

Abstract
Objective:
To identify features of primary progressive aphasia (PPA) associated with Alzheimer disease (AD) neuropathology. A related objective was to determine whether logopenic PPA is a clinical marker for AD.
Methods:
A total of 139 prospectively enrolled participants with a root diagnosis of PPA constituted the reference set. Those with autopsy or biomarker evidence of AD, and who had been evaluated at mild disease stages (Aphasia Quotient ≥85), were included (n = 19). All had quantitative language testing and APOE genotyping. Fifteen had MRI morphometry.
Results:
Impaired word-finding was the universal presenting complaint in the aphasic AD group. PPA clinical subtype was logopenic (n = 13) and agrammatic (n = 6). Fluency, repetition, naming, and grammaticality ranged from preserved to severely impaired. All had relative preservation of word comprehension. Eight of the 15 aphasic participants with AD showed no appreciable cortical atrophy at the individual level on MRI. As a group, atrophy was asymmetrically concentrated in the left perisylvian cortex. APOE ε4 frequency was not elevated.
Conclusions:
There is a close, but not obligatory, association between logopenic PPA and AD. No language measure, with the possible exception of word comprehension, can confirm or exclude AD in PPA. Biomarkers are therefore essential for diagnosis. Asymmetry of cortical atrophy and normal APOE ε4 prevalence constitute deviations from typical AD. These and additional neuropathologic features suggest that AD has biological subtypes, one of which causes PPA. Better appreciation of this fact should promote the inclusion of individuals with PPA and positive AD biomarkers into relevant clinical trials.
Primary progressive aphasia (PPA) is diagnosed when language impairment arises in relative isolation and progresses to become the primary obstacle to daily functioning. Frontotemporal lobar degeneration (FTLD) and Alzheimer disease (AD) are its most common neuropathologic correlates. The primary pathology is frequently FTLD-tau in agrammatic subtypes (PPA-G), FTLD-TDP in semantic subtypes (PPA-S), and AD in logopenic subtypes (PPA-L).1
The goal of this report is to characterize the features of PPA associated with AD. Previous investigations were based on samples of convenience with aphasias of variable severity and language testing of limited coverage, especially in the domain of grammar. The current report is based on 19 individuals with a clinical diagnosis of PPA and with postmortem verification or amyloid-PET scans consistent with AD pathology. All participants were enrolled into a prospective project where language measures are quantitatively and uniformly assessed and where cortical morphometry is used to identify regions of peak atrophy. Only participants initially studied at mild stages of aphasia were included to emphasize the characteristic features of the early language disturbance and their anatomical correlates as close to disease onset as possible.
Several specific questions were addressed. First, is there justification for equating PPA-L with AD pathology? Second, are there measures of language function characteristic/pathognomonic of PPA associated with AD? Third, is there a consistent pattern of atrophy distribution in PPA associated with AD? Fourth, is the APOE genotype frequency in this group different from that seen in more typical amnestic forms of AD?
METHODS
The reference set for this investigation consisted of 139 participants with PPA prospectively enrolled into a longitudinal project, with clinical subtype distribution of 32% PPA-G, 18% PPA-L, 23% PPA-S, and 27% unclassifiable by current diagnostic criteria.2 Twenty-seven have come to autopsy and an additional 61 have amyloid-PET determinations. Data from these 88 participants were reviewed to identify individuals who had postmortem or biomarker findings consistent with AD and mild aphasia (e.g., initial Western Aphasia Battery–Revised [WAB-R3] aphasia quotient [AQ] ≥85). The AQ threshold eliminated individuals where all language functions become compromised and the pattern of the aphasia loses its specificity. Data from the 19 participants who fulfilled these 2 criteria were included in the analyses (figure 1). The PPA diagnosis was based on 3 core criteria: (1) progressive language disorder of recent onset not attributable to elementary motor or perceptual deficits; (2) absence of consequential impairments in episodic memory, visuospatial skills, or comportment for approximately 2 years as ascertained by medical records, structured interviews with family members, and results of nonlanguage tests (table e-1 at Neurology.org); and (3) diagnostic investigations consistent with a neurodegenerative etiology.1,4,5
Figure 1. Flowchart showing the selection of participants included in the analysis.
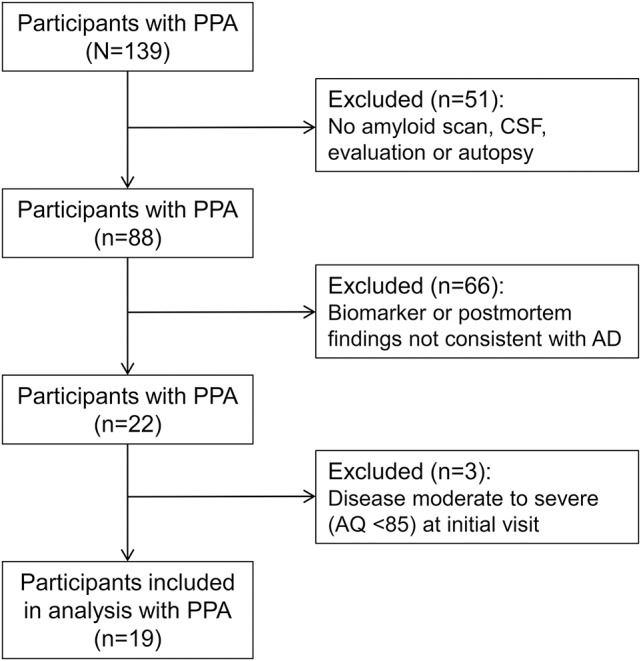
AD = Alzheimer disease; AQ = aphasia quotient; PPA = primary progressive aphasia.
Standard protocol approvals, registrations, and patient consents.
Northwestern University's Institutional Review Board approved this study. Informed consent was obtained from each participant.
Speech and language assessment.
Apraxia of speech was qualitatively rated during clinical assessment (M.-M.M.) by labored output, voice distortions, errors of syllabic stress or duration, mispronunciation of multisyllabic words, and errors of articulatory sequencing that could not be attributed to upper or lower motor neuron dysfunction.6 The WAB-R AQ served as a global measure of language impairment.3 Individual language domains were assessed quantitatively with tests previously used to characterize mild PPA.5 Fluency (i.e., words per minute [WPM]) was quantified from a recorded story narrative.7,8 Sentence production scores on 2 subsets of 15 noncanonical sentences, one from the Northwestern Anagram Test (NAT; flintbox.com/public/project/19927)9 and another from the Sentence Production Priming Test of the Northwestern Assessment of Verbs and Sentences (NAVS-SPPT),10 were averaged to derive a composite score of grammaticality of sentence production.5 The NAT does not require oral responses and was specifically designed to dissociate agrammatism from lack of fluency. The ability to understand the syntactic structure of sentences was assessed with the Sentence Comprehension Test of the NAVS, based on a subset of 15 noncanonical sentences of the same type as tested with the NAVS-SPPT and NAT. Repetition was quantitated with the 6 most difficult items from the repetition subtest of the WAB-R (items 10–15, which consist of phrases and sentences).3 Object naming was tested with the Boston Naming Test.11 Single word comprehension was tested with a subset of 36 moderately difficult items (157–192) of the Peabody Picture Vocabulary test.12 To control for differently scaled variables, quantitative performance scores were transformed into percentages. The WPM counts were transformed into percentages based on the control mean (132 ± 20).5 In all other tests, performance of neurologically intact participants of a similar age (n = 37) was ≥94%.5
Aphasia classification.
The 2011 classification guidelines for PPA-S, PPA-G, and PPA-L were followed with a few modifications.2,13 None of the 19 fit the PPA-S criteria. The differentiation of PPA-L from PPA-G is arguably the most challenging aspect of PPA classification.13,14 In this study, the diagnosis of PPA-G was made when 2 criteria were met: (1) noncanonical sentence production scores of ≤80% on the NAT-NAVS; and (2) presence of agrammatic sentences in recorded narrative or writing samples. The cutoff at 80% performance on the NAT-NAVS test was empirically chosen based on a prior investigation of PPA subtyping at mild impairment stages.5 Participants with word-finding deficits and hesitations in speech, whose single word comprehension was intact, who did not fulfill the 2 criteria for agrammatism described above, and whose repetition score was under 90% were classified as PPA-L. This left 2 unclassifiable patients who had all the other features of PPA-L (including logopenic speech with prominent word-finding failures) but whose repetition scores were 91 and 94. We included these patients in the PPA-L group based on our experience that such patients have clinicoanatomic features similar to those with repetition impairments and that they may represent an early stage of PPA-L.5,13
Quantitative MRI morphometry.
Structural MRI scans from the initial visit were acquired at Northwestern University's Center for Translational Imaging with a 3.0T Siemens (Munich, Germany) TIM Trio scanner and were reconstructed with the FreeSurfer image analysis suite (version 5.1.0) as previously described.15 FreeSurfer provides estimates of cortical thickness by measuring the distance between representations of the white-gray and pial-CSF boundaries across each point of the cortical surface.16 Geometric inaccuracies and topologic defects were corrected using manual and automatic methods based on FreeSurfer's validated guidelines.
Cortical thickness of the PPA group was statistically contrasted against 35 previously described right-handed, age- and education-matched healthy volunteers using spherical surface maps.17 Differences in cortical thickness between groups were calculated by conducting a general linear model on every vertex along the cortical surface. A stringent false discovery rate (FDR)18 threshold of 0.001 was used to detect areas of peak cortical thinning (i.e., atrophy) for the PPA group contrast compared to controls.
Neuropathologic and biomarker evidence of AD.
The 19 individuals with PPA in this study were selected to have postmortem or in vivo biomarker evidence consistent with Alzheimer pathology. Four of the participants had a primary postmortem diagnosis of AD by National Institute on Aging–Alzheimer's Association criteria.19 The remaining 15 had amyloid-PET with 18F-florbetapir, which showed elevated amyloid, defined as the mean cortical standardized uptake value ratio ≥1.10 (cerebral-to-cerebellar), using previously described methods for processing.20 This threshold was recommended for detecting neuritic plaques indicative of underlying AD.21,22
RESULTS
All participants were right-handed and Caucasian. There were 11 men and 8 women. Symptom onset was under age 65 years in 14 (74%), with 8 reporting onset in the 50s. Daily living activities that did not depend on language were mostly preserved. The mean AQ was 90 ± 4 and the mean Clinical Dementia Rating sum of boxes (SOB) was 0.8 (range 0–3), with 16 PPA participants having SOB scores of ≤1, indicating essentially preserved activities of daily living beyond those that had become difficult due to aphasia.
Individual language measures and subtypes.
Impaired word-finding was the presenting complaint and a prominent finding at the initial clinical assessment of all of the PPA participants (table 1). Word-finding hesitation lowered word output fluency in some participants, but not in those who reacted to word-finding failures with lengthy circumlocutions. In fact, 2 individuals had WPM (fluency) scores higher than control values. In the other language measures, performance ranged from severely impaired to nearly intact (table 1). The one exception was single word comprehension, which was universally preserved, with a group mean performance of 96% ± 5%. Symptoms of apraxia of speech were present in 6/19 individuals (4 PPA-L, 2 PPA-G) but at a level of prominence that was overshadowed by the aphasia.
Table 1.
Demographics and performance on language tasks
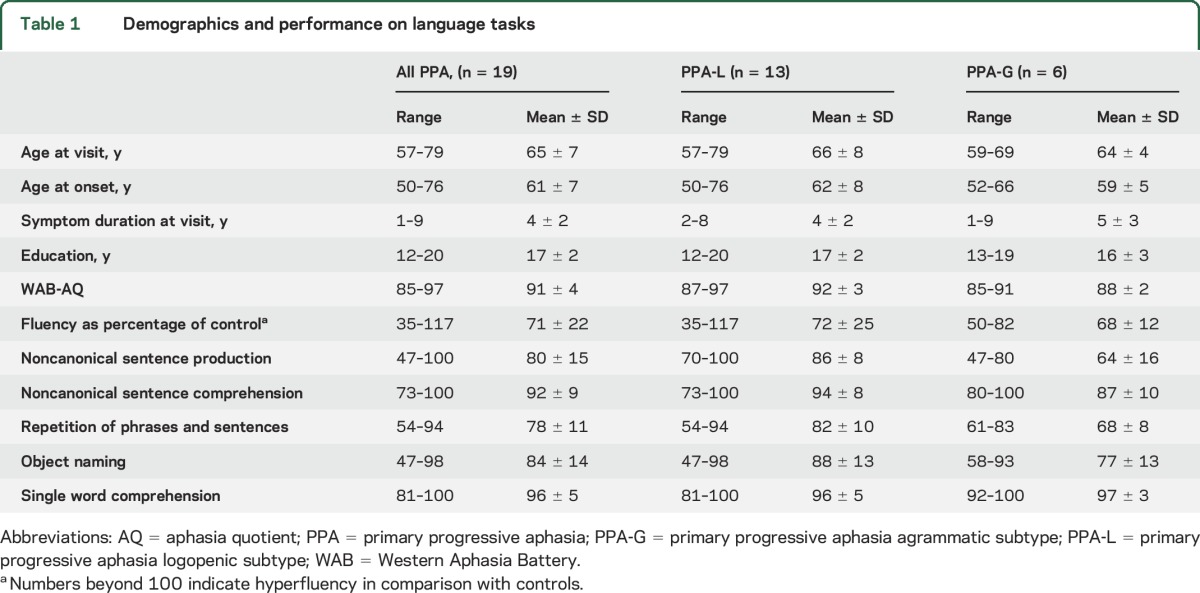
Thirteen of the participants (68%) were classified as PPA-L. This proportion would drop to 58% if the 2 patients with relatively preserved repetition but logopenic speech were considered unclassifiable. Six (32%) were classified as PPA-G. Table 2 provides examples of agrammatic statements from each of the 6 PPA-G participants and their performance scores on the noncanonical sentence production tasks (NAT-NAVS). Independent t tests showed that the PPA-G group was more impaired than the PPA-L group in sentence repetition and their AQ (ps ≤ 0.01, uncorrected) but these differences were not significant with Bonferroni correction. The other measures did not reveal significant group differences (table 1).
Table 2.
Examples of agrammatic statements from each of the 6 primary progressive aphasia agrammatic subtype participants and their performance scores on the noncanonical sentence production tasks (Northwestern Anagram Test–Northwestern Assessment of Verbs and Sentences) (% correct)

MRI morphometry.
Quantitative MRI morphometry was available for 15 participants (11 PPA-L, 4 PPA-G). The group atrophy map of the entire sample (n = 15) is shown in figure 2. Peak atrophy sites for the group as a whole were concentrated within the lateral temporal cortex, especially the superior temporal gyrus and the adjacent temporoparietal junction of the left hemisphere. Additional patchy atrophy was detected in dorsolateral and medial prefrontal cortex, precuneus, and inferior temporal cortex. Atrophy patterns for the clinical PPA-L and PPA-G subgroups yielded no significant differences in a direct group comparison (FDR = 0.05).
Figure 2. Group atrophy map (false discovery rate = 0.001) and quantitative morphometry for the 15 individuals with primary progressive aphasia.

DFC = dorsal frontal cortex; ITC = inferior temporal cortex; PC = precuneus; STG = superior temporal gyrus; TPJ = temporoparietal junction. Numbers below the heat map represent p values.
Scans were also qualitatively assessed (M.-M.M.) at the individual level. Eight out of 15 individual scans had no appreciable cortical atrophy. Each of the remaining 7 showed distinctly asymmetric atrophy mostly encompassing temporoparietal components of the left hemisphere language network.
APOE.
Five cases (1 PPA-G and 4 PPA-L) had the ε4 allele of APOE. Only one of these was homozygous. The remaining 14 (73%) had an ε3,3 genotype. At the Northwestern Alzheimer's Disease Brain Bank, 26% of the control population (n = 190) has at least one ε4 allele, whereas this frequency increases to 59% in those with an amnestic dementia during life and AD at autopsy (n = 75).1 The 27% frequency of ε4 allele carriers in the 19 PPA participants with biomarker evidence or autopsy-confirmed AD reported in the current investigation is similar to control values. These data are consistent with previous reports23,24 suggesting the ε4 allele of APOE is not a risk factor for clinical PPA or AD pathology in PPA.
DISCUSSION
This report is based on 19 participants with PPA with autopsy or biomarker evidence of Alzheimer pathology, examined in the mild stages of the disease at initial assessment. Results are therefore relevant to clinical decision-making at the time of initial evaluation.
The majority of the participants (68%) had been clinically classified as PPA-L, including 2 cases with logopenic speech but preserved repetition. The remaining participants were classified as PPA-G. These proportions are in line with those of an autopsy series of 58 PPA cases examined at various severity stages, in which 69% of all cases with AD pathology as the primary diagnosis had logopenic aphasia.1 These results seem at odds with another study, also of PPA participants with autopsy or biomarker evidence of AD, where a complete overlap with PPA-L was reported.25 However, in that study, grammatical ability was not reported and 5/14 participants presented with additional nonverbal memory impairments. Some of the participants in that study may, therefore, have qualified for a PPA-G diagnosis and others may have failed to fulfill the root diagnostic PPA criteria, which require a relative sparing of episodic memory.
The converse question of whether all PPA-L is associated with AD reveals equally complex relationships. In the series of 58 autopsies, for example, only 56% of PPA-L cases were associated with a primary neuropathologic diagnosis of AD.1 Investigations addressing this question with amyloid-PET scans have also yielded variable results. One study reported that 92% of PPA-L participants had positive amyloid scans.26 Two other studies reported lower rates of 66% and 69%, more in line with the autopsy results.27,28
While there is evidence that PPA associated with AD is more likely to present as logopenic aphasia and that logopenic PPA is more likely to be associated with AD, there is no one-to-one correspondence. Neither individual measures of language impairment nor clinical subtyping into logopenic vs agrammatic variants can resolve the differential diagnosis. Fluency, repetition, naming, and grammatical ability in individuals with PPA and AD neuropathology, at least in the early stages, may range from preserved to severely impaired and MRI morphometry may initially show no obvious atrophy. Even the presence of speech apraxia, albeit of lesser salience than the aphasia, does not rule out the presence of AD. At the individual patient level, therefore, the nature of neuropathology in a logopenic patient would be difficult to resolve without biomarker evidence.
In the 7 cases with appreciable cortical atrophy on quantitative MRI, the common denominators were profound asymmetry favoring the left hemisphere and greater atrophy of lateral temporal cortex compared to medial temporal areas or the precuneus. The peak atrophy pattern for the group (figure 2) showed a distribution similar to previous reports of atrophy and hypoperfusion in PPA participants with autopsy or biomarker evidence of AD.25,27 The concentration of atrophy in the temporoparietal junction and adjacent superior temporal gyrus mirrors atrophy patterns of PPA-L13 and is in keeping with the greater representation of logopenic participants in the group atrophy map shown in figure 2. The asymmetrical atrophy distribution in our PPA cohort differs from typical amnestic forms of AD,29 which tends to be symmetric. However, the small patches of atrophy in dorsal frontal cortex, precuneus, and inferior temporal cortex overlapped with areas considered part of the cortical atrophy signature of amnestic dementia with AD.29 The absence of detectable atrophy in the clinical scans of 7 patients reflects the early disease stages represented in this study. Conceivably, more powerful imaging methods could have revealed abnormalities in these cases as well. Practically, however, these results indicate that a clinically unremarkable MRI in an individual with PPA does not rule out underlying AD.
The inclusion of 2 individuals with logopenic speech and relatively preserved repetition deficits under the PPA-L variant could be viewed as a limitation since this designation does not strictly follow the 2011 consensus criteria.2 The alternative would have been to categorize these 2 individuals as unclassifiable, which would decrease the percentage of PPA-L associated with AD and increase the number of PPA phenotypes associated with AD pathology. All in all, the same conclusion could be made; there is no unique correspondence between a single PPA phenotype and AD neuropathology.
Another potential limitation of this study is the reliance on amyloid-PET with florbetapir as the AD biomarker in 15 of the 19 participants. In individuals who had been imaged within 1 year of death, however, the same ligand was shown to offer better than 95% sensitivity and specificity for the postmortem detection of neuritic plaques characteristic of AD.30 It is also conceivable that some of the PPA participants with positive amyloid scans could have an additional FTLD pathology. In 58 consecutive PPA autopsies, such a double diagnosis was encountered in only 1 patient whose postmortem examination met criteria for both AD and FTLD-TDP.1 The probability that the biomarker-positive PPA participants in the current study were misclassified pathologically is therefore quite low. Furthermore, the 4 autopsy-verified participants were evenly split between the PPA-L and PPA-G phenotypes and would, by themselves, support the conclusion that there is no exclusive relationship between AD and logopenic PPA.
Typical late-onset amnestic AD is commonly associated with language impairments, mostly in the form of anomia. However, such language impairments tend to arise later than the amnesia and play a much less conspicuous role in disrupting customary activities. The current study addresses the entirely different phenotype of PPA where the aphasia is the first and most salient feature. The Alzheimer pathology associated with this phenotype has prominent features that set it apart from the typical amnestic form of this disease. (1) Onset is most commonly before age 65, perhaps explaining why the female predominance of typical AD is not present. (2) Peak atrophy shows an asymmetric predilection for the language-dominant left hemisphere and displays only partial overlap with the atrophy signature of typical AD.29 (3) As previously reported in an autopsy series,1 the APOE ε4 allele is not a risk factor for the AD that causes PPA. (4) Learning disabilities, including dyslexia, are risk factors for PPA but not amnestic dementias.31,32 (5) Neurofibrillary tangles (NFT) can display atypical asymmetric distributions that violate the Braak and Braak pattern by favoring language cortex over mediotemporal limbic areas.23 (6) Neuritic amyloid plaques may be asymmetrically distributed, favoring the left hemisphere.20 (7) TDP-43 abnormalities, seen in at least 30% of typical AD cases, are not present in the AD pathology associated with PPA.33 These features indicate that the AD associated with PPA has temporal, anatomic, neuropathologic, and genetic factors that diverge from those of the far more common late-onset and amnestic forms of AD. As shown in table 3, AD can have several variants, some of which, like PPA, are nonamnestic.34–38 Individuals with these atypical nonamnestic manifestations tend to be excluded from AD clinical trials, where outcome measures are chosen to emphasize memory function. The advent of molecular biomarkers now makes it possible to identify a sizable contingent of such individuals with underlying AD pathology. Their inclusion in clinical trials will offer them equal access to novel agents and will require the introduction of new outcome measures designed to assess the relevant nonamnestic domain of the primary cognitive impairment.
Table 3.
Variants of Alzheimer disease (AD)

Supplementary Material
ACKNOWLEDGMENT
The authors thank Emmaleigh Loyer, Marie Saxon, Hannah McKenna, Kristen Whitney, Amanda Rezutek and Christina Wieneke, for neuropsychological testing of the primary progressive aphasia (PPA) participants and Mark Plantz, Allison Rainford, and Aneesha Nilakantan for their assistance fixing topologic defects in the MRI data.
GLOSSARY
- AD
Alzheimer disease
- AQ
aphasia quotient
- FDR
false discovery rate
- FTLD
frontotemporal lobar degeneration
- NAT
Northwestern Anagram Test
- NAVS-SPPT
Sentence Production Priming Test of the Northwestern Assessment of Verbs and Sentences
- NFT
neurofibrillary tangles
- PPA
primary progressive aphasia
- PPA-G
primary progressive aphasia agrammatic subtype
- PPA-L
primary progressive aphasia logopenic subtype
- PPA-S
primary progressive aphasia semantic subtype
- SOB
sum of boxes
- WAB-R
Western Aphasia Battery–Revised
- WPM
words per minute
Footnotes
Supplemental data at Neurology.org
AUTHOR CONTRIBUTIONS
M.-M.M. and E.R. contributed to conception and design of the study; A.M., B.R., J.S., K.C., D.C., S.W., C.T.K., E.B., M.-M.M., and E.R. collected and analyzed the data; M.M. and E.R. drafted the manuscript.
STUDY FUNDING
This project was supported by DC008552 from the National Institute on Deafness and Other Communication Disorders, M.-M.M.; AG13854 (Alzheimer Disease Center) from the National Institute on Aging, M.-M.M.; NS075075 from the National Institute of Neurological Disorders and Stroke (NINDS), E.R.; this is not an industry-sponsored study. PET imaging of the PPA group was performed at the Northwestern Memorial Hospital Department of Nuclear Medicine. Florbetapir F18 (18F-AV-45) doses for the PET imaging were provided by Avid Radiopharmaceuticals, a wholly owned subsidiary of Eli Lilly and Company. 18F-florbetapir doses were provided as nonfinancial support by Avid Radiopharmaceuticals (awarded to E.R.).
DISCLOSURE
E. Rogalski reports grants from NIH during the conduct of the study. J. Sridhar, B. Rader, A. Martersteck, and K. Chen report no disclosures relevant to the manuscript. D. Cobia reports grants from NIH during the conduct of the study. C. Thompson reports no disclosures relevant to the manuscript. S. Weintraub reports grants from NIH during the conduct of the study. E. Bigio reports grants from NIH during the conduct of the study. M. Mesulam reports no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Mesulam MM, Weintraub S, Rogalski EJ, Wieneke C, Geula C, Bigio EH. Asymmetry and heterogeneity of Alzheimer's and frontotemporal pathology in primary progressive aphasia. Brain 2014;137:1176–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology 2011;76:1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kertesz A. Western Aphasia Battery–Revised (WAB-r). Austin: Pro-Ed; 2006. [Google Scholar]
- 4.Mesulam MM. Primary progressive aphasia. Ann Neurol 2001;49:425–432. [PubMed] [Google Scholar]
- 5.Mesulam MM, Wieneke C, Thompson C, Rogalski E, Weintraub S. Quantitative classification of primary progressive aphasia at early and mild impairment stages. Brain 2012;135:1537–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Josephs KA, Duffy JR, Strand EA, et al. Characterizing a neurodegenerative syndrome: primary progressive apraxia of speech. Brain 2012;135:1522–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thompson CK, Ballard KJ, Tait ME, Weintraub S, Mesulam M. Patterns of language decline in non-fluent primary progressive aphasia. Aphasiology 1997;11:297–321. [Google Scholar]
- 8.Thompson CK, Cho S, Hsu CJ, et al. Dissociations between fluency and agrammatism in primary progressive aphasia. Aphasiology 2012;26:20–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Weintraub S, Mesulam M-M, Wieneke C, Rademaker A, Rogalski EJ, Thompson CK. The Northwestern Anagram Test: measuring sentence production in primary progressive aphasia. Am J Alzheimers Dis Other Demen 2009;24:408–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Thompson CK. Northwestern Assessment of Verbs and Sentences (NAVS). 2011. Available at: flintbox.com/public/project/9299. Accessed May 27, 2016. [Google Scholar]
- 11.Kaplan E, Goodglass H, Weintraub S. The Boston Naming Test. Philadelphia: Lea & Febiger; 1983. [Google Scholar]
- 12.Dunn LA, Dunn LM. Peabody Picture Vocabulary Test-4. Upper Saddle River, NJ: Pearson; 2006. [Google Scholar]
- 13.Mesulam MM, Rogalski EJ, Wieneke C, et al. Primary progressive aphasia and the evolving neurology of the language network. Nat Rev Neurol 2014;10:554–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sajjadi SA, Patterson K, Arnold RJ, Watson PC, Nestor PJ. Primary progressive aphasia: a tale of two syndromes and the rest. Neurology 2012;78:1670–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rogalski E, Cobia D, Harrison TM, et al. Anatomy of language impairments in primary progressive aphasia. J Neurosci 2011;31:3344–3350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fischl B, Sereno MI, Tootell RB, Dale AM. High-resolution intersubject averaging and a coordinate system for the cortical surface. Hum Brain Mapp 1999;8:272–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rogalski E, Cobia D, Martersteck A, et al. Asymmetry of cortical decline in subtypes of primary progressive aphasia. Neurology 2014;83:1184–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Genovese CR, Lazar NA, Nichols TE. Thresholding of statistical maps in functional imaging using the false discovery rate. Neuroimage 2002;15:870–878. [DOI] [PubMed] [Google Scholar]
- 19.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging–Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Alzheimers Dement 2012;8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martersteck A, Murphy C, Rademaker A, et al. Is in vivo amyloid distribution asymmetric in primary progressive aphasia? Ann Neurol 2016;79:496–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joshi AD, Pontecorvo MJ, Clark CM, et al. Performance characteristics of amyloid PET with florbetapir F 18 in patients with Alzheimer's disease and cognitively normal subjects. J Nucl Med 2012;53:378–384. [DOI] [PubMed] [Google Scholar]
- 22.Clark CM, Schneider JA, Bedell BJ, et al. Use of florbetapir-PET for imaging beta-amyloid pathology. JAMA 2011;305:275–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gefen T, Gasho K, Rademaker A, et al. Clinically concordant variations of Alzheimer pathology in aphasic versus amnestic dementia. Brain 2012;135:1554–1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rogalski EJ, Rademaker A, Harrison TM, et al. ApoE E4 is a susceptibility factor in amnestic but not aphasic dementias. Alzheimer Dis Assoc Disord 2011;25:159–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rohrer JD, Rossor MN, Warren J. Alzheimer pathology in primary progressive aphasia. Neurobiol Aging 2012;33:744–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Leyton CE, Villemange VL, Savage S, et al. Subtypes of progressive aphasia: application of the international consensus criteria and validation using β-amyloid imaging. Brain 2011;134:3030–3043. [DOI] [PubMed] [Google Scholar]
- 27.Teichmann M, Kas A, Boutet C, et al. Deciphering logopenic primary progressive aphasia: a clinical, imaging and biomarker investigation. Brain 2013;136:3474–3488. [DOI] [PubMed] [Google Scholar]
- 28.Matias-Guiu JA, Cabrera-Martin MN, Moreno-Ramos T, et al. Amyloid and FDG-PET study of logopenic primary progressive aphasia: evidence for the existence of two subtypes. J Neurol 2015;262:1463–1472. [DOI] [PubMed] [Google Scholar]
- 29.Dickerson BC, Bakkour A, Salat DH, et al. The cortical signature of Alzheimer's disease: regionally specific cortical thinning relates to symptom severity in very mild to mild AD dementia and is detectable in asymptomatic amyloid-positive individuals. Cereb Cortex 2009;19:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Clark CM, Pontecorvo MJ, Beach TG, et al. Cerebral PET with florbetapir compared with neuropathology at autopsy for detection of neuritic amyloid-beta plaques: a prospective cohort study. Lancet Neurol 2012;11:669–678. [DOI] [PubMed] [Google Scholar]
- 31.Rogalski E, Johnson N, Weintraub S, Mesulam M-M. Increased frequency of learning disability in patients with primary progressive aphasia and their first degree relatives. Arch Neurol 2008;65:244–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rogalski EJ, Rademaker A, Wieneke C, Bigio EH, Weintraub S, Mesulam MM. Association between the prevalence of learning disabilities and primary progressive aphasia. JAMA Neurol 2014;71:1576–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bigio E, Mishra M, Hatanpaa KJ, et al. TDP-43 pathology in primary progressive aphasia and frontotemporal dementia with pathologic Alzheimer disease. Acta Neuropathol 2010;120:43–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer's disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 2011;10:785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Braak H, Braak E. Evolution of the neuropathology of Alzheimer's disease. Acta Neurol Scand Suppl 1996;165:3–12. [DOI] [PubMed] [Google Scholar]
- 36.Benson DF, Davis RJ, Snyder BD. Posterior cortical atrophy. Arch Neurol 1988;45:789–793. [DOI] [PubMed] [Google Scholar]
- 37.Hof PR, Vogt BA, Bouras C, Morrison JH. Atypical form of Alzheimer's disease with prominent posterior cortical atrophy: a review of lesion distribution and circuit disconnection in cortical visual pathways. Vision Res 1997;37:3609–3625. [DOI] [PubMed] [Google Scholar]
- 38.Johnson JK, Head E, Kim R, Starr A, Cotman CW. Clinical and pathological evidence for a frontal variant of Alzheimer disease. Arch Neurol 1999;56:1233–1239. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.