

Abstract
Objective
In a cohort of patients with young‐onset Parkinson's disease (PD), the authors assessed (1) the prevalence of genetic mutations in those who enrolled in deep brain stimulation (DBS) programs compared with those who did not enroll DBS programs and (2) specific genetic and clinical predictors of DBS enrollment.
Methods
Subjects were participants from 3 sites (Columbia University, Rush University, and the University of Pennsylvania) in the Consortium on Risk for Early Onset Parkinson's Disease (CORE‐PD) who had an age at onset < 51 years. The analyses presented here focus on glucocerebrosidase (GBA), leucine‐rich repeat kinase 2 (LRRK2), and parkin (PRKN) mutation carriers. Mutation carrier status, demographic data, and disease characteristics in individuals who did and did not enroll in DBS were analyzed. The association between mutation status and DBS placement was assessed in logistic regression models.
Results
Patients who had PD with either GBA,LRRK2, or PRKN mutations were more common in the DBS group (n = 99) compared with the non‐DBS group (n = 684; 26.5% vs. 16.8%, respectively; P = 0.02). In a multivariate logistic regression model, GBA mutation status (odds ratio, 2.1; 95% confidence interval, 1.0–4.3; P = 0.05) was associated with DBS surgery enrollment. However, when dyskinesia was included in the multivariate logistic regression model, dyskinesia had a strong association with DBS placement (odds ratio, 3.8; 95% confidence interval, 1.9–7.3; P < 0.0001), whereas the association between GBA mutation status and DBS placement did not persist (P = 0.25).
Conclusions
DBS populations are enriched with genetic mutation carriers. The effect of genetic mutation carriers on DBS outcomes warrants further exploration.
Keywords: Parkinson's disease, glucocerebrosidase (GBA), leucine‐rich repeat kinase 2 (LRRK2), parkin (PRKN), deep brain stimulation (DBS)
Each year, approximately 9,000 patients with Parkinson's disease (PD) in the United States electively undergo deep brain stimulator placement with the goal of improving their motor symptoms.1 Up to 29% of patients who have PD and receive deep brain stimulation (DBS) have a mutation in 1 of 3 genes: glucocerebrosidase (GBA), leucine‐rich repeat kinase 2 (LRRK2), and parkin (PRKN).2 The reason for this may be that patients with PD who receive DBS tend to have a younger age at onset (AAO) than the general PD population, which leads to an overrepresentation of genetic forms of PD in this population.2 Specific mutations, like those in GBA, may be associated with more rapid disease progression3; whereas other mutations, like those in LRRK2, may be associated with slower disease progression.4 However, it is unknown whether specific mutations influence the need for DBS, especially in young patients.
The Consortium on Risk for Early Onset Parkinson's Disease (CORE‐PD)5, 6 recruited 1136 participants with early onset PD, defined as an AAO < 51 years, from 17 sites. We previously reported the prevalence of GBA, LRRK2, and PRKN mutations in this cohort.7 The aims of the current study were: (1) to determine the prevalence of GBA, LRRK2, or PRKN mutations in patients with PD who receive DBS compared with those who do not receive DBS in the CORE‐PD cohort; and (2) to determine which variables were predictors of DBS in the 3 largest sites participating in this study.
Patients and Methods
Details of the CORE‐PD study were described in previous reports.5, 8 In brief, patients with PD who were diagnosed by movement disorders specialists were recruited from 17 sites based solely on AAO < 51 years. A blood sample was sent to the National Institute of Neurologic Disorders and Stroke Human Genetics Resource Center DNA and Cell Line Repository (http://ccr.coriell.org) for DNA extraction.
In total, 1136 participants were genotyped for PRKN, GBA, LRRK2, phosphatase and tensin homolog‐induced putative kinase 1 (PINK1), and DJ‐1 (PD protein), as previously described.7, 8, 9, 10 The analyses presented here focus on the most common genetic forms of PD, namely, GBA, LRRK2, and PRKN mutation carriers because 20% of our patients were of Ashkenazi Jewish (AJ) ancestry. The frequency of DBS at individual sites ranged between 0% and 90.9%. To reduce site‐specific biases, data from the 3 largest recruiting sites (Columbia University, Rush University, and the University of Pennsylvania) are presented. These sites collectively enrolled 814 patients.
Demographic data and disease characteristics were collected including AAO of PD, age at evaluation, disease duration, AJ ancestry, ethnicity (white, African‐American, Hispanic, other), DBS status (at the time of entry into the CORE‐PD), age at DBS implantation, duration in years from AAO to DBS, and duration in years from DBS to entry into the CORE‐PD. Clinical evaluation included an assessment of the motor subsection of the Unified Parkinson's Disease Rating Scale (UPDRS‐III) in the defined on‐medication condition,11 the Mini‐Mental Status Examination (MMSE),12 and whether patients reported a history of dyskinesia (at the time of entry into the CORE‐PD). Only those who had MMSE scores > 24 were enrolled. In the majority of patients who received DBS, UPDRS‐III scores reflected postoperative assessments in 82 of 100 (82%).
Mutation carrier status, demographic data, disease characteristics, UPDRS‐III scores, and MMSE scores of patients in the DBS and non‐DBS groups were compared using the Student t test, the χ2 test, or the Mann–Whitney test, as appropriate. Within the DBS group, demographic and disease and characteristics of GBA, LRRK2, and PRKN mutation carriers and noncarriers were compared using analysis of variance (ANOVA), the χ2 test, and the Fisher exact test, as appropriate. A post‐hoc Bonferroni procedure was used to perform pair‐wise comparisons and identify the source of significance in the ANOVA. For replication purposes, nominal P values < 0.05 were considered statistically significant; and, for novel associations, Bonferroni‐corrected P values < (0.05/number of comparisons) were considered statistically significant.
The association between mutation status and DBS placement was assessed in logistic regression models. The association was assessed first in a univariate model and then in a multivariate model, adjusting for site, AJ ancestry, AAO, disease duration (dichotomized, <15 years and >15 years), UPDRS‐III score, and MMSE score. Additional multivariate analyses were performed with the addition of prior history of dyskinesia as a covariate.
Results
There was no significant difference in the frequency of genetic mutation carriers combined in the 3 sites compared with the remaining 14 sites (18.1% vs. 17.5%; P = 0.38). Comparisons of the individual genetic mutations in the DBS and non‐DBS groups are illustrated in Figure 1. GBA mutations were the most common mutations in the DBS and non‐DBS groups (12.1% vs. 8.0%, respectively; P = 0.17). In the DBS group, 8 patients carried the GBA asparagine to serine substitution at position 370 (N370S) variant, and 3 carried the GBA leucine to proline substitution at position 444 (L444P) variant. In the non‐DBS group, 65% of GBA mutations were of the N370s variant (n = 33), and 27% were of the L444P variant (n = 14). When examining mutations in all 3 genes combined, mutation carriers were more prevalent in the DBS group compared with the non‐DBS group (26.5% vs. 16.8%, respectively; P = 0.019) (Table 1). GBA, LRRK2, and PRKN mutations were each slightly more prevalent in the DBS group than in the non‐DBS group, although this difference did not reach statistical significance (Fig. 2).
Figure 1.
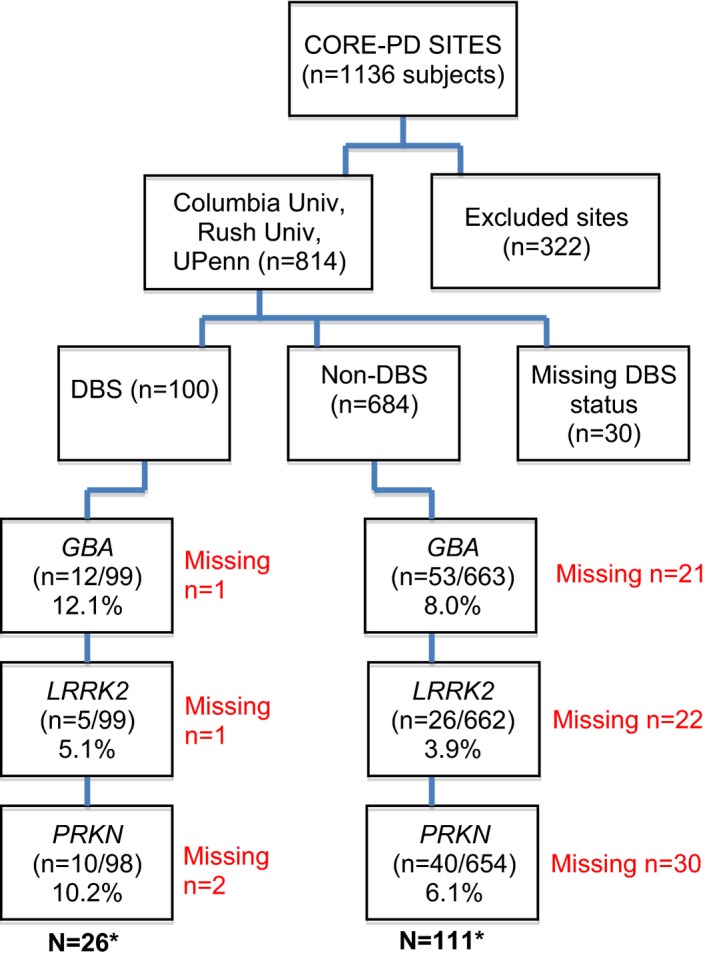
Consortium on Risk for Early Onset Parkinson's Disease (CORE‐PD) enrollment and frequency of mutation carriers in the deep brain stimulation (DBS) and non‐DBS groups. GBA, glucocerebrosidase; LRRK2, leucine‐rich repeat kinase 2 (all individuals with LRRK2 had the glycine‐to‐serine mutation at codon 2019 [G2019S]); PRKN, parkin. Asterisks indicate values that reflect the total number of unique subjects with mutations.
Table 1.
Demographic and Disease Characteristics of Subjects with and Without Deep Brain Stimulation. Values reported are mean ± SD
| Variable | DBS Group (n = 100) | Non‐DBS Group (n = 684) | P |
|---|---|---|---|
| Age of onset | 40.3 ± 7.1 | 41.7 ± 6.9 | 0.06 |
| Age at baseline evaluation | 56.7 ± 7.9 | 51.6 ± 9.1 | <0.0001 |
| Disease duration, y | 16.5 ± 6.8 | 9.9 ± 7.8 | <0.0001 |
| AJ ancestry, % of subjects | 15.0 | 15.8 | 0.81 |
| Ethnicity, %a | |||
| White | 83.0 | 82.0 | 0.51 |
| African‐American | 0.0 | 15.0 | |
| Hispanic | 13.0 | 12.3 | |
| Other | 4.0 | 3.51 | |
| UPDRS‐IIIb | 24.8 ± 13.1 | 20.3 ± 11.7 | 0.002 |
| MMSEc | 28.8 ± 1.9 | 28.9 ± 2.1 | 0.22 |
| Prior history of dyskinesia, % of subjectsd | 78.3 | 37.9 | <0.0001 |
| Prevalence of mutation carriers, %e | 26.5 | 16.8 | 0.019 |
Non‐DBS, n = 683.
DBS, n = 89; non‐DBS, n = 641.
DBS, n = 97; non‐DBS, n = 664.
DBS, n = 83; non‐DBS, n = 475.
DBS, n = 98; non‐DBS, n = 661.
SD, standard deviation; DBS, deep brain stimulation; AJ, Ashkenazi Jewish; MMSE, Mini‐Mental Status Examination; UPDRS‐III, the motor subsection of the Unified Parkinson's Disease Rating Scale.
Figure 2.
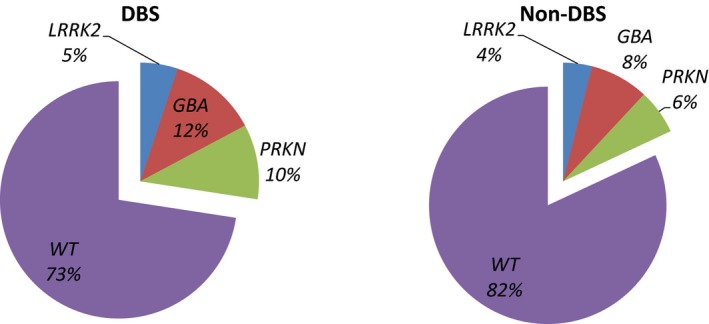
Frequency of mutation carriers in the deep brain stimulation (DBS) and non‐DBS groups. LRRK2, leucine‐rich repeat kinase 2; GBA, glucocerebrosidase; PRKN, parkin; WT, wild‐type (GBA: DBS vs. non‐DBS, P = 0.26; LRRK2: DBS vs. non‐DBS, P = 0.58; PRKN: DBS vs. non‐DBS, P = 0.13).
Demographic and disease characteristics of patients in the DBS and non‐DBS groups, including PD AAO, age at baseline evaluation, disease duration, AJ ancestry, ethnicity, UPDRS‐III score, MMSE score, and prior history of dyskinesia, are presented in Table 1. Compared with the non‐DBS group, patients in the DBS group were significantly older at baseline evaluation (56.7 ± 7.9 years vs. 51.6 ± 9.1 years; P < 0.0001) and had longer disease duration (16.5 ± 6.8 years vs. 9.9 ± 7.8 years; P < 0.001). A greater proportion of those in the DBS group, compared with the non‐DBS group, reported a history of dyskinesia (78% vs. 38%; P < 0.0001) and had a higher UPDRS‐III score (24.8 ± 13.1 vs. 20.3 ± 11.7; P = 0.002).
Demographic and disease characteristics of GBA, LRRK2, PRKN mutation carriers and nonmutation carriers with DBS were compared using ANOVA, χ 2, and Fisher exact tests, as appropriate (Table 2). Those who carried more than 1 mutation were excluded from the analysis (n = 1; a carrier of GBA and LRRK2 mutations). There was a significant difference in the AAO between the groups (P < 0.0001). PRKN mutation carriers had the youngest AAO, followed by GBA mutation carriers, while LRRK2 carriers had the oldest AAO. There was a significant difference in age at DBS implantation between the individual mutation carrier groups (P = 0.005). The largest mean difference in age at DBS implantation was between PRKN and LRRK2 carriers (−13.8 ± 4.4 years; P = 0.014). PRKN carriers had the youngest age at DBS implantation (47.0 ± 11.5 years), and LRRK2 carriers were the oldest group to undergo DBS surgery (60.8 ± 9.0 years).
Table 2.
Demographic and Disease Characteristics of Subjects with Deep Brain Stimulation.a Values reported are mean ± SD
| Variable | GBA (n = 11) | LRRK2 (n = 4) | PRKN (n = 10) | Nonmutation Carriers (n = 72) | P |
|---|---|---|---|---|---|
| Age of onset, y | 41.6 ± 5.3 | 47.5 ± 2.4 | 30.6 ± 9.1 | 41.2 ± 5.3 | <0.0001 |
| Age at baseline evaluation, y | 54.9 ± 2.6 | 64.3 ± 11.0 | 51.0 ± 10.6 | 57.4 ± 7.6 | 0.019 |
| Age at DBS implantation, y | 53.9 ± 2.6 (n = 9) | 60.8 ± 9.0 | 47.0 ± 11.5 | 55.2 ± 7.0 | 0.005 |
| Disease duration, y | 13.3 ± 4.9 | 16.8 ± 10.3 | 20.4 ± 7.3 | 16.1 ± 6.3 | 0.09 |
| AJ ancestry, % of subjects | 36.4 | 25.0 | 0.0 | 16.1 | 0.29 |
| UPDRS‐III score | 27.4 ± 14.5 | 30.8 ± 11.7 | 33.8 ± 20.5 (n = 6) | 23.4 ± 12.1 (n = 65) | 0.37 |
| MMSE score | 28.7 ± 1.3 | 28.3 ± 2.4 | 28.2 ± 1.8 | 28.6 ± 2.0 | 0.90 |
| Age of onset to DBS, ye | 10.9 ± 6.0 (n = 9) | 13.3 ± 8.5 | 16.4 ± 7.7 | 13.7 ± 5.7(n = 62) | 0.29 |
| DBS to current age, y | 1.6 ± 3.0 (n = 9) | 3.5 ± 2.4 | 4.0 ± 4.2 | 2.4 ± 2.6 (n = 62) | 0.28 |
| Prior report of dyskinesia, % of subjects | 100.0 (n = 6) | 66.6 (n = 3) | 80.0 (n = 8) | 75.4 (n = 61) | 0.55 |
One subject who carried both GBA and LRRK2 mutations was excluded.
SD, standard deviation; GBA, glucocerebrosidase; LRRK2, leucine‐rich repeat kinase 2; PKRN, parkin; DBS, deep brain stimulation; AJ, Ashkenazi Jewish; UPDRS‐III, the motor subsection of the Unified Parkinson's Disease Rating Scale; MMSE, Mini‐Mental Status Examination.
In a univariate logistic regression model, having a mutation in any of these 3 genes was associated with DBS placement (odds ratio [OR], 1.8; 95% confidence interval [CI], 1.1–2.9; P = 0.021). This association between mutation carrier status and DBS placement persisted when adjusted for site, AJ ancestry, AAO, disease duration, UPDRS‐III score, and MMSE score (OR, 1.8; 95% CI, 1.0–3.2; P = 0.036). Additional analyses for specific genes showed that GBA mutation status (OR, 2.1; 95% CI, 1.0–4.3; P = 0.05) and disease duration (OR, 1.1; 95% CI, 1.0–1.1; P < 0.001) were the main factors associated with DBS surgery enrollment (Table 3). Thus, GBA mutation carriers are 2‐fold more likely to be identified in the DBS group and are not more likely to be of AJ ancestry. Regarding disease duration, every year increase in PD duration increases the odds of DBS by a factor of 1.1. We repeated the same multivariate logistic regression model with history of dyskinesia as an additional covariate. Our data regarding dyskinesia were not complete, so the number of subjects included in the analyses was reduced from 690 to 479. In this new model, the association between GBA mutation status and DBS placement did not persist (P = 0.25), but disease duration (OR, 1.1; 95% CI, 1.0–1.1; P = 0.001) and history of dyskinesia (OR, 3.8; 95% CI, 1.9–7.3; P < 0.0001) were associated with DBS placement.
Table 3.
Multivariate Logistic Regression Model Predicting Deep Brain Stimulation Placement (N = 690)
| Variable | OR | 95% CI | P |
|---|---|---|---|
| GBA | 2.1 | 1.0‐4.3 | 0.05 |
| LRRK2 | 0.6 | 0.2‐2.0 | 0.50 |
| PRKN | 0.7 | 0.5‐1.2 | 0.29 |
| Site | 1.1 | 0.9‐1.7 | 0.17 |
| AJ ancestry | 0.9 | 0.5‐1.6 | 0.74 |
| Age of onset | 1.0 | 1.0‐1.1 | 0.19 |
| Disease duration | 1.1 | 1.0‐1.1 | <0.001 |
| UPDRS‐III | 1.0 | 0.9‐1.0 | 0.18 |
| MMSE | 1.0 | 0.9‐1.2 | 0.56 |
OR, odds ratio; CI, confidence interval; GBA, glucocerebrosidase; LRRK2, leucine‐rich repeat kinase 2; PKRN, parkin; AJ, Ashkenazi Jewish; UPDRS‐III, the motor subsection of the Unified Parkinson's Disease Rating Scale; MMSE, Mini‐Mental Status Examination.
Discussion
Our results demonstrate that carriers of mutations in specific genes present with characteristic phenotypes and provide insight into disease progression. In the multivariate logistic regression model, GBA mutation carrier status was significantly associated with DBS surgery. The reason for this may be that GBA mutations are the most common genetic risk factor for PD.13 Interestingly, the time from disease onset to DBS was shortest for GBA carriers (10.9 ± 6.0 years), which may suggest that GBA mutation carriers have a more rapid disease progression compared with other mutation carriers and nonmutation carriers,14, 15, 16 ultimately resulting in GBA mutation carriers pursuing DBS earlier. However, the difference in disease duration among GBA mutation carriers did not reach statistical significance compared with other groups, which may be due to the small sample size, so additional studies are needed to elucidate this finding. It is important to note that, when prior history of dyskinesia was included as a covariate, the association between GBA mutation status and DBS placement did not persist. Instead, a history of dyskinesia was strongly associated with DBS placement. This is not surprising considering that dyskinesia is 1 of the key reasons patients with PD obtain DBS. This highlights the need for additional studies to explore predictors of DBS and the correlation between mutation status and DBS outcomes.
Our results provide additional insight into disease progression based on genotype. We infer that all individuals who received DBS were not significantly cognitively impaired before surgery, because the inclusion criteria at the 3 assessment centers required successful cognitive screening to qualify for surgery and because inclusion criteria for the CORE‐PD required an MMSE score of 24. Because PRKN mutation carriers had long disease duration at the time of DBS, these data suggest that such mutation carriers may be resistant to cognitive decline. This observation is consistent with recent reports of long‐term cognitive outcomes in PRKN carriers.8
The strength of our study is that we have examined the association between genotype and DBS placement in the largest cohort to date. In these young‐onset PD subjects, we documented that the frequency of mutations in 3 genes (GBA, LRRK2, and PRKN), considered collectively, was higher in those who received DBS compared with those who did not. This finding is consistent with a prior study that reported a high prevalence of genetic mutation carriers in the DBS population.2 It has been hypothesized that, because PD genetic mutation carriers are relatively young in age, respond to dopaminergic therapy, and have motor fluctuations and/or dyskinesia, they are more likely to pursue DBS.2 Given the high prevalence of PD with mutations in these 3 genes in the DBS population (particularly in those with an AAO < 51 years), large studies are needed to assess whether particular genetic mutations are associated with DBS outcomes. However, our study had some limitations. Because this study was retrospective, we did not have pre‐DBS clinical data (such as UPDRS‐III scores), which precludes our ability to fully compare the impact of DBS in the different mutation carriers and nonmutation carriers. As such, compared with the non‐DBS group, patients in the DBS group had higher UPDRS‐III scores, which may be attributed to more advanced disease, but this remains unclear. Although all GBA carriers were initially screened for common mutations and then carriers of any of 10 mutations were subsequently fully sequenced to look for additional mutations,7 not all patients underwent full sequencing for GBA mutations in our study. Sidransky et al.22 reported that full sequencing of GBA can increase the detection rate by up to 50% in the non‐AJ population, a group that composed roughly 80% of our cohort. In the DBS group, the prevalence GBA mutation in non‐AJ patients was 9.5%. Full sequencing of GBA may increase this prevalence to as much as 18%. Thus, the prevalence of GBA mutation reported here may be an underestimate. Furthermore, our study was limited to the young‐onset PD population with an AAO < 51 years. Studies are needed to assess mutation prevalence in subjects with an AAO ≥ 51 years but < 70 years, because DBS is considered in individuals up to age 75 years.21, 23 Also, only patients with MMSE scores > 24 were included in the study; thus, it is possible that mutation carriers who developed dementia after DBS were missed in our cohort. A prospective study is needed to capture the prevalence of mutation carriers in the DBS candidate population and the impact of DBS in these individuals. Finally, there was a wide range in the frequency of DBS surgeries based on site, which may also be due to the retrospective nature of the study and to site‐specific practices regarding DBS. We tried to limit this site‐specific bias by selecting only the 3 largest recruiting sites from the CORE‐PD cohort.
Because there is increasing emphasis on performing DBS earlier in the course of PD, the enrichment of genetic forms of PD may increase, because patients will be younger, underscoring the urgent need to define the safety and efficacy of DBS in mutation carriers. Subjects in the EARLYSTIM trial had a mean age at DBS of 52 years,24 which is comparable to the age at DBS implantation in the GBA and LRRK2 groups in our cohort. GBA carriers are vulnerable to cognitive decline,13, 17, 19 and variants such as N370S are specifically associated with cognitive impairment.20, 25 The N370S variant was the most common GBA mutation identified in our cohort, regardless of DBS status. Thus, whether these patients have different cognitive outcomes after DBS based on the target (subthalamic nucleus vs. globus pallidus interna) remains to be explored18. There also are implications regarding DBS implantation in LRRK2 and PRKN carriers. Because these patients remain relatively cognitively intact despite long disease duration, they may have a longer window of opportunity regarding DBS and early implantation may not be critical. Our long‐term research goal is to define the best groups for DBS, and these genetic studies offer the potential to define candidates with a high likelihood of success and those at higher risk for long‐term decline.
Author Roles
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
G.D.P.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
D.H.: 1A, 1C, 2B, 2C, 3B
B.O.: 2B, 2C, 3B
J.P.: 3B
R.A.: 1A, 1B, 1C, 3B
M.W.P.: 3B
W.C.N.: 3B
L.C.: 3B
H.M.‐S.: 3B
L.B.: 3B
C.G.G.: 3B
C.C.: 3B
A.C.: 3B
Z.G.‐O.: 3B
G.A.R.: 3B
K.M.: 1A, 1B, 1C, 2A, 2B, 2C, 3A, 3B
Disclosures
Funding Sources and Conflicts of Interest: This study was funded by the National Institutes of Health (NIH R56NS0036630).
Financial disclosures for the previous 12 months: Gian D. Pal is supported by Medtronic, and he has received consultation fees from the Deerfield Institute, Huron Consulting Services, and Sermo. Deborah Hall reports funding from the National Institutes of Health (NIH), the Shapiro Foundation, and Pfizer. Roy Alcalay is supported by the NIH (National Institute of Neurologic Disorders and Stroke [NINDS] K02 NS080915), the Parkinson's Disease Foundation, the Michael J. Fox Foundation, and the Smart Foundation; he has received consultation fees from Sanofi/Genzyme and Prophase. Christopher G. Goetz receives consulting fees or advisory board membership with honoraria from Acadia (Deborah Wood Associates), AstraZeneca, Avanir, Boston Scientific, Clearview, Health Advances, Chelsea Pharmaceuticals (Link Medical Communications), ICON Pricespective LLC, MED‐IQ Educational Services, Neurocrine, Pfizer, Teva, and WPP Group Kantor Health LLC; he reports grant/research funding from the NIH and the Michael J. Fox Foundation; he directs the Rush Parkinson's Disease Research Center, which receives support from the Parkinson's Disease Foundation; he directs the translation program for the Movement Disorder Society (MDS)‐Unified Parkinson's Disease Rating Scale and the Unified Dyskinesia Rating Scale and receives funds from the MDS for this effort; he has received honoraria from the University of California, the University of Luxembourg, the University of Rochester, and the World Parkinson Coalition; he receives royalties from Oxford University Press, Elsevier Publishers, and Wolters Kluwer Health–Lippincott, Wilkins and Wilkins; and he receives salary from Rush University Medical Center; Cynthia Comella received research funding to her university from Merz Pharmaceuticals; Allergan, Inc.; and Ipsen Pharma; she received consulting fees from Merz Pharmaceuticals; Allergan, Inc.; Ipsen Pharma; and Medtronic; Amy Colcher is on the Speaker's Bureau for Lundbeck Pharmaceuticals, Teva Pharmaceuticals, and US World Meds; Ziv Gan‐Or is supported by the Michael J. Fox Foundation. Guy A. Rouleau is supported by the Michael J. Fox Foundation. Karen Marder reports grants from the Michael J. Fox Foundation, the NIH (NS036630), the Parkinson's Disease Foundation, the Huntington's Disease Society of America, CHDI, personal fees from Pfizer (invited lecture), and personal fees from Springer (section editor). The remaining authors report no sources of funding and no conflicts of interest.
Consortium on Risk for Early Onset Parkinson's Disease (CORE‐PD) Investigators
Additional authors who are part of the CORE‐PD Consortium: Madeleine E. Sharp, MD1; Elise Caccappolo, PhD1; Ming‐X. Tang, PhD1,2; Llency Rosado, MD1; Martha Orbe Reilly, MD1; Diana Ruiz, BSc1; Elan D. Louis MD, MSc1,2,3,4; Martha Nance, MD5; Susan Bressman, MD6,7; William K. Scott, PhD8; Caroline Tanner, MD, PhD9,10; Cheryl Waters, MD1; Stanley Fahn, MD1; Lucien Cote, MD1,3; Blair Ford, MD1; Michael Rezak, MD, PhD11; Kevin Novak, PhD12,13; Joseph H. Friedman, MD14,15; Ronald Pfeiffer, MD16; Haydeh Payami, PhD17; Eric Molho, MD18; Stuart A. Factor19; John Nutt, MD20; Carmen Serrano, MD21; and Maritza Arroyo, MD.21
1 Department of Neurology, College of Physicians and Surgeons, Columbia University, New York, New York, USA; 2 Taub Institute for Research on Alzheimer's Disease and the Aging Brain, College of Physicians and Surgeons, Columbia University, New York, New York, USA; 3 Gertrude H. Sergievsky Center, College of Physicians and Surgeons, Columbia University, New York, New York, USA; 4 Department of Epidemiology, Mailman School of Public Health, Columbia University, New York, New York, USA; 5 Struthers PRKNson's Center, Park Nicollet Clinic, Golden Valley, Minnesota, USA; 6 The Alan and Barbara Mirken Department of Neurology, Beth Israel Medical Center, New York, New York, USA; 7 Department of Neurology, Albert Einstein College of Medicine, Bronx, New York, USA; 8 Dr. John T Macdonald Foundation, Department of Human Genetics, Miami Institute for Human Genomics, Miller School of Medicine, University of Miami, Miami, Florida, USA; 9 PRKNson's Institute, Sunnyvale, California, USA; 10 San Francisco Veterans Affairs Medical Center and University of California–San Francisco, San Francisco, California, USA; 11 Central DuPage Hospital, Neurosciences Institute, Movement Disorders Center, Winfield, Illinois, USA; 12 Department of Neurology, at NorthShore University Health System, Evanston, Illinois, USA; 13 Department of Neurology, University of Chicago, Pritzker School of Medicine, Chicago, Illinois, USA; 14 Butler Hospital, Providence, Rhode Island, USA; 15 Department of Neurology, Alpert Medical School of Brown University, Providence, Rhode Island, USA; 16 Department of Neurology, College of Medicine, University of Tennessee Health Science Center, Memphis, Tennessee, USA; 17 New York State Department of Health Wadsworth Center, Albany, New York, USA; 18 PRKNson's Disease and Movement Disorders Center of Albany Medical Center, Albany, New York, USA; 19 Department of Neurology, Emory University, Atlanta, Georgia, USA; 20 Portland VA Medical Center PRKNson Disease Research, Education and Clinical Center; and Oregon Health & Science University, Portland, Oregon, USA; 21 University of Puerto Rico, Puerto Rico
Relevant disclosures and conflicts of interest are listed at the end of this article.
Contributor Information
Gian D. Pal, Email: gian_d_pal@rush.edu.
the Consortium on Risk for Early Onset Parkinson's Disease (CORE‐PD) Investigators:
Madeleine E. Sharp, Elise Caccappolo, Ming‐X. Tang, Llency Rosado, Martha Orbe Reilly, Diana Ruiz, Elan D. Louis, Martha Nance, Susan Bressman, William K. Scott, Caroline Tanner, Cheryl Waters, Stanley Fahn, Lucien Cote, Blair Ford, Michael Rezak, Kevin Novak, Joseph H. Friedman, Ronald Pfeiffer, Haydeh Payami, Eric Molho, Stuart A. Factor, John Nutt, Carmen Serrano, and Maritza Arroyo
References
- 1. Ponce FA, Lozano AM. Deep brain stimulation state of the art and novel stimulation targets. Prog Brain Res 2010;184:311–324. [DOI] [PubMed] [Google Scholar]
- 2. Angeli A, Mencacci NE, Duran R, et al. Genotype and phenotype in Parkinson's disease: lessons in heterogeneity from deep brain stimulation. Mov Disord 2013;28:1370–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Brockmann K, Srulijes K, Pflederer S, et al. GBA‐associated Parkinson's disease: reduced survival and more rapid progression in a prospective longitudinal study. Mov Disord 2015;30:407–411. [DOI] [PubMed] [Google Scholar]
- 4. Alcalay RN, Mejia‐Santana H, Mirelman A, et al. Neuropsychological performance in LRRK2 G2019S carriers with Parkinson's disease. Parkinsonism Relat Disord 2015;21:106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Marder KS, Tang MX, Mejia‐Santana H, et al. Predictors of parkin mutations in early‐onset Parkinson disease: the Consortium on risk for Early Onset Parkinson Disease study. Arch Neurol 2010;67:731–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Caccappolo E, Alcalay RN, Mejia‐Santana H, et al. Neuropsychological profile of parkin mutation carriers with and without Parkinson disease: the CORE‐PD study. J Int Neuropsychol Soc 2011;17:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alcalay RN, Caccappolo E, Mejia‐Santana H, et al. Frequency of known mutations in early‐onset Parkinson disease: implication for genetic counseling: the Consortium on Risk for Early Onset Parkinson Disease study. Arch Neurol 2010;67:1116–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Alcalay RN, Caccappolo E, Mejia‐Santana H, et al. Cognitive and motor function in long‐duration PARKIN‐associated Parkinson disease. JAMA Neurol 2014;71:62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clark LN, Wang Y, Karlins E, et al. Frequency of LRRK2 mutations in early‐ and late‐onset Parkinson disease. Neurology 2006;67:1786–1791. [DOI] [PubMed] [Google Scholar]
- 10. Clark LN, Ross BM, Wang Y, et al. Mutations in the glucocerebrosidase gene are associated with early‐onset Parkinson disease. Neurology 2007;69:1270–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fahn S, Elton R; Members of the UPDRS Development Committee . Unified Parkinson's Disease Rating Scale In: Fahn S, Marsden CD, Calne DB, Goldstein M, eds. Recent Developments in Parkinson's Disease, Vol 2. Florham Park, NJ: Macmillan Health Care Information; 1987:153–163, 293–304. [Google Scholar]
- 12. Folstein MF, Folstein SE, McHugh PR. “Mini‐Mental State”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 13. Alcalay RN, Mirelman A, Saunders‐Pullman R, et al. Parkinson disease phenotype in Ashkenazi Jews with and without LRRK2 G2019S mutations. Mov Disord 2013;28:1966–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Winder‐Rhodes SE, Evans JR, Ban M, et al. Glucocerebrosidase mutations influence the natural history of Parkinson's disease in a community‐based incident cohort. Brain 2013;136(pt 2):392–399. [DOI] [PubMed] [Google Scholar]
- 15. Gan‐Or Z, Bar‐Shira A, Mirelman A, et al. LRRK2 and GBA mutations differentially affect the initial presentation of Parkinson disease. Neurogenet 2010;11:121–125. [DOI] [PubMed] [Google Scholar]
- 16. Lesage S, Anheim M, Condroyer C, et al. Large‐scale screening of the Gaucher's disease‐related glucocerebrosidase gene in Europeans with Parkinson's disease. Hum Mol Genet 2011;20:202–210. [DOI] [PubMed] [Google Scholar]
- 17. Alcalay RN, Caccappolo E, Mejia‐Santana H, et al. Cognitive performance of GBA mutation carriers with early‐onset PD: the CORE‐PD study. Neurology 2012;78:1434–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Weaver FM, Follett KA, Stern M, et al. Randomized trial of deep brain stimulation for Parkinson disease: 36‐month outcomes. Neurology 2012;79:55–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brockmann K, Srulijes K, Hauser AK, et al. GBA‐associated PD presents with nonmotor characteristics. Neurology 2011;77:276–280. [DOI] [PubMed] [Google Scholar]
- 20. Altarescu G, Ioscovich D, Alcalay RN, Zimran A, Elstein D. α‐Synuclein rs356219 polymorphisms in patients with Gaucher disease and Parkinson disease. Neurosci Lett 2014;580:104–107. [DOI] [PubMed] [Google Scholar]
- 21. Rizzone MG, Fasano A, Daniele A, et al. Long‐term outcome of subthalamic nucleus DBS in Parkinson's disease: from the advanced phase towards the late stage of the disease? Parkinsonism Relat Disord 2014;20:376–381. [DOI] [PubMed] [Google Scholar]
- 22. Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009;361:1651–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. DeLong MR, Huang KT, Gallis J, et al. Effect of advancing age on outcomes of deep brain stimulation for Parkinson disease. JAMA Neurol 2014;71:1290–1295. [DOI] [PubMed] [Google Scholar]
- 24. Schuepbach WM, Rau J, Knudsen K, et al. Neurostimulation for Parkinson's disease with early motor complications. N Engl J Med 2013;368:610–622. [DOI] [PubMed] [Google Scholar]
- 25. Malec‐Litwinowicz M, Rudzińska M, Szubiga M, Michalski M, Tomaszewski T, Szczudlik A. Cognitive impairment in carriers of glucocerebrosidase gene mutation in Parkinson disease patients. Neurol Neurochir Pol 2014;48:258–261. [DOI] [PubMed] [Google Scholar]