaces. Appl. Phys. Lett. 98, 012106 (doi:10.1063/1.3529473) [Google Scholar]
- 56.Cheng J, Sprik M. 2010. Acidity of the aqueous rutile TiO2(110) surface from density functional theory based molecular dynamics. J. Chem. Theory Comput. 6, 880–889. (doi:10.1021/ct100013q) [DOI] [PubMed] [Google Scholar]
- 57.Kumar N, Neogi S, Kent PRC, Bandura AV, Kubicki JD, Wesolowski DJ, Cole D, Sofo JO. 2009. Hydrogen bonds and vibrations of water on (110) rutile. J. Phys. Chem. C 113, 13 732–13 740. (doi:10.1021/jp901665e) [Google Scholar]
- 58.Henderson MA. 1996. An HREELS and TPD study of water on TiO2 (110): the extent of molecular versus dissociative adsorption. Surf. Sci. 355, 151–166. (doi:10.1016/0039-6028(95)01357-1) [Google Scholar]
- 59.Cox PA, Edgell RG, Naylor PD. 1983. Hreels studies of adsorbates on polar solids: water on SrTiO3(100). J. El. Spectr. Rel. Phen. 29, 247–252. (doi:10.1016/0368-2048(83)80069-3) [Google Scholar]
- 60.Cheng J, Liu X, VandeVondele J, Sprik M. 2015. Reductive hydrogenation of the aqueous rutile TiO2(110) surface. Electrochim. Acta 179, 658–667. (doi:10.1016/j.electacta.2015.03.212) [Google Scholar]
- 61.Kumar N, Kent PRC, Kubicki AVBJD, Wesolowski DJ, Cole DR. 2011. Faster proton transfer dynamics of water on SnO2 compared to TiO2. J. Chem. Phys. 134, 044706 (doi:10.1063/1.3509386) [DOI] [PubMed] [Google Scholar]
- 62.Tuckerman ME, Marx D, Klein ML, Parrinello M. 1997. On the quantum nature of the shared proton in hydrogen bonds. Science 275, 817–820. (doi:10.1126/science.275.5301.817) [DOI] [PubMed] [Google Scholar]
- 63.Cowan AJ, Barnett CJ, Pendlebury SR, Barroso M, Sivula K, Grätzel M, Durrant JR, Klug DR. 2011. Activation energies for the rate-limiting step in water photooxidation by nanostructured α-Fe2O3 and TiO2. JACS 133, 10 134–10 140. (doi:10.1021/ja200800t) [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant information and data for reproducing and verifying the results of this work are contained in the manuscript.