

Abstract

Cross-linking/mass spectrometry has evolved into a robust technology that reveals structural insights into proteins and protein complexes. We leverage a new tribrid instrument with improved fragmentation capacities in a systematic comparison to identify which fragmentation method would be best for the identification of cross-linked peptides. Specifically, we explored three fragmentation methods and two combinations: collision-induced dissociation (CID), beam-type CID (HCD), electron-transfer dissociation (ETD), ETciD, and EThcD. Trypsin-digested, SDA-cross-linked human serum albumin (HSA) served as a test sample, yielding over all methods and in triplicate analysis in total 2602 matched PSMs and 1390 linked residue pairs at 5% false discovery rate, as confirmed by the crystal structure. HCD wins in number of matched peptide-spectrum-matches (958 PSMs) and identified links (446). CID is most complementary, increasing the number of identified links by 13% (58 links). HCD wins together with EThcD in cross-link site calling precision, with approximately 62% of sites having adjacent backbone cleavages that unambiguously locate the link in both peptides, without assuming any cross-linker preference for amino acids. Overall quality of spectra, as judged by sequence coverage of both peptides, is best for EThcD for the majority of peptides. Sequence coverage might be of particular importance for complex samples, for which we propose a data dependent decision tree, else HCD is the method of choice. The mass spectrometric raw data has been deposited in PRIDE (PXD003737).
Current methods of structural biology have left a systematic and large gap in our knowledge of protein structures.1 Cross-linking/mass spectrometry (CLMS) is an emerging tool that helps to gain structural information for challenging proteins and protein complexes. In CLMS experiments, protein complexes are chemically cross-linked, digested into peptides, and then analyzed via mass spectrometry and bioinformatics.2−5 Identifying a cross-linked peptide pair or the linked residues within, defines their maximal distance in the folded protein. The derived distance constraints can then be used to determine the low-resolution arrangement of protein complexes4,6,7 or even the high-resolution structure of a protein by the help of computational modeling.8
To identify cross-linked peptides, fragmentation spectra have to be matched with peptide sequences by database search. For this purpose, a number of tools have been developed,9,10 for example, pLINK,11 Protein Prospector,12,13 StavroX,14 xQuest,15 Kojak,16 Xi,6,17 or even search engines18 based on linear peptide identification search paradigms such as Mascot.19 One of the challenges in identifying cross-linked peptides is the unequal fragmentation of the two linked peptides,13,17 that is, often one of the two peptides is better fragmented and thus also better characterized by fragment ions. Under collision-induced dissociation (CID) conditions this has been investigated in more detail, revealing that the intensity of observed fragment ions is also affected.17 This is important for the scoring of cross-linked peptides since in general the number of identified fragment ions and their intensity is used for spectra matching. Despite the obvious disadvantage of the unequal fragmentation, scoring mechanisms managed to successfully exploit this fact: To judge the complete cross-linked peptide-spectrum match (PSM), the two individual peptide scores are weighted differently.13,16 However, this should only be an ad hoc solution; ideally the experimental setup can be changed in such a way that the sequence coverage for both peptides is increased. It is plausible that one of the available fragmentation methods performs better than the others, and a comparative analysis into the behavior of cross-linked peptides might reveal options for a refined acquisition strategy.
Throughout the manuscript we use CID for resonant excitation CID in the linear ion trap and HCD as the abbreviation for beam-type CID (HCD is also often referred to as higher-energy collisional dissociation). CID is one of the standard methods of fragmenting peptides in proteomics and has been used in many CLMS studies.6,20−24 The details of CID of cross-linked peptides have recently been systematically assessed,17 but a systematic comparison to other fragmentation methods such as HCD is lacking. HCD has also been used in many CLMS studies.11,13,25,26 Neither a systematic analysis of cross-linked peptides under HCD exists nor under electron-transfer dissociation (ETD). ETD-based fragmentation, that is, ETD with and without supplemental activation of CID (ETciD) or HCD (EThcD)27 has neither routinely been applied to cross-linked peptides nor investigated in much detail. A sequential fragmentation scheme of CID and ETD is reported to increase the identification and confidence levels of cross-linked peptides.28 Another study acquired sequential CID and ETD fragmentation spectra as an optimized method for CID cleavable cross-linkers with signature peaks. Both spectra are then matched with their appropriate ion types and scored together, yielding an improved sequence coverage compared to CID alone.29 Search strategies for noncleavable cross-linkers, however, do not rely on the detection of signature peaks, and thus, the time for the reisolation of the precursor can be saved by simply using ETD with supplemental activation. It was also shown that ETD alone can generate good ion coverage for both peptides using a novel cross-linker,30 albeit the effect on peptides cross-linked with another cross-linker remains to be investigated. In contrast, ETD has been used frequently for complete proteins31 or to characterize post-translational modifications (since it leaves the often labile peptide modifications intact32). Earlier studies stated that ETD fragment peptides with charge states higher than two, more extensively than CID.33 However, the underlying effect seems to correlate with the mass-to-charge ratio (m/z) of the precursors.34 For cross-linked peptides, we expect highly charged precursors6,15,18 and, thus, potentially well-suited targets for ETD.
High-sequence coverage is important to ensure selectivity during database search when trying to identify the two cross-linked peptides from the large choice of alternatives offered by the database. Good backbone fragmentation should also be beneficial to pinpoint the exact location of the cross-link. Despite being the intuitive expectation, sequence coverage and site calling precision do not necessarily have to be linked directly. Properties of the linkage site might direct fragmentation toward neighboring backbone bonds or away from them. Also, for amine-reactive cross-linkers, pinpointing the exact position of the cross-link is assisted by the restricted chemical reactivity toward lysine, serine, threonine, tyrosine, or the protein N-terminus. Hence, depending on the peptide sequence there might only be a single amino acid amenable to the cross-linker reaction. For highly reactive cross-linkers such as succinimidyl 4,4-azipentanoate (SDA) each residue in a peptide needs to be considered when locating the linkage site. Therefore, pinpointing the cross-link sites potentially requires more complete backbone fragmentation than for more specific cross-linkers.
In this study we compared three different fragmentation techniques and two combined fragmentation schemes available on a novel tribrid mass spectrometer (Orbitrap Fusion Lumos, Thermo Fisher Scientific), CID, HCD, ETD, ETciD, and EThcD, on cross-linked peptides obtained by tryptic cleavage of SDA-cross-linked human serum albumin (HSA). The three-dimensional structure of HSA has been resolved by X-ray crystallography35 and is used as ground-truth to evaluate the identification results. The right choice of fragmentation method allows the number of identified linkage sites to be increased; increasing the sequence coverage of both linked peptides boosts the confidence of the matches and also the correct localization of the cross-link site.
Methods
Sample Preparation
Purified HSA (Sigma-Aldrich, St. Louis, MO) was cross-linked using different cross-linker-to-protein, weight-to-weight (w/w) ratios: 0.152:1, 0.203:1, 0.303:1, 0.406:1, 0.606:1, 0.811:1, 1.21:1, and 1.62:1. Aliquots of purified HSA (15 μg, 0.75 mg/mL) in cross-linking buffer (20 mM HEPES–OH, 20 mM NaCl, 5 mM MgCl2, pH 7.8) were mixed with sulfo-SDA (Thermo Scientific Pierce, Rockford, IL) to initiate incomplete reaction of the protein with the sulfo-NHS ester component of the cross-linker. Human blood serum from a healthy donor (20 μg, 1.0 mg/mL) was cross-linked in a similar manner, using cross-linker-to-protein ratios (w/w) of 0.5:1, 1:1, 2:1, and 4:1. Total reaction volume in each case was 30 μL. For the second step of the cross-linking procedure, photoactivation of the diazirine group was carried out using UV irradiation from a UVP CL-1000 UV Cross-linker (UVP Inc.). Samples were irradiated for either 25 or 50 min for purified HSA samples, and either 10, 20, 40, or 60 min in the case of blood serum samples and separated using gel electrophoresis. Bands corresponding to monomeric HSA were excised from gels and the proteins reduced with DTT, alkylated using IAA, and digested using trypsin following standard protocols.18 Peptides were loaded onto self-made C18 StageTips36 and eluted using 80% acetonitrile and 20%, 0.1% TFA in water. The eluates from blood serum HSA and purified HSA digests were mixed 0.33:1 as a master mix to be used throughout this study. The two samples originally used in our structural analysis of HSA8 were mixed here to gain enough material to perform the experiments of this study in triplicates.
Data Acquisition
Peptides were loaded directly (2% B, 500 nL/min) onto a spray emitter analytical column (75 μm inner diameter, 8 μm opening, 250 mm length; New Objectives) packed with C18 material (ReproSil-Pur C18-AQ 3 μm; Dr Maisch GmbH, Ammerbuch-Entringen, Germany) using an air pressure pump (Proxeon Biosystems).37 The 0.1% formic acid served as mobile phase A and 0.1% formic acid/80% acetonitrile as mobile phase B. Peptides were eluted (200 nL/min, linear gradient of 2–40% B over 139 min) directly into an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific, San Jose, CA). Survey spectra were recorded in the Orbitrap at 120000 resolution. Spectra for all fragmentation methods were acquired using a scan range of 300–1700 m/z. Precursor ion isolation was performed with the quadrupole and an m/z window of 1.6 Th. The precursor automatic gain control (AGC) target value was 4 × 105, maximum injection time 50 ms. For CID only, CID collision energy was set to 30%. For HCD only, HCD collision energy was set to 35%. For ETD only, the option to inject ions for all available parallelizable time was selected (anion AGC 5 × 104, 60 ms maximum injection time). Supplemental activation (SA) collision energy was set to 10% for ETciD, and 25% for EThcD.
Data Analysis
Raw files were preprocessed with MaxQuant (v. 1.5.2.8) with “Top MS/MS peaks per 100 Da” set to 100.38 Resulting peak files (APL format) were subjected to Xi (ERI Edinburgh, v. 1.5.584) and searched with the following settings: MS accuracy, 6 ppm; MS/MS accuracy, 20 ppm; enzyme, trypsin; max. missed cleavages, 4; max. number of modifications, 3; fixed modification, none; variable modifications, carbamidomethylation on cysteine; oxidation on methionine; cross-linker, SDA (mass modification: 109.0396 Da). In addition, variable modifications by the hydrolyzed cross-linker (“SDA-hyd”, mass modification: 82.0413 Da) and loop-links (“SDA-loop”, mass modification: 83.0491 Da) were allowed. SDA cross-link reactions were assumed to connect lysine, serine, threonine, tyrosine, or the protein N-terminus on the one end of the spacer with any other amino acid on the other end. FDR was estimated using XiFDR (v. 1.0.6.14)39 on a 5% peptide spectrum match (PSM) level and 5% link-level only including unique PSMs. The reference database consisted of a single entry with the protein sequence of HSA (Uniprot: P02768). For further analysis, PSM information (precursor m/z, annotated fragments, score, peptide sequences, etc.) were extracted from a local PostgreSQL database. The annotated spectra are available in the Supporting Information (Figures S4–S8).
To derive a decision tree for an optimized fragmentation scheme for cross-linked peptides we divided the acquisition range into a grid of m/z bins of size 200 for each charge state from 3 to 7. After sorting all PSMs into this theoretical grid we assigned each cell the best performing and second best performing fragmentation method. The performance was assessed through the median achieved sequence coverage of the complete cross-linked peptide. Note, sequence coverage does not depend on the possible fragment ions but rather on the actual evidence (fragment ions) for specific n-terminal or c-terminal sequences. To decide whether or not a fragmentation method is favorable over another we conducted a simple, one-sided permutation test40 with label swaps and 10.000 iterations. P values lower than 0.05 were regarded as significant. Permutation tests were only performed if more than 15 observations were in the best performing class. If the best and second best were too similar to give significant results the best performing method was also compared to all other methods.
All raw files are available via the PRIDE repository41 (PDX: PXD003737) along with PSM results and the reference FASTA.
Results and Discussion
We investigated the impact of five fragmentation techniques (CID, HCD, ETD, EThcD, ETciD) on the analysis of cross-linked peptides using a latest generation Orbitrap mass spectrometer (Orbitrap Fusion Lumos, Thermo Fisher Scientific). HSA was used as a model protein with a known crystal structure. Cross-linking experiments suffer under CID conditions from the underrepresentation of fragment ions from one of the two peptides.13,17 Here we define the peptide with more intense ions among the ten most intense fragment ions as the α-peptide and the remaining peptide as the β-peptide.17 Note that the nomenclature for the two peptides in a cross-link is not standardized; other definitions using the achieved search score13 or the peptide’s chain length or mass4 are used. We hypothesized that the usage of other fragmentation techniques has an impact on the fragmentation pattern of cross-linked peptides and subsequently on the success rate of identification. In our analysis we applied two different FDR-levels according to the descriptive features that we evaluated.39 For the evaluation of identification results on the crystal structure, a link-level FDR is used. For the evaluation of PSM properties (e.g., sequence coverage), a regular PSM FDR is used. An overview is available in Table S1.
HCD Fragmentation Gives the Highest Number of Identified Cross-Links
We compared the number of identified cross-links that passed a 5% link-level FDR and a 5% PSM-level FDR to assess which fragmentation approach leads to the highest identification success. The results, accumulating the three technical replicates for all fragmentation techniques, show that HCD (958 PSMs) gives the highest number of identifications followed by CID (604 PSMs, Figure 1A). ETciD fragmentation achieves the lowest number of identified cross-links with 296 PSMs. This order is closely related to the number of acquired spectra in all replicates. While HCD is the fastest acquisition technique producing ∼109000 MS2 spectra ETciD and ETD only produce ∼80000 spectra (Table S1). While the number of PSMs is only a proxy for the success of CLMS experiments, the true value of CLMS data comes from the corresponding distance constraints. Therefore, for the comparison of cross-linking data it makes sense to compare the results on the link-level. For the comparison on the link-level only unique links are regarded for further analysis. A unique link is defined by the combination of residues involved in a cross-link, that is, a unique residue pair.
Figure 1.

Number of SDA-induced cross-links identified in HSA using different fragmentation techniques. (A) Identified PSMs and links were computed for 5% FDR-level on the respective category. (B) Evaluation of the identified cross-links against the crystal structure of HSA. The light gray distribution reflects the distance measurement between identified residues in a cross-link mapped to the crystal structure (the median is shown above the vertical line). The dark gray distribution reflects all pairwise combinations of cross-linkable residues in the crystal structure. The black vertical line at 25 Å is used to classify cross-links as long distance or not.
As is the case for PSMs, HCD fragmentation also returns the highest number of identified links (Figure 1A). In total 1390 links (972 unique) were identified with the various methods: Of the unique links HCD observed 446 links (46%), CID 297 links (31%), EThCD 240 links (25%), ETciD 205 links (21%), and ETD 202 (21%). Note, the comparison of the links is not straightforward if the cross-link site is ambiguous. We applied a simple heuristic that assigns the linkage site to the c-terminal residue in ambiguous linkage windows. As HSA’s three-dimensional structure has been resolved, it is possible to utilize it as ground truth and further evaluate the quality of the identified links. We used 25 Å here for SDA as the maximal α-carbon distance of two linkable amino acids in the three-dimensional structure. This provides a clear distinction between true positive and false positive identifications. Each identified link that lies within 25 Å in the crystal structure is plausibly a true positive. Accordingly, every link that is further than 25 Å apart is plausibly a false positive. This is a simplified approach, as links shorter than 25 Å will also contain false positives as a result of random matching, and conversely, longer links may be true and result from protein structural flexibility. Comparing the link information from all five fragmentation techniques shows that the overall quality of the results is comparable across all fragmentation modes and distinctly different from random results. The derived distance distributions have a median of 12–13 Å and are very distinct from the random distance distribution (Figure 1B). In addition, the results are comparable in meeting the approximated 5% FDR. FDR analysis for the HCD data and the ETciD data slightly underestimates the number of false positives by 1% and 2.5%, respectively (Figure S1). These can be partially explained by the definition of the FDR itself, which only gives an approximation of the true false discovery rate. Furthermore, the hard cutoff that was used has a large impact on the computed FDR. For example, the ETciD distances showed a larger peak just to the right of the desired distance cutoff, indicating that a small increase in the maximal allowed distance would give an FDR closer to the desired 5%. The HCD distance distribution looks similar to a small enrichment of false positives just outside the maximal allowed distance. Thus, accounting for more flexibility would change the FDR and suggests that the different methods lead to data of comparable quality but different quantity.
Having a preranking
of the individual fragmentation techniques
in terms of number of PSMs and unique cross-links is desirable to
maximize the information content in a single run. Depending on the
peptide properties, some fragmentation methods might be more suited
for a certain group of peptides, and thus, using two (or more) orthogonal
fragmentation techniques may increase the overall yield in peptide
identifications and thus distance constraints. Disregarding the link
information to focus first on the identified peptide pairs shows that
HCD fragmentation also yields the largest number of unique peptide
pairs (Figure 2A).
A total of 43% (201 peptide pairs) are shared between at least two
fragmentation techniques. The remaining 57% (269 peptide pairs) are
unique to one of the five fragmentation techniques. To maximize the
information content, HCD should be combined with CID fragmentation
to increase the number of unique links by 58 (Figure 2B). Interestingly, ETD fragmentation can
maximally increase the number of unique links by 41 by using EThcD.
We suspect that the difference in the number of acquired spectra and
actually identified PSMs is the main driver for this effect. We define
the identification rate, IR, as  , where Nid is
the number of identified unique PSMs and Nacq is the total number of acquired MS2 spectra (Table S1). The IR reveals that HCD not only acquires most
spectra, but also has the highest success rate of 0.88% compared to
CID (0.61%), EThcD (0.52%), and ETD/ETciD (0.38%). If speed and reliability
of ETD-based fragmentation should change in the future, this order
of complementarity may change. In comparison with linear peptide identifications,
where the IR reaches up to 54%42 (depending
on the instrumentation), the success rate of cross-link identification
is much lower. A contributing factor will be the generally low abundance
of cross-linked peptides when compared to linear peptides, which will
reduce their frequency of selection for MS2, especially in competition
with the linear peptides also present. Other factors will include
poorer database matching due to often lower intensity, but also more
complex spectra and a larger search space.
, where Nid is
the number of identified unique PSMs and Nacq is the total number of acquired MS2 spectra (Table S1). The IR reveals that HCD not only acquires most
spectra, but also has the highest success rate of 0.88% compared to
CID (0.61%), EThcD (0.52%), and ETD/ETciD (0.38%). If speed and reliability
of ETD-based fragmentation should change in the future, this order
of complementarity may change. In comparison with linear peptide identifications,
where the IR reaches up to 54%42 (depending
on the instrumentation), the success rate of cross-link identification
is much lower. A contributing factor will be the generally low abundance
of cross-linked peptides when compared to linear peptides, which will
reduce their frequency of selection for MS2, especially in competition
with the linear peptides also present. Other factors will include
poorer database matching due to often lower intensity, but also more
complex spectra and a larger search space.
Figure 2.
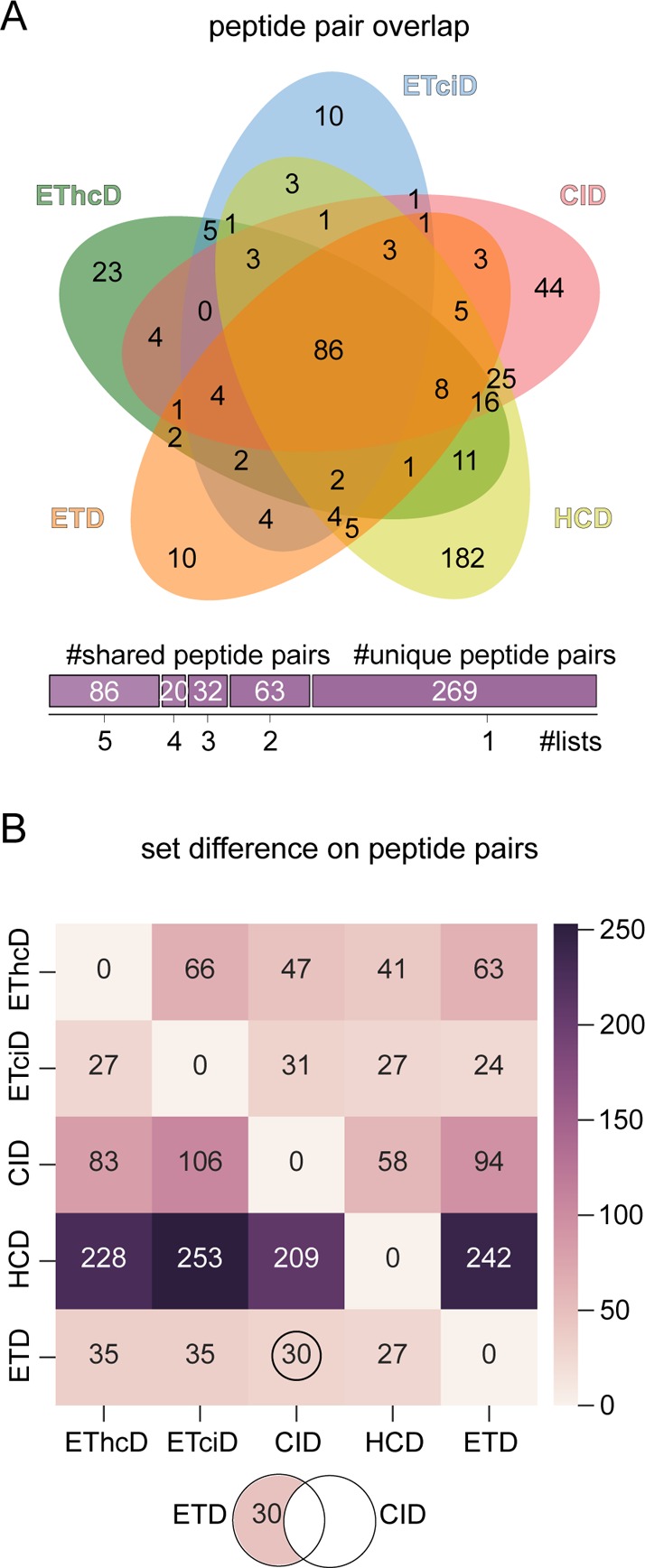
Pairwise result overlaps of fragmentation techniques. (A) Overlap of identified peptide pairs (disregarding link-site positions) between fragmentation techniques (Venn diagram generated with Jvenn49). (B) Set difference matrix shows the number of uniquely identified peptide pairs (disregarding link-site positions) by one fragmentation technique (y-axis) when compared to another one (x-axis).
ETD-Aided Fragmentation Improves the Coverage of the Second Peptide
The identification of cross-linked peptides poses two challenges: First, finding the correct peptide pair, and second, assigning the correct cross-link site. High peptide sequence coverage for both individual peptides should be beneficial to assigning the correct site. Site calling will be especially challenging when considering cross-linkers such as SDA, where the number of cross-link target sites is large.
Under HCD conditions the coverage distribution for the α-peptide is the lowest, with a mean coverage around 50% (Figure 3A). The other four fragmentation techniques perform very similar to only small improvements in the coverage of the α-peptide with CID or EThcD fragmentation. Interestingly, ETD involving fragmentation schemes do not increase the fragmentation efficiency (measured by the peptide coverage) much for the α-peptide. In fact, the highest coverage values for the α-peptide were observed with CID fragmentation. In contrast, the sequence coverage for the beta peptide largely depends on the fragmentation method (Figure 3B). ETD, ETciD, EThcD, and HCD show a much better fragmentation compared to CID. Previously, ETD was reported to improve the sequence coverage compared to CID.32,43 We observe here that for cross-linked peptides this effect is very pronounced for the β-peptide, but not for the α-peptide.
Figure 3.

Achieved sequence coverage comparison. Coverage distribution of the α-peptide (A; more matches among the 10 most intense fragment ions) and the β-peptide (B). The vertical line in (A)–(C) reflects a reference value of 50% sequence coverage, meaning fragments (b, c, y, or z) match to half of the backbone links between residues along the sequence of the peptide. (C) Coverage distribution for the complete cross-linked peptide. (D) Symmetry (absolute coverage difference between alpha and beta peptide) distributions for the different fragmentation techniques. The data in (A)–(D) were analyzed using a 5% PSM FDR.
In general, in cross-linked peptides, one peptide matches more and with higher intense fragment ions than the other. All fragmentation methods yield at least an average coverage of around 50% for the α-peptide. For the β-peptide, the average coverage lies between 39% and 50%. CID would be the method of choice for high α-peptide coverage. However, CID is systematically disadvantaging the β-peptide. For the β-peptide, the other fragmentation methods perform much better: EThcD and HCD almost reach the same fragmentation efficiency as for the α-peptide. In numbers, the largest discrepancy between α- and β-peptide coverage was observed with CID, with a mean coverage difference (MCD) of 19%. EThCD and HCD show the lowest MCD of 8%. The overall best coverage is observed with EThcD fragmentation (Figure 3C). ETciD seems to be less effective, presumably as ETD in the first stage leads to charge reduction, and CID then fragments a single precursor, while HCD fragments all. Nevertheless, ETciD greatly improves the coverage of the second peptide when compared to CID.
To compare the fragmentation efficiency on both peptides in a cross-link more systematically, we define the symmetry factor (SF) as
| 1 |
where covα and covβ refer to the sequence coverage of the α- and β-beta peptide, respectively. For convenience, we use the negation SF′ of SF defined as
| 2 |
A large SF′ means that α- and β-peptide coverage are very similar and vice versa. CID shows the smallest among the five fragmentation methods of ∼0.8. The other four methods perform better than CID, with a median of ∼0.9 (Figure 3D). In addition, ETD, ETciD, EThcD, and HCD have a smaller spread than CID. In summary, CID exasperates the second peptide problem. Nevertheless, CID still slightly outperforms HCD in overall cross-linked peptide sequence coverage. In order to maximize overall cross-linked peptide coverage ETD, ETciD, and EThcD are recommended, based on median coverage of the complete cross-linked peptide.
Precursor m/z Has a Large Effect on the Efficiency of the Fragmentation
To follow-up on the different fragmentation behavior of cross-linked peptides we investigated how the precursor properties influence the fragmentation efficiency. We first divided the m/z acquisition range into bins of m/z 150 (starting from m/z 550). For each bin we then collected the peptide identifications of all different fragmentation methods and investigated the sequence coverage based on the m/z of the precursor.
ETD and EThcD lead to the highest sequence coverage between m/z 500–800 (Figure 4A,B). However, ETD efficiency decreases steeply with higher m/z, making HCD and CID the better choice for precursors larger than m/z 1000. The same trend is observed for all ETD-based methods. These differences are more pronounced on the individual α- and β-peptides. When the complete peptide coverage is compared (Figure 4C), all methods stick more closely together but EThcD and ETD still outperform all other methods for precursors smaller than m/z 850. In higher m/z areas, only CID and HCD are able to still produce enough peptide identifications (data in the figure was limited to only include m/z bins with at least five observations).
Figure 4.
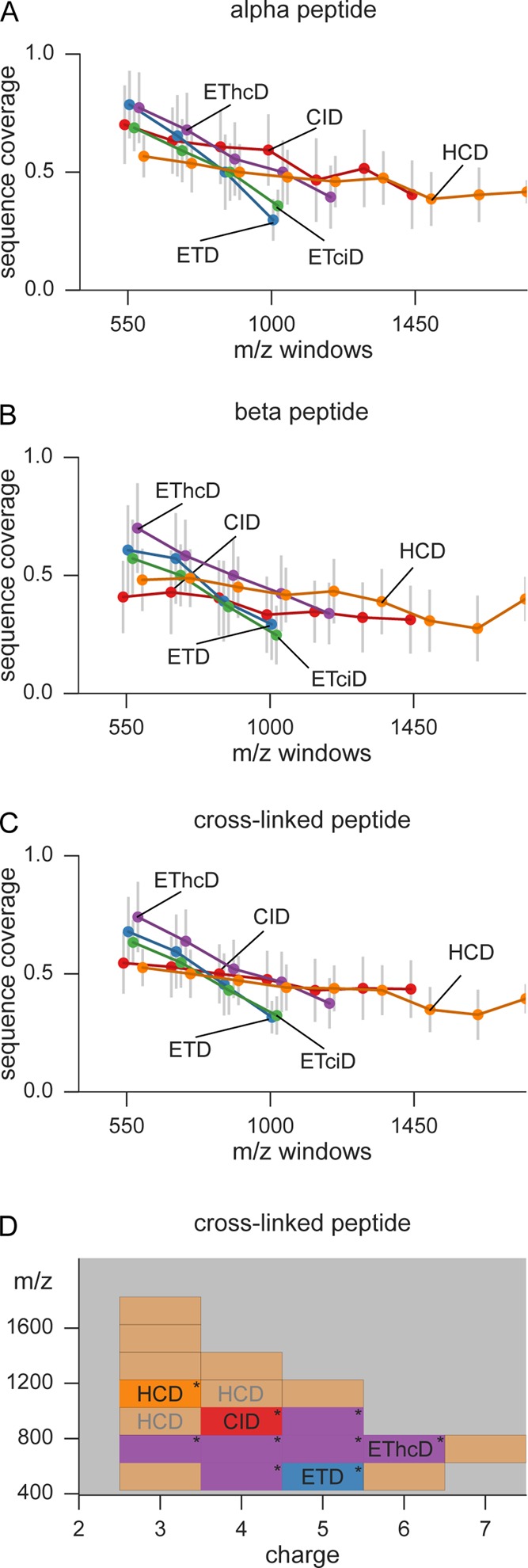
Sequence coverage depending on precursor m/z and charge. The average coverage values from (A) α-peptides, (B) β-peptides, and (C) the complete cross-linked peptide are plotted vs the precursor m/z. Each dot represents the median of all identified peptides in a window of m/z 150. Error bars show the standard deviation. (D) Decision surface to optimize the sequence coverage of cross-linked peptide. The acquisition range was divided into bins of 200 m/z per charge state. In each bin the best performing fragmentation method (judged by median achieved sequence coverage) is used to color that particular bin. The “*” denotes a significant improvement in sequence coverage by using the best performing fragmentation method over the second best. Areas with less than 15 observations are colored in light red, falling back to HCD as standard fragmentation technique. Gray annotations show areas where no significant improvement could be obtained by choosing one method over the others.
As demonstrated in the sections above, there are differences in the efficiency of the fragmentation of cross-linked peptides. In a more detailed comparison, we divided the acquisition range into a grid made of charge bins of size one and m/z bins of size 200. In each of these cells we then tested how well the five different fragmentation methods performed. The performance was evaluated on the cross-linked peptide sequence coverage. For the majority of peptides, EThcD achieved significantly higher sequence coverage (Figure 4D) than the second best method between 600 and 800 m/z (precursor charge 3–6). In addition, the m/z cells 400–600 (z = 4) and 800–1000 (z = 5) are also favored by EThcD fragmentation. Since the majority of cross-linked peptides (71%) lie within 600–1000 m/z, the most important area is dominated by EThcD fragmentation. However, evaluated by pure numbers of identifications, EThcD is not the best performing method. On average, ∼35 PSMs are missed if EThcD is chosen over the method that achieves the highest number of identifications. If the evaluation metric is changed to the highest number of identifications, HCD is outperforming the other fragmentation methods for all m/z bins (Figure S3). Therefore, HCD was selected as default method for regions where no significant improvement could be observed by any of the other methods (Figure 4D, HCD written in gray).
HCD, EThcD, and ETD fragmentation define the cross-link site most unambiguously
The overall sequence coverage is a valuable feature to assess the quality of peptide identifications. However, for cross-linked peptides those fragments flanking the cross-linked residues are important to define the linkage site. This resembles the localization of post-translational modifications such as phosphorylation, which greatly benefited from the usage of combined fragmentation methods.44 Limited information about the cross-link site is available when none of the fragments next to a cross-linked residue are observed; the cross-link site can then only be assigned by prior assumptions or to larger sequence windows, which becomes problematic if the site call is off by ±5 residues (at least in HSA and using current ab initio structure computation).8 Given the information from correct fragment identifications, a combination of one c-terminal and one n-terminal ion is enough to locate the cross-link site unambiguously. Utilizing the high-resolution/accurate mass measurement in our experimental design, we thus assumed that each assigned fragment is correct for peptides passing the specified FDR.
The cross-link site in α-peptides could be assigned to a single residue in ∼65% of all PSMs identified with EThcD or HCD (Figure 5A). The second best performing method was ETD, with approximately 60% of PSMs where the cross-link could be assigned to a single residue. CID and ETciD PSMs show the lowest number of accurate site localizations to a single residue (below 50% of all PSMs). All methods placed the cross-link site on average within the critical 5 residue window for 97.2% ± 1.17 (α-peptides) and 95.6% ± 1.3 (β-peptides) of all PSMs. For the β-peptide, this looks very similar; EThcD and HCD show the best fragmentation behavior to localize the cross-link site (Figure 5B). With approximately 50% of precisely localized cross-links in the β-peptide, the link-localization is less well for the β-peptide than for the α-peptide. However, this is not as pronounced as would be expected from the sequence coverage asymmetry. This is counterintuitive since the coverage distributions for HCD is among the lowest of all five fragmentation techniques for the β-peptide. For EThcD, the results for the determination of the cross-link site are more in line with the observed coverage distributions. Still, the large difference in the coverage distribution of the α- and β-peptides seems not to be as pronounced for the distribution of correct localizations of the cross-link site. One of the possible reasons is that the cleavage of the peptides before and after the cross-link site is preferred. For CID a statistical trend was reported that cross-linked fragments outnumber linear fragments and tend to have a higher intensity.17 We encounter the opposite for HCD, linear fragments visibly outnumber cross-linked fragments (Figure S8).
Figure 5.

Cross-link site localization precision. (A) Cumulative precision curve for the α-peptide. (B) Cumulative precision curve for the β-peptide. With a precision value of one the cross-link site is unambiguously located by adjacent backbone fragments (b, c, y, or z) in the peptide. A value of two limits the cross-link site to two eligible residues.
Data-Dependent Decision Tree for Optimized Acquisition of Cross-Linked Peptides
CLMS studies vary in the degree of complexity: single proteins, multiple protein complexes or complete proteomes can be analyzed to generate protein–protein interaction information or the three-dimensional structure. Depending on the specific case we propose two different acquisition strategies (Figure 6A): First, for single proteins or small protein complexes, we recommend HCD as the method of choice. Since the complexity of the sample is not very high, cross-linked peptides can often be matched by precursor mass alone. In addition, HCD fragmentation generates enough fragments to precisely localize the cross-link site in the majority of cases. For the second case, that is, complex samples with many proteins not only the search space becomes an issue but also the associated random matches. A fragmentation scheme that generates highly discriminative scores for target and decoy peptides will identify more peptides under the same FDR threshold. The optimal fragmentation scheme for such an experiment is shown in Figure 6B. Earlier studies on the development of data dependent decision trees (DDDT) for the acquisition of linear peptides mainly support our conclusions: HCD gives the highest number of identifications, but ETD gives higher search engine scores45 or, as in our case, higher sequence coverage. Compared to a DDDT for linear peptides our results are slightly different but still comparable. For example, linear DDDTs precursors with charge state 3+ have been analyzed with ETD up to 750 m/z46 or 650 m/z,45 we only use ETD from 600–800 m/z. In addition, instead of using ETD alone for 4+, 5+ precursors below 1000 m/z and 800 m/z, respectively, EThcD is used. In this study we investigated SDA-cross-linked, tryptic peptides. Other cross-linkers or enzymes may lead to peptide populations with distinct fragmentation behavior due to differences in size or amino acid composition. Note, however, that the proposed fragmentation scheme is similar to the decision tree for linear peptides45,46 and may therefore be of more general value.
Figure 6.

Acquisition strategy for cross-linked peptides. (A) Recommended acquisition scheme for cross-linking samples. (B) Data-dependent decision tree (DDDT) for cross-linked peptides. Depending on the precursor charge state (3+, 4+, 5+, 6+, and other) and the m/z, the appropriate fragmentation technique is selected.
Conclusion
For the majority of the peptides EThcD is the method of choice to achieve the highest sequence coverage. HCD is an important alternative because of its superior speed, with only somewhat reduced peptide sequence coverage. CID, ETD, and ETciD only play minor roles. We advise to adjust the acquisition scheme to follow the experimental setup: simple protein samples should be analyzed using only HCD to maximize number of observed links, which starts having value in protein structure determination.8,47 For complex samples, we propose a decision tree that is mainly based on EThcD and HCD to maximize search specificity.
Acknowledgments
The Wellcome Trust generously funded this work through a Senior Research Fellowship to J.R. (103139), a Centre Core Grant (092076), and an Instrument Grant (108504).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.6b02082.
Quality control results, details regarding the decision tree areas, the relative performance of the individual fragmentation techniques, and annotated spectra of all PSMs (PDF).
The authors declare no competing financial interest.
Supplementary Material
References
- Perdigão N.; Heinrich J.; Stolte C.; Sabir K. S.; Buckley M. J.; Tabor B.; Signal B.; Gloss B. S.; Hammang C. J.; Rost B.; et al. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 15898–15903. 10.1073/pnas.1508380112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J. J. Struct. Biol. 2011, 173, 530–540. 10.1016/j.jsb.2010.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holding A. N. Methods 2015, 89, 54–63. 10.1016/j.ymeth.2015.06.010. [DOI] [PubMed] [Google Scholar]
- Sinz A. Mass Spectrom. Rev. 2006, 25, 663–682. 10.1002/mas.20082. [DOI] [PubMed] [Google Scholar]
- Schmidt C.; Robinson C. V. Nat. Protoc. 2014, 9, 2224–2236. 10.1038/nprot.2014.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. A.; Jawhari A.; Fischer L.; Buchen C.; Tahir S.; Kamenski T.; Rasmussen M.; Lariviere L.; Bukowski-Wills J.-C.; Nilges M.; et al. EMBO J. 2010, 29, 717–726. 10.1038/emboj.2009.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walzthoeni T.; Leitner A.; Stengel F.; Aebersold R. Curr. Opin. Struct. Biol. 2013, 23, 252–260. 10.1016/j.sbi.2013.02.008. [DOI] [PubMed] [Google Scholar]
- Belsom A.; Schneider M.; Fischer L.; Brock O.; Rappsilber J. Mol. Cell. Proteomics 2016, 15, 1105–1116. 10.1074/mcp.M115.048504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayne S. L. N.; Patterton H.-G. Briefings Bioinf. 2011, 12, 660–671. 10.1093/bib/bbq087. [DOI] [PubMed] [Google Scholar]
- Sinz A.; Arlt C.; Chorev D.; Sharon M. Protein Sci. 2015, 24, 1193–1209. 10.1002/pro.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang B.; Wu Y.-J.; Zhu M.; Fan S.-B.; Lin J.; Zhang K.; Li S.; Chi H.; Li Y.-X.; Chen H.-F.; et al. Nat. Methods 2012, 9, 904–906. 10.1038/nmeth.2099. [DOI] [PubMed] [Google Scholar]
- Chalkley R. J.; Baker P. R.; Medzihradszky K. F.; Lynn A. J.; Burlingame A. L. Mol. Cell. Proteomics 2008, 7, 2386–2398. 10.1074/mcp.M800021-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trnka M. J.; Baker P. R.; Robinson P. J. J.; Burlingame a L.; Chalkley R. J. Mol. Cell. Proteomics 2014, 13, 420–434. 10.1074/mcp.M113.034009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götze M.; Pettelkau J.; Schaks S.; Bosse K.; Ihling C. H.; Krauth F.; Fritzsche R.; Kühn U.; Sinz A. J. Am. Soc. Mass Spectrom. 2012, 23, 76–87. 10.1007/s13361-011-0261-2. [DOI] [PubMed] [Google Scholar]
- Rinner O.; Seebacher J.; Walzthoeni T.; Mueller L. N.; Beck M.; Schmidt A.; Mueller M.; Aebersold R. Nat. Methods 2008, 5, 315–318. 10.1038/nmeth.1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoopmann M. R.; Zelter A.; Johnson R. S.; Riffle M.; MacCoss M. J.; Davis T. N.; Moritz R. L. J. Proteome Res. 2015, 14, 2190–2198. 10.1021/pr501321h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giese S. H.; Fischer L.; Rappsilber J. Mol. Cell. Proteomics 2016, 15, 1094–1104. 10.1074/mcp.M115.049296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiolica A.; Cittaro D.; Borsotti D.; Sennels L.; Ciferri C.; Tarricone C.; Musacchio A.; Rappsilber J. Mol. Cell. Proteomics 2007, 6, 2200–2211. 10.1074/mcp.M700274-MCP200. [DOI] [PubMed] [Google Scholar]
- Perkins D. N.; Pappin D. J.; Creasy D. M.; Cottrell J. S. Electrophoresis 1999, 20, 3551–3567. 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Fischer L.; Chen Z. A.; Rappsilber J. J. Proteomics 2013, 88, 120–128. 10.1016/j.jprot.2013.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson K. M.; Pannell L. K.; Fales H. M. Rapid Commun. Mass Spectrom. 2002, 16, 149–159. 10.1002/rcm.554. [DOI] [PubMed] [Google Scholar]
- Chu F.; Maynard J. C.; Chiosis G.; Nicchitta C. V.; Burlingame A. L. Protein Sci. 2006, 15, 1260–1269. 10.1110/ps.052065106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M. Q.; Dreiocker F.; Ihling C. H.; Schäfer M.; Sinz A. Anal. Chem. 2010, 82, 6958–6968. 10.1021/ac101241t. [DOI] [PubMed] [Google Scholar]
- Leitner A.; Reischl R.; Walzthoeni T.; Herzog F.; Bohn S.; Förster F.; Aebersold R. Mol. Cell. Proteomics 2012, 11, M111.014126. 10.1074/mcp.M111.014126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y.; Fernandez-Martinez J.; Tjioe E.; Pellarin R.; Kim S. J.; Williams R.; Schneidman-Duhovny D.; Sali A.; Rout M. P.; Chait B. T. Mol. Cell. Proteomics 2014, 13, 2927–2943. 10.1074/mcp.M114.041673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen-Huynh N.-T.; Sharov G.; Potel C.; Fichter P.; Trowitzsch S.; Berger I.; Lamour V.; Schultz P.; Potier N.; Leize-Wagner E. Protein Sci. 2015, 24, 1232–1246. 10.1002/pro.2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Altelaar A. F. M.; van den Toorn H.; Nolting D.; Griep-Raming J.; Heck A. J. R.; Mohammed S. Anal. Chem. 2012, 84, 9668–9673. 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- Chowdhury S. M.; Du X.; Tolić N.; Wu S.; Moore R. J.; Mayer M. U.; Smith R. D.; Adkins J. N. Anal. Chem. 2009, 81, 5524–5532. 10.1021/ac900853k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F.; Rijkers D. T. S.; Post H.; Heck A. J. R. Nat. Methods 2015, 12, 1179–1184. 10.1038/nmeth.3603. [DOI] [PubMed] [Google Scholar]
- Trnka M. J.; Burlingame A. L. Mol. Cell. Proteomics 2010, 9, 2306–2317. 10.1074/mcp.M110.003764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syka J. E. P.; Coon J. J.; Schroeder M. J.; Shabanowitz J.; Hunt D. F. Proc. Natl. Acad. Sci. U. S. A. 2004, 101, 9528–9533. 10.1073/pnas.0402700101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikesh L. M.; Ueberheide B.; Chi A.; Coon J. J.; Syka J. E. P.; Shabanowitz J.; Hunt D. F. Biochim. Biophys. Acta, Proteins Proteomics 2006, 1764, 1811–1822. 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.-S.; Pandey A. Proteomics 2012, 12, 530–542. 10.1002/pmic.201100517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Good D. M.; Wirtala M.; McAlister G. C.; Coon J. J. Mol. Cell. Proteomics 2007, 6, 1942–1951. 10.1074/mcp.M700073-MCP200. [DOI] [PubMed] [Google Scholar]
- Sugio S.; Kashima A.; Mochizuki S.; Noda M.; Kobayashi K. Protein Eng., Des. Sel. 1999, 12, 439–446. 10.1093/protein/12.6.439. [DOI] [PubMed] [Google Scholar]
- Rappsilber J.; Ishihama Y.; Mann M. Anal. Chem. 2003, 75, 663–670. 10.1021/ac026117i. [DOI] [PubMed] [Google Scholar]
- Ishihama Y.; Rappsilber J.; Andersen J. S.; Mann M. J. Chromatogr. A 2002, 979, 233–239. 10.1016/S0021-9673(02)01402-4. [DOI] [PubMed] [Google Scholar]
- Renard B. Y.; Kirchner M.; Monigatti F.; Ivanov A. R.; Rappsilber J.; Winter D.; Steen J. A. J.; Hamprecht F. A.; Steen H. Proteomics 2009, 9, 4978–4984. 10.1002/pmic.200900326. [DOI] [PubMed] [Google Scholar]
- Fischer L.; Rappsilber J.. 2016, in preparation.
- EDGINGTON E. S. J. Psychol. 1964, 57, 445–449. 10.1080/00223980.1964.9916711. [DOI] [PubMed] [Google Scholar]
- Vizcaíno J. A.; Côté R. G.; Csordas A.; Dianes J. A.; Fabregat A.; Foster J. M.; Griss J.; Alpi E.; Birim M.; Contell J.; et al. Nucleic Acids Res. 2013, 41, D1063–D1069. 10.1093/nar/gks1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert A. S.; Richards A. L.; Bailey D. J.; Ulbrich A.; Coughlin E. E.; Westphall M. S.; Coon J. J. Mol. Cell. Proteomics 2014, 13, 339–347. 10.1074/mcp.M113.034769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molina H.; Matthiesen R.; Kandasamy K.; Pandey A. Anal. Chem. 2008, 80, 4825–4835. 10.1021/ac8007785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Zhou H.; Taus T.; Altelaar A. F. M.; Mechtler K.; Heck A. J. R.; Mohammed S. J. Proteome Res. 2013, 12, 1520–1525. 10.1021/pr301130k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frese C. K.; Altelaar A. F. M.; Hennrich M. L.; Nolting D.; Zeller M.; Griep-Raming J.; Heck A. J. R.; Mohammed S. J. Proteome Res. 2011, 10, 2377–2388. 10.1021/pr1011729. [DOI] [PubMed] [Google Scholar]
- Swaney D. L.; McAlister G. C.; Coon J. J. Nat. Methods 2008, 5, 959–964. 10.1038/nmeth.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsom A.; Schneider M.; Brock O.; Rappsilber J. Trends Biochem. Sci. 2016, 41, 564–567. 10.1016/j.tibs.2016.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.