

Abstract
The immunologic processes involved in Graves' disease (GD) have one unique characteristic – the autoantibodies to the TSH receptor (TSHR) – which have both linear and conformational epitopes. Three types of TSHR antibodies (stimulating, blocking, and cleavage) with different functional capabilities have been described in GD patients, which induce different signaling effects varying from thyroid cell proliferation to thyroid cell death. The establishment of animal models of GD by TSHR antibody transfer or by immunization with TSHR antigen has confirmed its pathogenic role and, therefore, GD is the result of a breakdown in TSHR tolerance. Here we review some of the characteristics of TSHR antibodies with a special emphasis on new developments in our understanding of what were previously called “neutral” antibodies and which we now characterize as autoantibodies to the “cleavage” region of the TSHR ectodomain.
Keywords: Graves' disease, thyroid, TSH-receptor antibodies, TSH, signaling
Background
The TSH receptor antigen of Graves' Disease (GD)
In Graves' disease (GD), the main autoantigen is the thyroid stimulating hormone receptor (TSHR), which is expressed primarily in the thyroid but also in adipocytes, fibroblasts, bone cells, and a variety of additional sites including the heart [1]. The TSHR is a G-protein coupled receptor with 7 transmembrane-spanning domains (Fig. 1a). TSH, acting via the TSHR, regulates thyroid growth and thyroid hormone production and secretion. The TSHR undergoes complex post-translational processing involving dimerization and intramolecular cleavage; the latter modification leaves a 2-subunit structural form of the receptor which eventually undergoes degradation or shedding of the ectodomain [2–4] (Fig. 1b). Each of these post-translational events may influence the antigenicity of the receptor and, furthermore, this complex processing may contribute to a break in self-tolerance. For example, the affinity of TSHR antibodies for the TSHR ectodomain is greater than for the holoreceptor itself [2].
Fig. 1.
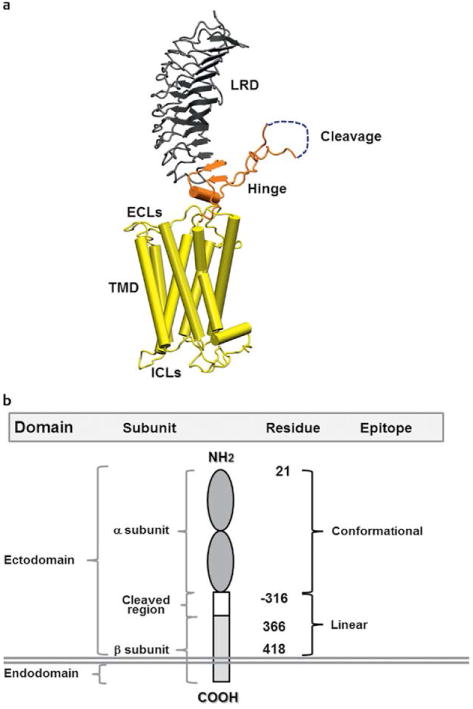
a A computer generated model of the TSHR based on the crystal structure of the ectodomain with the 7 transmembrane domain structure derived from the rhodopsin receptor crystal. The large ectodomain consists of 9 leucine-rich repeats (LRRs), which form a characteristic “horse shoe” shaped structure with a concave inner surface which harbors the major ligand and TSHR-Ab binding regions. The cleavage region and unique 50 amino acids cleaved region in the ectodomain, of unknown structure, is shown in gray and is a unique characteristic of the TSHR. b A schematic model of the TSHR to illustrate the multiple domains, subunits and epitope distributions containing different amino acid residues. TSHR stimulating and blocking antibodies are directed to the conformational epitopes whereas cleavage antibodies are directed at linear epitopes, mainly targeting the cleavage region.
Humoral immunity to the TSHR
One of the unique characteristics of GD, not found in normal individuals or in the rest of the animal kingdom, is the presence of TSHR antibodies (TSHR-Abs) easily detectable in the vast majority of patients [5]. In patients with GD, as for other antigens in other autoimmune diseases, TSHR-reactive B cells survive deletion and can potentially present thyroid autoantigen to T cells inducing proinflammatory cytokines [6]. Hence both B cells and T cells play a central role not [6] only in producing TSHR-antibodies but also in mediating chronic inflammatory changes of the disease seen in the thyroid gland, in the retro-orbit and in the skin (Fig. 2).
Fig. 2.
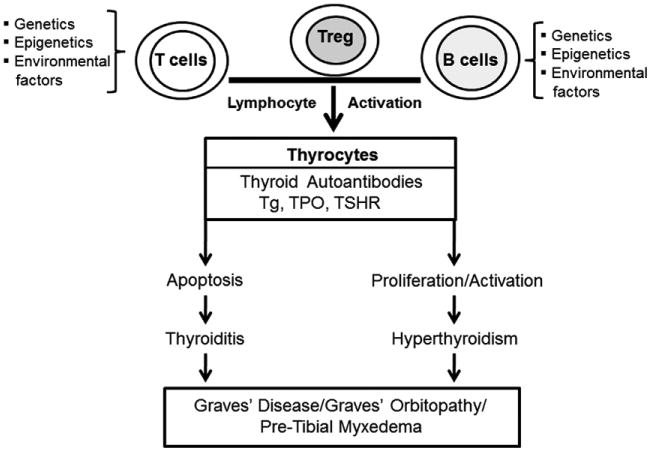
A simplified outline of the factors contributing to the development of Graves' disease on a background of thyroiditis.
The role of thyroid-specific B cells and their control
Insight into the contribution of autoreactive B cells to the normal human B cell repertoire has come from the analysis of monoclonal antibodies cloned from single purified B cells at different stages during their development [7]. Since diversity by V(D) J recombination and somatic hypermutation provides protective humoral immunity and also generates potentially harmful autoreactive B cell clones, several checkpoints ensure which developing autoreactive B cells are counter-selected. Thus, defects in central and peripheral checkpoints for B cell tolerance may be involved in the autoimmunity of GD. Furthermore, our recent mRNA-Seq pathway study of thyroid tissue from patients with GD indicated that B cells in the thyroid gland were hyperactive and B-cell receptor (BCR) signal transduction may prevail over T cell signaling [6]. These observations confirm that memory B cell generation or maturation takes place within the thyroid gland. B cell survival factors such as B cell-activating factor (BAFF) and a proliferation-inducing ligand (APRIL) have been shown to be important in an induced GD animal model [8]. Blockade of both BAFF and B cell maturation antigen (BCMA) using soluble decoy receptors ameliorated hyperthyroid GD in mice induced by TSHR immunization. Studies using gene silencing, targeting BAFF, inhibited proinflammatory cytokine expression, suppressed plasma cell generation and Th17 cells and caused marked amelioration in autoimmune arthritis [9]. Similar early clinical studies using B cell suppression in Graves' orbitopathy with monoclonal anti-CD20 add further support to this concept [10].
TSH receptor antibodies
Characterization of TSHR antibodies (TSHR-Abs)
Since the discovery and early analyses of TSHR-Abs [11] the most precise delineation of their characteristics has resulted from the analysis of monoclonal antibodies to the TSHR being derived from human, mouse and hamster sources: the mouse and hamster antibodies being secondary to TSHR immunization [12–14]. Three varieties of TSHR-Ab are now recognized amongst patients with autoimmune thyroid disease and immunized rodents; stimulating, blocking and so called “neutral” antibodies which are characteristically directed at the cleavage region of the TSHR ectodomain and are referred to in this report as “cleavage” antibodies since they have proven to be far from neutral in their biological activity (Fig. 3).
Fig. 3.
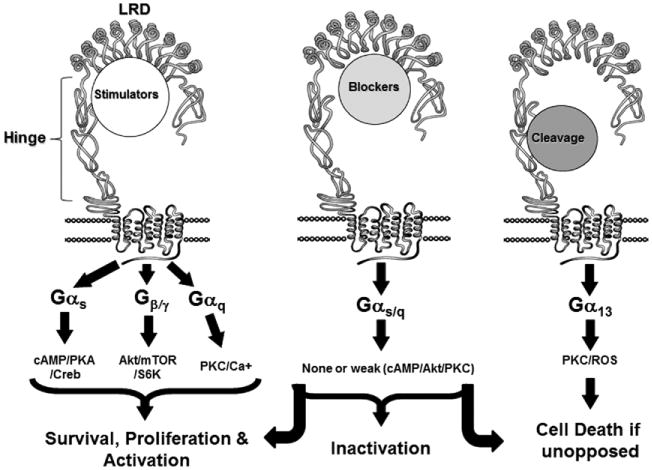
The 3 different varieties of TSHR-Abs and their signal transduction pathways and functional consequences.
Stimulating antibodies bind only to the naturally conformed TSHR and compete for TSH binding to the receptor site. These antibodies induce cyclic AMP generation, thyroid cell proliferation, and thyroid hormone synthesis and secretion. Stimulating antibodies bind exclusively to conformational epitopes in the TSHR ectodomain.
TSHR blocking antibodies also prevent TSH binding to the receptor. However, once bound they inhibit TSH action to such an extent that they may induce hypothyroidism although some blocking TSHR antibodies may act as weak TSHR agonists. TSHR blocking antibodies are usually conformationally dependent for TSH binding while others may bind to linear epitopes.
“Cleavage” TSHR antibodies neither block TSH binding nor block TSH action and they do not induce cyclic AMP generation. Cleavage TSHR-Abs bind exclusively with linear epitopes directed to the hinge region (aa 280–410) including the “unique region” of the receptor between amino acids 316–366 [15]. This region has been shown to be important in the signaling process [16, 17] and it is possible to demonstrate unique signaling activity of such cleavage region antibodies, which may induce thyroid cell apoptosis if unopposed [18–20].
The presence of differing proportions of high affinity TSHR-Abs with varied biological activity in patients with GD no doubt contributes to the clinical phenotype; varying from hyperthyroidism to hypothyroidism and vice versa. Thus, a classification of these antibodies based on function as suggested previously is more relevant than their ability or lack of ability in influencing TSH binding (Table 1).
Table 1.
Characteristics of different TSH receptor antibodies.
| %TSH Block | Conformational/Linear | Function | Signaling effectors | Ref. | |
|---|---|---|---|---|---|
| Stimulators | |||||
| MS1 (Hamster) | 80 | ± | Activation | cAMP/Akt/PKC/Ca + | [13, 15, 20, 26] |
| RSR-12 (Mouse) | ND | ± | Activation | cAMP/Akt/PKC/Ca + | [22, 24, 26] |
| M22 (Human) | 80 | ± | Activation | cAMP/Akt/PKC/Ca + | [22, 24, 26] |
| Blockers | |||||
| Tab-8 (Hamster) | 80 | ± | TSH antagonist | No cAMP | [13, 15, 26] |
| RSR-B2 (Mouse) | 80 | ± | Weak agonist | cAMP/ERK1/2 | [26] |
| K1–70 (Human) | 87 | ± | TSH antagonist | No cAMP | [28] |
| Cleavage | |||||
| Tab-16 (Hamster) aa 322–341 | 0 | ± | Apoptosis | ROS | [13, 15, 20, 26] |
| 7G10 (Hamster) aa 337–356 | 0 | ± | Apoptosis | ROS | [13, 15, 20, 26] |
Monoclonal antibodies (mAbs) to the TSHR
Only when immunizing with intact natural TSHR or TSHR cDNA was it possible to induce thyroid-stimulating antibodies and a successful animal model of hyperthyroidism [21–24]. Subsequently, the rare B cells secreting TSHR antibodies with stimulating activity were used to generate mAbs, including MS-1 raised in a hamster [13]. Later, mAbs were successfully derived directly from human peripheral blood including the widely used M-22, which is a high affinity stimulating antibody now used as the international standard [25]. In addition, a blocking mAb has been well characterized from a patient with Graves' disease and while human sera demonstrate the presence of cleavage antibodies, the cleavage mAbs used to date have been derived exclusively from immunized rodents [25, 26].
Stimulating TSHR-Ab epitopes
Part of the TSHR ectodomain has been crystallized with a human stimulating monoclonal TSHR Fab fragment bound in situ [27]. Several amino acids distributed along an extensive part of the concave surface of the leucine-rich repeat region (LRR) of the TSHR ectodomain were found to be important for antibody binding. Recent studies from our laboratory looking at conformational epitopes using mass spectrometry [28] have indicated that epitopes also exist outside the LRR for blocking as well as stimulating monoclonal antibodies. In addition, the importance of the N terminal region of the extracellular domain (ECD) has been well illustrated as well as residues in the “hinge” region [2]. These and other studies of TSHR antibodies raised the obvious question of whether epitope differences may be responsible for their different biological activities and this certainly would appear to be part of the answer. For example, our own recent study of 3 stimulating TSHR-mAbs showed variation in their patterns of signal transduction and is consistent with this conclusion [20, 29].
Blocking TSHR-Ab epitopes
Epitopes for TSHR blocking antibodies appear to be more widely distributed than for stimulating antibodies. Experimentally produced blocking TSHR-mAbs have been shown to bind to independent linear or conformational epitopes [15]. TSHR autoantibodies from patients with GD or HT have been shown to compete with a blocking TSHR-mAb to the N-terminus of the TSHR beta subunit (aa 382–415). Hence, blocking antibodies that cause hypothyroidism are also heterogeneous and there appear to be multiple epitopes involved in this repertoire of antibodies. Crystallization and modeling of human and mouse blocking TSHR-Abs have suggested their binding involves the N-terminal and leucine rich domain in agreement with this conclusion [30, 31].
Cleavage TSHR-Ab epitopes
Antibodies to the cleavage region (aa 316–366) have been demonstrated in patients with Graves' disease by peptide binding (ELISA) and mAb competition (competitive inhibition assay by FACS) and they bind with high affinity to the TSHR expressing cells [12, 15, 20, 29]. In animal models of GD, the major linear epitopes recognized are those in the cleavage region [32]. Such antibodies to the cleavage region do not compete for TSH binding and hence they are often referred to as “neutral”.
Signal transduction induced by TSHR antibodies (Table 1)
TSHR stimulating antibodies act mostly like TSH
Stimulating TSHR antibodies use signaling pathways similar to TSH for cell activation and growth [16, 29] (Fig. 4). Each of 3 stimulating mAbs we examined, activated Akt and PKC-ζ/λ. These antibodies induced a dose-dependent suppression of the c-Raf/ERK/p90RSK pathway as seen with TSH but there was heterogeneity of additional pathway signaling in relation to PKA activity and CREB induction.
Fig. 4.
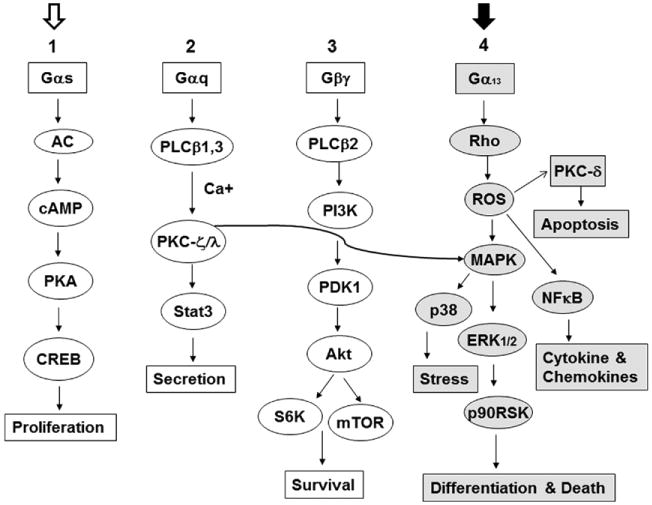
Although still highly complex, this is a simplified TSHR signaling model for the major G-protein activation pathways. In FRTL-5 thyrocytes, TSHR-stimulating antibodies and TSH itself activate the major signaling cascades numbered 1, 2, and 3. In contrast, cleavage TSHR-Abs activate mainly cascade #4 in grey (with the closed arrow at the top). Signaling cascade #4 appears to be exclusive to the cleavage antibodies and induces Rho, ROS and other down-stream signaling effectors such as NFκB and PKC-δ. In this proposed model system, Gαs activates cAMP/PKA/CREB via adenyl cyclase activation and Gαq induces PKC/Stat3 via activation of PLCβ1.3 kinases with Ca + [40]. Similarly, Gβγ activates PI3 kinase and downstream molecules PDK1/Akt/mTOR/S6k1 via PLCβ2 kinase. PKC (curved arrow) once activated can induce different MAPKs including ERK1/2/p38/p90RSK.
TSHR-blocking antibodies induce signaling cascades indicative of weak agonist activity
In our studies, TSHR-blocking antibodies have shown diverse effects on multiple signaling cascades in keeping with their known weak agonist or inverse agonist activity [29]. One such mAb (RSR-B2) induced low-level cyclic AMP generation and cell proliferation and increased Akt, CREB, c-Raf, and ERK whereas another (Tab-8) activated PKA and PKC in line with their mild proliferative effects [29]. These observations help explain how TSHR-Abs with variable activity may contribute to different clinical phenotypes in autoimmune thyroid disease. Most striking was that 2 of the blocking TSHR antibodies we examined resulted not only in signal initiation but they showed different pathway dominance.
TSHR-cleavage antibodies are not neutral
Cleavage TSHR mAbs do not induce cyclic AMP generation but may use the PKA II pathway. To examine the functional characteristics of cleavage TSHR-mAbs (C-TSHR-mAbs), we used a panel of mAbs generated from hamsters and mice [19, 20, 30]. As examples, one mAb showed modest activation of many signaling molecules such as Akt, PKC, PKA, CREB, ERK, and p90RSK, whereas another tended to consistently reduce constitutive signaling activities as seen with PKC, c-Raf, ERK, and p90RSK. The Akt/mTOR/S6K cascade was increased by 2 of the 4 mAbs tested and there were higher activities of PI3K-p110, PI3KR4, and PIP5K2 by proteomic analysis. The PI3Ks are a family of related intracellular signal transducer enzymes capable of phosphorylating the 3 position hydroxyl group of the inositol ring of phosphatidylinositol (PtdIns) [33, 34]. They are involved in cellular functions such as cell growth, proliferation, differentiation, motility, survival and intracellular trafficking. A significant increase in mTOR, a PI3K down-stream effector, activity was also observed but not seen with TSH in the FRTL5 model cells. These observations indicated that selected cleavage mAbs were capable of producing a robust effect on the PI3K/Akt/mTOR/S6K signaling cascade, an important arm of the (Gβ/γ) GPCR signaling unit (Fig. 4).
C-mAbs induce both PKC and ERK1/2/p38 pathway modules
The phosphorylation of the MAPKs (mitogen-activated protein kinases) is of major importance to cell function. The pathway via MAPK includes many proteins, including ERK (extracellular signal-regulated kinases), p38 and JNK (stress-activated protein kinases) proteins. MAPKs are involved in directing cellular responses to a diverse array of stimuli, such as mitogens, osmotic stress, heat-shock and pro-inflammatory cytokines. They regulate cell function, proliferation, gene expression, differentiation, mitosis, cell survival, and apoptosis [17]. These data show that the activation of MAPK/ERK induced by GPCRs such as the TSHR is mediated by Gαq or Gβ/γ subunits and involve a common signaling pathway with receptor-tyrosine kinases [29, 35, 36]. MAPK/ERK can also be induced by Rho via Gα13 activation [34]. As described earlier [20, 29], TSH and stimulating TSHR-mAbs do not induce the c-Raf/ERK1/2 module but 2 of the 4 cleavage mAbs we examined did activate c-Raf/ERK1/2. One of them activated Rho significantly while TSH suppressed it. Consistent with these results, activating upstream molecules of ERK1/2/p38 and their downstream effectors RSK/MNK1/MSK1/MAPKAPK2 were all activated. In contrast, TSH suppressed most of these elements. While JNK activity was reduced by both TSH and cleavage mAbs, Jun, an immediate early gene marker that binds with activator protein 1 (AP-1) and one of the downstream effectors of MAPK/JNK phosphorylation was activated only by C-mAb. Whereas TSH lacked such effect, it was able to activate Fos, another early gene product downstream to MAPK/ERK1/2. This suggested that these differences between C-mAb and TSH were indeed ligand dependent.
Cleavage mAbs activate NFκB signaling
In the nucleus, NFκB regulates genes encoding cytokines, cytokine receptors, cell adhesion molecules, proteins involved in coagulation, and genes involved in cell growth control. Both Gαq-coupled GPCRs (activating PKC) and Gαs-coupled GPCRs (activating PKA) interact with NFκB in regulating inflammation and cancer [37]. Activation of NFκB is initiated by the signal induced degradation of inhibitory κB (IκB) proteins. A variety of stimuli lead to the rapid nuclear accumulation of NFκB by the induced phosphorylation and subsequent degradation of IκB. We found C-mAbs which activated NFκB and 2 IκB kinases [20]. In contrast, TSH suppressed most of these signals.
Differential activation of JAK/STAT and SOCS-4 molecules by cleavage TSHR-mAbs
JAKs and STATs are critical components of many cytokine receptor systems, regulating growth, survival, differentiation, and pathogen resistance. They are mainly linked to MAPK and Akt signaling cascades [38]. TSH is known to activate STAT3 and so do some cleavage TSHR mAbs. However, the cleavage mAbs also activate STAT2 and STAT-5a phosphorylation, demonstrating a wider effect on cytokine production [20]. In addition to JAK/STAT pathway effectors, there are some negative regulators and SOCS is one of them [38]. In keeping with this, SOCS-4 was not activated. The fact that STAT and NFκB were activated allows us to conclude that certain thyroid cell responses produced by the TSHR signalosome will result in immune modulation. Among 18 cytokines and chemokines assessed by multiplex bead array, we found cleavage mAb induced responses in 6 including IL-2 and IL-10 which were induced by both TSH and cleavage TSHR-mAb, although the effect was small [20].
Apoptosis in GD
Background
It is now apparent that apoptosis plays an important role in the development and perpetuation of autoimmune thyroid disease. In early investigations, antibody- and T-cell-mediated death mechanisms were proposed as responsible for autoimmune thyrocyte depletion in thyroiditis but later, areas of apoptosis were recognized in thyroid tissue from patients with GD [39]. Subsequent studies on apoptosis have since provided new insights into autoimmune target destruction, indicating the involvement of death receptors and cytokine-regulated apoptotic pathways in the likely pathogenesis and perpetuation of thyroid autoimmunity. There is evidence that such thyrocyte apoptosis in GD may be antibody induced [40] or T cell mediated via defects in T regulatory cells which induce an abnormal production of cytokines [41] or changes in the expression of apoptotic molecules (Fas/FasL and caspase 8) on the surface of T lymphocytes and thyroid follicular cells [42, 43]. Eight tagged SNPs representing the majority of common variations in the programmed cell death 1 gene (PDCD1) within a large UK Caucasian GD data set revealed significant associations indicating that PDCD1 may also contribute toward the development of GD [44]. Clearly, death receptors/ligands appear to play a regulatory role in apoptosis, but caspase independent mechanisms may also coexist and contribute to thyroid cell death in GD. In fact, our own observations indicated that TSHR-Abs exerted diverse effects on thyroid cell apoptosis [18, 20].
Stimulating TSHR-Abs sustain survival of thyroid cells
In contrast to apoptosis, thyroid cell proliferation is induced by both TSH and stimulating TSHR-Abs. To first define the important signaling molecules involved in the thyroid cell fate decision process we detected increased PKA/CREB and AKT/mTOR/S6K activities induced by stimulating TSHR-mAbs (and TSH) and a dynamic change in cytoplasmic vs. nuclear accumulation of phosphorylated CREB causing phosphorylated CREB to be accumulated mostly in the nucleus [18]. These findings indicated that multiple signaling cascades, sustained by stimulating TSHR-Abs, are important in cell survival and proliferation.
Failure to sustain PKA/CREB and AKT/mTOR/S6K signaling cascades determines thyroid cell death
Cleavage TSHR-mAbs activated c-Raf/MEK/ERK1/2 and p38 but failed to sustain the activity of many of its signaling molecules while stimulating TSHR-mAbs sustained much of their activity. In agreement with this observation, the cleavage TSHR-mAbs had no influence on CREB which was detected only in the cell cytoplasm unlike nuclear phosphorylated CREB accumulation observed with stimulating TSHR-mAbs. In fact, with time, the cleavage TSH-mAbs, rather than inducing cell proliferation, induced thyroid cell death when unopposed [18]. These findings indicated that the ability to sustain such signaling cascades was vital to cell survival and proliferation, and therefore, these signaling cascades, or the lack thereof, underlie the thyroid cell fate decision under such conditions.
The effectors allowing cleavage TSHR-mAbs to induce thyroid cell apoptosis
To determine the key effectors induced by cleavage TSHR-Abs we found that both caspase and annexin V were highly induced by such antibodies confirming apoptosis as the main mechanism for thyroid cell death [18, 20]. Furthermore, we found that such cleavage antibodies were potent inducers of mitochondrial ROS (mROS) indicating the induction of cell stress and a mechanism for apoptosis induction (Fig. 5). In support of this conclusion, we found that both TSH and stimulating antibodies suppressed mROS induction and prevented apoptosis induced by the cleavage antibodies suggesting that cAMP/PKA generation was a mechanism for preventing apoptosis via suppression of mROS. Confirming the role of cAMP/PKA in the prevention of apoptosis via suppression of mROS, we found that PKA activators including IBMX and forskolin were also able to oppose the induction of mROS by cleavage TSHR-mAbs. These data indicated that a balance between negative and positive regulators of mROS was a key to maintaining thyrocyte homeostasis. Since apoptosis may be a major autoimmune stimulant [33, 45] this balance thus may play a vital role in GD initiation and perpetuation.
Fig. 5.
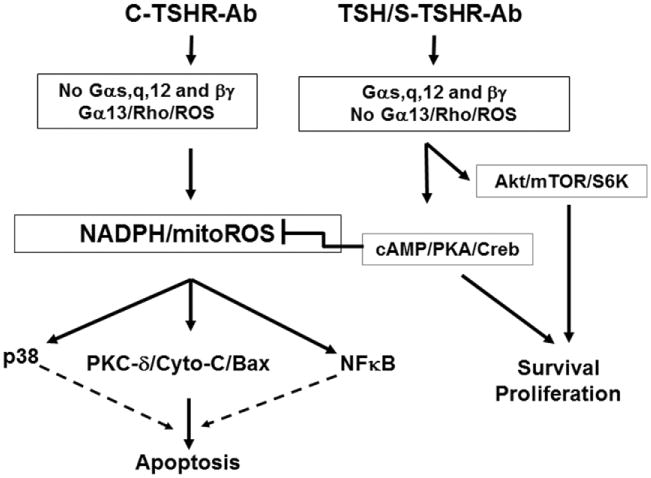
An outline of the proposed mechanisms involved in cleavage TSHR-Ab induced apoptosis (in FRTL-5) thyrocytes. While stimulating antibodies induce thyrocyte cell survival and proliferation via cAMP/PKA/CREB and Akt/mTOR/S6K, cleavage antibodies would do the opposite if unopposed and result in cell death via ROS induction. Since cleavage antibodies are unable to activate Gαs, Gαq, and Gβγ (unpublished), they may activate Gα13 and its downstream effectors Rho/ROS/PKC-δ leading to apoptosis via activation of the Cyt-C/Bax signaling cascade (unpublished).
Oxidative stress in GD
As described, cell stress induced by cleavage TSHR-Abs is a key regulatory component involved in determining thyroid cell survival or thyroid cell apoptosis via production of ROS [18]. ROS are highly reactive molecules induced by partially reduced forms of oxygen resulting from cellular metabolism. They include hydrogen peroxide (H2O2), hydroxyl radicals (OH•), superoxide anions (O2−) and lipid peroxides [46]. Antioxidant systems defend cells from ROS-induced cellular damage and, under physiological conditions, a balance between oxidant and antioxidant exists. Evaluation of human cellular defense systems (oxidant vs. antioxidant) in thyroid tissue from Graves' disease patients undergoing thyroidectomy has revealed increased levels of free radicals and their scavengers compared to normal thyroid [47]. Indeed when thyroid cells were exposed to cleavage TSHR-mAb for 24 h there was enhanced immunostaining of both mitochondrial (Mn-SOD and HSP60) and endoplasmic reticulum stress markers (HSP70) with perinuclear condensations indicating mitochondrial ROS (mROS) induction which was confirmed by MitoSOX red, a mitochondrial superoxide indicator dye (Fig. 6) [18].
Fig. 6.
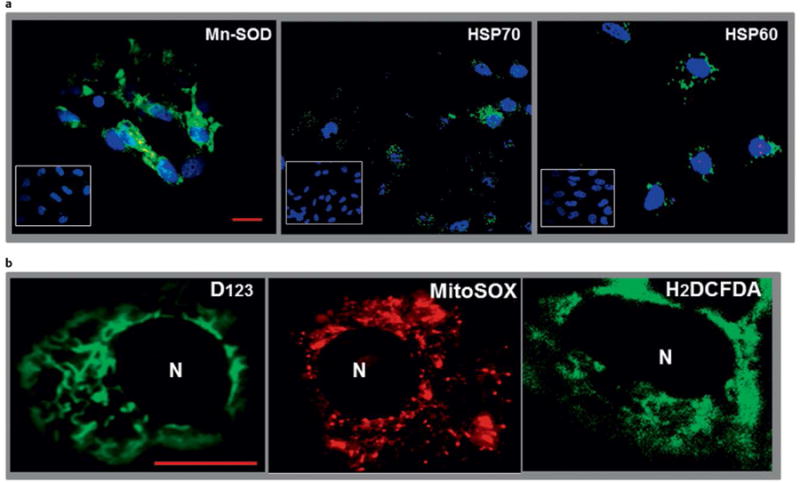
Immunohistochemistry and live imaging of stress markers in thyrocytes. Panel a Immunohistochemical detection of stress-induced proteins. Rabbit polyclonal primary antibody was used to detect Mn-SOD in C-TSHR-mAb (IC8 1 μg/ml) treated (24 h) FRTL-5 thyrocytes. The C-TSHR-mAb also induced heat shock proteins (HSP) 70 and 60 as detected by mAbs against each protein. HSP70 and 60 are normally localized within the endoplasmic reticulum and mitochondria, respectively. Such proteins were not induced by an isotype control mAb (see insets). Panel b Live imaging of ROS in thyrocytes using 3 independent dyes. Cells were treated for 24 h with C-TSHR-mAb (IC8 1 μg/ml). Representative images of both D123 and H2DCFDA showed diffuse staining throughout the cell cytoplasm. Some perinuclear condensations were also documented [16]. The distribution of staining patterns paralleled mitochondrial ROS (mROS). Scale bar on images corresponds to 20 micron. Nucleoli are indicated as N.
Conclusions
Autoimmunity represents a collection of heterogeneous disorders controlled by complex genetic and environmental factors with major stochastic contributions. In GD, the primary antigen is the TSHR and the reports of extrathyroidal TSHR expression in a variety of cell types including fibroblasts, bone cells and immune cells has added to the complexity of the disease and also introduced a variety of potential new mechanisms that may be involved. A common view of GD is that TSHR-Abs promote the disease by enhancing thyroid antigen expression. Stimulating TSHR-Abs are certainly capable of this role and may interact directly with the immune system including stimulation of maturing thymocytes [48]. However, T cell activation and subsequent thyroid infiltration in GD patients are not just the result of direct autoantibody induced mechanisms. In fact, GD appears to develop on a background of concurrent autoimmune thyroiditis. Therefore, the observations supporting the induction of apoptosis by cleavage TSHR-Abs suggests that such antibodies may be active very early in the disease and may serve to perpetuate the disorder once established. Indeed TSHR-Abs can even be detected in a small number of patients with Hashimoto's thyroiditis [49, 50]. Such TSHR-Abs have been reported as being of the blocking variety but, to date, cleavage antibodies have not been examined in that disorder.
Acknowledgments
This work was supported in part by NIH grants DK069713, DK052464 and the VA Merit Award Program.
Footnotes
Disclosure Statement: S.A.M. has nothing to declare. T.F.D. is a Member of the Board of Kronus Inc, Starr, Idaho, which markets diagnostic kits including those for thyroid autoantibodies.
References
- 1.Bahn RS, Dutton CM, Natt N, Joba W, Spitzweg C, Heufelder AE. Thyrotropin receptor expression in Graves' orbital adipose/connective tissues: potential autoantigen in Graves' ophthalmopathy. J Clin Endocrinol Metab. 1998;83:998–1002. doi: 10.1210/jcem.83.3.4676. [DOI] [PubMed] [Google Scholar]
- 2.Chazenbalk GD, Pichurin P, Chen CR, Latrofa F, Johnstone AP, McLachlan SM, Rapoport B. Thyroid-stimulating autoantibodies in Graves disease preferentially recognize the free A subunit, not the thyrotropin holoreceptor. J Clin Invest. 2002;110:209–217. doi: 10.1172/JCI15745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kajita Y, Rickards CR, Buckland PR, Howells RD, Rees SB. Analysis of thyrotropin receptors by photoaffinity labelling. Orientation of receptor subunits in the cell membrane. Biochem J. 1985;227:413–420. doi: 10.1042/bj2270413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Loosfelt H, Pichon C, Jolivet A, Misrahi M, Caillou B, Jamous M, Vannier B, Milgrom E. Two-subunit structure of the human thyrotropin receptor. Proc Natl Acad Sci USA. 1992;89:3765–3769. doi: 10.1073/pnas.89.9.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vlase H, Davies TF. Insights into the molecular mechanisms of the autoimmune thyroid diseases. In: Eisenbarth GS, editor. Endocrine and Organ Specific Autoimmunity. CA: R. G. Landes Co; 1999. pp. 98–132. [Google Scholar]
- 6.Bagriacik EU, Klein JR. The thyrotropin (thyroid-stimulating hormone) receptor is expressed on murine dendritic cells and on a subset of CD45RBhigh lymph node T cells: functional role for thyroid-stimulating hormone during immune activation. J Immunol. 2000;164:6158–6165. doi: 10.4049/jimmunol.164.12.6158. [DOI] [PubMed] [Google Scholar]
- 7.Meffre E, Wardemann H. B-cell tolerance checkpoints in health and autoimmunity. Curr Opin Immunol. 2008;20:632–638. doi: 10.1016/j.coi.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 8.Gilbert JA, Kalled SL, Moorhead J, Hess DM, Rennert P, Li Z, Khan MZ, Banga JP. Treatment of autoimmune hyperthyroidism in a murine model of Graves' disease with tumor necrosis factor-family ligand inhibitors suggests a key role for B cell activating factor in disease pathology. Endocrinology. 2006;147:4561–4568. doi: 10.1210/en.2006-0507. [DOI] [PubMed] [Google Scholar]
- 9.Lai Kwan LQ, King Hung KO, Zheng BJ, Lu L. Local BAFF gene silencing suppresses Th17-cell generation and ameliorates autoimmune arthritis. Proc Natl Acad Sci USA. 2008;105:14993–14998. doi: 10.1073/pnas.0806044105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salvi M, Vannucchi G, Campi I, Rossi S, Bonara P, Sbrozzi F, Guastella C, Avignone S, Pirola G, Ratiglia R, Beck-Peccoz P. Efficacy of rituximab treatment for thyroid-associated ophthalmopathy as a result of intraorbital B-cell depletion in one patient unresponsive to steroid immunosuppression. Eur J Endocrinol. 2006;154:511–517. doi: 10.1530/eje.1.02119. [DOI] [PubMed] [Google Scholar]
- 11.Adams DD, Purves HD. Abnormal responses in the assay of thyrotropin. Proc Univ Otago Med School. 1956;34:11–12. [Google Scholar]
- 12.Ando T, Davies TF. Monoclonal antibodies to the thyrotropin receptor. Clin Dev Immunol. 2005;12:137–143. doi: 10.1080/17402520500078238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ando T, Latif R, Pritsker A, Moran T, Nagayama Y, Davies TF. A monoclonal thyroid-stimulating antibody. J Clin Invest. 2002;110:1667–1674. doi: 10.1172/JCI16991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ando T, Latif R, Davies TF. Antibody-induced modulation of TSH receptor post-translational processing. J Endocrinol. 2007;195:179–186. doi: 10.1677/JOE-07-0058. [DOI] [PubMed] [Google Scholar]
- 15.Ando T, Latif R, Daniel S, Eguchi K, Davies TF. Dissecting linear and conformational epitopes on the native thyrotropin receptor. Endocrinology. 2004;145:5185–5193. doi: 10.1210/en.2004-0789. [DOI] [PubMed] [Google Scholar]
- 16.Mizutori Y, Chen CR, McLachlan SM, Rapoport B. The thyrotropin receptor hinge region is not simply a scaffold for the leucine-rich domain but contributes to ligand binding and signal transduction. Mol Endocrinol. 2008;22:1171–1182. doi: 10.1210/me.2007-0407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mueller S, Jaeschke H, Gunther R, Paschke R. The hinge region: an important receptor component for GPHR function. Trends Endocrinol Metab. 2010;21:111–122. doi: 10.1016/j.tem.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 18.Morshed SA, Ma R, Latif R, Davies TF. How one TSH receptor antibody induces thyrocyte proliferation while another induces apoptosis. J Autoimmun. 2013;47:17–24. doi: 10.1016/j.jaut.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Morshed SA, Latif R, Davies TF. Delineating the autoimmune mechanisms in Graves' disease. Immunol Res. 2012;54:191–203. doi: 10.1007/s12026-012-8312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Morshed SA, Ando T, Latif R, Davies TF. Neutral antibodies to the TSH receptor are present in Graves' disease and regulate selective signaling cascades. Endocrinology. 2010;151:5537–5549. doi: 10.1210/en.2010-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Costagliola S, Many MC, Denef JF, Pohlenz J, Refetoff S, Vassart G. Genetic immunization of outbred mice with thyrotropin receptor cDNA provides a model of Graves' disease. J Clin Invest. 2000;105:803–811. doi: 10.1172/JCI7665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanders J, Allen F, Jeffreys J, Bolton J, Richards T, Depraetere H, Nakatake N, Evans M, Kiddie A, Premawardhana LD, Chirgadze DY, Miguel RN, Blundell TL, Furmaniak J, Smith BR. Characteristics of a monoclonal antibody to the thyrotropin receptor that acts as a powerful thyroid-stimulating autoantibody antagonist. Thyroid. 2005;15:672–682. doi: 10.1089/thy.2005.15.672. [DOI] [PubMed] [Google Scholar]
- 23.Muehlberg T, Gilbert JA, Rao PV, McGregor AM, Banga JP. Dynamics of thyroid-stimulating and -blocking antibodies to the thyrotropin receptor in a murine model of Graves' disease. Endocrinology. 2004;145:1539–1545. doi: 10.1210/en.2003-1456. [DOI] [PubMed] [Google Scholar]
- 24.Sanders J, Allen F, Jeffreys J, Bolton J, Richards T, Depraetere H, Nakatake N, Evans M, Kiddie A, Premawardhana LD, Chirgadze DY, Miguel RN, Blundell TL, Furmaniak J, Smith BR. Characteristics of a monoclonal antibody to the thyrotropin receptor that acts as a powerful thyroid-stimulating autoantibody antagonist. Thyroid. 2005;15:672–682. doi: 10.1089/thy.2005.15.672. [DOI] [PubMed] [Google Scholar]
- 25.Sanders J, Jeffreys J, Depraetere H, Evans M, Richards T, Kiddie A, Brereton K, Premawardhana LD, Chirgadze DY, Nunez MR, Blundell TL, Furmaniak J, Rees SB. Characteristics of a human monoclonal autoantibody to the thyrotropin receptor: sequence structure and function. Thyroid. 2004;14:560–570. doi: 10.1089/1050725041692918. [DOI] [PubMed] [Google Scholar]
- 26.Nagy EV, Burch HB, Mahoney K, Lukes YG, Morris JC, III, Burman KD. Graves' IgG recognizes linear epitopes in the human thyrotropin receptor. Biochem Biophys Res Commun. 1992;188:28–33. doi: 10.1016/0006-291x(92)92345-x. [DOI] [PubMed] [Google Scholar]
- 27.Sanders J, Chirgadze DY, Sanders P, Baker S, Sullivan A, Bhardwaja A, Bolton J, Reeve M, Nakatake N, Evans M, Richards T, Powell M, Miguel RN, Blundell TL, Furmaniak J, Smith BR. Crystal Structure of the TSH Receptor in Complex with a Thyroid-Stimulating Autoantibody. Thyroid. 2007;17:395–410. doi: 10.1089/thy.2007.0034. [DOI] [PubMed] [Google Scholar]
- 28.Latif R, Teixeira A, Michalek K, Ali MR, Schlesinger M, Baliram R, Morshed SA, Davies TF. Antibody protection reveals extended epitopes on the human TSH receptor. PLoS One. 2012;7:e44669. doi: 10.1371/journal.pone.0044669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morshed SA, Latif R, Davies TF. Characterization of thyrotropin receptor antibody-induced signaling cascades. Endocrinology. 2009;150:519–529. doi: 10.1210/en.2008-0878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sanders P, Young S, Sanders J, Kabelis K, Baker S, Sullivan A, Evans M, Clark J, Wilmot J, Hu X, Roberts E, Powell M, Nunez Miguel R, Furmaniak J, Rees Smith B. Crystal structure of the TSH receptor (TSHR) bound to a blocking-type TSHR autoantibody. J Mol Endocrinol. 2011;46:81–99. doi: 10.1530/JME-10-0127. [DOI] [PubMed] [Google Scholar]
- 31.Evans M, Sanders J, Tagami T, Sanders P, Young S, Roberts E, Wilmot J, Hu X, Kabelis K, Clark J, Holl S, Richards T, Collyer A, Furmaniak J, Smith BR. Monoclonal autoantibodies to the TSH receptor, one with stimulating activity and one with blocking activity, obtained from the same blood sample. Clin Endocrinol (Oxf) 2010;73:404–412. doi: 10.1111/j.1365-2265.2010.03831.x. [DOI] [PubMed] [Google Scholar]
- 32.Davies TF, Ando T, Lin RY, Tomer Y, Latif R. Thyrotropin receptor-associated diseases: from adenomata to Graves disease. J Clin Invest. 2005;115:1972–1983. doi: 10.1172/JCI26031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vives-Pi M, Rodriguez-Fernandez S, Pujol-Autonell I. How apoptotic beta-cells direct immune response to tolerance or to autoimmune diabetes: a review. Apoptosis. 2015;20:263–272. doi: 10.1007/s10495-015-1090-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mao J, Yuan H, Xie W, Wu D. Guanine nucleotide exchange factor GEF115 specifically mediates activation of Rho and serum response factor by the G protein alpha subunit Galpha13. Proc Natl Acad Sci USA. 1998;95:12973–12976. doi: 10.1073/pnas.95.22.12973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Back CM, Stohr S, Schafer EA, Biebermann H, Boekhoff I, Breit A, Gudermann T, Buch TR. TSH induces metallothionein 1 in thyrocytes via Gq/11- and PKC-dependent signaling. J Mol Endocrinol. 2013;51:79–90. doi: 10.1530/JME-12-0200. [DOI] [PubMed] [Google Scholar]
- 36.Zaballos MA, Garcia B, Santisteban P. Gbetagamma dimers released in response to thyrotropin activate phosphoinositide 3-kinase and regulate gene expression in thyroid cells. Mol Endocrinol. 2008;22:1183–1199. doi: 10.1210/me.2007-0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye RD. Regulation of nuclear factor kappaB activation by G-protein-coupled receptors. J Leukoc Biol. 2001;70:839–848. [PubMed] [Google Scholar]
- 38.Aaronson DS, Horvath CM. A road map for those who don't know JAK-STAT. Science. 2002;296:1653–1655. doi: 10.1126/science.1071545. [DOI] [PubMed] [Google Scholar]
- 39.Stassi G, De MR. Autoimmune thyroid disease: new models of cell death in autoimmunity. Nat Rev Immunol. 2002;2:195–204. doi: 10.1038/nri750. [DOI] [PubMed] [Google Scholar]
- 40.Wang SH, Baker JR. The role of apoptosis in thyroid autoimmunity. Thyroid. 2007;17:975–979. doi: 10.1089/thy.2007.0208. [DOI] [PubMed] [Google Scholar]
- 41.Mao C, Wang S, Xiao Y, Xu J, Jiang Q, Jin M, Jiang X, Guo H, Ning G, Zhang Y. Impairment of regulatory capacity of CD4 + CD25 + regulatory T cells mediated by dendritic cell polarization and hyperthyroidism in Graves' disease. J Immunol. 2011;186:4734–4743. doi: 10.4049/jimmunol.0904135. [DOI] [PubMed] [Google Scholar]
- 42.Bossowski A, Czarnocka B, Bardadin K, Stasiak-Barmuta A, Urban M, Dadan J, Ratomski K, Bossowska A. Identification of apoptotic proteins in thyroid gland from patients with Graves' disease and Hashimoto's thyroiditis. Autoimmunity. 2008;41:163–173. doi: 10.1080/08916930701727749. [DOI] [PubMed] [Google Scholar]
- 43.Bossowski A, Czarnocka B, Bardadin K, Urban M, Niedziela M, Dadan J. Expression of Bcl-2 family proteins in thyrocytes from young patients with immune and nonimmune thyroid diseases. Horm Res. 2008;70:155–164. doi: 10.1159/000145017. [DOI] [PubMed] [Google Scholar]
- 44.Newby PR, Roberts-Davies EL, Brand OJ, Heward JM, Franklyn JA, Gough SCL, Simmonds MJ. Tag SNP screening of the PDCD1 gene for association with Graves' disease. Clinical Endocrinology. 2007;67:125–128. doi: 10.1111/j.1365-2265.2007.02848.x. [DOI] [PubMed] [Google Scholar]
- 45.Nagata S, Hanayama R, Kawane K. Autoimmunity and the clearance of dead cells. Cell. 2010;140:619–630. doi: 10.1016/j.cell.2010.02.014. [DOI] [PubMed] [Google Scholar]
- 46.Marchi S, Giorgi C, Suski JM, Agnoletto C, Bononi A, Bonora M, De Marchi E, Missiroli S, Patergnani S, Poletti F, Rimessi A, Duszynski J, Wieckowski MR, Pinton P. Mitochondria-ros crosstalk in the control of cell death and aging. J Signal Transduct. 2012;2012:329635. doi: 10.1155/2012/329635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mano T, Shinohara R, Iwase K, Kotake M, Hamada M, Uchimuro K, Hayakawa N, Hayashi R, Nakai A, Ishizuki Y, Nagasaka A. Changes in free radical scavengers and lipid peroxide in thyroid glands of various thyroid disorders. Horm Metab Res. 1997;29:351–354. doi: 10.1055/s-2007-979052. [DOI] [PubMed] [Google Scholar]
- 48.Gimenez-Barcons M, Colobran R, Gomez-Pau A, Marin-Sanchez A, Casteras A, Obiols G, Abella R, Fernandez-Doblas J, Tonacchera M, Lucas-Martin A, Pujol-Borrell R. Graves' Disease TSHR-Stimulating Antibodies (TSAbs) Induce the Activation of Immature Thymocytes: A Clue to the Riddle of TSAbs Generation? J Immunol. 2015;194:4199–4206. doi: 10.4049/jimmunol.1500183. [DOI] [PubMed] [Google Scholar]
- 49.Kamath C, Young S, Kabelis K, Sanders J, Adlan MA, Furmaniak J, Rees Smith B, Premawardhana LD. Thyrotrophin receptor antibody characteristics in a woman with long-standing Hashimoto's who developed Graves' disease and pretibial myxoedema. Clin Endocrinol (Oxf) 2012;77:465–470. doi: 10.1111/j.1365-2265.2012.04397.x. [DOI] [PubMed] [Google Scholar]
- 50.Takasu N, Yamada T, Sato A, Nakagawa M, Komiya I, Nagasawa Y, Asawa T. Graves' disease following hypothyroidism due to Hashimoto's disease: studies of eight cases. Clin Endocrinol (Oxf) 1990;33:687–698. doi: 10.1111/j.1365-2265.1990.tb03906.x. [DOI] [PubMed] [Google Scholar]